 Weiqi Hong
Weiqi Hong Fei Mo
Fei Mo Ziqi Zhang2
Ziqi Zhang2 Xiawei Wei
Xiawei Wei- 1Laboratory of Aging Research and Cancer Drug Target, State Key Laboratory of Biotherapy, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu, China
- 2West China Hospital and State Key Laboratory of Biotherapy, Sichuan University, Department of Biotherapy, Chengdu, China
NAD+, a co-enzyme involved in a great deal of biochemical reactions, has been found to be a network node of diverse biological processes. In mammalian cells, NAD+ is synthetized, predominantly through NMN, to replenish the consumption by NADase participating in physiologic processes including DNA repair, metabolism, and cell death. Correspondingly, aberrant NAD+ metabolism is observed in many diseases. In this review, we discuss how the homeostasis of NAD+ is maintained in healthy condition and provide several age-related pathological examples related with NAD+ unbalance. The sirtuins family, whose functions are NAD-dependent, is also reviewed. Administration of NMN surprisingly demonstrated amelioration of the pathological conditions in some age-related disease mouse models. Further clinical trials have been launched to investigate the safety and benefits of NMN. The NAD+ production and consumption pathways including NMN are essential for more precise understanding and therapy of age-related pathological processes such as diabetes, ischemia–reperfusion injury, heart failure, Alzheimer’s disease, and retinal degeneration.
Introduction
Nicotinamide adenine dinucleotide (NAD) is a vital metabolic redox co-enzyme found in eukaryotic cells and is necessary for over 500 enzymatic reactions. It plays a crucial role in various biological processes, including metabolism, aging, cell death, DNA repair, and gene expression (Rajman et al., 2018; Okabe et al., 2019). Thus, NAD+ is critical for human health and longevity.
The co-enzyme was first discovered by Harden and Young in 1906 as a component that enhanced the rate of alcohol fermentation in yeast extracts (Harden and Young, 1906). Over subsequent years, the chemical composition of the co-enzyme was established as an adenine, a reducing sugar group and a phosphate by Hans von Euler-Chelpin (von Euler and Myrback, 1930). Then, in 1936, Warburg suggested that NAD+ could play a role in redox reactions (Warburg and Christian, 1936). By 1960, it was assumed that all biochemical investigations on NAD+ had been exhausted. In 1963, Chambon and Mandel reported that NAD+ is a co-substrate for the addition of poly-ADP-ribose to proteins, and this prompted a series of studies on poly-ADP ribose and poly-ADP-ribose polymerases (PARPs) (Chambon et al., 1963; Yoshino et al., 2018). In the last decade, new interests in NAD+ emerged because of its association with sirtuins, a family of NAD-dependent protein deacylases (SIRT1–7) (Rajman et al., 2018). Roy Frye showed that mammalian sirtuins could metabolize NAD+ and that NAD+ had a protein ADP-ribosyltransferase activity (Frye, 1999). Guarente and Imai made a phenomenal discovery that yeast SIR2 (silent information regulator 2) and the mouse ortholog SIRT1 have NAD+-dependent protein deacetylase activity (Imai et al., 2000). Previously, several studies had shown that sirtuins play a critical role in regulating multiple cellular functions, such as cell growth, energy metabolism, stress resistance, inflammation, and circadian rhythm neuronal function, among others (Imai and Yoshino, 2013; Rajman et al., 2018). The deficiency of NAD+ is closely associated with diverse pathophysiologies, including type 2 diabetes (T2D), obesity, heart failure, Alzheimer’s disease (AD), and cerebral ischemia. The NAD+ levels decline in multiple organs with age, and this contributes to the development of various age-related diseases (Yoshino et al., 2011; Gomes et al., 2013; Mouchiroud et al., 2013; Mills et al., 2016). Therefore, NAD+ supplementation could be an effective therapy for the treatment of the conditions mentioned above.
Nicotinamide mononucleotide (NMN) is one of the intermediates in NAD+ biosynthesis and is a bioactive nucleotide formed by the reaction between a phosphate group and a nucleoside containing ribose and nicotinamide (NAM) (Poddar et al., 2019). NAM is directly converted to NMN by nicotinamide phosphoribosyltransferase (NAMPT). The molecular weight of NMN is 334.221 g/mol (Poddar et al., 2019). There are two anomeric forms of NMN named alpha and beta, and the latter is the active form (Poddar et al., 2019). NMN is found in various types of natural foods, such as vegetables, fruits, and meat. Edamame and broccoli contain 0.47–1.88 and 0.25–1.12 mg NMN/100 g, respectively, whereas avocado and tomato contain 0.36–1.60 and 0.26–0.30 mg NMN/100 g, respectively. However, raw beef only contains 0.06–0.42 mg NMN/100 g (Mills et al., 2016). Recent preclinical studies have demonstrated that the administration of NMN could compensate for the deficiency of NAD+, and NMN supplementation was able to effect diverse pharmacological activities in various diseases.
In this review, NAD+ biosynthesis pathways and the possible reason for its age-related decline are described. Also, a summary of studies on the role of NAD+ deprivation in causing human diseases and how the application of NMN could have positive effects on those diseases is provided.
NAD+ Biosynthesis Pathways
Three different NAD+ biosynthesis pathways have been described in mammalian cells (Figure 1): (1) Preiss–Handler, in which NAD is synthesized from nicotinic acid (NA); (2) de novo synthesis, which starts from tryptophan; and (3) salvage pathway, which is most predominant in mammalian cells.
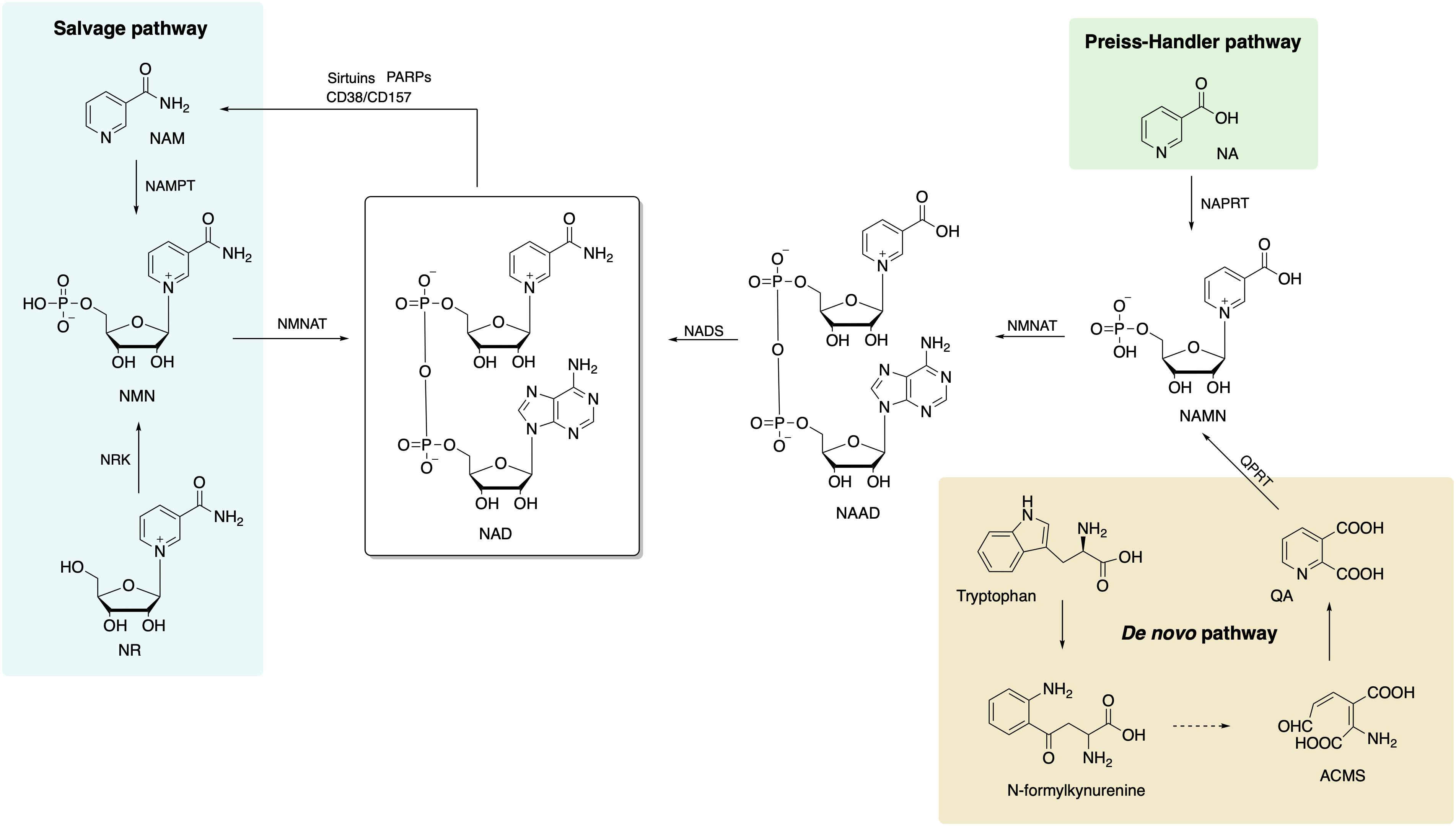
Figure 1. Biosynthetic pathways of NAD+ in mammalian cells includes de novo, Preiss–Handler, and salvage pathways, and the salvage pathway is the main source of NAD+. NAD, nicotinamide adenine dinucleotide; NA, nicotinic acid; NAPRT, nicotinic acid phosphoribosyltransferase; NAMN, nicotinic acid mononucleotide; NAAD, nicotinic acid adenine dinucleotide; NADS, NAD+ synthetase; NMNAT, nicotinamide/nicotinic acid mononucleotide adenylyltransferase; ACMS, 2-amino-3-carboxymuconate semialdehyde; QA, quinolinic acid; QPRT, quinolinate phosphoribosyltransferase; NAM, nicotinamide; NAMPT, nicotinamide phophoribosyltransferase; NMN, nicotinamide mononucleotide; NR, nicotinamide riboside; NRK, nicotinamide riboside kinase.
The Preiss–Handler Pathway
This pathway starts with conversion of NA to the nicotinic acid mononucleotide (NAMN) by the enzyme nicotinic acid phosphoribosyltransferase (NAPRT) (Preiss and Handler, 1958). Afterward, NAMN is used for nicotinic acid adenine dinucleotide (NAAD+) biosynthesis by nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT1/2/3). Finally, NAD+ synthetase (NADS) transforms NAAD+ to NAD+ with ammonia and ATP action as extra ingredients (Yang and Sauve, 2016).
De novo Synthesis From Tryptophan
The eight-step de novo synthesis pathway is initiated by indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO) that convert tryptophan to N-formylkynurenine (Salter et al., 1991). Through formamidase (KFase), N-formylkynurenine is then transformed to be kynurenine, to which hydroxyl is added via kynurenine 3-hydroxylase (K3H). The product, 3-hydroxy-kynurenine, is converted to 3-hydroxyanthranilate followed by 2-amino-3-carboxymuconate semialdehyde (ACMS) via kynureninase (Kyase) and 3-hydroxyanthranilate-3,4-dioxygenase. ACMS then cyclizes to form quinolinic acid (QA) that participates in NAMN biosynthesis with quinolinate phosphoribosyltransferase (QPRT) (Yang and Sauve, 2016). The last two steps are the same as the Preiss–Handler pathway that NAD+ is synthetized sequentially from NAMN and NAAD by NMNAT 1/2/3 and NADS.
The Salvage Pathway
The salvage pathway is the primary source of NAD+ in mammalian cells. The degradation of NAD+ and subsequent generation of NAM (as a by-product) are achieved by NAD-consuming enzymes, such as sirtuins, PARPs, CD38, CD157, and sterile alpha and TIR motif-containing protein 1 (SARM1) (Okabe et al., 2019). There are only two steps in the salvage pathway. The rate of NAD+ synthesis in this pathway is mostly determined by NAMPT that converts NAM and 5-phosphoribosyl-1-pyrophosphate (PRPP) to NMN in the first step. Then, NMN, the substrate for NAMNT, is conjugated to ATP and converted to NAD in the second step.
The NAMPT exists in two forms in mammals, that is, intracellular NAMPT (iNAMPT) in the cytoplasm and nucleus and extracellular NAMPT (eNAMPT) in the plasma or extracellular space (Revollo et al., 2007). The SIRT1-dependent deacetylation of iNAMPT predisposes the protein to secretion in adipocytes (Yoon et al., 2015). Various types of cells, including mature adipocytes, pancreatic β-cells, myocytes, epithelial cells, and hepatocytes (Revollo et al., 2007; Garten et al., 2010; Zhao et al., 2014) secrete and release eNAMPT to the plasma or extracellular space.
Also, another NAD precursor, nicotinamide riboside (NR), is incorporated into cells using equilibrative nucleoside transporters (ENTs) (Nikiforov et al., 2011) and phosphorylated to NMN by nicotinamide riboside kinase (NRK1/2) intracellularly (Ratajczak et al., 2016). Conversion of extracellular NMN to NR mediated by enzyme CD73 is required for cell uptake and intracellular synthesis of NAD+ (Grozio et al., 2013; Ratajczak et al., 2016). NAD+ biosynthesis in kidney and brown adipose tissue has been shown to decrease after administration of NMN in NRK1 knockout mice (Ratajczak et al., 2016). However, a recent study identified Slc12a8 as a specific transporter of NMN, which is highly expressed in the small intestine (Grozio et al., 2019). In the study, Slc12a8 expression was upregulated in the small intestines of aged mouse in response to a decrease of NAD+. These findings suggested that the uptake pathway of NMN could be via a cell- or tissue-specific manner.
Given that the salvage pathway is the main and the most efficient route for NAD+ biosynthesis, NMN or NR supplementation is becoming the preferred option of improving NAD+ levels that is devoid of side effects. Currently, increasing numbers of clinical trials using NMN and NR have been approved and are geared toward the treatment of various diseases, which further demonstrate that NMN is a suitable and safe drug for use in humans.
Effect of Aging on NAD+ Levels
NAD+ Biosynthetic Pathways Decline With Age
The decline in NAD+ biosynthetic pathways in the course of aging could be a possible explanation for the reduction of NAD+ levels. NAMPT controls NAD+ levels, thereby influencing the activity of NAD-dependent enzymes, including sirtuins and PARPs.
A study demonstrated that the NAD+ levels and NAMPT protein levels declined significantly in multiple organs, including the pancreas, white adipose tissue (WAT), and skeletal muscle of old mice (Yoshino et al., 2011). However, exercise training increased NAMPT expression in the skeletal muscles (Costford et al., 2010). NAD+ levels and exercise capacity were preserved in aged transgenic mice with muscle-specific NAMPT transgene expression (Frederick et al., 2016). These results suggested that the deficiency of NAMPT result in a reduction of NAD+ levels in the aged mice, and exercise may elevate NAMPT expression, thus restoring the NAD+ levels (Figure 2).
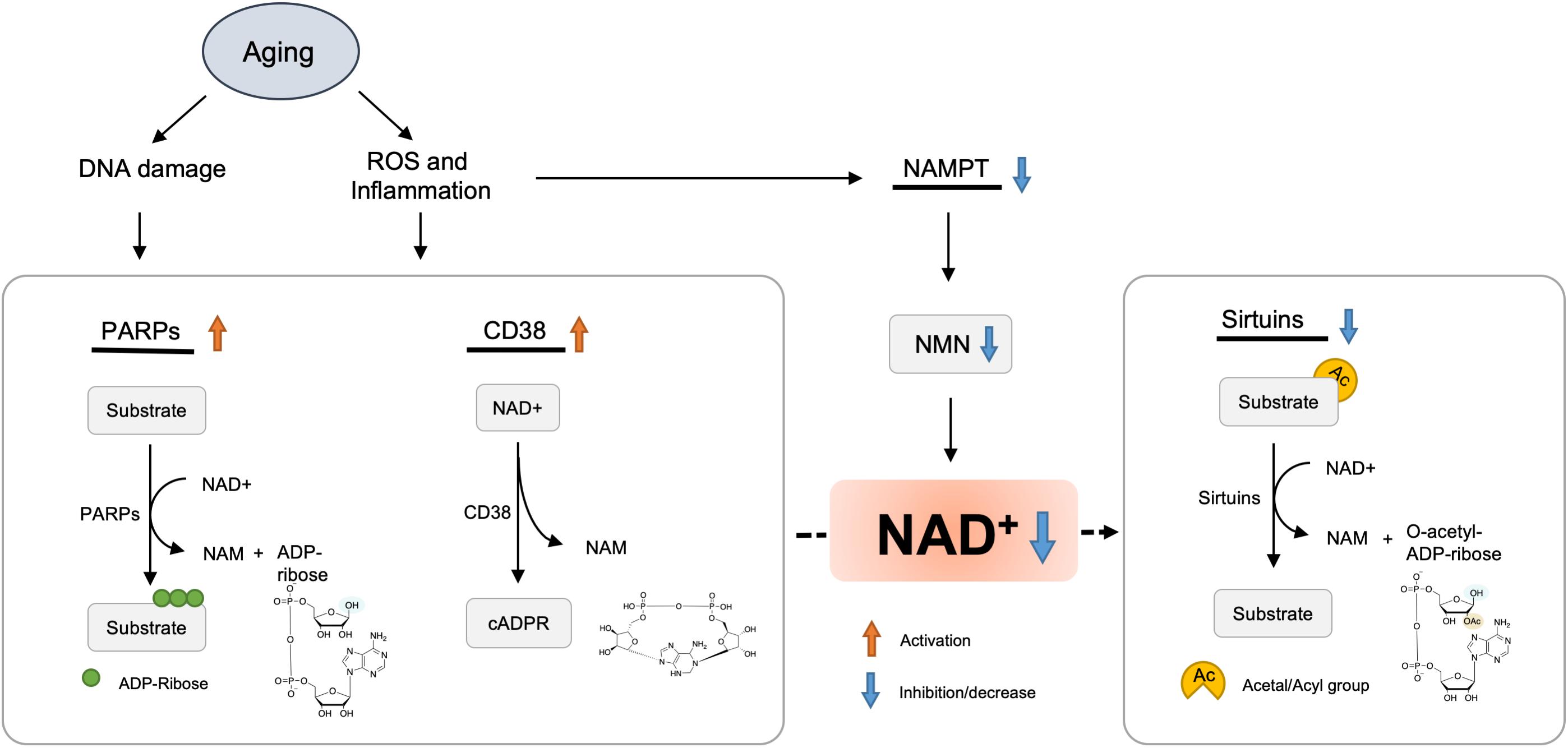
Figure 2. Hypothetic molecule mechanisms of NAD+ decreased with aging. Oxidative stress, DNA damage, and chronic inflammation are increased with aging, which results in accelerated NAD degradation via activation of CD38 and PARPs, or dysregulation of NAMPT. Finally, decreased levels of NAD+ lead to various metabolic and age-associated diseases.
Inflammation and oxidative stress caused by aging have been shown to reduce the NAMPT-mediated NAD+ biosynthesis (Yoshino et al., 2011; Figure 2). Besides, Nampt gene encoding is controlled by BMAL1/CLOCK complex, a heterodimeric complex of core circadian transcription factors, which is suppressed by inflammatory cytokines (Cavadini et al., 2007). Therefore, the development of chronic inflammation in the course of aging may contribute to the inhibition of NAMPT-mediated NAD+ biosynthesis and CLOCK/BMAL-mediated circadian machinery (Imai and Guarente, 2014).
