 Wanjun Ma1,2
Wanjun Ma1,2 Bikui Zhang
Bikui Zhang Wenqun Li
Wenqun Li- 1Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Institute of Clinical Pharmacy, Central South University, Changsha, China
Homeostatic regulation of cardiomyocytes plays a crucial role in maintaining the normal physiological activity of cardiac tissue. Severe cardiotoxicity results in cardiac diseases including but not limited to arrhythmia, myocardial infarction and myocardial hypertrophy. Drug-induced cardiotoxicity limits or forbids further use of the implicated drugs. Such drugs that are currently available in the clinic include anti-tumor drugs (doxorubicin, cisplatin, trastuzumab, etc.), antidiabetic drugs (rosiglitazone and pioglitazone), and an antiviral drug (zidovudine). This review focused on cardiomyocyte death forms and related mechanisms underlying clinical drug-induced cardiotoxicity, including apoptosis, autophagy, necrosis, necroptosis, pryoptosis, and ferroptosis. The key proteins involved in cardiomyocyte death signaling were discussed and evaluated, aiming to provide a theoretical basis and target for the prevention and treatment of drug-induced cardiotoxicity in the clinical practice.
Introduction
Cardiotoxicity commonly refers to toxicity that has a detrimental impact on the heart, which might finally lead to myocardiopathy such as arrhythmia, myocardial infarction and myocardial hypertrophy. These inevitable side effects, especially of anticancer drugs, are usually the main causes of treatment termination and drug development failure. In addition, modern cancer treatments recommend a combination of multiple agents, almost always leading to synergistic side-effects (Ewer and Ewer, 2015). In the past few decades, more than 10% of clinical drugs were forced out of the market due to cardiovascular side effects, which still hindered the drug development and seriously affected the improvement of patient health. Many studies have revealed that drug-induced myocardial damage may be a stepwise process accompanied by the increase of cardiac biomarkers and structural myocardial deformation, finally resulting in left ventricular ejection fraction (LVEF) decrease (Pistillucci et al., 2015; Patel and Cornell, 2019). Currently, the widely accepted definition of cardiotoxicity is the decline in LVEF of at least 10% to less than 55% (Nicol et al., 2019). Clinical data show that cardiomyocyte death or damage concomitantly takes place with the progression of cardiotoxicity, indicating that drug-induced cardiomyocyte death may be the main cause of cardiotoxicity.
Overview of Cell Death Forms
It is widely accepted that cell death, proliferation and differentiation are essential throughout the pathological and physiological processes. Although more than ten types of cell death have been discovered to date, the most common forms of cell death in drug-induced cardiotoxicity are apoptosis, autophagy and necrosis (Galluzzi et al., 2018). In addition, recently discovered cell death forms, such as necroptosis, pyroptosis and ferroptosis, are also involved in drug-induced cardiotoxicity.
Apoptosis
Morphologically, apoptosis is the most widely studied cell death form, exhibiting signs of cell shrinkage, increased cytoplasmic density, decreased mitochondrial membrane potential (MMP) disappearance and changes in permeability. Eventually, intact apoptotic bodies are formed to be efficiently absorbed and degraded by adjacent cells. Based on the underlying mechanisms, apoptosis is divided into intrinsic and extrinsic apoptosis. Intrinsic apoptosis is caused by microenvironment disorders such as DNA damage, excessive oxidative stress, mitotic disaster, loss of growth factor signaling and endoplasmic reticulum (ER) stress (Brumatti et al., 2010; Wu and Bratton, 2013; Czabotar et al., 2014; Roos et al., 2016). B cell lymphoma-2 (Bcl-2) family pro-apoptotic members, such as Bax, Bak, and BH3-only protein, primarily regulate the intrinsic cell apoptosis via their influence on mitochondria. Bcl-2 stimulates mitochondria translocation of Bax/Bak and causes mitochondrial membrane permeabilization, which eventually leads to the release of cytochrome C in the cytoplasm, where the apoptosome forms and caspases cascade reactions arise (Hutt, 2015). Unlike intrinsic apoptosis, the extrinsic apoptosis is mainly initiated by two kinds of plasma membrane receptors including Fas cell surface death receptor (Fas) and tumor necrosis factor receptor (TNF) super family member (TNFR1, TNFRSF10A, and TNFRSF10B) along with their respective homologous ligands. Death-inducing signaling complex (DISC), composed of death ligands and corresponding receptors on the cell membrane, is a receptor proximal protein complex that helps connect the death receptor signaling with caspases cascade reactions. The pro-apoptotic proteins caspase-8 and caspase-10 are recruited and cleaved by upstream caspase enzyme to initiate apoptosis (Barnhart et al., 2003; Yang, 2015).
Autophagy
As a pro-survival mechanism, autophagy occurs in destroying and recovering unwanted or damaged cellular components, playing an essential role in maintaining intracellular metabolic homeostasis (Yang et al., 2017). A series of evidences suggest that the autophagy-activating kinase 1 (ULK-1) initiates autophagy by phosphorylating Beclin1 and activating the vacuolar protein sorting 34 (VPS34) complex (Klionsky et al., 2016). As a critical signaling protein of autophagy, the mammalian target of rapamycin (mTOR) can be activated by nutritional deficiency, growth factor and receptor tyrosine kinase, which subsequently forms mammalian target of rapamycin complex 1 (mTORC1) with several other proteins. The mTORC1 further binds to the ULK-1 complex and blocks ULK-1-mediated Beclin 1 phosphorylation, thus inhibiting autophagy initiation (Jung et al., 2010). In contrast, the adenosine 5-monophosphate activated protein kinase (AMPK) mediates autophagy by decreasing mTOR-related autophagy suppression and phosphorylating the ULK-1 complex at Ser317 and Ser777 (Kim et al., 2011).
Necrosis
In the past few decades, necrosis is typically described as a form of passive and irreversible cell death that is always associated with pathology, usually accompanied by morphological characteristics such as increased membrane permeability, disintegration of organelles and cell swelling. As a traditional cell death form, necrosis occurs usually after the exposure to extreme physical or chemical insults, and therefore is regarded as an accident and unregulated cell death form. With in-depth study, more regulated cell death forms are proposed, including necroptosis, pryoptosis and ferroptosis, which share similar morphological characteristics with necrosis.
Other Regulated Cell Death Forms: Necroptosis, Pryoptosis, and Ferroptosis
In contrast to necrosis, necroptosis is regulated by specific transduction mechanism. Death receptor TNFR1 plays a key role in the development of necroptosis (Kaiser et al., 2013). Activation of TNFR1 can stimulate RIPK1 to further recruit RIPK3 that leads to necrosomes formation (Grootjans et al., 2017). In addition, sequential activation of RIPK3/MLKL is also crucial in necroptosis signaling (Song and Wang, 2013). Phosphorylated MLKL can destroy the plasma membrane and organelles to release inflammatory factors, and elicit an immune response, indicating the occurrence of necroptosis (Galluzzi et al., 2018). It is worth mentioning that caspase-8 plays a suppressing role in necroptosis since it inactivates RIPK1 and RIPK3 (Belmonte et al., 2015; Tummers and Green, 2017).
Pyroptosis, firstly proposed by Cooksonis, is widely recognized as inflammatory and regulated cell death form that usually occurs in defense of exogenous pathogens such as virus, bacteria and fungi (Cookson and Brennan, 2001; Jorgensen and Miao, 2015). Activation of caspases including caspase-1, caspase-3, caspase-4 and caspase-11 is necessary for initiating the pyroptosis, which further specifically cleaves GSDMD or GSDME to generate holes in the membrane and release interleukin-1beta (IL-1β) and IL-18, inducing pyroptosis (Shi et al., 2015; Man et al., 2017). Recent studies have identified a potential relationship between pyroptosis and myocardial injury (Chen et al., 2018).
Unlike pyroptosis, ferroptosis is firstly discovered in carcinoma cells and characterized by the accumulation of iron and lipid reactive oxygen species (ROS), which could deplete anti-oxidases and cause mitochondrial damages, leading to cell death (Sumneang et al., 2020). Moreover, Chen et al. (2019) found that the inhibition of Toll like receptor 4 (TLR4) and triphosphopyridine nucleotide oxidase 4 (NOX4) significantly alleviated ferroptosis. Glutathione peroxidase 4 (GPx4) can prevent erastin and RSL3-induced ferroptosis via suppressing lipid peroxidation (Imai et al., 2017).
Cardiomyocyte Death in Drug-Induced Cardiotoxicity
Multiple evidences have suggested that there is a strong correlation between drug-induced cardiomyocyte death and cardiotoxicity. Here, we will summarize and discuss cardiomyocyte death induced by the drugs listed in Table 1 and their underlying cell death mechanisms shown in Figure 1.
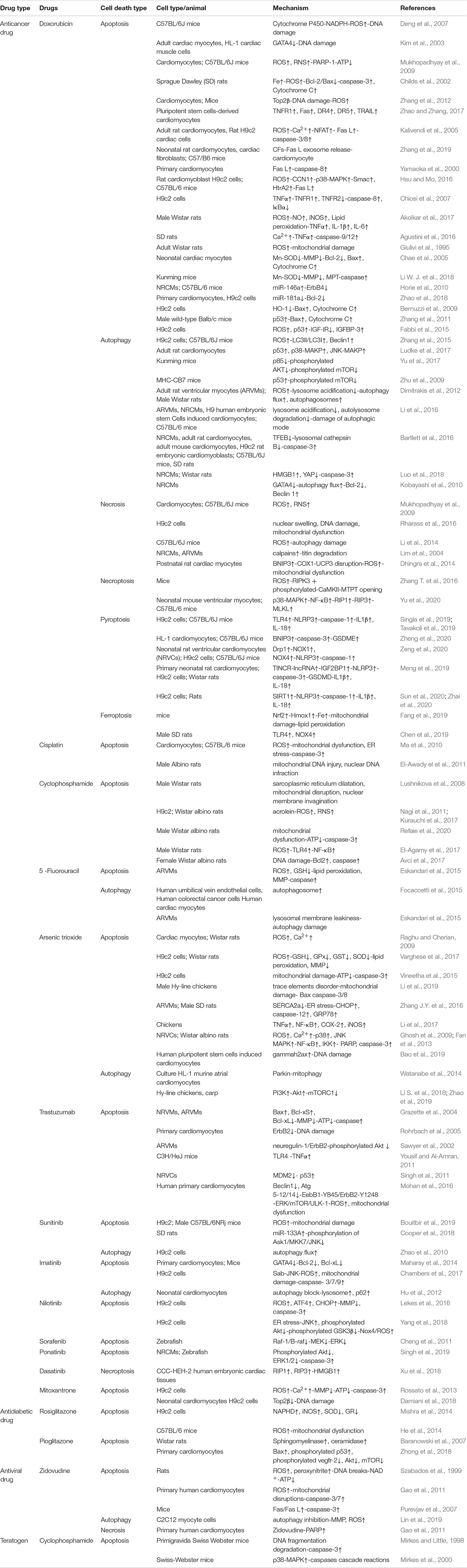
Table 1. Molecular mechanisms of cardiomyocyte death in drug-induced cardiotoxicity.
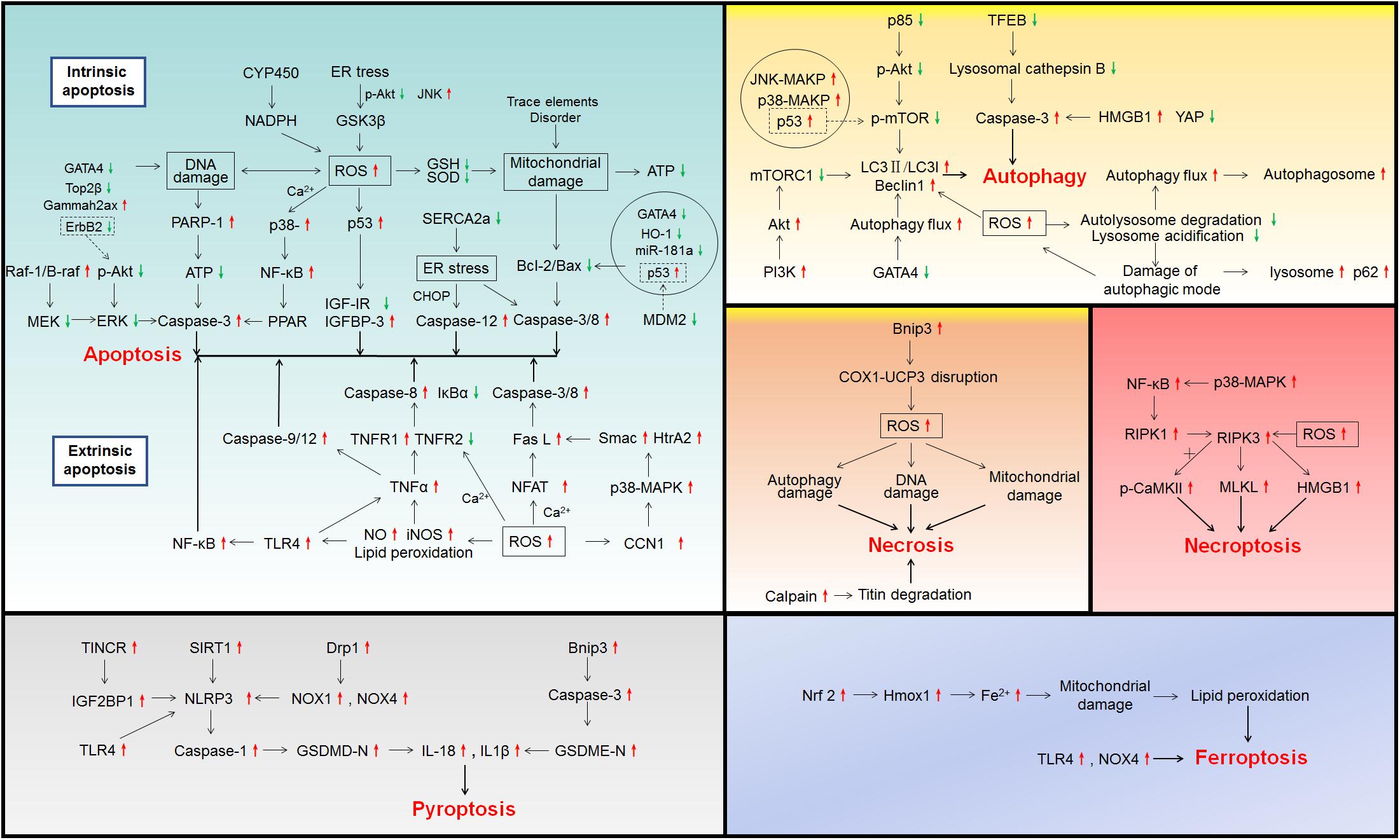
Figure 1. Signaling pathways involved in drug-induced cardiotoxicity.
