 Daniela Bakula
Daniela Bakula Morten Scheibye-Knudsen
Morten Scheibye-Knudsen- Department of Cellular and Molecular Medicine, Center for Healthy Aging, University of Copenhagen, Copenhagen, Denmark
Maintaining mitochondrial health is emerging as a keystone in aging and associated diseases. The selective degradation of mitochondria by mitophagy is of particular importance in keeping a pristine mitochondrial pool. Indeed, inherited monogenic diseases with defects in mitophagy display complex multisystem pathologies but particularly progressive neurodegeneration. Fortunately, therapies are being developed that target mitophagy allowing new hope for treatments for previously incurable diseases. Herein, we describe mitophagy and associated diseases, coin the term mitophaging and describe new small molecule interventions that target different steps in the mitophagic pathway. Consequently, several age-associated diseases may be treated by targeting mitophagy.
Mitochondrial Integrity Defines Organismal Health
Mitochondria, the powerhouses of eukaryotic cells, are the key organelles for energy production allowing organismal growth and survival. Besides serving as adenosine triphosphate generators, mitochondria act as signaling hubs for programmed cell death, regulate calcium homeostasis and are required for cholesterol, nucleotide and amino acid synthesis (Sun et al., 2016). To fulfil their broad range of biological roles, mitochondria contain more than 1,000 proteins that localize and function in four specialized compartments, the outer membrane, the inner membrane, the intermembrane space and the matrix. The minority of mitochondrial proteins are encoded by the circular mitochondrial genome, whereas the vast majority is encoded in the nuclear genome. However, mutations in both genomes can cause a heterogeneous group of disorders, known as mitochondrial diseases, which are characterized by severe metabolic and neurological defects. Due to their highly variable clinical features, the prevalence of mitochondrial diseases has likely been underestimated (Haas et al., 2007; Wallace, 2018). Nevertheless, advances in next generation sequencing technologies have simplified the clinical diagnosis and enabled molecular characterization of so far undescribed mitochondrial diseases (Calvo et al., 2012; Cui et al., 2013; Legati et al., 2016). Notably, computational approaches relying on phenotypic description of mitochondrial diseases can help to characterize new mitochondrial diseases of previously unknown pathogenesis (Scheibye-Knudsen et al., 2013).
Increased evidence indicates that mitochondrial integrity is disrupted during the aging process and contributes to the pathogenesis of age-related disorders in humans (Kauppila et al., 2017; Youle, 2019). In line with this, mice that carry a defective proof-reading mitochondrial DNA polymerase gamma show an accelerated aging phenotype that may be driven by the accumulation of mutations in the mitochondrial DNA (mtDNA) (Trifunovic et al., 2004). The described correlation between levels of mtDNA deletions in human brain and aging as well as the association between mtDNA haplogroups and diseases, further supports the direct influence of mitochondria on health- and lifespan in organisms (Cortopassi and Arnheim, 1990; Corral-Debrinski et al., 1992; Hudson et al., 2014; Wallace, 2015). Indeed, dysfunctional degradation of mitochondria through the process of mitophagy is increasingly associated with degenerative diseases and aging, a phenomenon we call mitophaging. Evidently, the maintenance of functional mitochondria is necessary to sustain cellular homeostasis and organismal health.
Mitochondrial Quality Control Mechanisms
Mitochondria have evolved multiple mechanisms ensuring mitochondrial quality. For instance, mitochondrial chaperones and proteases are constantly preventing the accumulation of misfolded and aggregated proteins by monitoring proteostasis through the mitochondrial unfolded protein stress response (UPRmt) (Melber and Haynes, 2018), a mechanism that has been shown to be critical for longevity in mammals (Houtkooper et al., 2013; Mouchiroud et al., 2013). Further, mitochondria are dynamic organelles existing in large tubular and highly dynamic networks that constantly undergo fission and fusion processes, thereby leading to the dilution of non-functional mitochondria (Youle and van der Bliek, 2012).
Nevertheless, autophagy is the only known pathway that mediates the turnover of whole mitochondria to avoid cellular damage and apoptosis. The degradation process is mediated by a double-membrane vesicle, known as the autophagosome, and it was first observed in mammalian cells by electron microscopy (De Duve and Wattiaux, 1966). For a long time, autophagy was considered a non-selective bulk degradation pathway, however, when the yeast mitochondrial protein Uth1p was found to be involved in the selective degradation of mitochondria (Kissová et al., 2004), the term “mitophagy” was subsequently introduced (Lemasters, 2005).
Herein, we discuss the role of mitophagy in impacting human disease development and the aging process itself. Further, interventions that target mitophagy will be discussed that may provide a promising strategy for the treatment of a broad spectrum of diseases.
What Is Mitophagy?
The process of mitophagy can act either as a response to various stress stimuli including nutrient starvation and oxidative stress or as a programmed removal of mitochondria (Palikaras et al., 2018; Pickles et al., 2018). Different pathways are known to regulate mitophagy, the best-studied pathway is mediated by the phosphatase and tensin homologue (PTEN)-induced putative kinase 1 (PINK1) and the E3-ubiquitin ligase Parkin (Figure 1A). Mutations in both genes encoding PINK1 and Parkin (PARK2), have been reported to cause autosomal recessive forms of Parkinson’s Disease (PD) (Kitada et al., 1998; Valente et al., 2004). Under un-stressed conditions, PINK1 is imported via the translocase of the outer membrane and translocase of the inner membrane (TOM/TIM) complex in a membrane potential dependent manner into mitochondria, leading to proteolytic cleavage of PINK1 (Jin et al., 2010; Deas et al., 2011; Meissner et al., 2011). The N-terminal truncated PINK1 is subsequently released to the cytosol, and degraded by the proteasome (Yamano and Youle, 2013). Loss of mitochondrial membrane potential disrupts the transport of PINK1 across the mitochondrial membrane leading to the accumulation of uncleaved PINK1 at the outer mitochondrial membrane. Subsequently, PINK1 regulates the recruitment and activation of the cytosolic Parkin via direct phosphorylation of the Parkin Ub-like (UBL) domain or via the phosphorylation of ubiquitin (Kondapalli et al., 2012; Shiba-Fukushima et al., 2012; Iguchi et al., 2013; Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Ordureau et al., 2014; Wauer et al., 2015). Once activated, Parkin drives the ubiquitination of multiple substrates, which leads to a positive feed forward mechanism through the generation of additional substrates for Pink1 (Ordureau et al., 2014).
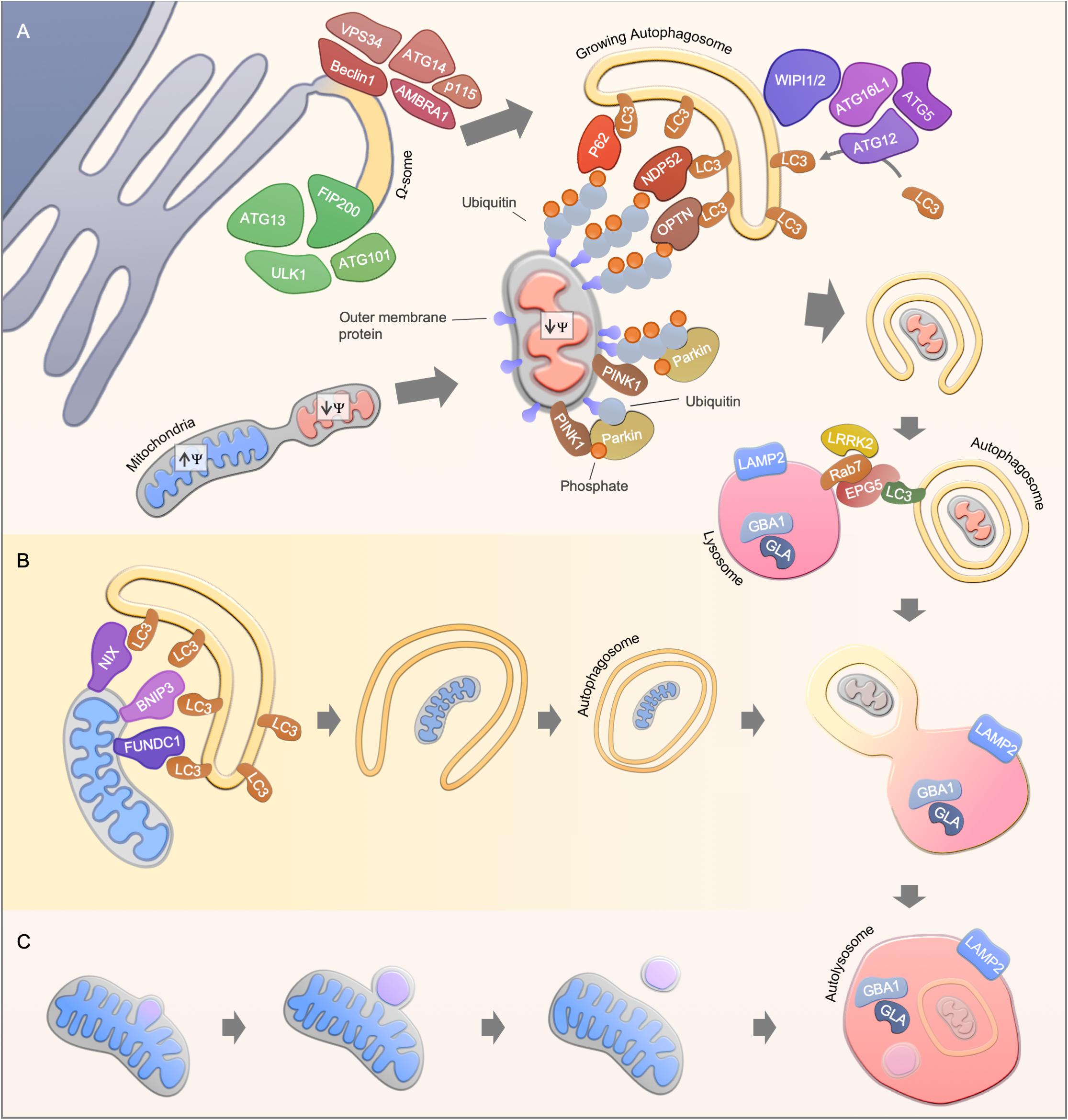
Figure 1. Mitophagy pathways. (A) Ubiquitin-dependent PINK1/Parkin-mediated mitophagy. Upon mitochondrial damage, PINK1 is stabilized at the outer mitochondrial membrane, leading to Parkin activation and subsequent ubiquitination of mitochondrial proteins. Finally, autophagy receptors such as NDP52, OPTN, and p62 are recruited to mediate the engulfment of mitochondria by the autophagosomal membrane through the interaction with LC3. A possible source of the autophagosomal membrane is provided by the endoplasmic reticulum, where the autophagy core complexes VPS34 and ULK1 initiate the membrane formation. The membrane formation is further mediated by WIPI1 and WIPI2, leading to the recruitment of the ATG16L1-complex and LC3, thereby facilitating the formation of autophagosomes. Finally, autophagosomes fuses with acidic lysosomes, a step that is regulated by concerted action of autophagosomal and lysosomal proteins. (B) Ubiquitin-independent receptor-mediated mitophagy. Ubiquitin-independent receptor mediated mitophagy is mediated by the recruitment of autophagy receptor proteins such as NIX, BNIP3, and FUNDC1 to the mitochondrial membrane. The receptor proteins recruit LC3, which enables the engulfment of mitochondria by autophagosomes. (C) Alternative degradation pathways. Piecemeal mitophagy and mitochondrial-derived vesicle degradation are cellular pathways that mediate localized degradation of mitochondria.
