 Yigang Wang
Yigang Wang Hui-Hui Liu
Hui-Hui Liu Yu-Ting Cao
Yu-Ting Cao Lei-Lei Zhang
Lei-Lei Zhang Fang Huang
Fang Huang Cong Yi
Cong Yi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 10 June 2020
Sec. Membrane Traffic and Organelle Dynamics
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00413
This article is part of the Research Topic Keeping in Touch: The Role of Organelle Dynamics and Contacts in Health and Disease View all 15 articles
Mitochondria are key cellular organelles and play vital roles in energy metabolism, apoptosis regulation and cellular homeostasis. Mitochondrial dynamics refers to the varying balance between mitochondrial fission and mitochondrial fusion that plays an important part in maintaining mitochondrial homeostasis and quality. Mitochondrial malfunction is involved in aging, metabolic disease, neurodegenerative disorders, and cancers. Mitophagy, a selective autophagy of mitochondria, can efficiently degrade, remove and recycle the malfunctioning or damaged mitochondria, and is crucial for quality control. In past decades, numerous studies have identified a series of factors that regulate mitophagy and are also involved in carcinogenesis, cancer cell migration and death. Therefore, it has become critically important to analyze signal pathways that regulate mitophagy to identify potential therapeutic targets. Here, we review recent progresses in mitochondrial dynamics, the mechanisms of mitophagy regulation, and the implications for understanding carcinogenesis, metastasis, treatment, and drug resistance.
Mitochondria are the energy-producing cellular compartment, also known as the cell’s “power house,” and act as the primary site for aerobic respiration (Giampazolias and Tait, 2016). Mitochondria not only have this core bioenergy function, but they also provide basic materials for tumor anabolism, tumor cell redox and calcium homeostasis control, transcription regulation and cell death control (Chen and Mellman, 2017). The effects of the host immune system on tumor development, progression, and response to therapy are similarly dependent on tumor mitochondrial metabolism (Wallace, 2012; Vyas et al., 2016). Consequently, mitochondria are expected to be an excellent target to exploit for new anticancer drugs (Porporato et al., 2018).
Mitophagy, the specific autophagy of mitochondria, plays a pivotal role in mitochondrial quality control by clearing damaged mitochondria. Severe defects in mitophagy, are associated with complete impairment of mitochondrial functions, oncogenesis and tumor progression in multiple tumors (Guo et al., 2013; Rosenfeldt et al., 2013; Rao et al., 2014). Hence, the study of mitophagy regulatory mechanisms in cancer-related events is of great significance. Mitochondria are highly dynamic structures. The coordinated cycle of mitochondrial division and fusion (“mitochondrial dynamics”; Tilokani et al., 2018) provides fast morphological adaptation of mitochondria and plays an important role in the regulation of the cell cycle, cellular immunity, apoptosis, and mitochondrial mass (Wai and Langer, 2016). Mitochondrial dynamic dysfunction can directly damage cells through inadequate ATP supply or high production of ROS and NOS. This in turn can lead to abnormalities, such as neurodegenerative diseases, cancer and autoimmune diseases (Vásquez-Trincado et al., 2016). Many human diseases are associated with mutations in mitochondrial core mechanical components and flaws in mitochondrial dynamics (Mishra and Chan, 2016; Chan, 2020).
This review covers the latest progress in mitochondrial dynamics and mechanisms of mitophagy regulation in the context of carcinogenesis, metastasis, cancer therapy and drug resistance, and proposes future directions for developing novel cancer therapy strategies focused on mitochondria.
Mitochondrial dynamics is the process of mitochondrial fusion and fission, and determines the shape, quality and quantity of mitochondria (Chan, 2012; Pernas and Scorrano, 2016). Mitochondrial dynamics is closely linked to mitochondrial functions, such as cell proliferation, cell metabolism, and cell migration (Chan, 2006) and is tightly regulated by a variety of proteins.
The mitochondrial fission process (mitofission) is mainly mediated by Drp1. Drp1 belongs to a class of GTP-binding proteins and can be recruited from cytoplasm to the mitochondrial membrane with the assistance of mitochondrial receptor proteins Fis1, MFF, MID49, and MID51 (Losón et al., 2013). Then Drp1 located on the mitochondrial membrane can form a ring structure that closely surrounds the mitochondrion (Ingerman et al., 2005), and induces a breakage of the mitochondrial membrane requiring hydrolysis of GTP (Ji et al., 2015). The phosphorylation, ubiquitination and sumoylation of Drp1 can regulate mitofission by influencing Drp1 stability and recruitment (Chang and Blackstone, 2010). Phosphorylation of Drp1 on Ser585 catalyzed by CDK 1/Cyclin B promotes mitofission in mitotic cells (Taguchi et al., 2007), while phosphorylation of Drp1 on Ser637 can inhibit mitofission.
The mitochondrial fusion process (mitofusion) can be divided into outer mitochondrial membrane (OMM) fusion mediated by mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) and inner mitochondrial membrane (IMM) fusion mediated by OPA1 (Ni et al., 2015). Mfn and OPA1 are dynamin-related GTPases. Mfn1 and Mfn2 expressed on the OMM can interact to mediate fusion between adjacent OMM (Cao et al., 2017). OPA1 located in the IMM together with Mfn1 mediates the fusion of IMM (Cipolat et al., 2004). At present, the exact mechanism of IMM fusion is not clear, but OPA1 has been proved to be necessary. Fusion-related proteins are regulated by posttranslational modifications that effect their abundance and activity (Senft and Ronai, 2016); Mfn1 and Mfn2 activities can be modified by specific phosphorylations, and ubiquitination of the proteins may lead to their degradation.
Mitochondrial dynamics are closely related to the occurrence and metastasis of tumors. Environmental alterations around a cell lead to mitochondrial dynamics change, which is also a mechanism for cancer adaption. There are disorders of mitochondrial dynamics in many cancers; up-regulation of fission-related proteins and down-regulation of fusion-related proteins is found in many types of cancer (Rehman et al., 2012; Zhao et al., 2013; Huang et al., 2017). For example, there is an increase of mitochondrial fragmentation and phosphorylation activation of Drp1 in brain tumor initiation cells (Xie et al., 2015). Additionally, the expression of Mfn2 in lung cancer is lower than that in normal tissues, and overexpression of Drp1/Mfn2 can affect mitochondrial dynamics and inhibit the proliferation of lung cancer cells (Rehman et al., 2012). The findings imply that Drp1/Mfn2 exerts different effects in different cancers.
Targeting therapy based on mitochondrial dynamic-related factors is becoming a potential method for cancer therapy. For instance, hypoxia increases the expression of Drp1 and mitofission in glioblastoma cells, enhances tumor migration, and treatment with Mdivi-1 (DRP1 inhibitor) reduces hypoxia-induced migration (Wan et al., 2014). Mdivi-1 is the best drug to use to characterize mitochondrial dynamics (Cassidy-Stone et al., 2008). It can induce proliferation arrest and apoptosis of tumor cells (Rehman et al., 2012; Wang et al., 2015), but it has some cytotoxicity to normal cells. The chemosensitivity of cancer cells can be improved by mitofission. By down-regulating the phosphorylation of Drp1 at Ser637, Piperlongumine can induce the apoptosis of cisplatin-resistant ovarian cancer cells (Farrand et al., 2013; Table 1). Moreover, treatment with a lectin, Sambucus nigra agglutinin, induces mitofission-mediated apoptosis by stimulating Drp1 translocation (Chowdhury et al., 2017). However, there is a reverse action for Drp1 in cancer treatment. The inhibition of Drp1-mediated mitochondrial fission results in the sensitization of ovarian cancer cells to cisplatin (Yang et al., 2017). Thus, the mechanisms of mitochondrial dynamics can rationally be used as a potential target for cancer therapy.

Table 1. Compounds targeting mitochondria in the treatment of cancer.
Mitophagy can selectively remove damaged or redundant mitochondria. Mitophagy is an essential part of mitochondrial stress response and homeostasis regulation, also playing a regulatory role in mitochondrial quality control (Abeliovich et al., 2013). Thus, when the mechanism of mitophagy is impaired, mitochondrial function is reduced or mitochondrial redundancy is caused, which will affect cell homeostasis and lead to the occurrence of related diseases.
Principally, mitophagy pathway can be divided into two types: ubiquitin-mediated mitophagy and receptor-mediated mitophagy. The well-studied pathways are PINK1-Parkin mediated ubiquitin pathway and FUNDC1 receptor-mediated pathway, respectively (Figure 1).
