 Maria Zachari
Maria Zachari Nicholas T. Ktistakis
Nicholas T. Ktistakis- 1MRC Protein Phosphorylation and Ubiquitylation Unit, University of Dundee, Dundee, United Kingdom
- 2Signalling Programme, Babraham Institute, Cambridge, United Kingdom
Mitophagy, a conserved intracellular process by which mitochondria are eliminated via the autophagic machinery, is a quality control mechanism which facilitates maintenance of a functional mitochondrial network and cell homeostasis, making it a key process in development and longevity. Mitophagy has been linked to multiple human disorders, especially neurodegenerative diseases where the long-lived neurons are relying on clearance of old/damaged mitochondria to survive. During the past decade, the availability of novel tools to study mitophagy both in vitro and in vivo has significantly advanced our understanding of the molecular mechanisms governing this fundamental process in normal physiology and in various disease models. We here give an overview of the known mitophagy pathways and how they are induced, with a particular emphasis on the early events governing mitophagosome formation.
Introduction
Macroautophagy (or simply here autophagy) is a conserved quality control pathway by which a double membrane structure, called an autophagosome, grows to engulf cytoplasmic components in order to deliver them to the lysosome for degradation. Different stimuli can induce either “bulk autophagy” which is a non-selective process degrading random portions of the cytoplasm or “selective autophagy” which is activated to specifically degrade cellular components such as whole organelles (e.g., mitochondria, ER, peroxisomes - to name a few) and protein aggregates (Kirkin, 2019). One of the best understood pathways of selective autophagy is mitophagy - the selective degradation of mitochondria via autophagy. Mitochondria are dynamic organelles whose main function is to produce energy to support the many intracellular processes our cells are constantly undertaking. Mitochondrial quality control is crucial as defects to these organelles can lead to apoptosis or tissue damage, and, unsurprisingly, such mitochondrial defects have been linked to numerous human diseases (Murphy and Hartley, 2018). Thus, elimination of damaged and potentially toxic mitochondria is of extreme importance in favor of homeostasis and survival. A major pathway for the clearance of these mitochondria - and maintenance of mitochondrial network integrity and quality control - is mitophagy. Autophagosomes engulfing mitochondria are called mitophagosomes. Mitophagy occurs in a basal level (with different cell types exhibiting different levels of basal mitophagy) but can also be induced upon stresses such as exercise and ischemia in vivo and by mitochondrial disrupting/damaging agents in cultured cells (Montava-Garriga and Ganley, 2019). Impaired mitophagy has been associated with aging and numerous human disorders such as Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), cancer, but also cardiovascular and liver diseases (Palikaras et al., 2017; Um and Yun, 2017; Palikaras et al., 2018). Pharmacological modulation of mitophagy has been suggested to have potential as a therapeutic strategy for the treatment of these diseases. Although there is evidence that mitophagy is involved in pathogenesis, the exact role of mitophagy and mitophagy-related genes in pathological conditions is yet unclear. Ongoing in vivo and in vitro studies are aiming to elucidate this as well as to explore whether mitophagy could make a good pharmacological target in the context of disease. Over the past two decades, key studies have significantly advanced our understanding of the molecular mechanisms governing mitophagy. Here, we will aim to review the main mitophagy pathways with a particular focus on the early signaling events.
Autophagy Machinery
The process of forming a double-membrane autophagosome depends on a series of hierarchical steps that bring together more than 30 proteins or protein complexes. Upon inactivation of mTOR (in pathways of non-selective autophagy) the ULK complex composed of the protein kinase ULK1 (or its homolog ULK2), and the adaptors FIP200, ATG13, and ATG101 translocates to endoplasmic reticulum (ER) tubulovesicular membranes that have been “marked” by the presence of ATG9-containing vesicles (Hara et al., 2008; Ganley et al., 2009; Hosokawa et al., 2009b,a; Karanasios et al., 2016). These membranes then recruit the VPS34 complex composed of the PI 3-kinase VPS34 [synthesizing phosphatidylinositol 3-phosphate (PI3P)] and the adaptors VPS15, ATG14, and Beclin-1 which generates PI3P on ER-associated membranes termed omegasomes (Axe et al., 2008). The PI3P-enriched omegasomes then recruit the WIPI effectors and DFCP1, with the former group responsible for bringing on site the lipidation machinery that mediates the covalent modification of ATG8 family members (LC3 and GABARAP families) with phosphatidylethanolamine (PE) (Dooley et al., 2014). These PE-modified ATG8 proteins become part of the autophagosomal membrane whereas all of the other proteins come off as the double membrane closes and travels to the lysosomes for degradation (Axe et al., 2008; Karanasios et al., 2013). One challenge specific to our topic is how this very complicated machinery for making the double membrane autophagosome co-ordinates with the machinery that selects damaged cargo during selective autophagy. We will address this question in later sections.
Main Mitophagy Triggers In Vitro and In Vivo
Induction of Mitophagy in vivo
It was recently shown that mitophagy occurs in vivo in multiple tissues of mice at steady state without the need of external stimuli. This so-called basal mitophagy occurs presumably to ensure quality control of mitochondria as a housekeeping mechanism (McWilliams et al., 2016; Sun et al., 2017; McWilliams et al., 2018). Apart from its basal occurrence, mitophagy is also induced to support many physiological processes in vivo during organismal development. For example, during early embryogenesis, mitophagy has been reported to be responsible for the degradation of paternal mitochondria from the fertilized oocyte and early embryo (Rojansky et al., 2016). Furthermore, during reticulocyte maturation, mitophagy is a key pathway in regulating elimination of mitochondria for the production of mature erythrocytes (Kundu et al., 2008; Sandoval et al., 2008). Mitophagy has been reported to trigger a metabolic switch from oxidative phosphorylation to glycolysis, which is required for retina ganglion cell (RGC) and M1 macrophage differentiation (Esteban-Martinez et al., 2017). Similarly, mitophagy is key in promoting a switch from glycolysis to oxidative phosphorylation in myoblast differentiation (Sin et al., 2016). Apart from its role during embryonic development, mitophagy induced in response to infection has been proposed to have a protective inhibitory effect on the inflammasome, to avoid an excessive immune response which can lead to tissue damage (Kim et al., 2016; Zhong et al., 2016). Multiple physiological stresses have been reported to induce mitophagy in mice, including exercise, starvation, a switch to high fat diet, ischemia and hypoxia. More specifically, acute exercise is a strong mitophagy inducer in heart and skeletal muscle to mediate mitochondrial remodeling (Moyzis et al., 2015; Laker et al., 2017; Drake et al., 2019). Starvation is well known to induce general autophagy in mice, but this stress has also been reported to induce mitophagy, and interestingly there is evidence for canonical and non-canonical mechanisms occurring during starvation-induced mitophagy (discussed below) (Mizushima et al., 2004; Nishida et al., 2009; Hirota et al., 2015; Saito et al., 2019). Cardiomyocytes from mice subjected to high fat diet were shown to exhibit elevated levels of mitophagy to prevent cytotoxicity (Tong et al., 2019), although this resulted in reduced mitophagy in liver (Sun et al., 2015). Myocardial ischemia and energy stress (48 h starvation) have been shown to induce mitophagy in cardiomyocytes of mice, whereas ischemic preconditioning as well as ischemia-reperfusion injury were shown to induce mitophagy in kidney and brain tissues (Tang et al., 2016; Livingston et al., 2019; Saito et al., 2019; Tang et al., 2019). Hypoxic conditions also result in mitophagy induction in multiple tissues in mice and in various cell lines in vitro (Zhang et al., 2008; Allen et al., 2013; Sun et al., 2015, Zhang W. et al., 2016).