NAD+-Consuming Enzymes Are Activated With Age
PARP1
PARPs were initially considered to be DNA damage repair agents in the 1960s (Chini et al., 2017). The accumulation of DNA damage during aging could activate PARP, among which PARP-1 acts as a major cellular NAD+-consuming enzyme (Imai and Guarente, 2014). Cockayne syndrome (CS) is an aging-related progressive neurodegeneration that occurs as a result of mutations in either Cockayne syndrome group A (CSA) or B (CSB) proteins (Gitiaux et al., 2015; Scheibye-Knudsen et al., 2014). In CS mice, PARP inhibitor or NAD+ supplementation reversed decline in SIRT1 activation and mitochondrial function caused by aberrant PARP activation (Scheibye-Knudsen et al., 2014). Consistently, another inhibitor of PARP, PJ34, or knockout boosted the levels of NAD+, SIRT1 activity, and oxidative metabolism (Bai et al., 2011).
CD38
The CD38 enzyme and its homolog CD157 were initially described as plasma membrane antigens on thymocytes and T lymphocytes. Their role in NAD+ consumption have been revealed; that is, CD157/BST-1 could hydrolyze NR (Preugschat et al., 2014) and CD38 hydrolyzes NAD+ to generate NAM, adenosine diphosphoribose (ADPR), and cyclic ADPR (cADPR). In addition, CD38 also hydrolyzes cADPR (De Flora et al., 2004) and NMN (Grozio et al., 2013).
In mammals, the level of NAD+ and mitochondrial function decreased partially through regulation of SIRT3 as the expression and activity of CD38 protein increased in various tissues during aging (Camacho-Pereira et al., 2016). Administration of CD38 inhibitors elevated intracellular NAD+ level (Escande et al., 2013; Boslett et al., 2017). Consistently, CD38 knockout mice displayed significantly higher NAD+ level in multiple organs (Young et al., 2006).
Sterile Alpha and TIR Motif-Containing 1 (SARM1) Protein
The toll/interleukin-1 receptor (TIR) domain of sterile alpha and TIR motif-containing 1 (SARM1) protein presents NADase activity (Rajman et al., 2018) that is involved in axonal degeneration after axon injury. In response to neuronal injury, the TIR domain of SARM1 cleaves NAD+ to generate ADP ribose (ADPR) and cyclic ADPR, which may contribute to axonal degeneration (Essuman et al., 2017). Paradoxically, overexpression of enzymes in NAD+ biosynthesis pathway or supplying NR could inhibit SARM1-induced axon destruction (Gerdts et al., 2015).
In summary, there are many ways of restoring NAD+ level depletion caused by aging or other diseases, including improving NAMPT expression, providing NAD+ precursors, or inhibiting NAD+, consuming enzymatic activities of PARP, CD38, and SARM1. Currently, supplementation with NMN or NR is considered a viable and highly efficient strategy of increasing NAD+ levels (Figure 3).
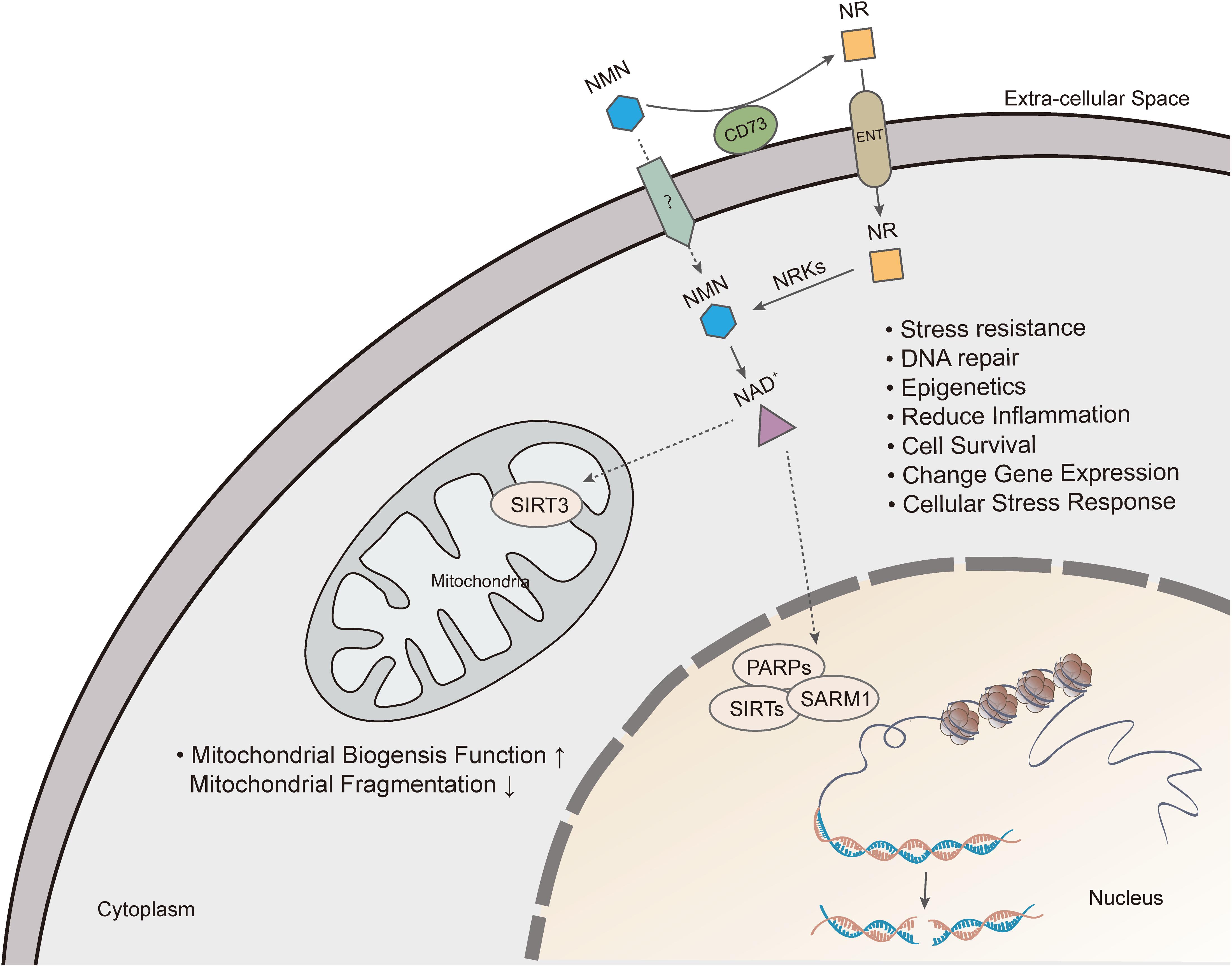
Figure 3. Nicotinamide mononucleotide exerts pharmacological effects by increasing intracellular NAD+ levels. Extracellular NMN is cleavage by CD73, which yields NR that is incorporated into cells using equilibrative nucleoside transporters (ENTs). NMN is converted to NAD+, which produces beneficial effects on cell, including mitochondrial function, DNA repair, gene expression, anti-inflammation and cell survival.
Diabetes
The global prevalence of diabetes has increased dramatically over the past four decades. According to the WHO report, the number of people with diabetes rose from 108 million in 1980 to 422 million in 2014. T2D is characterized by insulin resistance and subsequent impairment of insulin secretion (Okabe et al., 2019). The metabolism of NAD+ plays a crucial role in insulin sensitivity and secretion and is sometimes disrupted by obesity and aging.
Revollo et al. showed that eNAMPT was necessary for NAD+ biosynthesis (Revollo et al., 2007). Declined NAD+ levels and glucose-stimulated insulin secretion (GSIS) in pancreatic β cells and impaired glucose tolerance were observed in Nampt+/– mice. Similar blood glucose and plasma insulin levels were observed in Nampt+/– and control mice after NMN (i.p. 500 mg/kg) treatment. Also, FK866, an inhibitor of NAMPT, reduced NAD+ levels, and glucose-stimulated insulin secretion in primary islets, whereas NMN treatment reversed the defects (Revollo et al., 2007). These results demonstrated that Nampt-mediated NAD+ biosynthesis is critical for β cell function, and that NMN treatment can ameliorate the deficiency in NAD biosynthesis and glucose-stimulated insulin secretion.
Obesity and diabetes are inextricably linked. MicroRNAs (miRNAs) are key regulators of metabolism, by which SIRT1 expression is regulated in healthy conditions and metabolic diseases (Lee and Kemper, 2010). In the dietary obese mice, the elevation of hepatic microRNA-34a (miR-34a) inhibited the expression of NAMPT and SIRT1, which was responsible for the decrease in NAD+ levels and SIRT1 activity (Choi et al., 2013). The reduction of SIRT1 activity resulted in transcriptional responses of decreased fatty acid β-oxidation and increased lipogenesis and inflammation (Choi et al., 2013). Mice overexpressing miR-34a were intraperitoneally injected with NMN (500 mg/kg) for 10 days consecutively, the effects caused by hepatic overexpression of miR-34 were reversed, and glucose tolerance was enhanced (Choi et al., 2013). These results suggested that NMN could be a potential agent for the treatment of obesity-associated T2D involving SIRT1 dysfunction.
Because of the high intake of dietary sugar, the risk of metabolic syndrome and T2D has been on the increase in humans (Malik et al., 2010). Fructose consumption contributes to the development of pro-inflammatory effect in rodent models, which is involved in the process of insulin resistance and the onset of T2D (Roncal-Jimenez et al., 2011). Fructose-rich diet (FRD) results in T2D-like symptoms, including hyperglycemia, dyslipidemia, and inflammation (Roncal-Jimenez et al., 2011). FRD-fed mice showed an increased expression of IL-1b and TNF-α, which are pro-inflammatory phenotypes. The GSIS and leucine-stimulated insulin secretion (LSIS) were significantly reduced in FRD-fed mice, which was associated with islet dysfunction caused by a decrease of eNAMPT in plasma, whereas the administration of NMN at the dose of 500 mg/kg eliminated the adverse effects of FRD on GSIS and LSIS in mice (Caton et al., 2011).
FRD increased the expression of Inos (induces cellular stress and cell death) and Bax (pro-apoptotic gene) genes and reduced the expression of Pdx1, Glut2, and Gk genes, which are all essential for glucose detection and beta-cell differentiation. These changes in gene expression were restored by NMN treatment. Moreover, the decrease in expression of Sirt1 and Sirt3 genes in FDR mice were reversed by NMN treatment. These results suggested that NMN could improve islet function by influencing the expression of genes related to anti-inflammatory, islet beta-cell differentiation, and SIRT1 activation.
Yoshino et al. revealed that the administration of NMN was highly effective in combating diet- and age-induced T2D (Yoshino et al., 2011). In their study, mice fed with high-fat diet (HFD) displayed significantly reduced NAMPT protein and NAD+ levels in the liver and WAT and not in skeletal muscle as was expected. The HFD-induced male and female diabetic mice after receiving intraperitoneal administration of NMN (500 mg/kg/day) for 10 and 7 consecutive days, respectively, exhibited restored NAD+ levels in the liver and WAT. Impaired glucose and insulin tolerance were significantly improved in diabetic female mice. The effect of NMN in reversing impaired glucose tolerance was milder in males compared to females, and insulin tolerance remained unchanged in male mice.
Yoshino et al. also confirmed that NMN improves hepatic insulin sensitivity by reversing the expression of genes related to oxidative stress, inflammatory response, immune response, and lipid metabolism (Yoshino et al., 2011). For example, the expression of the glutathione S-transferase alpha two gene (Gsta2), which is crucial for the maintenance of hepatic insulin by protecting lipid peroxidation products, was suppressed by HFD but activated by NMN. Other genes that are related to insulin resistance were significantly influenced by NMN in HFD-induced mice, such as interleukin 1β, lipin1, and pyruvate dehydrogenase kinase 4 (Pdk4). Also, Yoshino et al. revealed that SIRT1 was responsible for gene expression dynamics, and its suppression by HFD was restored by NMN.
Aging is one of the highest risk factors for developing T2D (Moller et al., 2003). Previous studies have shown that progressive decline in β cell function in the course of aging contributes to the pathophysiology of T2D (Basu et al., 2003). As mentioned above, NAD+ and NAMPT levels declined during aging (Yoshino et al., 2011). Administration of NMN (500 mg/kg/day) for 11 consecutive days resulted in significant improvement in glucose tolerance and utilization in aged mice. Also, hyperlipidemia induced by HFD was also reversed by this treatment. Meanwhile, NMN did not have other effects on glucose homeostasis in non-diabetic old mice (Yoshino et al., 2011). Mills et al. (2016) also demonstrated that long-term (12 months) administration of NMN ameliorated age-associated decreased insulin sensitivity. Triglyceride levels in the liver were lower after 12-month NMN administration, which indicated a decline in insulin resistance. Mice that were put on long-term NMN intervention showed a tendency of plasma fatty acids (FFA), which is consistent with improved insulin sensitivity (Mills et al., 2016).
Moynihan et al. (2005) reported that an increase in Sirt1 dosage in pancreatic β cells improved GSIS and glucose tolerance in beta cell-specific Sirt1-overexpression (BESTO) transgenic mice at 3 and 8 months of age. However, the same cohort of BESTO mice did not show these beneficial effects at 18–24 months of age (Ramsey et al., 2008). It has been reported that Sirt1 improves glucose-stimulated insulin secretion through the repression of Ucp2 and improvement of ATP levels in pancreatic β cells (Bordone et al., 2006; Ramsey et al., 2008), which was abolished in aged mice, resulting in maintenance of a high level of Sirt1 protein. Also, NMN plasma levels declined significantly in aged BESTO mice, which suggested that a decrease in Sirt1 activity and loss of the glucose-responsiveness in BESTO mice resulted from the decline in systemic NAD biosynthesis. Thus, intraperitoneal injection of NMN (500 mg/kg) into 20-month-old BESTO mice led to an enhancement in GSIS and improvement in glucose tolerance in the aged BESTO females instead of males.
Generally, these discoveries demonstrated that NMN could be a promising drug for obese-associated and age-induced T2D through the role it plays in the enhancement of NAD+ biosynthesis and Sirt1 activity.
Obesity
Obesity is associated with insulin resistance in multiple organs. It is a systemic metabolic derangement, which is involved in the pathogenesis of many diseases such as T2D, non-alcoholic fatty liver disease (NAFLD), atherogenic dyslipidemia, and cardiovascular disease (Reaven, 1988). It has been reported that the dysfunction of adipose tissue could result in obesity-associated metabolic disorders in multiple organs because adipose tissue has effects on maintaining the functional integrity of whole-body metabolic health (Stromsdorfer et al., 2016). Significantly reduced iNAMPT has been manifested in adipocyte as a result of HFD (Yoshino et al., 2011; Chalkiadaki and Guarente, 2012), however, elevated eNAMPT was detected in obesity (Catalán et al., 2011). Compared with clear function of iNAMPT in NMN synthesis, the significance of eNAMPT is controversial. In response to cellular stress, nutritional cues, or inflammatory cytokines, adipocytes secreted eNAMPT through PI3K-AKT pathway (Haider et al., 2006), SIRT1-mediated pathway (Yoon et al., 2015), or other unknown pathway (Tanaka et al., 2007). The role of eNAMPT has been indicated to be inflammatory promotion, inflammatory suppression (Revollo et al., 2007; Li et al., 2008; Pillai et al., 2013; Zhao et al., 2013; Jing et al., 2014), enhancement of food intake (Brunetti et al., 2012), and regulation of insulin resistance as well as plasma free fatty acid concentration (Stromsdorfer et al., 2016; Nielsen et al., 2018). The function of eNAMPT remains elusive in recent research, demonstrating that low concentration of dimeric eNAMPT benefited for beta cell function through NAD+, however, a higher monomeric eNAMPT level presented hostile effect on beta cell function (Sayers et al., 2020). Obesity has also been associated with the dampened NAD+/SIRT pathway in adipose tissue (Jukarainen et al., 2016). These findings suggested that NAMPT-mediated NAD+ biosynthesis in adipose tissue could be involved in the regulation of whole-body glucose metabolism.
Adipocyte-specific Nampt knockout (ANKO) mice showed a severe multi-organ insulin resistance, including adipose tissue, liver, and skeletal muscle, independently with an increase in whole-body adiposity and weight. The plasma FFA availability and local adipose tissue inflammation were also increased in the ANKO mice (Stromsdorfer et al., 2016). The plasma concentration of two key adipokines, namely, adiponectin and adipsin, were significantly reduced in ANKO mice. It has been reported that adiponectin and adipsin regulate insulin sensitivity and glucose homeostasis (Kadowaki et al., 2006). Phosphorylation of cyclin-dependent kinase 5 (CDK5) and peroxisome proliferator-activated receptor γ (PPARγ) was increased in adipose tissue of the ANKO mice, which led to a significant decline in gene expression of obesity-linked specific targets of phosphorylated PPARγ, including adiponectin and adipsin. As was expected, NAD+ levels in adipose tissue from the ANKO mice were significantly increased after oral administration of NMN (500 mg/kg) for 4–6 weeks. The NMN treatment also improved multi-organ insulin sensitivity and normalized plasma insulin and FFA concentrations in the ANKO mice. Moreover, the phosphorylation of PPARγ (Ser273) and CDK5 in visceral adipose tissue (VAT) was reduced by the NMN treatment. Accordingly, the plasma concentrations and gene expression of adiponectin and adipsin in adipose tissue were enhanced (Stromsdorfer et al., 2016). Therefore, these results provided evidence that NMN could be a therapeutic molecule for obesity-associated systemic metabolic derangements, particularly multi-organ insulin resistance. ANKO and brown adipocyte-specific Nampt knockout (BANKO) mice both showed impaired gene programs involved in thermogenesis, mitochondrial biogenesis, and FFA metabolism in BAT (Yamaguchi et al., 2019). However, only ANKO mice have a blunted thermogenic response (lower temperature in rectal and BAT, and whole-body oxygen consumption) to acute cold exposure, fasting, and administration of β-adrenergic agonists. Altered function in WAT may contribute to this difference. Lack of NAMPT in WAT decreased adrenergic-mediated lipolysis through inactivation of the NAD+–SIRT1–caveolin-1 axis, which reduced the release of FFAs as fuel source for BAT thermogenesis. NMN administration normalized these metabolic abnormalities, including increasing BAT NAD+ levels, decreasing BAT weight and BAT whitening as well as restoring gene expression of caveolin-1 in WAT and those involved in thermogenesis, mitochondrial function, and FFA metabolism in BAT. Moreover, ANKO mice treated with NMN showed greater cold tolerance compared with ANKO mice (Yamaguchi et al., 2019).
Several studies have confirmed that physical exercises have various health benefits, especially in obesity-related cases (Vieira et al., 2009; Ross et al., 2015), which, in part, results from the upregulation of mitochondrial activity because of increased NAD levels. Mouse models showed that NAD+ levels in metabolic organs and mitochondrial biogenesis could be improved by physical exercises (Ross et al., 2015). Thus, taking physical exercises is thought to be an effective way of increasing NAD+ levels. To compare the efficiency of increasing NAD+ levels between NMN supplementation and exercise, HFD-induced obese mice were given NMN (500 mg/kg) for 17 days or treadmill running (45 min/day) was implemented for 6 days per week for 6 weeks. According to the results, NMN treatment increased NAD+ levels in muscles and the liver, but exercise increased NAD+ levels in muscles only. The liver mass and triglyceride content were significantly reduced, and citrate synthase activity was increased after NMN treatment in HFD-fed mice, which suggested that NMN could increase catabolism of fats. Exercises and NMN have a similar effect on glucose intolerance induced by obesity, however, these two interventions are tissue-specific with a different impact on mitochondrial function in muscles and the liver (Uddin et al., 2016). Uddin et al. (2017) also showed that NMN could reduce the effects of maternal obesity as compared to exercise. Maternal overnutrition is often associated with increased infant birth weight, adiposity, and high risk of long-term obesity in later life of the offspring (Castillo-Laura et al., 2015). Thus, it is important to investigate new strategies for reducing the risk in future generations. A study found that treadmill exercise (for 9 weeks) and NMN injection (for 15 days) both reduced adiposity and improved glucose tolerance and mitochondrial function. Moreover, NMN showed stronger effects on liver fat catabolism and synthesis than exercise. This study suggested that NMN treatment might be an effective option for reversing negative effects caused by maternal obesity (Uddin et al., 2017).