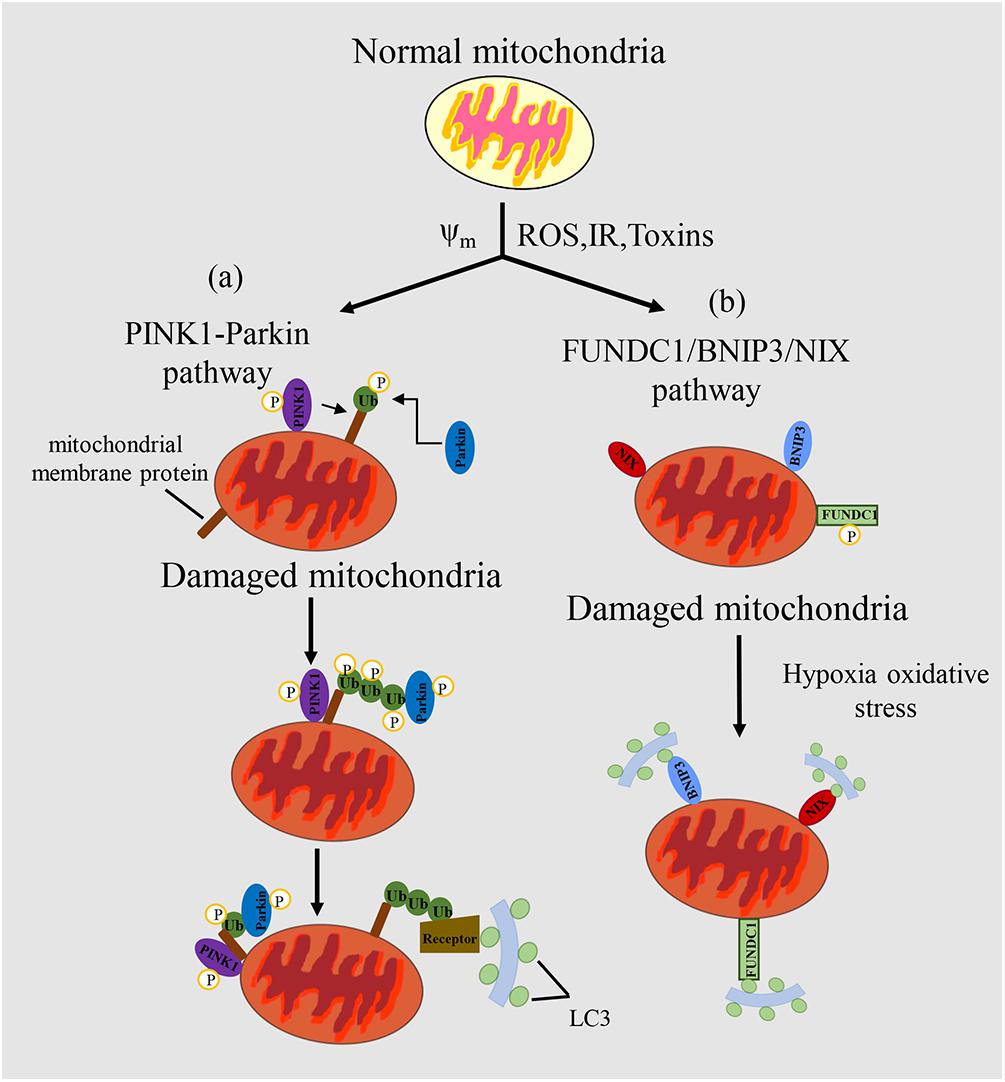
Figure 1. Mechanism of mitophagy regulation in mammals. (a) In the PINK/PARKIN pathway, upon mitochondrial impairment or loss of mitochondrial potential, PINK1 can phosphorylate various target proteins, such as ubiquitin. Then, PINK1 recruits Parkin, and Parkin can amplify the signal through ubiquitination of miyochondrial surface receptor proteins. Receptor proteins can recognize the ubiquitinated proteins, which promotes mitochondria to form autophagosomes and eventually degrade them. (b) In the FUNDC1/BNIP3/NIX pathway, LC3 can be recognized by receptor proteins of mitochondria to promote the complementation of phagophores, thus targeting the mitochondrion for mitophagy. Mitochondrial outer membrane proteins FUNDC1, BNIP3 or NIX bind to LC3-II through their cytoplasmic LIR motifs to promote selective clearance of mitochondria. Δψm, mitochondrial potential; ROS, reactive oxygen species; IR, ischemia/reperfusion; Toxins, poisons produced naturally by organisms.
The PINK1-Parkin mediated regulation of mitophagy is characterized by the ubiquitylation of some key mitochondrial proteins. Parkin, as an E3 ubiquitin ligase, and PINK1, as a serine/threonine kinase, were initially found to be closely related to Parkinson’s disease (Kitada et al., 1998). In normal cells, the PINK1 protein synthesized in the cytoplasm is firstly translocated into the mitochondrial inner membrane, in which is cleaved by PARL protease and degraded by the ubiquitin mediated proteasome pathway (Sekine and Youle, 2018). However, upon mitochondrial impairment or depolarization of mitochondrial membrane potential, the lysis of PINK1 is inhibited, which causes the selective accumulation of PINK1 in OMM (Kondapalli et al., 2012). PINK1 can be activated by autophosphorylation and then phosphorylate Ub at Ser65. Phosphorylated Ub can bind and recruit Parkin from cytoplasm to OMM where PINK1 phosphorylates Parkin at Ser65 and activates its ubiquitin ligase activity (Schubert et al., 2017). Parkin can ubiquitylate a few outer mitochondrial membrane proteins, such as Mfn1, Mfn2, VDAC, MIRO1 (Chan et al., 2011). Ubiquitylated protein such as VDAC can be recognized by LC3 junction proteins in the cytoplasm, including SQSTM1/P62, OPTN, NDP52, TAX1BP1, and NBR1, can bind with this ubiquitinated protein and induce mitophagy by recognizing and binding to LC3 on the autophagy membrane (Lazarou et al., 2015). During this process, Ambra1 is thought to be an interaction protein of Parkin, and its main function is to stimulate cellular autophagy and mitochondrial clearance by motivating the formation of new phagophore (Fimia et al., 2007). Thus, Ambra1 plays the crucial role in cell autophagy and mitophagy.
The main feature of the mitophagy pathway is that it is receptor-mediated, via not only protein receptors, but also some lipid molecules (Bingol et al., 2014). ATG32 (Kanki et al., 2009; Okamoto et al., 2009) in yeast and NIX (Novak et al., 2010), BNIP3 (Zhu et al., 2013), FUNDC1 (Liu et al., 2012), FKBP8 (Bhujabal et al., 2017), Bcl2L13 (Murakawa et al., 2015; Otsu et al., 2015), Ambra1 (Strappazzon et al., 2015), PHB2 (Wei et al., 2017), and NLRX1 (Zhang et al., 2019) in mammalian systems have been identified. The common characteristic of these receptor proteins is that they contain a conserved LC3-interacting receptor (LIR) domain, a key domain that binds directly to Atg8/LC3 and other proteins in the family.
Under the starvation conditions, mitophagy occurs in yeast cells. The occurrence may be related to the accumulation of mitochondrial oxidative damage (Farré et al., 2009). Yeast mitophagy is mainly mediated by Atg32, the first receptor that was found to mediate mitophagy (Kanki et al., 2009; Okamoto et al., 2009). Atg32 is a single transmembrane protein with the N-terminal and C-terminal exposed to cytoplasm and mitochondrial stroma respectively (Kondo-Okamoto et al., 2012). After mitochondrial damage, Protein kinase 2 can promote the phosphorylation of Atg32 in Ser114 and Ser119. Phosphorylated Atg32 forms a complex with Atg11, which is the first step of mitochondrial degradation (Hirota et al., 2012) and essential for the initiation of mitophagy in a pre-autophagosomal structure (PAS). Then Atg11 interacts with Atg8 which enables mitochondrial recognition by autophagy elements and recruitment into autophagy precursors. Atg32 can also directly interact with Atg8, to promote the engulfment of the mitochondria by the phagocytosis membrane. The interaction of Atg32 with Atg8 and Atg11 promotes the formation a mitophagy-initiating polymeric body (Kanki et al., 2011). Under nitrogen starvation conditions, the effect of Atg32-Atg11 is enhanced (Kanki et al., 2011). The knockout of Atg32 does not affect the non-selective autophagy, the cytosol-to-vacuole targeting (Cvt) pathway or the occurrence of peroxidase autophagy, but completely inhibits mitophagy. Atg11 is a binding protein in selective mitophagy that recognizes localized receptor proteins on autophagosomes. Atg8 is implicated in the expansion of phagocytic bubbles. LC3 is the Atg8 homologue in mammals, and is involved in the identification process between autophagosome membrane and target (Kabeya et al., 2000). When Atg32 is absent, cells grow normally and intracellular ROS levels remain unchanged when using non-fermentable carbon as a carbon source (Ashrafi and Schwarz, 2013), suggesting that there is another mitophagy pathway independent of Atg32.