In recent years, several substrates, in particular mitochondrial outer membrane proteins and autophagy receptors, have been identified to be ubiquitinated by the PINK1/Parkin-mediated signaling pathway (Sarraf et al., 2013). For instance, the mitochondrial fusion proteins mitofusin 1 and 2 (Mfn1 and Mfn2) are degraded in a PINK1/parkin dependent manner to make mitochondria accessible for degradation and to prevent fusion of damaged mitochondria with the healthy network (Gegg et al., 2010; Tanaka et al., 2010). However, conditional double-knockout of Mfn1 and Mfn2 in mice leads to mitochondrial dysfunction and, in line with this, Mfn2-depleted cardiomyocytes are deficient in Parkin recruitment to the mitochondrial outer membrane (Chen et al., 2011; Chen and Dorn, 2013). A similar priming function of mitochondria has been described for other mitochondrial proteins such as Miro1 and VDAC1 (Geisler et al., 2010; Wang et al., 2011; Sun et al., 2012; Safiulina et al., 2019). Recently, the apoptotic protein BAK has been identified as a Parkin target, further connecting Parkin-mediated mitophagy to the regulation of cellular apoptosis (Bernardini et al., 2019). The ubiquitination events driven by PINK1 and Parkin enable the recruitment of autophagy substrate receptors to the mitochondrial membrane including p62, Optineurin and NDP52, thereby promoting the engulfment of mitochondria by autophagosomes (Geisler et al., 2010; Wong and Holzbaur, 2014; Lazarou et al., 2015).
Notably, transcriptional regulation is a crucial process for functional PINK1-Parkin-mediated mitophagy. For instance, PINK1-Parkin-mediated mitophagy induction upon cellular stress such as through reactive oxygen species or ethanol exposure leads to the nuclear translocation of several transcription factors, including the transcription factor EB (TFEB) and the nuclear respiratory factors (NRFs), controlling the expression of mitochondrial, autophagy and lysosomal genes (Nezich et al., 2015; Ivankovic et al., 2016; Eid et al., 2019). Parkin expression itself has also been shown to be tightly controlled by stress pathways such as the unfolded protein response pathway and its activating transcription factor 4 (ATF4) (Bouman et al., 2011). Altogether, this highlights the great number of potential therapeutic avenues to target the PINK1-Parkin signaling pathway.
Pink1/Parkin-independent mitophagy pathways mainly rely on receptor proteins which mediate the recruitment of LC3/GABARAPs for the removal of mitochondria (Figure 1B). For instance, the BCL2-related protein NIX (also known as BNIP3L) mediates mitophagy in mammals during reticulocyte differentiation, a process that requires the elimination of mitochondria (Schweers et al., 2007; Sandoval et al., 2008; Novak et al., 2010). In line with this, NIX knockout mice develop anemia and reticulocytosis (Schweers et al., 2007; Sandoval et al., 2008). The interaction of NIX with LC3 protein members is mediated via the LC3-interacting (LIR) motif, however, re-expression of LIR-mutant NIX in NIX deficient reticulocytes partially rescued the observed phenotype, indicating LC3-independent or even autophagy-independent mechanisms for mitochondrial clearance in reticulocyte differentiation (Novak et al., 2010). Another LIR-motif containing protein, FUNDC1, regulates mitophagy under hypoxic conditions by promoting mitochondrial fission (Liu et al., 2012; Chen et al., 2016). During cardiac progenitor cell differentiation, FUNDC1 and NIX, but not Pink1 and Parkin, are upregulated to maintain a functional mitochondrial network (Lampert et al., 2019). Mitophagy is therefore also regulated in a lineage dependent fashion.
Localized removal of mitochondrial subdomains can be mediated by piecemeal mitophagy or mitochondrial-derived vesicles (Figure 1C). Mitochondrial-derived vesicle formation is thought to be dependent on PINK1/Parkin but independent of the canonical autophagy machinery (Soubannier et al., 2012; McLelland et al., 2014). Whereas, the accumulation of misfolded mitochondrial protein aggregates leads to localized recruitment of Parkin and autophagy proteins, thereby facilitating the degradation of mitochondrial subdomains (Burman et al., 2017). A PINK1/Parkin-independent piecemeal mitophagy has been recently reported that drives LC3C- and p62-mediated degradation of mitochondrial subregions (Le Guerroué et al., 2017). However, the protein machinery for these mitochondrial degradation pathways may overlap with the classic mitophagy pathways as well as their physiological relevance needs to be further investigated.
Mitophaging
A decline in mitochondrial function is a hallmark of the aging process and is connected to other aging hallmarks such as telomere dysfunction, genome instability and cellular senescence. However, it remains largely unclear how these processes are interconnected and finally provoke disruption of the cellular and tissue integrity (López-Otín et al., 2013). There is accumulating evidence that mitophagy impacts health- and lifespan in different model organisms. Using a transgenic mouse strain that expresses the fluorescent mitophagy reporter mt-Keima, a decreased mitophagy level was observed in the hippocampal dentate gyrus in 21-month old mice compared to 3-month old mice (Sun et al., 2015). A decline in mitophagy was also observed in aged mouse hearts, in line with this, altered mitophagy has been shown to influence different cardiac pathologies (Hoshino et al., 2013; Bravo-San Pedro et al., 2017). Other tissues that contribute to aging phenotypes are also characterized by defective mitophagy, as shown recently for aged skeletal muscle satellite cells isolated from humans or mice (García-Prat et al., 2016). Notably, decreased expression of mitophagy genes was observed in the skeletal muscle of physically inactive elderly women (Drummond et al., 2014).
The effect of changes in mitophagy on health- and lifespan has been particularly demonstrated by using the model organisms C. elegans and D. melanogaster. Several genetic studies in D. melanogaster revealed that the overexpression of mitochondrial and mitophagy genes leads to increased health- and/or lifespan. For instance, the overexpression of the mitochondrial fission protein dynamin-related protein 1 (DRP1) increased the lifespan along with a prolonged healthspan in flies (Rana et al., 2017). The importance of mitochondrial fission on drosophila lifespan was further demonstrated by the observation that lifespan extension caused by the overexpression of p62 was abrogated in DRP1 mutant flies (Aparicio et al., 2019). Lifespan extension in flies was also observed after overexpression of Parkin and Pink1, whereby, Parkin overexpression counteracted increased Mfn2 levels, which can be observed during aging (Todd and Staveley, 2012; Rana et al., 2013). These findings are consistent with studies in C. elegans, where mitophagy has been shown to contribute to lifespan regulation (Palikaras et al., 2015; Schiavi et al., 2015). Evidently, there is substantial data supporting a role of declining mitophagy, mitophaging, in aging.
What Happens When Mitophagy Goes Wrong?
Impaired mitophagy contributes to the pathogenesis of several human diseases, in particular to age-related sporadic disorders, such as Parkinson’s disease, Alzheimer’s disease, cardiomyopathies and cancer (Bernardini et al., 2017; Fivenson et al., 2017; Levine and Kroemer, 2019). While these observations yield interesting correlations between certain disease states and alterations in mitophagy it is difficult to deduct causation. Here, monogenic diseases with specific defects in mitophagy may give us mechanistic understanding of pathogenesis and biology (Table 1). Thus, monogenic disorders may provide valuable tools for studying molecular pathomechanisms that are driven by defective mitophagy. To explore the clinical phenotype of autophagy diseases, we identified the clinical descriptions in the literature of all the diseases in Table 1 and performed hierarchical clustering based on the prevalence of those features (Figure 2A; Scheibye-Knudsen et al., 2013; Andreassen et al., 2019). Although the clustering did connect clinically similar diseases (such as Charcot–Marie–Tooth 2A2 and 2B), it became immediately apparent that there is no good correlation between clinical outcome and the putative molecular function of the gene responsible for the disease. Indeed, principal component analysis also did not show any obvious separation of clinical groups based on proposed molecular functions (Figure 2B). This suggests that our knowledge of the pathogeneses of most of these disorders is quite limited. Nevertheless, when looking at the average prevalence of clinical features across all aging diseases there was a considerable overrepresentation of neurological features suggesting that defects in autophagy often leads to brain disease (Figure 2C). More specifically, the phenotype in the autophagy disorders show significant overlap with what is seen in mitochondrial diseases indicating that mitochondrial dysfunction may be driving diseases in many autophagy-related disorders ranging from lysosomal diseases to bonafide mitophagy deficiencies (Figures 2C,D). In the following we will examine a few key examples of these disorders.
