 Georgia R. Kafer
Georgia R. Kafer Anthony J. Cesare
Anthony J. Cesare- Genome Integrity Unit, Children’s Medical Research Institute, The University of Sydney, Westmead, NSW, Australia
Murine development demands that pluripotent epiblast stem cells in the peri-implantation embryo increase from approximately 120 to 14,000 cells between embryonic days (E) 4.5 and E7.5. This is possible because epiblast stem cells can complete cell cycles in under 3 h in vivo. To ensure conceptus fitness, epiblast cells must undertake this proliferative feat while maintaining genome integrity. How epiblast cells maintain genome health under such an immense proliferation demand remains unclear. To illuminate the contribution of genome stability pathways to early mammalian development we systematically reviewed knockout mouse data from 347 DDR and repair associated genes. Cumulatively, the data indicate that while many DNA repair functions are dispensable in embryogenesis, genes encoding replication stress response and homology directed repair factors are essential specifically during the peri-implantation stage of early development. We discuss the significance of these findings in the context of the unique proliferative demands placed on pluripotent epiblast stem cells.
Introduction
Overview
Pluripotent cells in early mammalian embryos proliferate at a phenomenal rate. This is necessary to maintain embryo growth and reach critical developmental milestones within defined temporal windows. Because all somatic tissues are derived from these early pluripotent precursors, it is critical that genome integrity is maintained during early development. Embryonic pluripotent stem cells are thus subjected to unique challenges to maintain their DNA health. To elucidate which genome stability pathways are essential for early development we probed the Mouse Genome Informatics Gene Ontology Project (MGI-GO) database (Bult et al., 2019). Within MGI-GO we identified 347 genes grouped within the ontologies of major DNA repair pathways (MGI-GO designations: DNA damage checkpoint, nucleotide excision repair, mismatch repair, base excision repair, homologous recombination, and non-homologous end joining). Of these genes, we identified 297 with a validated mouse knockout. From these 297 murine models, only 108 gene knockouts were lethal during embryonic development (Supplementary Table S1). Within the grouping of 108 embryonic lethal genes, 10 knockouts were lethal during preimplantation development prior to E4.5 (Table 1), and 36 knockouts were lethal during somite stages from E8.5 (Supplementary Table S1). Notably, most of the targeted genes that conferred embryonic lethality, 62 genes, did so specifically during the period of rapid cell proliferation occurring with peri-implantation development (E4.5 to E8.5) (Table 2). Below we briefly review pre- and peri-implantation murine development before considering the function of essential genome stability factors across the early stages of embryonic development. Finally, we discuss why the unique cells of the peri-implantation embryo appear to specifically require replication stress response factors for cell viability.

Table 1. Genome stability factors essential for preimplantation development.
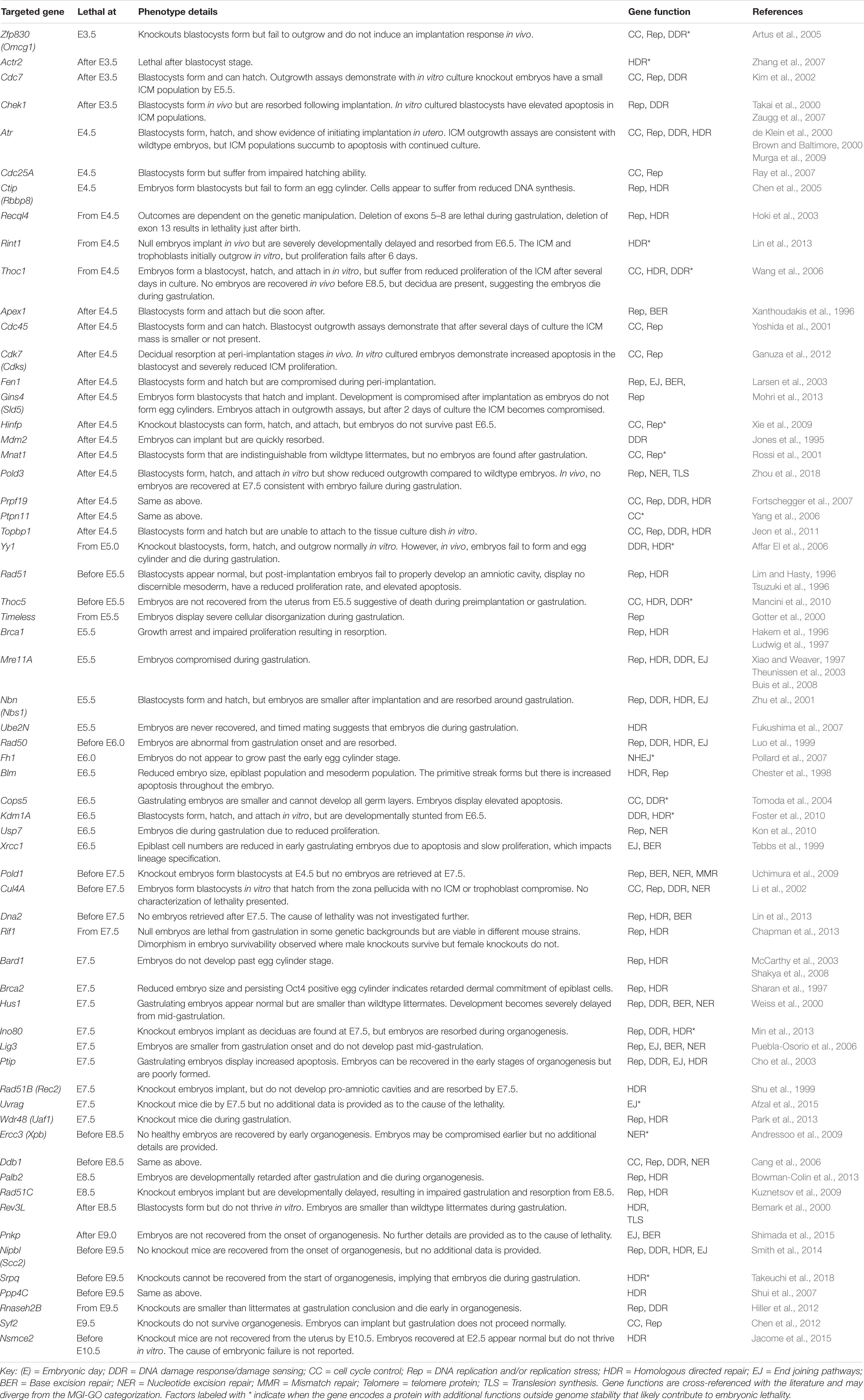
Table 2. Genome stability factors essential for peri-implantation development.
Early Murine Development
Embryonic development consists of a series of events occurring in chronological progression. Murine development takes 19 to 20 days depending on mouse strain (Figure 1; Murray et al., 2010). Preimplantation development occurs between fertilization (E0) and the initiation of embryo implantation in the uterine wall (around E4.0). Following fertilization, embryonic cells are uniformly totipotent and identical until formation of the morula at E2.5 (Condic, 2014) (see Box 1 for a detailed explanation of cell potency). Within the morula cells undergo polarization and compaction (Humięcka et al., 2017). By E3.0 the embryo forms a blastocyst structure containing two distinct cell populations; an outer layer of multipotent trophectoderm cells that eventually derive the placental tissues, and an inner group of pluripotent inner cell mass (ICM) cells which primarily serve as precursors for the embryo proper (Morris et al., 2010).
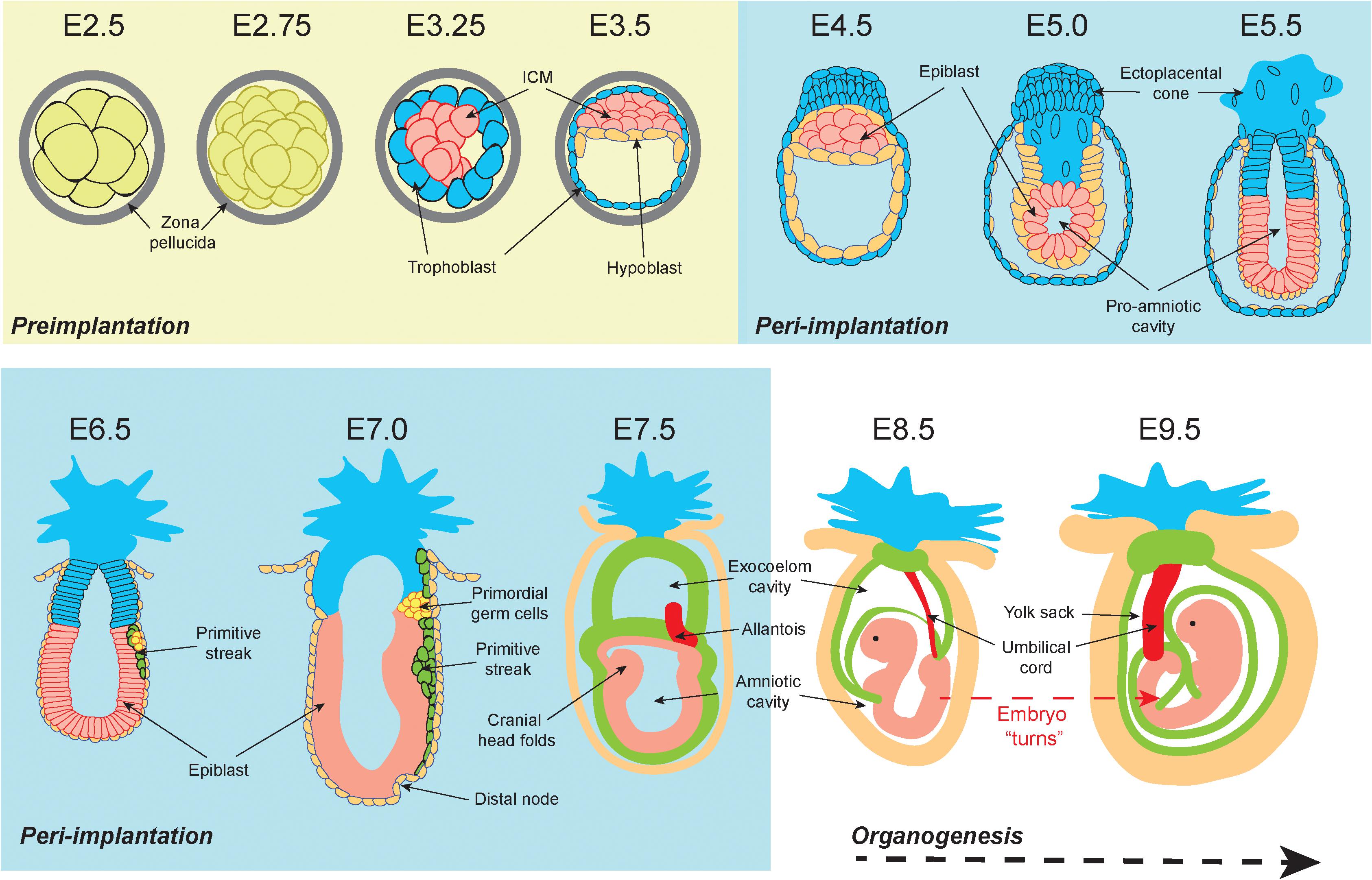
Figure 1. Graphical representation of the early stages of murine development. At embryonic day 2.5 (E2.5) totipotent cells within the preimplantation morula are encased within a protective zona pellucida shell. From E3.25 the blastocoel cavity opens, and the embryonic cells differentiate into the inner cell mass (ICM) (pink) and the outer trophoblast (blue). By E3.5 the hypoblast (orange) has developed. During peri-implantation the zona pellucida shell is lost by E4.5 and the ICM are now termed “epiblast cells” (pink). The outer trophoblast cell layer continues to encapsulate the embryo and expands to form the ecto-placental cone that is the precursor for placental tissues. From E5.0 a pro-amniotic cavity opens and elongates. By E6.5 the embryo has implanted, the primitive streak (green) emerges, and the primordial germ cell populations form (yellow). The primitive streak continues to elongate and is accompanied by the formation of the distal node at E7.0. At E7.5 the primitive streak derived mesoderm extends around the embryo and divides the pro-amniotic cavity into the exocoelom cavity and the amniotic cavity. At E8.5 the exocoelom becomes the yolk sack. Between E8.5 and E9.5 the embryo turns, resulting in complete envelopment of the embryo within the supporting extraembryonic tissues. Embryo depictions are not to scale.
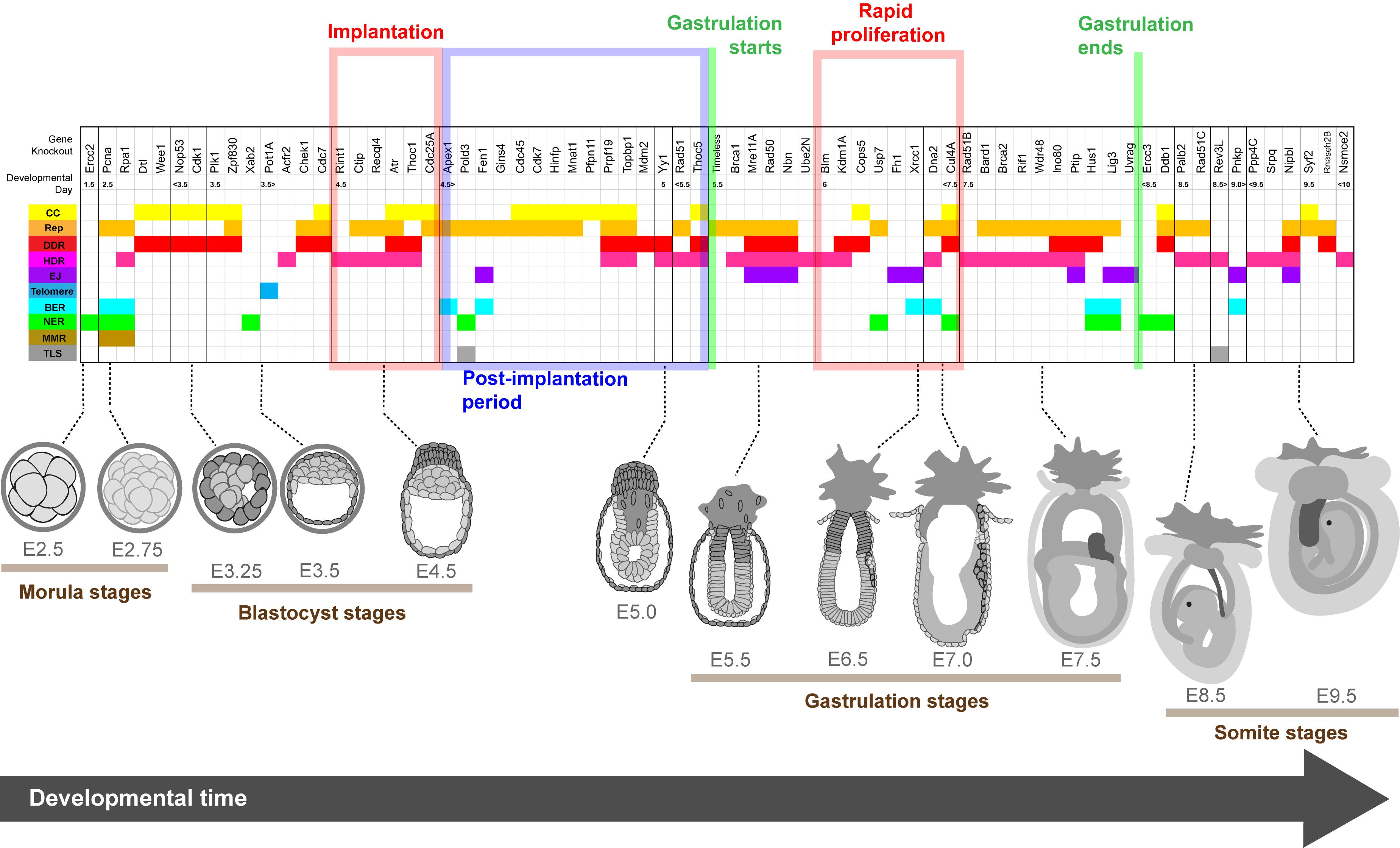
Figure 2. Lethality onset in embryos deleted for genome stability factors relative to developmental stage. Spatiotemporal heat map illustrating the timing of lethality for embryos lacking genome stability factors essential for early development. Genes are listed above and organized temporally relative to the reported timing of embryo compromise. Graphical depiction of normal embryo structure at the time of lethality is shown below and key developmental periods are highlighted. Gene functions according to the literature are color coded as follows: cell cycle regulation (CC, yellow); DNA replication and/or the replication stress response (Rep, orange); DNA damage response (DDR, red); Homology directed repair (HDR, pink); canonical and alternative non-homologous end joining (EJ, purple); telomere biology (Telomere, blue); base excision repair (BER, cyan); nucleotide excision repair (NER, green); mismatch repair (MMR, brown) and translesion synthesis (TLS, gray). Embryo depictions are not to scale.
