 Liming Wang
Liming Wang Guang Lu
Guang Lu Han-Ming Shen
Han-Ming Shen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 13 May 2020
Sec. Cell Death and Survival
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00299
This article is part of the Research Topic Mitophagy in Health and Disease View all 12 articles
Mitophagy is a key mitochondrial quality control mechanism for effective and selective elimination of damaged mitochondria through the autophagy-lysosome machinery. Defective mitophagy is associated with pathogenesis of important human diseases including neurodegenerative diseases, heart failure, innate immunity, and cancer. In the past two decades, the mechanistic studies of mitophagy have made many breakthroughs with the discoveries of phosphatase and tensin homolog (PTEN)-induced kinase protein 1 (PINK1)-parkin-mediated ubiquitin (Ub)-driven pathway and BCL2/adenovirus E1B 19 kDa protein-interacting proteins 3 (BNIP3)/NIX or FUN14 domain containing 1 (FUNDC1) mitochondrial receptor-mediated pathways. Recently, several isoforms of dual phosphatase PTEN, such as PTEN-long (PTEN-L), have been identified, and some of them are implicated in the mitophagy process via their protein phosphatase activity. In this review, we aim to discuss the regulatory roles of PTEN isoforms in mitophagy. These discoveries may provide new opportunities for development of novel therapeutic strategies for mitophagy-related diseases such as neurodegenerative disorders via targeting PTEN isoforms and mitophagy.
Autophagy is an evolutionarily conserved process to degrade or recycle intracellular materials through lysosomes or vacuoles (Mizushima, 2018). In mammalian cells, there exist three different types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Among them, macroautophagy (referred to as autophagy hereafter) is the most well-studied form, which is orchestrated by a group of proteins encoded by autophagy-related-genes (ATGs) and characterized by the formation of double-membraned autophagosomes (Zachari and Ganley, 2017; Dikic and Elazar, 2018; Mizushima, 2018). The formation of autophagosomes can be briefly divided into three main steps: (1) The initiation step is regulated by unc51-like activating kinase 1 (ULK1) complex comprised of ULK1, ATG13, FIP200, and ATG101 to form the phagophore; (2) the vesicle nucleation step is regulated by Beclin1-ATG14 and Vps34/class III phosphatidylinositol 3-kinases (PI3K) complex to generate phosphatidylinositol 3-phosphate (PI3P); and (3) the vesicle elongation step is mediated by two ubiquitination conjugation systems, ATG12-ATG5-ATG16L1 and LC3-PE (phosphatidylethanolamine) systems, as well as ATG9-containing vesicles to form the autophagosomes (Mizushima et al., 2011; Hurley and Young, 2017; Lahiri et al., 2019). Autophagy can be either a general non-selective process to randomly uptake cargos for degradation (bulk autophagy) or a selective process to remove or degrade specific organelles, aggregated proteins, DNA, and/or invading pathogens (selective autophagy). Up to date, several types of selective autophagy have been recognized, including mitophagy, ribophagy, xenophagy, reticulophagy, lysophagy, and aggrephagy (Rogov et al., 2014; Kirkin, 2020).
Among them, mitophagy represents the most well-studied form of selective autophagy to degrade dysfunctional or superfluous mitochondria through the autophagy-lysosome machinery, which is regulated by multiple factors with distinct posttranslational modifications (Montava-Garriga and Ganley, 2020; Wang et al., 2020). The phenomenon of mitophagy was first described by Christian De Duve and Robert Wattiaux in 1966 when they observed that mitochondria were engulfed by autophagic vacuoles (De Duve and Wattiaux, 1966). The term of “mitophagy” was coined by John J. Lemasters to distinguish this selective autophagy that degrades mitochondria from the bulk autophagy (Lemasters, 2005). Mitophagy is usually initiated by an “eat me” signal, such as labeling damaged mitochondria with ubiquitin (Ub) or autophagy receptors (Harper et al., 2018; Pickles et al., 2018; Wang et al., 2020). Owing to its critical role in maintaining mitochondrial homeostasis and close implication in multiple human diseases, such as Parkinson’s disease (PD) and Alzheimer’s disease (AD) (Williams and Ding, 2018; Lou et al., 2019), the machinery of mitophagy has drawn substantial attention in the past two decades. The discoveries of PINK1-Parkin-mediated Ub-driven pathway and BNIP3/NIX or FUNDC1 receptor-mediated pathways represent the milestones in the mitophagy field. In this review, we will discuss some of these key factors, especially the newly identified protein phosphatase, in the regulation of mitophagy.
One breakthrough in the understanding of the molecular mechanisms of mitophagy is the discovery of PINK1-Parkin-mediated pathway (Narendra et al., 2008, 2010; Vives-Bauza et al., 2010). PINK1 (encoded by the PARK6 gene) is a serine/threonine kinase, which was identified in 2001 (Unoki and Nakamura, 2001) and contains a mitochondrial targeting sequence (MTS) at its N-terminus as well as an outer mitochondrial localization signal (OMS) next to the transmembrane domain (TMD) (Okatsu et al., 2015a). Two homozygous mutations, including G→A in transition in exon 4 and G→A transitions in exon 7, in PINK1 were found in autosomal recessive early onset familial forms of PD patients (Valente et al., 2004). Parkin (encoded by the PARK2 gene) is an E3 Ub ligase, which was identified in 1998 and was named “Parkin” due to its important roles in the pathogenesis of autosomal recessive juvenile parkinsonism (AR-JP) (Kitada et al., 1998; Lucking et al., 1998; Abbas et al., 1999). Parkin contains a Ub-like (UBL) domain, a classic RING (RING1) domain, three zinc-coordinating domains termed in between RING (IBR) domain, a RING2 domain, and a RING0 domain that is a Parkin unique domain (Hristova et al., 2009; Trempe et al., 2013; Walden and Muqit, 2017). Numerous studies have reported that PINK1 and Parkin work in the same pathway to remove dysfunctional mitochondria and to maintain mitochondrial homeostasis, with the well-established feedforward model of PINK1-Parkin mitophagy activation (Harper et al., 2018; Pickles et al., 2018; Wang et al., 2020).
When mitochondria are healthy, PINK1 is constantly maintained at a low level due to mitochondrial import, protease cleavage, and proteasome degradation (Jin et al., 2010; Deas et al., 2011; Lazarou et al., 2012; Sekine et al., 2019). Upon mitochondrial damage and depolarization, PINK1 is rapidly accumulated on the outer mitochondrial membrane (OMM) and activated through dimerization and autophosphorylation (Okatsu et al., 2012, 2013; Aerts et al., 2015; Rasool et al., 2018). Therefore, PINK1 acts as a mitochondrial damage sensor to initiate mitophagy. Once activated, PINK1 phosphorylates mitochondrial pre-existing Ub at Ser 65 (pSer65-Ub) (Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Shiba-Fukushima et al., 2014). pSer65-Ub serves as a key receptor to recruit Parkin from cytosol to mitochondria through direct binding (Shiba-Fukushima et al., 2014; Okatsu et al., 2015b). Binding to pSer65-Ub releases the UBL domain of Parkin from its RING1 domain (Sauve et al., 2015; Wauer et al., 2015a; Aguirre et al., 2017), which promotes the phosphorylation of the UBL domain by PINK1 at Ser 65 (pSer65-Parkin) (Kondapalli et al., 2012; Shiba-Fukushima et al., 2012; Wauer et al., 2015a; McWilliams et al., 2018). Subsequently, the phospho-UBL domain rebinds to the RING0 domain of Parkin to release the catalytic RING2 domain to achieve full activation (Gladkova et al., 2018; Sauve et al., 2018). Activated Parkin then conjugates more Ub onto OMM proteins for PINK1 phosphorylation, which mediates further rounds of Parkin translocation to mitochondria; thus, PINK1, pSer65-Ub, and Parkin form a positive feedforward amplification loop to initiate mitophagy.