BNIP3 and BNIP3L/NIX are both OMM proteins that can trigger mitophagy under hypoxia through binding of their LIR sequences with LC3. NIX-mediated mitophagy was firstly found in the maturation process of red blood cells (Sandoval et al., 2008). The dimerization of BNIP3 is necessary for its interaction with LC3 that is promoted by the phosphorylation of two serine sites (Ser17 and Ser24) in BNIP3 promotes the interaction with LC3. BNIP3 and Nix have different tissue specificity. Nix is mainly expressed in hematopoietic tissue (Diwan et al., 2007), while BNIP3 is widely distributed in heart, liver and muscle. BNIP3 is regulated by RB, NF-κβ, FOX03, Ras and p53, while p53 can also regulate Nix (Mammucari et al., 2007). Hypoxia activates hypoxia inducible factor 1α (HIF1α), which can increases the expression of BNIP3 (Subarsky and Hill, 2003). Additionally, BNIP3 knockout can up-regulate the expression of NIX, but this upregulation cannot make up for the decrease of mitochondrial autophagy caused by BNIP3 knockout (Shi et al., 2014). Hypoxia-induced mitophagy can also be mediated by the outer mitochondrial membrane (OMM) protein termed FUN14 domain-containing protein 1 (FUNDC1) (Liu et al., 2012). FUNDC1 induces mitophagy in a manner independent of the Parkinson’s disease-associated E3 ubiquitin ligase Parkin and does so through the direct binding of its LIR motif to LC3. Phosphorylation or ubiquitin modification of FUNDC1 can affect mitophagy. PGAM5 and ULK1 catalyze de-phosphorylation and phosphorylation of FUNDC1 on Ser13 and Ser17 respectively, which transduces hypoxia signals and induces FUNDC1-mediated mitophagy (Chen et al., 2014). In addition, MARCH5 promotes the degradation of FUNDC1 by ubiquitin modification, and ultimately inhibits the occurrence of mitophagy (Chen Z. et al., 2017).
Mitochondria play important roles in cellular metabolism. Mitophagy maintains mitochondrial homeostasis and normal physiological function, while abnormal mitophagy will cause multiple diseases, such as heart disease, neurodegenerative disease and muscle disease. However, the role of mitophagy in tumor development and progression needs further study. More and more studies have found that there are different levels of mitophagy in diverse cancers including liver cancer, rectal cancer, breast cancer, lung cancer, which reflects the close relationship between mitophagy and cancer (Chourasia et al., 2015a). The changes in the complex functions of mitochondria have important impacts on the growth and progression of cancer (Panigrahi et al., 2019). The roles of mitophagy in cancer are shown in Figure 2.
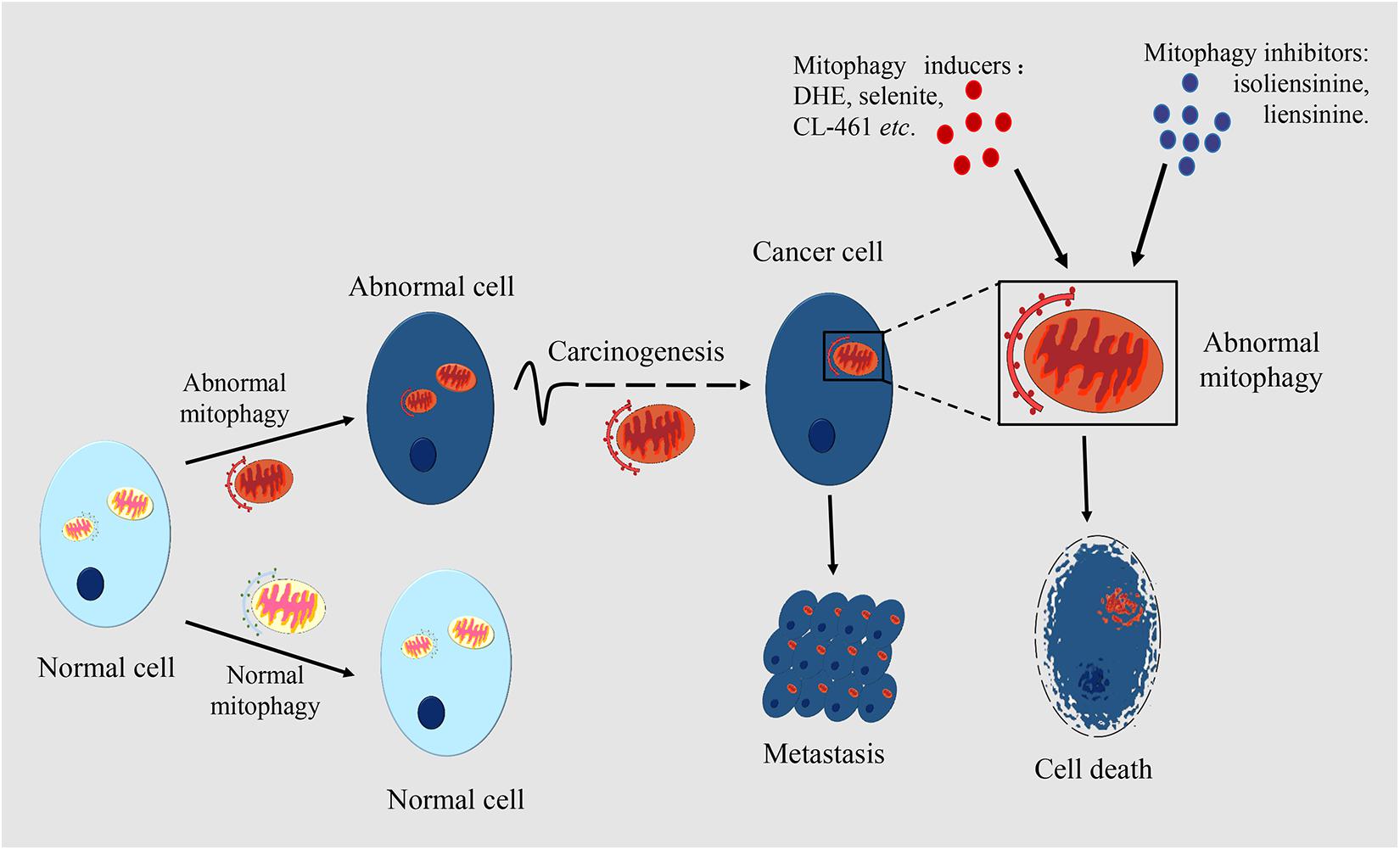
Figure 2. The roles of mitophagy in cancer. Mitophagy occurs in many types of cell. Mitophagic levels are usually enhanced or inhibited in cancer cells, which is different from normal cells. Mitophagy is associated with the occurrence and metastasis of cancers. In certain periods of tumorigenesis, limited mitophagy promotes the development of the tumor, while in established tumors, mitophagy can help the survival of tumor cells. Mitophagy also plays an important role in cancer metastasis. By making mitophagy as the target, inducers or inhibitors of mitophagy can have an anticancer effect through regulating the level of mitophagy. Damaged mitochondria in cancer cells are rapidly cleared through mitophagy which mediates the drug resistance of cancer cells.
Dysfunction in mitophagy has a close connection with tumorigenesis and tumor development (Chang et al., 2017). The role of mitophagy differs in different stages of tumor development. In the early stage of tumorigenesis, mitophagy maintains normal cell metabolism and inhibits tumorigenesis, while in the later stage of tumor development, the occurrence of mitophagy improves cell tolerance and promotes the development of the tumor.
The loss, mutation or functional change in diverse pivotal genes results in the accumulation of damaged mitochondria and suppression of mitophagy, which eventually stimulates tumorigenesis (Panigrahi et al., 2019). PINK1/Parkin pathway is known as the key pathway of mitophagy (Youle and Narendra, 2011). The loss of function of Parkin can inhibit mitophagy and promote carcinogenesis in various cancer models. The inhibition of mitophagy further results in an upsurge of reactive oxygen species which influence on the function of cells, tissues, organs and even whole systems. An early study found that mice with Parkin knock-out are more susceptible to spontaneous development of hepatocellular carcinoma (HCC) than the mock mice (Fujiwara et al., 2008). Moreover, the result also showed that Parkin-deficient hepatocytes continue to proliferate and exert an antiapoptotic effect by upregulating endogenous follistatin, which eventually promotes cancer progress.
PARK2 gene which is located on human chromosome 6 (FRA6E, 6q26), encodes Parkin protein and is very sensitive to gene mutation (Burrell et al., 2013). Parkin mutations are often detected in various tumors such as lung cancer (D’Amico et al., 2015), glioma (Maugeri et al., 2015), and colon cancer (Poulogiannis et al., 2010). The appearance of mutated forms of PARK2 gene is distinct in different cancers (Bernardini et al., 2017). For example, PARK2 amplification is common in sarcomas and uterine cancers, but damaging mutations of PARK2 such as deletion or loss of function often occur in other tumors. It was reported that up-regulation of Parkin expression can inhibit cancer cell growth in hepatocarcinoma, colon cancer, breast cancer and glioblastoma (Wang et al., 2004; Poulogiannis et al., 2010; Tay et al., 2010; Veeriah et al., 2010), while down-regulation of Parkin expression promotes cell proliferation and tumorigenesis in pancreatic cancer (Sun et al., 2013). When PARK2 knock-out mice were hybridized with colorectal cancer adenomatous polyposis mice, the development of intestinal adenoma in neonatal mice accelerated rapidly, and the diversity of polyps increased (Poulogiannis et al., 2010), indicating that PARK2 is a tumor suppressor gene.