Long-term administration of NMN can significantly reduce age-associated body weight gain in a dose-dependent manner. A study conducted a 12-month-long administration of NMN (from 5 to 17 months) in mice. The results indicated that the 100- and 300-mg/kg dose of NMN was able to reduce the mice’s weight by 4 and 9%, respectively, compared to the control mice. No difference was observed in body length between NMN-treated and control mice. NMN-treated mice also maintained higher levels of food and water consumption compared to control mice, which suggested that NMN did not cause severe side effects, such as growth defect and loss of appetite (Mills et al., 2016). In conclusion, the administration of NMN could be an effective option for maintaining body weight and reversing metabolic dysfunctions caused by obesity.
Ischemia–Reperfusion Injury
Ischemia is known to decrease oxygen and ATP levels in tissues, leading to cell necrosis. Reperfusion is a reoxygenation process whereby blood re-enters a previously ischemic tissue, and this often causes calcium overload and production of ROS (Sanada et al., 2011). Ischemia followed by reperfusion has been reported to trigger severe tissue damage, for which ischemic preconditioning (IPC) is a confirmed preventative strategy (Sanada et al., 2011).
It has been reported that activation of SIRT1 can protect the heart from ischemia and reperfusion (I/R)-induced injury (Hsu et al., 2010). SIRT1 upregulates cardioprotective molecules, such as MnSOD (antioxidants), Trx1 (antioxidants), and Bcl-xL (anti-apoptotic), while it decreases pro-apoptotic molecules, including Bax and cleaved caspase-3. FoxO1, a transcription factor deacetylated by Sirt1, partially mediates the SIRT1-induced upregulation of MnSOD (Hsu et al., 2010), which protects the heart from oxidative stress. Moreover, maintaining NAMPT expression is critical for prevention of myocardial injury caused by I/R (Hsu et al., 2009). The deacetylase activity of SIRT1 is dependent on NAD+. Thus, enhancing NAD+ may promote SIRT1-mediated IPC.
NMN has been shown to confer protection on the heart in ischemic and reperfusion conditions (Yamamoto et al., 2014). Yamamoto et al. found that IPC upregulates NAMPT and the protective effect of IPC on heart during I/R injury is attenuated in Nampt+/– mice, suggesting that NAMPT mediates the protective effect of IPC. The NAD+ levels in heart were decreased after 30 min of ischemia and transiently normalized by NMN administration. Implementation of two patterns of NMN administration, once 30 min before ischemia or 4 times repetitive administration just before and during reperfusion, reduced the infarct size by 44 and 29%, respectively. However, a single administration of NMN 12 h before ischemia and once immediately before reperfusion did not significantly reduce the infarct size. These findings suggest that NMN reduces infarct size after by I/R, and this effect is timing-dependent. Notably, NMN attenuated the increase in acetylation of FoxO1 during myocardial ischemia, but NMN failed to reduce the infarct size in Sirt1-KO mice, indicating that the protective effect of NMN on the heart following I/R injury is partly mediated by Sirt1.
NMN suppressed cardiomyocyte apoptosis in the peri-infarct area after I/R and significantly improves left ventricle (LV) systolic function caused by I/R. In addition, NMN activates autophagy during myocardial ischemia, which is consistent with the finding that NAMPT and SIRT1 promote autophagy in cardiomyocytes (Hsu et al., 2009). An ex vivo experiment showed that NMN attenuated myocardial I/R injury in aged rats (Hosseini et al., 2019b). Rats treated with NMN showed improved myocardial function and reduced infarct size. Besides, NMN exerted positive effects on mitochondrial function by improving the antioxidant system, restoring oxidative stress, and reducing mitochondrial ROS production and membrane depolarization. Importantly, the combination of NMN and melatonin could produce stronger cardioprotection.
Glycolytic stimulation during ischemia and enhanced acidosis during reperfusion are additional mechanisms for NMN-induced cardioprotection (Nadtochiy et al., 2018). Under normal conditions, cardiac ATP production is executed by β-oxidation of fatty acids (Stanley et al., 2005). Generating ATP via glycolysis is usually considered a manifestation of cardiac pathology and heart failure (Stanley et al., 2005). However, recent studies show that glycolysis has cardioprotective effects (Gohil et al., 2010). Nadtochiy et al. reported that NMN stimulates glycolysis and increases ATP production during ischemia, which partially contributes to NMN-induced cardioprotection (Nadtochiy et al., 2018). IR injury triggers the opening of the mitochondrial permeability transition pore (mPTP). Acid pH maintains the closed state of PT pores during ischemia, however, changes in pH during reperfusion promote mitochondrial pore opening (Griffiths and Halestrap, 1995). Addition of acidic media offers cardioprotection by maintaining the closed state of PT pore in early reperfusion (Cohen et al., 2007). It is worth noting that acidosis may promote mPTP in energetic mitochondria by stimulating Pi uptake (Kristian et al., 2001). NMN elevates cardiac lactate and pyruvate to induce acidosis, which protects against IR-induced injury (Nadtochiy et al., 2018).
Park et al. (2016) showed that NMN antagonizes global cerebral ischemia injury. Ischemic insults increase the production of free radicals, which causes DNA oxidative damage and PARP1 activation, and uncontrolled PARP1 activation decreases NAD+, which further decreases ATP synthesis, leading to cell death (Strosznajder et al., 2003). Conversely, NMN maintains normal cellular NAD+ levels by inhibiting the NAD+ catabolism of PARP1 to improve bioenergetics metabolism of post-ischemic tissue and ameliorate brain damage. NMN treatment at 30 min after initiation of reperfusion reduces hippocampal CA1 neuron cell death and improves ischemia-induced hippocampal dysfunction involved in spatial working memory (Park et al., 2016). The NAD+ levels in hippocampal tissue are significantly reduced after forebrain ischemia, and NMN inhibits this decrease in NAD+. Meanwhile, studies show that hippocampal PARP protein levels are significantly increased in mice with cerebral ischemia, accompanied by elevated ROS production. Interestingly, this effect is blocked by administration of NMN at the start of reperfusion. Notably, when administered at a dose of 62.5 mg/kg, NMN produced optimal treatment effects compared to other doses (500, 250, 125, and 31.25 mg/kg), suggesting that high dosage of NMN may introduce adverse effects on post-ischemic neurons. In fact, accumulation of NMN in nerve injury promotes axonal degeneration (Di Stefano et al., 2015) and NMN deamidase delays Wallerian degeneration and rescues axonal outgrowth defects (Di Stefano et al., 2017). Recently, Klimova et al. (2020) revealed a novel link between mitochondrial NAD+ metabolism, mitochondrial dynamics, and ROS production in cerebral ischemia. They showed that NMN treatment prevents post-ischemic depletion of mitochondrial NAD+, suppresses mitochondrial fragmentation, and reduces ROS generation via SIRT3-dependent mechanisms (Klimova et al., 2020). The activity of superoxide dismutase 2 (SOD2), a key mitochondrial antioxidant enzyme, was inhibited by an increase in its acetylation after ischemia, which can be reversed by NMN treatment. Moreover, NMN also prevents ischemia-induced phosphorylation of mitochondrial fission Dynamin-related protein (Drp1) (Klimova et al., 2020).
Wang P. et al. (2011) have already demonstrated that NAMPT protected against ischemic stroke through promoting neuronal survival via the SIRT1-dependent AMPK pathway. As the NAMPT enzymatic product, NMN alleviates cerebral infarction size, neurological deficit, and neuronal cell death (Wang P. et al., 2011). Furthermore, the important role of NAMPT–NAD cascade in regenerative neurogenesis after ischemic stroke has been underscored by Zhao et al. (2015), and delayed NMN supplementation for 7 days with the first administration at 12 h after cerebral ischemia improved post-ischemic regenerative neurogenesis.
Heart Failure and Cardiomyopathies
Heart failure is the end stage of heart disease development and refers to the inability of the heart to pump sufficient blood to match the metabolic requirements of tissues due to impaired systole and/or diastole. Generally, heat failure is always associated with enlarged heart and dilated ventricles (Pillai et al., 2005).
Heart failure is one of diseases that are associated with mitochondrial respiratory dysfunction (Karamanlidis et al., 2013). It was found that deletion of Ndufs4, a protein critical for assembly and/or stability of complex I, resulted in a significant loss of complex I function in the heart. The cardiac-specific Ndufs4 KO (cKO) mice not only showed normal longevity and cardiac function in unstressed condition, but also exhibited normal myocardial energetics and contractile function during baseline workload or acute increases of workload. However, after chronic increases of workload including pressure overload and repeated pregnancy, the cKO mice with complex I deficiency developed heart failure and high cell death, and could not be explained by oxidative stress mechanism. Complex I deficiency significantly decreased NAD+/NADH ratio, which inhibited sirt3 activity, increased mitochondrial protein acetylation, and sensitized mPTP. Additionally, it has been shown that NMN treatment rescues NAD+/NADH ratio and mitochondria protein acetylation in cKO hearts, and normalizes the sensitivity of the mPTP (Karamanlidis et al., 2013).
Lee et al. reported that mitochondrial protein hyperacetylation, which is caused by elevated NADH/NAD+, increases the risk of development of heart failure by two distinct mechanisms (Lee et al., 2016). Malate aspartate shuttle (MAS) modulates communication between cytosolic and mitochondrial NAD+ redox states, transferring electrons from cytosolic NADH generated from glycolysis into mitochondria for oxidative phosphorylation (LaNoue and Williamson, 1971; Lee et al., 2016). During mitochondrial dysfunction, hyperacetylation of MAS decreases cytosolic NAD+/NADH ratio, thereby inducing mPTP-related cell death and the development of heart failure (Elrod et al., 2010). Lee et al. (2016) also identified that the acetylation of lysine-70 on oligomycin-sensitive conferring protein (OSCP) sensitized mPTP opening by promoting its interaction with cyclophilin D (CypD), a regulator of mPTP. Thus, administration of NMN reverses hyperacetylation of these proteins by normalizing the NAD+ redox balance, thereby protecting mice from heart failure.
Kruppel-like factor 4 (KLF4) is critical for cardiac mitochondrial homeostasis. Accordingly, mice with cardiac-specific deficiency of KLF4 (CM-K4KO) are more sensitive to pressure overload-induced heart failure (Liao et al., 2015). Zhang et al. found that cardiac KLF4 deficiency led to hyperacetylation of mitochondrial proteins, including SOD2, CypD, and long-chain Acyl-CoA dehydrogenase (LCAD), which impaired mitochondrial metabolic function and predisposed the CM-K4KO hearts to stress-induced dysfunction (Zhang et al., 2017). Meanwhile, the expression of Sirt3, NAD+, and NAMPT were reduced in the KLF4-deficient heart, all of which decreased deacetylase activity in the mitochondria. Moreover, NMN increased NAD+ levels and normalized the mitochondrial protein acetylation levels in cardiac tissue. Remarkably, NMN preserved the cardiac contractile function and protected CM-K4KO mice from heart failure during pressure overload. LCAD is a fatty acid oxidation (FAO) enzyme, which oxidizes long-chain fatty acid, the main fuel of heart. The activity of this enzyme is dependent on Sirt3-NAD+ deacetylation (Hirschey et al., 2010), and acute NMN treatment increased mitochondrial FAO, indicating that NMN improves cardiac energetics and heart function. CM-K4KO hearts showed high rates of cell death in myocardium when subjected to stress, and NMN administration prevented cell death in pressure-overloaded hearts. Finally, NMN was found to preserve the mitochondrial ultrastructure and reduce ROS and inflammation in CM-K4KO myocardium partially through Sirt3-dependent deacetylation and activation of SOD2 in response to oxidative stress (Zhang et al., 2017). In summary, short-term administration of NMN may confer protection against cardiac mitochondrial homeostasis and prevent heart failure.
Martin et al. (2017) reported that NMN improves cardiac function and bioenergetics in a SIRT3-dependent manner in a Friedreich’s ataxia (FRDA) cardiomyopathy model, a mouse genetic cardiomyopathy model with frataxin-KO (Fxn–/–, FXN-KO). NMN (500 mg/kg) administered two times per week for 4–5 weeks improves diastolic function and normalizes the defective cardiac contractility in FXN-KO. A shortened ejection time (ET) represents impaired contractility or severe LV dysfunction in patients with heart failure or idiopathic-dilated cardiomyopathy (Dujardin et al., 1998). FXN-KO mice show a shortened ET, whereas NMN treatment significantly increases ET (Martin et al., 2017).
NMN supplementation also improves cardiac energy utilization and decreases energy wastage in the FXN-KO heart. Cardiac efficiency (CE), a ratio of energy used for mechanical work to overall energy available, is decreased in FXN-KO. Also, FXN-KO mice also shows an increased ventriculoarterial coupling (VC) ratio, another parameter representing the efficiency of converting ventricular mechanical energy (ME) into hydraulic energy in the aorta during systole (Suga, 1990; Walley, 2016). NMN treatment normalizes both CE and VC in the FXN-KO heart failure model. Pressure–volume area (PVA) is the sum of ME and potential energy (PE) (Suga, 1990). In FXN-KO hearts, a large proportion of PVA is consumed as PE, rather than ME, which indicates greater energy wastage in heart failure. However, NMN treatment improves myocardial energy utilization by normalizing the proportion of PE in PVA.
Additionally, the study reported that NMN-induced reduction in energy wastage paralleled the remarkable decrease in whole-body energy expenditure (EE) due to decreased FA metabolism and a SIRT3-dependent carbohydrate metabolism (Martin et al., 2017). FXN-KO mouse show elevated whole-body daily FA oxidation and serum triacylglycerides, while NMN supplementation blocks this elevation. Moreover, NMN reduces tissue lactate production and inhibits glycolysis by decreasing systemic and cardiac glucose utilization in the FXN-KO heart, thereby improving CE and function (Martin et al., 2017). Importantly, the therapeutic effects of NMN are mediated by deacetylation of SIRT3 in FXN-KO hearts (Martin et al., 2017). In summary, these findings provide preclinical evidence that NMN could be a promising drug for heart failure and cardiomyopathies.
Vascular Dysfunction
Cardiovascular disease (CVD) is the leading cause of mortality worldwide. Aging is a developmental risk factor for the disease. Vascular endothelial dysfunction and large elastic artery stiffness are two antecedents and predictors of clinical CVD (Mitchell, 2014). Vascular oxidative stress contributes to vascular endothelial dysfunction and large elastic artery stiffness (Bachschmid et al., 2013). Excessive superoxide reduces the bioavailability of nitric oxide (NO), a vasoprotective, and vasodilatory molecule, and thus causes alterations of structural proteins such as collagen and elastin, in large elastic arteries (Seals et al., 2014). Vascular endothelial dysfunction and large elastic artery stiffness are assessed by endothelium-dependent dilation (EDD) and aortic pulse wave velocity (aPWV), respectively (de Picciotto et al., 2016). Old mice showed impaired carotid artery EDD, and oral supplement of NMN (300 mg/kg) for 8 weeks restored EDD in the old mice by assisting in regaining NO-mediated dilation and reducing arterial oxidative stress (de Picciotto et al., 2016). Also, NMN restored the expression and activity of the SIRT1 in the arteries of old mice in accordance with previous studies, which revealed that the reduced expression and activity of SIRT1 contributes to impaired EDD in aging arteries (Donato et al., 2011; Gano et al., 2014) and by applying SIRT1 activator, the EDD was improved by partly reducing the oxidative stress (Gano et al., 2014).
An increase in stiffness of large elastic artery with age reduces the artery’s ability to buffer increase in pressure churned out by systolic ejection (de Picciotto et al., 2016). The type I collagen, a load-bearing protein in the arterial wall, is augmented during aging, while the main structural protein, elastin, diminishes in old arteries (Diez, 2007). Treatment using NMN also reduced the large elastic stiffness in aged mice by reversing the accumulation of whole-vessel type I collagen and enhancing arterial elastin (de Picciotto et al., 2016). A recent study showed that the protective effects of NMN supplement on vascular function are associated with increased anti-aging miRNA expression profile in the aorta of aged mice (Kiss et al., 2019b).
The brain, which is, a metabolically active organ, relies on the blood circulation to deliver nutrients and eliminate metabolic waste products (Tarantini et al., 2017). Cerebromicrovascular health is critical for brain perfusion, which helps in the maintenance of healthy cerebral function. Adjustment in cerebral blood flow maintains cellular homeostasis and function through neurovascular coupling (NVC), in response to the increased neuronal activity (Tarantini et al., 2017). Microvascular endothelium releases the vasodilator (NO) in response to increased neuronal and astrocytic activation (Toth et al., 2014). Several lines of evidence suggest that NVC responses are impaired in the course of aging, hence contributing to age-related cognitive impairment (Toth et al., 2014; Balbi et al., 2015). Emerging studies have revealed that an increase in mitochondrial oxidative stress and mitochondrial dysfunction results in impairment of neurovascular during the aging process (Springo et al., 2015).
A report by a recent study on aged mice showed that NMN could restore the function of cerebromicrovascular endothelium and NVC responses (Tarantini et al., 2019). NMN (i.p. 500 mg/kg) was injected in male aged mice (24 months old) for 14 consecutive days. The treatment restored the NO mediation of NVC in aged mice by reestablishing NO release, which resulted in endothelial NO-mediated vasodilation in the aortas of aged mice. The NO mediation of NVC responses is weakened by age-related mitochondrial oxidative stress. NMN increased the activation of SIRT1, which reversed mtDNA-encoded subunits, attenuated mtROS production, and improved mitochondrial bioenergetics. Thus, NMN can restore the protective effects of cerebromicrovascular by improving endothelial function, attenuating endothelial oxidative stress, and improving NVC responses in aged mice (Tarantini et al., 2019).
Age-related NAD+ depletion and consequential SIRT1 dysregulation are associated with impaired angiogenic processes in cerebromicrovascular. However, a study indicated that NMN could rescue angiogenic capacity in aged cerebromicrovascular endothelial cells (ECs) (Kiss et al., 2019a). Recently, the same team identified 590 genes differentially expressed in the aged neurovascular unit, 204 of which were shifted back toward youthful expression levels by NMN (Kiss et al., 2020). NMN supplementation reverses age-related changes in neurovascular gene expression, including SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic (Kiss et al., 2020).
A steady decline in tissue perfusion has been observed in humans from the age of 40, which often results in organ dysfunction and general body weakness in the last decades of life (Le Couteur and Lakatta, 2010). The number and function of ECs decline with age, resulting in increased apoptosis of ECs in the muscle, diminished neovascularization, and loss of blood vessels (Wang et al., 2014). These changes reduce muscle mass and endurance during aging (Prior et al., 2016). Abhirup et al. reported that SIRT1 in ECs is necessary for the response to pro-angiogenic signals secreted from myocytes and treatment with NMN improved blood flow and endurance in elderly mice by increasing capillary density (Das et al., 2018). In the same study, the number and density of ECs and capillaries in skeletal muscle for endurance were significantly decreased in the old mice, which could have been associated with impaired angiogenesis. Endothelial-specific SIRT1 knockout mice (ESKO) exhibited the same phenotype of aging, including reduced density of capillaries and decreased exercise endurance. Overexpression of endothelial SIRT1 sensitizes ECs to vascular endothelial growth factor, which improves the muscle neovascularization and hence increases capillary density and endurance. Moreover, NAD+/SIRT1 negatively regulates Notch signaling (Guarani et al., 2011), an indispensable signaling pathway for blood vessel formation (Blanco and Gerhardt, 2013).