Another important function of pSer65-Ub is to recruit autophagy receptors, such as NDP52 (CALCOCO2) and Optineurin (OPTN) to damaged mitochondria, a process that is TANK-binding kinase 1 (TBK1) dependent (Heo et al., 2015; Lazarou et al., 2015; Richter et al., 2016). TBK1 is a serine/threonine kinase and phosphorylates these autophagy receptors to promote their binding ability to various Ub chains (Heo et al., 2015; Richter et al., 2016). Interestingly, activation of TBK1 also requires OPTN binding to Ub chains in the presence of PINK1 and Parkin (Heo et al., 2015; Richter et al., 2016). In the prevailing model of mitophagy, after binding to the pSer65-Ub chains, OPTN and/or NDP52 recruit phagophore onto mitochondria by directly binding to LC3 through their LC3-interacting regions (LIR motifs) (Gatica et al., 2018; Palikaras et al., 2018). However, emerging studies suggest that LC3/GABARAP family proteins are dispensable in the selective recognition of damaged mitochondria, based on the observation that, in LC3/GABARAP knockout cells, mitochondria can still be engulfed by autophagosomes (Itakura et al., 2012; Nguyen et al., 2016; Padman et al., 2019). One very recent study has highlighted the role of NDP52 to recruit ULK1 complex to damaged mitochondria (Vargas et al., 2019). NDP52 directly interacts with FIP200 in a TBK1-dependent manner to recruit ULK1 complex, leading to autophagosome biogenesis on damaged mitochondria and initiation of autophagy machinery.
Interestingly, besides PINK1-mediated pSer65-Ub, several other PINK1-independent phosphorylation sites of Ub have been identified, including pThr7-Ub, pSer20-Ub, and pSer57-Ub (Wauer et al., 2015b). Among them, pSer57-Ub has been reported to hyperactivate Parkin (George et al., 2017). Obviously, more studies are needed to understand the functional implication of such Ub phosphorylation in mitophagy. In addition to Ub and Parkin as described above, a number of additional PINK1 substrates have been reported. For instance, PINK1 phosphorylates mitofusin 2 (MFN2) at Thr 111 and Ser 442, leading to Parkin mitochondrial recruitment through promoting the interaction between MFN2 and Parkin, suggesting that MFN2 may serve as a mitochondrial receptor for Parkin (Chen and Dorn, 2013). However, another study indicates that MFN2 antagonizes mitophagy through tethering mitochondria and endoplasmic reticulum (ER) and limiting the accessibility of other mitochondrial proteins to PINK1 and Parkin (McLelland et al., 2018). It is known that some OMM proteins such as MFN2 undergo ubiquitination and proteasomal degradation at the beginning of the mitophagy (Tanaka et al., 2010; Ding et al., 2012; McLelland et al., 2018). Therefore, it is possible that such a process may facilitate mitophagy by removing the barrier among PINK1, Parkin, and other mitochondrial proteins. PINK1 can also phosphorylate Miro (also called RhoT) at Ser156, which recruits Parkin onto mitochondria and results in ubiquitination and proteasomal degradation of Miro, and thus blocking mitochondrial motility (Wang et al., 2011; Shlevkov et al., 2016). Interestingly, a recent report found that Miro, through direct protein–protein interaction, recruits Parkin at healthy mitochondria independent of PINK1, and such pre-existing Parkin is essential for Parkin further recruitment and activation upon mitochondrial damage in a PINK1-dependent manner (Safiulina et al., 2019). In addition, in a phosphoproteomic screening study for PINK1 substrates, Lai and colleagues reported that the phosphorylation of Rab GTPases such as Rab8A at the conserved Ser 111 is indirectly regulated by PINK1, and this phosphorylation can block the phosphorylation of Rab8A at Thr72 by leucine-rich repeat kinase 2 (LRRK2), suggesting the interplay of PINK1 with other PD-related genes (Lai et al., 2015; Vieweg et al., 2019). Thus, identification of more PINK1 substrates will not only provide new insights into the molecular mechanisms of PINK1-Parkin-mediated mitophagy but also provide deeper understanding of the molecular mechanisms of important neurodegenerative disorders such as PD.
BNIP3, a member of prodeath BCL2 family proteins, was first found as an E1B 19-kDa interacting proteins (Boyd et al., 1994). NIX (also named BNIP3L) is a homolog of BNIP3 with ∼55% identical similar amino acid sequence (Matsushima et al., 1998). Both proteins contain an atypical BCL2-homology 3 (BH3) domain and C-terminal TMD, which is essential for their proapoptotic activity and mitochondrial localization (Yasuda et al., 1998; Imazu et al., 1999). Moreover, BNIP3 and NIX both contain an identical LIR motif, which makes them to interact with LC3s/GABARAP subfamilies and recruit autophagosomes to sequester damaged mitochondria, especially under hypoxia conditions (Novak et al., 2010; Hanna et al., 2012; Birgisdottir et al., 2013). Under hypoxia, the expression of BNIP3 and NIX are increased through the transcriptional regulation of hypoxia-inducible factor 1α (HIF-1α) or FOXO3 (Sowter et al., 2001; Mammucari et al., 2007; Zhang et al., 2008). Mutation of the LIR motif abolishes the interaction of BNIP3/NIX with LC3 and thereby attenuates mitochondrial clearance (Novak et al., 2010; Hanna et al., 2012; Zhu et al., 2013), while phosphorylation of the LIR motif enhances the interaction with LC3 and promotes mitophagy (Zhu et al., 2013; Rogov et al., 2017). However, the kinase(s) and phosphatase(s) regulating this phosphorylation of LIR remain to be identified.
It should be noted that NIX, but not BNIP3, plays an important role in the development of reticulocytes through the regulation of mitophagy. Mitochondria were not cleared in reticulocytes when NIX is deficient (Diwan et al., 2007; Schweers et al., 2007; Zhang and Ney, 2008; Zhang J. et al., 2012). Interestingly, treatment with mitochondrial uncoupling agents could restore the removal of mitochondria in the absence of NIX, suggesting that the regulatory effect of NIX on mitophagy was probably due to its role in regulating mitochondrial depolarization (Sandoval et al., 2008; Zhang and Ney, 2008). However, there is still no direct evidence to show that NIX could cause mitochondrial depolarization, and further studies are thus needed.
Intriguingly, several studies have revealed the crosstalk between BNIP3/NIX receptor-mediated pathway and PINK1-Parkin-mediated pathway. For instance, both BNIP3 and NIX can promote Parkin mitochondrial recruitment (Ding et al., 2010; Lee et al., 2011), while NIX can also be ubiquitinated by Parkin to promote autophagy receptor recruitment to damaged mitochondria (Gao et al., 2015). In addition, BNIP3 is able to inhibit PINK1 proteolytic degradation and stabilize PINK1 on OMM to facilitate Parkin mitochondrial recruitment and mitophagy (Zhang et al., 2016). These findings suggest that these pathways cooperate with each other to ensure efficient mitophagy.