Mitophagy has gradually become recognized as a critical adaptation in response to hypoxia. Just as cells can reduce the number of mitochondria and not only limit the production of reactive oxygen clusters, they can also maximize the use of available oxygen. Under hypoxic condition, BNIP3 and its ligand Nix are two main receptors in mitophagy. Like Parkin, Nix also can play an important role in tumorigenesis. Under hypoxia conditions, HIF-1 induces up-regulation of Nix and BNIP3 expression in tumor tissues (Sowter et al., 2001). Nix is highly expressed in hypoxic tumor cells, and Nix-mediated mitophagy promotes glioblastoma survival. The silencing of Nix impairs the clearance of ROS, cancer stem cell maintenance, and inhibits the survival of tumor cells (Jung et al., 2019). Oncogenic KRAS induces NIX-mediated mitophagy, and promotes pancreatic carcinogenesis, but the loss of NIX leads to the restoration of mitochondrial function, which significantly delays pancreatic cancer progression (Humpton et al., 2019). However, another report showed that up-regulated expression of NIX induced by p53 under hypoxia promotes tumor cells apoptosis, while knockout of Nix gene accelerates tumor growth (Fei et al., 2004).
Another mitophagy-related protein FUNDC1 also plays a vital role in carcinogenesis. In early-stage cervical cancer patients, it was found that FUNDC1 has a higher expression level than adjacent normal cells (Hou et al., 2017). Moreover, the deletion of FUNDC1 in tumor cells obviously inhibited cell proliferation, triggered cell apoptosis and increased the sensibility of tumor cell to chemo-radiotherapy such as cisplatin and ionizing radiation. FUNDC1 accumulates in most human HCC. In hepatocytes, FUNDC1 knock-out promotes the initiation and development of diethylnitrosamine (DEN)-induced primary HCC; in contrast when FUNDC1 is transferred into hepatocytes it inhibits hepatocarcinogenesis (Li et al., 2019). In this context, specific deletion of FUNDC1 in hepatocytes leads to the accumulation of dysfunctional mitochondria, and further stimulates a series of events including the activation of inflammasome and JAK/STAT signaling. This suggests that mitophagy can inhibit the occurrence and development of HCC by inhibiting the activation of inflammatory bodies.
During cancer progression, some cancer cells convert the aerobic respiration of mitochondria into glycolysis to maintain cancer cell’s energy needs. This is called the Warburg effect (Bensinger and Christofk, 2012). Cancer cells are typically characterized by a Warburg effect, abnormal quality control of mitochondria, production of ROS, regulation of cell redox state, and lack of apoptosis signals (Panigrahi et al., 2019). By eliminating dysfunctional mitochondria, mitophagy can promote tumor cell survival as an adaptation to stress. In the process of mitophagy, Parkin increases the level of oxidative metabolism and inhibits the Warburg effect downstream of the p53 tumor suppressor, which is likely to be achieved by enhancing the integrity of mitochondrion (Zhang et al., 2011). Thus, as a p53 target gene, Parkin may exhibit tumor suppressive activity. Another study further confirmed that in breast cancer, Parkin inhibited tumor migration and invasion through targeting HIF-1α for ubiquitination and degradation (Deng et al., 2017). When the Parkin gene was transferred into breast cancer cells with a deleted Parkin gene the progression of breast cancer and the rate of metastasis were significantly decreased. Notably, there is also emerging evidence that Parkin may promote cancer metastasis. Compared with normal dermatic tissues, pathological analysis showed that the expression of Parkin and cancer metastasis was obviously enhanced in melanoma (Lee et al., 2018). Further results indicated that the loss of Parkin inhibited tumor formation and metastasis of melanoma by suppressing Mfn2 ubiquitination (Lee et al., 2018). The cause of the opposite result for Parkin’s role in cancer metastasis could be due to different mechanisms for metastasis in several tumor types.
BNIP3 as a pro-apoptotic protein, can inhibit the fusion of damaged mitochondria and enhance mitophagy (Gustafsson, 2011). However, the role of BNIP3 in cancer metastasis is varied in different cancers. It is frequently found that the high expression of BNIP3 that occurs in many kinds of malignancies, such as salivary gland adenoid cystic carcinoma (Chen et al., 2015), endometrial cancer (Giatromanolaki et al., 2008), DCIS of breast and cervical cancer (Leo et al., 2006) is associated with the aggressiveness of the tumor and poor prognosis. The loss of BNIP3 makes mitophagy proceed abnormally and increases mitochondrial ROS levels, which is related with tumor metastasis in TNBC (Chourasia et al., 2015b). In DCIS, the expression of BNIP3 and Nix are increased, while BNIP3 is not expressed in invasive carcinoma, which is related to tumor cell proliferation index and lymph node metastasis (Sowter et al., 2003). It is also suggested that the higher expression of BNIP3 mediates excessive mitophagy, which inhibits HCC metastasis (Shi et al., 2018). As a small GTPase, Rheb can not only interact with BNIP3 and NIX, but also effectively acts on the upstream region of mTOR pathway to promote cell growth (Li et al., 2007). The inhibition of BNIP3 by Rheb under the interaction of Rheb and BNIP3 decreases the activity of mTOR signal, and suppresses cell growth, which corresponds to the role of BNIP3 in tumor inhibition (Ray et al., 2000).
However, similarly to PINK1/PARKIN, BNIP3 can not only serve as a tumor suppressor, but can also exert oncogenic activity. The epigenetic silencing of BNIP3 could enhance the aggressiveness and metastasis of pancreatic cancer cells, and contribute to resistance to hypoxia-induced cell death in pancreatic cancer (Okami et al., 2004). Besides, up-regulation of BNIP3 has also been reported to accelerate cancer cell migration and invasion (Maes et al., 2014; Wu H. et al., 2015). The dual roles of BNIP3 in cancer progress and metastasis could be due to different interact with different factors through its Bcl-2 homology 3 (BH3) domain, also to the heterogeneity of different tumors.
It is clear that mitophagy is beneficial in maintaining normal healthy physiological processes. Mitophagy shares common regulatory pathways with carcinogenesis and cancer cell death which converge at mitochondria (Lu et al., 2013). Thus, it provides therapeutic targets for removing cancer cells and inducing cancer cell apoptosis; therapies can aim at the crosstalk signals between the processes of mitophagy and cancer progression. Concurrently, mitochondria have a function on cell apoptosis and the ability to inhibit apoptosis and resist cell death is one of the well-established hallmarks of cancer (Hanahan and Weinberg, 2011), which implies the potential for mitochondria-mediated apoptosis in cancer therapy.
In a similar way to macroautophagy, mitophagy also plays dual roles in cancer therapy, including the induction of cell death and promotion of cell survival (Panigrahi et al., 2019). Generally, chemotherapy induces mitochondrial dysfunction and oxidative stress to generate the cytotoxic effects on cancer cells which result in the generation of mitophagy (Cao et al., 2019). Excessive mitophagy causes the loss of functional mitochondria, and further destruction of cellular energy requirement, leading to a form of cancer cell death called mitophagic cell death. However, another role of mitophagy in cancer is that of improving the cell internal environment by clearing abnormal mitochondria, which can result in better fitness in the aggressive environments (Araki et al., 2019).
Some compounds that activate mitophagy are gradually being identified as chemotherapeutic drugs. Dihydroergotamine tartrate (DHE) is a common drug for migraine. A study has reported that DHE induces lung cancer cell death by mitophagy and mitochondria-dependent cell apoptosis (Chang et al., 2016). Mechanistically, DHE could induce PINK1/PARKIN activation, ROS production and cell apoptosis, and reduce membrane permeability and destroy ATP synthesis, finally activating mitophagic cell death. This suggests that DHE might be a promising anticancer medicine for lung cancer. More recently, Zhu et al. (2020) reported a novel anticancer strategy based on excessive mitophagy. They used a novel nanomicelle that targets mitochondria to selectively damage mitochondria in tumor cells, thereby further activating excessive mitophagy pathway-driven lethal energy depletion and phototherapy. An in vivo study showed that CerS1/C18 pyridinium ceramide efficiently induces lethal mitophagy and abrogates growth of tumor xenograft in various solid cancers (Sentelle et al., 2012). In glioma cells, a novel BH3-mimetic AT101 (Meyer et al., 2018) and sodium selenite (Kim et al., 2007) induced an excessive mitophagic cell death that suggests lethal mitophagy as the tumor-suppressor mechanism. Similarly, LCL-461, a mitochondria-targeted ceramide analog, could also kill crenolanib-resistant acute myeloid leukemia (AML) cells through lethal mitophagy (Dany et al., 2016). However, other studies revealed that some compounds that inhibit mitophagy exert antitumor effects in breast cancer. Liensinine, a new inhibitor of autophagy and/or mitophagy, could enhance the sensitivity of breast cancer cells to chemotherapy via mitochondrial fission mediated by DNM1L (Zhou et al., 2015). Isoliensinine could induce cell apoptosis in TNBC by the production of ROS and the activation of p38 MAPK/JNK and improve anti-breast cancer effects by inhibiting mitophagy (Zhang et al., 2015).