Box 1.
Potency states. Totipotent cells are present in the embryo between fertilization and morula formation. They can give rise to any cell type from any stage of the animal’s life, including germ and placental cells. Pluripotent cells exist in the inner cell mass (ICM) and epiblast region of the developing embryo from the blastocyst stage to immediately prior to organogenesis. Pluripotent cells can give rise to any cell of dermal lineage (mesoderm, endoderm or ectoderm) but not placental or germ cells. Multipotent cells exist in newly formed tissues or organs and can develop into a limited number of cell types within their original dermal lineage.
Peri-implantation is the developmental period from implantation to organogenesis (E4.0 to E8.5). Implantation begins at E4.0 when the free-floating embryo contains 64 cells (Behringer et al., 2014). At this point, the embryo loses its glycoprotein zona pellucida shell and the outer trophoblast attaches to the uterine wall. Single cell transcriptomics demonstrate that at E4.0 the ICM has differentiated into two pluripotent cell types: the primitive endoderm (PrE, sometimes referred to as the hypoblast) and the epiblast (Mohammed et al., 2017). Peri-implantation development is associated with exceptionally rapid cell proliferation during “gastrulation,” which begins at E5.5 as the embryo elongates and a luminal pro-amniotic cavity opens in the center of the epiblast cell mass (Snow, 1976). At E6.5 the primitive streak emerges from the epiblast, and from the epiblast all three dermal lineages of the conceptus will arise (Tam et al., 1993). These are the ectoderm (nervous system and skin precursor), endoderm (gut precursor), and mesoderm (precursor for all other tissues) (Watson and Tam, 2001). Because continued gastrulation requires a critical mass of 1000 cells, delays in epiblast proliferation at E6.5 may result in the embryo forming a small primitive streak but developing no further (Snow and Tam, 1979; Tam, 1989; Power and Tam, 1993).
Between E6.5 and E7.5 the rate of epiblast proliferation is exceptionally pronounced, with some cell cycles completed in 2.2 h (Snow, 1977). Elongation of the primitive streak between E6.5 and E7.0 is associated with the first evidence of epithelial to mesenchymal transition (EMT) (Carver et al., 2001). EMT mobilizes epiblast cells to form the endoderm and mesoderm lineages, while cells that do not undergo EMT become the ectoderm (Acloque et al., 2009). At E7.5 the mouse embryo contains over 14,000 cells derived from epiblast progenitors. Proliferation slows at this point resulting in an increased average cell cycle duration of 8.1 h (Snow, 1977). There is evidence to suggest a that a specialized “proliferative zone” with sub 4-h cell cycles is maintained within the primitive streak after E7.5, however, definitive evidence remains to be substantiated (Tam et al., 2013).
Genome Instability Differentially Impacts Cell Viability in Pre- and Peri-Implantation Embryos
Embryonic lethality implies that one or more cell populations are compromised during development at the time of embryo failure. This may arise through DNA damage or genome instability, with multiple lines of evidence indicating that preimplantation embryos are more resistant to genome perturbations than peri-implantation embryos. For example, gamma irradiation-induced DNA damage at E6.5 or E7.5 confers apoptosis specifically within the epiblast (Heyer et al., 2000). Conversely, apoptosis levels are low in preimplantation embryos irradiated between E3.5 and E5.5, or during organogenesis from E8.5. It is therefore the epiblast that is particularly sensitive to these types of DNA lesions. Similarly, despite tetraploidy driving additional chromosomal instability in preimplantation cells (Paim and FitzHarris, 2019) chimeric mouse embryos with a mixture of diploid and tetraploid cells will develop through preimplantation before dying during peri-implantation (Horii et al., 2015). Notably, tetraploid sensitivity in vivo appears to be specific to the epiblast as embryos with tetraploid trophoblast cells and diploid epiblast cells can generate live pups (Wen et al., 2017). Mouse embryos containing a mixture of diploid and aneuploid cells will also develop to peri-implantation before the aneuploid cells are specifically depleted in the epiblast through apoptosis (Bolton et al., 2016).
As with somatic tissues, the tumor suppressor TP53 (p53) plays a central role regulating stem cell outcomes following genomic insult. p53 orchestrates growth arrest or apoptosis following activation of the DNA damage response (Mello and Attardi, 2018). Concordantly, inhibiting p53-dependant signaling pathways enables chimeric embryos made from tetraploid preimplantation murine embryonic stem cells (mESCs) to survive until birth (Horii et al., 2015). Deleting TP53 also reduced apoptosis levels in irradiated E6.5 embryos (Heyer et al., 2000) and extended the survival of embryos co-deleted for essential DNA repair factors (Jones et al., 1995; Haupt et al., 1997; Ludwig et al., 1997; Kim et al., 2002; McCarthy et al., 2003; Cang et al., 2006; Reinhardt and Schumacher, 2012). Not surprisingly, TP53 was identified as a critical mediator of apoptosis in the gastrulating epiblast (Laurent and Blasi, 2015). However, when activated in pluripotent stem cells, p53 also influences the expression of pluripotency factors to regulate differentiation (Lin et al., 2005; Li et al., 2012; Akdemir et al., 2014; Jain et al., 2016). p53 therefore functions through canonical and unique pathways in early development to regulate cellular outcomes. This highlights that our classic understanding of genome stability pathways may not strictly apply to early development or certain pluripotent cell types (Zaveri and Dhawan, 2018).
DNA Damage Response and Repair Pathways
Replication Stress Response
Somatic mammalian cells prepare for DNA replication in G1 phase by licensing replication origins and loading inactive Cdc45-MCM-GINS replicative helicase complexes (Bleichert, 2019; Miller et al., 2019). Cyclin dependent kinase activity promotes E2F transactivation to initiate replication at the G1/S transition (Kent and Leone, 2019). Replication then proceeds throughout the S-phase with origins firing in temporal coordination and DNA synthesis occurring across the entirety of the genome (Burgers and Kunkel, 2017; Limas and Cook, 2019). Intrinsic and extrinsic factors may disrupt replication fork processivity: a phenomenon known as “replication stress” (Zeman and Cimprich, 2014). Replication stress is sensed through the accumulation of RPA binding to its single strand DNA (ssDNA) substrate (Bhat and Cortez, 2018). When replication stress stalls DNA synthesis the replicative helicase continues to unwind its substrate exposing ssDNA for RPA coating (Byun et al., 2005). ATR kinase is the master regulator of the replication stress response (Saldivar et al., 2017). RPA coated ssDNA recruits ATR and its associated protein ATRIP (Cortez et al., 2001) to stalled replication forks through parallel pathways mediated by TopBP1 and ETAA1 (Kumagai et al., 2006; Bass et al., 2016; Haahr et al., 2016). Once localized to the stalled fork, ATR is activated and propagates a signaling cascade resulting in engagement of the replication stress response. This includes activation of the downstream effector CHK1 kinase to arrest S phase until replication stress is resolved (Zhang and Hunter, 2014). During the replication stress response, stalled replication forks are often remodeled into a four-way structure and protected before engaging one of many diverse repair mechanisms dependent upon the underlying stress the fork encountered (Quinet et al., 2017; Cortez, 2019).
If replicative stress is unresolved, arrested replication forks may collapse into one-ended double strand breaks (DSBs) (Ait Saada et al., 2018). Additionally, persistent replication stress can result in under-replicated DNA persisting through S-phase, the second growth (G2) phase, and into the mitotic (M) phase of the cell cycle (Mankouri et al., 2013). Specialized repair mechanisms address replication defects carried into mitosis (Minocherhomji et al., 2015), during which time the canonical DSB repair pathways are inhibited (Orthwein et al., 2014). Replication defects passed into mitosis can confer chromosome segregation errors resulting in aneuploidy (Burrell et al., 2013; Wilhelm et al., 2019), or if severe mitotic death (Masamsetti et al., 2019). If a replication stressed cell escapes mitosis this is often evident in the daughter cells where the under-replicated DNA is present as a scar in the first growth phase (G1) that is repaired in the subsequent S phase (Lukas et al., 2011; Spies et al., 2019).
Double Strand Break Repair
DSBs are a major threat to genome stability because a failure in repair may result in loss of an entire chromosome arm (Scully et al., 2019). Additionally, chromosome segregation errors resulting from inadequate DSB repair are rife with implications for genome instability, including chromothripsis and kataegis (Maciejowski et al., 2015; Ly and Cleveland, 2017). DSBs are sensed by Ku70/80 (Gottlieb and Jackson, 1993; Ochi et al., 2015), PARP1 (Ali et al., 2012; Liu C. et al., 2017), and the MRN (MRE11, RAD50 and NBS1) complex (Lee and Paull, 2005; Stracker and Petrini, 2011). MRN facilitates recruitment of the ATM kinase to DSBs (Uziel et al., 2003) while Ku70/80 recruits a related kinase DNA-PKcs (Hnizda and Blundell, 2019). Once recruited to the break, ATM is activated and engages downstream DDR pathways (Dupre et al., 2006; Maréchal and Zou, 2013). This includes phosphorylation of the histone variant H2AX (termed γ-H2AX when phosphorylated) within the break adjacent chromatin and assembly of factors at the break locus to facilitate repair (Rogakou et al., 1998). ATM activation also results in growth arrest, including activation of the p53 pathway (Banin et al., 1998). Subsequent repair of DNA breaks is then orchestrated by homology directed repair (HDR) or end-joining (EJ) pathways.
Homology directed repair utilizes the homologous sister chromatid as a template for repair and is thus limited to the S and G2 cell cycle phases (Hustedt and Durocher, 2016). HDR initiates with resection at the broken DNA end to provide a 3′ ssDNA overhang for insertion into homologous regions of the sister chromatid (Symington, 2016). Once resection occurs at a canonical two-ended DSB, a series of enzymatic steps facilitate strand-invasion of the broken end into the sister chromatid, formation of a displacement-loop, template copying from the invaded DNA end, potential formation of a “Holliday Junction,” and resolution of strand invasion (Scully et al., 2019). HDR factors also function within the replication stress response though diverse mechanisms including the remodeling and stabilization of stressed replication forks into four-way structures prior to repair (Neelsen and Lopes, 2015). Additionally, HDR factors participate in resolving one-ended DSBs that arise from collapsed replication forks to restart replication (Ait Saada et al., 2018). HDR is thus intrinsically linked to the replication stress response.
Conversely, EJ involves the covalent ligation of broken DNA ends to repair DSBs and thus does not require an additional chromatid template. End-joining mechanisms function in S and G2, but notably are the only DSB repair pathway available in G1 (Chang et al., 2017). Classical non-homologous end joining (c-NHEJ) directly ligates DNA ends, and if unregulated can also drive chromosome translocations (Ghezraoui et al., 2014) or telomere fusions (Celli et al., 2006; Van Ly et al., 2018). Cells can also engage alternative non-homologous end joining (alt-NHEJ), where DNA resection creates 3′ overhangs to align the DNA ends for repair through microhomology (Sfeir and Symington, 2015). This often requires a fill-in reaction by the error prone polymerase Pol θ which can introduce errors into the repaired sequence (Mateos-Gomez et al., 2015; Mateos-Gomez et al., 2017).
Nucleotide and Base Damage
In addition to DNA breaks, cells must also contend with damage to nucleotide bases, base mismatches, and bulky DNA lesions that distort the DNA double helix. Base excision repair (BER) mends small non-distorting DNA lesions such as oxidized DNA bases (Wallace, 2014), while nucleotide excision repair (NER) corrects bulky lesions such UV-induced pyrimidine dimers (Schärer, 2013). BER is mediated by DNA glycosylases that cleave and remove the damaged bases at the lesion. A correct base is then inserted by specialized DNA polymerases (Wallace, 2014). In NER, a 25–30 nucleotide patch of ssDNA containing the bulky lesion is excised from the double strand helix before specialized DNA polymerases fill in the ssDNA gap (Schärer, 2013). Mismatch repair (MMR) proofreads the newly replicated DNA to identify mis-incorporated bases (Liu D. et al., 2017). When a mismatch is identified, the newly synthesized strand is nicked and resected creating a patch of exposed ssDNA for fill in by a high-fidelity DNA polymerase.