Anticancer Drugs
Doxorubicin (DOX)
Apoptosis
Enhanced production of ROS is recognized as the classic mechanism of DOX induced cardiomyocyte death. ROS consists of both free radicals and non-free radicals derived from oxygen, including superoxide anions (O2–), hydrogen peroxide (H2O2), hydroxyl radicals (OH–), ozone (O3) and singlet oxygen (1O2) (Zorov et al., 2014). DOX can be reduced to semiquinone by the endothelial nitric oxide synthase (eNOS) and triphosphopyridine nucleotide (NADPH) oxidase, which in turn leads to production of O2–, a major free radical that can produce other ROS, such as H2O2 and hydroxyl radicals (OH⋅) (Vasquez-Vivar et al., 1997). This is the main pathway by which DOX treatment generates ROS. Moreover, Deng et al. (2007) found that reactions between DOX and NADPH could produce superoxide in the absence of any enzyme activity, suggesting ROS production may be caused by the chemical interaction of DOX and NADPH. The generated ROS further induced DNA damage, especially DNA single-strand breaks. Previous studies showed that DOX treatment suppressed the DNA binding activity of GATA binding protein 4 (GATA4), an oxidant-sensitive transcription factor that plays an important role in transducing nuclear events (Kim et al., 2003). In vitro, DNA breaks activated nuclear poly ADP-ribose polymerase 1 (PARP-1) to induce the synthesis of poly-ADP-ribose and cause ATP consumption by subsequent glycohydrolase reactions and ATP conversion, leading to the collapse of heart energy metabolism (Mukhopadhyay et al., 2009). Scaffold protein Sirt6 was proved to be protective against DOX-induced DNA damage (Brito et al., 2016). In addition to DNA damage, DOX-induced intracellular ion disorder also contributes to ROS production.
A previous study demonstrated that under aerobic conditions, DOX could automatically combine with iron to form DOX-iron complex, which increased the contents of OH– by self-reduction, contributing to subsequent lipid peroxidation through membrane interactions (Malisza and Hasinoff, 1995). Another study also discovered that the levels of iron and ROS were up-regulated in DOX-treated cardiomyocytes, which finally induced the mitochondrial apoptosis through caspase-3 activation and cytochrome C release (Childs et al., 2002). The participation of intracellular iron in the degradation of hypoxia-inducible factors (HIF) has been demonstrated (Peyssonnaux et al., 2008). Latter research indicated that iron/HIF signaling mediated the cardio-protective effect of dexrazoxane, the unique authorized protectant for DOX-induced cardiotoxicity (Spagnuolo et al., 2011). Paradoxically, DOX-induced ROS increased the synthesis of ferritin and mediated the protective effect of DOX against iron-induced cardiotoxicity (Corna et al., 2004).
In addition, topoisomerase 2 beta (Top2β) was found to involve in ROS formation during DOX treatment (Vejpongsa and Yeh, 2014). Topoisomerase 2 (Top2), consisting of Top2α and Top2β, played a crucial role in DNA replication, transcription, and repair (Wang, 2002). Top2 was considered a target for the anticancer effect of anthracyclines (Zhu et al., 2016). Top2β interaction with DOX caused DNA double-strand breaks and further triggered transcriptome changes in cardiomyocytes (Zhang et al., 2012). The DOX-Top2β combination may inhibit the transcription of peroxisome proliferator-activated receptor gamma coactivator-1 (PGC1) including PGC1α and PGC1β, which play critical roles in mitochondrial biogenesis as antioxidant (Finkel, 2006; Finck and Kelly, 2007). Moreover, mice with cardiomyocyte-specific Top2β conditional knockout (Top2β–/–) presented less DNA damage and mitochondrial dysfunction as well as oxidative phosphorylation after DOX exposure (Zhang et al., 2012).
Doxorubicin-induced oxidative stress can also stimulate death reporters to combine with corresponding cognate ligands, thereby inducing assembly of the DISC complex, which in turn caused caspase cascades activations and substrates cleavages. A recent study showed that the four death reporters (Fas, TNFR1, DR4, and DR5) were significant increased at the mRNA and protein levels after DOX treatment in induced pluripotent stem cells (iPS) -derived cardiomyocytes (Zhao and Zhang, 2017). One study suggested that ROS-induced up-regulation of cytosolic calcium concentration further elevated the expression of Fas Ligand (Fas L) by stimulating the nuclear factor of activated T-cells (NFAT) signaling (Kalivendi et al., 2005). Moreover, the calcium and calmodulin can conversely bind with eNOS electrons to increase superoxide formation, exacerbating cardiotoxicity (Vasquez-Vivar et al., 1998). Additionally, cardiac fibroblasts exacerbated cardiomyocyte apoptosis by releasing exosomes carrying Fas L in a paracrine manner during DOX treatment. Rosmarinic acid was shown to decrease Fas L secretion by suppressing the level of NFAT activation and metalloproteinase 7 (MMP7) expressions in cardiac fibroblasts, and served a protective role (Zhang et al., 2019). Preclinical experiments demonstrated a decrease in cardiomyocyte death of rats treated with anti-Fas L antibody (Nakamura et al., 2000). Moreover, caspase-8 inhibition blocked Fas L-induced apoptosis, indicating that downstream signaling of apoptosis was mediated by Fas L (Yamaoka et al., 2000). The matricellular protein CCN1 triggered by DOX reacted with integrin α6β1 to promote the activation of p38 mitogen-activated protein kinase (p38-MAPK), which stimulated the release of second mitochondrial activator of caspase (SMAC) and high-temperature requirement protein A2 (HtrA2), synergizing with Fas L to induce cardiomyocytes apoptosis (Hsu and Mo, 2016). As an important receptor in extrinsic apoptosis, TNFR1 was also involved in DOX-related cardiomyocyte death. DOX changed the level of TNFα in H9c2 cells, leading to an increase in TNFR1 expression and a decrease in TNFR2 expression, accompanied by the activation of caspase-8 and suppression of IκBα (Chiosi et al., 2007). It is noteworthy that vitamin C, a well-known reductant, can evidently decline the levels of TNFα, IL-1β and IL-6 in DOX-treated mice, indicating that oxidation/nitrosation stress may be one of the targets of cardiac protection during DOX treatment (Akolkar et al., 2017). In addition, the intracellular calcium homeostasis protective agent mangiferin was also verified to be able to relieve the up-regulation of TNFα and caspase-9 induced by DOX and stimulates the calcium regulatory gene, preventing myocarditis and apoptosis (Agustini et al., 2016).
Another plausible mechanism is that DOX-induced oxidative stress breaks the oxido-reduction balance of mitochondria in cardiomyocytes. DOX treatment promotes ROS overproduction, which remains inside the mitochondrial membrane and induces mitochondrial dysfunction (Giulivi et al., 1995). DOX caused a down-regulation of antioxidant enzymes such as copper, manganese and zinc superoxide dismutases (SODs), glutathione peroxidase (GSH-Px) and catalase (Costa et al., 2013). This imbalance between oxidation and antioxidation aggravates mitochondrial damage. Therefore, overexpression of antioxidant enzymes can reduce DOX-induced cardiotoxicity. In vitro, γ-ray pre-irradiation increased manganese superoxide dismutase (Mn-SOD) levels in neonatal rat ventricular myocytes (NRCMs), which up-regulated MMP, Bcl-2 expressions, and decreased the Bax expression and cytochrome C release (Childs et al., 2002; Chae et al., 2005). Nevertheless, polysaccharide elevated MMP and restrained mitochondrial permeability transition (MPT) by activating manganese superoxide dismutase (Mn-SOD) and suppressing the subsequent caspases cascade reactions (Li W. J. et al., 2018).
Recent studies demonstrated that DOX could indirectly target some receptors or anti-apoptosis factors by regulating miRNAs. DOX increased the level of miR-146a and induced the targeted inhibition of human epidermal growth factor receptor-4 (ErbB4), which caused cardiomyocyte apoptosis and acute cardiotoxicity (Horie et al., 2010). The miR-181a directly targeted the Bcl-2 transcript and negatively regulated Bcl-2 expression, which mediates the protective effect of propofol against DOX-induced cardiotoxicity in vitro and in vivo (Zhao et al., 2018). Moreover, miR-29b was found to target 3’ untranslated region of Bax and restrained Bax expression, hence alleviating DOX-induced cardiomyocyte apoptosis (Jing et al., 2018).
Several studies showed that varying DOX dosages caused apoptosis through different pathways. A study reported that treatment with a high concentration of DOX (2 μM) tended to promote ROS accumulation, while a lower concentration (0.25 M) was more likely to suppress the expression of haem oxygenase 1 (HO-1). HO-1 down-regulation induced cardiomyocyte apoptosis by activating caspase-3 and the release of mitochondrial cytochrome C (Bernuzzi et al., 2009). Another study found that a high concentration of DOX (1 μM) tended to cause DNA damage, PARP-1 dissociation and grievous apoptosis, and a low concentration of DOX (0.5 μM) could activate the p53-related mitochondrial apoptosis pathway (Cunha-Oliveira et al., 2018). Furthermore, DOX dose-dependently increased p53 expression in H9c2 cells, which inhibits type 1 insulin-like growth factor receptor (IGF-1R) transcription and induces IGF binding protein-3 (IGFBP-3) transcription, resulting in resistance to IGF-1 and contributing to apoptosis (Fabbi et al., 2015). More in-depth study indicated that the regulation of DOX on p53 may involve Sirtuin 1 (SIRT1) -mediated deacetylation of p53 (Zhang et al., 2011).
Autophagy
Autophagy is commonly considered as a conservative and beneficial regulatory process that maintains intracellular homeostasis, which is initially activated to resist DOX-induced cardiotoxicity. Oxidative stress is considered the main inducement for autophagy. As reported, during DOX treatment, ROS increased the ratio of LC3II/LC3I and the level of Beclin 1, both being the bio-markers of autophagy (Zhang et al., 2015). In addition, Dox up-regulated the levels of pro-autophagy factors (p53, p38-MAPK, and JNK-MAPK), and down-regulated the p85 expression, the catalytic subunit of phosphoinosmde-3-kinase (PI3K) as well as Akt phosphorylation (Ludke et al., 2017; Yu et al., 2017).
Even though the autophagy process is indeed initiated by DOX to serve a protective role, it somehow fails to finish the process since overwhelming oxidative stress blocks the degradation of lysosomes and even causes autophagic cell death, which in fact turns the original protective effect into damage. Under these circumstances, the normal protein degradation of cardiomyocytes was disrupted, and the subsequent increase in ubiquitinated proteins resulted in the accumulation of autophagy flux and autophagosomes (Dimitrakis et al., 2012). Meanwhile, DOX suppressed lysosome acidification and autolysosome degradation, which blocked the autophagic flux and augmented the damage (Li et al., 2016). Moreover, DOX-induced up-regulation of histone deacetylase 6 (HDAC6) decreased α-tubulin acetylation level, giving rise to mitochondrial dysfunction and autophagy flux damage (Song et al., 2018). Lysosome dysfunction was found to involve in the depletion of transcription factor EB (TFEB). DOX can suppress the expression of TFEB and induce the impairment of lysosomal cathepsin B, which subsequently inhibited lysosomal autophagy, increasing the levels of ROS and caspase-3 cleavage (Bartlett et al., 2016).