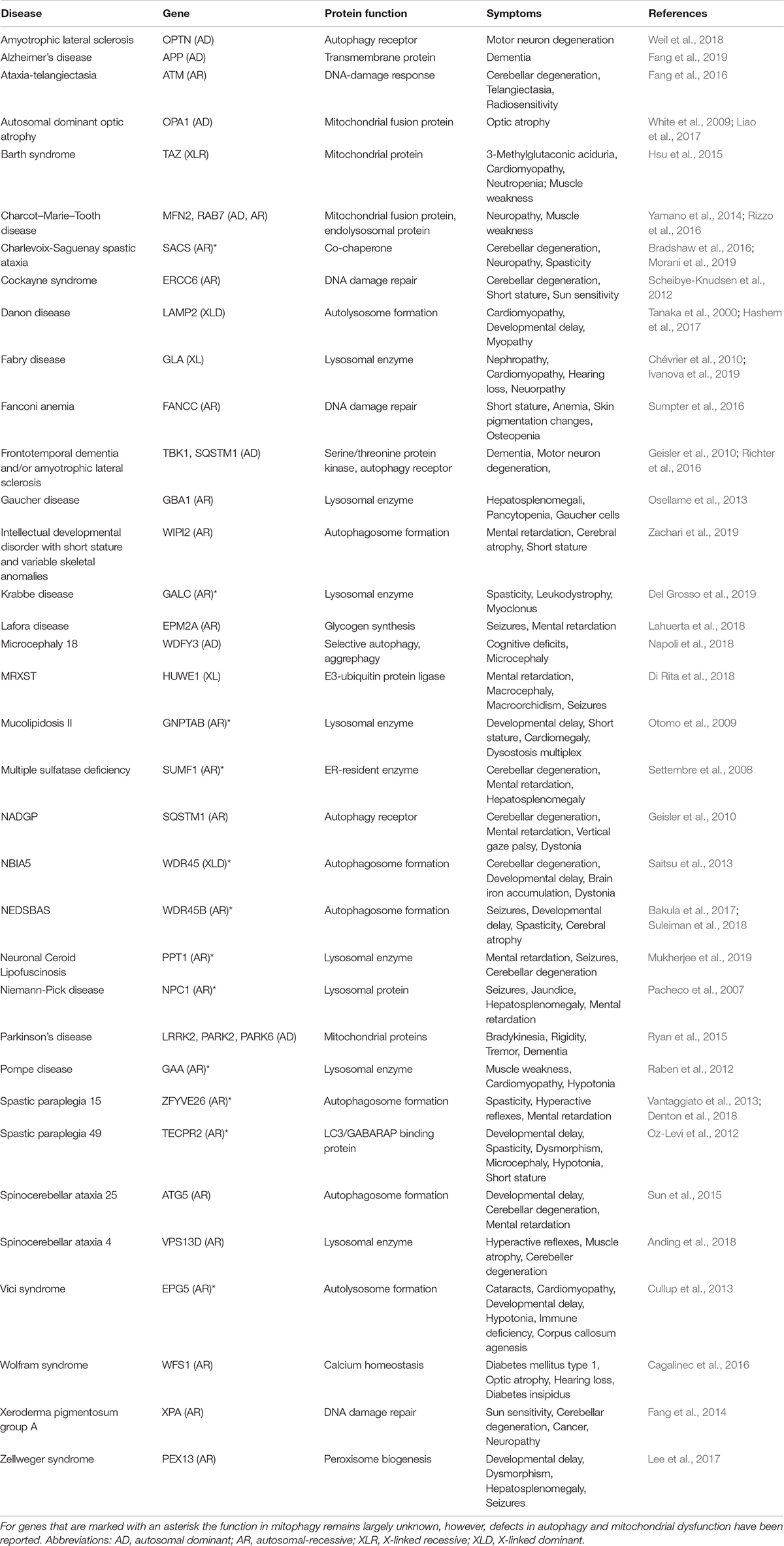
Table 1. Examples of autophagy/mitophagy-related monogenic disorders.
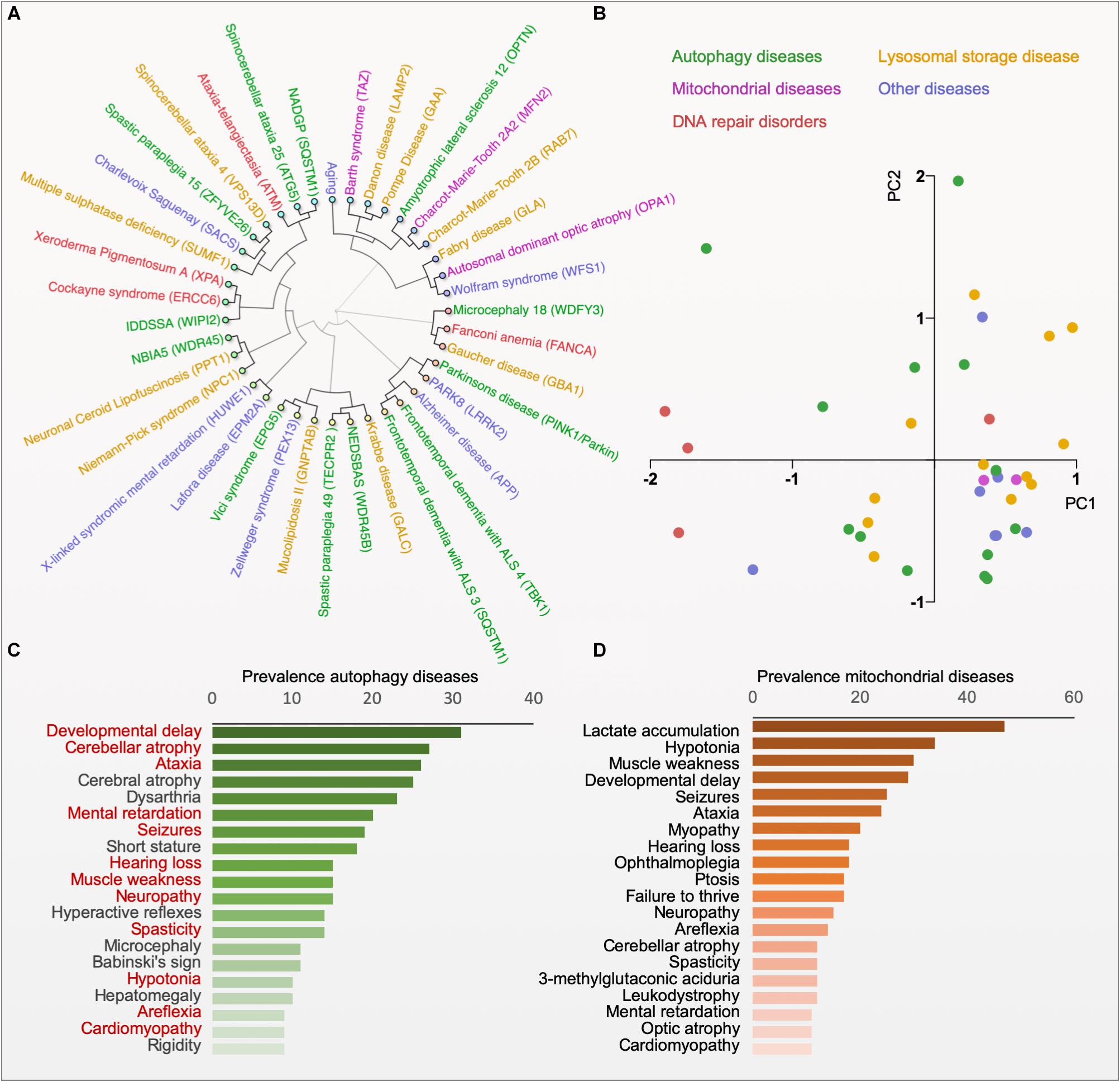
Figure 2. Phenotype clustering of autophagy diseases. (A) Hierarchical clustering of diseases based on the published prevalence of clinical features in the diseases (for data and references see www.mitodb.com). (B) Principal component analysis of diseases based on the prevalence of clinical features. (C) The average prevalence of top-20 clinical features in all autophagy-related disorders (Red, shared with the top-20 features in mitochondrial disorders). (D) The average prevalence of clinical features in mitochondrial diseases.
Defects in the Autophagic Machinery
To date, only a few monogenic diseases caused by single mutations in the autophagy core machinery have been reported. One of them, spinocerebellar ataxia-25 (SCAR25), is caused by a mutation in the autophagy-related 5 gene (ATG5), encoding a protein that is part of the ATG12-ATG5-ATG16L1 complex, which facilitates LC3/GABARAP conjugation (Mizushima, 2020). So far, two siblings have been identified with SCAR25, presenting with clinical symptoms such as truncal ataxia and intellectual disability (Kim et al., 2016). In line with the neurological phenotypes, a neuron-specific knockout of ATG5 in mice causes neuronal degeneration, by contrast, a complete ATG5 knockout is neonatal lethal (Kuma et al., 2004; Hara et al., 2006). Ataxia is a common feature of many mitochondrial disorders (Scheibye-Knudsen et al., 2013), however, mitochondrial viability in SCAR25 has not been investigated so far. Thus, the contribution of mitochondrial defects to the reported clinical features in SCAR25 remains speculative, since ATG5-independent mitophagy pathways have been reported (Honda et al., 2014; Hirota et al., 2015).
Mutations in members of the human WD-repeat protein interacting with phosphoinositides (WIPI) family are known to cause neurological deficits. The WIPI protein family consists of four members, WIPI1–WIPI4, that contribute to the early steps of autophagosome formation (Proikas-Cezanne et al., 2004). The family member WIPI2 localizes in a phosphatidylinositol 3-phosphate-dependent manner to the autophagosomal membrane, where it facilitates ATG16L1 recruitment and LC3 lipidation (Dooley et al., 2014; Bakula et al., 2017). Recently, patients with mutations in the WIPI2 gene have been described with multisystemic clinical features, primarily, neurological and skeletal deficiencies that are characterized by severe mental retardation and short stature (Jelani et al., 2019). Notably, WIPI2 overexpression prevents age-related autophagy decline in dorsal root ganglion neurons (Stavoe et al., 2019). Patients with mutations in the genes WIPI3 (WDR45B) or WIPI4 (WDR45) show severe and progressive neurodegenerative phenotypes (Haack et al., 2012; Hayflick et al., 2013; Saitsu et al., 2013; Suleiman et al., 2018). Notably, WIPI4 mutations result in degeneration of the substantia nigra, a target area of the brain affected in Parkinson’s disease (Mann et al., 1992). In line with these observations, WIPI3 or WIPI4 knockout mice show neurological defects, possibly caused by defective neuronal autophagy (Zhao et al., 2015; Ji et al., 2019). WIPI3 and −4 knockout mice display mitochondrial dysmorphology, which was also evident in WIPI4 mutant human fibroblast cells (Zhao et al., 2015; Seibler et al., 2018; Ji et al., 2019). The patient phenotypes caused by mutations in the WIPI genes highlight the importance of the WIPI protein members for neuronal function, however, the contribution of WIPI-mediated clearance of mitochondria in neurodegeneration remains unclear.