Essentiality of DNA Repair During Embryogenesis
Not all DNA repair activities are essential for early development (Supplementary Table S1). The most striking example is the dichotomy between the major DSB repair pathways. While many HDR factors are essential, the core c-NHEJ factors are either dispensable for development [Ku70 (Ouyang et al., 1997), Ku80 (Nussenzweig et al., 1996; Gu et al., 1997), Prkdc (DNA-PKcs) (Kurimasa et al., 1999), Nhej1 (XLF) (Li et al., 2008)] or only required for organogenesis [Lig4 (Frank et al., 1998), Xrcc4 (Dickinson et al., 2016)]. Some alt-NHEJ components are required for early development. Alt-NHEJ is promoted by the polymerase Pol θ which primes the DNA ends for repair by removing RPA from exposed ssDNA regions (Kent et al., 2015; Mateos-Gomez et al., 2017). Alt-NHEJ is also dependent on PARP-1, XRCC1 and LIG3 (Audebert et al., 2004), with PARP-1 having a critical role in synapsis, and XRCC1 and LIG3 mediating DNA ligation (Audebert et al., 2004). While Lig3 and Xrcc1 knockout embryos die during gastrulation (Tebbs et al., 1999; Puebla-Osorio et al., 2006), both genes encode factors that have functions in other DNA metabolic activities outside EJ (Wallace, 2014). Conversely Pol θ knockout mice are viable (Shima et al., 2004; Masuda et al., 2005), as are Parp1 knockouts (Wang et al., 1995), suggesting that alt-NHEJ is not essential for embryogenesis.
Preference for HDR over NHEJ is further evident in mechanistic studies of DDR engagement within embryonic stem cells (Tichy et al., 2010). Following DSB induction, ATM-dependent γ-H2AX phosphorylation at the break locus recruits the scaffolding protein MDC1 (Stewart et al., 2003). MDC1 subsequently recruits the ubiquitin ligases RNF8 and RNF168 that signal downstream recruitment of 53BP1 (Jackson and Durocher, 2013). 53BP1 then establishes a physical domain at the repair locus in coordination with RIF1 to inhibit end resection and HDR in favor of c-NHEJ (Chapman et al., 2013; Zimmermann et al., 2013; Lou et al., 2019; Ochs et al., 2019). Notably, many factors that promote c-NHEJ are dispensable for embryonic development, including Atm (Barlow et al., 1996; Xu et al., 1996), H2ax (Celeste et al., 2002), Mdc1 (Lou et al., 2006), Rnf8 (Valnegri et al., 2017), Rfn168 (Zong et al., 2019), and Trp53bp1 (53BP1) (Ward et al., 2003). Additionally, multiple reports demonstrate that 53BP1 does not localize to DSB foci in the ICM of preimplantation embryos or mESCs grown in culture (Ziegler-Birling et al., 2009; Kafer et al., 2016). In vivo, 53BP1 localizes to damage in E5.5 embryos, but only in the epiblast cells and not the endoderm or trophoblast cells (Laurent and Blasi, 2015). While Trp53bp1 expression is unchanged in early development, Rnf168 expression is limited prior to the epiblast stage which may explain differential engagement of DDR pathways in pre- and peri-implantation cells (Laurent and Blasi, 2015).
Genes Essential for Preimplantation Development
Consistent with a greater tolerance of genome instability in preimplantation cells, only 10 knockouts of genome stability factors that we reviewed were lethal during preimplantation (Table 1 and Supplementary Table S1). Factors encoded by many of these preimplantation essential genes also possessed critical functions outside DDR or repair. Additionally, preimplantation embryos commonly survive to the morula stage even if a targeted gene is essential for viability. This is because embryonic genomes are not immediately active following fertilization while the inherited maternal mRNA guides protein translation (Jukam et al., 2017).
Multifunctional Factors Essential for Preimplantation Development
Of the 10 genome stability factors required for preimplantation development, only Ercc2 deletion did not permit morula generation. Ercc2–/– embryos fail to develop past the 2-cell stage in vitro and there is no evidence of embryo implantation in vivo (de Boer et al., 1998). Ercc2 encodes the XPD helicase that functions both in NER and as a core component of the general transcription factor IIH (TFIIH) complex. TFIIH opens DNA and anchors the cdk-activating kinase (CAK) complex to facilitate transcription (Egly, 2001). Given that Ercc2 knockouts are compromised during embryonic genome activation (EGA), a reasonable conclusion is that preimplantation lethality is associated with a loss of XPD transcriptional activity. Likewise, Xab2 is a TFIIH and NER component and Xab2 deletion is lethal prior to the blastocyst stage (Yonemasu et al., 2005). However, Xab2 null embryos survive past the 2-cell stage suggesting Xab2 is not required for EGA.
Pcna and Rpa1 both function in multiple genome maintenance pathways and deleting either gene confers preimplantation lethality (Wang et al., 2005; Roa et al., 2008). PCNA (proliferating cell nuclear antigen) forms a homotrimeric sliding clamp that encircles DNA. The protein clamp travels with the replication fork to promote replicative polymerase processivity and mediate diverse functions in DNA replication and the replication stress response (Mailand et al., 2013). PCNA also functions in NER and MMR (Strzalka and Ziemienowicz, 2011). Rpa1 (Replication Protein A 1) codes for the 70 kDa subunit of the heterotrimeric RPA ssDNA binding complex that participates in replication, but which also functions in ATR activation, replication fork repair, HDR and NER (Bhat and Cortez, 2018). Analysis of Pcna mRNA showed a significant reduction of maternal transcripts from the zygote to the 2-cell stage consistent with the observed early embryonic lethality (Hamatani et al., 2004; Roa et al., 2008; Bult et al., 2019). Rpa1 null embryos form blastocysts at E3.5 that are smaller than their heterozygous littermates and which fail in blastocyst outgrowth assays, indicating Rpa1 is required for trophectoderm and ICM growth (Wang et al., 2005). While both Rpa1 and Pcna function in diverse aspects of genome maintenance, both genes are also required for DNA replication. Preimplantation failure of Pcna or Rpa1 null embryos likely stems from an inability to polymerize nascent DNA coupled with simultaneous attenuation of multiple DNA repair pathways.
Two additional genes classified as DNA repair factors in the MGI-GO database are also essential during preimplantation. Nop53 encodes a nucleolar protein with suggested roles regulating p53, DNA repair, and the cellular response to mitochondrial stress (Lee et al., 2012; Yoon et al., 2014). Nop53 null embryos fail to develop past the blastocyst stage, while Nop53–/– mESCs are viable, suggesting that Nop53 functions in morula to blastocyst maturation (Sasaki et al., 2011). In somatic cells, Pot1a encodes a telomere specific ssDNA binding protein that suppresses ATR and p53 activation by the chromosome end (Hockemeyer et al., 2005, 2006; Denchi and de Lange, 2007). Loss of Pot1a likely confers robust ATR activation which explains the failure of Pot1a–/– embryos to form an ICM (Hockemeyer et al., 2006).
Cell Cycle and Checkpoint Factors Essential for Preimplantation Viability
In the event of replication stress or DSBs, checkpoints slow the cell cycle to provide time for DNA repair. Deletion of several genes that encode checkpoint regulatory factors confer preimplantation embryo failure at the morula stage. These include Wee1 (Tominaga et al., 2006), Cdk1 (Diril et al., 2012), and Plk1 (Lu et al., 2008; Wachowicz et al., 2016). CDK1 promotes the G2/M transition through interaction with cyclin B, and CDK1-cyclin B activity is regulated by WEE1 (Harvey et al., 2005; Gavet and Pines, 2010). ATR and CHK1 activate the G2/M checkpoint by stimulating WEE1 to inhibit CDK1 (O’Connell et al., 1997; Nigg, 2001). Polo-like kinase 1 (PLK1) controls numerous mitotic activities, participates in the G2/M transition, may function in DNA replication, and is implicated in HDR through phosphorylation of RAD51 (Watanabe et al., 2004; Mandal and Strebhardt, 2013; Yata et al., 2014; Pintard and Archambault, 2018). Dtl is also required for preimplantation development (Liu et al., 2007). Dtl encodes a component of the CUL4A-DDB1 E3-ubiquitin ligase complex that degrades the replication licensing factor CDT1 to stimulate replication and S phase progression (Nishitani et al., 2001; Li and Blow, 2005). Because these essential cell cycle genes function in DDR-regulated checkpoints and normal cell cycle transitions, the mechanism of embryonic lethality following their deletion likely involves deregulation of both the DDR and normal cell cycles.
Genes Essential for Peri-Implantation Development
Peri-implantation is the period of embryonic development most associated with lethality following deletion of genome stability factors. Within the MGI-GO derived gene list, it stands out that 54 of the 62 genes essential for peri-implantation development have direct or indirect functions in DNA replication, the replication stress response, and/or HDR (Table 2 and Supplementary Table S1). Below we focus specifically on these pathways.
Genes Regulating Replication Initiation, DNA Polymerization, and the Replication Stress Response Are Essential for Peri-Implantation Development
Because of the rate of cell proliferation in peri-implantation embryos, it is perhaps unsurprising that factors which regulate the G1/S transition and initiate DNA replication are essential for peri-implantation development. This includes Cdc7 (Kim et al., 2002), Cdc45 (Yoshida et al., 2001), Cdk7 (Ganuza et al., 2012), Mnat1 (Rossi et al., 2001), Gins4 (Mohri et al., 2013), Cul4A (Li et al., 2002), and Ddb1 (Cang et al., 2006). Additionally, the Pold3 subunit of the replicative polymerase Polδ is required for peri-implantation development (Uchimura et al., 2009; Zhou et al., 2018), as is Fen1, which plays a central role in processing Okazaki fragments on the lagging strand of DNA synthesis (Larsen et al., 2003). Lethality following deletion of most of these genes occurs on or near E4.5 and is associated with proliferative failure within the ICM after blastocyst formation but prior to gastrulation. A likely explanation is that peri-implantation development is rapidly compromised when fundamental functions necessary for cellular proliferation and/or DNA replication are dysregulated.
Even in the absence of exogenous threats to replication, the exceptionally fast cell cycles within the developing epiblast are expected to confer intrinsic replication stress. For example, blastocyst derived mESCs display replication stress in unperturbed cultures in vitro (Ahuja et al., 2016). mESCs appear to manage this replication stress by effectively coupling replication and repair activities to facilitate near continuous DNA synthesis punctuated by brief G1 and M phases (Ahuja et al., 2016). Epiblast cell cycles in vivo are more rapid than cell cycles in cultured mESCs (Snow, 1977; Mohammed et al., 2017) suggesting that a similar reliance on efficient coupling of DNA replication and the repair activity is required to manage replication stress in peri-implantation embryos.
The replication stress response begins when ATR/ATRIP localizes to RPA-coated ssDNA, and ATR is activated through TOPBP1 and the RAD9-RAD1-HUS1 (9-1-1) protein complex (Kumagai et al., 2006; Navadgi-Patil and Burgers, 2009; Choi et al., 2010). 9-1-1 is a sliding clamp loaded onto DNA (Bermudez et al., 2003). TOPBP1 bridges ATR/ATRIP and 9-1-1, thereby stabilizing ATR at the fork. Once stabilized at the site of replication stress, ATR regulates downstream replication stress repair and cell cycle arrest via CHK1 kinase and CDC25A (Liu et al., 2000; Xiao et al., 2003; Delacroix et al., 2007). TIMELESS, a component of the replication fork protection complex (Kemp et al., 2010; Couch et al., 2013; Buisson et al., 2017), and the ubiquitin ligase PRP19 (Prpf19) (Maréchal et al., 2014), also function in ATR and CHK1 activation. Embryos null for Atr (Brown and Baltimore, 2000; de Klein et al., 2000; Murga et al., 2009), Topbp1 (Jeon et al., 2011), Hus1 (Weiss et al., 2000), Chek1 (CHK1) (Takai et al., 2000; Zaugg et al., 2007), Cdc25A (Ray et al., 2007), Timeless (Gotter et al., 2000), and Prpf19 (Fortschegger et al., 2007) are all compromised during peri-implantation.
In somatic tissues, ATR or CHK1 inhibition coupled with replication stress induces replication catastrophe and S-phase apoptosis (Myers et al., 2009; Toledo et al., 2013; van Harten et al., 2019). Similarly, Atr null embryos form blastocysts that in vitro display widespread apoptosis within the ICM but not in the trophoblast, and which are eventually compromised by day three of culture (Brown and Baltimore, 2000; de Klein et al., 2000; Murga et al., 2009). ATR activity is therefore essential in the pluripotent and rapidly proliferating epiblast cells that will become the embryo proper, but not in the multipotent placental precursors. Chek1 null embryos also form blastocysts, but unlike Atr null embryos, are unable to attach to the culture vessels in outgrowth experiments (Takai et al., 2000). The more severe embryonic response to a loss of CHK1 activity compared to ATR inhibition is consistent with observations in somatic cells (Buisson et al., 2015).
HDR Genes Are Essential for Peri-Implantation Development
Common HDR factors manipulate DNA substrates at two-ended DNA breaks and stressed replication forks (Ait Saada et al., 2018). During replication stress, this entails regressing and protecting stalled forks in 4-way DNA structures to facilitate repair (Quinet et al., 2017). HDR factors essential for peri-implantation that function in canonical DSB repair and replication fork protection include: Brca1 (Hakem et al., 1996; Ludwig et al., 1997; Taglialatela et al., 2017), Brca2 (Sharan et al., 1997; Lemaçon et al., 2017; Mijic et al., 2017), Rad51 (Lim and Hasty, 1996; Tsuzuki et al., 1996; Zellweger et al., 2015), Bard1 (McCarthy et al., 2003; Shakya et al., 2008; Daza-Martin et al., 2019), Palb2 (Bowman-Colin et al., 2013), and Rad51C (Kuznetsov et al., 2009; Somyajit et al., 2015). Similarly, common factors promote resection during HDR repair of canonical two-ended DSBs and at stalled replication forks (Ait Saada et al., 2018). This includes the essential peri-implantation genes: Ctip (Chen et al., 2005; Przetocka et al., 2018); Ptip (Cho et al., 2003; Ray Chaudhuri et al., 2016); Dna2 (Lin et al., 2013; Thangavel et al., 2015), and the MRN complex [Mre11, Rad50, and Nbs1 (Xiao and Weaver, 1997; Luo et al., 1999; Zhu et al., 2001; Theunissen et al., 2003; Buis et al., 2008; Schlacher et al., 2011).
Homology directed repair activity in replication fork remodeling and protection is an emerging topic in the DNA repair field, and it remains unclear which HDR functions are essential for cell viability. However, lethality in Brca2 null mESCs is rescued when fork protection is facilitated by inhibiting PARP activity to protect replication forks from MRE11 nuclease (Ding et al., 2016). For BRCA2, fork protection may be the essential function. In vivo, deleting factors that participate in both canonical HDR-dependent DSB repair and fork remodeling or resection, typically confers reduced cell proliferation and embryo failure during gastrulation. For example, Brca1–/– embryos are severely malformed with an underdeveloped pro-amniotic cavity and mesoderm (Hakem et al., 1996; Ludwig et al., 1997). Concordantly, Brca1 null embryos display a significant reduction of DNA synthesis by E6.5, increased p21 [a factor induced by p53 transactivation to mediate cell cycle arrest (Georgakilas et al., 2017)], and a high incidence of G1 arrested cells (Hakem et al., 1996). Similarly, Rad51 null embryos suffer from reduced proliferation and begin to degenerate during gastrulation by E7.5, but development can be extended to E9.5 in TP53/Rad51 double knockouts (Lim and Hasty, 1996). Loss of HDR factors therefore appears to primarily confer proliferative failure through p53-induced growth arrest and subsequently through accumulated genome instability if p53 function is compromised.