In addition to ROS-related autophagy, DOX also regulates autophagy-related factors and cause autophagic cell death. High mobility group box 1 (HMGB1) plays a vital role in the process of autophagy. DOX increased HMGB1expression, while silencing HMGB1 could reverse cardiomyocyte damage by attenuating autophagy (Luo et al., 2018). In addition, inhibition of the transcription factor GATA4 was observed in DOX-treated cardiomyocytes, and GATA4 induces the expression of Bcl2, which can interact with Beclin 1 to silence autophagy, decreases the cardiotoxicity (Kobayashi et al., 2010). Moreover, rats treated with 3-methyladenine, a specific inhibitor of autophagy, showed fewer autophagic vacuoles and mitochondrial MPT, but higher levels of Na+-K+ ATPase activity and MMP as compared with DOX treatment alone (Lu et al., 2009).
It has been reported that starvation or caloric restriction prior to DOX insult can suppress cardiotoxicity. Caloric restriction attenuated DOX-induced ATP exhaustion and enhances the activity of AMPK, which eventually corrected the harmful autophagy caused by DOX, demonstrating a protective role (Chen et al., 2011). On the contrary, prior starvation mitigated acute DOX-induced cardiotoxicity via further augmenting autophagy (Kawaguchi et al., 2012). In addition, Astragalus polysaccharide and resveratrol can restore autophagy in mice and H9c2 cells through the AMPK/mTOR signaling pathway, alleviating cardiotoxicity (Gu et al., 2016; Cao et al., 2017).
Necrosis
Unlike apoptosis and autophagy, emerging evidence has indicated that cardiomyocyte necrosis is triggered by a high dosage or prolonged exposure to DOX treatment. Dose-dependently elevated by DOX, the accumulation of ROS and peroxynitrite increase the rate of necrosis in cardiomyocyte death (Mukhopadhyay et al., 2009; Fulbright et al., 2015). The commonly used dosages of DOX are ≤20 mg/kg in vivo and 1 μM in vitro. A single intraperitoneal injection of DOX at 25 mg/kg in mice could immediately cause necrotic death and cardiac insufficiency (Li et al., 2014), and 2 μM DOX can directly induce cardiomyocyte necrosis in vitro (Bernuzzi et al., 2009). Moreover, when the cardiomyocytes are exposed to DOX for a long period, initial apoptosis develops into necrosis as the cells preferentially exhibits early DNA impairment and nuclear swelling (Rharass et al., 2016). As discussed above, DOX can destroy the function of lysosomes and disrupt normal autophagy as a result of oxidative stress. Consequently, the delayed autophagy in cardiomyocytes causes more severe apoptotic secondary necrosis (Dimitrakis et al., 2012; Li et al., 2014). These studies further confirm the notion that necrosis arose with extended exposure to DOX treatment.
Moreover, DOX initiates necrotic cell death by regulating necrosis-related intracellular factors. The degradation of titin, a myofilament protein associated with myocardial damage, was induced by DOX, and finally induced cardiomyocyte necrotic death (Lim et al., 2004). In addition, BH3-only protein BNIP3 was activated by DOX to destroy the combination of respiratory chain complex IV subunit 1 (COX1) and uncoupling protein 3 (UCP3), which disrupted respiratory efficiency, eventually leading to necrotic cell death (Dhingra et al., 2014).
Necroptosis
It has been demonstrated that RIPK3 is activated by DOX to bind with phosphorylated-CaMKII, causing the opening of mitochondrial permeability transition pore (MTPT), and resulting in necroptosis (Zhang T. et al., 2016). More importantly, dexrazoxane alleviated Dox-induced inflammation and cardiomyocyte necroptosis through inhibiting p38-MAPK/nuclear factor kappa-B (NF-κB) signal (Yu et al., 2020).
Pyroptosis
The increased secretion of IL-1β and IL-18, activation of TLR4, NLRP3 inflammasome and caspases were found in DOX-treated H9c2 cells, suggesting the occurrence of pyroptosis (Singla et al., 2019). BNIP3, the upstream regulator of cardiomyocyte pyroptosis, can activate caspase-3 and lead to subsequent GSDME cleavage (Zheng et al., 2020). DRP1/NOX signaling was activated to cause mitochondrial damage, which involved in DOX-induced pyrotosis (Zeng et al., 2020). Moreover, up-regulated lncRNA TINCR recruited IGF2BP1 to enhance the NLRP3 expression that mediated Dox-induced pyrotosis (Meng et al., 2019). However, embryonic stem cells-derived exosomes and Heat-shock Protein 22 can reverse the Dox-induced cardiomyocytes pyrotosis via inhibiting TLR4/NLRP3/caspase-1 signaling (Tavakoli et al., 2019; Lan et al., 2020). Moreover, suppression of ROS was also reported to be able to alleviate Dox-induced cardiomyocyte pyrotosis, whose mechanism involved the inhibition of sirtuin 1/NLRP3 signaling pathway (Sun et al., 2020; Zhai et al., 2020).
Ferroptosis
Doxorubicin-induced accumulation of ROS and lipid peroxidation can lead to cardiomyocyte ferroptosis (Koleini et al., 2019). Activation of TLR 4 and NOX 4 has also been proven to promote DOX-induced cardiomyocyte ferroptosis (Chen et al., 2019). Administering DOX to mice induced cardiomyopathy with a rapid, systemic accumulation of nonheme iron via heme degradation by NF-E2-related factor 2 (Nrf2)-mediated up-regulation of heme oxygenase-1 (HMOX1), indicating the cardio-protective role of targeting ferroptosis for cardiomyopathy prevention (Fang et al., 2019).
Cisplatin
Cisplatin is a chemotherapeutic agent for a vast spectrum of cancers. However, its acute and cumulative cardiotoxicity partially limits anti-tumor treatment and clinical applications (Ma et al., 2010). Cisplatin-treated cardiomyocytes showed mitochondrial abnormalities such as mitochondrial membrane depolarization, inflammatory responses and increased ER stress, which finally stimulated the activity of caspase-3 and induced apoptosis (Chowdhury et al., 2016). In addition, emerging evidences demonstrated a close connection between oxidative stress and cisplatin-induced cardiomyocyte apoptosis. El-Awady el et al. (2011) discovered that cisplatin improved lipid peroxidation, decreased GSH content and suppressed SOD activity, implying oxidative stress induced by cisplatin. Moreover, mitochondrial DNA injury and nuclear DNA damage were observed. Antioxidant natural products such as tutin (vitamin P1), zingerone and cyanidin can inhibit cisplatin-induced inflammatory infiltration, DNA damage, and mitochondrial dysfunction, indicating the key role of oxidative stress in cisplatin-induced cardiomyocyte apoptosis (Qian et al., 2018; Soliman et al., 2018; Topal et al., 2018).
Cyclophosphamide
Cyclophosphamide is commonly applied in the treatment of malignant tumors such as leukemia and lymphoma, and it is also adopted to treat systemic lupus erythaematosus and polymyositis as an immunosuppressor. Because of the dose-dependent manner, cyclophosphamide-induced cardiotoxicity basically coincides with high-dose treatment (Nishikawa et al., 2015; Wadia, 2015). Acrolein, the active metabolite of cyclophosphamide, was confirmed to be mainly responsible for cardiomyocyte death (Conklin et al., 2015; Nishikawa et al., 2015; Kurauchi et al., 2017). The cardiomyocyte injuries caused by cyclophosphamide treatment included sarcoplasmic reticulum dilatation, mitochondrial disruption and nuclear membrane invagination (Lushnikova et al., 2008). Further studies attributed these injuries to oxidative stress, clarifying that acrolein caused oxidative and nitrite stress through the suppression of intracellular GSH and SOD and increase of MDA (Nagi et al., 2011; Kurauchi et al., 2017; Omole et al., 2018). Corresponding lipid peroxidation initiated mitochondrial function damage, which further led to a collapse in APT production and the activation of caspase-3, resulting in apoptosis (Nagi et al., 2011; Refaie et al., 2020). In addition, cyclophosphamide was verified to stimulate TLR4, through which it initiated the TLR4/NF-κB signaling to trigger an inflammatory reaction, and eventually apoptosis (El-Agamy et al., 2017). Furthermore, DNA damage was observed in cyclophosphamide-treated rats, which were accompanied with activation of caspase-3 and inhibition of Bcl-2 expression (Avci et al., 2017). Antioxidant drugs such as silymarin and curcumin can inhibit cyclophosphamide-induced cardiotoxicity via decreasing the fragments of mitochondrial DNA and nuclear DNA, suggesting that excessive ROS might be responsible for cyclophosphamide-induced DNA injuries.
With the exception of the evidence discussed above, cyclophosphamide acted as a teratogen to injure cardiomyocytes. Cyclophosphamide affected cardiomyocytes developing via DNA fragmentation degradation, caspase-3 activation and PARP cleavage (Mirkes and Little, 1998). Moreover, cyclophosphamide was found to activate the apoptotic pathways, culminating in abnormality of the heart via the p38-MAPK signaling (Mirkes et al., 2000).
Fluorouracil and Capecitabin
5-Fluorouracil, a pyrimidine antimetabolite, is used widely in clinical practice as an anti-tumor treatment for cancers such as intestinal cancer and liver cancer. Capecitabin, a tumor-targeting drug that takes effect after the intracellular transformation into 5-fluorouracil, was regarded as having similar cardiotoxicity. Multiple studies confirmed the cardiotoxicity induced by 5-fluorouracil, which was attributed to the coronary arteries injury (Clavel et al., 1988; Herrmann et al., 2016). Further study suggested that the cardiomyocyte apoptosis might also play an important role in 5-fluorouracil-induced cardiotoxicity (Tsibiribi et al., 2006). 5-Fluorouracil stimulated intracellular oxidative stress by O2– generation, which eventually activated the caspases cascade reactions, leading to apoptosis (Lamberti et al., 2012). To further determine the underlying mechanism, Eskandari et al. (2015) discovered that 5-fluorouracil-induced ROS increase was accompanied by the depletion of GSH, a ROS scavenger. Next, the generated ROS mediated lipid peroxidation on mitochondria to decrease MMP, causing mitochondrial dysfunction and caspase-3 activation. Fluoroacetate, the metabolite of 5-fluorouracil, can restrain aconitase to block the tricarboxylic acid cycle, resulting in a mitochondrial energy metabolism crisis (Lischke et al., 2015). Moreover, accumulation of autophagosomes and lysosomal membrane leakiness were observed in 5-fluorouracil-treated human cardiomyocytes, indicating the involvement of autophagic cell death in 5-fluorouracil-induced cardiotoxicity (Eskandari et al., 2015; Focaccetti et al., 2015).
Arsenic Trioxide
Arsenic trioxide represents a breakthrough in the field of acute promyelocytic leukemia therapeutic, but its cardiotoxicity remains an unresolved problem. In the past few decades, numerous studies have proposed that cardiomyocyte apoptosis is induced by ROS, and mitochondrial damage might be the main reason for arsenic trioxide-induced cardiotoxicity (Zhao et al., 2008; Raghu and Cherian, 2009; Vineetha et al., 2015). Arsenic trioxide can increase ROS level and calcium concentration, which was accompanied with cardiomyocyte apoptosis (Raghu and Cherian, 2009). Moreover, ROS generation depleted intracellular antioxidants such as GSH, GSH-Px, glutathione s-transferase (GST) and SOD, which caused lipid peroxidation and decreased MMP (Varghese et al., 2017). In addition, mitochondrial damage such as MTP and mitochondrial swelling was also observed under arsenic trioxide exposure, following by a decrease in oxygen consumption as well as ATP production, resulting in caspase-3 activation and apoptosis (Vineetha et al., 2015). Recently, Li et al. (2019) found that arsenic trioxide interfered with the dynamic balance of trace elements in chicken cardiomyocytes to break mitochondrial cristae and mitochondrial vacuoles, increasing the expressions of Bax and caspase-3/8. In addition to ROS and mitochondrial damage, calcium imbalance is also involved in arsenic trioxide-induced cardiotoxicity. Arsenic trioxide suppressed the activity of sarcoplasmic reticulum Ca2+-ATPase2a, by which cytoplasmic calcium was taken back to the sarcoplasmic reticulum (Zhang J.Y. et al., 2016). Consequently, the imbalance of calcium homeostasis and ER stress activated C/EBP-homologous protein (CHOP), caspase-12 and GRP78, leading to apoptosis.
By increasing the levels of the inflammatory cytokines TNFα, NF-κB, cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), arsenic trioxides promoted the inflammatory reactions and brought ultrastructural damage to cardiomyocytes (Li et al., 2017). These inflammatory responses partly contributed to heavy metal-related cardiotoxicity (Lakkur et al., 2015). Additionally, it has been determined that arsenic trioxides up-regulates the content of phosphorylated p38 and JNK by oxidative stress stimulation and calcium overload, which further induces NF-κB phosphorylation, caspase-3 activation and PARP cleavage (Ghosh et al., 2009; Fan et al., 2013). Suppression of ROS apparently inhibited the activation of JNK, extracellular regulated protein kinases (ERK), and p38, which eventually reversed cardiomyocyte apoptosis (Miao et al., 2013; Zhang et al., 2017). One recent study reported that arsenic trioxides might induce cardiomyocyte apoptosis through DNA damage, since it dose-dependently increased the content of γH2AX, a sensitive biomarker for DNA breaks (Bao et al., 2019). Moreover, activation of Parkin, an E3 ubiquitin ligase, can inhibit arsenic trioxide-induced cardiotoxicity via the maintenance of mitochondrial as well as cellular homeostasis (Watanabe et al., 2014). It is well known that Parkin-induced ubiquitination of mitochondrial substrates finally leads to mitophagy. Therefore, mitophagy displays a positive role in maintaining cardiac homeostasis during arsenic trioxide exposure. However, a subsequent study demonstrated that arsenic trioxide-induced oxidative stress led to the formation of autophagosomes through PI3K/Akt/mTOR signaling, which resulted in myocardial damage (Li S. et al., 2018; Zhao et al., 2019). Moreover, cardiomyocyte necrosis was also observed under arsenic trioxide exposure (Raghu and Cherian, 2009; Vineetha et al., 2015), but the underlying mechanisms remained unclear.