Deficiency in the late stage of autophagy is observed in the autosomal recessive neurological disorder, Vici syndrome. The disease is caused by mutations in the ectopic P-granules autophagy protein 5 gene (EPG5), encoding for a Rab7 effector protein that is required for the fusion of late autophagosomes with lysosomes (Cullup et al., 2013; Wang et al., 2016). The disease is characterized by multisystemic defects that show some overlap with mitochondrial diseases, such as agenesis of corpus callosum, cardiomyopathy, immunodeficiency, cataracts and hypopigmentation (Cullup et al., 2013). Mitochondria with abnormal shape and distribution were observed in muscle tissue biopsies from patients with Vici syndrome or EPG5 knockout mice (Cullup et al., 2013; Zhao et al., 2013). The importance of EPG5 in mitochondrial homeostasis was further highlighted by a study showing deficient mitochondrial clearance during spermatogenesis in an EPG5-deficient medaka fish line (Herpin et al., 2015).
Cargo recognition and degradation in selective autophagy is mediated by autophagy receptor proteins, such as optineurin and p62. Both proteins are associated with the progressive neurological disorder amyotrophic lateral sclerosis (ALS), which is primarily caused by loss of motor neurons (Maruyama et al., 2010; Fecto et al., 2011). Around 10% of ALS cases are caused by inherited single gene mutations and frequently show comorbidity with frontotemporal dementia (FTD). Interestingly, optineurin and p62 are phosphorylated by tank-binding kinase 1 (TBK1), a serine/threonine kinase that has also been implicated in ALS-FTD disease development (Cirulli et al., 2015; Freischmidt et al., 2015; Pottier et al., 2015). Thus, there is a striking correlation with mutations in multiple mitophagy players leading to ALS.
Defects in Mitochondrial Quality Control
Proteins involved in the regulation of mitochondrial quality control are essential modulators of mitophagy, consequently, understanding their molecular mechanisms may give important insights into the consequences of impaired mitophagy. In recent years, mitochondrial dysfunction has been extensively discussed as an important contributor to neurodegeneration in familial Parkinson’s disease, as well as in idiopathic forms (Bose and Beal, 2016). Early onset recessive familial Parkinson’s disease can be caused by mutations in the genes Park2 (Parkin), Park6 (Pink1), or Park7 (DJ-1). All three proteins localize to mitochondria and loss of each of them leads to increased sensitivity toward oxidative stress along with mitochondrial and energetic dysfunction (Dodson and Guo, 2007). Pink1 and Parkin are directly involved in the mitophagy pathway, whereas, the precise function of DJ-1 remains under discussion. Interestingly, overexpression of Pink1 and Parkin rescues the observed phenotype caused by DJ-1 deficiency, suggesting partial redundancies in the mitophagic apparatus (Irrcher et al., 2010).
Mitochondrial fission and fusion are critical events for controlled degradation of damaged mitochondria. Optic atrophy 1 (OPA1) is an inner mitochondrial membrane protein that regulates the fusion of mitochondria, together with MFN1 and MFN2. Mutation in the OPA1 gene has been observed to cause autosomal dominant optic atrophy (ADOA) often accompanied by myopathy and progressive ataxia (Yu-Wai-Man et al., 2010). Myopathy and neurodegeneration is also observed in patients with Charcot–Marie–Tooth syndrome caused by loss of the MFN2 gene (Calvo et al., 2009), underscoring the importance of mitochondrial function in muscle and brain tissues. For both diseases impaired mitophagy has been reported, suggesting that dysfunctional mitophagy may contribute to the described disease pathology (White et al., 2009; Rizzo et al., 2016; Liao et al., 2017).
Defects in Lysosomal Function
Another group of diseases that may be partial driven by deficient mitophagy, are lysosomal storage disorders, a heterogenous group of more than 60 rare monogenic diseases that are caused by defects in lysosomal function (Platt et al., 2018). Some of the most well described are Gaucher disease and Niemann–Pick type C. Gaucher disease is caused by mutations in the glucocerebrosidase (GBA) gene, encoding a lysosomal enzyme required to hydrolyze the glycolipid glucosylceramide. Patients with Gaucher disease display features in multiple organs caused by lysosomal accumulation of glucosylceramide with a subset of patients display progressive neurodegeneration. Notably, the GBA gene represents a major risk locus for inherited Parkinson’s disease supporting the idea that mitophagy is important in this disease (Goker-Alpan et al., 2004; Lwin et al., 2004). Reduced mitochondrial respiration, increased ROS production and increased alpha-synuclein accumulation can be observed in various GBA deficiency models, cellular changes that are also described to be central drivers of neuronal loss in Parkinson’s disease (Osellame et al., 2013; Chen et al., 2019). Nieman Pick type C is caused by mutations in the NPC1 gene and is characterized by developmental delay, progressive neurodegeneration, dysphagia and vertical gaze palsy, a combination of phenotypes that can also be observed in mitochondrial disorders. In patient-derived fibroblast cells and NPC1-deficient neuronal cells impaired autophagy and an accumulation of mitochondrial fragments have been observed upon lysosomal cholesterol accumulation (Pacheco et al., 2007; Elrick et al., 2012; Ordonez et al., 2012).
Secondary Defects in Mitophagy
In addition to diseases with primary defects in mitophagy, several diseases have been described with secondary mitophagic dysfunction. In the context of monogenic diseases displaying premature aging, loss of mitophagy was first described in Cockayne syndrome, a disease characterized by progressive neurodegeneration reminiscent of mitochondrial disorders (Scheibye-Knudsen et al., 2012). The pathogenesis likely involves dysregulation of uncoupling proteins (U) due to decreased activity of the PGC-1alpha transcription factor. UCPs regulate mitochondrial membrane potential and consequently a reduction in UCPs lead to increased mitochondrial membrane potential and loss of PINK1 mediated mitophagy. Accordingly, overexpression of UCP2 can rescue mitochondrial and mitophagic defects in Cockayne syndrome. Notably, the same pathogenesis is found in related DNA repair disorders xeroderma pigmentosum group A and ataxia-telangiectasia (Fang et al., 2014, 2016).
Another disease that is characterized by mitochondrial deficiency is Zellweger syndrome, which belongs to a subgroup of peroxisome biogenesis disorders (Salpietro et al., 2015). Zellweger syndrome is caused by mutations in one of 14 human PEX genes, encoding for peroxin proteins that are required for the maintenance of peroxisomes (Waterham and Ebberink, 2012). Zellweger syndrome patients show dysmorphic features and suffer from severe neurological symptoms. Recently, PEX13 was shown to be required for mitophagy, but interestingly, dispensable for starvation-induced autophagy (Lee et al., 2017). Similarly, PEX5, an interaction partner of PEX13, has been shown to modulate autophagy via regulation of the mTOR signaling pathway (Eun et al., 2018), in line with this, mitochondrial defects can be observed in PEX5 knockout models (Baumgart et al., 2001). However, it is still unclear, to what extent the clinical features of Zellweger syndrome are driven by mitophagic defects.
Is Mitophagy a Therapeutic Target?
An increasing number of human diseases have been associated with impaired mitophagy, thus, interventions that modulate mitophagy may provide the possibility of counteracting disease development or progression (Figure 3). In recent years, multiple small molecules as well as lifestyle interventions have been shown to modulate autophagy, thereby causing health- and lifespan benefits in different organisms (Galluzzi et al., 2017). Due to the dependency on core autophagy regulators, mitophagy is modulated by most of the classic autophagy inducers such as the mTOR inhibitor rapamycin, the AMP-activated protein kinase (AMPK) activator AICAR as well as caloric restriction and exercise. In particular, the effectiveness of rapamycin and rapalogs has been intensively studied in the context of lifespan regulation and human disease development and rapamycin remain the most well documented compound for life- and healthspan extension in laboratory animals (Saxton and Sabatini, 2017). Further connections between longevity and mitophagy comes from work on the metabolite NAD+ and the NAD+-dependent acetylase Sirtuin 1 (SIRT1). Here, it has been shown that stimulation of SIRT1 through NAD+ augmentation or small molecules leads to activation of the energy responsive kinase AMPK that in turns regulates a central autophagy regulator, Unc-51-like kinase 1 (ULK1) (Egan et al., 2011; Price et al., 2012). Further, SIRT1 and AMPK also regulate the transcription factor PGC-1alpha, a key regulator of mitochondrial function that was initially found to control UCP levels and thereby mitochondrial membrane potential (Puigserver et al., 1998; Cantó et al., 2009). Indeed, SIRT1 activation leads to UCP-2 upregulation, stimulation of mitophagy and rescue of aging features in models of premature aging (Fang et al., 2014; Scheibye-Knudsen et al., 2014). Notably, direct stimulation of AMPK through the AMP-mimetic compound AICAR regulates mitochondrial dynamics via the induction of mitochondrial fission, further highlighting the broad effect of AMPK on mitochondrial function (Toyama et al., 2016).