Thus, NMN could restore angiogenesis in old mice through SIRT1-mediated inhibition of Notch signaling. NMN (400 mg/kg/day) was administered to old mice through drinking water for 2 months. The drug restored the number and density of capillaries, increased soluble oxygen (sO2) levels, and improved endurance in the old mice. However, these effects were abolished in SIRT1 knockout mice. Also, exogenous hydrogen sulfide (H2S) augmented the effect of NMN.
All these studies suggest that NMN could be a favorable agent for therapy against various diseases related to a decrease in blood flow brought about by cardiovascular dysfunction.
Intracerebral Hemorrhage
Intracerebral hemorrhage (ICH) is a primary brain injury resulting from mechanical damage that causes a hematoma. It can sometimes progress to a secondary injury, which is mainly subsequent pathophysiologic changes, including blood–brain barrier (BBB) destruction, hemoglobin-induced iron overload, neural cell death, neuroinflammation, and oxidative stress (Zhou et al., 2014; Wei et al., 2017b). A study suggested that NMN could be a promising agent for the treatment of ICH (Wei et al., 2017b). A single dose of NMN (300 mg/kg) was injected into CD1 mice 30 min after treatment with collagen-induced intracerebral hemorrhage (cICH). The NMN treatment relieved the edema that had been induced by cICH and improved neurological function. Treatment using NMN reduced cell death and oxidative stress in hemorrhage area. Moreover, it suppressed the cICH-induced microglia activation and neutrophil infiltration and inhibited inflammatory-associated factors, including TNF-α and IL-6. The intercellular adhesion molecule-1 (ICAM-1) protein, an adhesion molecule, is essential for the process of neuroinflammation activation after ICH. The NMN treatment also inhibited an increase in ICAM-1 after cICH (Sozzani et al., 2015; Wei et al., 2017b).
The secondary stage of ICH mainly results from toxicity caused by hemin, which is a breakdown product of hemoglobin. It causes cell death and induces extensive local inflammation/oxidative stress. Hemin can induce HO-1, a ubiquitous enzyme, which oxidatively cleaves pro-oxidant heme to produce biliverdin and carbon monoxide. NMN also elevates the nuclear Nrf2 protein expression to upregulate HO-1 protein expression in brain tissues, thereby inhibiting neuroinflammation and oxidative stress and contributing to the neuroprotection in ICH (Mylroie et al., 2015; Wei et al., 2017b). Finally, a prolonged NMN treatment for 7 days significantly arrested body weight decline and neurological deficit caused by ICH.
Carrie et al. also reported that NMN supplement preserved mitochondrial function, mitigated inflammation, and increased survival rate in hemorrhagic shock (Sims et al., 2018). Lactic acidosis, which is a by-product of anaerobic metabolism, is used to estimate the success of resuscitation in injured patients (Broder and Weil, 1964). High lactate levels reflect tissue hypoperfusion, which is, in turn, related to the severity and survivability of the shock (Rixen and Siegel, 2005). Pretreatment (400 mg/kg/day for 5 days, oral) and resuscitation (400 mg/kg, intravenous) with NMN significantly reduced serum lactate levels during a fixed pressure hemorrhagic shock.
The NAD+ concentration in various tissue decreases rapidly with the severity of the injury during hemorrhagic shock (Wurth et al., 1973), which can result in dysfunction of vital organs. Under hypoxic conditions, the mitochondrial electron transport chain becomes inefficient in reoxidizing NADH to the NAD (Wheaton and Chandel, 2011). The NMN treatment can compensate for the decrease in NAD+ levels and preserved the NAD/NADH ratio in both kidneys and livers. Adenosine triphosphate reserves are also depleted following hemorrhagic shock and resuscitation, and NMN treatment also restores the reserves in the kidney, but, not the liver. This could be caused by elevated ATP consumption in the liver. Treatment with NMN preserves the impaired complex I–dependent (CI-dependent) respiration following hemorrhagic shock and resuscitation.
Hemorrhagic shock and resuscitation are usually accompanied by increased cytokine levels, oxidative stress, and insulin-resistant hyperglycemia, which indicates the existence of inflammation (Sims et al., 2018). Circulating levels of the cytokine IL-6 are as predictors of mortality in human patients (Stensballe et al., 2009). Treatment with NMN significantly reduces serum IL-6 cytokine levels and ameliorated shock-induced hyperglycemia. The most important is that NMN can enhance physiologic reserve and improve survival after hemorrhagic shock. Pretreatment with NMN increases the ability of animals to tolerate longer periods of hypoperfusion. Resuscitation with NMN significantly improves the survival.
Tissue plasminogen activator (tPA) is used for therapy in case of acute brain ischemia. The window of tPA therapy is within 0–4.5 h. A delayed tPA therapy dose not decrease infarction but worsens hemorrhagic transformation (Wei et al., 2017a; Zhang et al., 2009). Wei et al. (2017a) suggested that NMN could attenuate delayed tPA-induced hemorrhagic transformation after cerebral ischemia without changing tPA thrombolytic activity. In the study, delayed tPA therapy resulted in a high mortality rate in middle cerebral artery occlusion (MCAO) mice. However, a single dose of NMN (300 mg/kg) injection (i.p.) reduced the mortality rate. NMN also attenuated the delayed tPA-induced severe infarction volume and brain edema. The cerebral hemorrhage caused by delayed tPA was fatal, but NMN significantly decreased the cerebral hemorrhage and higher hemoglobin level in the ipsilateral hemisphere of delayed tPA mice. The administration of NMN also inhibited neural apoptosis and ameliorated microglial activation and reduced neuroinflammation in delayed tPA-treatment mice.
It has been reported that disruption of BBB could be a reason for tPA-induced hemorrhagic transformation (Kastrup et al., 2008). The tPA activates matrix metalloproteinases (MMPs), which destroys tight junction proteins (TJPs) (Rempe et al., 2016). Consistent with previous findings, the protein levels of TJPs (claudins, occludin, and zonular occludens-1) were downregulated, and the activities of MMP9 and MMP2 were enhanced in delayed tPA treatment, which implied that the BBB integrity had been compromised. The NMN treatment significantly reversed these changes and protected BBB integrity. Thus, NMN could be a potential agent for the therapy of tPA-induced hemorrhagic transformation.
It was also reported that NMN is able to protect brain in both the early and the chronic phase of cryoinjury (Zhang et al., 2015). These studies demonstrated that NMN could be a remedy for ICH of various causes.
Neuroprotective and Cognitive Function
Cognitive decline is one of the many symptoms of aging, and regulation of adult neurogenesis could be a therapeutic strategy in overcoming the condition. There are two distinct populations of neural stem cells (NSCs) in the brain, which are found in the subgranular zone (SGZ) and subventricular zone (SVZ). They can self-renew and differentiate into transient amplifying progenitors, that is, neural stem/progenitor cells (NSPCs). The NSPCs undergo finite, lineage-restricted cell divisions to differentiate into the major cell types of the brain, such as neurons, oligodendrocytes, and astrocytes (Artegiani and Calegari, 2012; Jadasz et al., 2012). Studies have revealed that aging is a negative regulator of NSPC proliferation, whereas NSPCs could be reactivated in the aged brain (Artegiani and Calegari, 2012). Thus, restoring the function of NSPCs could effectively prevent age-associated cognitive decline.
Liana and Shin-ichiro established that, in response to insult-induced demyelination, NAMPT-mediated NAD+ biosynthesis was critical for NSPC self-renewal, proliferation, and differentiation into oligodendrocytes (Stein and Imai, 2014). The study found that levels of NAD+ and expression of NAMPT in hippocampus were declining with age. However, the long-term NMN administration was able to maintain the NSPC pool. Moreover, they found that NMN administration reduced defects in oligodendrogenesis caused by decrease in NAD+ levels. Thus, NMN could be a promising agent for maintaining the NSPC pool and reactivating NSPCs, which can improve remyelination caused by aging and neurodegenerative diseases (Stein and Imai, 2014). Also, Zhao et al. (2015) reported that NMN was able to induce NSC proliferation (via SIRT1 and SIRT2) and promote NSC differentiation (via SIRT1, SIRT2, and SIRT6).
Mental disorders such as anxiety are prevalent among older people and account for 10–20% of the total mental diseases in the population, most of which is dementia or major depressive disorder (Regier et al., 1988). Late-life anxiety disorder results in substantial financial burden on individuals and society. Sean et al. discovered that old mice developed cognitive hypersensitivity in response to aversive stimulation during contextual fear condition tests, which was associated with the age-related changes in emotionality and sensory processing (Johnson et al., 2018). Moreover, specific knockdown of NAMPT in the hippocampal CA1 region recapitulated the same age-associated cognitive hypersensitivity, whereas dentate gyrus specific NAMPT knockdown mice did not show cognitive hypersensitivity.
Sean et al. found that calcium/calmodulin-dependent serine protein kinase (Cask), a crucial multidomain scaffold protein at the synapse and in cell junctions (Hsueh, 2006), is the downstream effector in response to the reduction in NAD+ and is also downregulated in the hippocampus during aging (Johnson et al., 2018). Therefore, Sean et al. proposed a model to demonstrate that the levels of NAMPT and NAD+ in hippocampal neurons, particularly in CA1 neurons, decline during aging, leading to a decrease in SIRT1 activity and subsequent downregulation of Cask expression in the aged hippocampus. Cask interacts with the GluN2B subunit of N-methyl-D-aspartate receptor (NMDAR) for the transport of GluN2B-containing vesicles (Jeyifous et al., 2009), and the reduction in Cask could cause dysfunction of GluN2B-containing NMDARs at the synapse, which could contribute to a wide range of cognitive and behavioral impairments during aging (Johnson et al., 2018). The NMN supplementation, even in a short time, could reduce cognitive hypersensitivity and improve the sensory processing aspect of some aversive stimuli and possibly other related behaviors (Johnson et al., 2018). Therefore, the NMN administration could prevent and treat such cognitive impairments and enhance the quality of life in old age.
NMN also prevented aging-induced cognitive impairment by improving cerebrovascular (Kiss et al., 2019a; Tarantini et al., 2019) and mitochondrial function, and reducing apoptosis in the prefrontal cortex and hippocampus of aged animals (Hosseini et al., 2019a). Lu et al. (2014) reported that NMN improved energy activity and survival rate in an in vitro model of Parkinson’s disease. Fang et al. (2016) also demonstrated that NR and NMN were able to normalize neuromuscular function and memory by regulation of mitophagy and enhancement of DNA repair in mice and worm models of ataxia-telangiectasia (AT), an autosomal recessive disease with progressive neurodegeneration.
A study indicated that NAMPT enzymatic activity enhancer P7C3 could have a neuroprotective effect in a mouse model of amyotrophic lateral sclerosis (ALS) (Tesla et al., 2012), and the levels of iNAMPT in ALS patients were lower than those of age-matched controls (Wang et al., 2017). Wang et al. (2017) silenced NAMPT in the projection neurons of adult mice and created Thy1-YFP-Nampt–/– cKO mice that notably exhibited general motor abnormalities, muscle atrophy, progressive motor function deficits, and a shorter lifespan, some of which are the key features of ALS. Loss of NAMPT in projection neurons led to mitochondrial metabolic dysfunction and destroyed the mitochondrial homeostasis. Wang et al. reported that NAMPT knockdown disrupts the balance between mitochondrial fission/fusion and causes more fragmentation, which could lead to subsequent neuronal degeneration. The thy1-YFP-Nampt–/– cKO mice exhibited neurodegeneration in the brain, especially in the motor cortex and increased acetylation proteins, which implied that NAD+-related perturbations induced a reduction in Sirt3 activity. Also, many lines of evidence have shown that increasing Sirt1 and Sirt3 deacetylation activities have a neuroprotective role in motor neuron degeneration diseases, such as ALS (Fu et al., 2012; Watanabe et al., 2014).
The thy1-YFP-Nampt–/– cKO mice also showed widespread abnormalities of the neuromuscular junctions (NMJs), which is known to disrupt synaptic connectivity and cause defective synaptic transmission. Remarkably, NMN (400 mg/kg) treatment lessened disease severity, restored motor function, and extended the life span of Thy1-YFP-Nampt–/– cKO mice. Recently, this same team reported that loss of NAMPT in projection neurons cause detrimental effects on the function and structure of NJM, including impaired synaptic vesicle cycling, morphological changes to the motor endplate, alterations of skeletal muscle contractile responses, and noticeable sarcomere misalignment (Lundt et al., 2020). These detrimental effects were reverse by administration of NMN (400 mg/kg/day) for 14 days. However, NMN treatment did not restore skeletal muscle mitochondria morphology change caused by deletion of Nampt (Lundt et al., 2020). Thus, NMN is a potential therapeutic drug for motor neuron (MN) degenerative diseases, including ALS. Increasing the NAD+ salvage pathway could reduce the symptoms of neurodegenerative diseases.
Alzheimer’s Disease
AD is progressive dementia characterized by memory loss at an early stage of the disease progression. Currently, there is no effective therapy for AD treatment. Also, its molecular basis has not been elucidated (Gong et al., 2003). Thus, there is a need to focus on finding effective therapeutic drugs for the disease.
Mitochondrial dysfunction is a feature of many neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) (Hroudova et al., 2014). Morphological and functional abnormalities of mitochondria may lead defects in the electron transport chain and ATP production, which are associated with AD (Hroudova et al., 2014; Long et al., 2015). The NAD+ as a cofactor is crucial for the tricarboxylic acid cycle, mitochondrial oxidative phosphorylation, and glycolysis-related enzymatic reactions, and the levels of NAD+ in the cell are critical for neuronal survival (Liu et al., 2008). Thus, preventing NAD+ depletion and boosting cellular energy could be a therapy for neurodegenerative disease (Long et al., 2015).
A study reported that NMN improved NAD+ catabolism and changed mitochondrial morphological dynamics in mice with AD (Long et al., 2015). In the study, AD chimeric amyloid precursor protein APP(swe)/PS1(ΔE9) double transgenic (AD-Tg) mice showed deficits in mitochondrial oxygen consumption rates (OCRs) in the brain and muscle at 3 months of age. The NMN (100 mg/kg) was subcutaneously injected to AD-Tg female and male mice consecutively for 28 days, which reversed mitochondrial OCR deficiencies in the AD model. Still, in the same study, full-length mutant human APP levels were significantly increased in the brain of AD-Tg mice compared to non-transgenic (NTG) mice. The administration of NMN decreased full-length mutant APP expression in AD-Tg mice. Immunoreactivity of SIRT1 and CD38 was also significantly increased in AD-Tg compared to NTG mice, which could be attributed to NAD+ catabolism. The AD mice pre-treated with NMN showed a lower SIRT1 immunoreactivity compared to Ag-TD mice, but still had a higher SIRT1 compared to NTG mice.
The mitochondrial morphology is vital for its functions, such as mitochondrial respiration and calcium homeostasis, among others. Fusion and fission are two fundamental processes in mitochondria, which are essential for cellular survival and disease vulnerability. The protection of the nervous system is associated with the fusion, and fission processes, both of which contribute to the damaged organelles’ elimination (Escobar-Henriques and Anton, 2013). NMN administration increased the length of mitochondria and decreased fragmentation in the hippocampal sub-region (Long et al., 2015). A study reported that NMN alters mitochondrial dynamics by SIRT3-dependent mechanism (Klimova et al., 2019). Nina et al. also showed that the levels of the active form of fission protein, phosphorylated Drp1 (S616) (pDrp1), were significantly decreased in hippocampal mitochondria after a single-dose NMN treatment, and the same result was observed in AD-Tg mice (Long et al., 2015). NMN also decreased the acetylation of mitochondrial proteins, such as mitochondrial SOD2, which is an important antioxidant enzyme and one of the targets of SIRT3. The SOD2 catalyzes superoxide into hydrogen peroxide and oxygen, which is then converted to water by glutathione peroxidase (Chen et al., 2011). Studies have shown that mitochondrial morphologies are associated with the generation of ROS, and the accumulation of ROS could induce more mitochondrial fragmentation (Willems et al., 2015). Thus, NMN treatment reduced hippocampal ROS through SIRT3-dependent deacetylation of SOD2, which resulted in lowering mitochondrial fragmentation via pDrp1 (S616).
The Aβ oligomers have been widely reported to be responsible for AD pathology. They usually accumulate in AD frontal cortex levels of up to 70-fold compared to normal brains (Gong et al., 2003). Wistar rats with an intracerebroventricular infusion of Aβ1–42 oligomer were used as AD mouse models. Intraperitoneal NMN (500 mg/kg) treatment improved their cognitive and memory functions, which might have been as a result of NMN reducing oxidative stress actions (Wang et al., 2016). Long-term potentiation (LPT), a mechanism for memory and learning functions, is significantly inhibited by Aβ oligomers, but NMN treatment was able to prevent this inhibition in organotypic hippocampal slice cultures (OHCs). Oxidative stress and a high concentration of ROS have toxic effects on synaptic transmission, which is essential in neurodegeneration (Guidi et al., 2006). Administration of NMN prevented the accumulation of ROS induced by Aβ1–42 oligomer in OHCs, which could have been the reason for memory and learning improvement. The NMN treatment also attenuated the neuronal cell death and reduction of NAD+ and ATP in Aβ1–42 oligomer-treated hippocampal slices (Wang et al., 2016).
The c-Jun N-terminal kinases (JNKs) are a family of multifunction-signaling protein kinases that respond to various cellular stresses and inflammatory mediators (Mehan et al., 2011). However, aberrant activation of JNK is associated with pathogenesis of Alzheimer’s disease. A study demonstrated that NMN could reverse AD by inhibiting JNK activation in the hippocampus and cerebral cortex of AD-Tg mice (Yao et al., 2017). Administration of NMN ameliorated amyloid-induced synaptic loss and dysfunction, and reversed cognitive impairments in AD-Tg mice, including severe impairment of spatial learning, spatial memory and contextual memory.
Also, NMN decreased β-amyloid production and amyloid plaque burden (Yao et al., 2017). The NMN regulated the expression of APP cleavage secretase in AD-Tg mice, including elevation of sAPPα and reduction of sAPPβ. A previous study indicated that phosphorylation of APP on threonine 668 could promote β-secretase cleavage of APP, resulting in more Aβ generation (Colombo et al., 2009). SIRT1 activation promoted non-amyloidogenic α-secretase processing of the amyloid precursor protein by inhibiting rho-associated kinase (ROCK1) expression (Qin et al., 2006). These results showed that NMN might enhance non-amyloidogenic APP processing and hence decrease Aβ pathology in AD-Tg mice. Neuroinflammatory, a crucial response for the development of AD (Morales et al., 2014), was shown to be inhibited by NMN treatment in AD-Tg mice (Yao et al., 2017), including decrease in IL-6, IL-1β, and TNFα. These proinflammatory cytokines stimulate β-secretase and γ-secretase to generate more Aβ in the brain through a JNK-dependent MAPK pathway (Liao et al., 2004). In conclusion, NMN improved cognitive abilities and decreased amyloid plaque, loss of synapse, β-secretase, and neuroinflammation, at least partially by inhibition of JNK activation.
These studies demonstrate that NMN could be a viable intervention for managing AD.