In addition to the use of mitophagy-associated compounds, the most prominent approach for mitophagy-mediated cell death is by targeting regulation of mitophagy associated proteins by a selective reduction in mitochondrial compartments (Zhou et al., 2015; Panigrahi et al., 2019). Several mitophagy-related proteins PINK1, Parkin, BNIP3, CDKN2A, PUMA, ULK1, and TR3 could induce mitochondrial damage and activate mitophagy, and ultimately lead to cell death. Activated TR3 receptor could result in irreversible mitophagic cell death and excessive clearance of mitochondria in the melanoma cells to TR3-specific compounds (Wang et al., 2014). Mitochondria-targeting drugs (Mito-CP and Mito-Metformin) trigger mitophagy and inhibit the proliferation of colon cancer cell through an AMPK-mTOR-ULK1-dependent pathway (Boyle et al., 2018). Moreover, lethal mitophagy also happens through activation of ceramide stress in human cancer cells (Sentelle et al., 2012), and PUMA-mediated mitophagy by Abrus agglutinin promotes cell apoptosis by the generation of C18 ceramide or the overexpression of CerS1 (Panda et al., 2018).
Inhibition of the tumor promotion role of mitophagy is another promising strategy for cancer therapy. Many studies reported that mitophagy-associated molecules PINK1, Parkin, BNIP3, FUNDC1 promote tumor development, and provide targets for therapeutic inhibition in multiple cancers (Tan et al., 2007; D’Amico et al., 2015; Liu et al., 2018; Hui et al., 2019). For example, the silencing of PINK1 can not only inhibit the proliferation and migration of lung cancer cells, but can also induce cell apoptosis (Liu et al., 2018). Furthermore, high expression of PINK1 in ESCC patients receiving neoadjuvant therapy is related to low chemotherapy effects and poor prognosis (Yamashita et al., 2017). PINK1-mediated excessive mitophagy promotes survival of cancer cells resistant to chemotherapy, implying that suppression of mitophagy can restore the chemo-sensitivity of ESCC cells (Yamashita et al., 2017). Moreover, overexpression of FUNDC1 promotes growth of cervical cancer cells, and inhibition of expression enhances the sensitivity to cisplatin treatment and ionizing radiation (Hou et al., 2017). This shows that the regulation of mitophagy-associated molecules could affect the outcome of cancer therapy.
Tumor resistance raises complex questions involving multiple steps and factors. Recent study has shown that both the efficacy of tumor chemotherapy and the degree of drug resistance are influenced by mitophagy, probably because cancer cells rapidly sweep away damaged mitochondria through mitophagy to mediate their own drug resistance (Yamashita et al., 2017). Consistent with this, the toxicity of most chemotherapeutics is at least partly due to the induction of mitochondrial dysfunction and oxidative stress (Hockenbery, 2010).
The common chemotherapeutic drugs such as cisplatin, paclitaxel, doxorubicin (Dox) and 5-fluorouracil (5-FU) can kill cancer cells in early treatment of multiple solid cancers. However, the occurrence of drug resistance often leads to treatment failure due to autophagy or mitophagy (Oun et al., 2018). For instance, activation of autophagy triggered by overexpression of galectin-1 leads to chemotherapy resistance of epithelial ovarian cancer to cisplatin (Chen L. et al., 2017) and a similar result has been observed in liver cancer (Su et al., 2016). In addition, hypoxia-induced mitophagy accounts for lung cancer cells resisting cisplatin in an HIF1α- and HIF2α-dependent manner (Wu H. et al., 2015). Notably, an E3 ubiquitin ligase ARIH1 highly expressed in cancer cells could trigger mitophagy in a PINK1-dependent and Parkin-defective way; this countered the chemotherapy-induced cell death in breast cancer and lung adenocarcinomas, eventually resulting in cancer cell resistance (Villa et al., 2017).
However, the desired anticancer results can be achieved through regulation of autophagy or mitophagy. On one hand, suppression of autophagy by Atg gene knockdown could activate the BNIP3-mediated cancer cell death pathway after treatment with cisplatin (Wu H. et al., 2015). Similarly, enhanced sensitivity of multidrug-resistant cancer cells to betulinic acid analog B5G1 that was induced by the inhibition of PINK1-Parkin dependent mitophagy resulted in liver cancer cell death (Yao et al., 2019). On the other hand, inducement of mitophagy could improve drug sensitivity of cancer cells. A recent study found that mitophagy-dependent necrosis and ferroptosis can be induced through the suppression of mitochondrial respiratory chain by BAY 87-2243 in melanoma cells (Basit et al., 2017). In addition, through DNM1L-mediated mitochondrial fusion, mitophagy can enhance the sensitivity of breast cancer cells to liensinine (Zhou et al., 2015). These results implicate mitophagy in drug resistance call for further exploration in different cancers.
CSCs are usually resistant to chemotherapy. Interestingly, mitophagy participates in CSCs-mediated drug resistance. In OSCC, the CD44+/ABCB1+/ADAM17+cells behave with the characteristics of CSCs and exhibit chemo-resistance through the modulation of mitophagy (Naik et al., 2018). BNIP3/NIX-mediated mitophagy contributes to the dox-resistance to CSCs in colorectal cancer and the silencing of BNIP3L both prevents mitophagy and increases the sensitivity to doxorubicin therapy (Yan et al., 2017), which suggests the importance of mitophagy in drug resistance for cancer therapy targeting CSCs.
Furthermore, drug resistance regulated by mitophagy also restricts the efficiency of radiotherapy. A recent study has shown that mitophagy induced by p53/BNIP3 exerts an important effect on the viability of HNSCC cancer cells following radiotherapy by maintaining the integrity of mitochondrial (Chang et al., 2019). BNIP3-dependent mitophagy in the radio-resistant cancer cells relies on wild type p53 status to limit glycolytic remodeling, highlighting the potential use of drugs targeting glycolysis as an alternative strategy for overcoming radio-resistant cancers.
Mitophagy appears to be a pivotal cellular event that disposes of dysfunctional mitochondria by autophagic degradation. Although significant advances have been made in delineating mitophagy regulation and the role of mitophagy in carcinogenesis and cancer therapeutics, it is still an intractable problem to understand how mitophagy acts either as a suppressor or an inducer in cancer therapy. Moreover, the homeostasis and regulation of mitochondrial dynamics and mitophagy retain a complexity that requires more exploration. For example, what is the relationship of mitochondrial dysfunction to the maintenance of CSCs self-renewal in tumor progression? What are the molecular crosstalks and interactions between mitophagy and key oncogenic signaling pathways? Can the regulation of mitophagy can be beneficial to molecular targeting in cancer therapy, for example with checkpoint inhibitors (anti-PD-1, PD-L1, and CTLA4), oncolytic virotherapy, molecular immunotherapy? How might we adjust cancer cell mitophagy behavior in relation to specific oncometabolites in various cellular stresses, including hypoxia, nutrient deficiency, and elevated ROS level? How might we best exploit the targeting therapy strategies based on mitochondria and mitophagy by enhancing drug uptake into cancer cells? How does mitophagy further orchestrate the crosstalk that constitutes the metabolic regulation system of cancer cells, which is the key step for maintaining the balance between glycolysis and OXPHOS? In consideration of such questions, further studies will undoubtedly focus on the exploration of the molecular mechanisms of mitophagy in cancer events, the identification of novel mitophagic modulators and the development of promising mitophagy-based cancer treatments.
YW, Y-TC, H-HL, and L-LZ performed the literature searches and wrote the manuscript. FH received a grant for this project, conceived the idea, and reviewed the drafts. YW contributed to the conceptual idea and supervised the writing process. CY gave suggestions and significantly refined the manuscript.
This work was supported by the Zhejiang Provincial Natural Science Foundation (No. LY18C070002), the National Natural Science Foundation of China (No. 81803069), Zhejiang Medical and Health Science and Technology project (No. 2019337459) and the Grant for 521 talent project of ZSTU to YW, and National Natural Science Foundation of China (Nos. 91754107 and 31771528) to CY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ABCB1, P-glycoprotein; ADAM17, A disintegrin and metalloproteinase; Ambra1, Autophagy and Beclin 1 Regulator 1; ARIH1, Ariadne RBR E3 Ubiquitin Protein Ligase 1; AT 101, (-)-gossypol; Atg, autophagy-related protein; ATP, adenosine triphosphate; Bcl2L13, BCL2 Like 13; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; BNIP3L/NIX, BNIP3-like; CDK 1, cyclin dependent kinase 1; CDKN2A, cyclin-dependent kinase inhibitor 2A; CSC, cancer stem cell; CTLA4, cytotoxic T lymphocyte-associated antigen-4; Cvt, cytoplasm to vacuole targeting; DCIS, ductal carcinoma in situ; DNM1L, dynamin-1-like protein; Drp1, dynamin-related protein 1; ESCC, esophageal squamous cell carcinoma; Fis1, fission, mitochondrial 1; FKBP8, FK506-binding protein 8; FOX03, Forkhead Box O3; FUNDC1, FUN14 domain-containing protein 1; GTP, guanosine triphosphate; GTPases, guanosine triphosphatase; HCT8, Human Colon Tumor (HCT8) Cell Line; HNSCC, head and neck squamous cell carcinoma; JNK, c-JunN-terminal kinase; KRAS, Kirsten Rat Sarcoma Viral Oncogene; LC3, microtubule-associated proteins 1A/1B light chain 3A; LIR, LC3-interacting domain; MAPK, mitogen-activated protein kinase; MARCH5, membrane-associated RING finger protein 5; Mdivi-1, mitochondrial division inhibitor 1; MID49, mitochondrial dynamics protein of 49 kDa; MID51, mitochondrial dynamics protein of 51 kDa; MIRO1, Mitochondrial Rho GTPase 1; Mito-CP, 3-carboxyl proxyl (CP) nitroxide; mTOR, mammalian target of rapamycin; NBR1, neighbor of BRCA1 gene protein; NDP52, nuclear dot protein 52; NF- κβ, nuclear factor kappa beta; NLRX1, NLR (Nucleotide-binding domain and leucine-rich repeat-containing receptors) family member X1; NOS, nitric oxide synthase; OPA1, optic atrophy 1; OPTN, optineurin; OSCC, oral squamous cell carcinoma; OXPHOS, oxidative phosphorylation; PARK2, gene encoding Parkin; PARL, presenilin-associated rhomboid-like; PD-L1, Programmed Cell Death 1 Ligand 1; PGAM5, hosphoglycerate mutase-5; PHB2, Prohibitin 2; PINK1, mitochondrial PTEN-induced kinase 1; PUMA, p53 upregulated modulator of apoptosis; Ras, Rat Sarcoma protein, Small GTP Binding Protein; RB, retinoblastoma; Rheb, Ras homolog enriched in brain protein; ROS, reactive oxygen species; TAX1BP1, TAX1 binding protein 1; TNBC, triple negative breast cancer; TR3, orphan nuclear receptor; ULK1, Unc-51 Like Autophagy Activating Kinase 1; VDAC, Voltage Dependent Anion Channel 1; Yme1L, YME1-Like Protein 1.