Induction of Mitophagy in vitro
Apart from the above more physiological ways to trigger mitophagy, chemical inducers of mitophagy have proved to be great tools and have allowed the dissection of the molecular mechanisms of this pathway in vitro (mostly in tissue cultured cells). Multiple compounds have been reported to induce mitophagy and have been recently reviewed in detail (Georgakopoulos et al., 2017). For the purpose of this review we will mention a few recent and widely used mitophagy inducers for which some mechanistic detail is known. The mitochondrial uncouplers CCCP and FCCP are proton ionophores which cause severe loss of mitochondrial membrane potential, leading to a robust activation of mitophagy. A combination of the F0F1-ATPase inhibitor oligomycin and the complex III inhibitor antimycin A (symbolized as O/A treatment) also causes membrane depolarization and is known to be a strong mitophagy inducer in cultured cells (Georgakopoulos et al., 2017). The iron chelators depheriprone (DFP) and deferoxamine (DFO) are hypoxia mimicking agents and robustly induce mitophagy in a HIF1α-dependent manner (Allen et al., 2013).
Celastrol, a plant-derived pentacyclic triterpene with reported anti-inflammatory, anti-cancer and anti-obesity effects, was shown to induce mitophagy via Nur77 in a pathway that will be described later on in more detail (Hu et al., 2017). Lastly, we recently reported that the lactone Ivermectin acutely induces a strong mitophagic response upon fragmentation of mitochondria and is the fastest mitophagy inducer to our knowledge (Zachari et al., 2019). The mechanism by which mitophagy is induced by Ivermectin is currently unknown, but it is unlikely that its action is similar to CCCP or FCCP as it reduces oxygen consumption rate (OCR) whereas CCCP and FCCP are well known to dramatically increase OCR (Georgakopoulos et al., 2017; Juarez et al., 2018; Zachari et al., 2019). On the other hand, treatment with Oligomycin/Antimycin A results in decreased OCR as Ivermectin does, although in this case the timings of the two responses are very different with Ivermectin causing a much faster induction of mitophagy. Furthermore, it is unlikely that it acts through a HIF1α-dependent pathway, as this type of mitophagy stimulation requires transcriptional activity and usually takes longer time to occur. Thus, the above compounds appear to act through different mechanisms in inducing mitophagy.
Mitophagy Signals Before Engaging the Autophagy Machinery: Ubiquitin Dependent and Independent Pathways
Different pathways can regulate mitophagy and one of the main differences among them is their dependency or not on ubiquitin. The best studied mitophagy pathway is regulated by the Parkinson’s disease related proteins PINK1 and Parkin (Bingol and Sheng, 2016). An illustration of the PINK1/Parkin pathway is shown in Figure 1A. PINK1 is a serine/threonine kinase which contains a mitochondrial-targeting signal. In healthy mitochondria, PINK1 is imported into the mitochondria via the TOM and TIM complexes, following cleavage by mitochondrial proteases including PARL (Jin et al., 2010; Greene et al., 2012). Cleaved PINK1 is exported back to the cytoplasm in an unstable form which is subjected to proteasomal degradation (Yamano and Youle, 2013). Upon mitochondrial depolarization, a loss of mitochondrial membrane potential inhibits import of PINK1 triggering its stabilization on the outer mitochondrial membrane (OMM), which results in its autophosphorylation and dimerization (Okatsu et al., 2012, 2013). PINK1 then regulates E3 ligase Parkin recruitment by phosphorylating ubiquitin at serine 65 attached on multiple OMM proteins (Narendra et al., 2010; Kane et al., 2014; Kazlauskaite et al., 2014). Phosphorylated ubiquitin acts as a key signal for the recruitment and activation of Parkin (Wauer et al., 2015). Parkin activation also requires direct phosphorylation by PINK1 at serine 65 (Kondapalli et al., 2012; Shiba-Fukushima et al., 2012). Parkin then further conjugates ubiquitin on OMM proteins, marking the mitochondria for degradation by the autophagic machinery (Narendra et al., 2008). It is worth mentioning here that phosphorylation of ubiquitin by PINK1 has been reported to be sufficient to recruit mitophagy cargo receptor proteins (primarily optineurin and NDP52) independently of Parkin but this is enhanced by Parkin activity (Lazarou et al., 2015). Recent work revealed that the phosphatase PPEF2 is responsible for dephosphorylating ubiquitin at serine 65 to oppose the PINK1 effect and inhibit mitophagy (Wall et al., 2019). The deubiquitinating enzyme that reverses this process by removing ubiquitin from the OMM is USP30 (Bingol et al., 2014). Another E3 ligase that has been reported to act in parallel with Parkin, is MUL1 (Yun et al., 2014; Rojansky et al., 2016). The upstream signals that regulate MUL1 recruitment and activity require further study, as the process has been reported to be both dependent and also independent of PINK1, raising the possibility of context dependent regulation of this protein (Yun et al., 2014; Rojansky et al., 2016). Multiple other proteins have been reported to regulate PINK1/Parkin-mediated mitophagy. More specifically, choline dehydrogenase (CHCD), a mitochondrial enzyme regulating methionine synthesis, was shown to act as a ubiquitin “eat me” signal and to associate with p62 for phagophore recruitment (Park et al., 2014). Depletion of CHCD was reported to impair PINK1/Parkin mitophagy induced by CCCP (Park et al., 2014). Similarly, two mitochondrial matrix proteins required for PINK1/Parkin mitophagy, NIPSNAP1 and NIPSNAP2, were recently reported to accumulate on mitochondria upon CCCP or hypoxia treatment, to act as “eat me” signals by directly recruiting mitophagy adaptor proteins (NDP52 and p62) as well as LC3/GABARAPs (Princely Abudu et al., 2019). Apart from receptor proteins, cardiolipin, a phospholipid normally localized in the inner mitochondrial membrane (IMM), translocates to the OMM upon Rotenone (a complex I inhibitor) and FCCP/CCCP treatment to act as a receptor for LC3 and thus mediate autophagosomal engulfment of damaged mitochondria (Chu et al., 2013). Cardiolipin potentially acts downstream of PINK1/Parkin signaling, although further research is required to confirm this scenario (Chu et al., 2013). An inner membrane mitochondrial protein, prohibitin 2, was also reported to mediate mitophagy by binding to LC3 upon proteasomal-dependent OMM rupture caused by O/A treatment (Wei et al., 2017). The presence of prohibitin 2 is important for PINK1/Parkin mitophagy but also for elimination of paternal mitochondria upon oocyte fertilization in C. elegans (Wei et al., 2017). It was recently suggested that prohibitin 2 might be involved in PINK1/Parkin mitophagy by positively regulating PINK1 stabilization onto mitochondria (Yan et al., 2019).