Retinal Degeneration and Corneal Injury
The photoreceptor is crucial for light transduction, which is necessary for vison, and there are two classes of photoreceptors: rods and cones. Rod and cone photoreceptors mediate dim and precise central vision in ambient light, respectively (Lin et al., 2016). Death of photoreceptors results in vision loss, and this occurs in many diseases, including age-related macular degeneration (AMD), retinitis pigmentosa (RP), and Leber congenital amaurosis (LCA) (Wright et al., 2010). It has been reported that mutations in NMNAT1 cause LCA, as a result of reduced NAD+ biosynthesis and impaired protein folding (Koenekoop et al., 2012). Thus, NAD+ biosynthesis has a role in photoreceptor function and survival.
Knockout mice lacking Nampt in rod photoreceptors (Nampt–rod/–rod) and cone photoreceptors (Nampt–cone/–cone) showed a decreased retinal NAD+ level and a degenerative phenotype, including vascular attenuation, optic nerve atrophy, outer nuclear layer thickness reduction, and retinal function impairment (Lin et al., 2016). Retinal NAD+ deficiency was observed in multiple mouse models with retinal dysfunction, including light-induced degeneration, streptozotocin (STZ)-induced diabetic retinopathy, and aging-associated retinal dysfunction. NAD+ deficiency in photoreceptors causes significant glycolytic and mitochondrial dysfunction under basal conditions and impairs the normal response to moderate metabolic stresses, which results in photoreceptor cell death and retinal degeneration (Lin et al., 2016). Besides, photoreceptors are susceptible to defects in energy homeostasis because of their limited mitochondrial reserve (Kooragayala et al., 2015). Lin et al. (2016) demonstrated that SIRT3 and SIRT5 are both critical for photoreceptor survival and retinal homeostasis. It is through SIRT3 that NAD+ deficiency causes aberrant hyperacetylation of mitochondrial proteins, hence contributing to mitochondrial dysfunction.
Nampt is critical for energy metabolism in retinal cells and its deficiency results in impairment of retinal homeostasis. Downstream production of Nampt is a potential approach to relieve the impairment. Intraperitoneal injections of NMN (150 mg/kg) for 4 weeks improved scotopic and photopic retinal function and reduced photoreceptor death in Nampt–rod/–rod and Nampt–cone/–cone mice. Also, intraperitoneal injections of NMN (300 mg/kg) for 10 days were able to protect the retina from light-induced injury (Lin et al., 2016).
Mills et al. reported that long-term NMN administration ameliorated age-associated pathological changes in the eyes (Mills et al., 2016). The mutation in rd8 of C57BL/6N mice induced accumulation of subretinal microglia and macrophages with age, which was consistent with an increase in light-colored spots in the fundus of the eyes (Aredo et al., 2015; Mills et al., 2016). The C57BL/6N mice at 17 months of age showed several light-colored spots in the fundus. However, the spots were significantly reduced in the fundus of the aged mice, which were on a long-term NMN supplementation. The long-term supplementation prevented rod cell dysfunction and improved scotopic b and photopic b waves in aged C57BL/6N, which suggested that the function of Muller/bipolar cell and cone cell was enhanced by NMN supplementation (Mills et al., 2016). The functioning of the lacrimal gland in humans and rodents gradually decreases with age (Zoukhri, 2006). Notably, long-term NMN administration significantly increased tear production in aged mice (Mills et al., 2016).
The cornea is one of the most densely innervated tissues in the human body and corneal innervations play a critical role in the regulation of epithelial homeostasis (Bonini et al., 2003). Neurotrophic keratopathy, a degenerative corneal disease with an impairment of trigeminal nerve, shows corneal epithelial defects, ulcer, and even perforation (Bonini et al., 2003). Li et al. demonstrated that corneal denervation impaired the epithelial NAD+ levels by reducing the expression of NAMPT. This process led to deactivation of SIRT1, pAKR, and pCREB, and caused the apoptosis of corneal epithelial cells (Li et al., 2019). NMN treatment significantly reduced the wound area and slowed down the corneal nerve fiber degeneration in the denervated mice. The supplement of NMN restored the activation levels of SIRT1, AKT, and CREB, and reversed the cell apoptosis and epithelial defects (Li et al., 2019). A recent study also reported that subconjunctival injection of NMN or other NAD+ precursors effectively prevented ultraviolet B light (UVB)-induced tissue damage and EC apoptosis in the mouse cornea through reactivating AKT signaling (Zhao et al., 2020). These findings demonstrate that NMN could be a therapeutic agent for treating diverse diseases that are associated with blindness.
Acute Kidney Injury (AKI)
Approximately 1.33 million people worldwide suffer from AKI every year, resulting in a substantial economic and social burden on patients and society. The condition is associated with a high mortality rate exceeding 50% of those affected and the development of chronic kidney disease and other types of organ dysfunctions (Lewington et al., 2013). Aging is an independent risk factor for AKI (Kane-Gill et al., 2015), and various age-related factors contribute to the increased susceptibility to AKI (Fan et al., 2013), including diabetes, hypertension, vascular diseases, and some iatrogenic factors (Kane-Gill et al., 2015).
Studies have revealed that SIRT1 and SIRT3 are critical in protecting the kidney from injury (Fan et al., 2013; Morigi et al., 2015). Guan et al. reported that the administration of NMN could prevent age-associated susceptibility to AKI by restoring renal SIRT1 activity (Guan et al., 2017). The study found that kidneys of aged mice were more susceptible to cisplatin-induced AKI, and the NAD+ levels and SIRT1 expression were low in aged kidney. NAMPT and NMNAT were significantly lower in the kidney cortex of the old mice compared with those of the young mice. Four days of NMN treatment restored NAD+ levels in the kidneys and protected them from age-associated AKI. However, this protective effect of NMN was inhibited by SIRT1 deficiency, indicating that the renal protective effect of NMN was dependent on SIRT1. NMN treatment also protects the kidney from ischemia–reperfusion injury. Mice with NMN administration showed less ischemia–reperfusion injury and better kidney function compared with mice treated with PBS, which includes decreased blood urea nitrogen (BUN) and lower serum creatinine levels, and ameliorated tubular damage (Guan et al., 2017). Moreover, it has been shown that NMN could alleviate diabetic nephropathy nephritic fibrosis by inhibiting endogenous Nampt (Chen et al., 2017).
In conclusion, the findings of these studies show that NMN could be a potential therapeutic agent for AKI because of its ability to restore NAD+ and SIRT levels in the kidney.
Alcoholic Liver Disease
As the most common chronic liver disease, alcoholic liver disease (ALD) is caused by chronic alcohol consumption and can develop from alcoholic fatty liver (AFL) to alcoholic steatohepatitis (ASH) (Seitz et al., 2018). Ethanol-induced NAD+ depletion is involved in the development of ethanol-induced steatosis, oxidative stress, steatohepatitis, and insulin resistance (Luo et al., 2017). However, NMN treatment maintained the NAD+ levels and restored the alterations of TCA cycle metabolites that is induced by ethanol (Assiri et al., 2019). NMN also successfully prevented an ethanol-induced increase in plasma levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), two damage biomarkers of liver. RNA-seq analysis revealed that ethanol changed the expression of 1778 genes, 25% of which were altered by NMN treatment (Assiri et al., 2019). The mitogen-activated protein kinases (MAPK) pathway was one of the signaling pathways that were significantly affected by NMN. Previous studies have demonstrated that activating transcription factor 3 (Atf3) is associated with NAD+/NADH ratios and can be induced by ethanol (Mohammadnia et al., 2015). Elevated Atf3 also correlated with increased ALT and AST (Allen-Jennings et al., 2002). Moreover, Atf3 overexpression has been found in patients with alcoholic steatohepatitis (Mohammadnia et al., 2015). Interestingly, NMN normalized extracellular signal-regulated kinase1/2 (Erk1/2) signaling and reduced the expression of Atf3 (Assiri et al., 2019).
Other Diseases and Improvement of Physical Function
Associated with the loss of oocyte quality, reproductive aging in female mammals is an irreversible process that accompanies decreased levels of NAD+ (Bertoldo et al., 2020). However, administration of NMN can hopefully restore oocyte quality and fertility in aged mice and reverse the adverse effect of maternal age on developing embryo, suggesting that NMN can rescue female reproductive function in mammals (Bertoldo et al., 2020). Multigenerational obesity-induced perturbations in oocyte-secreted factor signal can also be normalized by NMN supplementation (Bertoldo et al., 2018). However, NMN does not protect the ovarian reserve from radiotherapy and chemotherapy, such as γ-irradiation or cyclophosphamide (Stringer et al., 2019).
Depression is a major mental health problem and has especially large effects on individual health and social burden. Previous studies have suggested that mitochondrial dysfunction and decreased ATP production contribute to depression (Allen et al., 2018). Recently, Xie et al. reported that in a corticosterone (CORT)-induced depressed mouse model, NMN could alleviate depression-like behaviors by improving mitochondrial energy metabolism via enhancing the activity of SIRT3 (Xie et al., 2020). Transcriptome and metabolome analysis demonstrated that NMN inhibited CORT-induced lipid synthesis, stimulated β-oxidation and glycolysis, and improved the TCA cycle to enhance ATP production in mitochondria.
Besides, NMN has also been reported to enhance skeletal muscle mitochondrial oxidative metabolism in aged mice (Gomes et al., 2013), improve hepatic mitochondrial function in circadian mutant mice (Peek et al., 2013), reduce DNA damage, and protect against irradiation-induced alterations in white blood cell counts, lymphocytes, and hemoglobin (Li et al., 2017). Recently, a study reported that NMN treatment improved mesenchymal stromal cells self-renewal with promoted osteogenesis and reduced adipogenesis via the SIRT1 pathway in aged and irradiated adult mice (Song et al., 2019). Moreover, NMN supplement can alleviate aluminum-induced bone injuries via suppression of the thioredoxin-interacting protein (TXNIP)–NLRP3 inflammasome pathway (Liang et al., 2019). These findings further demonstrate that NMN can be used to treat a variety of diseases involving NAD+ decline (Figure 4 and Table 1).
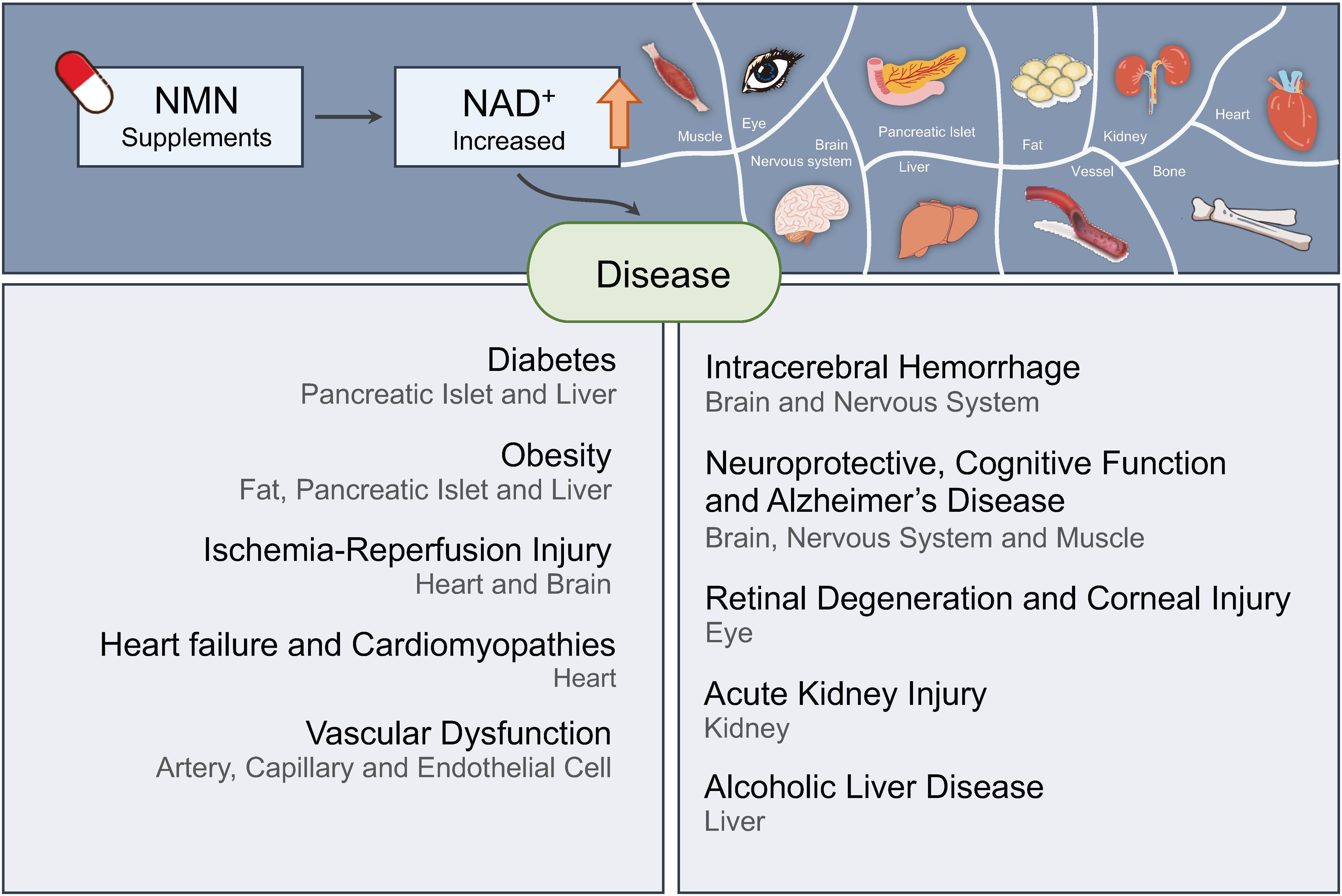
Figure 4. Nicotinamide mononucleotide ameliorates various diseases by increasing NAD+ levels in human. NMN is a promising molecule for therapy of diverse diseases, including diabetes, obesity, ischemia–reperfusion injury, heart failure, Alzheimer’s disease, retinal degeneration, acute kidney injury, and so on.
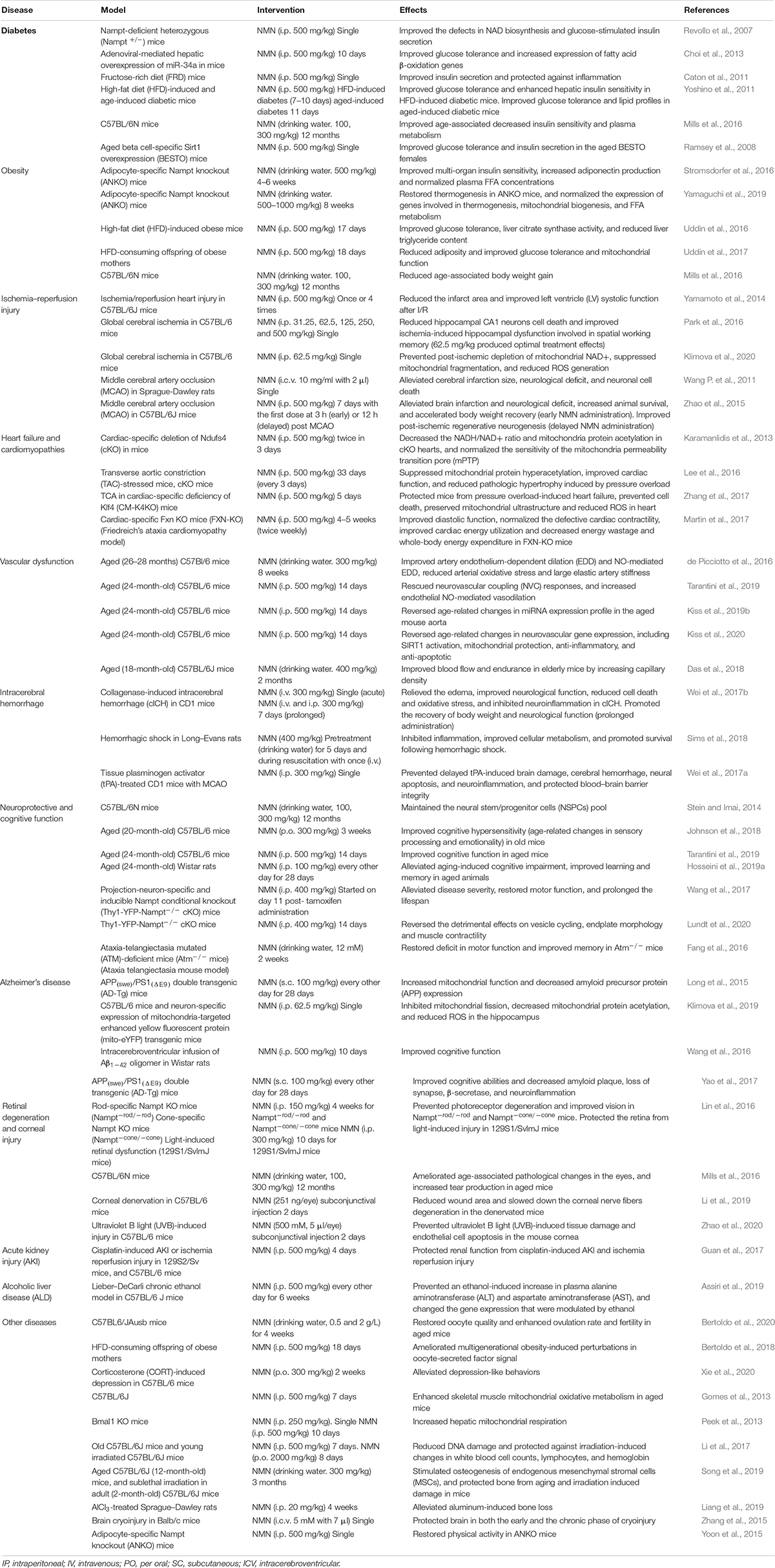
Table 1. Therapeutic effects of NMN administration in vivo.
Possible Detrimental Effects of NMN Supplementary
Although the benefits of NMN are obvious, some studies suggest that people need to use it with caution. Keeping NMN at a low level was beneficial to the axon survival (Di Stefano et al., 2017). NMN accumulation could promote Wallerian or Wallerian-like degeneration, which was involved with the physical neurite injury. Inhibiting NAMPT or maintaining NMNAT activity could preserve the structure and function of the transected axons and the distal axon stump by preventing the increase of NMN. It is worth noting that since a long-term NAD+ depletion would become harmful for protection of axons, a time window of inhibiting the NMN accumulation was shown after the acute injury (Di Stefano et al., 2015). To decrease the level of NMN and maintain the production of NAD+, nicotinic acid riboside (NAR) was introduced in treating chemotherapy-induced axon degeneration by generating NAD+ through an alternative pathway that bypassed NMN formation (Liu et al., 2018).
It was reported that a lower dosage of NMN could be more effective and safer, for example, the brain may be more sensitive to the concentration of NMN (Park et al., 2016). A lower-dose administration of NMN resulted in improved female infertility with enhanced oocyte quality. Overdose of NMN may have some adverse effects on other aspects of fertility (Bertoldo et al., 2020). NMN administered by intraperitoneal injection increased oxidative stress of sperm and reduced sperm quality in male offspring of obese mothers with HFD, whereas oral administration of NMN did not have these effects (Youngson et al., 2019). These data indicated that the effects of NMN were complex and the therapeutic dose or mode of administration needs more investigation in the future.
Carefully, NAD turnover in tumor cells is higher than nontumor cells. The expression levels of NAMPT were found to be elevated and have a positive relationship with the stage of tumor progression in many types of tumor, indicating that downregulation of NAMPT/NAD+ may become a strategy for anti-cancer therapy (Wang B. et al., 2011; Gujar et al., 2016). It is noteworthy that SIRT1, as downstream targets, has both tumor-suppressor and oncogenic roles under different circumstances (Chalkiadaki and Guarente, 2015). Thus, efficiency and long-term safety of NMN should be precisely assessed in further preclinical and clinical studies.