Trastuzumab
A novel and widely used monoclonal antibody drug, Trastuzumab, is also reported to cause cardiotoxicity, such as cardiac insufficiency and heart failure. Under physiological condition, neuregulin-1 interacted with epidermal growth factor receptor-2 (ErbB2) to allow for the formation of ErbB4/ErbB2 heterodimer, which blocked cell death through an Akt-dependent signaling in cardiomyocytes (Sawyer et al., 2002; Lemmens et al., 2007). Functioning as an inhibitor of ErbB2 receptor, trastuzumab interrupted ErbB4/ErbB2 heterodimerization, thereby leading to apoptosis (Holbro and Hynes, 2004; Rohrbach et al., 2005). Interaction between Trastuzumab and ErbB2 triggered the downstream signal transduction pathways, such as increased levels of Bax and Bcl-xS, decreased Bcl-xL level and caspases cascade activations. Furthermore, the decreased MMP caused by Bcl-xL suppression and subsequent mitochondrial energy catastrophe also contributed to trastuzumab-induced apoptosis (Grazette et al., 2004). Singh et al. (2011) proposed that the cardiotoxicity induced by trastuzumab might stem from its negative regulation of murine double minute 2 (MDM2) and p53. In addition, trastuzumab-induced cardiomyocyte apoptosis was found to be related to inflammatory infiltration (Coppola et al., 2016), and the TLR4-mediated chemokine expressions of TNFα, MCP-1 and ICAM-1 contributed to inflammatory responses induced by trastuzumab (Yousif and Al-Amran, 2011).
A recent study reported that trastuzumab suppressed autophagy, resulting in mitochondrial dysfunction and ROS accumulation (Mohan et al., 2017). Trastuzumab insult inhibited the expressions of Beclin 1, autophagy related gene (Atg) 5-12 and Atg14. Moreover, Trastuzumab stimulated EebB1-Y845 and ErbB2-Y1248 and activated the ERK/mTOR/ULK-1 signaling to supress autophagy (Mohan et al., 2016).
Currently, drug combination therapies with trastuzumab are quite popular among cancer treatment plans. However, the combination therapies show more serious cardiotoxicity compared with single drug remedies. Trastuzumab combining with DOX exacerbated the exhaustion of antioxidant enzymes as well as damage to the mitochondrial structure (Xu et al., 2004). Trastuzumab-induced ErbB2 inhibition further suppressed c-Abl and Arg, which plays an antioxidant role by activating GSH-Px and catalase, thereby increasing DOX-induced cytotoxicity (Belmonte et al., 2015). Additionally, inhibition of the neuregulin-ErbB signaling by trastuzumab restrained the phosphorylation of ERK1/2 and Akt, and eventually exacerbated DOX-induced cardiomyocyte apoptosis and myocardial fiber injury (Sawyer et al., 2002). The underlying mechanism might also include the increased level of iNOS, which contributed to oxidative stress and inflammatory cell infiltration (Milano et al., 2020). Moreover, DOX-trastuzumab combination synergistically repressed Top2β, and eventually resulted in DNA double strand breaks and the ROS overproduction (Jiang et al., 2018). N-Acetyl Cysteine Amide, a ROS scavenger, can attenuate the DOX and trastuzumab-induced cardiac dysfunction (Goyal et al., 2016). Furthermore, paclitaxel-trastuzumab combination was reported to have a worsening effect on cardiomyocyte function, which might result from the inhibition of phosphorylated-ERK1/2. However, no cell death was observed during treatment in addition to myofibrillar structure changes (Pentassuglia et al., 2007).
Tyrosine-Kinase Inhibitors (Sunitinib, Imatinib, Nilotinib, Sorafenib, Ponatinib and Dasatinib)
Tyrosine-kinase inhibitors such as sunitinib, imatinib, nilotinib, sorafenib, ponatinib, and dasatinib are widely applied for chronic myelogenous leukemia and solid tumors, however, an increasing number of studies have reported cardiotoxicity associated with tyrosine-kinase inhibitors. Clinical research observed aberrantly shaped, swollen mitochondria in cardiomyocytes of patients who developed congestive heart after sunitinib treatment (Chu et al., 2007). Bouitbir et al. (2019) demonstrated that the oxidative stress caused by sunitinib was mainly responsible for the mitochondrial damage and final apoptosis of cardiomyocytes. By suppressing mitochondrial electron transport chain enzyme complexes, sunitinib induced ROS accumulation, which further decreased MMP and destroyed the mitochondrial structure to initiate caspases cascade reactions. In addition, sunitinib was confirmed to increase the expression of miR-133A, and suppress the apoptosis signal-regulating kinase 1 (ASK1)/mitogen activated kinase kinase 7 (MKK7)/JNK signaling to induce apoptosis and myocardial damage (Cooper et al., 2018). The elevation of autophagy flux was observed in sunitinib-treated H9c2 cells, (Zhao et al., 2010), and inhibition of autophagy was demonstrated to attenuate sunitinib-induced cardiotoxicity (Kimura et al., 2017), indicating involvement of autophagy in the cytotoxicity.
Mitochondrial damage was also observed in imatinib-treated cardiomyocytes. Imatinib down-regulates the expression of Bcl-2 and Bcl-xL by suppressing the content of the transcription factor GATA4, resulting in apoptosis. Moreover, aging was verified to be a risk factor during imatinib treatment because of its positive impact on the oxidative stress (Maharsy et al., 2014). In addition, Chambers et al. (2017) showed that imatinib induced oxidative stress and mitochondrial dysfunction via mediating JNK-related mitochondrial signaling and activating caspase-3/7/9. Autophagic death was also reported to be associated with imatinib-induced cardiotoxicity (Hu et al., 2012). Imatinib blocked the autophagic process as indicated by an increased level of lysosomes, which was consistent with the accumulation of p62, a protein degraded by autophagic clearance.
As a second-generation Bcr-Abl inhibitor, nilotinib mediated apoptosis mainly through the accumulation of ROS and ER stress (Doherty et al., 2013). The increase of ROS and ER stress biomarkers (ATF4 and CHOP), was observed in nilotinib-treated H9c2 cells, which subsequently decreased MMP and activated caspase-3, markers of apoptosis (Lekes et al., 2016). Recently, Yang et al. (2018) made further efforts to demonstrate that nilotinib induced ER stress to activate JNK and restrain Akt phosphorylation, which in turn suppressed glycogen synthase kinase-3 beta (GSK3β) phosphorylation and activated NOX4/ROS signaling, resulting in apoptosis.
Few studies reported potential apoptosis mechanisms associated with sorafenib, ponatinib and dasatinib. Cheng et al. (2011) demonstrated that sorafenib induced cardiomyocyte apoptosis and cardiotoxicity by inhibiting the Raf/MEK/ERK signaling. In addition, cardiomyocytes necrosis was observed during high-dose sorafenib treatment with unclear mechanism (Duran et al., 2014). The most recent study reported that ponatinib inhibited phosphorylation of Akt and ERK1/2, which contributed to the activation of pro-apoptotic caspase-3 (Singh et al., 2019). Xu et al. (2018) found that dasatinib dose-dependently up-regulated intracellular HMGB1 to induce cardiomyocyte necroptosis.
Mitoxantrone
With similar structure to DOX, mitoxantrone was thought to be an alternative to DOX with less cardiotoxicity (Herman et al., 1997). However, several studies have also reported mitoxantrone-induced cardiotoxicity. Mitoxantrone induced cardiomyocyte apoptosis associated with oxidative stress and mitochondrial dysfunction. With a time-dependent increase in the content of ROS, mitoxantrone promoted the accumulation of calcium and decreased the MMP, which further caused mitochondrial energy deficiency and activated caspase-3 (Rossato et al., 2013). In addition, DNA strand breaks were observed in apoptotic H9c2 cells treated with mitoxantrone, which was probably mediated by a Top2β-dependent signaling (Wu et al., 2013; Damiani et al., 2018).
Antidiabetic Drugs (Rosiglitazone and Pioglitazone)
Rosiglitazone and pioglitazone, as thiazolidinedione antidiabetic agents, were regarded as protective agents in diabetic cardiomyopathy and ischaemia-reperfusion injury (Cao et al., 2007; Baraka and Abdelgawad, 2010). However, with further expansion of their clinical applications, rosiglitazone and pioglitazone are also found to cause serious side effects such as myocardial hypertrophy and heart failure. It has been reported that rosiglitazone exerts both protective and detrimental effects in rats treated by ischaemia reperfusion (Palee et al., 2013). Although rosiglitazone was known as a PPARγ agonist, it caused cardiotoxicity via oxidative stress-induced mitochondrial dysfunction independent of PPARγ. By increasing the levels of NAPHD and iNOS, rosiglitazone induced oxidative stress accompanied by the exhaustion of antioxidant enzymes such as SOD and glutathione reductase, resulting in cardiotoxicity apoptosis (Mishra et al., 2014). In addition, the oxidative effect caused mitochondrial dysfunction, followed by cardiomyocyte energy deficiency (He et al., 2014). For pioglitazone, a study once reported that pioglitazone up-regulated the levels of sphingomyelinase and ceramidase, a mediator of cardiomyocyte apoptosis (Baranowski et al., 2007). Moreover, by activating Bax and phosphorylated p53 as well as suppressing phosphorylated Akt and mTOR, pioglitazone could induce apoptosis in a VEGFR-2 dependent manner (Zhong et al., 2018).
Antiviral Drug (Zidovudine)
Zidovudine, like other nucleoside reverse transcriptase inhibitors, is widely used for human immunodeficiency virus type 1 (HIV-1) infection. However, side effects such as hypertension and cardiomyopathy limit its long-term application. Zidovudine stimulated the accumulation of ROS and peroxynitrite, which in turn gives rise to single-strand DNA breaks, and eventually results in mitochondrial energy depletion in a NAD+-dependent manner (Szabados et al., 1999). Zidovudine induced the transport of protein kinase C δ (PKCδ) from the cytosol to the membrane, which promoted the activation of NADPH oxidases (Papparella et al., 2007). Meanwhile, mitochondrial damage is observed after zidovudine treatment (Fiala et al., 2004). Mitochondrial ROS caused by zidovudine played a significant role in mitochondrial disruption-induced apoptosis by activating caspase-3/7 (Ry et al., 2011). Fas/Fas L was also involved in zidovudine-induced cardiomyocytes apoptosis (Purevjav et al., 2007). Besides the above effects, zidovudine was shown to inhibit autophagosome maturation and decrease autophagic flux, leading to mitochondrial membrane polarization and ROS accumulation (Lin et al., 2019). Zidovudine-induced cardiomyocyte necrosis involved PARP activation (Gao et al., 2011).
Perspectives and Conclusion
Cardiotoxicity is a major concern when evaluating whether drugs can be put on the market during preclinical research and is an important reason for post-approval drug withdrawal. Even for widely used drugs, cardiotoxicity limits their clinical applications. Fortunately, the mechanisms of cardiotoxicity have gradually come to light in recent years. ROS serves a main driver in drug-induced cardiotoxicity, and thereby many antioxidants have undergone preclinical development or clinically research for cardiotoxicity. For example, dexrazoxane, an iron chelating agent against iron-mediated oxidative stress, is the cardio-protective medicine approved by FDA in July 1995 for preventing anthracycline-induced cardiotoxicity, and now has been widely applied in the clinical practice (Padegimas et al., 2020). In addition, numerous natural antioxidants serve as adjuvant therapies to reduce drug-induced cardiotoxicity, such as berberine, epigallocatechin-3-gallate, and resveratrol (Coelho et al., 2017; Yu et al., 2018).
Recently, immune checkpoint inhibitors including anti-PD-1, anti-PD-L1 and CTLA-4 blockade have attracted a substantial amount of attention, which may revolutionize the treatment of cancer. Ever since the first case of cardiotoxicity induced by ipilimumab (CTLA-4 blockade) was reported in 2013 (Voskens et al., 2013), more and more case reports have indicated immune checkpoint inhibitor-induced cardiotoxicity. Worse still is the underlying mechanism remains unknown, possibly due to the lack of suitable animal models. Immune inflammation and ROS accumulation may play key roles in the immune checkpoint inhibitor-induced cardiotoxicity, which needs to be confirmed by future studies.
To achieve a better treatment effect, combinations of anticancer drugs have been widely applied in the clinical practice, but unfortunately lead to greater cardiotoxicity than with individual drug. Much attention has been paid to trastuzumab combined with DOX for treating women with ErbB2-positive breast cancer. Addition of trastuzumab to adjuvant DOX chemotherapy increases the incidence of cardiotoxicity, and few studies have been conducted to explore the underlying mechanisms, with iNOS or Top2β-mediated oxidative stress being the only acceptable mechanism (Jiang et al., 2018; Milano et al., 2020). To sum up, the mechanisms of drug-induced cardiomyocyte death are not absolutely independent, with the crosstalk and overlap of signaling pathways perplexing and complicating the cardiotoxicity. Therefore, further in-depth mechanisms deserve urgent investigation to avoid synergistic cardiotoxicity.