FUNDC1 is another important hypoxia-induced mitophagy receptor (Liu et al., 2012). As a mitochondrial outer membrane protein, FUNDC1 contains three TMDs and an LIR motif in its N-terminus exposed to the cytosol that interacts with LC3 to recruit autophagosome (Liu et al., 2012; Wu et al., 2016). Mutation or deletion of LIR motif of FUNDC1 significantly reduces or blocks mitophagy (Liu et al., 2012). Similar to the cases of other mitophagy key factors, the activity of FUNDC1 is also regulated by phosphorylation and dephosphorylation. Under normal conditions, FUNDC1 is phosphorylated by Src and CK2 at the sites of Tyr18 and Ser13, which blocks the interaction of FUNDC1 with LC3 (Liu et al., 2012; Chen et al., 2014). Another study showed that FUNDC1 can be phosphorylated by ULK1 at Ser17 to promote mitophagy (Wu et al., 2014). However, upon induction of hypoxia, Src and CK2 are inhibited, then phosphoglycerate mutase family member 5 (PGAM5), one unique mitochondrial phosphatase, dephosphorylates FUNDC1 at Ser13, which in turn promotes the interaction between FUNDC1 and LC3 to facilitate mitophagy (Chen et al., 2014). Interestingly, the same group reported that FUNDC1 is accumulated at the ER-mitochondrial contact site in response to hypoxia, which is essential for the mitochondrial recruitment of DRP1 to facilitate mitochondrial fission prior to mitophagy (Wu et al., 2016).
PTEN is a powerful tumor suppressor with both lipid phosphatase and protein phosphatase activity, which was identified in 1997 (Li and Sun, 1997; Li et al., 1997; Steck et al., 1997). PTEN contains 403 amino acids with a N-terminal phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2]-binding domain (PBD), a catalytic phosphatase domain, a C2 domain, a C-tail domain, and a PDZ-binding motif (Figure 1A; Lee et al., 1999). Loss of PTEN leads to cancer, neurological disorders, metabolic diseases, and tissue homeostasis defects (Backman et al., 2001; Kwon et al., 2006; Chen et al., 2018; Lee et al., 2018). PTEN is also vital for embryonic development, as its homozygous deletion causes lethality in mice (Di Cristofano et al., 1998; Stumpf and den Hertog, 2016). All these findings reveal that PTEN’s function is not only important for tumor suppression but also vital for other biological processes.
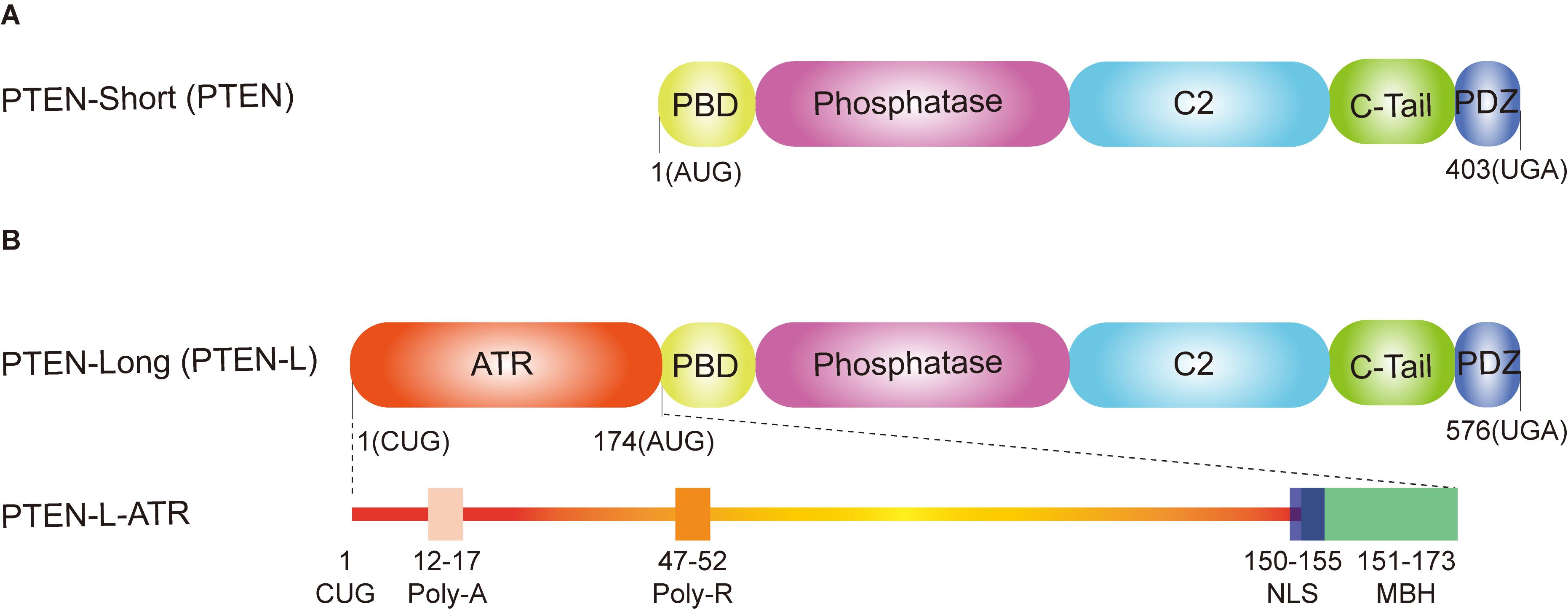
Figure 1. Domain structure of phosphatase and tensin homolog (PTEN) isoforms. (A) PTEN-short (canonical PTEN), translated from an AUG start codon, contains five functional domains: a N-terminal PtdIns (4,5) P2 (PIP2)-binding domain (PBD), a dual phosphatase domain, a C2 domain, a C-tail domain, and PDZ-binding motif. (B) PTEN-long (PTEN-L) is translated from a CUG start codon upstream from the classic AUG start codon. In addition to the same five functional domains with the canonical PTEN, PTEN-L contains an alternatively translated region (ATR) adding 173 amino acids at the N-terminus. The extended ATR is composed of a secreted polyalanine signal sequence (Poly-A, residues 12–17), a cell permeable polyarginine motif (Poly-R, residues 47–52), a nuclear localization sequence (NLS, QKKPRH, residues 150–155) as well as a membrane-binding α-helix (MBH, residues 151–173).
The probably most important function of PTEN is to block the activation of pro-oncogenic class I PI3K–AKT–mTOR signaling pathway through its lipid phosphatase activity (Cantley and Neel, 1999). PI3K phosphorylates PI(4,5)P2 to generate phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3], which recruits AKT at the cell membrane, and then AKT is phosphorylated via PDK1 and mTORC2 to indirectly activate mTORC1 (King et al., 2015). PTEN opposes this pathway through dephosphorylating PI(3,4,5)P3 to PI(4,5)P2 via its lipid phosphatase activity, leading to reduced AKT phosphorylation and inactivation (Worby and Dixon, 2014). Thus, the phosphorylation level of AKT has been widely used as an indicator for PTEN activity.