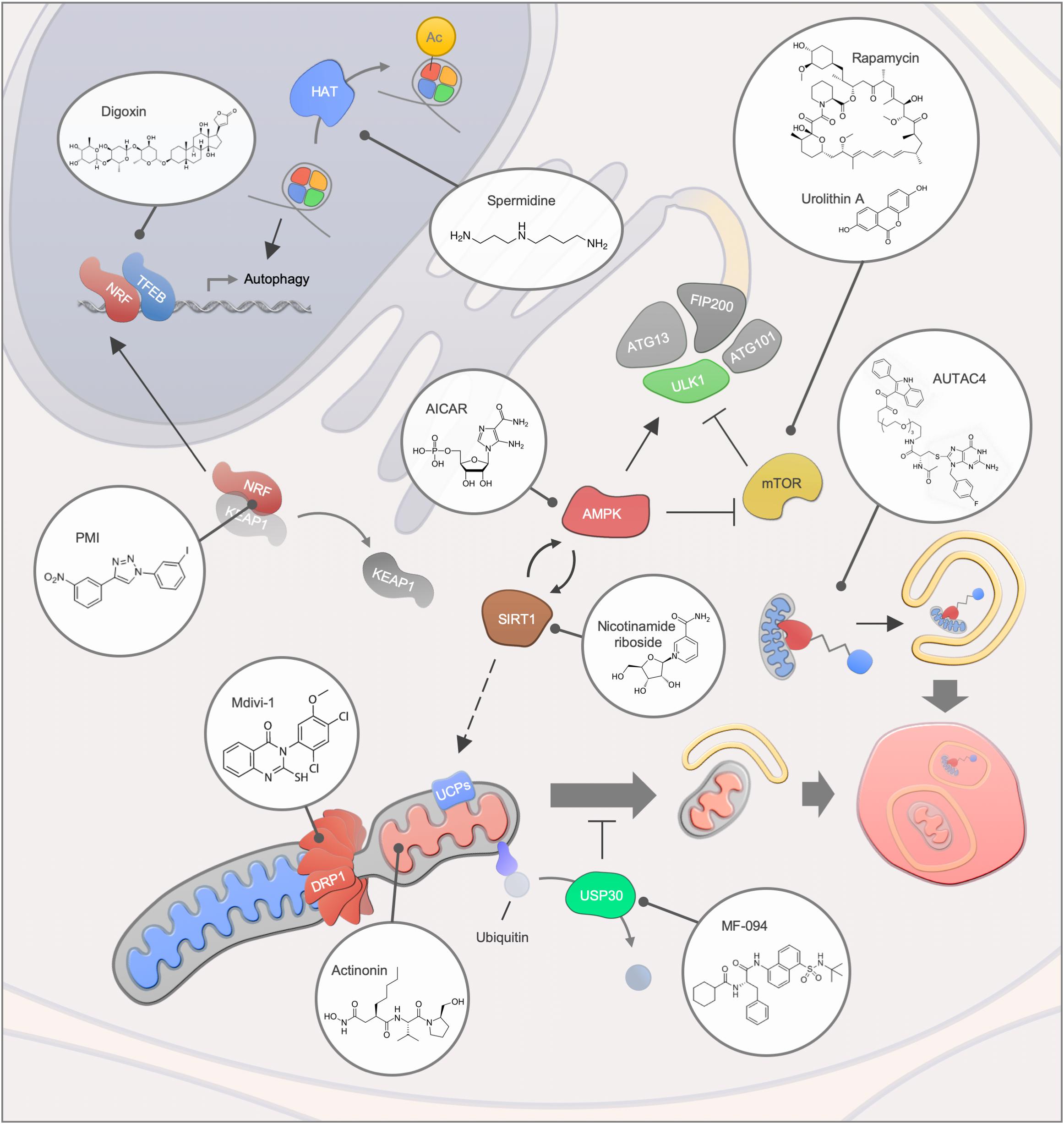
Figure 3. Mitophagy interventions. An overview of different mitophagy modulating compounds and their targets. Abbreviations: Ac, Acetylation; HAT, Histone acetyltransferase.
Due to their great diversity, natural compounds are a tremendous source for novel mitophagy modulators. Urolithin A, a gut metabolite of ellagic acid, extends health- and lifespan in C. elegans as well as improving muscle function in rodent models via the induction of mitophagy (Ryu et al., 2016). The effectiveness of urolithin A was further highlighted in animal models of Alzheimer’s disease, where the disease pathology was ameliorated in the group of urolithin A-treated mice (Fang et al., 2019). In a human clinical trial study, the safety of urolithin A was evaluated, and signatures of improved mitochondrial function were demonstrated (Andreux et al., 2019). Similar to Urolithin A, the potency of antibacterial compound actinonin was demonstrated in Alzheimer’s disease models (Fang et al., 2019). Actinonin inhibits mitochondrial translation, thereby inducing mitophagy via the activation of the PINK1/Parkin-regulated signaling pathway (Richter et al., 2013; Sun et al., 2015; Burman et al., 2017). Another natural compound that has been suggested as a potential intervention for aging and diseases is the polyamine spermidine (Eisenberg et al., 2009; Madeo et al., 2018; Schwarz et al., 2018). The administration of spermidine leads to an induction of mitophagy in cardiomyocytes, along with cardio protection in mice (Eisenberg et al., 2016). The induction of autophagy via spermidine has been associated, among others, with the inhibition of the acetyltransferase EP300 and the ATM-driven activation of the PINK1/Parkin-regulated mitophagy pathway (Pietrocola et al., 2015; Qi et al., 2016).
Transcriptional regulation of mitophagy has also been shown as a viable pathway for increased mitochondrial health. An example is the synthetic compound PMI that stimulates mitophagy via the activation of the transcription factor Nrf2, which controls the expression of mitophagy genes including p62 (East et al., 2014; Bertrand et al., 2015). PMI treatment facilitates LC3 recruitment and mitochondrial ubiquitination in a PINK1/Parkin-independent manner, notably without disrupting the mitochondrial membrane potential (East et al., 2014).
Besides targeting mitophagy core proteins, intervention strategies targeting mitochondrial proteins may present a useful approach for disorders that are characterized by abnormal mitochondrial dynamic. Mdivi-1, has been identified in a yeast screen for mitochondrial fission inhibitors and several studies indicate its therapeutic potential for the treatment of neurological disorders (Cassidy-Stone et al., 2008; Cui et al., 2010; Solesio et al., 2012). However, the specificity of Mdivi-1 toward its putative target Drp1 has recently been questioned and needs to be further clarified (Bordt et al., 2017). USP30, a deubiquitinase that targets mitochondrial proteins, may present another promising target to facilitate mitophagy, since improved mitochondrial function was obtained upon USP30 depletion in different Parkinson’s disease models (Bingol et al., 2014). Notably, MF-094 has been recently identified as a selective inhibitor of USP30 that may thereby facilitate mitophagy through increased ubiquitination of outer membrane proteins (Kluge et al., 2018). Thus, a number of mitophagy modulators have been identified, yet the main goal will be the precise and specific targeting of damaged mitochondria. One possible way is to apply chimeric molecules such as the recently generated autophagy-targeting chimeric molecule (AUTAC4) that selectively targets the mitochondrial membrane for ubiquitination and subsequent degradation (Takahashi et al., 2019). These approaches may be particularly efficacious in conditions of mitophaging where the mitophagy apparatus is likely intact but mitophagy occurs at suboptimal levels.
In diseases characterized by dysfunctional lysosomes, stimulation of mitophagy may be detrimental due to an accumulation of undigested cargo material. In this regard, the inhibition of mitophagy is considered as a therapeutic strategy. In a mouse model of Pompe disease autophagy inhibition next to an enzyme replacement therapy has been proposed as a potential intervention (Raben et al., 2010). In line with this, knockdown of the mTOR pathway inhibitor TSC2 in muscle of Pompe disease mice reduced accumulation of autophagy markers and a decline in muscle atrophy was osberved (Lim et al., 2017). However, strategies to facilitate the fusion of autophagosomes and lysosomes in lysosomal storage disorders are also proposed for the treatment of several lysosomal storage disorders (Spampanato et al., 2013; Bartolomeo et al., 2017). TFEB, which controls the expression of autophagy as well as lysosomal genes and longevity (Napolitano and Ballabio, 2016), may provide a promising target since its agonists, such as the clinically approved cardiac drug digoxin or the natural compound ikarugamycin, improve metabolic function in mice and extend lifespan in C. elegans (Wang et al., 2017). The therapeutic potential of TFEB in Parkinson’s disease was further highlighted by a recent study that showed restored TFEB and improved neurological function upon rapamycin treatment in Q311X mutant parkin mice independently of the parkin E3 ligase (Siddiqui et al., 2015).
In summary, great progress has been made in recent years, however, the clinical safety of mitophagy modulating drugs needs to be further clarified. More refined tools that allow the distinction between mitophagy and general macroautophagy may be beneficial and could accelerate future discoveries. Altogether, this will enable us to step closer toward clinical validation of mitophagy modulators.
Concluding Remarks
Mitophagy is emerging as a central process preserving organismal and, especially, neurological health. Since most trials targeting age-associated neurodegeneration in the last decades have been disappointing, new pharmaceutical avenues are direly needed. Here, mitophagy stimulators could play a key role. Indeed, several clinical trials are underway testing the efficacy of mitophagy modulating compounds and the outcome of these studies will undoubtedly prove critical for the future translatability of the field. Nonetheless, the regulatory mechanism of mitophagy and its contribution to age-associated diseases still remains elusive and potential issues with artificially augmenting mitophagy have not been considered. However, given the central role of mitophaging in multiple age-related pathologies it appears highly likely that these new promising approaches may present possible interventions in age-associated diseases. The future is bright!
Author Contributions
DB and MS-K wrote the manuscript and made the figures.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past collaboration with one of the authors, MS-K.
Funding
DB is supported by the German Research Foundation (Forschungsstipendium; BA 6276/1-1) and the Lundbeck foundation (# R303-2018-3159). MS-K is funded through the Nordea Foundation (#02-2017-1749), the Novo Nordisk Foundation (#NNF17OC0027812), the Danish Cancer Society (#R167-A11015_001), the Independent Research Fund Denmark (#7016-00230B), the Neye Foundation, the Lundbeck Foundation (#R324-2019-1492) and Insilico Medicine.
Acknowledgments
We would like to express our deep gratitude to Dr. Brenna Osborne for her critical comments to our manuscript.