Abeliovich, H., Zarei, M., Rigbolt, K. T. G., Youle, R. J., and Dengjel, J. (2013). Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat. Commun. 4:2789. doi: 10.1038/ncomms3789
Araki, K., Kawauchi, K., Sugimoto, W., Tsuda, D., Oda, H., Yoshida, R., et al. (2019). Mitochondrial protein E2F3d, a distinctive E2F3 product, mediates hypoxia-induced mitophagy in cancer cells. Commun. Biol. 2:3. doi: 10.1038/s42003-018-0246-9
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. doi: 10.1038/cdd.2012.81
Basit, F., Van Oppen, L. M., Schöckel, L., Bossenbroek, H. M., Van Emst-De Vries, S. E., Hermeling, J. C., et al. (2017). Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 8:e2716. doi: 10.1038/cddis.2017.133
Bensinger, S. J., and Christofk, H. R. (2012). New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 23, 352–361. doi: 10.1016/j.semcdb.2012.02.003
Bernardini, J. P., Lazarou, M., and Dewson, G. (2017). Parkin and mitophagy in cancer. Oncogene 36, 1315–1327. doi: 10.1038/onc.2016.302
Bhujabal, Z., Birgisdottir, ÅB., Sjøttem, E., Brenne, H. B., Øvervatn, A., Habisov, S., et al. (2017). FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 18, 947–961. doi: 10.15252/embr.201643147
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Boyle, K. A., Van Wickle, J., Hill, R. B., Marchese, A., Kalyanaraman, B., and Dwinell, M. B. (2018). Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Biol. Chem. 293, 14891–14904. doi: 10.1074/jbc.RA117.001469
Burrell, R. A., Mcclelland, S. E., Endesfelder, D., Groth, P., Weller, M.-C., Shaikh, N., et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. doi: 10.1038/nature11935
Cao, S., Shen, Z., Wang, C., Zhang, Q., Hong, Q., He, Y., et al. (2019). Resveratrol improves intestinal barrier function, alleviates mitochondrial dysfunction and induces mitophagy in diquat challenged piglets. Food Funct. 10, 344–354. doi: 10.1039/c8fo02091d
Cao, Y.-L., Meng, S., Chen, Y., Feng, J.-X., Gu, D.-D., Yu, B., et al. (2017). MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 542, 372–376. doi: 10.1038/nature21077
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204. doi: 10.1016/j.devcel.2007.11.019
Chan, D. C. (2006). Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99. doi: 10.1146/annurev.cellbio.22.010305.104638
Chan, D. C. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265–287. doi: 10.1146/annurev-genet-110410-132529
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L. J., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chang, C.-R., and Blackstone, C. (2010). Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 1201, 34–39. doi: 10.1111/j.1749-6632.2010.05629.x
Chang, H. W., Kim, M. R., Lee, H. J., Lee, H. M., Kim, G. C., Lee, Y. S., et al. (2019). p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 38, 3729–3742. doi: 10.1038/s41388-019-0697-6
Chang, J. Y., Yi, H.-S., Kim, H.-W., and Shong, M. (2017). Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim. Biophys. Acta. Bioenerg. 1858, 633–640. doi: 10.1016/j.bbabio.2016.12.008
Chang, S.-H., Lee, A. Y., Yu, K.-N., Park, J., Kim, K. P., and Cho, M.-H. (2016). Dihydroergotamine tartrate induces lung cancer cell death through apoptosis and mitophagy. Chemotherapy 61, 304–312. doi: 10.1159/000445044
Chen, D. S., and Mellman, I. (2017). Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330. doi: 10.1038/nature21349
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 54, 362–377. doi: 10.1016/j.molcel.2014.02.034
Chen, L., Yao, Y., Sun, L., and Tang, J. (2017). Galectin-1 promotes tumor progression via NF-κB signaling pathway in epithelial ovarian cancer. J. Cancer 8, 3733–3741. doi: 10.7150/jca.20814
Chen, Z., Liu, L., Cheng, Q., Li, Y., Wu, H., Zhang, W., et al. (2017). Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 18, 495–509. doi: 10.15252/embr.201643309
Chen, Z., Wu, H., Huang, S., Li, W., Zhang, S., Zheng, P., et al. (2015). Expression of BNIP3 and its correlations to hypoxia-induced autophagy and clinicopathological features in salivary adenoid cystic carcinoma. Cancer Biomark. 15, 467–475. doi: 10.3233/CBM-150474
Chourasia, A. H., Boland, M. L., and Macleod, K. F. (2015a). Mitophagy and cancer. Cancer Metab. 3:4. doi: 10.1186/s40170-015-0130-8
Chourasia, A. H., Tracy, K., Frankenberger, C., Boland, M. L., Sharifi, M. N., Drake, L. E., et al. (2015b). Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 16, 1145–1163. doi: 10.15252/embr.201540759
Chowdhury, S. R., Ray, U., Chatterjee, B. P., and Roy, S. S. (2017). Targeted apoptosis in ovarian cancer cells through mitochondrial dysfunction in response to Sambucus nigra agglutinin. Cell Death Dis. 8:e2762. doi: 10.1038/cddis.2017.77
Cipolat, S., Martins De Brito, O., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U.S.A. 101, 15927–15932. doi: 10.1073/pnas.0407043101
D’Amico, A. G., Maugeri, G., Magro, G., Salvatorelli, L., Drago, F., and D’agata, V. (2015). Expression pattern of parkin isoforms in lung adenocarcinomas. Tumour Biol. 36, 5133–5141. doi: 10.1007/s13277-015-3166-z
Dany, M., Gencer, S., Nganga, R., Thomas, R. J., Oleinik, N., Baron, K. D., et al. (2016). Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128, 1944–1958. doi: 10.1182/blood-2016-04-708750
Deng, Y., Wu, W., Guo, S., Chen, Y., Liu, C., Gao, X., et al. (2017). Altered mTOR and Beclin-1 mediated autophagic activation during right ventricular remodeling in monocrotaline-induced pulmonary hypertension. Respir Res. 18:53. doi: 10.1186/s12931-017-0536-7
Diwan, A., Koesters, A. G., Odley, A. M., Pushkaran, S., Baines, C. P., Spike, B. T., et al. (2007). Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 104, 6794–6799. doi: 10.1073/pnas.0610666104
Farrand, L., Kim, J. Y., Im-Aram, A., Suh, J.-Y., Lee, H. J., and Tsang, B. K. (2013). An improved quantitative approach for the assessment of mitochondrial fragmentation in chemoresistant ovarian cancer cells. PLoS One 8:e74008. doi: 10.1371/journal.pone.0074008
Farré, J.-C., Krick, R., Subramani, S., and Thumm, M. (2009). Turnover of organelles by autophagy in yeast. Curr. Opin. Cell Biol. 21, 522–530. doi: 10.1016/j.ceb.2009.04.015
Fei, P., Wang, W., Kim, S.-H., Wang, S., Burns, T. F., Sax, J. K., et al. (2004). Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 6, 597–609. doi: 10.1016/j.ccr.2004.10.012
Fimia, G. M., Stoykova, A., Romagnoli, A., Giunta, L., Di Bartolomeo, S., Nardacci, R., et al. (2007). Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125. doi: 10.1038/nature05925
Fujiwara, M., Marusawa, H., Wang, H. Q., Iwai, A., Ikeuchi, K., Imai, Y., et al. (2008). Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27, 6002–6011. doi: 10.1038/onc.2008.199
Giampazolias, E., and Tait, S. W. G. (2016). Mitochondria and the hallmarks of cancer. FEBS J. 283, 803–814. doi: 10.1111/febs.13603
Giatromanolaki, A., Koukourakis, M. I., Gatter, K. C., Harris, A. L., and Sivridis, E. (2008). BNIP3 expression in endometrial cancer relates to active hypoxia inducible factor 1alpha pathway and prognosis. J. Clin. Pathol. 61, 217–220. doi: 10.1136/jcp.2007.046680
Guo, J. Y., Karsli-Uzunbas, G., Mathew, R., Aisner, S. C., Kamphorst, J. J., Strohecker, A. M., et al. (2013). Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 27, 1447–1461. doi: 10.1101/gad.219642.113
Gustafsson, A. B. (2011). Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr. Cardiol. 32, 267–274. doi: 10.1007/s00246-010-9876-5
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Hirota, Y., Kang, D., and Kanki, T. (2012). The physiological role of mitophagy: new insights into phosphorylation events. Int. J. Cell Biol. 2012:354914. doi: 10.1155/2012/354914
Hockenbery, D. M. (2010). Targeting mitochondria for cancer therapy. Environ. Mol. Mutagen. 51, 476–489. doi: 10.1002/em.20552
Hou, H., Er, P., Cheng, J., Chen, X., Ding, X., Wang, Y., et al. (2017). High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med. 6, 1871–1881. doi: 10.1002/cam4.1112
Huang, Q., Cao, H., Zhan, L., Sun, X., Wang, G., Li, J., et al. (2017). Mitochondrial fission forms a positive feedback loop with cytosolic calcium signaling pathway to promote autophagy in hepatocellular carcinoma cells. Cancer Lett. 403, 108–118. doi: 10.1016/j.canlet.2017.05.034
Hui, L., Wu, H., Wang, T. W., Yang, N., Guo, X., and Jang, X. J. (2019). Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 21, 596–606. doi: 10.1007/s12094-018-1958-5
Humpton, T. J., Alagesan, B., Denicola, G. M., Lu, D., Yordanov, G. N., Leonhardt, C. S., et al. (2019). Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 9, 1268–1287. doi: 10.1158/2159-8290.CD-18-1409
Ingerman, E., Perkins, E. M., Marino, M., Mears, J. A., Mccaffery, J. M., Hinshaw, J. E., et al. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027. doi: 10.1083/jcb.200506078
Ji, W.-K., Hatch, A. L., Merrill, R. A., Strack, S., and Higgs, H. N. (2015). Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 4:e11553. doi: 10.7554/eLife.11553
Jung, J., Zhang, Y., Celiku, O., Zhang, W., Song, H., Williams, B. J., et al. (2019). Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 79, 5218–5232. doi: 10.1158/0008-5472.CAN-19-0198
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Kanki, T., Klionsky, D. J., and Okamoto, K. (2011). Mitochondria autophagy in yeast. Antioxid. Redox Signal. 14, 1989–2001.