Additional Peri-Implantation Genes Linked to HDR or Replication
Peri-implantation development additionally requires several genes that are functionally linked to HDR or replication stress mitigation. Such genes include Apex1 (Xanthoudakis et al., 1996), Rnaseh2B (Hiller et al., 2012), Rif1 (Chapman et al., 2013), Blm (Chester et al., 1998), Rev3L (Bemark et al., 2000), Usp7 (Kon et al., 2010), and Ino80 (Min et al., 2013). Apex1 does not directly function in replication, but is critical for the repair of abasic DNA sites that if left unresolved confer replication stress (Boiteux and Guillet, 2004). Similarly, Rnaseh2B resolves RNA within RNA-DNA hybrids, including mis-incorporated ribonucleotides, which if not resolved can impede replication (Pizzi et al., 2015). In addition to functioning in DSB repair pathway choice, Rif1 also coordinates replication timing (Yamazaki et al., 2012; Foti et al., 2016). Blm encodes a helicase that functions in homologous recombination, the replication stress response (Davies et al., 2007; Machwe et al., 2011), promotes replication at telomeres (Barefield and Karlseder, 2012), and participates in a specialized form of break induced replication at chromosome ends (Sobinoff et al., 2017). Rev3L encodes the catalytic subunit of POL ζ which facilitates translesion synthesis to enable replication forks to negotiate lesions within DNA without stalling (Bhat et al., 2013). Usp7 is a deubiquitylating enzyme that promotes efficient DNA replication (Lecona et al., 2016) and Ino80 encodes a chromatin remodeling factor that functions in both replication and HDR (Poli et al., 2017).
Maintaining Genome Stability During Peri-Implantation Development
While HDR and replication associated genes are not the only genome stability factors required for peri-implantation development, their predominance on the list of genes essential for this developmental window is compelling. Conditions enabling in vitro culture of peri-implantation murine epiblast stem cells are now established (Brons et al., 2007; Tesar et al., 2007). However, there are no published studies focusing on genome maintenance in isolated epiblast cells. It is thus necessary to extrapolate from studies of other rapidly dividing cells why HDR and replication factors may be essential during peri-implantation development.
Replication Stress in Peri-Implantation Cells
The fastest cell cycles in the lifetime of a mouse are likely during the brief window of epiblast development. Outside of the embryo, rapid proliferation contributes to replication stress in hematopoietic and cancerous tissues by robbing cells of the time to properly execute DNA polymerization (Pilzecker et al., 2017). For example, during a somatic G1-phase replication is facilitated by increasing the abundance of MCM proteins and loading these factors at replication origins in preparation for S-phase (Boos et al., 2012). Late G1 cells also upregulate dNTP production for the oncoming burst of DNA synthesis (Hirschi et al., 2010; Dick and Rubin, 2013). When origins are not established properly, or dNTP production is dysregulated, cancer and aging hematopoietic cells are susceptible to replication stress (Flach et al., 2014; Aird and Zhang, 2015; Alvarez et al., 2015; Garzon et al., 2017).
Pluripotent cells in the early embryo display almost non-existent G1 phases and appear to use unique mechanisms to ensure S-phase progression is not impeded. For example, human ESCs counter G1 brevity by loading MCM proteins faster than cultured non-pluripotent cells (Matson et al., 2017). Retinoblastoma protein is also constitutively hyperphosphorylated in mouse and human ESCs (Conklin et al., 2012; Ter Huurne et al., 2017), which maintains high E2F transactivation to promote unperturbed dNTP synthesis (Hirschi et al., 2010; Dick and Rubin, 2013). To ensure rapid S-phase progression, mouse ESCs also license more dormant origins than non-embryonic progenitor cells (Ge et al., 2015). This increases the ability of ESCs to complete replication during a short S phase (Courtot et al., 2018). ESCs also demonstrate an increased ability to restart stalled replication forks (Ahuja et al., 2016; Zhao et al., 2018). Human induced pluripotent stem cells (iPSC) also license replication origins rapidly in their abbreviated G1 phase (Matson et al., 2017), and dormant origins are critical to maintain rapid proliferation in hematopoietic stem cell pools (Alvarez et al., 2015). Although not yet directly tested, we anticipate that epiblast cells employ similar countermeasures to expedite DNA synthesis.
Despite these efforts, human and mouse ESCs display endogenous replication stress in unperturbed cultures (Ahuja et al., 2016; Lamm et al., 2016). It is not clear why replication is stressed in pluripotent cells, however in proliferating cancer cells transcription commonly drives replication stress through collisions between RNA and DNA polymerases, or persistent RNA/DNA hybrid molecules termed R-loops (Crossley et al., 2019). Primary somatic cells mitigate transcription and R-loop interference by temporally coordinating transcription and replication during S-phase (Meryet-Figuiere et al., 2014). It is probable that peri-implantation epiblast cells facilitate rapid genome duplication by simultaneously engaging large numbers of origins. Such activity could increase the probability of replisome and transcriptome collisions and/or R-loop induced replication stress.
Under-replication due to endogenous stress is described in mESCs and human iPSCs (Ahuja et al., 2016; Vallabhaneni et al., 2018). Lengthening the G1 phase reduces replication defects in mESCs consistent with the notion that replication stress is driven by rushed cell cycles (Ahuja et al., 2016). Pluripotent cells are reported to have enhanced DNA repair capacities and it is possible this translates to highly effectively management of inefficient replication in embryos (Maynard et al., 2008; Luo et al., 2012). Alternatively, pluripotent blastocyst cells persist for a limited number of cell divisions in vivo, and it is feasible that pre-implantation embryos simply tolerate under-replication for this brief period. Tolerance of replication defects is consistent with observations that preimplantation embryonic cells are more resistant to genomic insult than peri-implantation cells. Compromising the replication stress response should confer similar DDR activation, chromosomal segregation errors, and aneuploidy in both pre- and peri-implantation cells. However, cells with supernumerary chromosomes persist in the blastocyst before elimination during peri-implantation (Bolton et al., 2016). This may explain why many genome stability factors become essential specifically during peri-implantation development. In vivo, activating p53 through deletion of its negative regulator Mdm2 leads to lethality during peri-implantation (Jones et al., 1995). This suggests that p53 signaling during peri-implantation is a conduit to remove genomically unstable cells in vivo.
Additionally, it is important to recognize that genome stability factors may possesses cryptic essential functions during early development. For example, PARP1 is DDR factor involved in multiple aspects of genome integrity (Ray Chaudhuri and Nussenzweig, 2017). However, in mESCs, Parp1 has roles preventing the trans-differentiation of extraembryonic trophoblast cells (Nozaki et al., 1999; Hemberger et al., 2003). This pluripotency-specific function is linked to the DNA binding ability of PARP1 which confers an epigenetic-like regulation of pluripotency (Roper et al., 2014). Additionally, pluripotency-specific repair factors may provide essential functions in early development. The Filia-Floped protein complex is abundantly expressed in pluripotent but not somatic cells, and is suggested to increase the abundance of essential repair factors including BLM and promote ATR activation to encourage fork restart (Zhao et al., 2015; Zhao et al., 2018; Zheng, 2020).
Essential Roles for HDR Factors During Peri-Implantation
It is possible the canonical G1-phase c-NHEJ DSB repair pathway is dispensable during peri-implantation because cells rapidly transition through their brief G1 phase and engage alt-NHEJ or HDR in S-phase. Conversely, c- and alt-NHEJ remain active in HDR-deficient embryos, and in principle EJ functions could provide redundant DSB repair activity. An explanation why c- or alt-NHEJ cannot rescue HDR during peri-implantation may stem from the diversity of substrates repaired by HDR. While two-ended DSBs are readily repaired by EJ, collapsed replication forks present as one-ended DNA breaks which c- or alt-NHEJ cannot process (Feng and Jasin, 2017; Scully et al., 2019). One-ended breaks, however, can be repaired through a specialized HDR mechanism termed break-induced replication (BIR). During BIR, the exposed DNA end is resected, and following strand-invasion to form a displacement loop on the sister chromatid, DNA is polymerized from the invaded strand in a conservative manner (Kramara et al., 2018). While BIR is commonly studied in yeast, the analogous mechanism in vertebrates remains poorly defined. Notwithstanding, Blm promotes BIR at chromosome ends in human cancer cells that maintain telomere length through the alternative lengthening of telomeres mechanism suggesting that Blm may promote BIR elsewhere in the genome following replication stress (Sobinoff et al., 2017). Additionally, Pold3 is also implicated in vertebrate BIR (Costantino et al., 2014). Conversely, the non-essential gene Rad52 also functions in vertebrate BIR casting doubt on the requirement for BIR in early development and indicating the need for future studies (Sotiriou et al., 2016).
Multiple HDR factors essential for peri-implantation also play central roles in replication stress management. This includes HDR factors that regress, stabilize, or resect stalled replication forks including Brca1/2, Bard1, Rad51 and Rad51C, Ctip, Ptip, Dna2, and genes encoding the MRN complex. Interestingly, the helicases and translocases that promote fork regression, including Smarcal1, are not essential for peri-implantation (Baradaran-Heravi et al., 2012). This may reflect redundancy in the mechanisms that drive formation of 4-strand structures at stalled replication forks (Quinet et al., 2017). Given the essentiality of the replication stress response for epiblast development, it stands to reason that replication fork remodeling and protective functions encoded by HDR factors play a role in the viability of peri-implantation embryos. Failure to facilitate these activities would confer genome instability, chromosome segregation errors, and molecular outcomes consistent with unrepaired replication stress. As described above, this would activate p53 surveillance systems during peri-implantation. In the future it will be interesting to determine which specific HDR functions are essential for embryogenesis.
Conclusion and Future Directions
While most DDR and repair factors are non-essential, this does not exclude non-essential genome stability pathways from playing an important role in healthy development. Deletion of non-essential DDR and repair genes commonly results in the birth of live pups exhibiting a wide array of deleterious phenotypes (Supplementary Table S1). Additionally, cancer predisposition and a reduced lifespan are common in mice lacking non-essential genome stability factors (Supplementary Table S1). The difference between essential and non-essential genome stability genes is likely derived from the necessity of the targeted repair pathway to resolve lethal genome instability that arises during a specific window of development.
The critical genome stability pathways required for cell survival are determined by the type of genomic lesions the cell encounters and when in the cell cycle those lesions arise. Early embryonic development is largely protected from exogenous influence. Preimplantation embryos are wholly contained, and maternal blood supply begins at E9.5 (Behringer et al., 2014). The critical early stages of development likely benefit from sequestration from external threats to genome stability. We suggest the imminent threat to genome stability in early development stems from the need to mitigate endogenous replication stress within the epiblast to sustain rapid cell cycles. If left unresolved, replication stress drives genome instability, chromosome segregation errors, growth arrest and/or cell death. Dwindling cell numbers of epiblast cells that fail to effectively replicate their genome will progressively lead to embryo compromise. This premise is supported by the timing of embryo failure associated with deletion of essential DDR and repair genes. Consistent with systemic proliferative failure, deletion of factors required for basal DNA replication, or up-stream signaling in the replication stress response, typically induce an early embryonic demise (E5.0 or before). Whereas deleting HDR factors induces death at a subsequent time (E5.5 or later), potentially as the additive outcome of progressive genome instability.
Another caveat of reports reviewed here is that many were pioneering studies that utilized the newly developed technology of gene targeting in mice. In the intervening decades, our understanding of early development and experimental capability has blossomed. Development of new stem cell culture technologies now enables mechanistic study of peri-implantation development within three-dimensional gastrula structures in vitro. Co-culture of ICM derived ESCs, trophectoderm derived trophoblast stem cells, and extraembryonic endoderm stem cells results in the spontaneous self-assembly of a structure remarkably like the gastrulating embryo. These structures, termed “ETX embryos” will enable directed gene deletion within specific embryonic cell types (Sozen et al., 2018). Coupled with precise mechanistic investigations, ETX embryo models will reveal new insights on the mechanisms of cell compromise during embryogenesis. It will be exciting to learn in the coming years why a limited number of DDR and repair pathways are essential for development, and the underlying reasons for their importance within the peri-implantation embryo.
Data Availability Statement
MGI-GO designations discussed in this review can be found at the following. DNA damage checkpoint: http://www.informatics.jax.org/go/term/GO:0000077; nucleotide excision repair: http://www.informatics.jax.org/go/term/GO:0006289; mismatch re- pair: http://www.informatics.jax.org/go/term/GO:0006298; base excision repair: http://www.informatics.jax.org/go/term/GO:0006284; DSB repair via homologous recombination: http://www.informatics.jax.org/go/term/GO:0000724; DSB repair via non-homologous end joining: http://www.informatics.jax.org/go/term/GO:0006303.
Author Contributions
GK and AC conceived of the review manuscript. GK compiled the information presented in Tables 1, 2 and Supplementary Table S1 from the MGI-GO database as indicated. GK made the figures. GK and AC composed the manuscript.