Human Clinical Study
Given that NMN has shown high efficacy and benefits in various mouse models of human disease, several clinical trials of NMN have been conducted to investigate its clinical applicability (Table 2). This has led to some capsule formulations of NMN being approved and put on the market as health supplements (Poddar et al., 2019).
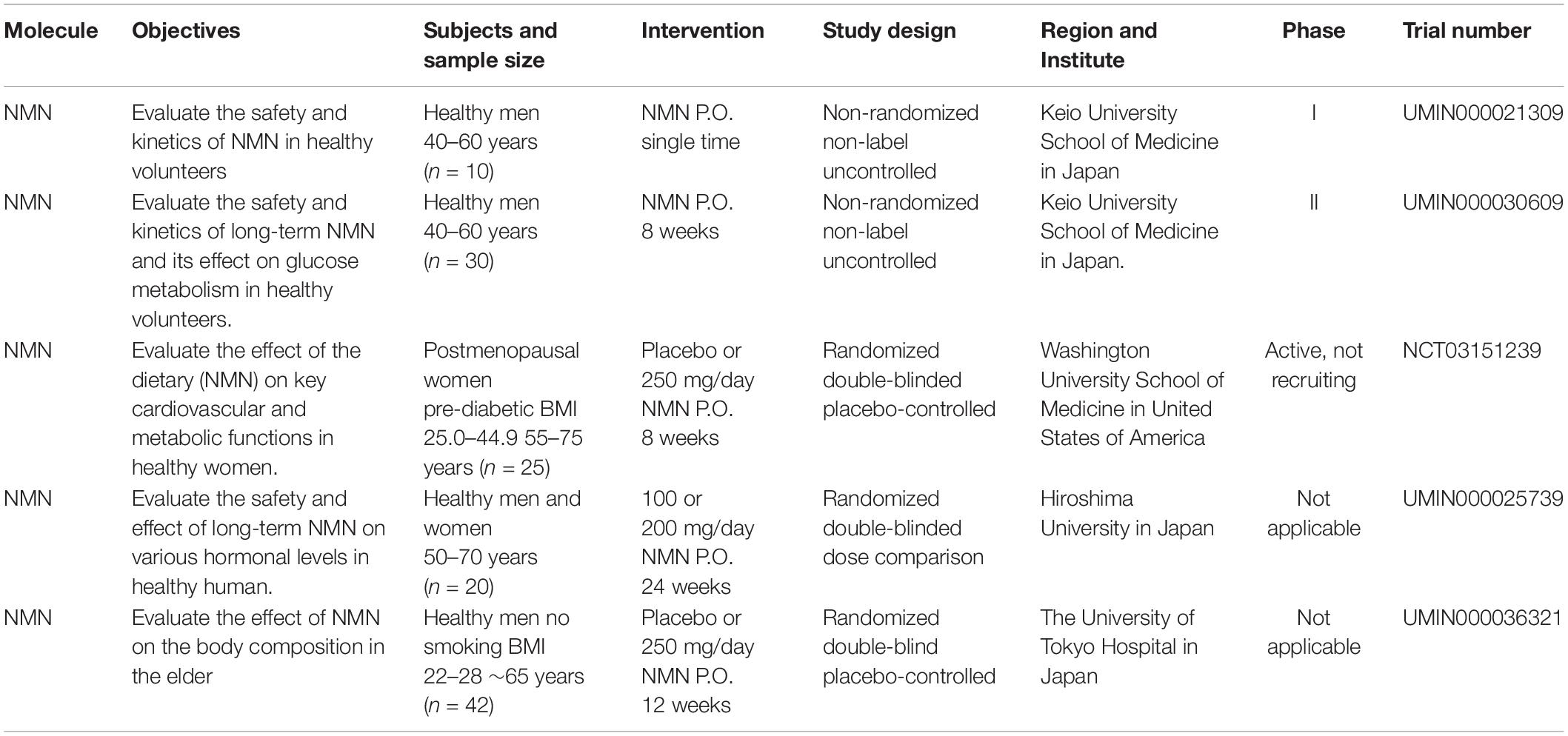
Table 2. Human clinical trials of NMN.
The first phase I human clinical study (UMIN000021309) for NMN has been initiated by an international collaborative team between Keio University School of Medicine in Tokyo and Washington University School of Medicine in St. Louis. The aim of this study is to examine the safety and bioavailability of NMN in human bodies. This research is led by Hiroshi Itoh, a professor of Endocrinology, Metabolism and Nephrology, Tokyo and Shin-ichiro Imai, a professor of Developmental Biology, at Washington University (Tsubota, 2016). Recently, they reported that a single oral administration of NMN up to 500 mg was safe and effectively metabolized in healthy subjects without causing severe adverse events. The major final metabolites of NMN were significantly increased in a dose-dependent manner by NMN administration. However, the team failed to detect NMN in plasma samples in this study. In a further study, they will detect the levels of NMN in plasma and NAD+ in peripheral blood mononuclear cells (Irie et al., 2019).
Itoh is also conducting a phase II study (UMIN000030609) to assess the safety of long-term NMN in healthy subjects, the kinetics of NMN and metabolites of NAM, and the effect of daily administration of NMN on glucose metabolism. Other clinical trials of NMN are ongoing at Washington University to examine the effect of NMN on insulin sensitivity, endothelial function, blood lipids, body fat and liver fat, and fat tissue and muscle tissue markers of cardiovascular and metabolic health. Additionally, a study (UMIN000025739) has been initiated at Hiroshima University, Institute of Biomedical and Health Sciences to evaluate the effect of long-term oral administration of NMN on various hormones in healthy volunteers. Recently, a new clinical study (UMIN000036321) was initiated at the University of Tokyo Hospital to evaluate the effect of NMN oral administration on the body composition in elderly persons.
In summary, despite the tremendous research efforts aimed at exploiting the therapeutic potential of NMN to treat metabolic and aging-related diseases, the clinical and toxicological evidence to support its utility is currently insufficient (Poddar et al., 2019). Thus, further research is needed to increase the prospects of developing drugs based on NMN.
Perspective
Among various NAD+ precursors, NMN and NR seem to increase NAD levels more effectively than NAM in rodents (Rajman et al., 2018). Since NAM acts as a feedback inhibitor to suppress sirtuins and PARPs, the benefits of increase in NAD+ levels may be compromised (Bitterman et al., 2002). Additionally, because of the shorter resident time, high-dose administration, and some side effects of NAM, it was not the preferred choice compared with NMN and NR (Kawamura et al., 2016).
It is difficult to compare NMN with NR, both of which are subjected to first-pass metabolism and the quick conversion to other NAD+ intermediates before uptake in vivo (Frederick et al., 2016; Ratajczak et al., 2016). NR, instead of NMN, is unstable and quickly converted into NAM in murine plasma (Ratajczak et al., 2016). Some differences regarding the pharmacological effects of NMN and NR should be noted. As mentioned above, NMN improves cardiac function in a SIRT3-dependent manner in an FRDA cardiomyopathy model (Martin et al., 2017), whereas NR does not (Stram et al., 2017). Exploring the pharmacokinetics of NMN and NR in vivo could help determine the optimal concentration of them in different regions and facilitate understanding the mechanisms of their pharmacological actions. Head-to-head study should be performed to compare NMN and NR in the future.
Besides making a choice between NMN and NR, more precise data about the dosage and more detailed mechanism are needed. Some researches attempted to utilize novel methods to achieve integral understanding (Assiri et al., 2019; Kiss et al., 2020; Xie et al., 2020). In order to understand the complex NMN-related network changes, high-throughput methods are indispensable, such as methylation profiling, transcriptome, proteome, and metabolomics. In addition, the multifunctional role of NAD+ indicates that NAD+ might function in various organelles. It would be intriguing to decipher NAD+-related mechanism in subcellular compartment.
Further researches are compelling to reveal links between NAD+-related pathway and other disease-related pathway. More importantly, efficiency and safety should be precisely assessed in NMN clinical application.
Conclusion
NAD+ metabolism has been proven to be an essential part of biochemical reaction that acts as a link between various physiologic processes. During aging, weakened NAD+ biosynthesis and accelerated NAD+ consumption lead to dysfunction in multiple tissues. Depressed NAD+ levels disturb many biochemical processes, for instance, abnormal deacetylation activity of sirtuins. Downstream alterations of abnormal sirtuin activity include transcription pattern, mitochondrial permeability, mtROS production, and oxidative stress response. As an intermediate in NAD+ biosynthesis, NMN is a promising agent to reinforce NAD+ metabolism and alleviate age-related pathologic processes in vivo, which has promoted NMN to stage of clinical trial. More details of NAD+ metabolism pathway and applications of NMN are fascinating for further researches.
Author Contributions
WH wrote the initial manuscript. MH and ZZ created the figures. FM contributed writing material and new ideas. XW revised the manuscript and approved the final version. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81602492), the National Key Research and Development Program of China (No. 2016YFA0201402), the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001), and the Excellent Youth Foundation of Sichuan Scientific Committee Grant in China (No. 2019JDJQ0008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allen, J., Romay-Tallon, R., Brymer, K. J., Caruncho, H. J., and Kalynchuk, L. E. (2018). Mitochondria and mood: mitochondrial dysfunction as a key player in the manifestation of depression. Front. Neurosci. 12:386. doi: 10.3389/fnins.2018.00386
Allen-Jennings, A. E., Hartman, M. G., Kociba, G. J., and Hai, T. (2002). The roles of ATF3 in liver dysfunction and the regulation of phosphoenolpyruvate carboxykinase gene expression. J. Biol. Chem. 277, 20020–20025. doi: 10.1074/jbc.M200727200
Aredo, B., Zhang, K., Chen, X., Wang, C. X., Li, T., and Ufret-Vincenty, R. L. (2015). Differences in the distribution, phenotype and gene expression of subretinal microglia/macrophages in C57BL/6N (Crb1 rd8/rd8) versus C57BL6/J (Crb1 wt/wt) mice. J. Neuroinflammation 12:6. doi: 10.1186/s12974-014-0221-4
Artegiani, B., and Calegari, F. (2012). Age-related cognitive decline: can neural stem cells help us? Aging 4, 176–186. doi: 10.18632/aging.100446
Assiri, M. A., Ali, H. R., Marentette, J. O., Yun, Y., Liu, J., Hirschey, M. D., et al. (2019). Investigating RNA expression profiles altered by nicotinamide mononucleotide therapy in a chronic model of alcoholic liver disease. Hum. Genomics 13:65. doi: 10.1186/s40246-019-0251-1
Bachschmid, M. M., Schildknecht, S., Matsui, R., Zee, R., Haeussler, D., Cohen, R. A., et al. (2013). Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann. Med. 45, 17–36. doi: 10.3109/07853890.2011.645498
Bai, P., Canto, C., Oudart, H., Brunyanszki, A., Cen, Y., Thomas, C., et al. (2011). PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468. doi: 10.1016/j.cmet.2011.03.004
Balbi, M., Ghosh, M., Longden, T. A., Jativa Vega, M., Gesierich, B., Hellal, F., et al. (2015). Dysfunction of mouse cerebral arteries during early aging. J. Cereb. Blood Flow Metab. 35, 1445–1453. doi: 10.1038/jcbfm.2015.107
Basu, R., Breda, E., Oberg, A. L., Powell, C. C., Dalla Man, C., Basu, A., et al. (2003). Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes Metab. Res. Rev. 52, 1738–1748. doi: 10.2337/diabetes.52.7.1738
Bertoldo, M. J., Listijono, D. R., Ho, W. J., Riepsamen, A. H., Goss, D. M., Richani, D., et al. (2020). NAD(+) repletion rescues female fertility during reproductive aging. Cell Rep. 30, 1670–1681.e7. doi: 10.1016/j.celrep.2020.01.058
Bertoldo, M. J., Uddin, G. M., Youngson, N. A., Agapiou, D., Walters, K. A., Sinclair, D. A., et al. (2018). Multigenerational obesity-induced perturbations in oocyte-secreted factor signalling can be ameliorated by exercise and nicotinamide mononucleotide. Hum. Reprod. Open 2018:hoy010. doi: 10.1093/hropen/hoy010
Bitterman, K. J., Anderson, R. M., Cohen, H. Y., Latorre-Esteves, M., and Sinclair, D. A. (2002). Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277, 45099–45107. doi: 10.1074/jbc.M205670200
Blanco, R., and Gerhardt, H. (2013). VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 3:a006569. doi: 10.1101/cshperspect.a006569
Bonini, S., Rama, P., Olzi, D., and Lambiase, A. (2003). Neurotrophic keratitis. Eye 17, 989–995. doi: 10.1038/sj.eye.6700616
Bordone, L., Motta, M. C., Picard, F., Robinson, A., Jhala, U. S., Apfeld, J., et al. (2006). Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 4:e31. doi: 10.1371/journal.pbio.0040031
Boslett, J., Hemann, C., Zhao, Y. J., Lee, H. C., and Zweier, J. L. (2017). Luteolinidin protects the postischemic heart through CD38 inhibition with preservation of NAD(P)(H). J. Pharmacol. Exp. Ther. 361, 99–108. doi: 10.1124/jpet.116.239459
Broder, G., and Weil, M. H. (1964). Excess lactate: an index of reversibility of shock in human patients. Science 143, 1457–1459. doi: 10.1126/science.143.3613.1457
Brunetti, L., Recinella, L., Di Nisio, C., Chiavaroli, A., Leone, S., Ferrante, C., et al. (2012). Effects of visfatin/PBEF/NAMPT on feeding behaviour and hypothalamic neuromodulators in the rat. J. Biol. Regul. Homeost. Agents 26, 295–302.
Camacho-Pereira, J., Tarrago, M. G., Chini, C. C. S., Nin, V., Escande, C., Warner, G. M., et al. (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23, 1127–1139. doi: 10.1016/j.cmet.2016.05.006
Castillo-Laura, H., Santos, I. S., Quadros, L. C., and Matijasevich, A. (2015). Maternal obesity and offspring body composition by indirect methods: a systematic review and meta-analysis. Cad. Saude. Publica 31, 2073–2092. doi: 10.1590/0102-311x00159914
Catalán, V., Gómez-Ambrosi, J., Rodríguez, A., Ramírez, B., Silva, C., Rotellar, F., et al. (2011). Association of increased visfatin/PBEF/NAMPT circulating concentrations and gene expression levels in peripheral blood cells with lipid metabolism and fatty liver in human morbid obesity. Nutr. Metab. Cardiovasc. Dis. 21, 245–253. doi: 10.1016/j.numecd.2009.09.008
Caton, P. W., Kieswich, J., Yaqoob, M. M., Holness, M. J., and Sugden, M. C. (2011). Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia 54, 3083–3092. doi: 10.1007/s00125-011-2288-0
Cavadini, G., Petrzilka, S., Kohler, P., Jud, C., Tobler, I., Birchler, T., et al. (2007). TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. U.S.A. 104, 12843–12848. doi: 10.1073/pnas.0701466104
Chalkiadaki, A., and Guarente, L. (2012). High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 16, 180–188. doi: 10.1016/j.cmet.2012.07.003
Chalkiadaki, A., and Guarente, L. (2015). The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 15, 608–624. doi: 10.1038/nrc3985
Chambon, P., Weill, J. D., and Mandel, P. (1963). Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 11, 39–43. doi: 10.1016/0006-291x(63)90024-x
Chen, Y., Liang, Y., Hu, T., Wei, R., Cai, C., Wang, P., et al. (2017). Endogenous Nampt upregulation is associated with diabetic nephropathy inflammatory-fibrosis through the NF-κB p65 and Sirt1 pathway; NMN alleviates diabetic nephropathy inflammatory-fibrosis by inhibiting endogenous Nampt. Exp. Ther. Med. 14, 4181–4193. doi: 10.3892/etm.2017.5098
Chen, Y., Zhang, J., Lin, Y., Lei, Q., Guan, K. L., Zhao, S., et al. (2011). Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 12, 534–541. doi: 10.1038/embor.2011.65
Chini, C. C. S., Tarrago, M. G., and Chini, E. N. (2017). NAD and the aging process: role in life, death and everything in between. Mol. Cell. Endocrinol. 455, 62–74. doi: 10.1016/j.mce.2016.11.003
Choi, S. E., Fu, T., Seok, S., Kim, D. H., Yu, E., Lee, K. W., et al. (2013). Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 12, 1062–1072. doi: 10.1111/acel.12135
Cohen, M. V., Yang, X. M., and Downey, J. M. (2007). The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation 115, 1895–1903. doi: 10.1161/circulationaha.106.675710
Colombo, A., Bastone, A., Ploia, C., Sclip, A., Salmona, M., Forloni, G., et al. (2009). JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol. Dis. 33, 518–525. doi: 10.1016/j.nbd.2008.12.014
Costford, S. R., Bajpeyi, S., Pasarica, M., Albarado, D. C., Thomas, S. C., Xie, H., et al. (2010). Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 298, E117–E126. doi: 10.1152/ajpendo.00318.2009
Das, A., Huang, G. X., Bonkowski, M. S., Longchamp, A., Li, C., Schultz, M. B., et al. (2018). Impairment of an endothelial NAD(+)-H2S signaling network is a reversible cause of vascular aging. Cell 173, 74–89.e20. doi: 10.1016/j.cell.2018.02.008
De Flora, A., Zocchi, E., Guida, L., Franco, L., and Bruzzone, S. (2004). Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N. Y. Acad. Sci. 1028, 176–191. doi: 10.1196/annals.1322.021
de Picciotto, N. E., Gano, L. B., Johnson, L. C., Martens, C. R., Sindler, A. L., Mills, K. F., et al. (2016). Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15, 522–530. doi: 10.1111/acel.12461
Di Stefano, M., Loreto, A., Orsomando, G., Mori, V., Zamporlini, F., Hulse, R. P., et al. (2017). NMN deamidase delays wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr. Biol. 27, 784–794. doi: 10.1016/j.cub.2017.01.070
Di Stefano, M., Nascimento-Ferreira, I., Orsomando, G., Mori, V., Gilley, J., Brown, R., et al. (2015). A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death. Differ. 22, 731–742. doi: 10.1038/cdd.2014.164
Diez, J. (2007). Arterial stiffness and extracellular matrix. Adv. Cardiol. 44, 76–95. doi: 10.1159/000096722
Donato, A. J., Magerko, K. A., Lawson, B. R., Durrant, J. R., Lesniewski, L. A., and Seals, D. R. (2011). SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 589(Pt 18), 4545–4554. doi: 10.1113/jphysiol.2011.211219
Dujardin, K. S., Tei, C., Yeo, T. C., Hodge, D. O., Rossi, A., and Seward, J. B. (1998). Prognostic value of a Doppler index combining systolic and diastolic performance in idiopathic-dilated cardiomyopathy. Am. J. Cardiol. 82, 1071–1076. doi: 10.1016/s0002-9149(98)00559-1
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. doi: 10.1172/jci43171
Escande, C., Nin, V., Price, N. L., Capellini, V., Gomes, A. P., Barbosa, M. T., et al. (2013). Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes Metab. Res. Rev. 62, 1084–1093. doi: 10.2337/db12-1139
Escobar-Henriques, M., and Anton, F. (2013). Mechanistic perspective of mitochondrial fusion: tubulation vs. fragmentation. Biochim. Biophys. Acta 1833, 162–175. doi: 10.1016/j.bbamcr.2012.07.016
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 Toll/Interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334–1343.e5. doi: 10.1016/j.neuron.2017.02.022
Fan, H., Yang, H. C., You, L., Wang, Y. Y., He, W. J., and Hao, C. M. (2013). The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int. 83, 404–413. doi: 10.1038/ki.2012.394
Fang, E. F., Kassahun, H., Croteau, D. L., Scheibye-Knudsen, M., Marosi, K., Lu, H., et al. (2016). NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581. doi: 10.1016/j.cmet.2016.09.004
Frederick, D. W., Loro, E., Liu, L., Davila, A. Jr., Chellappa, K., Silverman, I. M., et al. (2016). Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 24, 269–282. doi: 10.1016/j.cmet.2016.07.005
Frye, R. A. (1999). Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260, 273–279. doi: 10.1006/bbrc.1999.0897
Fu, J., Jin, J., Cichewicz, R. H., Hageman, S. A., Ellis, T. K., Xiang, L., et al. (2012). trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J. Biol. Chem. 287, 24460–24472. doi: 10.1074/jbc.M112.382226
Gano, L. B., Donato, A. J., Pasha, H. M., Hearon, C. M. Jr., Sindler, A. L., and Seals, D. R. (2014). The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 307, H1754–H1763. doi: 10.1152/ajpheart.00377.2014
Garten, A., Petzold, S., Barnikol-Oettler, A., Korner, A., Thasler, W. E., Kratzsch, J., et al. (2010). Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem. Biophys. Res. Commun. 391, 376–381. doi: 10.1016/j.bbrc.2009.11.066