In this review, we summarized and discussed six cardiomyocyte death forms associated with drug-induced cardiotoxicity, including apoptosis, autophagy, necrosis, necroptosis, pyroptosis and ferroptosis. However, most of studies focused on the apoptosis, and whether the coexistence of multiple cardiomyocyte death forms was a common phenomenon of drug-induced cardiotoxicity remains to be explored. A recent study found that caspase-8 was the molecular switch that controls apoptosis, necroptosis and pyroptosis, and prevented tissue damage during embryonic development and adulthood (Fritsch et al., 2019). Therefore, it may be an interesting and important research topic to explore the contribution of each form and conversion of different forms in drug-induced cardiomyocyte death.
Author Contributions
WM and SW wrote the manuscript. BZ and WL revised the manuscript. All authors read and approved the final version of the manuscript for publication.
Funding
This work was supported by grants of the National Natural Scientific Foundation of China (Nos. 81703518 and 81973406), the Hunan Provincial Natural Scientific Foundation (No. 2019JJ50849), and the Scientific Research Project of Hunan Provincial Health and Family Planning Commission (No. B20180253).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agustini, F. D., Arozal, W., Louisa, M., Siswanto, S., Soetikno, V., Nafrialdi, N., et al. (2016). Cardioprotection mechanism of mangiferin on doxorubicin-induced rats: focus on intracellular calcium regulation. Pharm. Biol. 54, 1289–1297. doi: 10.3109/13880209.2015.1073750
Akolkar, G., Da Silva, Dias, D., Ayyappan, P., Bagchi, A. K., Jassal, D. S., et al. (2017). Vitamin C mitigates oxidative/nitrosative stress and inflammation in doxorubicin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 313, H795–H809.
Avci, H., Epikmen, E. T., Ipek, E., Tunca, R., Birincioglu, S. S., Aksit, H., et al. (2017). Protective effects of silymarin and curcumin on cyclophosphamide-induced cardiotoxicity. Exp. Toxicol. Pathol. 69, 317–327. doi: 10.1016/j.etp.2017.02.002
Bao, Z., Han, Z., Zhang, B., Yu, Y., Xu, Z., Ma, W., et al. (2019). Arsenic trioxide blocked proliferation and cardiomyocyte differentiation of human induced pluripotent stem cells: implication in cardiac developmental toxicity. Toxicol. Lett. 309, 51–58. doi: 10.1016/j.toxlet.2019.03.008
Baraka, A., and Abdelgawad, H. (2010). Targeting apoptosis in the heart of streptozotocin-induced diabetic rats. J. Cardiovasc. Pharmacol. Ther. 15, 175–181. doi: 10.1177/1074248409356557
Baranowski, M., Blachnio, A., Zabielski, P., and Gorski, J. (2007). Pioglitazone induces de novo ceramide synthesis in the rat heart. Prostaglandins Other Lipid Mediat. 83, 99–111. doi: 10.1016/j.prostaglandins.2006.10.004
Barnhart, B. C., Alappat, E. C., and Peter, M. E. (2003). The CD95 type I/type II model. Semin. Immunol. 15, 185–193. doi: 10.1016/s1044-5323(03)00031-9
Bartlett, J. J., Trivedi, P. C., Yeung, P., Kienesberger, P. C., and Pulinilkunnil, T. (2016). Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 473, 3769–3789. doi: 10.1042/bcj20160385
Belmonte, F., Das, S., Sysa-Shah, P., Sivakumaran, V., Stanley, B., Guo, X., et al. (2015). ErbB2 overexpression upregulates antioxidant enzymes, reduces basal levels of reactive oxygen species, and protects against doxorubicin cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 309, H1271–H1280.
Bernuzzi, F., Recalcati, S., Alberghini, A., and Cairo, G. (2009). Reactive oxygen species-independent apoptosis in doxorubicin-treated H9c2 cardiomyocytes: role for heme oxygenase-1 down-modulation. Chem. Biol. Interact 177, 12–20. doi: 10.1016/j.cbi.2008.09.012
Bouitbir, J., Alshaikhali, A., Panajatovic, M. V., Abegg, V. F., Paech, F., and Krahenbuhl, S. (2019). Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: running title: sunitinib and oxidative stress in hearts. Toxicology 426:152281. doi: 10.1016/j.tox.2019.152281
Brito, V. B., Nascimento, L. V., Nunes, R. B., Moura, D. J., Lago, P. D., and Saffi, J. (2016). Exercise during pregnancy decreases doxorubicin-induced cardiotoxic effects on neonatal hearts. Toxicology 36, 46–57. doi: 10.1016/j.tox.2016.08.017
Brumatti, G., Salmanidis, M., and Ekert, P. G. (2010). Crossing paths: interactions between the cell death machinery and growth factor survival signals. Cell Mol. Life Sci. 67, 1619–1630. doi: 10.1007/s00018-010-0288-8
Cao, Y., Shen, T., Huang, X., Lin, Y., Chen, B., Pang, J., et al. (2017). Astragalus polysaccharide restores autophagic flux and improves cardiomyocyte function in doxorubicin-induced cardiotoxicity. Oncotarget 8, 4837–4848. doi: 10.18632/oncotarget.13596
Cao, Z., Ye, P., Long, C., Chen, K., Li, X., and Wang, H. (2007). Effect of pioglitazone, a peroxisome proliferator-activated receptor gamma agonist, on ischemia-reperfusion injury in rats. Pharmacology 79, 184–192. doi: 10.1159/000100870
Chae, H. J., Kim, H. R., Lee, W. G., Kwak, Y. K., and Kim, W. H. (2005). Radiation protects adriamycin-induced apoptosis. Immunopharmacol. Immunotoxicol. 27, 211–232. doi: 10.1081/iph-200067715
Chambers, T. P., Santiesteban, L., Gomez, D., and Chambers, J. W. (2017). Sab mediates mitochondrial dysfunction involved in imatinib mesylate-induced cardiotoxicity. Toxicology 382, 24–35. doi: 10.1016/j.tox.2017.03.006
Chen, G., Chelu, M. G., Dobrev, D., and Li, N. (2018). Cardiomyocyte inflammasome signaling in cardiomyopathies and atrial fibrillation: mechanisms and potential therapeutic implications. Front. Physiol. 9:1115. doi: 10.3389/fphys.2018.01115
Chen, K., Xu, X., Kobayashi, S., Timm, D., Jepperson, T., and Liang, Q. (2011). Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J. Biol. Chem. 286, 21993–22006. doi: 10.1074/jbc.m111.225805
Chen, X., Xu, S., Zhao, C., and Liu, B. (2019). Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 516, 37–43. doi: 10.1016/j.bbrc.2019.06.015
Cheng, H., Kari, G., Dicker, A. P., Rodeck, U., Koch, W. J., and Force, T. (2011). A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ. Res. 109, 1401–1409. doi: 10.1161/circresaha.111.255695
Childs, A. C., Phaneuf, S. L., Dirks, A. J., Phillips, T., and Leeuwenburgh, C. (2002). Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res. 62, 4592–4598.
Chiosi, E., Spina, A., Sorrentino, A., Romano, M., Sorvillo, L., Senatore, G., et al. (2007). Change in TNF-alpha receptor expression is a relevant event in doxorubicin-induced H9c2 cardiomyocyte cell death. J. Interferon Cytokine Res. 27, 589–597.
Chowdhury, S., Sinha, K., Banerjee, S., and Sil, P. C. (2016). Taurine protects cisplatin induced cardiotoxicity by modulating inflammatory and endoplasmic reticulum stress responses. Biofactors 42, 647–664. doi: 10.1002/biof.1301
Chu, T. F., Rupnick, M. A., Kerkela, R., Dallabrida, S. M., Zurakowski, D., Nguyen, L., et al. (2007). Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370, 2011–2019.
Clavel, M., Simeone, P., and Grivet, B. (1988). [Cardiac toxicity of 5-fluorouracil. review of the literature, 5 new cases]. Presse Med. 17, 1675–1678.
Coelho, A. R., Martins, T. R., Couto, R., Deus, C., Pereira, C. V., Simoes, R. F., et al. (2017). Berberine-induced cardioprotection and Sirt3 modulation in doxorubicin-treated H9c2 cardiomyoblasts. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 2904–2923. doi: 10.1016/j.bbadis.2017.07.030
Conklin, D. J., Haberzettl, P., Jagatheesan, G., Baba, S., Merchant, M. L., Prough, R. A., et al. (2015). Glutathione S-transferase P protects against cyclophosphamide-induced cardiotoxicity in mice. Toxicol. Appl. Pharmacol. 285, 136–148. doi: 10.1016/j.taap.2015.03.029
Cookson, B. T., and Brennan, M. A. (2001). Pro-inflammatory programmed cell death. Trends Microbiol. 9, 113–114. doi: 10.1016/s0966-842x(00)01936-3
Cooper, S. L., Sandhu, H., Hussain, A., Mee, C., and Maddock, H. (2018). Involvement of mitogen activated kinase kinase 7 intracellular signalling pathway in Sunitinib-induced cardiotoxicity. Toxicology 394, 72–83. doi: 10.1016/j.tox.2017.12.005
Coppola, C., Riccio, G., Barbieri, A., Monti, M. G., Piscopo, G., Rea, D., et al. (2016). Antineoplastic-related cardiotoxicity, morphofunctional aspects in a murine model: contribution of the new tool 2D-speckle tracking. Onco Targets Ther. 9, 6785–6794. doi: 10.2147/ott.s106528
Corna, G., Santambrogio, P., Minotti, G., and Cairo, G. (2004). Doxorubicin paradoxically protects cardiomyocytes against iron-mediated toxicity: role of reactive oxygen species and ferritin. J. Biol. Chem. 279, 13738–13745. doi: 10.1074/jbc.m310106200
Costa, V. M., Carvalho, F., Duarte, J. A., Bastos Mde, L., and Remiao, F. (2013). The heart as a target for xenobiotic toxicity: the cardiac susceptibility to oxidative stress. Chem. Res. Toxicol. 26, 1285–1311. doi: 10.1021/tx400130v
Cunha-Oliveira, T., Ferreira, L. L., Coelho, A. R., Deus, C. M., and Oliveira, P. J. (2018). Doxorubicin triggers bioenergetic failure and p53 activation in mouse stem cell-derived cardiomyocytes. Toxicol. Appl. Pharmacol. 348, 1–13. doi: 10.1016/j.taap.2018.04.009
Czabotar, P. E., Lessene, G., Strasser, A., and Adams, J. M. (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63. doi: 10.1038/nrm3722
Damiani, R. M., Moura, D. J., Viau, C. M., Brito, V., Moras, A. M., Henriques, J. P., et al. (2018). Influence of PARP-1 inhibition in the cardiotoxicity of the topoisomerase 2 inhibitors doxorubicin and mitoxantrone. Toxicol. In Vitro 52, 203–213. doi: 10.1016/j.tiv.2018.06.013
Deng, S., Kruger, A., Kleschyov, A. L., Kalinowski, L., Daiber, A., and Wojnowski, L. (2007). Gp91phox-containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Free Radic. Biol. Med. 42, 466–473. doi: 10.1016/j.freeradbiomed.2006.11.013
Dhingra, R., Margulets, V., Chowdhury, S. R., Thliveris, J., Jassal, D., Fernyhough, P., et al. (2014). Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Natl. Acad. Sci. U.S.A. 111, E5537–E5544.
Dimitrakis, P., Romay-Ogando, M. I., Timolati, F., Suter, T. M., and Zuppinger, C. (2012). Effects of doxorubicin cancer therapy on autophagy and the ubiquitin-proteasome system in long-term cultured adult rat cardiomyocytes. Cell Tissue Res. 350, 361–372. doi: 10.1007/s00441-012-1475-8
Doherty, K. R., Wappel, R. L., Talbert, D. R., Trusk, P. B., Moran, D. M., Kramer, J. W., et al. (2013). Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol. 272, 245–255. doi: 10.1016/j.taap.2013.04.027
Duran, J. M., Makarewich, C. A., Trappanese, D., Gross, P., Husain, S., Dunn, J., et al. (2014). Sorafenib cardiotoxicity increases mortality after myocardial infarction. Circ. Res. 114, 1700–1712. doi: 10.1161/circresaha.114.303200
El-Agamy, D. S., Elkablawy, M. A., and Abo-Haded, H. M. (2017). Modulation of cyclophosphamide-induced cardiotoxicity by methyl palmitate. Cancer Chemother. Pharmacol. 79, 399–409. doi: 10.1007/s00280-016-3233-1
El-Awady el, S. E., Moustafa, Y. M., Abo-Elmatty, D. M., and Radwan, A. (2011). Cisplatin-induced cardiotoxicity: mechanisms and cardioprotective strategies. Eur. J. Pharmacol 650, 335–341. doi: 10.1016/j.ejphar.2010.09.085
Eskandari, M. R., Moghaddam, F., Shahraki, J., and Pourahmad, J. (2015). A comparison of cardiomyocyte cytotoxic mechanisms for 5-fluorouracil and its pro-drug capecitabine. Xenobiotica 45, 79–87. doi: 10.3109/00498254.2014.942809
Ewer, M. S., and Ewer, S. M. (2015). Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 12, 547–558. doi: 10.1038/nrcardio.2015.65
Fabbi, P., Spallarossa, P., Garibaldi, S., Barisione, C., Mura, M., Altieri, P., et al. (2015). Doxorubicin impairs the insulin-like growth factor-1 system and causes insulin-like growth factor-1 resistance in cardiomyocytes. PLoS One 10:e0124643. doi: 10.1371/journal.pone.0124643
Fan, Y., Wang, C., Zhang, Y., Hang, P., Liu, Y., Pan, Z., et al. (2013). Genistein ameliorates adverse cardiac effects induced by arsenic trioxide through preventing cardiomyocytes apoptosis. Cell Physiol. Biochem. 31, 80–91. doi: 10.1159/000343351
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 116, 2672–2680. doi: 10.1073/pnas.1821022116
Fiala, M., Murphy, T., Macdougall, J., Yang, W., Luque, A., Iruela-Arispe, L., et al. (2004). HAART drugs induce mitochondrial damage and intercellular gaps and gp120 causes apoptosis. Cardiovasc. Toxicol. 4, 327–337.