Funding
The Cesare laboratory is supported by grants from the Australian National Health and Medical Research Council (1162886 and 1185870) and The Neil and Norma Hill Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank members of the Cesare and Tam laboratories at the Children’s Medical Research Institute for their helpful discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.00416/full#supplementary-material
References
Acloque, H., Adams, M. S., Fishwick, K., Bronner-Fraser, M., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J. Clin. Invest. 119, 1438–1449. doi: 10.1172/JCI38019
Affar El, B., Gay, F., Shi, Y., Liu, H., Huarte, M., Wu, S., et al. (2006). Essential dosage-dependent functions of the transcription factor yin yang 1 in late embryonic development and cell cycle progression. Mol. Cell. Biol. 26, 3565–3581. doi: 10.1128/MCB.26.9.3565-3581.2006
Afzal, S., Hao, Z., Itsumi, M., Abouelkheer, Y., Brenner, D., Gao, Y., et al. (2015). Autophagy-independent functions of UVRAG are essential for peripheral naive T-cell homeostasis. Proc. Natl. Acad. Sci. U.S.A. 112, 1119–1124. doi: 10.1073/pnas.1423588112
Ahuja, A. K., Jodkowska, K., Teloni, F., Bizard, A. H., Zellweger, R., Herrador, R., et al. (2016). A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 7:10660. doi: 10.1038/ncomms10660
Aird, K. M., and Zhang, R. (2015). Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. 356, 204–210. doi: 10.1016/j.canlet.2014.01.017
Ait Saada, A., Lambert, S. A. E., and Carr, A. M. (2018). Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 71, 135–147. doi: 10.1016/j.dnarep.2018.08.017
Akdemir, K. C., Jain, A. K., Allton, K., Aronow, B., Xu, X., Cooney, A. J., et al. (2014). Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 42, 205–223. doi: 10.1093/nar/gkt866
Ali, A. A. E., Timinszky, G., Arribas-Bosacoma, R., Kozlowski, M., Hassa, P. O., Hassler, M., et al. (2012). The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 19, 685–692. doi: 10.1038/nsmb.2335
Alvarez, S., Diaz, M., Flach, J., Rodriguez-Acebes, S., Lopez-Contreras, A. J., Martinez, D., et al. (2015). Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat. Commun. 6:8548. doi: 10.1038/ncomms9548
Andressoo, J. O., Weeda, G., De Wit, J., Mitchell, J. R., Beems, R. B., Van Steeg, H., et al. (2009). An Xpb mouse model for combined xeroderma pigmentosum and cockayne syndrome reveals progeroid features upon further attenuation of DNA repair. Mol. Cell. Biol. 29, 1276–1290. doi: 10.1128/MCB.01229-08
Artus, J., Vandormael-Pournin, S., Frodin, M., Nacerddine, K., Babinet, C., and Cohen-Tannoudji, M. (2005). Impaired mitotic progression and preimplantation lethality in mice lacking OMCG1, a new evolutionarily conserved nuclear protein. Mol. Cell. Biol. 25, 6289–6302. doi: 10.1128/MCB.25.14.6289-6302.2005
Audebert, M., Salles, B., and Calsou, P. (2004). Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 279, 55117–55126. doi: 10.1074/jbc.M404524200
Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C. W., Chessa, L., et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677. doi: 10.1126/science.281.5383.1674
Baradaran-Heravi, A., Cho, K. S., Tolhuis, B., Sanyal, M., Morozova, O., Morimoto, M., et al. (2012). Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Hum. Mol. Genet. 21, 2572–2587. doi: 10.1093/hmg/dds083
Barefield, C., and Karlseder, J. (2012). The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 40, 7358–7367. doi: 10.1093/nar/gks407
Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Collins, F., et al. (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159–171. doi: 10.1016/s0092-8674(00)80086-0
Bass, T. E., Luzwick, J. W., Kavanaugh, G., Carroll, C., Dungrawala, H., Glick, G. G., et al. (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 18, 1185–1195. doi: 10.1038/ncb3415
Behringer, R., Gertsenstein, M., Nagy, K. V., and Nagy, A. (2014). Manipulating the Mouse Embryo. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Bemark, M., Khamlichi, A. A., Davies, S. L., and Neuberger, M. S. (2000). Disruption of mouse polymerase zeta (Rev3) leads to embryonic lethality and impairs blastocyst development in vitro. Curr. Biol. 10, 1213–1216. doi: 10.1016/s0960-9822(00)00724-7
Bermudez, V. P., Lindsey-Boltz, L. A., Cesare, A. J., Maniwa, Y., Griffith, J. D., Hurwitz, J., et al. (2003). Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 1633–1638. doi: 10.1073/pnas.0437927100
Bhat, A., Andersen, P. L., Qin, Z., and Xiao, W. (2013). Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res. 41, 2328–2339. doi: 10.1093/nar/gks1442
Bhat, K. P., and Cortez, D. (2018). RPA and RAD51: fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 25, 1–8. doi: 10.1038/s41594-018-0075-z
Bleichert, F. (2019). Mechanisms of replication origin licensing: a structural perspective. Curr. Opin. Struct. Biol. 59, 195–204. doi: 10.1016/j.sbi.2019.08.007
Boiteux, S., and Guillet, M. (2004). Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair. 3, 1–12. doi: 10.1016/j.dnarep.2003.10.002
Bolton, H., Graham, S. J. L., Van Der Aa, N., Kumar, P., Theunis, K., Fernandez Gallardo, E., et al. (2016). Mouse model of chromosome mosaicism reveals lineage-specific depletion of aneuploid cells and normal developmental potential. Nat. Commun. 7:11165. doi: 10.1038/ncomms11165
Boos, D., Frigola, J., and Diffley, J. F. (2012). Activation of the replicative DNA helicase: breaking up is hard to do. Curr. Opin. Cell Biol. 24, 423–430. doi: 10.1016/j.ceb.2012.01.011
Bowman-Colin, C., Xia, B., Bunting, S., Klijn, C., Drost, R., Bouwman, P., et al. (2013). Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proc. Natl. Acad. Sci. U.S.A. 110, 8632–8637. doi: 10.1073/pnas.1305362110
Brons, I. G., Smithers, L. E., Trotter, M. W., Rugg-Gunn, P., Sun, B., Chuva De Sousa Lopes, S. M., et al. (2007). Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195. doi: 10.1038/nature05950
Brown, E. J., and Baltimore, D. (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 14, 397–402.
Buis, J., Wu, Y., Deng, Y., Leddon, J., Westfield, G., Eckersdorff, M., et al. (2008). Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 135, 85–96. doi: 10.1016/j.cell.2008.08.015
Buisson, R., Boisvert, J. L., Benes, C. H., and Zou, L. (2015). Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S Phase. Mol. Cell 59, 1011–1024. doi: 10.1016/j.molcel.2015.07.029
Buisson, R., Niraj, J., Rodrigue, A., Ho, C. K., Kreuzer, J., Foo, T. K., et al. (2017). Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 65, 336–346. doi: 10.1016/j.molcel.2016.12.007
Bult, C. J., Blake, J. A., Smith, C. L., Kadin, J. A., Richardson, J. E., and Mouse Genome Database, G. (2019). Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 47, D801–D806. doi: 10.1093/nar/gky1056
Burgers, P. M. J., and Kunkel, T. A. (2017). Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 86, 417–438. doi: 10.1146/annurev-biochem-061516-044709
Burrell, R. A., Mcclelland, S. E., Endesfelder, D., Groth, P., Weller, M. C., Shaikh, N., et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. doi: 10.1038/nature11935
Byun, T. S., Pacek, M., Yee, M.-C., Walter, J. C., and Cimprich, K. A. (2005). Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 19, 1040–1052. doi: 10.1101/gad.1301205
Cang, Y., Zhang, J., Nicholas, S. A., Bastien, J., Li, B., Zhou, P., et al. (2006). Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell 127, 929–940. doi: 10.1016/j.cell.2006.09.045
Carver, E. A., Jiang, R., Lan, Y., Oram, K. F., and Gridley, T. (2001). The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol. 21, 8184–8188. doi: 10.1128/MCB.21.23.8184-8188.2001
Celeste, A., Petersen, S., Romanienko, P. J., Fernandez-Capetillo, O., Chen, H. T., Sedelnikova, O. A., et al. (2002). Genomic instability in mice lacking histone H2AX. Science 296, 922–927. doi: 10.1126/science.1069398
Celli, G. B., Denchi, E. L., and De Lange, T. (2006). Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 8, 885–890. doi: 10.1038/ncb1444
Chang, H. H. Y., Pannunzio, N. R., Adachi, N., and Lieber, M. R. (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506. doi: 10.1038/nrm.2017.48
Chapman, J. R., Barral, P., Vannier, J. B., Borel, V., Steger, M., Tomas-Loba, A., et al. (2013). RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871. doi: 10.1016/j.molcel.2013.01.002
Chen, C. H., Chu, P. C., Lee, L., Lien, H. W., Lin, T. L., Fan, C. C., et al. (2012). Disruption of murine mp29/Syf2/Ntc31 gene results in embryonic lethality with aberrant checkpoint response. PLoS One 7:e33538. doi: 10.1371/journal.pone.0033538
Chen, P.-L., Liu, F., Cai, S., Lin, X., Li, A., Chen, Y., et al. (2005). Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol. Cell. Biol. 25, 3535–3542. doi: 10.1128/MCB.25.9.3535-3542.2005
Chester, N., Kuo, F., Kozak, C., O’hara, C. D., and Leder, P. (1998). Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 12, 3382–3393. doi: 10.1101/gad.12.21.3382
Cho, E. A., Prindle, M. J., and Dressler, G. R. (2003). BRCT domain-containing protein PTIP is essential for progression through mitosis. Mol. Cell. Biol. 23, 1666–1673. doi: 10.1128/mcb.23.5.1666-1673.2003
Choi, J.-H., Lindsey-Boltz, L. A., Kemp, M., Mason, A. C., Wold, M. S., and Sancar, A. (2010). Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 13660–13665. doi: 10.1073/pnas.1007856107
Condic, M. L. (2014). Totipotency: what it is and what it is not. Stem Cells Dev. 23, 796–812. doi: 10.1089/scd.2013.0364
Conklin, J. F., Baker, J., and Sage, J. (2012). The RB family is required for the self-renewal and survival of human embryonic stem cells. Nat. Commun. 3:1244. doi: 10.1038/ncomms2254
Cortez, D. (2019). Replication-Coupled DNA Repair. Mol. Cell 74, 866–876. doi: 10.1016/j.molcel.2019.04.027
Cortez, D., Guntuku, S., Qin, J., and Elledge, S. J. (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. doi: 10.1126/science.1065521
Costantino, L., Sotiriou, S. K., Rantala, J. K., Magin, S., Mladenov, E., Helleday, T., et al. (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91. doi: 10.1126/science.1243211
Couch, F. B., Bansbach, C. E., Driscoll, R., Luzwick, J. W., Glick, G. G., Betous, R., et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623. doi: 10.1101/gad.214080.113
Courtot, L., Hoffmann, J. S., and Bergoglio, V. (2018). The protective role of dormant origins in response to replicative stress. Int. J. Mol. Sci. 19:E3569. doi: 10.3390/ijms19113569
Crossley, M. P., Bocek, M., and Cimprich, K. A. (2019). R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 73, 398–411. doi: 10.1016/j.molcel.2019.01.024
Davies, S. L., North, P. S., and Hickson, I. D. (2007). Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 14, 677–679. doi: 10.1038/nsmb1267
Daza-Martin, M., Starowicz, K., Jamshad, M., Tye, S., Ronson, G. E., Mackay, H. L., et al. (2019). Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 571, 521–527. doi: 10.1038/s41586-019-1363-4
de Boer, J., Donker, I., De Wit, J., Hoeijmakers, J. H., and Weeda, G. (1998). Disruption of the mouse xeroderma pigmentosum group D DNA repair/basal transcription gene results in preimplantation lethality. Cancer Res. 58, 89–94.
de Klein, A., Muijtjens, M., Van Os, R., Verhoeven, Y., Smit, B., Carr, A. M., et al. (2000). Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 10, 479–482. doi: 10.1016/s0960-9822(00)00447-4
Delacroix, S., Wagner, J. M., Kobayashi, M., Yamamoto, K., and Karnitz, L. M. (2007). The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 21, 1472–1477. doi: 10.1101/gad.1547007
Denchi, E. L., and de Lange, T. (2007). Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071. doi: 10.1038/nature06065
Dick, F. A., and Rubin, S. M. (2013). Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306. doi: 10.1038/nrm3567
Dickinson, M. E., Flenniken, A. M., Ji, X., Teboul, L., Wong, M. D., White, J. K., et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. doi: 10.1038/nature19356
Ding, X., Ray Chaudhuri, A., Callen, E., Pang, Y., Biswas, K., Klarmann, K. D., et al. (2016). Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 7:12425. doi: 10.1038/ncomms12425
Diril, M. K., Ratnacaram, C. K., Padmakumar, V. C., Du, T., Wasser, M., Coppola, V., et al. (2012). Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. U S.A. 109, 3826–3831. doi: 10.1073/pnas.1115201109
Dupre, A., Boyer-Chatenet, L., and Gautier, J. (2006). Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 13, 451–457. doi: 10.1038/nsmb1090
Egly, J. M. (2001). The 14th Datta Lecture. TFIIH: from transcription to clinic. FEBS Lett. 498, 124–128. doi: 10.1016/s0014-5793(01)02458-9
Feng, W., and Jasin, M. (2017). BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 8:525. doi: 10.1038/s41467-017-00634-0
Flach, J., Bakker, S. T., Mohrin, M., Conroy, P. C., Pietras, E. M., Reynaud, D., et al. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202. doi: 10.1038/nature13619
Fortschegger, K., Wagner, B., Voglauer, R., Katinger, H., Sibilia, M., and Grillari, J. (2007). Early embryonic lethality of mice lacking the essential protein SNEV. Mol. Cell. Biol. 27, 3123–3130. doi: 10.1128/MCB.01188-06
Foster, C. T., Dovey, O. M., Lezina, L., Luo, J. L., Gant, T. W., Barlev, N., et al. (2010). Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol. Cell. Biol. 30, 4851–4863. doi: 10.1128/MCB.00521-10
Foti, R., Gnan, S., Cornacchia, D., Dileep, V., Bulut-Karslioglu, A., Diehl, S., et al. (2016). Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 61, 260–273. doi: 10.1016/j.molcel.2015.12.001
Frank, K. M., Sekiguchi, J. M., Seidl, K. J., Swat, W., Rathbun, G. A., Cheng, H. L., et al. (1998). Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396, 173–177. doi: 10.1038/24172
Fukushima, T., Matsuzawa, S., Kress, C. L., Bruey, J. M., Krajewska, M., Lefebvre, S., et al. (2007). Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc. Natl. Acad. Sci. U.S.A. 104, 6371–6376. doi: 10.1073/pnas.0700548104
Ganuza, M., Sáiz-Ladera, C., Cañamero, M., Gómez, G., Schneider, R., Blasco, M. A., et al. (2012). Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. EMBO J. 31, 2498–2510. doi: 10.1038/emboj.2012.94
Garzon, J., Rodriguez, R., Kong, Z., Chabes, A., Rodriguez-Acebes, S., Mendez, J., et al. (2017). Shortage of dNTPs underlies altered replication dynamics and DNA breakage in the absence of the APC/C cofactor Cdh1. Oncogene 36, 5808–5818. doi: 10.1038/onc.2017.186
Gavet, O., and Pines, J. (2010). Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533–543. doi: 10.1016/j.devcel.2010.02.013
Ge, X. Q., Han, J., Cheng, E. C., Yamaguchi, S., Shima, N., Thomas, J. L., et al. (2015). Embryonic Stem Cells License a High Level of Dormant Origins to Protect the Genome against Replication Stress. Stem Cell Rep. 5, 185–194. doi: 10.1016/j.stemcr.2015.06.002
Georgakilas, A. G., Martin, O. A., and Bonner, W. M. (2017). p21: a two-faced genome guardian. Trends Mol. Med. 23, 310–319. doi: 10.1016/j.molmed.2017.02.001
Ghezraoui, H., Piganeau, M., Renouf, B., Renaud, J.-B., Sallmyr, A., Ruis, B., et al. (2014). Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 55, 829–842. doi: 10.1016/j.molcel.2014.08.002
Gotter, A. L., Manganaro, T., Weaver, D. R., Kolakowski, LF Jr, Possidente, B., Sriram, S., et al. (2000). A time-less function for mouse timeless. Nat. Neurosci. 3, 755–756.