References
Anding, A. L., Wang, C., Chang, T.-K., Sliter, D. A., Powers, C. M., Hofmann, K., et al. (2018). Vps13D encodes a ubiquitin-binding protein that is required for the regulation of mitochondrial size and clearance. Curr. Biol. CB 28:287–295.e6. doi: 10.1016/j.cub.2017.11.064
Andreassen, S. N., Ben Ezra, M., and Scheibye-Knudsen, M. (2019). A defined human aging phenome. Aging 11, 5786–5806. doi: 10.18632/aging.102166
Andreux, P. A., Blanco-Bose, W., Ryu, D., Burdet, F., Ibberson, M., Aebischer, P., et al. (2019). The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 1, 595–603. doi: 10.1038/s42255-019-0073-4
Aparicio, R., Rana, A., and Walker, D. W. (2019). Upregulation of the autophagy adaptor p62/SQSTM1 prolongs health and lifespan in middle-aged drosophila. Cell Rep. 28:1029–1040.e5. doi: 10.1016/j.celrep.2019.06.070
Bakula, D., Müller, A. J., Zuleger, T., Takacs, Z., Franz-Wachtel, M., Thost, A.-K., et al. (2017). WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat. Commun. 8:15637. doi: 10.1038/ncomms15637
Bartolomeo, R., Cinque, L., De Leonibus, C., Forrester, A., Salzano, A. C., Monfregola, J., et al. (2017). mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J. Clin. Invest. 127, 3717–3729. doi: 10.1172/JCI94130
Baumgart, E., Vanhorebeek, I., Grabenbauer, M., Borgers, M., Declercq, P. E., Fahimi, H. D., et al. (2001). Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am. J. Pathol. 159, 1477–1494. doi: 10.1016/S0002-9440(10)62534-5
Bernardini, J. P., Brouwer, J. M., Tan, I. K., Sandow, J. J., Huang, S., Stafford, C. A., et al. (2019). Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. 38:e99916. doi: 10.15252/embj.201899916
Bernardini, J. P., Lazarou, M., and Dewson, G. (2017). Parkin and mitophagy in cancer. Oncogene 36, 1315–1327. doi: 10.1038/onc.2016.302
Bertrand, H. C., Schaap, M., Baird, L., Georgakopoulos, N. D., Fowkes, A., Thiollier, C., et al. (2015). Design, synthesis, and evaluation of triazole derivatives that induce Nrf2 dependent gene products and inhibit the Keap1-Nrf2 protein-protein interaction. J. Med. Chem. 58, 7186–7194. doi: 10.1021/acs.jmedchem.5b00602
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Bordt, E. A., Clerc, P., Roelofs, B. A., Saladino, A. J., Tretter, L., Adam-Vizi, V., et al. (2017). The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 40:583–594.e6. doi: 10.1016/j.devcel.2017.02.020
Bose, A., and Beal, M. F. (2016). Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 139(Suppl. 1), 216–231. doi: 10.1111/jnc.13731
Bouman, L., Schlierf, A., Lutz, A. K., Shan, J., Deinlein, A., Kast, J., et al. (2011). Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death. Differ 18, 769–782. doi: 10.1038/cdd.2010.142
Bradshaw, T. Y., Romano, L. E. L., Duncan, E. J., Nethisinghe, S., Abeti, R., Michael, G. J., et al. (2016). A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 25, 3232–3244. doi: 10.1093/hmg/ddw173
Bravo-San Pedro, J. M., Kroemer, G., and Galluzzi, L. (2017). Autophagy and mitophagy in cardiovascular disease. Circ. Res. 120, 1812–1824. doi: 10.1161/CIRCRESAHA.117.311082
Burman, J. L., Pickles, S., Wang, C., Sekine, S., Vargas, J. N. S., Zhang, Z., et al. (2017). Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 216, 3231–3247. doi: 10.1083/jcb.201612106
Cagalinec, M., Liiv, M., Hodurova, Z., Hickey, M. A., Vaarmann, A., Mandel, M., et al. (2016). Role of mitochondrial dynamics in neuronal development: mechanism for wolfram syndrome. PLoS Biol. 14:e1002511. doi: 10.1371/journal.pbio.1002511
Calvo, J., Funalot, B., Ouvrier, R. A., Lazaro, L., Toutain, A., De Mas, P., et al. (2009). Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch. Neurol. 66, 1511–1516. doi: 10.1001/archneurol.2009.284
Calvo, S. E., Compton, A. G., Hershman, S. G., Lim, S. C., Lieber, D. S., Tucker, E. J., et al. (2012). Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 4:118ra10. doi: 10.1126/scitranslmed.3003310
Cantó, C., Gerhart-Hines, Z., Feige, J. N., Lagouge, M., Noriega, L., Milne, J. C., et al. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060. doi: 10.1038/nature07813
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204. doi: 10.1016/j.devcel.2007.11.019
Chen, C., Turnbull, D. M., and Reeve, A. K. (2019). Mitochondrial dysfunction in Parkinson’s disease-cause or consequence? Biology 8:20038. doi: 10.3390/biology8020038
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, Y., and Dorn, G. W. (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Chen, Y., Liu, Y., and Dorn, G. W. (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331. doi: 10.1161/CIRCRESAHA.111.258723
Chévrier, M., Brakch, N., Céline, L., Genty, D., Ramdani, Y., Moll, S., et al. (2010). Autophagosome maturation is impaired in Fabry disease. Autophagy 6, 589–599. doi: 10.4161/auto.6.5.11943
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., Flint Beal, M., and Wallace, D. C. (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 2, 324–329. doi: 10.1038/ng1292-324
Cortopassi, G. A., and Arnheim, N. (1990). Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 18, 6927–6933. doi: 10.1093/nar/18.23.6927
Cui, H., Li, F., Chen, D., Wang, G., Truong, C. K., Enns, G. M., et al. (2013). Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet. Med. Off. J. Am. Coll. Med. Genet. 15, 388–394. doi: 10.1038/gim.2012.144
Cui, M., Tang, X., Christian, W. V., Yoon, Y., and Tieu, K. (2010). Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 285, 11740–11752. doi: 10.1074/jbc.M109.066662
Cullup, T., Kho, A. L., Dionisi-Vici, C., Brandmeier, B., Smith, F., Urry, Z., et al. (2013). Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 45, 83–87. doi: 10.1038/ng.2497
De Duve, C., and Wattiaux, R. (1966). Functions of lysosomes. Annu. Rev. Physiol. 28, 435–492. doi: 10.1146/annurev.ph.28.030166.002251
Deas, E., Plun-Favreau, H., Gandhi, S., Desmond, H., Kjaer, S., Loh, S. H. Y., et al. (2011). PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879. doi: 10.1093/hmg/ddq526
Del Grosso, A., Angella, L., Tonazzini, I., Moscardini, A., Giordano, N., Caleo, M., et al. (2019). Dysregulated autophagy as a new aspect of the molecular pathogenesis of Krabbe disease. Neurobiol. Dis. 129, 195–207. doi: 10.1016/j.nbd.2019.05.011
Denton, K., Mou, Y., Xu, C.-C., Shah, D., Chang, J., Blackstone, C., et al. (2018). Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum. Mol. Genet. 27, 2517–2530. doi: 10.1093/hmg/ddy156
Di Rita, A., Peschiaroli, A., Acunzo, P. D., Strobbe, D., Hu, Z., Gruber, J., et al. (2018). HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun. 9:3755. doi: 10.1038/s41467-018-05722-3
Dodson, M. W., and Guo, M. (2007). Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr. Opin. Neurobiol. 17, 331–337. doi: 10.1016/j.conb.2007.04.010
Dooley, H. C., Razi, M., Polson, H. E. J., Girardin, S. E., Wilson, M. I., and Tooze, S. A. (2014). WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 55, 238–252. doi: 10.1016/j.molcel.2014.05.021
Drummond, M. J., Addison, O., Brunker, L., Hopkins, P. N., McClain, D. A., LaStayo, P. C., et al. (2014). Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison. J. Gerontol. A. Biol. Sci. Med. Sci. 69, 1040–1048. doi: 10.1093/gerona/glu004
East, D. A., Fagiani, F., Crosby, J., Georgakopoulos, N. D., Bertrand, H., Schaap, M., et al. (2014). PMI: a ΔΨm independent pharmacological regulator of mitophagy. Chem. Biol. 21, 1585–1596. doi: 10.1016/j.chembiol.2014.09.019
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Eid, N., Ito, Y., Horibe, A., Otsuki, Y., and Kondo, Y. (2019). Ethanol-induced mitochondrial damage in sertoli cells is associated with parkin overexpression and activation of mitophagy. Cells 8:30283. doi: 10.3390/cells8030283
Eisenberg, T., Abdellatif, M., Schroeder, S., Primessnig, U., Stekovic, S., Pendl, T., et al. (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438. doi: 10.1038/nm.4222
Eisenberg, T., Knauer, H., Schauer, A., Büttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314. doi: 10.1038/ncb1975
Elrick, M. J., Yu, T., Chung, C., and Lieberman, A. P. (2012). Impaired proteolysis underlies autophagic dysfunction in Niemann–Pick type C disease. Hum. Mol. Genet. 21, 4876–4887. doi: 10.1093/hmg/dds324
Eun, S. Y., Lee, J. N., Nam, I.-K., Liu, Z.-Q., So, H.-S., Choe, S.-K., et al. (2018). PEX5 regulates autophagy via the mTORC1-TFEB axis during starvation. Exp. Mol. Med. 50:4. doi: 10.1038/s12276-017-0007-8
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Fang, E. F., Kassahun, H., Croteau, D. L., Scheibye-Knudsen, M., Marosi, K., Lu, H., et al. (2016). NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581. doi: 10.1016/j.cmet.2016.09.004
Fang, E. F., Scheibye-Knudsen, M., Brace, L. E., Kassahun, H., SenGupta, T., Nilsen, H., et al. (2014). Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157, 882–896. doi: 10.1016/j.cell.2014.03.026
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Fivenson, E. M., Lautrup, S., Sun, N., Scheibye-Knudsen, M., Stevnsner, T., Nilsen, H., et al. (2017). Mitophagy in neurodegeneration and aging. Neurochem. Int. 109, 202–209. doi: 10.1016/j.neuint.2017.02.007
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Galluzzi, L., Pedro, J. M. B.-S., Levine, B., Green, D. R., and Kroemer, G. (2017). Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 16, 487–511. doi: 10.1038/nrd.2017.22
García-Prat, L., Martínez-Vicente, M., Perdiguero, E., Ortet, L., Rodríguez-Ubreva, J., Rebollo, E., et al. (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. doi: 10.1038/nature16187
Gegg, M. E., Cooper, J. M., Chau, K.-Y., Rojo, M., Schapira, A. H. V., and Taanman, J.-W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Goker-Alpan, O., Schiffmann, R., LaMarca, M. E., Nussbaum, R. L., McInerney-Leo, A., and Sidransky, E. (2004). Parkinsonism among Gaucher disease carriers. J. Med. Genet. 41, 937–940. doi: 10.1136/jmg.2004.024455
Haack, T. B., Hogarth, P., Kruer, M. C., Gregory, A., Wieland, T., Schwarzmayr, T., et al. (2012). Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 91, 1144–1149. doi: 10.1016/j.ajhg.2012.10.019
Haas, R. H., Parikh, S., Falk, M. J., Saneto, R. P., Wolf, N. I., Darin, N., et al. (2007). Mitochondrial disease: a practical approach for primary care physicians. Pediatrics 120, 1326–1333. doi: 10.1542/peds.2007-0391