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 348, 453–457. doi: 10.1126/science.1258366
Gitiaux, C., Blin-Rochemaure, N., Hully, M., Echaniz-Laguna, A., Calmels, N., Bahi-Buisson, N., et al. (2015). Progressive demyelinating neuropathy correlates with clinical severity in Cockayne syndrome. Clin. Neurophysiol. 126, 1435–1439. doi: 10.1016/j.clinph.2014.10.014
Gohil, V. M., Sheth, S. A., Nilsson, R., Wojtovich, A. P., Lee, J. H., Perocchi, F., et al. (2010). Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 28, 249–255. doi: 10.1038/nbt.1606
Gomes, A. P., Price, N. L., Ling, A. J., Moslehi, J. J., Montgomery, M. K., Rajman, L., et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638. doi: 10.1016/j.cell.2013.11.037
Gong, Y., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., et al. (2003). Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422. doi: 10.1073/pnas.1834302100
Griffiths, E. J., and Halestrap, A. P. (1995). Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 307(Pt 1), 93–98. doi: 10.1042/bj3070093
Grozio, A., Mills, K. F., Yoshino, J., Bruzzone, S., Sociali, G., Tokizane, K., et al. (2019). Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 1, 47–57. doi: 10.1038/s42255-018-0009-4
Grozio, A., Sociali, G., Sturla, L., Caffa, I., Soncini, D., Salis, A., et al. (2013). CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 288, 25938–25949. doi: 10.1074/jbc.M113.470435
Guan, Y., Wang, S. R., Huang, X. Z., Xie, Q. H., Xu, Y. Y., Shang, D., et al. (2017). Nicotinamide mononucleotide, an NAD(+) precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-Dependent manner. J. Am. Soc. Nephrol. 28, 2337–2352. doi: 10.1681/ASN.2016040385
Guarani, V., Deflorian, G., Franco, C. A., Kruger, M., Phng, L. K., Bentley, K., et al. (2011). Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 473, 234–238. doi: 10.1038/nature09917
Guidi, I., Galimberti, D., Lonati, S., Novembrino, C., Bamonti, F., Tiriticco, M., et al. (2006). Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 27, 262–269. doi: 10.1016/j.neurobiolaging.2005.01.001
Gujar, A. D., Le, S., Mao, D. D., Dadey, D. Y. A., Turski, A., Sasaki, Y., et al. (2016). An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 113, E8247–E8256. doi: 10.1073/pnas.1610921114
Haider, D. G., Schaller, G., Kapiotis, S., Maier, C., Luger, A., and Wolzt, M. (2006). The release of the adipocytokine visfatin is regulated by glucose and insulin. Diabetologia 49, 1909–1914. doi: 10.1007/s00125-006-0303-7
Harden, A., and Young, W. (1906). The alcoholic ferment of yeast-juice. Part II. The conferment of yeast-juice. Proc. R. Soc. Lond. Ser. B 78, 369–375. doi: 10.1098/rspb.1906.0070
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. doi: 10.1038/nature08778
Hosseini, L., Farokhi-Sisakht, F., Badalzadeh, R., Khabbaz, A., Mahmoudi, J., and Sadigh-Eteghad, S. (2019a). Nicotinamide mononucleotide and melatonin alleviate aging-induced cognitive impairment via modulation of mitochondrial function and apoptosis in the prefrontal cortex and hippocampus. Neuroscience 423, 29–37. doi: 10.1016/j.neuroscience.2019.09.037
Hosseini, L., Vafaee, M. S., and Badalzadeh, R. (2019b). Melatonin and nicotinamide mononucleotide attenuate myocardial ischemia/reperfusion injury via modulation of mitochondrial function and hemodynamic parameters in aged rats. J. Cardiovasc. Pharmacol. Ther. 25, 240–250. doi: 10.1177/1074248419882002
Hroudova, J., Singh, N., and Fisar, Z. (2014). Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer’s disease. Biomed. Res. Int. 2014:175062. doi: 10.1155/2014/175062
Hsu, C. P., Oka, S., Shao, D., Hariharan, N., and Sadoshima, J. (2009). Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 105, 481–491. doi: 10.1161/CIRCRESAHA.109.203703
Hsu, C. P., Zhai, P., Yamamoto, T., Maejima, Y., Matsushima, S., Hariharan, N., et al. (2010). Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 122, 2170–2182. doi: 10.1161/CIRCULATIONAHA.110.958033
Hsueh, Y. P. (2006). The role of the MAGUK protein CASK in neural development and synaptic function. Curr. Med. Chem. 13, 1915–1927. doi: 10.2174/092986706777585040
Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. doi: 10.1038/35001622
Imai, S., and Guarente, L. (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471. doi: 10.1016/j.tcb.2014.04.002
Imai, S., and Yoshino, J. (2013). The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 15(Suppl. 3), 26–33. doi: 10.1111/dom.12171
Irie, J., Inagaki, E., Fujita, M., Nakaya, H., Mitsuishi, M., Yamaguchi, S., et al. (2019). Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 67, 153–160. doi: 10.1507/endocrj.EJ19-0313
Jadasz, J. J., Aigner, L., Rivera, F. J., and Kury, P. (2012). The remyelination Philosopher’s Stone: stem and progenitor cell therapies for multiple sclerosis. Cell Tissue Res. 349, 331–347. doi: 10.1007/s00441-012-1331-x
Jeyifous, O., Waites, C. L., Specht, C. G., Fujisawa, S., Schubert, M., Lin, E. I., et al. (2009). SAP97 and CASK mediate sorting of NMDA receptors through a previously unknown secretory pathway. Nat. Neurosci. 12, 1011–1019. doi: 10.1038/nn.2362
Jing, Z., Xing, J., Chen, X., Stetler, R. A., Weng, Z., Gan, Y., et al. (2014). Neuronal NAMPT is released after cerebral ischemia and protects against white matter injury. J. Cereb. Blood Flow Metab. 34, 1613–1621. doi: 10.1038/jcbfm.2014.119
Johnson, S., Wozniak, D. F., and Imai, S. (2018). CA1 Nampt knockdown recapitulates hippocampal cognitive phenotypes in old mice which nicotinamide mononucleotide improves. NPJ Aging Mech. Dis. 4:10. doi: 10.1038/s41514-018-0029-z
Jukarainen, S., Heinonen, S., Ramo, J. T., Rinnankoski-Tuikka, R., Rappou, E., Tummers, M., et al. (2016). Obesity is associated with low NAD(+)/SIRT pathway expression in adipose tissue of BMI-discordant monozygotic twins. J. Clin. Endocrinol. Metab. 101, 275–283. doi: 10.1210/jc.2015-3095
Kadowaki, T., Yamauchi, T., Kubota, N., Hara, K., Ueki, K., and Tobe, K. (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116, 1784–1792. doi: 10.1172/jci29126
Kane-Gill, S. L., Sileanu, F. E., Murugan, R., Trietley, G. S., Handler, S. M., and Kellum, J. A. (2015). Risk factors for acute kidney injury in older adults with critical illness: a retrospective cohort study. Am. J. Kidney Dis. 65, 860–869. doi: 10.1053/j.ajkd.2014.10.018
Karamanlidis, G., Lee, C. F., Garcia-Menendez, L., Kolwicz, S. C. Jr., Suthammarak, W., Gong, G., et al. (2013). Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18, 239–250. doi: 10.1016/j.cmet.2013.07.002
Kastrup, A., Groschel, K., Ringer, T. M., Redecker, C., Cordesmeyer, R., Witte, O. W., et al. (2008). Early disruption of the blood-brain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke 39, 2385–2387. doi: 10.1161/strokeaha.107.505420
Kawamura, T., Mori, N., and Shibata, K. (2016). β-Nicotinamide mononucleotide, an anti-aging candidate compound, is retained in the body for longer than nicotinamide in rats. J. Nutr. Sci. Vitaminol. 62, 272–276. doi: 10.3177/jnsv.62.272
Kiss, T., Balasubramanian, P., Valcarcel-Ares, M. N., Tarantini, S., Yabluchanskiy, A., Csipo, T., et al. (2019a). Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angiogenic capacity in aged cerebromicrovascular endothelial cells: a potential mechanism for the prevention of vascular cognitive impairment. Geroscience 41, 619–630. doi: 10.1007/s11357-019-00074-2
Kiss, T., Giles, C. B., Tarantini, S., Yabluchanskiy, A., Balasubramanian, P., Gautam, T., et al. (2019b). Nicotinamide mononucleotide (NMN) supplementation promotes anti-aging miRNA expression profile in the aorta of aged mice, predicting epigenetic rejuvenation and anti-atherogenic effects. Geroscience 41, 419–439. doi: 10.1007/s11357-019-00095-x
Kiss, T., Nyul-Toth, A., Balasubramanian, P., Tarantini, S., Ahire, C., Yabluchanskiy, A., et al. (2020). Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience doi: 10.1007/s11357-020-00165-5 [Epub ahead of print].
Klimova, N., Fearnow, A., Long, A., and Kristian, T. (2020). NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 325:113144. doi: 10.1016/j.expneurol.2019.113144
Klimova, N., Long, A., and Kristian, T. (2019). Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3-dependent mechanism in male mice. J. Neurosci. Res. 97, 975–990. doi: 10.1002/jnr.24397
Koenekoop, R. K., Wang, H., Majewski, J., Wang, X., Lopez, I., Ren, H., et al. (2012). Mutations in NMNAT1 cause leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 44, 1035–1039. doi: 10.1038/ng.2356
Kooragayala, K., Gotoh, N., Cogliati, T., Nellissery, J., Kaden, T. R., French, S., et al. (2015). Quantification of oxygen consumption in retina ex vivo demonstrates limited reserve capacity of photoreceptor mitochondria. Invest. Ophthalmol. Vis. Sci. 56, 8428–8436. doi: 10.1167/iovs.15-17901
Kristian, T., Bernardi, P., and Siesjö, B. K. (2001). Acidosis promotes the permeability transition in energized mitochondria: implications for reperfusion injury. J. Neurotrauma 18, 1059–1074. doi: 10.1089/08977150152693755
LaNoue, K. F., and Williamson, J. R. (1971). Interrelationships between malate-aspartate shuttle and citric acid cycle in rat heart mitochondria. Metabolism 20, 119–140. doi: 10.1016/0026-0495(71)90087-4
Le Couteur, D. G., and Lakatta, E. G. (2010). A vascular theory of aging. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1025–1027. doi: 10.1093/gerona/glq135
Lee, C. F., Chavez, J. D., Garcia-Menendez, L., Choi, Y., Roe, N. D., Chiao, Y. A., et al. (2016). Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134, 883–894. doi: 10.1161/CIRCULATIONAHA.116.022495
Lee, J., and Kemper, J. K. (2010). Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging 2, 527–534. doi: 10.18632/aging.100184
Lewington, A. J., Cerda, J., and Mehta, R. L. (2013). Raising awareness of acute kidney injury: a global perspective of a silent killer. Kidney Int. 84, 457–467. doi: 10.1038/ki.2013.153
Li, J., Bonkowski, M. S., Moniot, S., Zhang, D., Hubbard, B. P., Ling, A. J., et al. (2017). A conserved NAD(+) binding pocket that regulates protein-protein interactions during aging. Science 355, 1312–1317. doi: 10.1126/science.aad8242
Li, Y., Ma, X., Li, J., Yang, L., Zhao, X., Qi, X., et al. (2019). Corneal denervation causes epithelial apoptosis through inhibiting NAD+ biosynthesis. Invest. Ophthalmol. Vis. Sci. 60, 3538–3546. doi: 10.1167/iovs.19-26909
Li, Y., Zhang, Y., Dorweiler, B., Cui, D., Wang, T., Woo, C. W., et al. (2008). Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J. Biol. Chem. 283, 34833–34843. doi: 10.1074/jbc.M805866200
Liang, H., Gao, J., Zhang, C., Li, C., Wang, Q., Fan, J., et al. (2019). Nicotinamide mononucleotide alleviates Aluminum induced bone loss by inhibiting the TXNIP-NLRP3 inflammasome. Toxicol. Appl. Pharmacol. 362, 20–27. doi: 10.1016/j.taap.2018.10.006
Liao, X., Zhang, R., Lu, Y., Prosdocimo, D. A., Sangwung, P., Zhang, L., et al. (2015). Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J. Clin. Invest. 125, 3461–3476. doi: 10.1172/jci79964
Liao, Y. F., Wang, B. J., Cheng, H. T., Kuo, L. H., and Wolfe, M. S. (2004). Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 279, 49523–49532. doi: 10.1074/jbc.M402034200
Lin, J. B., Kubota, S., Ban, N., Yoshida, M., Santeford, A., Sene, A., et al. (2016). NAMPT-Mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep. 17, 69–85. doi: 10.1016/j.celrep.2016.08.073
Liu, D., Pitta, M., and Mattson, M. P. (2008). Preventing NAD(+) depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann. N. Y. Acad. Sci. 1147, 275–282. doi: 10.1196/annals.1427.028
Liu, H.-W., Smith, C. B., Schmidt, M. S., Cambronne, X. A., Cohen, M. S., Migaud, M. E., et al. (2018). Pharmacological bypass of NAD(+) salvage pathway protects neurons from chemotherapy-induced degeneration. Proc. Natl. Acad. Sci. U.S.A. 115, 10654–10659. doi: 10.1073/pnas.1809392115
Long, A. N., Owens, K., Schlappal, A. E., Kristian, T., Fishman, P. S., and Schuh, R. A. (2015). Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 15:19. doi: 10.1186/s12883-015-0272-x
Lu, L., Tang, L., Wei, W., Hong, Y., Chen, H., Ying, W., et al. (2014). Nicotinamide mononucleotide improves energy activity and survival rate in an in vitro model of Parkinson’s disease. Exp. Ther. Med. 8, 943–950. doi: 10.3892/etm.2014.1842
Lundt, S., Zhang, N., Wang, X., Polo-Parada, L., and Ding, S. (2020). The effect of NAMPT deletion in projection neurons on the function and structure of neuromuscular junction (NMJ) in mice. Sci. Rep. 10:99. doi: 10.1038/s41598-019-57085-4
Luo, G., Huang, B., Qiu, X., Xiao, L., Wang, N., Gao, Q., et al. (2017). Resveratrol attenuates excessive ethanol exposure induced insulin resistance in rats via improving NAD(+)/NADH ratio. Mol. Nutr. Food Res. 61:1700087. doi: 10.1002/mnfr.201700087
Malik, V. S., Popkin, B. M., Bray, G. A., Despres, J. P., Willett, W. C., and Hu, F. B. (2010). Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care 33, 2477–2483. doi: 10.2337/dc10-1079
Martin, A. S., Abraham, D. M., Hershberger, K. A., Bhatt, D. P., Mao, L., Cui, H., et al. (2017). Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2:93885. doi: 10.1172/jci.insight.93885
Mehan, S., Meena, H., Sharma, D., and Sankhla, R. (2011). JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neurodegenerative abnormalities. J. Mol. Neurosci. 43, 376–390. doi: 10.1007/s12031-010-9454-6
Mills, K. F., Yoshida, S., Stein, L. R., Grozio, A., Kubota, S., Sasaki, Y., et al. (2016). Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 24, 795–806. doi: 10.1016/j.cmet.2016.09.013
Mitchell, G. F. (2014). Arterial stiffness and hypertension. Hypertension 64, 13–18. doi: 10.1161/hypertensionaha.114.00921
Mohammadnia, A., Yaqubi, M., and Fallahi, H. (2015). Predicting transcription factors in human alcoholic hepatitis from gene regulatory network. Eur. Rev. Med. Pharmacol. Sci. 19, 2246–2253.
Moller, N., Gormsen, L., Fuglsang, J., and Gjedsted, J. (2003). Effects of ageing on insulin secretion and action. Horm. Res. 60(Suppl. 1), 102–104. doi: 10.1159/000071233
Morales, I., Guzman-Martinez, L., Cerda-Troncoso, C., Farias, G. A., and Maccioni, R. B. (2014). Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 8:112. doi: 10.3389/fncel.2014.00112
Morigi, M., Perico, L., Rota, C., Longaretti, L., Conti, S., Rottoli, D., et al. (2015). Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest. 125, 715–726. doi: 10.1172/JCI77632
Mouchiroud, L., Houtkooper, R. H., Moullan, N., Katsyuba, E., Ryu, D., Canto, C., et al. (2013). The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. doi: 10.1016/j.cell.2013.06.016
Moynihan, K. A., Grimm, A. A., Plueger, M. M., Bernal-Mizrachi, E., Ford, E., Cras-Meneur, C., et al. (2005). Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2, 105–117. doi: 10.1016/j.cmet.2005.07.001
Mylroie, H., Dumont, O., Bauer, A., Thornton, C. C., Mackey, J., Calay, D., et al. (2015). PKCepsilon-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc. Res. 106, 509–519. doi: 10.1093/cvr/cvv131
Nadtochiy, S. M., Wang, Y. T., Nehrke, K., Munger, J., and Brookes, P. S. (2018). Cardioprotection by nicotinamide mononucleotide (NMN): involvement of glycolysis and acidic pH. J. Mol. Cell Cardiol. 121, 155–162. doi: 10.1016/j.yjmcc.2018.06.007
Nielsen, K. N., Peics, J., Ma, T., Karavaeva, I., Dall, M., Chubanava, S., et al. (2018). NAMPT-mediated NAD(+) biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol. Metab. 11, 178–188. doi: 10.1016/j.molmet.2018.02.014
Nikiforov, A., Dolle, C., Niere, M., and Ziegler, M. (2011). Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 286, 21767–21778. doi: 10.1074/jbc.M110.213298
Okabe, K., Yaku, K., Tobe, K., and Nakagawa, T. (2019). Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 26:34. doi: 10.1186/s12929-019-0527-8
Park, J. H., Long, A., Owens, K., and Kristian, T. (2016). Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 95, 102–110. doi: 10.1016/j.nbd.2016.07.018
Peek, C. B., Affinati, A. H., Ramsey, K. M., Kuo, H. Y., Yu, W., Sena, L. A., et al. (2013). Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342:1243417. doi: 10.1126/science.1243417
Pillai, J. B., Isbatan, A., Imai, S., and Gupta, M. P. (2005). Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 280, 43121–43130. doi: 10.1074/jbc.M506162200
Pillai, V. B., Sundaresan, N. R., Kim, G., Samant, S., Moreno-Vinasco, L., Garcia, J. G. N., et al. (2013). Nampt secreted from cardiomyocytes promotes development of cardiac hypertrophy and adverse ventricular remodeling. Am. J. Physiol. Heart Circ. Physiol. 304, H415–H426. doi: 10.1152/ajpheart.00468.2012
Poddar, S. K., Sifat, A. E., Haque, S., Nahid, N. A., Chowdhury, S., and Mehedi, I. (2019). Nicotinamide mononucleotide: exploration of diverse therapeutic applications of a potential molecule. Biomolecules 9:34. doi: 10.3390/biom9010034
Preiss, J., and Handler, P. (1958). Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J. Biol. Chem. 233, 488–492.