Gottlieb, T. M., and Jackson, S. P. (1993). The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72, 131–142. doi: 10.1016/0092-8674(93)90057-w
Gu, Y., Seidl, K. J., Rathbun, G. A., Zhu, C., Manis, J. P., Van Der Stoep, N., et al. (1997). Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 7, 653–665. doi: 10.1016/s1074-7613(00)80386-6
Haahr, P., Hoffmann, S., Tollenaere, M. A., Ho, T., Toledo, L. I., Mann, M., et al. (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 18, 1196–1207. doi: 10.1038/ncb3422
Hakem, R., De La Pompa, J. L., Sirard, C., Mo, R., Woo, M., Hakem, A., et al. (1996). The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 85, 1009–1023. doi: 10.1016/s0092-8674(00)81302-1
Hamatani, T., Carter, M. G., Sharov, A. A., and Ko, M. S. H. (2004). Dynamics of global gene expression changes during mouse preimplantation development. Dev. Cell 6, 117–131. doi: 10.1016/s1534-5807(03)00373-3
Harvey, S. L., Charlet, A., Haas, W., Gygi, S. P., and Kellogg, D. R. (2005). Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 122, 407–420. doi: 10.1016/j.cell.2005.05.029
Haupt, Y., Maya, R., Kazaz, A., and Oren, M. (1997). Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299. doi: 10.1038/387296a0
Hemberger, M., Nozaki, T., Winterhager, E., Yamamoto, H., Nakagama, H., Kamada, N., et al. (2003). Parp1-deficiency induces differentiation of ES cells into trophoblast derivatives. Dev. Biol. 257, 371–381. doi: 10.1016/s0012-1606(03)00097-6
Heyer, B. S., Macauley, A., Behrendtsen, O., and Werb, Z. (2000). Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev. 14, 2072–2084.
Hiller, B., Achleitner, M., Glage, S., Naumann, R., Behrendt, R., and Roers, A. (2012). Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med. 209, 1419–1426. doi: 10.1084/jem.20120876
Hirschi, A., Cecchini, M., Steinhardt, R. C., Schamber, M. R., Dick, F. A., and Rubin, S. M. (2010). An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol. 17, 1051–1057. doi: 10.1038/nsmb.1868
Hnizda, A., and Blundell, T. L. (2019). Multicomponent assemblies in DNA-double-strand break repair by NHEJ. Curr. Opin. Struct. Biol. 55, 154–160. doi: 10.1016/j.sbi.2019.03.026
Hockemeyer, D., Daniels, J.-P., Takai, H., and De Lange, T. (2006). Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. Cell 126, 63–77. doi: 10.1016/j.cell.2006.04.044
Hockemeyer, D., Sfeir, A. J., Shay, J. W., Wright, W. E., and De Lange, T. (2005). POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 24, 2667–2678. doi: 10.1038/sj.emboj.7600733
Hoki, Y., Araki, R., Fujimori, A., Ohhata, T., Koseki, H., Fukumura, R., et al. (2003). Growth retardation and skin abnormalities of the Recql4-deficient mouse. Hum. Mol. Genet. 12, 2293–2299. doi: 10.1093/hmg/ddg254
Horii, T., Yamamoto, M., Morita, S., Kimura, M., Nagao, Y., and Hatada, I. (2015). p53 suppresses tetraploid development in mice. Sci. Rep. 5:8907. doi: 10.1038/srep08907
Humięcka, M., Szpila, M., Kłoś, P., Maleszewski, M., and Szczepańska, K. (2017). Mouse blastomeres acquire ability to divide asymmetrically before compaction. PLoS One 12:e0175032. doi: 10.1371/journal.pone.0175032
Hustedt, N., and Durocher, D. (2016). The control of DNA repair by the cell cycle. Nat. Cell Biol. 19, 1–9. doi: 10.1038/ncb3452
Jackson, S. P., and Durocher, D. (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807. doi: 10.1016/j.molcel.2013.01.017
Jacome, A., Gutierrez-Martinez, P., Schiavoni, F., Tenaglia, E., Martinez, P., Rodriguez-Acebes, S., et al. (2015). NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. EMBO J. 34, 2604–2619. doi: 10.15252/embj.201591829
Jain, A. K., Xi, Y., Mccarthy, R., Allton, K., Akdemir, K. C., Patel, L. R., et al. (2016). LncPRESS1 is a p53-Regulated LncRNA that safeguards pluripotency by disrupting SIRT6-mediated de-acetylation of histone H3K56. Mol. Cell 64, 967–981. doi: 10.1016/j.molcel.2016.10.039
Jeon, Y., Ko, E., Lee, K. Y., Ko, M. J., Park, S. Y., Kang, J., et al. (2011). TopBP1 deficiency causes an early embryonic lethality and induces cellular senescence in primary cells. J. Biol. Chem. 286, 5414–5422. doi: 10.1074/jbc.M110.189704
Jones, S. N., Roe, A. E., Donehower, L. A., and Bradley, A. (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208. doi: 10.1038/378206a0
Jukam, D., Shariati, S. A. M., and Skotheim, J. M. (2017). Zygotic genome activation in vertebrates. Dev. Cell 42, 316–332.
Kafer, G. R., Li, X., Horii, T., Suetake, I., Tajima, S., Hatada, I., et al. (2016). 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep. 14, 1283–1292. doi: 10.1016/j.celrep.2016.01.035
Kemp, M. G., Akan, Z., Yilmaz, S., Grillo, M., Smith-Roe, S. L., Kang, T. H., et al. (2010). Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J. Biol. Chem. 285, 16562–16571. doi: 10.1074/jbc.M110.110304
Kent, L. N., and Leone, G. (2019). The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 19, 326–338. doi: 10.1038/s41568-019-0143-7
Kent, T., Chandramouly, G., Mcdevitt, S. M., Ozdemir, A. Y., and Pomerantz, R. T. (2015). Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat. Struct. Mol. Biol. 22, 230–237. doi: 10.1038/nsmb.2961
Kim, J. M., Nakao, K., Nakamura, K., Saito, I., Katsuki, M., Arai, K.-I., et al. (2002). Inactivation of Cdc7 kinase in mouse ES cells results in S-phase arrest and p53-dependent cell death. EMBO J. 21, 2168–2179. doi: 10.1093/emboj/21.9.2168
Kon, N., Kobayashi, Y., Li, M., Brooks, C. L., Ludwig, T., and Gu, W. (2010). Inactivation of HAUSP in vivo modulates p53 function. Oncogene 29, 1270–1279. doi: 10.1038/onc.2009.427
Kramara, J., Osia, B., and Malkova, A. (2018). Break-induced replication: the Where. The Why, and The How. Trends Genet. 34, 518–531. doi: 10.1016/j.tig.2018.04.002
Kumagai, A., Lee, J., Yoo, H. Y., and Dunphy, W. G. (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955.
Kurimasa, A., Ouyang, H., Dong, L. J., Wang, S., Li, X., Cordon-Cardo, C., et al. (1999). Catalytic subunit of DNA-dependent protein kinase: impact on lymphocyte development and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 96, 1403–1408. doi: 10.1073/pnas.96.4.1403
Kuznetsov, S. G., Haines, D. C., Martin, B. K., and Sharan, S. K. (2009). Loss of Rad51c leads to embryonic lethality and modulation of Trp53-dependent tumorigenesis in mice. Cancer Res. 69, 863–872. doi: 10.1158/0008-5472.CAN-08-3057
Lamm, N., Ben-David, U., Golan-Lev, T., Storchova, Z., Benvenisty, N., and Kerem, B. (2016). Genomic instability in human pluripotent stem cells arises from replicative stress and chromosome condensation defects. Cell Stem Cell 18, 253–261. doi: 10.1016/j.stem.2015.11.003
Larsen, E., Gran, C., Saether, B. E., Seeberg, E., and Klungland, A. (2003). Proliferation failure and gamma radiation sensitivity of Fen1 null mutant mice at the blastocyst stage. Mol. Cell. Biol. 23, 5346–5353. doi: 10.1128/mcb.23.15.5346-5353.2003
Laurent, A., and Blasi, F. (2015). Differential DNA damage signalling and apoptotic threshold correlate with mouse epiblast-specific hypersensitivity to radiation. Development 142, 3675–3685. doi: 10.1242/dev.125708
Lecona, E., Rodriguez-Acebes, S., Specks, J., Lopez-Contreras, A. J., Ruppen, I., Murga, M., et al. (2016). USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol. 23, 270–277. doi: 10.1038/nsmb.3185
Lee, J. H., and Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554. doi: 10.1126/science.1108297
Lee, S., Kim, J. Y., Kim, Y. J., Seok, K. O., Kim, J. H., Chang, Y. J., et al. (2012). Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ. 19, 1613–1622. doi: 10.1038/cdd.2012.40
Lemaçon, D., Jackson, J., Quinet, A., Brickner, J. R., Li, S., Yazinski, S., et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8:860. doi: 10.1038/s41467-017-01180-5
Li, A., and Blow, J. J. (2005). Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J. 24, 395–404. doi: 10.1038/sj.emboj.7600520
Li, B., Ruiz, J. C., and Chun, K. T. (2002). CUL-4A is critical for early embryonic development. Mol. Cell. Biol. 22, 4997–5005. doi: 10.1128/mcb.22.14.4997-5005.2002
Li, G., Alt, F. W., Cheng, H. L., Brush, J. W., Goff, P. H., Murphy, M. M., et al. (2008). Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol. Cell 31, 631–640. doi: 10.1016/j.molcel.2008.07.017
Li, M., He, Y., Dubois, W., Wu, X., Shi, J., and Huang, J. (2012). Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell 46, 30–42. doi: 10.1016/j.molcel.2012.01.020
Lim, D. S., and Hasty, P. (1996). A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol. 16, 7133–7143. doi: 10.1128/mcb.16.12.7133
Limas, J. C., and Cook, J. G. (2019). Preparation for DNA replication: the key to a successful S phase. FEBS Lett. 593, 2853–2867. doi: 10.1002/1873-3468.13619
Lin, T., Chao, C., Saito, S., Mazur, S. J., Murphy, M. E., Appella, E., et al. (2005). p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 7, 165–171. doi: 10.1038/ncb1211
Lin, W., Sampathi, S., Dai, H., Liu, C., Zhou, M., Hu, J., et al. (2013). Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 32, 1425–1439. doi: 10.1038/emboj.2013.88
Liu, C., Vyas, A., Kassab, M. A., Singh, A. K., and Yu, X. (2017). The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 45, 8129–8141. doi: 10.1093/nar/gkx565
Liu, C.-L., Yu, I. S., Pan, H.-W., Lin, S.-W., and Hsu, H.-C. (2007). L2dtl is essential for cell survival and nuclear division in early mouse embryonic development. J. Biol. Chem. 282, 1109–1118. doi: 10.1074/jbc.M606535200
Liu, D., Keijzers, G., and Rasmussen, L. J. (2017). DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 773, 174–187. doi: 10.1016/j.mrrev.2017.07.001
Liu, Q., Guntuku, S., Cui, X. S., Matsuoka, S., Cortez, D., Tamai, K., et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459.
Lou, J., Scipioni, L., Wright, B. K., Bartolec, T. K., Zhang, J., Masamsetti, V. P., et al. (2019). Phasor histone FLIM-FRET microscopy quantifies spatiotemporal rearrangement of chromatin architecture during the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 116, 7323–7332. doi: 10.1073/pnas.1814965116
Lou, Z., Minter-Dykhouse, K., Franco, S., Gostissa, M., Rivera, M. A., Celeste, A., et al. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 21, 187–200. doi: 10.1016/j.molcel.2005.11.025
Lu, L. Y., Wood, J. L., Minter-Dykhouse, K., Ye, L., Saunders, T. L., Yu, X., et al. (2008). Polo-like kinase 1 is essential for early embryonic development and tumor suppression. Mol. Cell. Biol. 28, 6870–6876. doi: 10.1128/MCB.00392-08
Ludwig, T., Chapman, D. L., Papaioannou, V. E., and Efstratiadis, A. (1997). Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 11, 1226–1241. doi: 10.1101/gad.11.10.1226
Lukas, C., Savic, V., Bekker-Jensen, S., Doil, C., Neumann, B., Pedersen, R. S., et al. (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253. doi: 10.1038/ncb2201
Luo, G., Yao, M. S., Bender, C. F., Mills, M., Bladl, A. R., Bradley, A., et al. (1999). Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. U.S.A. 96, 7376–7381. doi: 10.1073/pnas.96.13.7376
Luo, L. Z., Gopalakrishna-Pillai, S., Nay, S. L., Park, S. W., Bates, S. E., Zeng, X., et al. (2012). DNA repair in human pluripotent stem cells is distinct from that in non-pluripotent human cells. PLoS One 7:e30541. doi: 10.1371/journal.pone.0030541
Ly, P., and Cleveland, D. W. (2017). Rebuilding chromosomes after catastrophe: emerging mechanisms of chromothripsis. Trends Cell Biol. 27, 917–930. doi: 10.1016/j.tcb.2017.08.005
Machwe, A., Karale, R., Xu, X., Liu, Y., and Orren, D. K. (2011). The Werner and Bloom syndrome proteins help resolve replication blockage by converting (regressed) holliday junctions to functional replication forks. Biochemistry 50, 6774–6788. doi: 10.1021/bi2001054
Maciejowski, J., Li, Y., Bosco, N., Campbell, P. J., and De Lange, T. (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654. doi: 10.1016/j.cell.2015.11.054
Mailand, N., Gibbs-Seymour, I., and Bekker-Jensen, S. (2013). Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 14, 269–282. doi: 10.1038/nrm3562
Mancini, A., Niemann-Seyde, S. C., Pankow, R., El Bounkari, O., Klebba-Farber, S., Koch, A., et al. (2010). THOC5/FMIP, an mRNA export TREX complex protein, is essential for hematopoietic primitive cell survival in vivo. BMC Biol 8:1. doi: 10.1186/1741-7007-8-1
Mandal, R., and Strebhardt, K. (2013). Plk1: unexpected roles in DNA replication. Cell Res. 23, 1251–1253. doi: 10.1038/cr.2013.130
Mankouri, H. W., Huttner, D., and Hickson, I. D. (2013). How unfinished business from S-phase affects mitosis and beyond. EMBO J. 32, 2661–2671. doi: 10.1038/emboj.2013.211
Maréchal, A., Li, J.-M., Ji, X. Y., Wu, C.-S., Yazinski, S. A., Nguyen, H. D., et al. (2014). PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 53, 235–246. doi: 10.1016/j.molcel.2013.11.002
Maréchal, A., and Zou, L. (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 5:a012716. doi: 10.1101/cshperspect.a012716
Masamsetti, V. P., Low, R. R. J., Mak, K. S., O’connor, A., Riffkin, C. D., Lamm, N., et al. (2019). Replication stress induces mitotic death through parallel pathways regulated by WAPL and telomere deprotection. Nat. Commun. 10:4224. doi: 10.1038/s41467-019-12255-w
Masuda, K., Ouchida, R., Takeuchi, A., Saito, T., Koseki, H., Kawamura, K., et al. (2005). DNA polymerase theta contributes to the generation of C/G mutations during somatic hypermutation of Ig genes. Proc. Natl. Acad. Sci. U.S.A. 102, 13986–13991. doi: 10.1073/pnas.0505636102
Mateos-Gomez, P. A., Gong, F., Nair, N., Miller, K. M., Lazzerini-Denchi, E., and Sfeir, A. (2015). Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257. doi: 10.1038/nature14157
Mateos-Gomez, P. A., Kent, T., Deng, S. K., Mcdevitt, S., Kashkina, E., Hoang, T. M., et al. (2017). The helicase domain of Polθ counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol. 24, 1116–1123. doi: 10.1038/nsmb.3494
Matson, J. P., Dumitru, R., Coryell, P., Baxley, R. M., Chen, W., Twaroski, K., et al. (2017). Rapid DNA replication origin licensing protects stem cell pluripotency. eLife 6:e30473.