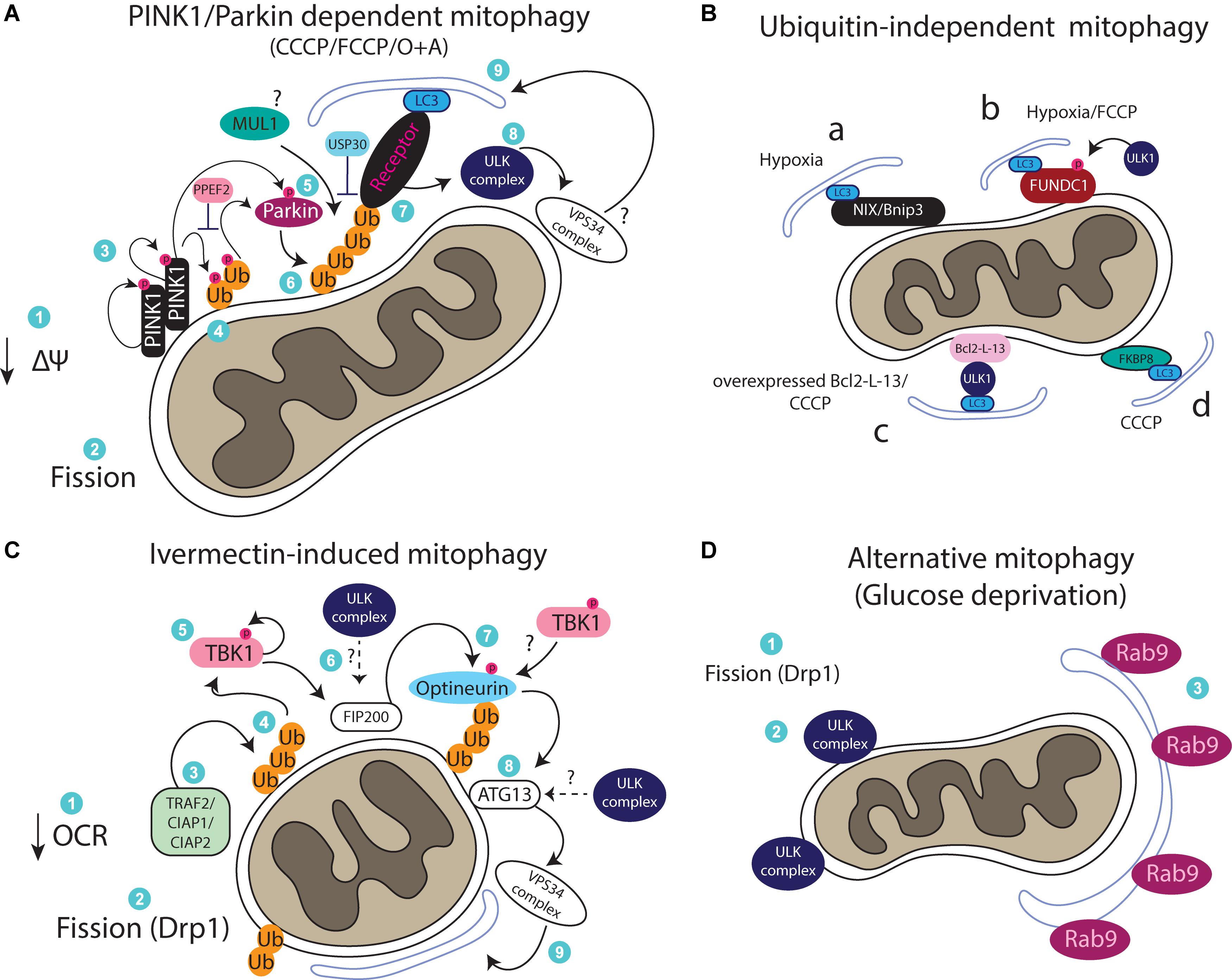
Figure 1. (A) Cartoon of the PINK1/Parkin pathway. Upon loss of membrane potential (ΔΨ), PINK1 stabilizes on the OMM, dimerizes and autophosphorylates. PINK1 next phosphorylates Ubiquitin attached onto OMM proteins, leading to the recruitment of Parkin, which also gets phosphorylated and activated by PINK1. Parkin further ubiquitinates OMM proteins leading to the recruitment of receptor proteins for the generation/recruitment of an autophagosome and subsequent degradation of the mitochondrion. (B) Representation of mitophagy pathways which do not rely on ubiquitin. Various mitochondrial proteins can act as mitophagy receptors including: (a) NIX/Bnip3, (b) FUNDC1, (c) Bcl2-L-13, and (d) FKBP8. (C) Ivermectin-induced mitophagy relies on mitochondrial fragmentation and ubiquitylation via TRAF2/CIAP1/CIAP2. Upon ubiquitylation, TBK1 is required for FIP200 recruitment, which results in optineurin recruitment and downstream activation of ATG13 and the rest of the autophagic machinery for mitophagy. (D) An alternative autophagy pathway which does not rely on LC3 lipidation can also mediate mitophagy. This pathway requires mitochondrial fission, ULK1 and Rab9-possitive membranes.
We recently reported a second ubiquitin-dependent mitophagy pathway which is rapidly induced by the lactone Ivermectin (Zachari et al., 2019). Ivermectin treatment results in acute mitochondrial damage as observed by a decrease in OCR and severe fragmentation of the mitochondrial network. This leads to induction of mitophagy, independently of PINK1 and Parkin, but dependent on the E3 ligases TRAF2, CIAP1, and CIAP2 which work synergistically (and potentially in complex) to conjugate ubiquitin onto fragmented mitochondria. Upon ubiquitination, TBK1 becomes activated in order to regulate recruitment of the adaptor protein optineurin, to mediate mitophagy in a pathway which will be discussed later in more detail (Zachari et al., 2019) (Figure 1C). The DUB enzyme(s) reversing this process is yet to be identified. Apart from optineurin and NDP52, several other adaptor proteins are involved in mitophagy of ubiquitinated mitochondria including TAX1BP, p62, and NBR1 (Geisler et al., 2010; Moore and Holzbaur, 2016; Zachari et al., 2019). Another TRAF2/ubiquitin-dependent mitophagy pathway occurs in the presence of celastrol and the cytokine TNFa (Hu et al., 2017). More specifically, under these conditions, celastrol binds to Nur77 (a transcription factor nuclear receptor protein which can translocate to mitochondria to induce apoptosis), resulting in its translocation to mitochondria. When on mitochondria, Nur77 recruits TRAF2 resulting in mitochondrial ubiquitination and p62 recruitment for autophagic elimination in an anti-inflammatory mechanism (Hu et al., 2017).