Preugschat, F., Carter, L. H., Boros, E. E., Porter, D. J., Stewart, E. L., and Shewchuk, L. M. (2014). A pre-steady state and steady state kinetic analysis of the N-ribosyl hydrolase activity of hCD157. Arch. Biochem. Biophys. 564, 156–163. doi: 10.1016/j.abb.2014.09.008
Prior, S. J., Ryan, A. S., Blumenthal, J. B., Watson, J. M., Katzel, L. I., and Goldberg, A. P. (2016). Sarcopenia is associated with lower skeletal muscle capillarization and exercise capacity in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 71, 1096–1101. doi: 10.1093/gerona/glw017
Qin, W., Yang, T., Ho, L., Zhao, Z., Wang, J., Chen, L., et al. (2006). Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 281, 21745–21754. doi: 10.1074/jbc.M602909200
Rajman, L., Chwalek, K., and Sinclair, D. A. (2018). Therapeutic potential of NAD-Boosting molecules: the in vivo evidence. Cell Metab. 27, 529–547. doi: 10.1016/j.cmet.2018.02.011
Ramsey, K. M., Mills, K. F., Satoh, A., and Imai, S. (2008). Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 7, 78–88. doi: 10.1111/j.1474-9726.2007.00355.x
Ratajczak, J., Joffraud, M., Trammell, S. A., Ras, R., Canela, N., Boutant, M., et al. (2016). NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 7:13103. doi: 10.1038/ncomms13103
Reaven, G. M. (1988). Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37, 1595–1607. doi: 10.2337/diab.37.12.1595
Regier, D. A., Boyd, J. H., Burke, J. D. Jr., Rae, D. S., Myers, J. K., Kramer, M., et al. (1988). One-month prevalence of mental disorders in the United States. Based on five epidemiologic catchment area sites. Arch. Gen. Psychiatry 45, 977–986. doi: 10.1001/archpsyc.1988.01800350011002
Rempe, R. G., Hartz, A. M. S., and Bauer, B. (2016). Matrix metalloproteinases in the brain and blood-brain barrier: versatile breakers and makers. J. Cereb. Blood Flow Metab. 36, 1481–1507. doi: 10.1177/0271678x16655551
Revollo, J. R., Korner, A., Mills, K. F., Satoh, A., Wang, T., Garten, A., et al. (2007). Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6, 363–375. doi: 10.1016/j.cmet.2007.09.003
Rixen, D., and Siegel, J. H. (2005). Bench-to-bedside review: oxygen debt and its metabolic correlates as quantifiers of the severity of hemorrhagic and post-traumatic shock. Crit. Care 9, 441–453. doi: 10.1186/cc3526
Roncal-Jimenez, C. A., Lanaspa, M. A., Rivard, C. J., Nakagawa, T., Sanchez-Lozada, L. G., Jalal, D., et al. (2011). Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism 60, 1259–1270. doi: 10.1016/j.metabol.2011.01.008
Ross, R., Hudson, R., Stotz, P. J., and Lam, M. (2015). Effects of exercise amount and intensity on abdominal obesity and glucose tolerance in obese adults: a randomized trial. Ann. Intern. Med. 162, 325–334. doi: 10.7326/m14-1189
Salter, M., Beams, R. M., Critchley, M. A., Hodson, H. F., Iyer, R., Knowles, R. G., et al. (1991). Effects of tryptophan 2,3-dioxygenase inhibitors in the rat. Adv. Exp. Med. Biol. 294, 281–288. doi: 10.1007/978-1-4684-5952-4_25
Sanada, S., Komuro, I., and Kitakaze, M. (2011). Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 301, H1723–H1741. doi: 10.1152/ajpheart.00553.2011
Sayers, S. R., Beavil, R. L., Fine, N. H. F., Huang, G. C., Choudhary, P., Pacholarz, K. J., et al. (2020). Structure-functional changes in eNAMPT at high concentrations mediate mouse and human beta cell dysfunction in type 2 diabetes. Diabetologia 63, 313–323. doi: 10.1007/s00125-019-05029-y
Scheibye-Knudsen, M., Mitchell, S. J., Fang, E. F., Iyama, T., Ward, T., Wang, J., et al. (2014). A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855. doi: 10.1016/j.cmet.2014.10.005
Seals, D. R., Kaplon, R. E., Gioscia-Ryan, R. A., and LaRocca, T. J. (2014). You’re only as old as your arteries: translational strategies for preserving vascular endothelial function with aging. Physiology 29, 250–264. doi: 10.1152/physiol.00059.2013
Seitz, H. K., Bataller, R., Cortez-Pinto, H., Gao, B., Gual, A., Lackner, C., et al. (2018). Alcoholic liver disease. Nat. Rev. Dis. Primers 4:16. doi: 10.1038/s41572-018-0014-7
Sims, C. A., Guan, Y., Mukherjee, S., Singh, K., Botolin, P., Davila, A., et al. (2018). Nicotinamide mononucleotide preserves mitochondrial function and increases survival in hemorrhagic shock. JCI Insight 3:e120182. doi: 10.1172/jci.insight.120182
Song, J., Li, J., Yang, F., Ning, G., Zhen, L., Wu, L., et al. (2019). Nicotinamide mononucleotide promotes osteogenesis and reduces adipogenesis by regulating mesenchymal stromal cells via the SIRT1 pathway in aged bone marrow. Cell Death Dis. 10:336. doi: 10.1038/s41419-019-1569-2
Sozzani, S., Del Prete, A., Bonecchi, R., and Locati, M. (2015). Chemokines as effector and target molecules in vascular biology. Cardiovasc. Res. 107, 364–372. doi: 10.1093/cvr/cvv150
Springo, Z., Tarantini, S., Toth, P., Tucsek, Z., Koller, A., Sonntag, W. E., et al. (2015). Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1355–1359. doi: 10.1093/gerona/glu244
Stanley, W. C., Recchia, F. A., and Lopaschuk, G. D. (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85, 1093–1129. doi: 10.1152/physrev.00006.2004
Stein, L. R., and Imai, S. (2014). Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 33, 1321–1340. doi: 10.1002/embj.201386917
Stensballe, J., Christiansen, M., Tonnesen, E., Espersen, K., Lippert, F. K., and Rasmussen, L. S. (2009). The early IL-6 and IL-10 response in trauma is correlated with injury severity and mortality. Acta Anaesthesiol. Scand. 53, 515–521. doi: 10.1111/j.1399-6576.2008.01801.x
Stram, A. R., Pride, P. M., and Payne, R. M. (2017). NAD+ replacement therapy with nicotinamide riboside does not improve cardiac function in a model of mitochondrial heart disease. FASEB J 31, 602–602.615. doi: 10.1096/fasebj.31.1_supplement.602.15
Stringer, J., Groenewegen, E., Liew, S. H., and Hutt, K. J. (2019). Nicotinamide mononucleotide does not protect the ovarian reserve from cancer treatments. Reproduction doi: 10.1530/rep-19-0337 [Epub ahead of print].
Stromsdorfer, K. L., Yamaguchi, S., Yoon, M. J., Moseley, A. C., Franczyk, M. P., Kelly, S. C., et al. (2016). NAMPT-Mediated NAD(+) biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Rep. 16, 1851–1860. doi: 10.1016/j.celrep.2016.07.027
Strosznajder, R. P., Gadamski, R., Czapski, G. A., Jesko, H., and Strosznajder, J. B. (2003). Poly(ADP-ribose) polymerase during reperfusion after transient forebrain ischemia: its role in brain edema and cell death. J. Mol. Neurosci. 20, 61–72. doi: 10.1385/jmn:20:1:61
Suga, H. (1990). Ventricular energetics. Physiol. Rev. 70, 247–277. doi: 10.1152/physrev.1990.70.2.247
Tanaka, M., Nozaki, M., Fukuhara, A., Segawa, K., Aoki, N., Matsuda, M., et al. (2007). Visfatin is released from 3T3-L1 adipocytes via a non-classical pathway. Biochem. Biophys. Res. Commun. 359, 194–201. doi: 10.1016/j.bbrc.2007.05.096
Tarantini, S., Tran, C. H. T., Gordon, G. R., Ungvari, Z., and Csiszar, A. (2017). Impaired neurovascular coupling in aging and Alzheimer’s disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 94, 52–58. doi: 10.1016/j.exger.2016.11.004
Tarantini, S., Valcarcel-Ares, M. N., Toth, P., Yabluchanskiy, A., Tucsek, Z., Kiss, T., et al. (2019). Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 24:101192. doi: 10.1016/j.redox.2019.101192
Tesla, R., Wolf, H. P., Xu, P., Drawbridge, J., Estill, S. J., Huntington, P., et al. (2012). Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 109, 17016–17021. doi: 10.1073/pnas.1213960109
Toth, P., Tarantini, S., Tucsek, Z., Ashpole, N. M., Sosnowska, D., Gautam, T., et al. (2014). Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 306, H299–H308. doi: 10.1152/ajpheart.00744.2013
Tsubota, K. (2016). The first human clinical study for NMN has started in Japan. NPJ Aging Mech. Dis. 2:16021. doi: 10.1038/npjamd.2016.21
Uddin, G. M., Youngson, N. A., Doyle, B. M., Sinclair, D. A., and Morris, M. J. (2017). Nicotinamide mononucleotide (NMN) supplementation ameliorates the impact of maternal obesity in mice: comparison with exercise. Sci. Rep. 7:15063. doi: 10.1038/s41598-017-14866-z
Uddin, G. M., Youngson, N. A., Sinclair, D. A., and Morris, M. J. (2016). Head to head comparison of short-term treatment with the NAD(+) precursor nicotinamide mononucleotide (NMN) and 6 weeks of exercise in obese female mice. Front. Pharmacol. 7:258. doi: 10.3389/fphar.2016.00258
Vieira, V. J., Valentine, R. J., Wilund, K. R., Antao, N., Baynard, T., and Woods, J. A. (2009). Effects of exercise and low-fat diet on adipose tissue inflammation and metabolic complications in obese mice. Am. J. Physiol. Endocrinol. Metab. 296, E1164–E1171. doi: 10.1152/ajpendo.00054.2009
von Euler, H., and Myrback, K. (1930). Co-zynase, XVII. Hoppe Seylers Zeitschrift Fur Physiologische Chemie 190, 93–100. doi: 10.1515/bchm2.1930.190.3-6.93
Walley, K. R. (2016). Left ventricular function: time-varying elastance and left ventricular aortic coupling. Crit. Care 20:270. doi: 10.1186/s13054-016-1439-6
Wang, B., Hasan, M. K., Alvarado, E., Yuan, H., Wu, H., and Chen, W. Y. (2011). NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 30, 907–921. doi: 10.1038/onc.2010.468
Wang, P., Xu, T.-Y., Guan, Y.-F., Tian, W.-W., Viollet, B., Rui, Y.-C., et al. (2011). Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 69, 360–374. doi: 10.1002/ana.22236
Wang, H., Listrat, A., Meunier, B., Gueugneau, M., Coudy-Gandilhon, C., Combaret, L., et al. (2014). Apoptosis in capillary endothelial cells in ageing skeletal muscle. Aging Cell 13, 254–262. doi: 10.1111/acel.12169
Wang, X., Hu, X., Yang, Y., Takata, T., and Sakurai, T. (2016). Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 1643, 1–9. doi: 10.1016/j.brainres.2016.04.060
Wang, X., Zhang, Q., Bao, R., Zhang, N., Wang, Y., Polo-Parada, L., et al. (2017). Deletion of nampt in projection neurons of adult mice leads to motor dysfunction, neurodegeneration, and death. Cell Rep. 20, 2184–2200. doi: 10.1016/j.celrep.2017.08.022
Warburg, O., and Christian, W. (1936). Pyridine, the hydrogen transferring element of fermentation enzymes. (Pyridine-nucleotide.). Biochemische Zeitschrift 287, 291–328.
Watanabe, S., Ageta-Ishihara, N., Nagatsu, S., Takao, K., Komine, O., Endo, F., et al. (2014). SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 7:62. doi: 10.1186/s13041-014-0062-1
Wei, C. C., Kong, Y. Y., Hua, X., Li, G. Q., Zheng, S. L., Cheng, M. H., et al. (2017a). NAD replenishment with nicotinamide mononucleotide protects blood-brain barrier integrity and attenuates delayed tissue plasminogen activator-induced haemorrhagic transformation after cerebral ischaemia. Br. J. Pharmacol. 174, 3823–3836. doi: 10.1111/bph.13979
Wei, C. C., Kong, Y. Y., Li, G. Q., Guan, Y. F., Wang, P., and Miao, C. Y. (2017b). Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 7:717. doi: 10.1038/s41598-017-00851-z
Wheaton, W. W., and Chandel, N. S. (2011). Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 300, C385–C393. doi: 10.1152/ajpcell.00485.2010
Willems, P. H., Rossignol, R., Dieteren, C. E., Murphy, M. P., and Koopman, W. J. (2015). Redox homeostasis and mitochondrial dynamics. Cell Metab. 22, 207–218. doi: 10.1016/j.cmet.2015.06.006
Wright, A. F., Chakarova, C. F., Abd El-Aziz, M. M., and Bhattacharya, S. S. (2010). Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 11, 273–284. doi: 10.1038/nrg2717
Wurth, M. A., Sayeed, M. M., and Baue, A. E. (1973). Nicotinamide adenine dinucleotide (n.d.) content of liver with hemorrhagic shock. Proc. Soc. Exp. Biol. Med. 144, 654–658. doi: 10.3181/00379727-144-37656
Xie, X., Yu, C., Zhou, J., Xiao, Q., Shen, Q., Xiong, Z., et al. (2020). Nicotinamide mononucleotide ameliorates the depression-like behaviors and is associated with attenuating the disruption of mitochondrial bioenergetics in depressed mice. J. Affect. Disord. 263, 166–174. doi: 10.1016/j.jad.2019.11.147
Yamaguchi, S., Franczyk, M. P., Chondronikola, M., Qi, N., Gunawardana, S. C., Stromsdorfer, K. L., et al. (2019). Adipose tissue NAD(+) biosynthesis is required for regulating adaptive thermogenesis and whole-body energy homeostasis in mice. Proc. Natl. Acad. Sci. U.S.A. 116, 23822–23828. doi: 10.1073/pnas.1909917116
Yamamoto, T., Byun, J., Zhai, P., Ikeda, Y., Oka, S., and Sadoshima, J. (2014). Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One 9:e98972. doi: 10.1371/journal.pone.0098972
Yang, Y., and Sauve, A. A. (2016). NAD(+) metabolism: bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 1864, 1787–1800. doi: 10.1016/j.bbapap.2016.06.014
Yao, Z., Yang, W., Gao, Z., and Jia, P. (2017). Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 647, 133–140. doi: 10.1016/j.neulet.2017.03.027
Yoon, M. J., Yoshida, M., Johnson, S., Takikawa, A., Usui, I., Tobe, K., et al. (2015). SIRT1-Mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. 21, 706–717. doi: 10.1016/j.cmet.2015.04.002
Yoshino, J., Baur, J. A., and Imai, S. I. (2018). NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 27, 513–528. doi: 10.1016/j.cmet.2017.11.002
Yoshino, J., Mills, K. F., Yoon, M. J., and Imai, S. (2011). Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536. doi: 10.1016/j.cmet.2011.08.014
Young, G. S., Choleris, E., Lund, F. E., and Kirkland, J. B. (2006). Decreased cADPR and increased NAD+ in the Cd38-/- mouse. Biochem. Biophys. Res. Commun. 346, 188–192. doi: 10.1016/j.bbrc.2006.05.100
Youngson, N. A., Uddin, G. M., Das, A., Martinez, C., Connaughton, H. S., Whiting, S., et al. (2019). Impacts of obesity, maternal obesity and nicotinamide mononucleotide supplementation on sperm quality in mice. Reproduction 158, 169–179. doi: 10.1530/REP-18-0574
Zhang, L., Chopp, M., Jia, L., Cui, Y., Lu, M., and Zhang, Z. G. (2009). Atorvastatin extends the therapeutic window for tPA to 6 h after the onset of embolic stroke in rats. J. Cereb. Blood Flow Metab. 29, 1816–1824. doi: 10.1038/jcbfm.2009.105
Zhang, R., Shen, Y., Zhou, L., Sangwung, P., Fujioka, H., Zhang, L., et al. (2017). Short-term administration of nicotinamide mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell Cardiol. 112, 64–73. doi: 10.1016/j.yjmcc.2017.09.001
Zhang, X. Q., Lu, J. T., Jiang, W. X., Lu, Y. B., Wu, M., Wei, E. Q., et al. (2015). NAMPT inhibitor and metabolite protect mouse brain from cryoinjury through distinct mechanisms. Neuroscience 291, 230–240. doi: 10.1016/j.neuroscience.2015.02.007
Zhao, B., Zhang, M., Han, X., Zhang, X.-Y., Xing, Q., Dong, X., et al. (2013). Cerebral ischemia is exacerbated by extracellular nicotinamide phosphoribosyltransferase via a non-enzymatic mechanism. PLoS One 8:e85403. doi: 10.1371/journal.pone.0085403
Zhao, C., Li, W., Duan, H., Li, Z., Jia, Y., Zhang, S., et al. (2020). NAD(+) precursors protect corneal endothelial cells from UVB-induced apoptosis. Am. J. Physiol. Cell Physiol. 318, C796–C805. doi: 10.1152/ajpcell.00445.2019
Zhao, Y., Guan, Y. F., Zhou, X. M., Li, G. Q., Li, Z. Y., Zhou, C. C., et al. (2015). Regenerative neurogenesis after ischemic stroke promoted by nicotinamide phosphoribosyltransferase-nicotinamide adenine dinucleotide cascade. Stroke 46, 1966–1974. doi: 10.1161/STROKEAHA.115.009216
Zhao, Y., Liu, X. Z., Tian, W. W., Guan, Y. F., Wang, P., and Miao, C. Y. (2014). Extracellular visfatin has nicotinamide phosphoribosyltransferase enzymatic activity and is neuroprotective against ischemic injury. CNS Neurosci. Ther. 20, 539–547. doi: 10.1111/cns.12273
Zhou, Y., Wang, Y., Wang, J., Anne Stetler, R., and Yang, Q. W. (2014). Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog. Neurobiol. 115, 25–44. doi: 10.1016/j.pneurobio.2013.11.003
Keywords: nicotinamide adenine dinucleotide (NAD), nicotinamide mononucleotide (NMN), aging, diabetes, obesity, Alzheimer’s disease
Citation: Hong W, Mo F, Zhang Z, Huang M and Wei X (2020) Nicotinamide Mononucleotide: A Promising Molecule for Therapy of Diverse Diseases by Targeting NAD+ Metabolism. Front. Cell Dev. Biol. 8:246. doi: 10.3389/fcell.2020.00246
Received: 21 December 2019; Accepted: 24 March 2020;
Published: 28 April 2020.
Edited by:
Mario Antonio Bianchet, School of Medicine, Johns Hopkins University, United StatesReviewed by:
Tibor Kristian, University of Maryland, Baltimore, United StatesPei Wang, Second Military Medical University, China
Takashi Nakagawa, University of Toyama, Japan
Copyright © 2020 Hong, Mo, Zhang, Huang and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiawei Wei, d2VpeGlhd2Vpc2N1QDEyNi5jb20=; eGlhd2Vpd2VpQHNjdS5lZHUuY24=
†These authors have contributed equally to this work