Maynard, S., Swistowska, A. M., Lee, J. W., Liu, Y., Liu, S. T., Da Cruz, A. B., et al. (2008). Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells 26, 2266–2274. doi: 10.1634/stemcells.2007-1041
McCarthy, E. E., Celebi, J. T., Baer, R., and Ludwig, T. (2003). Loss of Bard1, the heterodimeric partner of the Brca1 tumor suppressor, results in early embryonic lethality and chromosomal instability. Mol. Cell. Biol. 23, 5056–5063. doi: 10.1128/mcb.23.14.5056-5063.2003
Mello, S. S., and Attardi, L. D. (2018). Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol. 51, 65–72. doi: 10.1016/j.ceb.2017.11.005
Meryet-Figuiere, M., Alaei-Mahabadi, B., Ali, M. M., Mitra, S., Subhash, S., Pandey, G. K., et al. (2014). Temporal separation of replication and transcription during S-phase progression. Cell Cycle 13, 3241–3248. doi: 10.4161/15384101.2014.953876
Mijic, S., Zellweger, R., Chappidi, N., Berti, M., Jacobs, K., Mutreja, K., et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 8:859. doi: 10.1038/s41467-017-01164-5
Miller, T. C. R., Locke, J., Greiwe, J. F., Diffley, J. F. X., and Costa, A. (2019). Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature 575, 704–710. doi: 10.1038/s41586-019-1768-0
Min, J.-N., Tian, Y., Xiao, Y., Wu, L., Li, L., and Chang, S. (2013). The mINO80 chromatin remodeling complex is required for efficient telomere replication and maintenance of genome stability. Cell Res. 23, 1396–1413. doi: 10.1038/cr.2013.113
Minocherhomji, S., Ying, S., Bjerregaard, V. A., Bursomanno, S., Aleliunaite, A., Wu, W., et al. (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. doi: 10.1038/nature16139
Mohammed, H., Hernando-Herraez, I., Savino, A., Scialdone, A., Macaulay, I., Mulas, C., et al. (2017). Single-cell landscape of transcriptional heterogeneity and cell fate decisions during mouse early gastrulation. Cell Rep. 20, 1215–1228. doi: 10.1016/j.celrep.2017.07.009
Mohri, T., Ueno, M., Nagahama, Y., Gong, Z.-Y., Asano, M., Oshima, H., et al. (2013). Requirement of SLD5 for early embryogenesis. PLoS One 8:e78961. doi: 10.1371/journal.pone.0078961
Morris, S. A., Teo, R. T., Li, H., Robson, P., Glover, D. M., and Zernicka-Goetz, M. (2010). Origin and formation of the first two distinct cell types of the inner cell mass in the mouse embryo. Proc. Natl. Acad. Sci. U.S.A. 107, 6364–6369. doi: 10.1073/pnas.0915063107
Murga, M., Bunting, S., Montaña, M. F., Soria, R., Mulero, F., Cañamero, M., et al. (2009). A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 41, 891–898. doi: 10.1038/ng.420
Murray, S. A., Morgan, J. L., Kane, C., Sharma, Y., Heffner, C. S., Lake, J., et al. (2010). Mouse gestation length is genetically determined. PLoS One 5:e12418. doi: 10.1371/journal.pone.0012418
Myers, K., Gagou, M. E., Zuazua-Villar, P., Rodriguez, R., and Meuth, M. (2009). ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress. PLoS Genet. 5:e1000324. doi: 10.1371/journal.pgen.1000324
Navadgi-Patil, V. M., and Burgers, P. M. (2009). A tale of two tails: activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair 8, 996–1003. doi: 10.1016/j.dnarep.2009.03.011
Neelsen, K. J., and Lopes, M. (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220. doi: 10.1038/nrm3935
Nigg, E. A. (2001). Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2, 21–32. doi: 10.1038/35048096
Nishitani, H., Taraviras, S., Lygerou, Z., and Nishimoto, T. (2001). The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J. Biol. Chem. 276, 44905–44911. doi: 10.1074/jbc.M105406200
Nozaki, T., Masutani, M., Watanabe, M., Ochiya, T., Hasegawa, F., Nakagama, H., et al. (1999). Syncytiotrophoblastic giant cells in teratocarcinoma-like tumors derived from Parp-disrupted mouse embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 96, 13345–13350. doi: 10.1073/pnas.96.23.13345
Nussenzweig, A., Chen, C., Da Costa Soares, V., Sanchez, M., Sokol, K., Nussenzweig, M. C., et al. (1996). Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 382, 551–555. doi: 10.1038/382551a0
Ochi, T., Blackford, A. N., Coates, J., Jhujh, S., Mehmood, S., Tamura, N., et al. (2015). DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 347, 185–188. doi: 10.1126/science.1261971
Ochs, F., Karemore, G., Miron, E., Brown, J., Sedlackova, H., Rask, M. B., et al. (2019). Stabilization of chromatin topology safeguards genome integrity. Nature 574, 571–574. doi: 10.1038/s41586-019-1659-4
O’Connell, M. J., Raleigh, J. M., Verkade, H. M., and Nurse, P. (1997). Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 16, 545–554. doi: 10.1093/emboj/16.3.545
Orthwein, A., Fradet-Turcotte, A., Noordermeer, S. M., Canny, M. D., Brun, C. M., Strecker, J., et al. (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193. doi: 10.1126/science.1248024
Ouyang, H., Nussenzweig, A., Kurimasa, A., Soares, V. C., Li, X., Cordon-Cardo, C., et al. (1997). Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J. Exp. Med. 186, 921–929. doi: 10.1084/jem.186.6.921
Paim, L. M. G., and FitzHarris, G. (2019). Tetraploidy causes chromosomal instability in acentriolar mouse embryos. Nat. Commun. 10:4834. doi: 10.1038/s41467-019-12772-8
Park, E., Kim, J. M., Primack, B., Weinstock, D. M., Moreau, L. A., Parmar, K., et al. (2013). Inactivation of Uaf1 causes defective homologous recombination and early embryonic lethality in mice. Mol. Cell. Biol. 33, 4360–4370. doi: 10.1128/MCB.00870-13
Pilzecker, B., Buoninfante, O. A., Van Den Berk, P., Lancini, C., Song, J. Y., Citterio, E., et al. (2017). DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc. Natl. Acad. Sci. U.S.A. 114, E6875–E6883. doi: 10.1073/pnas.1706508114
Pintard, L., and Archambault, V. (2018). A unified view of spatio-temporal control of mitotic entry: Polo kinase as the key. Open Biol. 8:180114. doi: 10.1098/rsob.180114
Pizzi, S., Sertic, S., Orcesi, S., Cereda, C., Bianchi, M., Jackson, A. P., et al. (2015). Reduction of hRNase H2 activity in Aicardi-Goutieres syndrome cells leads to replication stress and genome instability. Hum. Mol. Genet. 24, 649–658. doi: 10.1093/hmg/ddu485
Poli, J., Gasser, S. M., and Papamichos-Chronakis, M. (2017). The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160290. doi: 10.1098/rstb.2016.0290
Pollard, P. J., Spencer-Dene, B., Shukla, D., Howarth, K., Nye, E., El-Bahrawy, M., et al. (2007). Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell 11, 311–319. doi: 10.1016/j.ccr.2007.02.005
Power, M. A., and Tam, P. P. (1993). Onset of gastrulation, morphogenesis and somitogenesis in mouse embryos displaying compensatory growth. Anat. Embryol. 187, 493–504. doi: 10.1007/BF00174425
Przetocka, S., Porro, A., Bolck, H. A., Walker, C., Lezaja, A., Trenner, A., et al. (2018). CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 72, doi: 10.1016/j.molcel.2018.09.014
Puebla-Osorio, N., Lacey, D. B., Alt, F. W., and Zhu, C. (2006). Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol. Cell. Biol. 26, 3935–3941. doi: 10.1128/MCB.26.10.3935-3941.2006
Quinet, A., Lemaçon, D., and Vindigni, A. (2017). Replication fork reversal: players and guardians. Mol. Cell 68, 830–833. doi: 10.1016/j.molcel.2017.11.022
Ray, D., Terao, Y., Nimbalkar, D., Hirai, H., Osmundson, E. C., Zou, X., et al. (2007). Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res. 67, 6605–6611. doi: 10.1158/0008-5472.CAN-06-4815
Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J. E., et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. doi: 10.1038/nature18325
Ray Chaudhuri, A., and Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. doi: 10.1038/nrm.2017.53
Reinhardt, H. C., and Schumacher, B. (2012). The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 28, 128–136. doi: 10.1016/j.tig.2011.12.002
Roa, S., Avdievich, E., Peled, J. U., Maccarthy, T., Werling, U., Kuang, F. L., et al. (2008). Ubiquitylated PCNA plays a role in somatic hypermutation and class-switch recombination and is required for meiotic progression. Proc. Natl. Acad. Sci. U.S.A. 105, 16248–16253. doi: 10.1073/pnas.0808182105
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., and Bonner, W. M. (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868. doi: 10.1074/jbc.273.10.5858
Roper, S. J., Chrysanthou, S., Senner, C. E., Sienerth, A., Gnan, S., Murray, A., et al. (2014). ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acids Res. 42, 8914–8927. doi: 10.1093/nar/gku591
Rossi, D. J., Londesborough, A., Korsisaari, N., Pihlak, A., Lehtonen, E., Henkemeyer, M., et al. (2001). Inability to enter S phase and defective RNA polymerase II CTD phosphorylation in mice lacking Mat1. EMBO J. 20, 2844–2856. doi: 10.1093/emboj/20.11.2844
Saldivar, J. C., Cortez, D., and Cimprich, K. A. (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 18, 622–636. doi: 10.1038/nrm.2017.67
Sasaki, M., Kawahara, K., Nishio, M., Mimori, K., Kogo, R., Hamada, K., et al. (2011). Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 17, 944–951. doi: 10.1038/nm.2392
Schärer, O. D. (2013). Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 5:a012609. doi: 10.1101/cshperspect.a012609
Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H., and Jasin, M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. doi: 10.1016/j.cell.2011.03.041
Scully, R., Panday, A., Elango, R., and Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714. doi: 10.1038/s41580-019-0152-0
Sfeir, A., and Symington, L. S. (2015). Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 40, 701–714. doi: 10.1016/j.tibs.2015.08.006
Shakya, R., Szabolcs, M., Mccarthy, E., Ospina, E., Basso, K., Nandula, S., et al. (2008). The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 105, 7040–7045. doi: 10.1073/pnas.0711032105
Sharan, S. K., Morimatsu, M., Albrecht, U., Lim, D. S., Regel, E., Dinh, C., et al. (1997). Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 386, 804–810. doi: 10.1038/386804a0
Shima, N., Munroe, R. J., and Schimenti, J. C. (2004). The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol. Cell. Biol. 24, 10381–10389. doi: 10.1128/MCB.24.23.10381-10389.2004
Shimada, M., Dumitrache, L. C., Russell, H. R., and Mckinnon, P. J. (2015). Polynucleotide kinase-phosphatase enables neurogenesis via multiple DNA repair pathways to maintain genome stability. EMBO J. 34, 2465–2480. doi: 10.15252/embj.201591363
Shu, Z., Smith, S., Wang, L., Rice, M. C., and Kmiec, E. B. (1999). Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can Be partially rescued in a p53(-/-) background. Mol. Cell. Biol. 19, 8686–8693. doi: 10.1128/mcb.19.12.8686
Shui, J. W., Hu, M. C., and Tan, T. H. (2007). Conditional knockout mice reveal an essential role of protein phosphatase 4 in thymocyte development and pre-T-cell receptor signaling. Mol. Cell. Biol. 27, 79–91. doi: 10.1128/MCB.00799-06
Smith, T. G., Laval, S., Chen, F., Rock, M. J., Strachan, T., and Peters, H. (2014). Neural crest cell-specific inactivation of Nipbl or Mau2 during mouse development results in a late onset of craniofacial defects. Genesis 52, 687–694. doi: 10.1002/dvg.22780
Snow, M. H., and Tam, P. P. (1979). Is compensatory growth a complicating factor in mouse teratology? Nature 279, 555–557. doi: 10.1038/279555a0
Snow, M. H. L. (1976). “Embryo Growth during the Immediate Postimplantation Period,” in Embryogenesis in Mammals: Ciba Foundation Symposium 40 - Embryogenesis in Mammals, eds K., Elliott, and M., O’Connor (Amsterdam: Elsevier), 53–70.