Mitophagy can also occur independently of the presence of ubiquitin on the mitochondria (Figure 1B). These mitophagy pathways are regulated by adaptor proteins containing mitochondrial-targeting domains and LC3-interacting region (LIR) motifs. Whereas only one yeast mitophagy receptor has been identified so far (the protein ATG32) (Kanki et al., 2009), multiple mitochondrial receptors have been identified in mammals, namely: BNIP3, NIX (or BNIP3L), FUNDC1, and Bcl2-L-13. BNIP3 and NIX are two very similar proteins with pro-apoptotic functions that are transcriptionally upregulated upon hypoxia in a HIF1α-dependent manner to mediate hypoxia-induced mitophagy (Zhang et al., 2008; Hanna et al., 2012, Ney, 2015; Yuan et al., 2017). NIX is also required for mitophagy to mediate mitochondrial removal during reticulocyte maturation (Schweers et al., 2007) and was recently reported to mediate ischemia-reperfusion-induced mitophagy in the brain (Yuan et al., 2017). Furthermore, NIX overexpression can rescue mitophagy induced upon CCCP treatment in cells deficient for Parkin-mediated mitophagy (Koentjoro et al., 2017). The interaction of NIX with LC3 during mitophagy has been suggested to be regulated by phosphorylation of NIX (at serine residues 34 and 35), although the kinase mediating these events is not yet identified (Rogov et al., 2017). Furthermore, NIX was recently reported to be a substrate of Parkin during mitophagy (Gao et al., 2015). Parkin-mediated ubiquitination of NIX results in the recruitment of NBR1 to mitochondria to mediate their autophagosomal engulfment and removal (Gao et al., 2015). Interestingly, BNIP3 has been reported to promote PINK1-dependent mitophagy as well, by stabilizing PINK1 levels onto mitochondria in hypoxic conditions (Zhang T. et al., 2016). Of note, overexpression of BNIP3 alone causes membrane potential loss and is sufficient to induce mitophagy, highlighting its significant role in activating this pathway (Rikka et al., 2011). The OMM protein FUNDC1 has been reported to regulate mitophagy induced upon hypoxia, but also upon mitochondrial depolarization with FCCP (Liu et al., 2012; Chen et al., 2014). Basal phosphorylation of FUNDC1 by Src (at tyrosine 18) and CK2 (at serine 13) inhibit its interaction with LC3 and concomitantly mitophagy, whereas hypoxia-induced dephosphorylation increases its interaction with LC3 to promote mitophagy (Liu et al., 2012; Chen et al., 2014). FUNDC1 has also been reported to be regulated by direct phosphorylation by ULK1 (at serine 17) to promote mitophagy both in the context of hypoxia and of FCCP treatment (Wu et al., 2014). Recently, NIX and FUNDC1 have been reported to be essential for mitophagy during cardiac progenitor cell differentiation, mediating mitochondrial network remodeling (Lampert et al., 2019). Bcl2-L-13 has been suggested to be the mammalian homolog of the yeast Atg32 due to sequence similarity and because it can rescue mitophagy in yeast upon loss of Atg32 (Murakawa et al., 2015). Bcl2-L-13 is thought to regulate mitophagy both by binding to LC3 but also by mediating mitochondrial fission (Murakawa et al., 2015). Another recently discovered mitophagy adaptor, FKBP8 with a preferred binding to LC3A, induces mitochondrial fragmentation and mitophagy when overexpressed in cells, independently of the PINK1/Parkin pathway (Bhujabal et al., 2017) (Figure 1B).
Recruitment of the Autophagic Machinery
Since receptor proteins are central in mediating mitophagy and can bind both mitochondria and LC3/GABARAP proteins, it was thought until recently that they mediate recruitment of the autophagic machinery by binding to LC3/GABARAP-positive forming phagophores. Recent evidence suggests that both ubiquitin dependent and independent mitophagy adaptor/receptor proteins can also act as hubs for the recruitment of proteins involved in early autophagy events and working upstream of LC3/GABARAPs such as ULK1 complex components and the PI3P-binding proteins WIPIs and DFCP1. These data reveal that mitophagy can occur not only by utilizing already forming autophagosomes but also by direct initiation of autophagosome formation via activation of early components. A study by Itakura et al. showed that during Parkin-mediated mitophagy, early autophagy proteins such as ULK1, ATG14, DFCP1, WIPI1, and ATG16L1 translocated onto damaged mitochondria even in the absence of LC3 conjugation on membranes (Itakura et al., 2012). It was later shown by the Youle laboratory that this is mediated in a receptor-dependent manner (Lazarou et al., 2015). More specifically, upon mitochondria depolarization, phospho-ubiquitin produced as a result of PINK1 activation, recruits the receptor proteins NDP52 and optineurin, which are required for the recruitment of ULK1 and the omegasome markers WIPI1 and DFCP1 (Lazarou et al., 2015). Interestingly, later work showed that ectopic targeting of NDP52 onto mitochondria is sufficient to recruit the ULK1 complex (via a direct interaction of NDP52 with FIP200), ATG14 and ATG16L1 and concomitantly induce mitophagy in both Parkin-dependent and independent mechanisms (Vargas et al., 2019). In the same study it was shown that targeting of ULK1 to mitochondria was sufficient to induce mitophagy (even when NDP52 or TBK1 are absent), supporting the notion that the function of early ubiquitin/receptor signals is to recruit the autophagy initiation machinery for the de novo formation of an autophagosome on the target mitochondrion. The key event regulated by NDP52-mediated recruitment of the ULK1 complex is a direct interaction between NDP52 and FIP200 which requires the presence and activation of TBK1 (Vargas et al., 2019). Binding of receptor proteins to FIP200 is not a mitophagy specific mechanism. It was recently reported to be the key event in other types of selective autophagy as well including ER-phagy, aggrephagy, xenophagy, and pexophagy (Smith et al., 2018; Ravenhill et al., 2019; Turco et al., 2019) highlighting that different selective autophagy pathways share common features. In addition, it was recently shown that LC3/GABARAP proteins can mediate recruitment of the adaptor proteins optineurin and NDP52 to mitochondria, in a positive feedback loop in order to accelerate PINK1/Parkin mediated mitophagy (Padman et al., 2019). Interestingly, LC3/GABARAPs likely play different roles during PINK1/Parkin mitophagy as GABARAPs appear to be more essential than the LC3s (Nguyen et al., 2016). Furthermore, loss of all LC3/GABARAPs did not abolish autophagosome formation (as seen by EM and recruitment of earlier autophagy markers such as ULK1 on mitochondria) but it rather caused defects in mitophagosome-lysosome fusion (Nguyen et al., 2016). Independently of PINK1 and Parkin, we recently showed that ubiquitination of fragmented mitochondria induced by Ivermectin leads to activation of TBK1 which is required for optineurin recruitment and mitophagy (Zachari et al., 2019). In this mitophagy pathway, recruitment of optineurin required FIP200 but not ULK1/2 or ATG13, revealing that the activity of the ULK1/2 kinases is not essential for certain mitophagy pathways. [It is worth mentioning here that the FIP200 and ATG13 proteins have been previously reported to act independently of the complex during autophagy (Alers et al., 2011) although the exact mechanisms underlying these functions are poorly understood.] Furthermore, TBK1 activation was hierarchically earlier than FIP200, and was required for optineurin recruitment. Given the recently published evidence discussed above, it is possible that a direct interaction between optineurin (and possibly more adaptors) and FIP200 is key also for the progression of Ivermectin-induced mitophagy. ATG13 was required for the formation of the omegasomes, within which the LC3-positive mitophagosomes formed (Zachari et al., 2019). ATG9 vesicles - essential components of the autophagic machinery - have been involved in both PINK1/Parkin and Ivermectin-induced mitophagy, although their exact role during mitophagy remains elusive (Itakura et al., 2012; Yamano et al., 2018, Zachari et al., 2019).
Mitophagy induced via Bcl2-L-13 was also recently reported to involve recruitment of ULK1 to mitochondria, even though this was shown to occur in an indirect manner whereby Bcl2-L-13 recruits LC3B, leading to recruitment of ULK1 which binds to LC3B via a LIR motif (Murakawa et al., 2019). As mentioned earlier, FUNDC1-dependent mitophagy also involves recruitment of ULK1 to the mitochondria. In this case FUNDC1 is phosphorylated by ULK1 which causes enhanced binding to LC3 during mitophagy (Wu et al., 2014). Predictably, ULK1 kinase activity is required for this pathway (Wu et al., 2014) (Figure 1B).