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hashem, S. I., Murphy, A. N., Divakaruni, A. S., Klos, M. L., Nelson, B. C., Gault, E. C., et al. (2017). Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell. Cardiol. 108, 86–94. doi: 10.1016/j.yjmcc.2017.05.007
Hayflick, S. J., Kruer, M. C., Gregory, A., Haack, T. B., Kurian, M. A., Houlden, H. H., et al. (2013). β-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain J. Neurol. 136, 1708–1717. doi: 10.1093/brain/awt095
Herpin, A., Englberger, E., Zehner, M., Wacker, R., Gessler, M., and Schartl, M. (2015). Defective autophagy through epg5 mutation results in failure to reduce germ plasm and mitochondria. FASEB J. 29, 4145–4161. doi: 10.1096/fj.14-265462
Hirota, Y., Yamashita, S., Kurihara, Y., Jin, X., Aihara, M., Saigusa, T., et al. (2015). Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 11, 332–343. doi: 10.1080/15548627.2015.1023047
Honda, S., Arakawa, S., Nishida, Y., Yamaguchi, H., Ishii, E., and Shimizu, S. (2014). Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun. 5:4004. doi: 10.1038/ncomms5004
Hoshino, A., Mita, Y., Okawa, Y., Ariyoshi, M., Iwai-Kanai, E., Ueyama, T., et al. (2013). Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 4:2308. doi: 10.1038/ncomms3308
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. doi: 10.1038/nature12188
Hsu, P., Liu, X., Zhang, J., Wang, H.-G., Ye, J.-M., and Shi, Y. (2015). Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–652. doi: 10.1080/15548627.2015.1023984
Hudson, G., Gomez-Duran, A., Wilson, I. J., and Chinnery, P. F. (2014). Recent Mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet. 10:e1004369. doi: 10.1371/journal.pgen.1004369
Iguchi, M., Kujuro, Y., Okatsu, K., Koyano, F., Kosako, H., Kimura, M., et al. (2013). Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 288, 22019–22032. doi: 10.1074/jbc.M113.467530
Irrcher, I., Aleyasin, H., Seifert, E. L., Hewitt, S. J., Chhabra, S., Phillips, M., et al. (2010). Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 19, 3734–3746. doi: 10.1093/hmg/ddq288
Ivankovic, D., Chau, K.-Y., Schapira, A. H. V., and Gegg, M. E. (2016). Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 136, 388–402. doi: 10.1111/jnc.13412
Ivanova, M. M., Changsila, E., Iaonou, C., and Goker-Alpan, O. (2019). Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS One 14:e0210617. doi: 10.1371/journal.pone.0210617
Jelani, M., Dooley, H. C., Gubas, A., Mohamoud, H. S. A., Khan, M. T. M., Ali, Z., et al. (2019). A mutation in the major autophagy gene. WIPI2, associated with global developmental abnormalities. Brain J. Neurol. 142, 1242–1254. doi: 10.1093/brain/awz075
Ji, C., Zhao, H., Li, D., Sun, H., Hao, J., Chen, R., et al. (2019). Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy 0, 1–11. doi: 10.1080/15548627.2019.1632621
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kauppila, T. E. S., Kauppila, J. H. K., and Larsson, N.-G. (2017). Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71. doi: 10.1016/j.cmet.2016.09.017
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/BJ20140334
Kim, M., Sandford, E., Gatica, D., Qiu, Y., Liu, X., Zheng, Y., et al. (2016). Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 5:e12245. doi: 10.7554/eLife.12245
Kissová, I., Deffieu, M., Manon, S., and Camougrand, N. (2004). Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 279, 39068–39074. doi: 10.1074/jbc.M406960200
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kluge, A. F., Lagu, B. R., Maiti, P., Jaleel, M., Webb, M., Malhotra, J., et al. (2018). Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 28, 2655–2659. doi: 10.1016/j.bmcl.2018.05.013
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., et al. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036. doi: 10.1038/nature03029
Lahuerta, M., Aguado, C., Sánchez-Martín, P., Sanz, P., and Knecht, E. (2018). Degradation of altered mitochondria by autophagy is impaired in Lafora disease. FEBS J. 285, 2071–2090. doi: 10.1111/febs.14468
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., et al. (2019). BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15, 1182–1198. doi: 10.1080/15548627.2019.1580095
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Le Guerroué, F., Eck, F., Jung, J., Starzetz, T., Mittelbronn, M., Kaulich, M., et al. (2017). Autophagosomal content profiling reveals an LC3C-dependent piecemeal mitophagy pathway. Mol. Cell 68:786–796.e6. doi: 10.1016/j.molcel.2017.10.029
Lee, M. Y., Sumpter, R., Zou, Z., Sirasanagandla, S., Wei, Y., Mishra, P., et al. (2017). Peroxisomal protein PEX13 functions in selective autophagy. EMBO Rep 18, 48–60. doi: 10.15252/embr.201642443
Legati, A., Reyes, A., Nasca, A., Invernizzi, F., Lamantea, E., Tiranti, V., et al. (2016). New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta 1857, 1326–1335. doi: 10.1016/j.bbabio.2016.02.022
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Levine, B., and Kroemer, G. (2019). Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. doi: 10.1016/j.cell.2018.09.048
Liao, C., Ashley, N., Diot, A., Morten, K., Phadwal, K., Williams, A., et al. (2017). Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 88, 131–142. doi: 10.1212/WNL.0000000000003491
Lim, J.-A., Li, L., Shirihai, O. S., Trudeau, K. M., Puertollano, R., and Raben, N. (2017). Modulation of mTOR signaling as a strategy for the treatment of Pompe disease. EMBO Mol. Med. 9, 353–370. doi: 10.15252/emmm.201606547
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lwin, A., Orvisky, E., Goker-Alpan, O., LaMarca, M. E., and Sidransky, E. (2004). Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 81, 70–73. doi: 10.1016/j.ymgme.2003.11.004
Madeo, F., Eisenberg, T., Pietrocola, F., and Kroemer, G. (2018). Spermidine in health and disease. Science 359:aan2788. doi: 10.1126/science.aan2788
Mann, V. M., Cooper, J. M., Krige, D., Daniel, S. E., Schapira, A. H., and Marsden, C. D. (1992). Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain J. Neurol. 115(Pt 2), 333–342. doi: 10.1093/brain/115.2.333
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
McLelland, G.-L., Soubannier, V., Chen, C. X., McBride, H. M., and Fon, E. A. (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. doi: 10.1002/embj.201385902
Meissner, C., Lorenz, H., Weihofen, A., Selkoe, D. J., and Lemberg, M. K. (2011). The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 117, 856–867. doi: 10.1111/j.1471-4159.2011.07253.x
Melber, A., and Haynes, C. M. (2018). UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28, 281–295. doi: 10.1038/cr.2018.16
Mizushima, N. (2020). The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 63, 1–10. doi: 10.1016/j.ceb.2019.12.001
Morani, F., Doccini, S., Sirica, R., Paterno, M., Pezzini, F., Ricca, I., et al. (2019). Functional transcriptome analysis in ARSACS KO cell model reveals a role of sacsin in autophagy. Sci. Rep. 9:11878. doi: 10.1038/s41598-019-48047-x
Mouchiroud, L., Houtkooper, R. H., Moullan, N., Katsyuba, E., Ryu, D., Cantó, C., et al. (2013). The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. doi: 10.1016/j.cell.2013.06.016
Mukherjee, A. B., Appu, A. P., Sadhukhan, T., Casey, S., Mondal, A., Zhang, Z., et al. (2019). Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 14:4. doi: 10.1186/s13024-018-0300-6
Napoli, E., Song, G., Panoutsopoulos, A., Riyadh, M. A., Kaushik, G., Halmai, J., et al. (2018). Beyond autophagy: a novel role for autism-linked Wdfy3 in brain mitophagy. Sci. Rep. 8:11348. doi: 10.1038/s41598-018-29421-7
Napolitano, G., and Ballabio, A. (2016). TFEB at a glance. J. Cell Sci. 129, 2475–2481. doi: 10.1242/jcs.146365
Nezich, C. L., Wang, C., Fogel, A. I., and Youle, R. J. (2015). MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 210, 435–450. doi: 10.1083/jcb.201501002
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Ordonez, M. P., Roberts, E. A., Kidwell, C. U., Yuan, S. H., Plaisted, W. C., and Goldstein, L. S. B. (2012). Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum. Mol. Genet. 21, 2651–2662. doi: 10.1093/hmg/dds090
Ordureau, A., Sarraf, S. A., Duda, D. M., Heo, J.-M., Jedrychowski, M. P., Sviderskiy, V. O., et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360–375. doi: 10.1016/j.molcel.2014.09.007
Osellame, L. D., Rahim, A. A., Hargreaves, I. P., Gegg, M. E., Richard-Londt, A., Brandner, S., et al. (2013). Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson’s disease. Cell Metab. 17, 941–953. doi: 10.1016/j.cmet.2013.04.014
Otomo, T., Higaki, K., Nanba, E., Ozono, K., and Sakai, N. (2009). Inhibition of autophagosome formation restores mitochondrial function in mucolipidosis II and III skin fibroblasts. Mol. Genet. Metab. 98, 393–399. doi: 10.1016/j.ymgme.2009.07.002
Oz-Levi, D., Ben-Zeev, B., Ruzzo, E. K., Hitomi, Y., Gelman, A., Pelak, K., et al. (2012). Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 91, 1065–1072. doi: 10.1016/j.ajhg.2012.09.015
Pacheco, C. D., Kunkel, R., and Lieberman, A. P. (2007). Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 16, 1495–1503. doi: 10.1093/hmg/ddm100
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Pickles, S., Vigié, P., and Youle, R. J. (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. CB 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Pietrocola, F., Lachkar, S., Enot, D. P., Niso-Santano, M., Bravo-San Pedro, J. M., Sica, V., et al. (2015). Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death. Differ. 22, 509–516. doi: 10.1038/cdd.2014.215
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F., and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Primer 4:27. doi: 10.1038/s41572-018-0025-4
Pottier, C., Bieniek, K. F., Finch, N., van de Vorst, M., Baker, M., Perkersen, R., et al. (2015). Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92. doi: 10.1007/s00401-015-1436-x
Price, N. L., Gomes, A. P., Ling, A. J. Y., Duarte, F. V., Martin-Montalvo, A., North, B. J., et al. (2012). SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15, 675–690. doi: 10.1016/j.cmet.2012.04.003
Proikas-Cezanne, T., Waddell, S., Gaugel, A., Frickey, T., Lupas, A., and Nordheim, A. (2004). WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 23, 9314–9325. doi: 10.1038/sj.onc.1208331
Puigserver, P., Wu, Z., Park, C. W., Graves, R., Wright, M., and Spiegelman, B. M. (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839. doi: 10.1016/s0092-8674(00)81410-5
Qi, Y., Qiu, Q., Gu, X., Tian, Y., and Zhang, Y. (2016). ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci. Rep. 6:24700. doi: 10.1038/srep24700