Snow, M. H. L. (1977). Gastrulation in the mouse: growth and regionalization of the epiblast. Development 42, 293–303. doi: 10.1111/dgd.12568
Sobinoff, A. P., Allen, J. A., Neumann, A. A., Yang, S. F., Walsh, M. E., Henson, J. D., et al. (2017). BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 36, 2907–2919. doi: 10.15252/embj.201796889
Somyajit, K., Saxena, S., Babu, S., Mishra, A., and Nagaraju, G. (2015). Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 43, 9835–9855. doi: 10.1093/nar/gkv880
Sotiriou, S. K., Kamileri, I., Lugli, N., Evangelou, K., Da-Re, C., Huber, F., et al. (2016). Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol. Cell 64, 1127–1134. doi: 10.1016/j.molcel.2016.10.038
Sozen, B., Amadei, G., Cox, A., Wang, R., Na, E., Czukiewska, S., et al. (2018). Self-assembly of embryonic and two extra-embryonic stem cell types into gastrulating embryo-like structures. Nat. Cell Biol. 20, 979–989. doi: 10.1038/s41556-018-0147-7
Spies, J., Lukas, C., Somyajit, K., Rask, M. B., Lukas, J., and Neelsen, K. J. (2019). 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 21, 487–497. doi: 10.1038/s41556-019-0293-6
Stewart, G. S., Wang, B., Bignell, C. R., Taylor, A. M., and Elledge, S. J. (2003). MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966. doi: 10.1038/nature01446
Stracker, T. H., and Petrini, J. H. J. (2011). The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103. doi: 10.1038/nrm3047
Strzalka, W., and Ziemienowicz, A. (2011). Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann. Bot. 107, 1127–1140. doi: 10.1093/aob/mcq243
Symington, L. S. (2016). Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 51, 195–212. doi: 10.3109/10409238.2016.1172552
Taglialatela, A., Alvarez, S., Leuzzi, G., Sannino, V., Ranjha, L., Huang, J. W., et al. (2017). Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 68, 414–430.e8. doi: 10.1016/j.molcel.2017.09.036
Takai, H., Tominaga, K., Motoyama, N., Minamishima, Y. A., Nagahama, H., Tsukiyama, T., et al. (2000). Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 14, 1439–1447.
Takeuchi, A., Iida, K., Tsubota, T., Hosokawa, M., Denawa, M., Brown, J. B., et al. (2018). Loss of Sfpq Causes Long-Gene Transcriptopathy in the Brain. Cell Rep. 23, 1326–1341. doi: 10.1016/j.celrep.2018.03.141
Tam, P. P. (1989). Regionalisation of the mouse embryonic ectoderm: allocation of prospective ectodermal tissues during gastrulation. Development 107, 55–67.
Tam, P. P., Williams, E. A., and Chan, W. Y. (1993). Gastrulation in the mouse embryo: ultrastructural and molecular aspects of germ layer morphogenesis. Microsc. Res. Tech. 26, 301–328. doi: 10.1002/jemt.1070260405
Tam, P. P. L., Nelson, J., and Rossant, J. (2013). Mammalian Development. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Tebbs, R. S., Flannery, M. L., Meneses, J. J., Hartmann, A., Tucker, J. D., Thompson, L. H., et al. (1999). Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev. Biol. 208, 513–529. doi: 10.1006/dbio.1999.9232
Ter Huurne, M., Chappell, J., Dalton, S., and Stunnenberg, H. G. (2017). Distinct cell-cycle control in two different states of mouse pluripotency. Cell Stem Cell 21, 449–455.e4. doi: 10.1016/j.stem.2017.09.004
Tesar, P. J., Chenoweth, J. G., Brook, F. A., Davies, T. J., Evans, E. P., Mack, D. L., et al. (2007). New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199. doi: 10.1038/nature05972
Thangavel, S., Berti, M., Levikova, M., Pinto, C., Gomathinayagam, S., Vujanovic, M., et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562. doi: 10.1083/jcb.201406100
Theunissen, J. W., Kaplan, M. I., Hunt, P. A., Williams, B. R., Ferguson, D. O., Alt, F. W., et al. (2003). Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol. Cell 12, 1511–1523. doi: 10.1016/s1097-2765(03)00455-6
Tichy, E. D., Pillai, R., Deng, L., Liang, L., Tischfield, J., Schwemberger, S. J., et al. (2010). Mouse embryonic stem cells, but not somatic cells, predominantly use homologous recombination to repair double-strand DNA breaks. Stem Cells Dev. 19, 1699–1711. doi: 10.1089/scd.2010.0058
Toledo, L. I., Altmeyer, M., Rask, M. B., Lukas, C., Larsen, D. H., Povlsen, L. K., et al. (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. doi: 10.1016/j.cell.2013.10.043
Tominaga, Y., Li, C., Wang, R.-H., and Deng, C.-X. (2006). Murine Wee1 plays a critical role in cell cycle regulation and pre-implantation stages of embryonic development. Int. J. Biol. Sci. 2, 161–170. doi: 10.7150/ijbs.2.161
Tomoda, K., Yoneda-Kato, N., Fukumoto, A., Yamanaka, S., and Kato, J. Y. (2004). Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J. Biol. Chem. 279, 43013–43018. doi: 10.1074/jbc.M406559200
Tsuzuki, T., Fujii, Y., Sakumi, K., Tominaga, Y., Nakao, K., Sekiguchi, M., et al. (1996). Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 6236–6240. doi: 10.1073/pnas.93.13.6236
Uchimura, A., Hidaka, Y., Hirabayashi, T., Hirabayashi, M., and Yagi, T. (2009). DNA polymerase delta is required for early mammalian embryogenesis. PLoS One 4:e4184. doi: 10.1371/journal.pone.0004184
Uziel, T., Lerenthal, Y., Moyal, L., Andegeko, Y., Mittelman, L., and Shiloh, Y. (2003). Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22, 5612–5621. doi: 10.1093/emboj/cdg541
Vallabhaneni, H., Lynch, P. J., Chen, G., Park, K., Liu, Y., Goehe, R., et al. (2018). High basal levels of gammaH2AX in human induced pluripotent stem cells are linked to replication-associated DNA damage and repair. Stem Cells 36, 1501–1513. doi: 10.1002/stem.2861
Valnegri, P., Huang, J., Yamada, T., Yang, Y., Mejia, L. A., Cho, H. Y., et al. (2017). RNF8/UBC13 ubiquitin signaling suppresses synapse formation in the mammalian brain. Nat. Commun. 8:1271. doi: 10.1038/s41467-017-01333-6
van Harten, A. M., Buijze, M., Van Der Mast, R., Rooimans, M. A., Martens-De Kemp, S. R., Bachas, C., et al. (2019). Targeting the cell cycle in head and neck cancer by Chk1 inhibition: a novel concept of bimodal cell death. Oncogenesis 8:38. doi: 10.1038/s41389-019-0147-x
Van Ly, D., Low, R. R. J., Frolich, S., Bartolec, T. K., Kafer, G. R., Pickett, H. A., et al. (2018). Telomere loop dynamics in chromosome end protection. Mol. Cell 71, 510–525.e6. doi: 10.1016/j.molcel.2018.06.025
Wachowicz, P., Fernández-Miranda, G., Marugán, C., Escobar, B., and De Cárcer, G. (2016). Genetic depletion of Polo-like kinase 1 leads to embryonic lethality due to mitotic aberrancies. Bioessays 38, (Suppl. 1), S96–S106. doi: 10.1002/bies.201670908
Wallace, S. S. (2014). Base excision repair: a critical player in many games. DNA Repair 19, 14–26. doi: 10.1016/j.dnarep.2014.03.030
Wang, X., Chang, Y., Li, Y., Zhang, X., and Goodrich, D. W. (2006). Thoc1/Hpr1/p84 is essential for early embryonic development in the mouse. Mol. Cell. Biol. 26, 4362–4367. doi: 10.1128/MCB.02163-05
Wang, Y., Putnam, C. D., Kane, M. F., Zhang, W., Edelmann, L., Russell, R., et al. (2005). Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 37, 750–755. doi: 10.1038/ng1587
Wang, Z. Q., Auer, B., Stingl, L., Berghammer, H., Haidacher, D., Schweiger, M., et al. (1995). Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 9, 509–520. doi: 10.1101/gad.9.5.509
Ward, I. M., Minn, K., Van Deursen, J., and Chen, J. (2003). p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23, 2556–2563. doi: 10.1128/mcb.23.7.2556-2563.2003
Watanabe, N., Arai, H., Nishihara, Y., Taniguchi, M., Watanabe, N., Hunter, T., et al. (2004). M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. U.S.A. 101, 4419–4424. doi: 10.1073/pnas.0307700101
Watson, C. M., and Tam, P. P. (2001). Cell lineage determination in the mouse. Cell Struct. Funct. 26, 123–129.
Weiss, R. S., Enoch, T., and Leder, P. (2000). Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev. 14, 1886–1898.
Wen, B., Li, R., Cheng, K., Li, E., Zhang, S., Xiang, J., et al. (2017). Tetraploid embryonic stem cells can contribute to the development of chimeric fetuses and chimeric extraembryonic tissues. Sci. Rep. 7:3030. doi: 10.1038/s41598-017-02783-0
Wilhelm, T., Olziersky, A.-M., Harry, D., De Sousa, F., Vassal, H., Eskat, A., et al. (2019). Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 10:3585. doi: 10.1038/s41467-019-11584-0
Xanthoudakis, S., Smeyne, R. J., Wallace, J. D., and Curran, T. (1996). The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. U.S.A. 93, 8919–8923. doi: 10.1073/pnas.93.17.8919
Xiao, Y., and Weaver, D. T. (1997). Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 25, 2985–2991. doi: 10.1093/nar/25.15.2985
Xiao, Z., Chen, Z., Gunasekera, A. H., Sowin, T. J., Rosenberg, S. H., Fesik, S., et al. (2003). Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 278, 21767–21773. doi: 10.1074/jbc.M300229200
Xie, R., Medina, R., Zhang, Y., Hussain, S., Colby, J., Ghule, P., et al. (2009). The histone gene activator HINFP is a nonredundant cyclin E/CDK2 effector during early embryonic cell cycles. Proc. Natl. Acad. Sci. U.S.A. 106, 12359–12364. doi: 10.1073/pnas.0905651106
Xu, Y., Ashley, T., Brainerd, E. E., Bronson, R. T., Meyn, M. S., and Baltimore, D. (1996). Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 10, 2411–2422. doi: 10.1101/gad.10.19.2411
Yamazaki, S., Ishii, A., Kanoh, Y., Oda, M., Nishito, Y., and Masai, H. (2012). Rif1 regulates the replication timing domains on the human genome. EMBO J. 31, 3667–3677. doi: 10.1038/emboj.2012.180
Yang, W., Klaman, L. D., Chen, B., Araki, T., Harada, H., Thomas, S. M., et al. (2006). An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev. Cell 10, 317–327. doi: 10.1016/j.devcel.2006.01.002
Yata, K., Bleuyard, J. Y., Nakato, R., Ralf, C., Katou, Y., Schwab, R. A., et al. (2014). BRCA2 coordinates the activities of cell-cycle kinases to promote genome stability. Cell Rep. 7, 1547–1559. doi: 10.1016/j.celrep.2014.04.023
Yonemasu, R., Minami, M., Nakatsu, Y., Takeuchi, M., Kuraoka, I., Matsuda, Y., et al. (2005). Disruption of mouse XAB2 gene involved in pre-mRNA splicing, transcription and transcription-coupled DNA repair results in preimplantation lethality. DNA Repair 4, 479–491. doi: 10.1016/j.dnarep.2004.12.004
Yoon, J. C., Ling, A. J., Isik, M., Lee, D. Y., Steinbaugh, M. J., Sack, L. M., et al. (2014). GLTSCR2/PICT1 links mitochondrial stress and Myc signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 3781–3786. doi: 10.1073/pnas.1400705111
Yoshida, K., Kuo, F., George, E. L., Sharpe, A. H., and Dutta, A. (2001). Requirement of CDC45 for postimplantation mouse development. Mol. Cell. Biol. 21, 4598–4603. doi: 10.1128/MCB.21.14.4598-4603.2001
Zaugg, K., Su, Y. W., Reilly, P. T., Moolani, Y., Cheung, C. C., Hakem, R., et al. (2007). Cross-talk between Chk1 and Chk2 in double-mutant thymocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 3805–3810. doi: 10.1073/pnas.0611584104
Zaveri, L., and Dhawan, J. (2018). Cycling to meet fate: connecting pluripotency to the cell cycle. Front. Cell Dev. Biol. 6:57. doi: 10.3389/fcell.2018.00057
Zellweger, R., Dalcher, D., Mutreja, K., Berti, M., Schmid, J. A., Herrador, R., et al. (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 208, 563–579. doi: 10.1083/jcb.201406099
Zeman, M. K., and Cimprich, K. A. (2014). Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9. doi: 10.1038/ncb2897
Zhang, Y., and Hunter, T. (2014). Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 134, 1013–1023. doi: 10.1002/ijc.28226
Zhang, Y., Zolov, S. N., Chow, C. Y., Slutsky, S. G., Richardson, S. C., Piper, R. C., et al. (2007). Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc. Natl. Acad. Sci. U.S.A. 104, 17518–17523. doi: 10.1073/pnas.0702275104
Zhao, B., Zhang, W., Cun, Y., Li, J., Liu, Y., Gao, J., et al. (2018). Mouse embryonic stem cells have increased capacity for replication fork restart driven by the specific Filia-Floped protein complex. Cell Res. 28, 69–89. doi: 10.1038/cr.2017.139
Zhao, B., Zhang, W. D., Duan, Y. L., Lu, Y. Q., Cun, Y. X., Li, C. H., et al. (2015). Filia Is an ESC-Specific Regulator of DNA Damage Response and Safeguards Genomic Stability. Cell Stem Cell 16, 684–698. doi: 10.1016/j.stem.2015.03.017
Zheng, P. (2020). Maintaining genomic stability in pluripotent stem cells. Genome Instabil. Dis. 1, 92–97.
Zhou, Z., Wang, L., Ge, F., Gong, P., Wang, H., Wang, F., et al. (2018). Pold3 is required for genomic stability and telomere integrity in embryonic stem cells and meiosis. Nucleic Acids Res. 46, 3468–3486. doi: 10.1093/nar/gky098
Zhu, J., Petersen, S., Tessarollo, L., and Nussenzweig, A. (2001). Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 11, 105–109. doi: 10.1016/s0960-9822(01)00019-7
Ziegler-Birling, C., Helmrich, A., Tora, L., and Torres-Padilla, M.-E. (2009). Distribution of p53 binding protein 1 (53BP1) and phosphorylated H2A.X during mouse preimplantation development in the absence of DNA damage. Int. J. Dev. Biol. 53, 1003–1011. doi: 10.1387/ijdb.082707cz
Zimmermann, M., Lottersberger, F., Buonomo, S. B., Sfeir, A., and De Lange, T. (2013). 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339, 700–704. doi: 10.1126/science.1231573
Keywords: early development, embryology, pluripotency, DNA damage response, DNA repair, DNA replication, replication stress response
Citation: Kafer GR and Cesare AJ (2020) A Survey of Essential Genome Stability Genes Reveals That Replication Stress Mitigation Is Critical for Peri-Implantation Embryogenesis. Front. Cell Dev. Biol. 8:416. doi: 10.3389/fcell.2020.00416
Received: 10 February 2020; Accepted: 05 May 2020;
Published: 29 May 2020.
Edited by:
Liz Caldon, Garvan Institute of Medical Research, AustraliaReviewed by:
Fumiko Esashi, University of Oxford, United KingdomYibo Luo, The University of Toledo, United States
Copyright © 2020 Kafer and Cesare. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony J. Cesare, dGNlc2FyZUBjbXJpLm9yZy5hdQ==