Even though mitophagy generally is reported to occur via the canonical autophagic machinery that we described earlier, a non-canonical mitophagy pathway which does not require LC3 lipidation was reported to occur in cardiomyocytes upon ischemic stress and starvation as well as in HeLa cells upon starvation and hypoxia (Hirota et al., 2015; Saito et al., 2019). In cardiomyocytes, this pathway depends on ULK1 but not on ATG5 and does not involve LC3-positive autophagosomes; instead it is mediated by Rab9 positive membranes/vesicles which drive lysosomal degradation of mitochondria. It is currently unknown whether ubiquitin signals and receptor proteins are involved in ULK1-Rab9 dependent mitophagy (Saito et al., 2019) (Figure 1D). Rab9-mediated autophagy has been previously reported as a non-canonical/alternative non-selective autophagy pathway in cells lacking ATG5, where it was also suggested that it plays a role in mitochondrial clearance during erythrocyte differentiation (Nishida et al., 2009).
It is important to mention here that mitochondrial fission (the separation from the network or fragmentation), appears to be an essential (but not sufficient) step for mitophagy (Twig et al., 2008; Williams and Ding, 2018). A key regulator of mitochondrial fission is Drp1 - a member of the dynamin family of large GTPases (Labrousse et al., 1999). Although multiple reports have shown that Drp1 is required for both PINK1-dependent and independent mitophagy pathways (Lee et al., 2011; Kageyama et al., 2014; Wu et al., 2016; Li et al., 2019; Zachari et al., 2019), other studies suggest that it might be dispensable for mitophagy (Murakawa et al., 2015; Yamashita et al., 2016; Burman et al., 2017). Thus, mitophagy dependency on Drp1 could be potentially context specific and further investigation is needed to understand its role in this pathway. A generalized illustration of our current understanding of mitophagy signaling is shown in Figure 2.
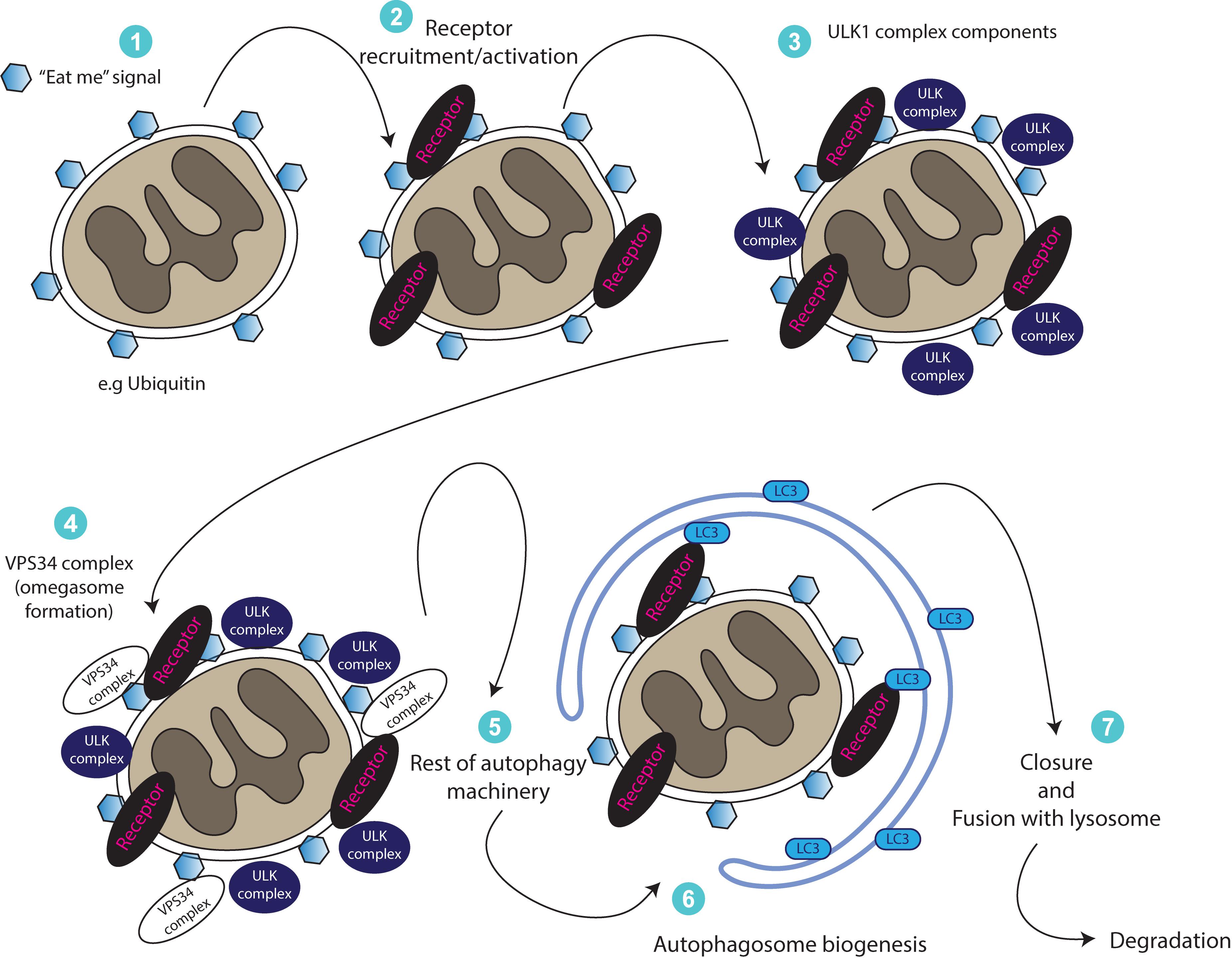
Figure 2. A generalized illustration of mitophagy. Upon damage or other stimulus, mitochondria become positive for molecules resulting in an “eat me” signal. This results in recruitment (or activation of already existing) receptor proteins, which regulate the recruitment of forming autophagosomes or that of early autophagy machinery proteins (ULK complex, VPS34 complex) for the generation of a double membrane mitophagosome. Ultimately, mitophagosomes fuse with the lysosomes to degrade themselves.
Does mTORC1 Regulate Mitophagy?