Raben, N., Schreiner, C., Baum, R., Takikita, S., Xu, S., Xie, T., et al. (2010). Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder–murine Pompe disease. Autophagy 6, 1078–1089. doi: 10.4161/auto.6.8.13378
Raben, N., Wong, A., Ralston, E., and Myerowitz, R. (2012). Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am. J. Med. Genet. C Semin. Med. Genet. 160C, 13–21. doi: 10.1002/ajmg.c.31317
Rana, A., Oliveira, M. P., Khamoui, A. V., Aparicio, R., Rera, M., Rossiter, H. B., et al. (2017). Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 8:448. doi: 10.1038/s41467-017-00525-4
Rana, A., Rera, M., and Walker, D. W. (2013). Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. U.S.A. 110, 8638–8643. doi: 10.1073/pnas.1216197110
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U.S.A. 113, 4039–4044. doi: 10.1073/pnas.1523926113
Richter, U., Lahtinen, T., Marttinen, P., Myöhänen, M., Greco, D., Cannino, G., et al. (2013). A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr. Biol. 23, 535–541. doi: 10.1016/j.cub.2013.02.019
Rizzo, F., Ronchi, D., Salani, S., Nizzardo, M., Fortunato, F., Bordoni, A., et al. (2016). Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot-Marie-Tooth 2A motor neurons. Hum. Mol. Genet. 25, 4266–4281. doi: 10.1093/hmg/ddw258
Ryan, B. J., Hoek, S., Fon, E. A., and Wade-Martins, R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. doi: 10.1016/j.tibs.2015.02.003
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Félix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888. doi: 10.1038/nm.4132
Safiulina, D., Kuum, M., Choubey, V., Gogichaishvili, N., Liiv, J., Hickey, M. A., et al. (2019). Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 38:e99384. doi: 10.15252/embj.201899384
Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K., et al. (2013). De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449. doi: 10.1038/ng.2562
Salpietro, V., Phadke, R., Saggar, A., Hargreaves, I. P., Yates, R., Fokoloros, C., et al. (2015). Zellweger syndrome and secondary mitochondrial myopathy. Eur. J. Pediatr. 174, 557–563. doi: 10.1007/s00431-014-2431-2
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi: 10.1038/nature12043
Saxton, R. A., and Sabatini, D. M. (2017). mTOR Signaling in Growth. Metabolism, and Disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Scheibye-Knudsen, M., Mitchell, S. J., Fang, E. F., Iyama, T., Ward, T., Wang, J., et al. (2014). A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855. doi: 10.1016/j.cmet.2014.10.005
Scheibye-Knudsen, M., Ramamoorthy, M., Sykora, P., Maynard, S., Lin, P.-C., Minor, R. K., et al. (2012). Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 209, 855–869. doi: 10.1084/jem.20111721
Scheibye-Knudsen, M., Scheibye-Alsing, K., Canugovi, C., Croteau, D. L., and Bohr, V. A. (2013). A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging. Aging 5, 192–208. doi: 10.18632/aging.100546
Schiavi, A., Maglioni, S., Palikaras, K., Shaik, A., Strappazzon, F., Brinkmann, V., et al. (2015). Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr. Biol. CB 25, 1810–1822. doi: 10.1016/j.cub.2015.05.059
Schwarz, C., Stekovic, S., Wirth, M., Benson, G., Royer, P., Sigrist, S. J., et al. (2018). Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging 10, 19–33. doi: 10.18632/aging.101354
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 19500–19505. doi: 10.1073/pnas.0708818104
Seibler, P., Burbulla, L. F., Dulovic, M., Zittel, S., Heine, J., Schmidt, T., et al. (2018). Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 141, 3052–3064. doi: 10.1093/brain/awy230
Settembre, C., Fraldi, A., Jahreiss, L., Spampanato, C., Venturi, C., Medina, D., et al. (2008). A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17, 119–129. doi: 10.1093/hmg/ddm289
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2:1002. doi: 10.1038/srep01002
Siddiqui, A., Bhaumik, D., Chinta, S. J., Rane, A., Rajagopalan, S., Lieu, C. A., et al. (2015). Mitochondrial quality control via the PGC1α-TFEB signaling pathway is compromised by parkin Q311X mutation but independently restored by rapamycin. J. Neurosci. Off. J. Soc. Neurosci. 35, 12833–12844. doi: 10.1523/JNEUROSCI.0109-15.2015
Solesio, M. E., Saez-Atienzar, S., Jordán, J., and Galindo, M. F. (2012). Characterization of mitophagy in the 6-hydoxydopamine Parkinson’s disease model. Toxicol. Sci. Off. J. Soc. Toxicol. 129, 411–420. doi: 10.1093/toxsci/kfs218
Soubannier, V., McLelland, G.-L., Zunino, R., Braschi, E., Rippstein, P., Fon, E. A., et al. (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. CB 22, 135–141. doi: 10.1016/j.cub.2011.11.057
Spampanato, C., Feeney, E., Li, L., Cardone, M., Lim, J.-A., Annunziata, F., et al. (2013). Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 5, 691–706. doi: 10.1002/emmm.201202176
Stavoe, A. K., Gopal, P. P., Gubas, A., Tooze, S. A., and Holzbaur, E. L. (2019). Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. eLife 8:e44219. doi: 10.7554/eLife.44219
Suleiman, J., Allingham-Hawkins, D., Hashem, M., Shamseldin, H. E., Alkuraya, F. S., and El-Hattab, A. W. (2018). WDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: a consistent neurodevelopmental syndrome. Clin. Genet. 93, 360–364. doi: 10.1111/cge.13054
Sumpter, R., Sirasanagandla, S., Fernández, ÁF., Wei, Y., Dong, X., Franco, L., et al. (2016). Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 165, 867–881. doi: 10.1016/j.cell.2016.04.006
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028
Sun, N., Yun, J., Liu, J., Malide, D., Liu, C., Rovira, I. I., et al. (2015). Measuring In Vivo Mitophagy. Mol. Cell 60, 685–696. doi: 10.1016/j.molcel.2015.10.009
Sun, Y., Vashisht, A. A., Tchieu, J., Wohlschlegel, J. A., and Dreier, L. (2012). Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 287, 40652–40660. doi: 10.1074/jbc.M112.419721
Takahashi, D., Moriyama, J., Nakamura, T., Miki, E., Takahashi, E., Sato, A., et al. (2019). AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell 76:797–810.e10. doi: 10.1016/j.molcel.2019.09.009
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D.-F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tanaka, Y., Guhde, G., Suter, A., Eskelinen, E. L., Hartmann, D., Lüllmann-Rauch, R., et al. (2000). Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906. doi: 10.1038/35022595
Todd, A. M., and Staveley, B. E. (2012). Expression of Pink1 with α-synuclein in the dopaminergic neurons of Drosophila leads to increases in both lifespan and healthspan. Genet. Mol. Res. GMR 11, 1497–1502. doi: 10.4238/2012.May.21.6
Toyama, E. Q., Herzig, S., Courchet, J., Lewis, T. L., Losón, O. C., Hellberg, K., et al. (2016). AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. doi: 10.1126/science.aab4138
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Vantaggiato, C., Crimella, C., Airoldi, G., Polishchuk, R., Bonato, S., Brighina, E., et al. (2013). Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain J. Neurol. 136, 3119–3139. doi: 10.1093/brain/awt227
Wallace, D. C. (2015). Mitochondrial DNA variation in human radiation and disease. Cell 163, 33–38. doi: 10.1016/j.cell.2015.08.067
Wallace, D. C. (2018). Mitochondrial genetic medicine. Nat. Genet. 50, 1642–1649. doi: 10.1038/s41588-018-0264-z
Wang, C., Niederstrasser, H., Douglas, P. M., Lin, R., Jaramillo, J., Li, Y., et al. (2017). Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat. Commun. 8, 1–14. doi: 10.1038/s41467-017-02332-3
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Wang, Z., Miao, G., Xue, X., Guo, X., Yuan, C., Wang, Z., et al. (2016). The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol. Cell 63, 781–795. doi: 10.1016/j.molcel.2016.08.021
Waterham, H. R., and Ebberink, M. S. (2012). Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1822, 1430–1441. doi: 10.1016/j.bbadis.2012.04.006
Wauer, T., Swatek, K. N., Wagstaff, J. L., Gladkova, C., Pruneda, J. N., Michel, M. A., et al. (2015). Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 34, 307–325. doi: 10.15252/embj.201489847
Weil, R., Laplantine, E., Curic, S., and Génin, P. (2018). Role of optineurin in the mitochondrial dysfunction: potential implications in neurodegenerative diseases and Cancer. Front. Immunol. 9:1243. doi: 10.3389/fimmu.2018.01243
White, K. E., Davies, V. J., Hogan, V. E., Piechota, M. J., Nichols, P. P., Turnbull, D. M., et al. (2009). OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest. Ophthalmol. Vis. Sci. 50, 2567–2571. doi: 10.1167/iovs.08-2913
Wong, Y. C., and Holzbaur, E. L. F. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U.S.A. 111, E4439–E4448. doi: 10.1073/pnas.1405752111
Yamano, K., Fogel, A. I., Wang, C., van der Bliek, A. M., and Youle, R. J. (2014). Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3:e01612. doi: 10.7554/eLife.01612
Yamano, K., and Youle, R. J. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi: 10.4161/auto.24633
Youle, R. J. (2019). Mitochondria-Striking a balance between host and endosymbiont. Science 365:aaw9855. doi: 10.1126/science.aaw9855
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Yu-Wai-Man, P., Griffiths, P. G., Gorman, G. S., Lourenco, C. M., Wright, A. F., Auer-Grumbach, M., et al. (2010). Multi-system neurological disease is common in patients with OPA1 mutations. Brain J. Neurol. 133, 771–786. doi: 10.1093/brain/awq007
Zachari, M., Gudmundsson, S. R., Li, Z., Manifava, M., Shah, R., Smith, M., et al. (2019). Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 50:627–643.e5. doi: 10.1016/j.devcel.2019.06.016
Zhao, H., Zhao, Y. G., Wang, X., Xu, L., Miao, L., Feng, D., et al. (2013). Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J. Cell Biol. 200, 731–741. doi: 10.1083/jcb.201211014
Keywords: autophagy, mitophagy, aging, mitophaging, monogenic disorders, interventions
Citation: Bakula D and Scheibye-Knudsen M (2020) MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 8:239. doi: 10.3389/fcell.2020.00239
Received: 29 January 2020; Accepted: 23 March 2020;
Published: 15 April 2020.
Edited by:
Konstantinos Palikaras, Foundation for Research and Technology Hellas, GreeceReviewed by:
Nabil Eid, United Arab Emirates University, United Arab EmiratesCarsten Culmsee, University of Marburg, Germany
Copyright © 2020 Bakula and Scheibye-Knudsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Bakula, YmFrdWxhQHN1bmQua3UuZGs=; Morten Scheibye-Knudsen, bXNjaGVpYnllQHN1bmQua3UuZGs=; bW9ydGVuQGZvcnNvZWdzcGVyc29uLmRr