Kanki, T., Wang, K., Cao, Y., Baba, M., and Klionsky, D. J. (2009). Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 17, 98–109. doi: 10.1016/j.devcel.2009.06.014
Kim, E. H., Sohn, S., Kwon, H. J., Kim, S. U., Kim, M.-J., Lee, S.-J., et al. (2007). Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 67, 6314–6324. doi: 10.1158/0008-5472.CAN-06-4217
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Kondo-Okamoto, N., Noda, N. N., Suzuki, S. W., Nakatogawa, H., Takahashi, I., Matsunami, M., et al. (2012). Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J. Biol. Chem. 287, 10631–10638. doi: 10.1074/jbc.M111.299917
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, Y. S., Jung, Y. Y., Park, M. H., Yeo, I. J., Im, H. S., Nam, K. T., et al. (2018). Deficiency of parkin suppresses melanoma tumor development and metastasis through inhibition of MFN2 ubiquitination. Cancer Lett. 433, 156–164. doi: 10.1016/j.canlet.2018.07.007
Leo, C., Horn, L. C., and Höckel, M. (2006). Hypoxia and expression of the proapoptotic regulator BNIP3 in cervical cancer. Int. J. Gynecol. Cancer 16, 1314–1320. doi: 10.1111/j.1525-1438.2006.00394.x
Li, W., Li, Y., Siraj, S., Jin, H., Fan, Y., Yang, X., et al. (2019). FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology (Baltimore, Md.) 69, 604–621. doi: 10.1002/hep.30191
Li, Y., Wang, Y., Kim, E., Beemiller, P., Wang, C.-Y., Swanson, J., et al. (2007). Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 282, 35803–35813. doi: 10.1074/jbc.M705231200
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, L., Zuo, Z., Lu, S., Wang, L., Liu, A., and Liu, X. (2018). Silencing of PINK1 represses cell growth, migration and induces apoptosis of lung cancer cells. Biomed. Pharmacother. 106, 333–341. doi: 10.1016/j.biopha.2018.06.128
Losón, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lu, H., Li, G., Liu, L., Feng, L., Wang, X., and Jin, H. (2013). Regulation and function of mitophagy in development and cancer. Autophagy 9, 1720–1736. doi: 10.4161/auto.26550
Maes, H., Van Eygen, S., Krysko, D. V., Vandenabeele, P., Nys, K., Rillaerts, K., et al. (2014). BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 5:e1127. doi: 10.1038/cddis.2014.94
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi: 10.1016/j.cmet.2007.11.001
Maugeri, G., D’amico, A. G., Magro, G., Salvatorelli, L., Barbagallo, G. M. V., Saccone, S., et al. (2015). Expression profile of parkin isoforms in human gliomas. Int. J. Oncol. 47, 1282–1292. doi: 10.3892/ijo.2015.3105
Meyer, N., Zielke, S., Michaelis, J. B., Linder, B., Warnsmann, V., Rakel, S., et al. (2018). AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy 14, 1693–1709. doi: 10.1080/15548627.2018.1476812
Mishra, P., and Chan, D. C. (2016). Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387. doi: 10.1083/jcb.201511036
Murakawa, T., Yamaguchi, O., Hashimoto, A., Hikoso, S., Takeda, T., Oka, T., et al. (2015). Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6:7527. doi: 10.1038/ncomms8527
Naik, P. P., Mukhopadhyay, S., Panda, P. K., Sinha, N., Das, C. K., Mishra, R., et al. (2018). Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 51:e12411. doi: 10.1111/cpr.12411
Ni, H.-M., Williams, J. A., and Ding, W.-X. (2015). Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. doi: 10.1016/j.redox.2014.11.006
Novak, I., Kirkin, V., Mcewan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Okami, J., Simeone, D. M., and Logsdon, C. D. (2004). Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 64, 5338–5346. doi: 10.1158/0008-5472.CAN-04-0089
Okamoto, K., Kondo-Okamoto, N., and Ohsumi, Y. (2009). Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 17, 87–97. doi: 10.1016/j.devcel.2009.06.013
Otsu, K., Murakawa, T., and Yamaguchi, O. (2015). BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy 11, 1932–1933. doi: 10.1080/15548627.2015.1084459
Oun, R., Moussa, Y. E., and Wheate, N. J. (2018). The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 47, 6645–6653. doi: 10.1039/c8dt90088d
Panda, P. K., Naik, P. P., Meher, B. R., Das, D. N., Mukhopadhyay, S., Praharaj, P. P., et al. (2018). PUMA dependent mitophagy by Abrus agglutinin contributes to apoptosis through ceramide generation. Biochim. Biophys. Acta. Mol. Cell Res. 1865, 480–495. doi: 10.1016/j.bbamcr.2017.12.002
Panigrahi, D. P., Praharaj, P. P., Bhol, C. S., Mahapatra, K. K., Patra, S., Behera, B. P., et al. (2019). The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2019.07.015 [Epub ahead of print].
Pernas, L., and Scorrano, L. (2016). Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Phys. 78, 505–531. doi: 10.1146/annurev-physiol-021115-105011
Porporato, P. E., Filigheddu, N., Pedro, J. M. B.-S., Kroemer, G., and Galluzzi, L. (2018). Mitochondrial metabolism and cancer. Cell Res. 28, 265–280. doi: 10.1038/cr.2017.155
Poulogiannis, G., Mcintyre, R. E., Dimitriadi, M., Apps, J. R., Wilson, C. H., Ichimura, K., et al. (2010). PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. U.S.A. 107, 15145–15150. doi: 10.1073/pnas.1009941107
Rao, S., Tortola, L., Perlot, T., Wirnsberger, G., Novatchkova, M., Nitsch, R., et al. (2014). A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 5:3056. doi: 10.1038/ncomms4056
Ray, R., Chen, G., Vande Velde, C., Cizeau, J., Park, J. H., Reed, J. C., et al. (2000). BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J. Biol. Chem. 275, 1439–1448. doi: 10.1074/jbc.275.2.1439
Rehman, J., Zhang, H. J., Toth, P. T., Zhang, Y., Marsboom, G., Hong, Z., et al. (2012). Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 26, 2175–2186. doi: 10.1096/fj.11-196543
Rosenfeldt, M. T., O’prey, J., Morton, J. P., Nixon, C., Mackay, G., Mrowinska, A., et al. (2013). p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300. doi: 10.1038/nature12865
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Schubert, A. F., Gladkova, C., Pardon, E., Wagstaff, J. L., Freund, S. M. V., Steyaert, J., et al. (2017). Structure of PINK1 in complex with its substrate ubiquitin. Nature 552, 51–56. doi: 10.1038/nature24645
Sekine, S., and Youle, R. J. (2018). PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 16:2. doi: 10.1186/s12915-017-0470-7
Senft, D., and Ronai, Z. E. A. (2016). Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 39, 43–52. doi: 10.1016/j.ceb.2016.02.001
Sentelle, R. D., Senkal, C. E., Jiang, W., Ponnusamy, S., Gencer, S., Selvam, S. P., et al. (2012). Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8, 831–838. doi: 10.4161/auto.22739
Shi, C., Cai, Y., Li, Y., Li, Y., Hu, N., Ma, S., et al. (2018). Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 14, 59–71. doi: 10.1016/j.redox.2017.08.013
Shi, R.-Y., Zhu, S.-H., Li, V., Gibson, S. B., Xu, X.-S., and Kong, J.-M. (2014). BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 20, 1045–1055. doi: 10.1111/cns.12325
Sowter, H. M., Ferguson, M., Pym, C., Watson, P., Fox, S. B., Han, C., et al. (2003). Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J. Pathol. 201, 573–580. doi: 10.1002/path.1486
Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H., and Harris, A. L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673.