As mentioned earlier, one of the master regulators of bulk autophagy is mTORC1, which when active, suppresses autophagy via direct phosphorylation of the ULK complex components ULK1 and ATG13 (Ganley et al., 2009; Hosokawa et al., 2009a; Kim et al., 2011; Puente et al., 2016). Even though mTORC1 has a well-established role during starvation-induced autophagy, its role during mitophagy is unclear. Inhibition of mTORC1 with rapamycin has been reported to improve mitochondrial health (Nacarelli et al., 2018). Studies with CCCP and FCCP in a non-mitophagy context, have reported that these proton uncouplers cause a reduction in mTORC1 activity, suggesting that PINK1/Parkin mitophagy might require mTOR inactivation as bulk autophagy does (Inoki et al., 2003; Kim et al., 2013; Bartolome et al., 2017). Furthermore, recent reports suggest that mTORC1 hyperactivation can have an inhibitory effect on PINK1/Parkin mediated mitophagy as well as general autophagy (Bartolome et al., 2014, 2017; Bordi et al., 2019). Importantly, hypoxia leads to a reduction in mTORC1 activity as well, meaning that mTORC1 might play a role in hypoxia-induced mitophagy (Vadysirisack and Ellisen, 2012). Ivermectin did not appear to affect mTORC1 activity in the time points and concentrations we used to induce mitophagy (although treatments of 24–48 h have been shown to have an inhibitory effect on mTORC1) (Dou et al., 2016; Liu et al., 2019; Zachari et al., 2019). This might mean that the ULK complex can mediate mitophagosome formation even if it is phosphorylated by mTORC1 or that mitophagy is mediated by a pool of ULK complex that has escaped inhibition of mTORC1. Vargas et al. (2019), showed that mitophagy-induced due to ULK1 targeting to mitochondria was unaffected by mTOR overexpression and concomitant increase in ULK1 phosphorylation. It is important to mention here that amino acid (or full nutrient) starvation has been shown to block mitophagy in multiple cell lines as a result of reduced mitochondrial fission, even though mTORC1 activity is lost in these conditions (Gomes et al., 2011; Rambold et al., 2011). Thus, mTORC1 inactivation does not necessarily lead to mitophagy induction. Most of the mitophagy studies do not evaluate mTORC1 activity and our knowledge on this is limited. In conclusion further research is required to understand its involvement during mitophagy. Of note, AMPK appears to play an important (but not fully understood) role during mitophagy in multiple contexts and it is worth mentioning that mTORC1 regulates AMPK and that both these kinases can potentially influence mitophagy via ULK1. The literature on this topic has recently been reviewed (Herzig and Shaw, 2018).
Is There a Membrane Source Specific for Mitophagy?
The origin of the autophagosomal membrane during bulk autophagy induced by amino acid starvation is a long-standing question: autophagosomes form de novo and the membranes contributing to autophagosomal growth are devoid of protein-markers of the donor membranous compartments, making it hard to identify their provenance. Different organelles and membrane compartments have been proposed to contribute membrane to the autophagosome forming upon amino acid starvation, and these include the ER, Golgi, plasma membrane, mitochondria, and endosomes (Ktistakis and Tooze, 2016; Wei et al., 2018). As discussed earlier, during amino acid starvation autophagosomes form in ER platforms called omegasomes which are marked by the omegasome marker DFCP1 (Axe et al., 2008). Thus, a candidate membrane donor has been proposed to be the ER, with contributions from virtually all intracellular membranes in a way not entirely clear (Ktistakis, 2020). When it comes to mitophagy and other types of selective autophagy our understanding of the membrane source(s), and whether this differs from bulk autophagy, is even less clear. For Ivermectin-induced mitophagy we recently showed that at an early step of the process mitochondria became entrapped within ER strands prior to omegasome formation and mitophagosome generation (Zachari et al., 2019). In our analysis by both live imaging and electron tomography it appeared that the autophagosomal membrane engulfing the mitochondria grew as an extension of the neighboring ER strands-such close apposition of ER with targeted mitochondria was evident in some older publications studying PINK1/Parkin-induced mitophagy (discussed in Zachari et al., 2019). Since observations from other research groups support the notion that mitophagosomes (independently of induction mode) form within omegasomes (Yang and Yang, 2013; Wong and Holzbaur, 2014; Gelmetti et al., 2017; Hsieh and Yang, 2019) it is possible that the ER plays a key role as a membrane source for mitophagy in general. It will be interesting to determine if the different ER proteins that have been reported to be involved in autophagosome biogenesis upon amino acid starvation are also important for mitophagy as well (Walker and Ktistakis, 2019; Ktistakis, 2020). It was recently reported that in yeast mitochondria-ER contact sites are crucial for mitophagy (and pexophagy) to occur (Bockler and Westermann, 2014; Liu et al., 2018). Given the importance of ER-mitochondrial contacts in autophagosome formation in general, it is likely that they are crucial for mitophagy in mammals too, although this is yet to be confirmed experimentally (Hamasaki et al., 2013).
Conclusion and Future Perspectives
Our understanding of mitophagy signaling process and function has significantly advanced in the past decade. It is now clear that multiple proteins are involved in the regulation of mitophagy and various physiological events or types of stress can induce different mitophagy pathways both in vivo and in vitro. The PINK1/Parkin pathway has attracted a lot of attention, partially due to the importance of these proteins in Parkinson’s disease and undoubtedly as a result of the detailed mechanistic studies that have dissected it. However, it is now evident that this pathway is not required for the regulation of basal mitophagy (as well as other types of mitophagy) in mice and it might be more relevant in clearing mitochondria following particular damage/stress. Thus, more work is required to understand its physiological relevance (McWilliams et al., 2018; Drake et al., 2019). In terms of other types of mitophagy, there are still a lot of unanswered questions remaining in the field. For example, what kind of damage exactly do the mitophagy-inducing agents cause to the mitochondria? How can this relate to mitochondrial stress in humans (e.g., exposure to environmental hazards or aging)? How can this lead to disease? Can mitophagy be targeted for the development of therapeutics? Related to the last question, biochemical advances in the autophagy field, led to the generation of therapeutically promising compounds called “AUTACs” which can bind to mitochondria causing their ubiquitylation and induction of mitophagy (Takahashi et al., 2019). This way, AUTACs were shown to successfully drive mitochondria to the autophagosomal lumen for degradation, opening up an exciting new era in preclinical research on selective autophagy and its potential in treatment development.
Author Contributions
MZ and NK wrote and edited the manuscript.
Funding
Our work is supported by the BBSRC and the MRC. Work in the NK lab was funded by the Biotechnology and Biological Sciences Research Council (Grant Number: BB/K019155/1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Ian Ganley and his laboratory (especially Dr. Lambert Montava Garriga) for critical discussions on the topic of mitophagy.
References
Alers, S., Loffler, A. S., Paasch, F., Dieterle, A. M., Keppeler, H., Lauber, K., et al. (2011). Atg13 and Fip200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy 7, 1423–1433.
Allen, G. F., Toth, R., James, J., and Ganley, I. G. (2013). Loss of iron triggers Pink1/Parkin-independent mitophagy. EMBO Rep. 14, 1127–1135. doi: 10.1038/embor.2013.168
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi: 10.1083/jcb.200803137
Bartolome, A., Garcia-Aguilar, A., Asahara, S. I., Kido, Y., Guillen, C., Pajvani, U. B., et al. (2017). Mtorc1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol. Cell. Biol. 37, e441–e417.
Bartolome, A., Kimura-Koyanagi, M., Asahara, S., Guillen, C., Inoue, H., Teruyama, K., et al. (2014). Pancreatic beta-cell failure mediated by mtorc1 hyperactivity and autophagic impairment. Diabetes 63, 2996–3008. doi: 10.2337/db13-0970
Bhujabal, Z., Birgisdottir, A. B., Sjottem, E., Brenne, H. B., Overvatn, A., Habisov, S., et al. (2017). Fkbp8 recruits Lc3A to mediate Parkin-independent mitophagy. EMBO Rep. 18, 947–961. doi: 10.15252/embr.201643147
Bingol, B., and Sheng, M. (2016). Mechanisms of mitophagy: Pink1, Parkin, Usp30 and beyond. Free Radic. Biol. Med. 100, 210–222. doi: 10.1016/j.freeradbiomed.2016.04.015
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase Usp30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Bockler, S., and Westermann, B. (2014). Mitochondrial Er contacts are crucial for mitophagy in yeast. Dev. Cell 28, 450–458. doi: 10.1016/j.devcel.2014.01.012
Bordi, M., Darji, S., Sato, Y., Mellen, M., Berg, M. J., Kumar, A., et al. (2019). mtor hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 10:563.