Finck, B. N., and Kelly, D. P. (2007). Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 115, 2540–2548. doi: 10.1161/circulationaha.107.670588
Finkel, T. (2006). Cell biology: a clean energy programme. Nature 444, 151–152. doi: 10.1038/444151a
Focaccetti, C., Bruno, A., Magnani, E., Bartolini, D., Principi, E., Dallaglio, K., et al. (2015). Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS One 10:e0115686. doi: 10.1371/journal.pone.0115686
Fritsch, M., Gunther, S. D., Schwarzer, R., Albert, M. C., Schorn, F., Werthenbach, J. P., et al. (2019). Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575, 683–687. doi: 10.1038/s41586-019-1770-6
Fulbright, J. M., Egas-Bejar, D. E., Huh, W. W., and Chandra, J. (2015). Analysis of redox and apoptotic effects of anthracyclines to delineate a cardioprotective strategy. Cancer Chemother. Pharmacol. 76, 1297–1307. doi: 10.1007/s00280-015-2879-4
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death. Differ. 25, 486–541.
Gao, R. Y., Mukhopadhyay, P., Mohanraj, R., Wang, H., Horvath, B., Yin, S., et al. (2011). Resveratrol attenuates azidothymidine-induced cardiotoxicity by decreasing mitochondrial reactive oxygen species generation in human cardiomyocytes. Mol. Med. Rep. 4, 151–155.
Ghosh, J., Das, J., Manna, P., and Sil, P. C. (2009). Taurine prevents arsenic-induced cardiac oxidative stress and apoptotic damage: role of NF-kappa B, p38 and JNK MAPK pathway. Toxicol. Appl. Pharmacol. 240, 73–87. doi: 10.1016/j.taap.2009.07.008
Giulivi, C., Boveris, A., and Cadenas, E. (1995). Hydroxyl radical generation during mitochondrial electron transfer and the formation of 8-hydroxydesoxyguanosine in mitochondrial DNA. Arch. Biochem. Biophys. 316, 909–916. doi: 10.1006/abbi.1995.1122
Goyal, V., Bews, H., Cheung, D., Premecz, S., Mandal, S., Shaikh, B., et al. (2016). The Cardioprotective role of N-Acetyl cysteine amide in the prevention of doxorubicin and trastuzumab-mediated cardiac dysfunction. Can. J. Cardiol. 32, 1513–1519. doi: 10.1016/j.cjca.2016.06.002
Grazette, L. P., Boecker, W., Matsui, T., Semigran, M., Force, T. L., Hajjar, R. J., et al. (2004). Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. J. Am. Coll. Cardiol. 44, 2231–2238. doi: 10.1016/j.jacc.2004.08.066
Grootjans, S., Vanden Berghe, T., and Vandenabeele, P. (2017). Initiation and execution mechanisms of necroptosis: an overview. Cell Death. Differ. 24, 1184–1195. doi: 10.1038/cdd.2017.65
Gu, J., Hu, W., Song, Z. P., Chen, Y. G., Zhang, D. D., and Wang, C. Q. (2016). Resveratrol-induced autophagy promotes survival and attenuates doxorubicin-induced cardiotoxicity. Int. Immunopharmacol. 32, 1–7. doi: 10.1016/j.intimp.2016.01.002
He, H., Tao, H., Xiong, H., Duan, S. Z., Mcgowan, F. X., Mortensen, R. M., et al. (2014). Rosiglitazone causes cardiotoxicity via peroxisome proliferator-activated receptor gamma-independent mitochondrial oxidative stress in mouse hearts. Toxicol. Sci. 138, 468–481. doi: 10.1093/toxsci/kfu015
Herman, E. H., Zhang, J., Hasinoff, B. B., Clark, J. R., and Ferrans, V. J. (1997). Comparison of the structural changes induced by doxorubicin and mitoxantrone in the heart, kidney and intestine and characterization of the Fe(III)-mitoxantrone complex. J. Mol. Cell Cardiol. 29, 2415–2430. doi: 10.1006/jmcc.1997.0477
Herrmann, J., Yang, E. H., Iliescu, C. A., Cilingiroglu, M., Charitakis, K., Hakeem, A., et al. (2016). Vascular toxicities of cancer therapies: the old and the new–an evolving avenue. Circulation 133, 1272–1289. doi: 10.1161/circulationaha.115.018347
Holbro, T., and Hynes, N. E. (2004). ErbB receptors: directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 44, 195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440
Horie, T., Ono, K., Nishi, H., Nagao, K., Kinoshita, M., Watanabe, S., et al. (2010). Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc. Res. 87, 656–664. doi: 10.1093/cvr/cvq148
Hsu, P. L., and Mo, F. E. (2016). Matricellular protein CCN1 mediates doxorubicin-induced cardiomyopathy in mice. Oncotarget 7, 36698–36710. doi: 10.18632/oncotarget.9162
Hu, W., Lu, S., Mcalpine, I., Jamieson, J. D., Lee, D. U., Marroquin, L. D., et al. (2012). Mechanistic investigation of imatinib-induced cardiac toxicity and the involvement of c-Abl kinase. Toxicol. Sci. 129, 188–199. doi: 10.1093/toxsci/kfs192
Hutt, K. J. (2015). The role of BH3-only proteins in apoptosis within the ovary. Reproduction 149, R81–R89.
Imai, H., Matsuoka, M., Kumagai, T., Sakamoto, T., and Koumura, T. (2017). Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 403, 143–170. doi: 10.1007/82_2016_508
Jiang, J., Mohan, N., Endo, Y., Shen, Y., and Wu, W. J. (2018). Type IIB DNA topoisomerase is downregulated by trastuzumab and doxorubicin to synergize cardiotoxicity. Oncotarget 9, 6095–6108. doi: 10.18632/oncotarget.23543
Jing, X., Yang, J., Jiang, L., Chen, J., and Wang, H. (2018). MicroRNA-29b regulates the mitochondria-dependent apoptotic pathway by targeting bax in doxorubicin cardiotoxicity. Cell Physiol. Biochem. 48, 692–704. doi: 10.1159/000491896
Jorgensen, I., and Miao, E. A. (2015). Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265, 130–142. doi: 10.1111/imr.12287
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., and Kim, D. H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295.
Kaiser, W. J., Sridharan, H., Huang, C., Mandal, P., Upton, J. W., Gough, P. J., et al. (2013). Toll-like receptor 3-mediated necrosis via TRIF. RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279. doi: 10.1074/jbc.m113.462341
Kalivendi, S. V., Konorev, E. A., Cunningham, S., Vanamala, S. K., Kaji, E. H., Joseph, J., et al. (2005). Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem. J. 389, 527–539. doi: 10.1042/bj20050285
Kawaguchi, T., Takemura, G., Kanamori, H., Takeyama, T., Watanabe, T., Morishita, K., et al. (2012). Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc. Res. 96, 456–465. doi: 10.1093/cvr/cvs282
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–134.
Kim, Y., Ma, A. G., Kitta, K., Fitch, S. N., Ikeda, T., Ihara, Y., et al. (2003). Anthracycline-induced suppression of GATA-4 transcription factor: implication in the regulation of cardiac myocyte apoptosis. Mol. Pharmacol. 63, 368–377. doi: 10.1124/mol.63.2.368
Kimura, T., Uesugi, M., Takase, K., Miyamoto, N., and Sawada, K. (2017). Hsp90 inhibitor geldanamycin attenuates the cytotoxicity of sunitinib in cardiomyocytes via inhibition of the autophagy pathway. Toxicol. Appl. Pharmacol 329, 282–292. doi: 10.1016/j.taap.2017.06.015
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222.
Kobayashi, S., Volden, P., Timm, D., Mao, K., Xu, X., and Liang, Q. (2010). Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 285, 793–804. doi: 10.1074/jbc.m109.070037
Koleini, N., Nickel, B. E., Edel, A. L., Fandrich, R. R., Ravandi, A., and Kardami, E. (2019). Oxidized phospholipids in Doxorubicin-induced cardiotoxicity. Chem. Biol. Interact 303, 35–39. doi: 10.1016/j.cbi.2019.01.032
Kurauchi, K., Nishikawa, T., Miyahara, E., Okamoto, Y., and Kawano, Y. (2017). Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res Notes 10:406. doi: 10.1186/s13104-017-2726-2
Lakkur, S., Judd, S., Bostick, R. M., Mcclellan, W., Flanders, W. D., Stevens, V. L., et al. (2015). Oxidative stress, inflammation, and markers of cardiovascular health. Atherosclerosis 243, 38–43. doi: 10.1016/j.atherosclerosis.2015.08.032
Lamberti, M., Porto, S., Marra, M., Zappavigna, S., Grimaldi, A., Feola, D., et al. (2012). 5-Fluorouracil induces apoptosis in rat cardiocytes through intracellular oxidative stress. J. Exp. Clin. Cancer Res. 31:60. doi: 10.1186/1756-9966-31-60
Lan, Y., Wang, Y., Huang, K., and Zeng, Q. (2020). Heat shock protein 22 attenuates doxorubicin-induced cardiotoxicity via regulating inflammation and apoptosis. Front. Pharmacol. 11:257. doi: 10.3389/fphar.2020.00257
Lekes, D., Szadvari, I., Krizanova, O., Lopusna, K., Rezuchova, I., Novakova, M., et al. (2016). Nilotinib induces ER stress and cell death in H9c2 cells. Physiol. Res. 65, S505–S514.
Lemmens, K., Doggen, K., and De Keulenaer, G. W. (2007). Role of neuregulin-1/ErbB signaling in cardiovascular physiology and disease: implications for therapy of heart failure. Circulation 116, 954–960. doi: 10.1161/circulationaha.107.690487
Li, D. L., Wang, Z. V., Ding, G., Tan, W., Luo, X., Criollo, A., et al. (2016). Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133, 1668–1687. doi: 10.1161/circulationaha.115.017443
Li, S., Wang, W., Niu, T., Wang, H., Li, B., Shao, L., et al. (2014). Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid Med. Cell Longev 2014:748524.
Li, S., Zhao, H., Wang, Y., Shao, Y., Liu, J., and Xing, M. (2019). Arsenic-induced cardiotoxicity correlates with mitochondrial damage and trace elements imbalance in broiler chickens. Poult. Sci. 98, 734–744. doi: 10.3382/ps/pey469
Li, S., Zhao, H., Wang, Y., Shao, Y., Wang, B., Wang, Y., et al. (2018). Regulation of autophagy factors by oxidative stress and cardiac enzymes imbalance during arsenic or/and copper induced cardiotoxicity in Gallus gallus. Ecotoxicol. Environ. Saf. 148, 125–134. doi: 10.1016/j.ecoenv.2017.10.018
Li, W. J., Zhang, X. Y., Wu, R. T., Song, Y. H., and Xie, M. Y. (2018). Ganoderma atrum polysaccharide improves doxorubicin-induced cardiotoxicity in mice by regulation of apoptotic pathway in mitochondria. Carbohydr. Polym. 202, 581–590. doi: 10.1016/j.carbpol.2018.08.144
Li, S. W., Sun, X., He, Y., Guo, Y., Zhao, H. J., Hou, Z. J., et al. (2017). Assessment of arsenic trioxide in the heart of Gallus gallus: alterations of oxidative damage parameters, inflammatory cytokines, and cardiac enzymes. Environ. Sci. Pollut. Res. Int. 24, 5781–5790. doi: 10.1007/s11356-016-8223-7
Lim, C. C., Zuppinger, C., Guo, X., Kuster, G. M., Helmes, M., Eppenberger, H. M., et al. (2004). Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J. Biol. Chem. 279, 8290–8299. doi: 10.1074/jbc.m308033200
Lin, H., Stankov, M. V., Hegermann, J., Budida, R., Panayotova-Dimitrova, D., Schmidt, R. E., et al. (2019). Zidovudine-mediated autophagy inhibition enhances mitochondrial toxicity in muscle cells. Antimicrob. Agents Chemother 63:e01443-18. doi: 10.1128/AAC.01443-18
Lischke, J., Lang, C., Sawodny, O., and Feuer, R. (2015). Impairment of energy metabolism in cardiomyocytes caused by 5-FU catabolites can be compensated by administration of amino acids. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2015, 5363–5366.