Due to the inhibitory effects of PTEN on the PI3K–AKT–mTOR signaling pathway, several studies have shown that PTEN can positively regulate autophagy (Arico et al., 2001; Ueno et al., 2008; Cai et al., 2018). Intriguingly, two independent groups reported that inhibition of AKT signaling impaired PINK1 accumulation, Parkin recruitment, and subsequent efficient mitophagy in response to mitochondrial depolarization (McCoy et al., 2014; Soutar et al., 2018). However, the role of PTEN in the regulation of mitophagy is still largely unclear. Harper and colleagues reported that RAB7A could be directly phosphorylated by TBK1 at Ser 72 (pSer72-RAB7A) to facilitate the efficient recruitment of ATG9A vesicles to damaged mitochondria and promote PINK1-Parkin-mediated mitophagy, and non-phosphorylated RAB7A failed to support this process (Heo et al., 2018). Importantly, PTEN has been found to dephosphorylate pSer72-RAB7A via its protein phosphatase activity (Shinde and Maddika, 2016; Hanafusa et al., 2019), thus suggesting a potential role of PTEN in regulating mitophagy. A more direct study showed that deletion of PTEN increased MFN2 expression and rescued mitophagic flux via the AMP-activated protein kinase (AMPK)–cAMP response element-binding protein (CREB) pathways (Li et al., 2019). Interestingly, both PTEN and MFN2 have a distribution at ER-mitochondrial contact site (de Brito and Scorrano, 2008; Bononi et al., 2013; Naon et al., 2016). As discussed above, MFN2 can be phosphorylated by PINK1 and serves as a mitochondrial receptor for Parkin (Chen and Dorn, 2013). Moreover, phosphorylated MFN2 dissociates mitochondria from ER to initiate mitophagy (McLelland et al., 2018). Thus, it will be interesting to explore whether PTEN can dephosphorylate MFN2 at the ER-mitochondrial contact site to suppress mitophagy. In addition, overexpression of PTEN inhibits mitophagy via blockage of Toll-like receptor 4 (TLR4)–c-JUN N-terminal kinase (JNK)–BNIP3 pathway (Li M. et al., 2018).
Moreover, several in vivo studies have highlighted that PTEN deletion in dopamine neurons provides neuroprotective effects in both genetic and neurotoxin-induced PD mouse models (Diaz-Ruiz et al., 2009; Domanskyi et al., 2011; Zhang Y. et al., 2012). Another study showed that the protein level of PTEN is significantly increased in neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-hydrochloride (MPTP)-treated mice and 1-methyl-4-phenylpyridinium (MPP+)-treated SH-SY5Y cells, leading to enhanced neurotoxicity and apoptosis (Zhao et al., 2020). In addition, inhibition of PTEN is able to attenuate amyloid-β (Aβ)-induced synaptic toxicity and rescue cognitive function in AD models (Knafo et al., 2016). Consistently, a PTEN inhibitor, bisperoxovanadium-pic [bpV(pic)], provides neuroprotective effects in Aβ-induced neurotoxicity in a human neuroblastoma cell model (Liu et al., 2017). Apparently, more studies are needed to explore whether the above processes are due to the regulative effects of PTEN on mitophagy.
PTEN-L is the first characterized isoform of canonical PTEN, which was identified in 2013 (Hopkins et al., 2013). PTEN-L and PTEN shares the same mRNA, but PTEN-L translates from a non-AUG start codon (CUG start codon), adding an alternatively translated region (ATR) at the N-terminus of PTEN (Hopkins et al., 2013). PTEN-L can be secreted from one cell and taken up by other neighboring cells to inhibit PI3K–AKT signaling pathway both in vitro and in vivo (Hopkins et al., 2013). Intriguingly, Liang et al. reported that PTEN-L (also termed as PTENα) is a mitochondrial protein to regulate mitochondrial energy metabolism (Liang et al., 2014). They found that somatic deletion of PTEN-L resulted in much smaller mitochondria with irregular shape and led to mitochondrial depolarization (Liang et al., 2014). It is known that, in addition to the same domains with canonical PTEN (PTEN-short), the extended ATR of PTEN-L contains a secreted polyalanine signal sequence (Poly-A), a cell permeable polyarginine motif (Poly-R), a nuclear localization sequence (NLS, QKKPRH) as well as a membrane-binding α-helix (MBH) (Figure 1B; Hopkins et al., 2013; Malaney et al., 2013; Masson et al., 2016; Shen et al., 2019). In addition, most parts of the ATR are intrinsically disordered and probably contain various postmodification sites and protein-binding motifs (Malaney et al., 2013; Masson et al., 2016), indicating that PTEN-L may modify distinct substrates compared with PTEN.
Recently, our group has revealed that PTEN-L functions as a protein phosphatase for Ub and antagonizes the PINK1-Parkin-mediated mitophagy pathway (Wang et al., 2018a, b). First, topology assay and immunogold electron microscopy revealed that a significant proportion of PTEN-L was associated with the mitochondrial outer membrane. Second, PTEN-L overexpression blocked mitophagy induced by mitochondrial damage agents including carbonyl cyanide 3-chlorophenylhydrazone (CCCP), combination of oligomycin and antimycin A (O/A), and valinomycin, whereas PTEN-L knockout accelerated mitophagic flux. Third, PTEN-L overexpression was able to strongly prevent Parkin mitochondrial recruitment, autoubiquitination, and subsequent activation of its E3 ligase activity. Finally, PTEN-L could dephosphorylate various types of pSer65-Ub chains in vivo and in vitro via its protein phosphatase activity but independent of its lipid phosphatase activity, leading to the disruption of the feedforward amplification loops formed by PINK1, Parkin, and pSer65-Ub chains. Since Ub modification is a vital posttranslational process in mitophagy, deubiquitinating enzymes (DUBs) become potential regulators to maintain the mitochondrial homeostasis, especially in the PINK1-Parkin-mediated Ub-driven mitophagy pathway. There are more than 100 putative DUB genes in humans, which can be grouped into two classes: cysteine proteases and metalloproteases. Among them, ubiquitin-specific proteases (USPs), which are encoded by 58 different genes, such as USP30, USP15, and USP8, have been widely studied in the field of mitophagy (Bingol et al., 2014; Cornelissen et al., 2014; Durcan et al., 2014; Marcassa et al., 2018; Ordureau et al., 2020). Recently, USP36 has been reported as a positive regulator of mitophagy; knockdown of USP36 impairs Parkin mitochondrial translocation, leading to blockage of mitophagy (Geisler et al., 2019). Interestingly, they also found that the protein level of PTEN-L was increased after USP36 knockdown, which was associated with reduced pSer65-Ub level and consistent with our findings (Geisler et al., 2019).
Intriguingly, Li et al. demonstrated that PTEN-L promotes mitophagy through interaction with Parkin by its MBH motif to promote Parkin self-association and mitochondrial localization (Li G. et al., 2018). Further studies are thus needed to examine the precise role of PTEN-L in this pathway and more importantly to explore whether PTEN-L is implicated in the pathology of mitophagy-related diseases, such as PD and AD.
Mitochondria are one of the essential organelles in eukaryotic cells, with critical functions including energy (ATP) production, cell survival/cell death, cell signaling, and immune response. Dysfunctional mitochondria are implicated in many pathological processes and diseases such as cell death, inflammation, neurodegenerative diseases, and cancer. Thus, removal of damaged mitochondria by mitophagy has been shown to be an important mitochondrial quality control mechanism to maintain the mitochondrial homeostasis. However, this process must be restricted to dysfunctional mitochondria. Excessive degradation of essential mitochondria will cause cell death (Ordureau and Harper, 2014; Shi et al., 2014; Guo et al., 2016; Sharma et al., 2019). In addition, during the mitochondria fission process, the membrane potential of healthy mitochondria is temporarily compromised (Twig et al., 2008), which possibly activates PINK1-Parkin pathway to remove healthy mitochondria. Therefore, the mitophagy machinery is orchestrated by key mitophagy effectors with reversible posttranslational modifications, such as phosphorylation and dephosphorylation, to determine a finely tuned mitophagic activity in response to diverse stresses (Figure 2).