Burman, J. L., Pickles, S., Wang, C., Sekine, S., Vargas, J. N. S., Zhang, Z., et al. (2017). Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 216, 3231–3247. doi: 10.1083/jcb.201612106
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the Pgam5 phosphatase and Ck2 controls receptor-mediated mitophagy. Mol. Cell 54, 362–377. doi: 10.1016/j.molcel.2014.02.034
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Dooley, H. C., Razi, M., Polson, H. E., Girardin, S. E., Wilson, M. I., and Tooze, S. A. (2014). Wipi2 links Lc3 conjugation with Pi3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 55, 238–252. doi: 10.1016/j.molcel.2014.05.021
Dou, Q., Chen, H. N., Wang, K., Yuan, K., Lei, Y., Li, K., et al. (2016). Ivermectin induces cytostatic autophagy by blocking the Pak1/Akt axis in breast cancer. Cancer Res. 76, 4457–4469. doi: 10.1158/0008-5472.can-15-2887
Drake, J. C., Laker, R. C., Wilson, R. J., Zhang, M., and Yan, Z. (2019). Exercise-induced mitophagy in skeletal muscle occurs in the absence of stabilization of Pink1 on mitochondria. Cell Cycle 18, 1–6. doi: 10.1080/15384101.2018.1559556
Esteban-Martinez, L., Sierra-Filardi, E., Mcgreal, R. S., Salazar-Roa, M., Marino, G., Seco, E., et al. (2017). Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 36, 1688–1706. doi: 10.15252/embj.201695916
Ganley, I. G., Lam, Du, H., Wang, J., Ding, X., Chen, S., et al. (2009). Ulk1.Atg13.Fip200 complex mediates mtor signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305. doi: 10.1074/jbc.m900573200
Gao, F., Chen, D., Si, J., Hu, Q., Qin, Z., Fang, M., et al. (2015). The mitochondrial protein Bnip3L is the substrate of Park2 and mediates mitophagy in Pink1/Park2 pathway. Hum. Mol. Genet. 24, 2528–2538. doi: 10.1093/hmg/ddv017
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). Pink1/Parkin-mediated mitophagy is dependent on Vdac1 and p62/Sqstm1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Gelmetti, V., De Rosa, P., Torosantucci, L., Marini, E. S., Romagnoli, A., Di Rienzo, M., et al. (2017). Pink1 and Becn1 relocalize at mitochondria-associated membranes during mitophagy and promote Er-mitochondria tethering and autophagosome formation. Autophagy 13, 654–669. doi: 10.1080/15548627.2016.1277309
Georgakopoulos, N. D., Wells, G., and Campanella, M. (2017). The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 13, 136–146. doi: 10.1038/nchembio.2287
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. doi: 10.1038/ncb2220
Greene, A. W., Grenier, K., Aguileta, M. A., Muise, S., Farazifard, R., Haque, M. E., et al. (2012). Mitochondrial processing peptidase regulates Pink1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385. doi: 10.1038/embor.2012.14
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at Er-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Hanna, R. A., Quinsay, M. N., Orogo, A. M., Giang, K., Rikka, S., and Gustafsson, A. B. (2012). Microtubule-associated protein 1 light chain 3 (Lc3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104. doi: 10.1074/jbc.m111.322933
Hara, T., Takamura, A., Kishi, C., Iemura, S., Natsume, T., Guan, J. L., et al. (2008). Fip200, a Ulk-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181, 497–510. doi: 10.1083/jcb.200712064
Herzig, S., and Shaw, R. J. (2018). Ampk: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135. doi: 10.1038/nrm.2017.95
Hirota, Y., Yamashita, S., Kurihara, Y., Jin, X., Aihara, M., Saigusa, T., et al. (2015). Mitophagy is primarily due to alternative autophagy and requires the Mapk1 and Mapk14 signaling pathways. Autophagy 11, 332–343. doi: 10.1080/15548627.2015.1023047
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009a). Nutrient-dependent mtorc1 association with the Ulk1-Atg13-Fip200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991. doi: 10.1091/mbc.e08-12-1248
Hosokawa, N., Sasaki, T., Iemura, S., Natsume, T., Hara, T., and Mizushima, N. (2009b). Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5, 973–979. doi: 10.4161/auto.5.7.9296
Hsieh, C. W., and Yang, W. Y. (2019). Omegasome-proximal PtdIns(4,5)P2 couples F-actin mediated mitoaggregate disassembly with autophagosome formation during mitophagy. Nat. Commun. 10:969.
Hu, M., Luo, Q., Alitongbieke, G., Chong, S., Xu, C., Xie, L., et al. (2017). Celastrol-induced Nur77 interaction with Traf2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol. Cell 66, 141–153.e6. doi: 10.1016/j.molcel.2017.03.008
Inoki, K., Zhu, T., and Guan, K. L. (2003). Tsc2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/s0092-8674(03)00929-2
Itakura, E., Kishi-Itakura, C., Koyama-Honda, I., and Mizushima, N. (2012). Structures containing Atg9A and the Ulk1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 125, 1488–1499. doi: 10.1242/jcs.094110
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates Pink1 import and proteolytic destabilization by Parl. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Juarez, M., Schcolnik-Cabrera, A., and Duenas-Gonzalez, A. (2018). The multitargeted drug ivermectin: from an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 8, 317–331.
Kageyama, Y., Hoshijima, M., Seo, K., Bedja, D., Sysa-Shah, P., Andrabi, S. A., et al. (2014). Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 33, 2798–2813. doi: 10.15252/embj.201488658
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). Pink1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kanki, T., Wang, K., Cao, Y., Baba, M., and Klionsky, D. J. (2009). Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 17, 98–109. doi: 10.1016/j.devcel.2009.06.014
Karanasios, E., Stapleton, E., Manifava, M., Kaizuka, T., Mizushima, N., Walker, S. A., et al. (2013). Dynamic association of the Ulk1 complex with omegasomes during autophagy induction. J. Cell Sci. 126, 5224–5238. doi: 10.1242/jcs.132415
Karanasios, E., Walker, S. A., Okkenhaug, H., Manifava, M., Hummel, E., Zimmermann, H., et al. (2016). Autophagy initiation by Ulk complex assembly on Er tubulovesicular regions marked by Atg9 vesicles. Nat. Commun. 7:12420.
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by Pink1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139.
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). Ampk and mtor regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, M. J., Yoon, J. H., and Ryu, J. H. (2016). Mitophagy: a balance regulator of Nlrp3 inflammasome activation. BMB Rep. 49, 529–535. doi: 10.5483/bmbrep.2016.49.10.115
Kim, S. G., Hoffman, G. R., Poulogiannis, G., Buel, G. R., Jang, Y. J., Lee, K. W., et al. (2013). Metabolic stress controls mtorc1 lysosomal localization and dimerization by regulating the Ttt-Ruvbl1/2 complex. Mol. Cell 49, 172–185. doi: 10.1016/j.molcel.2012.10.003
Kirkin, V. (2019). History of the selective autophagy research: how did it begin and where does it stand today? J. Mol. Biol. 432, 3–27. doi: 10.1016/j.jmb.2019.05.010
Koentjoro, B., Park, J. S., and Sue, C. M. (2017). Nix restores mitophagy and mitochondrial function to protect against Pink1/Parkin-related Parkinson’s disease. Sci. Rep. 7:44373.