Lu, L., Wu, W., Yan, J., Li, X., Yu, H., and Yu, X. (2009). Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int. J. Cardiol. 134, 82–90. doi: 10.1016/j.ijcard.2008.01.043
Ludke, A., Akolkar, G., Ayyappan, P., Sharma, A. K., and Singal, P. K. (2017). Time course of changes in oxidative stress and stress-induced proteins in cardiomyocytes exposed to doxorubicin and prevention by vitamin C. PLoS One 12:e0179452. doi: 10.1371/journal.pone.0179452
Luo, P., Zhu, Y., Chen, M., Yan, H., Yang, B., Yang, X., et al. (2018). HMGB1 contributes to adriamycin-induced cardiotoxicity via up-regulating autophagy. Toxicol. Lett. 292, 115–122. doi: 10.1016/j.toxlet.2018.04.034
Lushnikova, E. L., Nepomnyashchikh, L. M., Sviridov, E. A., and Klinnikova, M. G. (2008). Ultrastructural signs of cyclophosphamide-induced damage to cardiomyocytes. Bull. Exp. Biol. Med. 146, 366–371. doi: 10.1007/s10517-008-0287-z
Ma, H., Jones, K. R., Guo, R., Xu, P., Shen, Y., and Ren, J. (2010). Cisplatin compromises myocardial contractile function and mitochondrial ultrastructure: role of endoplasmic reticulum stress. Clin. Exp. Pharmacol. Physiol. 37, 460–465. doi: 10.1111/j.1440-1681.2009.05323.x
Maharsy, W., Aries, A., Mansour, O., Komati, H., and Nemer, M. (2014). Ageing is a risk factor in imatinib mesylate cardiotoxicity. Eur. J. Heart Fail. 16, 367–376. doi: 10.1002/ejhf.58
Malisza, K. L., and Hasinoff, B. B. (1995). Production of hydroxyl radical by iron(III)-anthraquinone complexes through self-reduction and through reductive activation by the xanthine oxidase/hypoxanthine system. Arch. Biochem. Biophys. 321, 51–60. doi: 10.1006/abbi.1995.1367
Man, S. M., Karki, R., and Kanneganti, T. D. (2017). Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75. doi: 10.1111/imr.12534
Meng, L., Lin, H., Zhang, J., Lin, N., Sun, Z., Gao, F., et al. (2019). Doxorubicin induces cardiomyocyte pyroptosis via the TINCR-mediated posttranscriptional stabilization of NLR family pyrin domain containing 3. J. Mol. Cell Cardiol. 136, 15–26. doi: 10.1016/j.yjmcc.2019.08.009
Miao, X., Tang, Z., Wang, Y., Su, G., Sun, W., Wei, W., et al. (2013). Metallothionein prevention of arsenic trioxide-induced cardiac cell death is associated with its inhibition of mitogen-activated protein kinases activation in vitro and in vivo. Toxicol. Lett. 220, 277–285. doi: 10.1016/j.toxlet.2013.04.025
Milano, G., Biemmi, V., Lazzarini, E., Balbi, C., Ciullo, A., Bolis, S., et al. (2020). Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovasc. Res. 116, 383–392.
Mirkes, P. E., and Little, S. A. (1998). Teratogen-induced cell death in postimplantation mouse embryos: differential tissue sensitivity and hallmarks of apoptosis. Cell Death. Differ. 5, 592–600. doi: 10.1038/sj.cdd.4400390
Mirkes, P. E., Wilson, K. L., and Cornel, L. M. (2000). Teratogen-induced activation of ERK, JNK, and p38 MAP kinases in early postimplantation murine embryos. Teratology 62, 14–25. doi: 10.1002/1096-9926(200007)62:1<14::aid-tera6>3.0.co;2-9
Mishra, P., Singh, S. V., Verma, A. K., Srivastava, P., Sultana, S., and Rath, S. K. (2014). Rosiglitazone induces cardiotoxicity by accelerated apoptosis. Cardiovasc. Toxicol. 14, 99–119. doi: 10.1007/s12012-013-9234-y
Mohan, N., Jiang, J., and Wu, W. J. (2017). Implications of autophagy and oxidative stress in trastuzumab-mediated cardiac toxicities. Austin. Pharmacol. Pharm. 2:1005.
Mohan, N., Shen, Y., Endo, Y., Elzarrad, M. K., and Wu, W. J. (2016). Trastuzumab, but not pertuzumab, dysregulates HER2 signaling to mediate inhibition of autophagy and increase in reactive oxygen species production in human cardiomyocytes. Mol. Cancer Ther. 15, 1321–1331. doi: 10.1158/1535-7163.mct-15-0741
Mukhopadhyay, P., Rajesh, M., Batkai, S., Kashiwaya, Y., Hasko, G., Liaudet, L., et al. (2009). Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 296, H1466–H1483.
Nagi, M. N., Al-Shabanah, O. A., Hafez, M. M., and Sayed-Ahmed, M. M. (2011). Thymoquinone supplementation attenuates cyclophosphamide-induced cardiotoxicity in rats. J. Biochem. Mol. Toxicol. 25, 135–142. doi: 10.1002/jbt.20369
Nakamura, T., Ueda, Y., Juan, Y., Katsuda, S., Takahashi, H., and Koh, E. (2000). Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: in vivo study. Circulation 102, 572–578. doi: 10.1161/01.cir.102.5.572
Nicol, M., Baudet, M., and Cohen-Solal, A. (2019). Subclinical left ventricular dysfunction during chemotherapy. Cardiac Fail. Rev. 5, 31–36.
Nishikawa, T., Miyahara, E., Kurauchi, K., Watanabe, E., Ikawa, K., Asaba, K., et al. (2015). Mechanisms of fatal cardiotoxicity following high-dose cyclophosphamide therapy and a method for its prevention. PLoS One 10:e0131394. doi: 10.1371/journal.pone.0131394
Omole, J. G., Ayoka, O. A., Alabi, Q. K., Adefisayo, M. A., Asafa, M. A., Olubunmi, B. O., et al. (2018). Protective effect of kolaviron on cyclophosphamide-induced cardiac toxicity in rats. J. Evid. Based Integr. Med. 23:2156587218757649.
Padegimas, A., Clasen, S., and Ky, B. (2020). Cardioprotective strategies to prevent breast cancer therapy-induced cardiotoxicity. Trends Cardiovasc. Med. 30, 22–28. doi: 10.1016/j.tcm.2019.01.006
Palee, S., Weerateerangkul, P., Chinda, K., Chattipakorn, S. C., and Chattipakorn, N. (2013). Mechanisms responsible for beneficial and adverse effects of rosiglitazone in a rat model of acute cardiac ischaemia-reperfusion. Exp. Physiol. 98, 1028–1037. doi: 10.1113/expphysiol.2012.070433
Papparella, I., Ceolotto, G., Berto, L., Cavalli, M., Bova, S., Cargnelli, G., et al. (2007). Vitamin C prevents zidovudine-induced NAD(P)H oxidase activation and hypertension in the rat. Cardiovasc. Res. 73, 432–438. doi: 10.1016/j.cardiores.2006.10.010
Patel, V. G., and Cornell, R. F. (2019). Cardiovascular complications associated with multiple myeloma therapies: incidence, pathophysiology, and management. Curr. Oncol. Rep. 21:29.
Pentassuglia, L., Timolati, F., Seifriz, F., Abudukadier, K., Suter, T. M., and Zuppinger, C. (2007). Inhibition of ErbB2/neuregulin signaling augments paclitaxel-induced cardiotoxicity in adult ventricular myocytes. Exp. Cell Res. 313, 1588–1601. doi: 10.1016/j.yexcr.2007.02.007
Peyssonnaux, C., Nizet, V., and Johnson, R. S. (2008). Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 7, 28–32. doi: 10.4161/cc.7.1.5145
Pistillucci, G., Ciorra, A. A., Sciacca, V., Raponi, M., Rossi, R., and Veltri, E. (2015). Troponin I and B-type natriuretic peptide (BNP) as biomarkers for the prediction of cardiotoxicity in patients with breast cancer treated with adjuvant anthracyclines and trastuzumab. Clin. Terapeutica 166, E67–E71.
Purevjav, E., Nelson, D. P., Varela, J. J., Jimenez, S., Kearney, D. L., Sanchez, X. V., et al. (2007). Myocardial fas ligand expression increases susceptibility to AZT-induced cardiomyopathy. Cardiovasc. Toxicol. 7, 255–263. doi: 10.1007/s12012-007-9004-9
Qian, P., Yan, L. J., Li, Y. Q., Yang, H. T., Duan, H. Y., Wu, J. T., et al. (2018). Cyanidin ameliorates cisplatin-induced cardiotoxicity via inhibition of ROS-mediated apoptosis. Exp. Ther. Med. 15, 1959–1965.
Raghu, K. G., and Cherian, O. L. (2009). Characterization of cytotoxicity induced by arsenic trioxide (a potent anti-APL drug) in rat cardiac myocytes. J. Trace Elem. Med. Biol. 23, 61–68. doi: 10.1016/j.jtemb.2008.10.001
Refaie, M. M. M., Shehata, S., El-Hussieny, M., Abdelraheem, W. M., and Bayoumi, A. M. A. (2020). Role of ATP-sensitive potassium channel (KATP) and eNOS in mediating the protective effect of nicorandil in cyclophosphamide-induced cardiotoxicity. Cardiovasc. Toxicol. 20, 71–81. doi: 10.1007/s12012-019-09535-8
Rharass, T., Gbankoto, A., Canal, C., Kursunluoglu, G., Bijoux, A., Panakova, D., et al. (2016). Oxidative stress does not play a primary role in the toxicity induced with clinical doses of doxorubicin in myocardial H9c2 cells. Mol. Cell. Biochem. 413, 199–215. doi: 10.1007/s11010-016-2653-x
Rohrbach, S., Muller-Werdan, U., Werdan, K., Koch, S., Gellerich, N. F., and Holtz, J. (2005). Apoptosis-modulating interaction of the neuregulin/erbB pathway with anthracyclines in regulating Bcl-xS and Bcl-xL in cardiomyocytes. J. Mol. Cell Cardiol. 38, 485–493. doi: 10.1016/j.yjmcc.2004.12.013
Roos, W. P., Thomas, A. D., and Kaina, B. (2016). DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 16, 20–33. doi: 10.1038/nrc.2015.2
Rossato, L. G., Costa, V. M., Vilas-Boas, V., De Lourdes Bastos, M., Rolo, A., Palmeira, C., et al. (2013). Therapeutic concentrations of mitoxantrone elicit energetic imbalance in H9c2 cells as an earlier event. Cardiovasc. Toxicol. 13, 413–425. doi: 10.1007/s12012-013-9224-0
Ry, G., Mukhopadhyay, P., Mohanraj, R., Wang, H., Horváth, B., and Yin, S. (2011). Resveratrol attenuates azidothymidine-induced cardiotoxicity by decreasing mitochondrial reactive oxygen species generation in human cardiomyocytes. Mol. Med. Rep. 4, 151–155.
Sawyer, D. B., Zuppinger, C., Miller, T. A., Eppenberger, H. M., and Suter, T. M. (2002). Modulation of anthracycline-induced myofibrillar disarray in rat ventricular myocytes by neuregulin-1beta and anti-erbB2: potential mechanism for trastuzumab-induced cardiotoxicity. Circulation 105, 1551–1554. doi: 10.1161/01.cir.0000013839.41224.1c
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Singh, A. P., Glennon, M. S., Umbarkar, P., Gupte, M., Galindo, C. L., Zhang, Q., et al. (2019). Ponatinib-induced cardiotoxicity: delineating the signalling mechanisms and potential rescue strategies. Cardiovasc. Res. 115, 966–977. doi: 10.1093/cvr/cvz006
Singh, K. K., Shukla, P. C., Quan, A., Lovren, F., Pan, Y., Wolfstadt, J. I., et al. (2011). Herceptin, a recombinant humanized anti-ERBB2 monoclonal antibody, induces cardiomyocyte death. Biochem. Biophys. Res. Commun. 411, 421–426. doi: 10.1016/j.bbrc.2011.06.169
Singla, D. K., Johnson, T. A., and Tavakoli Dargani, Z. (2019). Exosome treatment enhances anti-inflammatory M2 macrophages and reduces inflammation-induced pyroptosis in doxorubicin-induced cardiomyopathy. Cells 8:1224. doi: 10.3390/cells8101224
Soliman, A. F., Anees, L. M., and Ibrahim, D. M. (2018). Cardioprotective effect of zingerone against oxidative stress, inflammation, and apoptosis induced by cisplatin or gamma radiation in rats. Naunyn Schmiedebergs Arch. Pharmacol. 391, 819–832. doi: 10.1007/s00210-018-1506-4
Song, B. W., and Wang, L. (2013). [Necroptosis: a programmed cell necrosis]. Sheng Li Ke Xue Jin Zhan 44, 281–286.