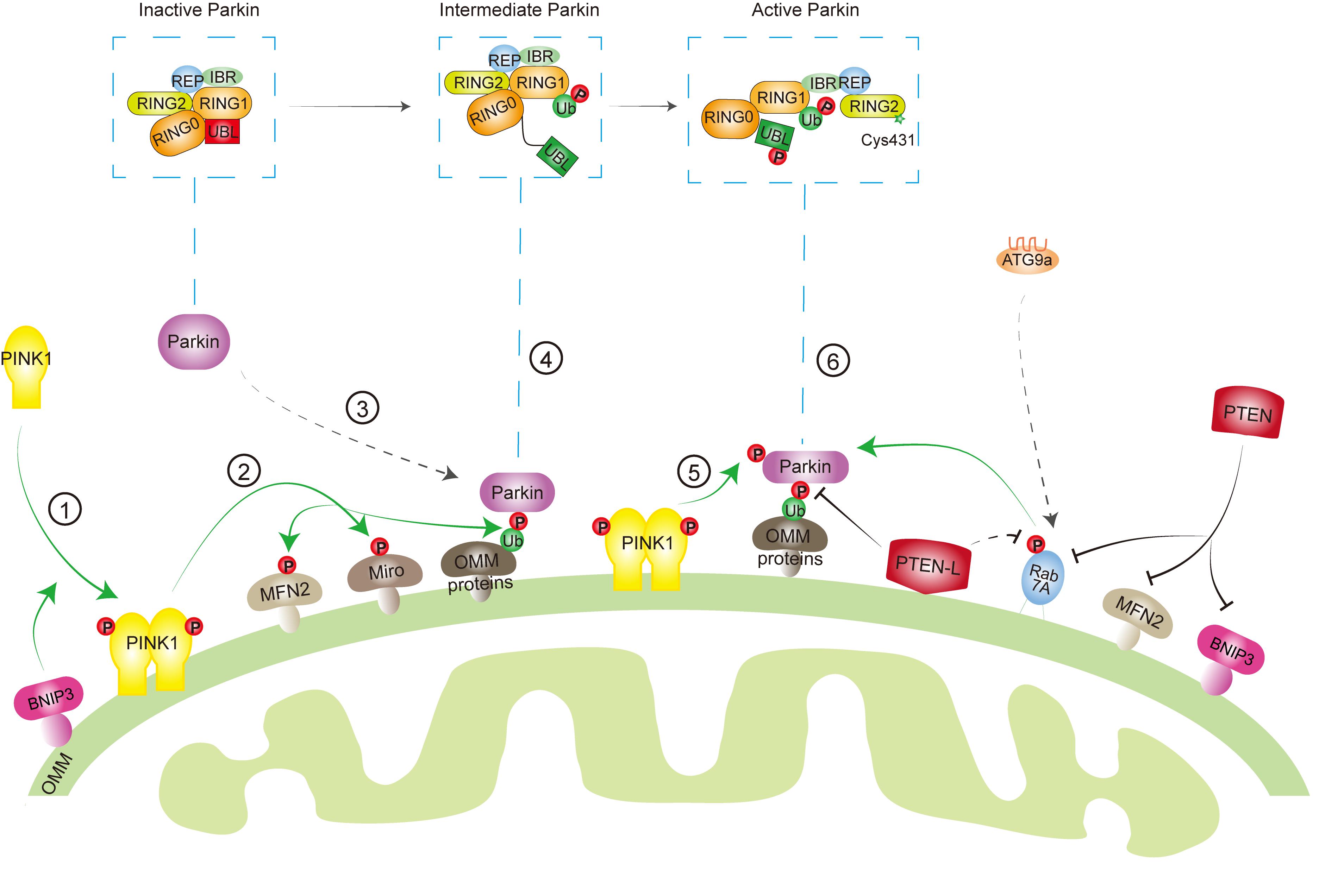
Figure 2. Key effectors involved in mitophagy machinery. When mitochondria are healthy, phosphatase and tensin homolog-induced kinase protein 1 (PINK1) is imported into the mitochondria, cleaved by protease, and degraded by proteasome, while Parkin keeps in an inactive conformation in the cytosol through intradomain–domain interactions. Upon mitochondrial damage or depolarization, PINK1 is stabilized and activated at the outer mitochondrial membrane (OMM) ➀, which leads to the phosphorylation of its downstream targets, such as ubiquitin (Ub) ➁. Parkin has a high affinity to phosphorylated Ub (pSer65-Ub), which recruits Parkin from cytosol to mitochondria ➂. Several other factors, such as mitofusin 2 (MFN2), Miro, Rab7A, as well as BCL2/adenovirus E1B 19 kDa protein-interacting proteins 3 (BNIP3) are also involved in Parkin mitochondrial recruitment. Binding to pSer65-Ub releases the Ub-like (UBL) domain of Parkin from RING1 domain, partially activating Parkin ➃. Then, PINK1 phosphorylates the UBL domain at Ser65 ➄, which drives the phospho-UBL to rebind to the RING0 domain of Parkin to expose RING2′ catalytic site (Cys431) and fully activate Parkin ➅. On the other hand, phosphatase and tensin homolog long (PTEN-L) located at OMM dephosphorylates Ub to inhibit mitophagy, whereas PTEN in the cytosol suppresses mitophagy through targeting Rab7A, MFN2, or BNIP3.
We now appreciate that phosphorylation of Ub by PINK1 (pSer65-Ub) plays central roles in the regulation of Ub-dependent mitophagy pathway. pSer65-Ub levels are very low in healthy mitochondria, but dramatically increased after mitochondrial damage and also increased during aging or in PD patient brain, which highlights its roles in diseases (Fiesel et al., 2015; Hou et al., 2018). Although PINK1 is the only reported kinase to generate pSer65-Ub, pSer65-Ub could be detected in PINK knockout cells (Ordureau et al., 2014) and in PINK1-deficient yeast (Swaney et al., 2015), suggesting another kinase exists to phosphorylate Ub at Ser 65. However, the function of PINK1-independent pSer65-Ub remains largely unclear. Another question is whether pSer65-Ub can be involved in other selective autophagy, such as xenophagy, which shares several key factors with mitophagy, including TBK1, NDP52, OPTN, and SQSTM1.
Recent studies have indicated that PTEN family proteins are involved in the regulation of both PINK1-Parkin-mediated Ub-driven and BNIP3 receptor-mediated mitophagy. Some important questions need to be further addressed. First is how the cells determine the expression level of different PTEN isoforms to function under different conditions. Second is whether there is a specific recruitment of PTEN-L and PTEN to mitochondria in response to mitochondrial damage. Third and more importantly is whether PTEN isoforms can serve as molecular targets for development of novel interventional approaches in the regulation of mitophagy to benefit mitophagy-related human diseases.
LW and H-MS designed the outline of the review and wrote the draft of the manuscript. GL wrote part of the review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to colleagues whose work could not be cited owing to space limitations. The work was supported by Singapore National Medical Research Council (NMRC/CIRG/1490/2018) and Singapore Ministry of Education (MOE) Tier 2 (MOE2018-T2-1-060) to H-MS. LW was supported by NUSMed Post-Doctoral Fellowship and Swee Liew-Wadsorth Research Grant.
Abbas, N., Lucking, C. B., Ricard, S., Durr, A., Bonifati, V., De Michele, G., et al. (1999). A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s disease genetics study group and the european consortium on genetic susceptibility in Parkinson’s Disease. Hum. Mol. Genet. 8, 567–574.
Aerts, L., Craessaerts, K., De Strooper, B., and Morais, V. A. (2015). PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 290, 2798–2811. doi: 10.1074/jbc.M114.620906
Aguirre, J. D., Dunkerley, K. M., Mercier, P., and Shaw, G. S. (2017). Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc. Natl. Acad. Sci. U.S.A. 114, 298–303. doi: 10.1073/pnas.1613040114
Arico, S., Petiot, A., Bauvy, C., Dubbelhuis, P. F., Meijer, A. J., Codogno, P., et al. (2001). The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 276, 35243–35246.
Backman, S. A., Stambolic, V., Suzuki, A., Haight, J., Elia, A., Pretorius, J., et al. (2001). Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 29, 396–403.