Strappazzon, F., Nazio, F., Corrado, M., Cianfanelli, V., Romagnoli, A., Fimia, G. M., et al. (2015). AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 22, 419–432. doi: 10.1038/cdd.2014.139
Su, Y.-C., Davuluri, G. V. N., Chen, C.-H., Shiau, D.-C., Chen, C.-C., Chen, C.-L., et al. (2016). Galectin-1-induced autophagy facilitates cisplatin resistance of hepatocellular carcinoma. PLoS One 11:e0148408. doi: 10.1371/journal.pone.0148408
Subarsky, P., and Hill, R. P. (2003). The hypoxic tumour microenvironment and metastatic progression. Clin. Exp. Metastasis 20, 237–250. doi: 10.1023/a:1022939318102
Sun, X., Liu, M., Hao, J., Li, D., Luo, Y., Wang, X., et al. (2013). Parkin deficiency contributes to pancreatic tumorigenesis by inducing spindle multipolarity and misorientation. Cell Cycle 12, 1133–1141. doi: 10.4161/cc.24215
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi: 10.1074/jbc.M607279200
Tan, E. Y., Campo, L., Han, C., Turley, H., Pezzella, F., Gatter, K. C., et al. (2007). BNIP3 as a progression marker in primary human breast cancer; opposing functions in in situ versus invasive cancer. Clin. Cancer Res. 13, 467–474. doi: 10.1158/1078-0432.CCR-06-1466
Tay, S.-P., Yeo, C. W. S., Chai, C., Chua, P.-J., Tan, H.-M., Ang, A. X. Y., et al. (2010). Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J. Biol. Chem. 285, 29231–29238. doi: 10.1074/jbc.M110.108241
Tilokani, L., Nagashima, S., Paupe, V., and Prudent, J. (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360. doi: 10.1042/EBC20170104
Vásquez-Trincado, C., García-Carvajal, I., Pennanen, C., Parra, V., Hill, J. A., Rothermel, B. A., et al. (2016). Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 594, 509–525. doi: 10.1113/JP271301
Veeriah, S., Taylor, B. S., Meng, S., Fang, F., Yilmaz, E., Vivanco, I., et al. (2010). Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 42, 77–82. doi: 10.1038/ng.491
Villa, E., Proïcs, E., Rubio-Patiño, C., Obba, S., Zunino, B., Bossowski, J. P., et al. (2017). Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. 20, 2846–2859. doi: 10.1016/j.celrep.2017.08.087
Vyas, S., Zaganjor, E., and Haigis, M. C. (2016). Mitochondria and Cancer. Cell 166, 555–566. doi: 10.1016/j.cell.2016.07.002
Wai, T., and Langer, T. (2016). Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 27, 105–117. doi: 10.1016/j.tem.2015.12.001
Wan, Y.-Y., Zhang, J.-F., Yang, Z.-J., Jiang, L.-P., Wei, Y.-F., Lai, Q.-N., et al. (2014). Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol. Rep. 32, 619–626. doi: 10.3892/or.2014.3235
Wang, F., Denison, S., Lai, J.-P., Philips, L. A., Montoya, D., Kock, N., et al. (2004). Parkin gene alterations in hepatocellular carcinoma. Genes Chromosomes Cancer 40, 85–96. doi: 10.1002/gcc.20020
Wang, J., Li, J., Santana-Santos, L., Shuda, M., Sobol, R. W., Van Houten, B., et al. (2015). A novel strategy for targeted killing of tumor cells: induction of multipolar acentrosomal mitotic spindles with a quinazolinone derivative mdivi-1. Mol. Oncol. 9, 488–502. doi: 10.1016/j.molonc.2014.10.002
Wang, W.-J., Wang, Y., Chen, H.-Z., Xing, Y.-Z., Li, F.-W., Zhang, Q., et al. (2014). Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat. Chem. Biol. 10, 133–140. doi: 10.1038/nchembio.1406
Wei, Y., Chiang, W.-C., Sumpter, R., Mishra, P., and Levine, B. (2017). Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168, 224–238.e10. doi: 10.1016/j.cell.2016.11.042
Wu, H., Huang, S., Chen, Z., Liu, W., Zhou, X., and Zhang, D. (2015). Hypoxia-induced autophagy contributes to the invasion of salivary adenoid cystic carcinoma through the HIF-1α/BNIP3 signaling pathway. Mol. Med. Rep. 12, 6467–6474. doi: 10.3892/mmr.2015.4255
Wu, H.-M., Jiang, Z.-F., Ding, P.-S., Shao, L.-J., and Liu, R.-Y. (2015). Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 5:12291. doi: 10.1038/srep12291
Xie, Q., Wu, Q., Horbinski, C. M., Flavahan, W. A., Yang, K., Zhou, W., et al. (2015). Mitochondrial control by DRP1 in brain tumor initiating cells. Nature Neurosci. 18, 501–510. doi: 10.1038/nn.3960
Yamashita, K., Miyata, H., Makino, T., Masuike, Y., Furukawa, H., Tanaka, K., et al. (2017). High expression of the mitophagy-related protein pink1 is associated with a poor response to chemotherapy and a poor prognosis for patients treated with neoadjuvant chemotherapy for esophageal squamous cell carcinoma. Ann. Surg. Oncol. 24, 4025–4032. doi: 10.1245/s10434-017-6096-8
Yan, C., Luo, L., Guo, C.-Y., Goto, S., Urata, Y., Shao, J.-H., et al. (2017). Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 388, 34–42. doi: 10.1016/j.canlet.2016.11.018
Yang, Z., Feng, Z., Gu, J., Li, X., Dong, Q., Liu, K., et al. (2017). microRNA-488 inhibits chemoresistance of ovarian cancer cells by targeting Six1 and mitochondrial function. Oncotarget 8, 80981–80993. doi: 10.18632/oncotarget.20941
Yao, N., Wang, C., Hu, N., Li, Y., Liu, M., Lei, Y., et al. (2019). Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis. 10:232. doi: 10.1038/s41419-019-1470-z
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Zhang, C., Lin, M., Wu, R., Wang, X., Yang, B., Levine, A. J., et al. (2011). Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. U.S.A. 108, 16259–16264. doi: 10.1073/pnas.1113884108
Zhang, X., Wang, X., Wu, T., Li, B., Liu, T., Wang, R., et al. (2015). Isoliensinine induces apoptosis in triple-negative human breast cancer cells through ROS generation and p38 MAPK/JNK activation. Sci. Rep. 5:12579. doi: 10.1038/srep12579
Zhang, Y., Yao, Y., Qiu, X., Wang, G., Hu, Z., Chen, S., et al. (2019). Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 20, 433–446. doi: 10.1038/s41590-019-0324-2
Zhao, J., Zhang, J., Yu, M., Xie, Y., Huang, Y., Wolff, D. W., et al. (2013). Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814–4824. doi: 10.1038/onc.2012.494
Zhou, J., Li, G., Zheng, Y., Shen, H.-M., Hu, X., Ming, Q.-L., et al. (2015). A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 11, 1259–1279. doi: 10.1080/15548627.2015.1056970
Zhu, Y., Massen, S., Terenzio, M., Lang, V., Chen-Lindner, S., Eils, R., et al. (2013). Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 288, 1099–1113. doi: 10.1074/jbc.M112.399345
Keywords: mitochondria, mitochondrial dynamics, mitophagy, carcinogenesis, therapy
Citation: Wang Y, Liu H-H, Cao Y-T, Zhang L-L, Huang F and Yi C (2020) The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 8:413. doi: 10.3389/fcell.2020.00413
Received: 07 February 2020; Accepted: 04 May 2020;
Published: 10 June 2020.
Edited by:
Du Feng, Guangzhou Medical University, ChinaReviewed by:
Paolo Remondelli, University of Salerno, ItalyCopyright © 2020 Wang, Liu, Cao, Zhang, Huang and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Huang, aHVhbmdmYW5nMDc5NEAxNjMuY29t; Cong Yi, eWljb25nbGFiQHpqdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.