Song, R., Yang, Y., Lei, H., Wang, G., Huang, Y., Xue, W., et al. (2018). HDAC6 inhibition protects cardiomyocytes against doxorubicin-induced acute damage by improving alpha-tubulin acetylation. J. Mol. Cell Cardiol. 124, 58–69. doi: 10.1016/j.yjmcc.2018.10.007
Spagnuolo, R. D., Recalcati, S., Tacchini, L., and Cairo, G. (2011). Role of hypoxia-inducible factors in the dexrazoxane-mediated protection of cardiomyocytes from doxorubicin-induced toxicity. Br. J. Pharmacol. 163, 299–312. doi: 10.1111/j.1476-5381.2011.01208.x
Sumneang, N., Siri-Angkul, N., Kumfu, S., Chattipakorn, S. C., and Chattipakorn, N. (2020). The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 680:108241. doi: 10.1016/j.abb.2019.108241
Sun, Z., Lu, W., Lin, N., Lin, H., Zhang, J., Ni, T., et al. (2020). Dihydromyricetin alleviates doxorubicin-induced cardiotoxicity by inhibiting NLRP3 inflammasome through activation of SIRT1. Biochem. Pharmacol. 175:113888. doi: 10.1016/j.bcp.2020.113888
Szabados, E., Fischer, G. M., Toth, K., Csete, B., Nemeti, B., Trombitas, K., et al. (1999). Role of reactive oxygen species and poly-ADP-ribose polymerase in the development of AZT-induced cardiomyopathy in rat. Free Radic. Biol. Med. 26, 309–317. doi: 10.1016/s0891-5849(98)00199-3
Tavakoli, R., Dargani, Z., and Singla, D. K. (2019). Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am. J. Physiol. Heart Circ. Physiol. 317, H460–H471.
Topal, I., Ozbek Bilgin, A., Keskin Cimen, F., Kurt, N., Suleyman, Z., Bilgin, Y., et al. (2018). The effect of rutin on cisplatin-induced oxidative cardiac damage in rats. Anatol. J. Cardiol. 20, 136–142.
Tsibiribi, P., Bui-Xuan, C., Bui-Xuan, B., Lombard-Bohas, C., Duperret, S., Belkhiria, M., et al. (2006). Cardiac lesions induced by 5-fluorouracil in the rabbit. Hum. Exp. Toxicol. 25, 305–309. doi: 10.1191/0960327106ht628oa
Tummers, B., and Green, D. R. (2017). Caspase-8: regulating life and death. Immunol. Rev. 277, 76–89. doi: 10.1111/imr.12541
Varghese, M. V., Abhilash, M., Paul, M. V., Alex, M., and Nair, R. H. (2017). Omega-3 fatty acid protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Cardiovasc. Toxicol. 17, 109–119. doi: 10.1007/s12012-016-9361-3
Vasquez-Vivar, J., Kalyanaraman, B., Martasek, P., Hogg, N., Masters, B. S., Karoui, H., et al. (1998). Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc. Natl. Acad. Sci. U.S.A. 95, 9220–9225. doi: 10.1073/pnas.95.16.9220
Vasquez-Vivar, J., Martasek, P., Hogg, N., Masters, B. S., Pritchard, K. A., and Kalyanaraman, B. (1997). Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry 36, 11293–11297. doi: 10.1021/bi971475e
Vejpongsa, P., and Yeh, E. T. (2014). Topoisomerase 2beta: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin. Pharmacol. Ther. 95, 45–52. doi: 10.1038/clpt.2013.201
Vineetha, V. P., Soumya, R. S., and Raghu, K. G. (2015). Phloretin ameliorates arsenic trioxide induced mitochondrial dysfunction in H9c2 cardiomyoblasts mediated via alterations in membrane permeability and ETC complexes. Eur. J. Pharmacol. 754, 162–172. doi: 10.1016/j.ejphar.2015.02.036
Voskens, C. J., Goldinger, S. M., Loquai, C., Robert, C., Kaehler, K. C., Berking, C., et al. (2013). The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One 8:e53745. doi: 10.1371/journal.pone.0053745
Wadia, S. (2015). Acute cyclophosphamide hemorrhagic myopericarditis: dilemma case report. Literature review and proposed diagnostic criteria. J. Clin. Diagn Res. 9, Oe01–Oe03.
Wang, J. C. (2002). Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 3, 430–440. doi: 10.1038/nrm831
Watanabe, M., Funakoshi, T., Unuma, K., Aki, T., and Uemura, K. (2014). Activation of the ubiquitin-proteasome system against arsenic trioxide cardiotoxicity involves ubiquitin ligase Parkin for mitochondrial homeostasis. Toxicology 322, 43–50. doi: 10.1016/j.tox.2014.04.008
Wu, C.-C., and Bratton, S. B. (2013). Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 19, 546–558. doi: 10.1089/ars.2012.4905
Wu, C. C., Li, Y. C., Wang, Y. R., Li, T. K., and Chan, N. L. (2013). On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res. 41, 10630–10640. doi: 10.1093/nar/gkt828
Xu, Y., He, B., Wang, Y. J., and Fu, Q. (2004). [Effect of herceptin combined with Doxorubicin on rat cardiotoxicity]. Ai Zheng 23, 367–371.
Xu, Z., Jin, Y., Yan, H., Gao, Z., Xu, B., Yang, B., et al. (2018). High-mobility group box 1 protein-mediated necroptosis contributes to dasatinib-induced cardiotoxicity. Toxicol. Lett. 296, 39–47. doi: 10.1016/j.toxlet.2018.08.003
Yamaoka, M., Yamaguchi, S., Suzuki, T., Okuyama, M., Nitobe, J., Nakamura, N., et al. (2000). Apoptosis in rat cardiac myocytes induced by Fas ligand: priming for Fas-mediated apoptosis with doxorubicin. J. Mol. Cell Cardiol. 32, 881–889. doi: 10.1006/jmcc.2000.1132
Yang, J. K. (2015). Death effecter domain for the assembly of death-inducing signaling complex. Apoptosis 20, 235–239. doi: 10.1007/s10495-014-1060-6
Yang, Q., Wen, L., Meng, Z., and Chen, Y. (2018). Blockage of endoplasmic reticulum stress attenuates nilotinib-induced cardiotoxicity by inhibition of the Akt-GSK3beta-Nox4 signaling. Eur. J. Pharmacol 822, 85–94. doi: 10.1016/j.ejphar.2018.01.011
Yang, S., Liu, J., Qu, C., Sun, J., Zhang, B. Q., Sun, Y. R., et al. (2017). [Potassium channels and autophagy]. Sheng Li Xue Bao 69, 509–514.
Yousif, N. G., and Al-Amran, F. G. (2011). Novel Toll-like receptor-4 deficiency attenuates trastuzumab (Herceptin) induced cardiac injury in mice. BMC Cardiovasc. Disord. 11:62. doi: 10.1186/1471-2261-11-62
Yu, J., Wang, C., Kong, Q., Wu, X., Lu, J. J., and Chen, X. (2018). Recent progress in doxorubicin-induced cardiotoxicity and protective potential of natural products. Phytomedicine 40, 125–139. doi: 10.1016/j.phymed.2018.01.009
Yu, W., Sun, H., Zha, W., Cui, W., Xu, L., Min, Q., et al. (2017). apigenin attenuates adriamycin-induced cardiomyocyte apoptosis via the PI3K/AKT/mTOR pathway. Evid Based Complement. Alternat. Med. 2017:2590676.
Yu, X., Ruan, Y., Huang, X., Dou, L., Lan, M., Cui, J., et al. (2020). Dexrazoxane ameliorates doxorubicin-induced cardiotoxicity by inhibiting both apoptosis and necroptosis in cardiomyocytes. Biochem. Biophys. Res. Commun. 523, 140–146. doi: 10.1016/j.bbrc.2019.12.027
Zeng, C., Duan, F., Hu, J., Luo, B., Huang, B., Lou, X., et al. (2020). NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 101523. doi: 10.1016/j.redox.2020.101523 [Epub ahead of print].
Zhai, J., Tao, L., Zhang, S., Gao, H., Zhang, Y., Sun, J., et al. (2020). Calycosin ameliorates doxorubicin-induced cardiotoxicity by suppressing oxidative stress and inflammation via the sirtuin 1-NOD-like receptor protein 3 pathway. Phytother. Res. 34, 649–659. doi: 10.1002/ptr.6557
Zhang, C., Feng, Y., Qu, S., Wei, X., Zhu, H., Luo, Q., et al. (2011). Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc. Res. 90, 538–545. doi: 10.1093/cvr/cvr022
Zhang, J. Y., Sun, G. B., Luo, Y., Wang, M., Wang, W., Du, Y. Y., et al. (2017). Salvianolic acid A protects H9c2 Cells from arsenic trioxide-induced injury via inhibition of the MAPK signaling pathway. Cell Physiol. Biochem. 41, 1957–1969. doi: 10.1159/000472409
Zhang, J. Y., Sun, G. B., Wang, M., Liao, P., Du, Y. Y., Yang, K., et al. (2016). Arsenic trioxide triggered calcium homeostasis imbalance and induced endoplasmic reticulum stress-mediated apoptosis in adult rat ventricular myocytes. Toxicol. Res. 5, 682–688. doi: 10.1039/c5tx00463b
Zhang, T., Zhang, Y., Cui, M., Jin, L., Wang, Y., Lv, F., et al. (2016). CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 22, 175–182. doi: 10.1038/nm.4017
Zhang, S., Liu, X., Bawa-Khalfe, T., Lu, L. S., Lyu, Y. L., Liu, L. F., et al. (2012). Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 18, 1639–1642. doi: 10.1038/nm.2919
Zhang, X., Zhu, J. X., Ma, Z. G., Wu, H. M., Xu, S. C., Song, P., et al. (2019). Rosmarinic acid alleviates cardiomyocyte apoptosis via cardiac fibroblast in doxorubicin-induced cardiotoxicity. Int. J. Biol. Sci. 15, 556–567. doi: 10.7150/ijbs.29907
Zhang, Y. Y., Meng, C., Zhang, X. M., Yuan, C. H., Wen, M. D., Chen, Z., et al. (2015). Ophiopogonin D attenuates doxorubicin-induced autophagic cell death by relieving mitochondrial damage in vitro and in vivo. J. Pharmacol. Exp. Ther. 352, 166–174. doi: 10.1124/jpet.114.219261
Zhao, H., Wang, Y., Liu, J., Guo, M., Fei, D., Yu, H., et al. (2019). The cardiotoxicity of the common carp (Cyprinus carpio) exposed to environmentally relevant concentrations of arsenic and subsequently relieved by zinc supplementation. Environ. Pollut. 253, 741–748. doi: 10.1016/j.envpol.2019.07.065
Zhao, H., Zhang, X., Zheng, Y., Li, Y., Wang, X., Hu, N., et al. (2018). Propofol protects rat cardiomyocytes from anthracycline-induced apoptosis by regulating MicroRNA-181a In vitro and in vivo. Oxid Med. Cell Longev 2018:2109216.
Zhao, L., and Zhang, B. (2017). Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 7:44735.
Zhao, X. Y., Li, G. Y., Liu, Y., Chai, L. M., Chen, J. X., Zhang, Y., et al. (2008). Resveratrol protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Br. J. Pharmacol. 154, 105–113. doi: 10.1038/bjp.2008.81
Zhao, Y., Xue, T., Yang, X., Zhu, H., Ding, X., Lou, L., et al. (2010). Autophagy plays an important role in sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol. Appl. Pharmacol. 248, 20–27. doi: 10.1016/j.taap.2010.07.007
Zheng, X., Zhong, T., Ma, Y., Wan, X., Qin, A., Yao, B., et al. (2020). Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. 242:117186. doi: 10.1016/j.lfs.2019.117186
Zhong, W., Jin, W., Xu, S., Wu, Y., Luo, S., Liang, M., et al. (2018). Pioglitazone induces cardiomyocyte apoptosis and inhibits cardiomyocyte hypertrophy Via VEGFR-2 signaling pathway. Arq. Bras. Cardiol. 111, 162–169.
Zhu, H., Sarkar, S., Scott, L., Danelisen, I., Trush, M. A., Jia, Z., et al. (2016). Doxorubicin redox biology: redox cycling, topoisomerase inhibition, and oxidative stress. React. Oxyg Species 1, 189–198. doi: 10.20455/ros.2016.835
Zhu, W., Soonpaa, M. H., Chen, H., Shen, W., Payne, R. M., Liechty, E. A., et al. (2009). Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119, 99–106. doi: 10.1161/circulationaha.108.799700
Keywords: cardiotoxicity, cardiomyocytes, cell death, apoptosis, autophagy, necrosis
Citation: Ma W, Wei S, Zhang B and Li W (2020) Molecular Mechanisms of Cardiomyocyte Death in Drug-Induced Cardiotoxicity. Front. Cell Dev. Biol. 8:434. doi: 10.3389/fcell.2020.00434
Received: 01 March 2020; Accepted: 08 May 2020;
Published: 03 June 2020.
Edited by:
Lawrence H. Boise, Emory University, United StatesReviewed by:
Darren Finlay, Sanford-Burnham Institute for Medical Research, United StatesChunying Li, Georgia State University, United States
Copyright © 2020 Ma, Wei, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bikui Zhang, NTA1OTk1QGNzdS5lZHUuY24=; Wenqun Li, bGl3cTEyMDRAY3N1LmVkdS5jbg==