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Birgisdottir, A. B., Lamark, T., and Johansen, T. (2013). The LIR motif - crucial for selective autophagy. J. Cell Sci. 126, 3237–3247. doi: 10.1242/jcs.126128
Bononi, A., Bonora, M., Marchi, S., Missiroli, S., Poletti, F., Giorgi, C., et al. (2013). Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 20, 1631–1643. doi: 10.1038/cdd.2013.77
Boyd, J. M., Malstrom, S., Subramanian, T., Venkatesh, L. K., Schaeper, U., Elangovan, B., et al. (1994). Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 79, 341–351.
Cai, J., Li, R., Xu, X., Zhang, L., Lian, R., Fang, L., et al. (2018). CK1α suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nat. Cell Biol. 20, 465–478. doi: 10.1038/s41556-018-0065-8
Cantley, L. C., and Neel, B. G. (1999). New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 4240–4245.
Chen, C.-Y., Chen, J., He, L., and Stiles, B. L. (2018). PTEN: tumor suppressor and metabolic regulator. Front. Endocrinol. (Lausanne) 9:338. doi: 10.3389/fendo.2018.00338
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 54, 362–377. doi: 10.1016/j.molcel.2014.02.034
Chen, Y., and Dorn, G. W. II (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Cornelissen, T., Haddad, D., Wauters, F., Van Humbeeck, C., Mandemakers, W., Koentjoro, B., et al. (2014). The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 23, 5227–5242. doi: 10.1093/hmg/ddu244
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610.
Deas, E., Plun-Favreau, H., Gandhi, S., Desmond, H., Kjaer, S., Loh, S. H., et al. (2011). PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879. doi: 10.1093/hmg/ddq526
Di Cristofano, A., Pesce, B., Cordon-Cardo, C., and Pandolfi, P. P. (1998). Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355.
Diaz-Ruiz, O., Zapata, A., Shan, L., Zhang, Y., Tomac, A. C., Malik, N., et al. (2009). Selective deletion of PTEN in dopamine neurons leads to trophic effects and adaptation of striatal medium spiny projecting neurons. PLoS ONE 4:e7027. doi: 10.1371/journal.pone.0007027
Dikic, I., and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364. doi: 10.1038/s41580-018-0003-4
Ding, W.-X., Guo, F., Ni, H.-M., Bockus, A., Manley, S., Stolz, D. B., et al. (2012). Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J. Biol. Chem. 287, 42379–42388. doi: 10.1074/jbc.M112.413682
Ding, W.-X., Ni, H.-M., Li, M., Liao, Y., Chen, X., Stolz, D. B., et al. (2010). Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 285, 27879–27890. doi: 10.1074/jbc.M110.119537
Diwan, A., Koesters, A. G., Odley, A. M., Pushkaran, S., Baines, C. P., Spike, B. T., et al. (2007). Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 104, 6794–6799.
Domanskyi, A., Geissler, C., Vinnikov, I. A., Alter, H., Schober, A., Vogt, M. A., et al. (2011). Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J. 25, 2898–2910. doi: 10.1096/fj.11-181958
Durcan, T. M., Tang, M. Y., Perusse, J. R., Dashti, E. A., Aguileta, M. A., McLelland, G. L., et al. (2014). USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 33, 2473–2491. doi: 10.15252/embj.201489729
Fiesel, F. C., Ando, M., Hudec, R., Hill, A. R., Castanedes-Casey, M., Caulfield, T. R., et al. (2015). (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 16, 1114–1130. doi: 10.15252/embr.201540514
Gao, F., Chen, D., Si, J., Hu, Q., Qin, Z., Fang, M., et al. (2015). The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 24, 2528–2538. doi: 10.1093/hmg/ddv017
Gatica, D., Lahiri, V., and Klionsky, D. J. (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242. doi: 10.1038/s41556-018-0037-z
Geisler, S., Jäger, L., Golombek, S., Nakanishi, E., Hans, F., Casadei, N., et al. (2019). Ubiquitin-specific protease USP36 knockdown impairs Parkin-dependent mitophagy via downregulation of Beclin-1-associated autophagy-related ATG14L. Exp. Cell Res. 384, 111641–111641. doi: 10.1016/j.yexcr.2019.111641
George, S., Wang, S. M., Bi, Y., Treidlinger, M., Barber, K. R., Shaw, G. S., et al. (2017). Ubiquitin phosphorylated at Ser57 hyper-activates parkin. Biochim. Biophys. Acta Gen. Subj. 1861, 3038–3046. doi: 10.1016/j.bbagen.2017.06.023
Gladkova, C., Maslen, S. L., Skehel, J. M., and Komander, D. (2018). Mechanism of parkin activation by PINK1. Nature 559, 410–414. doi: 10.1038/s41586-018-0224-x
Guo, X., Sun, X., Hu, D., Wang, Y.-J., Fujioka, H., Vyas, R., et al. (2016). VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 7, 12646–12646. doi: 10.1038/ncomms12646
Hanafusa, H., Yagi, T., Ikeda, H., Hisamoto, N., Nishioka, T., Kaibuchi, K., et al. (2019). LRRK1 phosphorylation of Rab7 at S72 links trafficking of EGFR-containing endosomes to its effector RILP. J. Cell Sci. 132, jcs228809. doi: 10.1242/jcs.228809
Hanna, R. A., Quinsay, M. N., Orogo, A. M., Giang, K., Rikka, S., and Gustafsson, ÅB. (2012). Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104.
Harper, J. W., Ordureau, A., and Heo, J. M. (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108. doi: 10.1038/nrm.2017.129
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN MITOCHONDRIAL UBIQUITYLATION PATHWAY DRIVES A PROGRAm of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell. 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Heo, J. M., Ordureau, A., Swarup, S., Paulo, J. A., Shen, K., Sabatini, D. M., et al. (2018). RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 4:eaav0443. doi: 10.1126/sciadv.aav0443
Hopkins, B. D., Fine, B., Steinbach, N., Dendy, M., Rapp, Z., Shaw, J., et al. (2013). A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341, 399–402. doi: 10.1126/science.1234907
Hou, X., Fiesel, F. C., Truban, D., Castanedes Casey, M., Lin, W. L., Soto, A. I., et al. (2018). Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 14, 1404–1418.
Hristova, V. A., Beasley, S. A., Rylett, R. J., and Shaw, G. S. (2009). Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J. Biol. Chem. 284, 14978–14986. doi: 10.1074/jbc.M808700200
Hurley, J. H., and Young, L. N. (2017). Mechanisms of autophagy initiation. Annu. Rev. Biochem. 86, 225–244. doi: 10.1146/annurev-biochem-061516-044820
Imazu, T., Shimizu, S., Tagami, S., Matsushima, M., Nakamura, Y., Miki, T., et al. (1999). Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 18, 4523–4529.
Itakura, E., Kishi-Itakura, C., Koyama-Honda, I., and Mizushima, N. (2012). Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 125, 1488–1499. doi: 10.1242/jcs.094110
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/BJ20140334
King, D., Yeomanson, D., and Bryant, H. E. (2015). PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J. Pediat. Hematol. Oncol. 37, 245–251. doi: 10.1097/MPH.0000000000000329
Kirkin, V. (2020). History of the selective autophagy research: how did it begin and where does it stand today? J. Mol. Biol. 432, 3–27. doi: 10.1016/j.jmb.2019.05.010
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608.
Knafo, S., Sanchez-Puelles, C., Palomer, E., Delgado, I., Draffin, J. E., Mingo, J., et al. (2016). PTEN recruitment controls synaptic and cognitive function in Alzheimer’s models. Nat. Neurosci. 19, 443–453. doi: 10.1038/nn.4225
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kwon, C.-H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388.
Lahiri, V., Hawkins, W. D., and Klionsky, D. J. (2019). Watch what you (self-) eat: autophagic mechanisms that modulate metabolism. Cell Metab. 29, 803–826. doi: 10.1016/j.cmet.2019.03.003
Lai, Y. C., Kondapalli, C., Lehneck, R., Procter, J. B., Dill, B. D., Woodroof, H. I., et al. (2015). Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J. 34, 2840–2861. doi: 10.15252/embj.201591593
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, J. O., Yang, H., Georgescu, M. M., Di Cristofano, A., Maehama, T., Shi, Y., et al. (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334.
Lee, Y., Lee, H.-Y., Hanna, R. A., and Gustafsson, ÅB. (2011). Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931.
Lee, Y. R., Chen, M., and Pandolfi, P. P. (2018). The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 19, 547–562. doi: 10.1038/s41580-018-0015-0
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuven. Res. 8, 3–5.
Li, D. M., and Sun, H. (1997). TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 57, 2124–2129.
Li, G., Yang, J., Yang, C., Zhu, M., Jin, Y., McNutt, M. A., et al. (2018). PTENα regulates mitophagy and maintains mitochondrial quality control. Autophagy 14, 1742–1760.
Li, M., Yang, X., and Wang, S. (2018). PTEN enhances nasal epithelial cell resistance to TNFalphainduced inflammatory injury by limiting mitophagy via repression of the TLR4JNKBnip3 pathway. Mol. Med. Rep. 18, 2973–2986. doi: 10.3892/mmr.2018.9264
Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S. I., et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (New York, NY) 275, 1943–1947.
Li, P., Wang, J., Zhao, X., Ru, J., Tian, T., An, Y., et al. (2019). PTEN inhibition attenuates endothelial cell apoptosis in coronary heart disease via modulating the AMPK-CREB-Mfn2-mitophagy signaling pathway. J. Cell. Physiol. 235, 4878–4889. doi: 10.1002/jcp.29366
Liang, H., He, S., Yang, J., Jia, X., Wang, P., Chen, X., et al. (2014). PTENalpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 19, 836–848. doi: 10.1016/j.cmet.2014.03.023
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, X. Y., Zhang, L. J., Chen, Z., and Liu, L. B. (2017). The PTEN inhibitor bpV(pic) promotes neuroprotection against amyloid beta-peptide (25-35)-induced oxidative stress and neurotoxicity. Neurol. Res. 39, 758–765. doi: 10.1080/01616412.2017.1317916
Lou, G., Palikaras, K., Lautrup, S., Scheibye-Knudsen, M., Tavernarakis, N., and Fang, E. F. (2019). Mitophagy and Neuroprotection. Trends Mol. Med. 26, 8–20.
Lucking, C. B., Abbas, N., Durr, A., Bonifati, V., Bonnet, A. M., de Broucker, T., et al. (1998). Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson’s Disease and the French Parkinson’s Disease Genetics Study Group. Lancet (London, England) 352, 1355–1356.
Malaney, P., Uversky, V. N., and Davé, V. (2013). The PTEN Long N-tail is intrinsically disordered: increased viability for PTEN therapy. Mol. Biosyst. 9, 2877–2888. doi: 10.1039/c3mb70267g
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471.
Marcassa, E., Kallinos, A., Jardine, J., Rusilowicz-Jones, E. V., Martinez, A., Kuehl, S., et al. (2018). Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep. 19:e45595.
Masson, G. R., Perisic, O., Burke, J. E., and Williams, R. L. (2016). The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity. Biochem. J. 473, 135–144. doi: 10.1042/BJ20150931
Matsushima, M., Fujiwara, T., Takahashi, E., Minaguchi, T., Eguchi, Y., Tsujimoto, Y., et al. (1998). Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer 21, 230–235.
McCoy, M. K., Kaganovich, A., Rudenko, I. N., Ding, J., and Cookson, M. R. (2014). Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum. Mol. Genet. 23, 145–156. doi: 10.1093/hmg/ddt407
McLelland, G.-L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 7:e32866.
McWilliams, T. G., Barini, E., Pohjolan-Pirhonen, R., Brooks, S. P., Singh, F., Burel, S., et al. (2018). Phosphorylation of Parkin at serine 65 is essential for its activation in vivo. Open Biol. 8:180108. doi: 10.1098/rsob.180108
Mizushima, N. (2018). A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527. doi: 10.1038/s41556-018-0092-5
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Montava-Garriga, L., and Ganley, I. G. (2020). Outstanding questions in mitophagy: what we do and do not know. J. Mol. Biol. 432, 206–230. doi: 10.1016/j.jmb.2019.06.032
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U.S.A. 113, 11249–11254.
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Nguyen, T. N., Padman, B. S., Usher, J., Oorschot, V., Ramm, G., and Lazarou, M. (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874.
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Okatsu, K., Kimura, M., Oka, T., Tanaka, K., and Matsuda, N. (2015a). Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 128, 964–978. doi: 10.1242/jcs.161000
Okatsu, K., Koyano, F., Kimura, M., Kosako, H., Saeki, Y., Tanaka, K., et al. (2015b). Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 209, 111–128. doi: 10.1083/jcb.201410050
Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3:1016. doi: 10.1038/ncomms2016
Okatsu, K., Uno, M., Koyano, F., Go, E., Kimura, M., Oka, T., et al. (2013). A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem. 288, 36372–36384. doi: 10.1074/jbc.M113.509653
Ordureau, A., Paulo, J. A., Zhang, J., An, H., Swatek, K. N., Cannon, J. R., et al. (2020). Global landscape and dynamics of parkin and USP30-dependent ubiquitylomes in iNeurons during mitophagic signaling. Mol. Cell. 77, 1124–1142. e1110.
Ordureau, A., Sarraf, S. A., Duda, D. M., Heo, J. M., Jedrychowski, M. P., Sviderskiy, V. O., et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell. 56, 360–375. doi: 10.1016/j.molcel.2014.09.007
Padman, B. S., Nguyen, T. N., Uoselis, L., Skulsuppaisarn, M., Nguyen, L. K., and Lazarou, M. (2019). LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 10:408. doi: 10.1038/s41467-019-08335-6
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022.
Pickles, S., Vigie, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Rasool, S., Soya, N., Truong, L., Croteau, N., Lukacs, G. L., and Trempe, J. F. (2018). PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 19:e44981.
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U.S.A. 113, 4039–4044. doi: 10.1073/pnas.1523926113
Rogov, V., Dötsch, V., Johansen, T., and Kirkin, V. (2014). Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell. 53, 167–178. doi: 10.1016/j.molcel.2013.12.014
Rogov, V. V., Suzuki, H., Marinkovic, M., Lang, V., Kato, R., Kawasaki, M., et al. (2017). Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 7:1131. doi: 10.1038/s41598-017-01258-6
Safiulina, D., Kuum, M., Choubey, V., Gogichaishvili, N., Liiv, J., Hickey, M. A., et al. (2019). Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 38:e99384.
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Sauve, V., Lilov, A., Seirafi, M., Vranas, M., Rasool, S., Kozlov, G., et al. (2015). A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 34, 2492–2505. doi: 10.15252/embj.201592237
Sauve, V., Sung, G., Soya, N., Kozlov, G., Blaimschein, N., Miotto, L. S., et al. (2018). Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol. 25, 623–630.
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 19500–19505.
Sekine, S., Wang, C., Sideris, D. P., Bunker, E., Zhang, Z., and Youle, R. J. (2019). Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol. Cell. 73, 1028–1043.e1025.
Sharma, M., Jarquín, U. N. R., Rivera, O., Kazantzis, M., Eshraghi, M., Shahani, N., et al. (2019). Rhes, a striatal-enriched protein, promotes mitophagy via Nix. Proc. Natl. Acad. Sci. U.S.A. 116, 23760–23771. doi: 10.1073/pnas.1912868116
Shen, S.-M., Zhang, C., Ge, M.-K., Dong, S.-S., Xia, L., He, P., et al. (2019). PTENα and PTENβ promote carcinogenesis through WDR5 and H3K4 trimethylation. Nat. Cell Biol. 21, 1436–1448.
Shi, R. Y., Zhu, S. H., Li, V., Gibson, S. B., Xu, X. S., and Kong, J. M. (2014). BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 20, 1045–1055. doi: 10.1111/cns.12325
Shiba-Fukushima, K., Arano, T., Matsumoto, G., Inoshita, T., Yoshida, S., Ishihama, Y., et al. (2014). Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet. 10:e1004861. doi: 10.1371/journal.pgen.1004861
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2:1002. doi: 10.1038/srep01002
Shinde, S. R., and Maddika, S. (2016). PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 7:10689. doi: 10.1038/ncomms10689
Shlevkov, E., Kramer, T., Schapansky, J., LaVoie, M. J., and Schwarz, T. L. (2016). Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. U.S.A. 113, E6097–E6106.
Soutar, M. P. M., Kempthorne, L., Miyakawa, S., Annuario, E., Melandri, D., Harley, J., et al. (2018). AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 8:8855. doi: 10.1038/s41598-018-26949-6
Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H., and Harris, A. L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673.
Steck, P. A., Pershouse, M. A., Jasser, S. A., Yung, W. K., Lin, H., Ligon, A. H., et al. (1997). Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362.
Stumpf, M., and den Hertog, J. (2016). Differential requirement for pten lipid and protein phosphatase activity during zebrafish embryonic development. PLoS ONE 11:e0148508. doi: 10.1371/journal.pone.0148508
Swaney, D. L., Rodriguez-Mias, R. A., and Villen, J. (2015). Phosphorylation of ubiquitin at Ser65 affects its polymerization, targets, and proteome-wide turnover. EMBO Rep. 16, 1131–1144. doi: 10.15252/embr.201540298
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D.-F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Trempe, J.-F., Sauvé, V., Grenier, K., Seirafi, M., Tang, M. Y., Ménade, M., et al. (2013). Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science (New York, NY) 340, 1451–1455.
Twig, G., Hyde, B., and Shirihai, O. S. (2008). Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta 1777, 1092–1097. doi: 10.1016/j.bbabio.2008.05.001
Ueno, T., Sato, W., Horie, Y., Komatsu, M., Tanida, I., Yoshida, M., et al. (2008). Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy 4, 692–700.
Unoki, M., and Nakamura, Y. (2001). Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 20, 4457–4465.
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160.
Vargas, J. N. S., Wang, C., Bunker, E., Hao, L., Maric, D., Schiavo, G., et al. (2019). Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol. Cell. 74, 347–362. e6.
Vieweg, S., Mulholland, K., Bräuning, B., Kachariya, N., Lai, Y.-C., Toth, R., et al. (2019). PINK1-dependent phosphorylation of Serine111 within the SF3 motif of Rab GTPases impairs effector interactions and LRRK2 mediated phosphorylation at Threonine72. bioRxiv [Preprint] doi: 10.1101/764019
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383. doi: 10.1073/pnas.0911187107
Walden, H., and Muqit, M. M. K. (2017). Ubiquitin and Parkinson’s disease through the looking glass of genetics. Biochem. J. 474, 1439–1451. doi: 10.1042/BCJ20160498
Wang, L., Cho, Y. L., Tang, Y., Wang, J., Park, J. E., Wu, Y., et al. (2018a). PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 28, 787–802.
Wang, L., Wang, J., Tang, Y., and Shen, H.-M. (2018b). PTEN-L puts a brake on mitophagy. Autophagy 14, 2023–2025. doi: 10.1080/15548627.2018.1502565
Wang, L., Qi, H., Tang, Y., and Shen, H.-M. (2020). Post-translational modifications of key machinery in the control of mitophagy. Trends Biochem. Sci. 45, 58–75. doi: 10.1016/j.tibs.2019.08.002
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Wauer, T., Simicek, M., Schubert, A., and Komander, D. (2015a). Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374.
Wauer, T., Swatek, K. N., Wagstaff, J. L., Gladkova, C., Pruneda, J. N., Michel, M. A., et al. (2015b). Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 34, 307–325. doi: 10.15252/embj.201489847
Williams, J. A., and Ding, W. X. (2018). Mechanisms, pathophysiological roles and methods for analyzing mitophagy – recent insights. Biol. Chem. 399, 147–178. doi: 10.1515/hsz-2017-0228
Worby, C. A., and Dixon, J. E. (2014). PTEN. Annu. Rev. Biochem. 83, 641–669. doi: 10.1146/annurev-biochem-082411-113907
Wu, W., Lin, C., Wu, K., Jiang, L., Wang, X., Li, W., et al. (2016). FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 35, 1368–1384. doi: 10.15252/embj.201593102
Wu, W., Tian, W., Hu, Z., Chen, G., Huang, L., Li, W., et al. (2014). ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15, 566–575. doi: 10.1002/embr.201438501
Yasuda, M., Theodorakis, P., Subramanian, T., and Chinnadurai, G. (1998). Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem. 273, 12415–12421.
Zachari, M., and Ganley, I. G. (2017). The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 61, 585–596. doi: 10.1042/EBC20170021
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. doi: 10.1074/jbc.M800102200
Zhang, J., Loyd, M. R., Randall, M. S., Waddell, M. B., Kriwacki, R. W., and Ney, P. A. (2012). A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy 8, 1325–1332. doi: 10.4161/auto.20764
Zhang, Y., Granholm, A. C., Huh, K., Shan, L., Diaz-Ruiz, O., Malik, N., et al. (2012). PTEN deletion enhances survival, neurite outgrowth and function of dopamine neuron grafts to MitoPark mice. Brain 135, 2736–2749. doi: 10.1093/brain/aws196
Zhang, J., and Ney, P. A. (2008). NIX induces mitochondrial autophagy in reticulocytes. Autophagy 4, 354–356.
Zhang, T., Xue, L., Li, L., Tang, C., Wan, Z., Wang, R., et al. (2016). BNIP3 Protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J. Biol. Chem. 291, 21616–21629.
Zhao, J., Geng, L., Chen, Y., and Wu, C. (2020). SNHG1 promotes MPP(+)-induced cytotoxicity by regulating PTEN/AKT/mTOR signaling pathway in SH-SY5Y cells via sponging miR-153-3p. Biol. Res. 53, 1–1. doi: 10.1186/s40659-019-0267-y
Keywords: mitophagy, PINK1, Parkin, BNIP3, PTEN, PTEN-L
Citation: Wang L, Lu G and Shen H-M (2020) The Long and the Short of PTEN in the Regulation of Mitophagy. Front. Cell Dev. Biol. 8:299. doi: 10.3389/fcell.2020.00299
Received: 13 February 2020; Accepted: 06 April 2020;
Published: 13 May 2020.
Edited by:
Konstantinos Palikaras, Foundation for Research and Technology Hellas, GreeceReviewed by:
Michael Lazarou, Monash University, AustraliaCopyright © 2020 Wang, Lu and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liming Wang, cGhzd2xAbnVzLmVkdS5zZw==; Han-Ming Shen, cGhzc2htQG51cy5lZHUuc2c=; aG1zaGVuQHVtLmVkdS5tbw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.