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). Pink1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Ktistakis, N. T. (2020). Er platforms mediating autophagosome generation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1865, S1388–S1981.
Ktistakis, N. T., and Tooze, S. A. (2016). Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 26, 624–635. doi: 10.1016/j.tcb.2016.03.006
Kundu, M., Lindsten, T., Yang, C. Y., Wu, J., Zhao, F., Zhang, J., et al. (2008). Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112, 1493–1502. doi: 10.1182/blood-2008-02-137398
Labrousse, A. M., Zappaterra, M. D., Rube, D. A., and Van Der Bliek, A. M. (1999). C. elegans dynamin-related protein Drp-1 controls severing of the mitochondrial outer membrane. Mol. Cell 4, 815–826. doi: 10.1016/s1097-2765(00)80391-3
Laker, R. C., Drake, J. C., Wilson, R. J., Lira, V. A., Lewellen, B. M., Ryall, K. A., et al. (2017). Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8:548.
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., et al. (2019). Bnip3L/Nix and Fundc1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15, 1182–1198. doi: 10.1080/15548627.2019.1580095
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase Pink1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, Y., Lee, H. Y., Hanna, R. A., and Gustafsson, A. B. (2011). Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931.
Li, S., Lin, Q., Shao, X., Zhu, X., Wu, J., Wu, B., et al. (2019). Drp1-regulated Park2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic. Biol. Med doi: 10.1016/j.freeradbiomed.2019.12.005 [Epub ahead of print].
Liu, J., Liang, H., Chen, C., Wang, X., Qu, F., Wang, H., et al. (2019). Ivermectin induces autophagy-mediated cell death through the Akt/mtor signaling pathway in glioma cells. Biosci. Rep. 39:BSR20192489.
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein Fundc1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, X., Wen, X., and Klionsky, D. J. (2018). Er-mitochondria contacts are required for pexophagy in Saccharomyces cerevisiae. Contact 2. doi: 10.1177/2515256418821584
Livingston, M. J., Wang, J., Zhou, J., Wu, G., Ganley, I. G., Hill, J. A., et al. (2019). Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 15, 2142–2162. doi: 10.1080/15548627.2019.1615822
McWilliams, T. G., Prescott, A. R., Allen, G. F., Tamjar, J., Munson, M. J., Thomson, C., et al. (2016). mito-Qc illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 214, 333–345. doi: 10.1083/jcb.201603039
McWilliams, T. G., Prescott, A. R., Montava-Garriga, L., Ball, G., Singh, F., Barini, E., et al. (2018). Basal mitophagy occurs independently of Pink1 in mouse tissues of high metabolic demand. Cell Metab. 27, 4395–449.e5.
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T., and Ohsumi, Y. (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 15, 1101–1111. doi: 10.1091/mbc.e03-09-0704
Montava-Garriga, L., and Ganley, I. G. (2019). Outstanding questions in mitophagy: what we do and do not know. J. Mol. Biol. 432, 206–230. doi: 10.1016/j.jmb.2019.06.032
Moore, A. S., and Holzbaur, E. L. (2016). Spatiotemporal dynamics of autophagy receptors in selective mitophagy. Autophagy 12, 1956–1957. doi: 10.1080/15548627.2016.1212788
Moyzis, A. G., Sadoshima, J., and Gustafsson, A. B. (2015). Mending a broken heart: the role of mitophagy in cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 308, H183–H192.
Murakawa, T., Okamoto, K., Omiya, S., Taneike, M., Yamaguchi, O., and Otsu, K. (2019). A mammalian mitophagy receptor, Bcl2-L-13, recruits the Ulk1 complex to induce mitophagy. Cell Rep. 26, 338–345.e6. doi: 10.1016/j.celrep.2018.12.050
Murakawa, T., Yamaguchi, O., Hashimoto, A., Hikoso, S., Takeda, T., Oka, T., et al. (2015). Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6:7527.
Murphy, M. P., and Hartley, R. C. (2018). Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 17, 865–886. doi: 10.1038/nrd.2018.174
Nacarelli, T., Azar, A., Altinok, O., Orynbayeva, Z., and Sell, C. (2018). Rapamycin increases oxidative metabolism and enhances metabolic flexibility in human cardiac fibroblasts. Geroscience [Epub ahead of print].
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). Pink1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Ney, P. A. (2015). Mitochondrial autophagy: origins, significance, and role of Bnip3 and Nix. Biochim. Biophys. Acta 1853, 2775–2783. doi: 10.1016/j.bbamcr.2015.02.022
Nguyen, T. N., Padman, B. S., Usher, J., Oorschot, V., Ramm, G., and Lazarou, M. (2016). Atg8 family Lc3/Gabarap proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during Pink1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874. doi: 10.1083/jcb.201607039
Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., et al. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. doi: 10.1038/nature08455
Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., et al. (2012). Pink1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3:1016.
Okatsu, K., Uno, M., Koyano, F., Go, E., Kimura, M., Oka, T., et al. (2013). A dimeric Pink1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem. 288, 36372–36384. doi: 10.1074/jbc.m113.509653
Padman, B. S., Nguyen, T. N., Uoselis, L., Skulsuppaisarn, M., Nguyen, L. K., and Lazarou, M. (2019). Lc3/Gabaraps drive ubiquitin-independent recruitment of optineurin and Ndp52 to amplify mitophagy. Nat. Commun. 10:408.
Palikaras, K., Daskalaki, I., Markaki, M., and Tavernarakis, N. (2017). Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 178, 157–174. doi: 10.1016/j.pharmthera.2017.04.005
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Park, S., Choi, S. G., Yoo, S. M., Son, J. H., and Jung, Y. K. (2014). Choline dehydrogenase interacts with Sqstm1/p62 to recruit Lc3 and stimulate mitophagy. Autophagy 10, 1906–1920. doi: 10.4161/auto.32177
Princely Abudu, Y., Pankiv, S., Mathai, B. J., Hakon Lystad, A., Bindesboll, C., et al. (2019). Nipsnap1 and Nipsnap2 Act as “Eat Me” signals for mitophagy. Dev. Cell 49:e12.
Puente, C., Hendrickson, R. C., and Jiang, X. (2016). Nutrient-regulated phosphorylation of Atg13 inhibits starvation-induced autophagy. J. Biol. Chem. 291, 6026–6035. doi: 10.1074/jbc.m115.689646
Rambold, A. S., Kostelecky, B., Elia, N., and Lippincott-Schwartz, J. (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U.S.A. 108, 10190–10195. doi: 10.1073/pnas.1107402108
Ravenhill, B. J., Boyle, K. B., Von Muhlinen, N., Ellison, C. J., Masson, G. R., Otten, E. G., et al. (2019). The cargo receptor Ndp52 initiates selective autophagy by recruiting the ulk complex to cytosol-invading bacteria. Mol. Cell 74, 320–329.e6. doi: 10.1016/j.molcel.2019.01.041
Rikka, S., Quinsay, M. N., Thomas, R. L., Kubli, D. A., Zhang, X., Murphy, A. N., et al. (2011). Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 18, 721–731. doi: 10.1038/cdd.2010.146
Rogov, V. V., Suzuki, H., Marinkovic, M., Lang, V., Kato, R., Kawasaki, M., et al. (2017). Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with Lc3 proteins. Sci. Rep. 7:1131.
Rojansky, R., Cha, M. Y., and Chan, D. C. (2016). Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on Parkin and Mul1. Elife 5:e17896.
Saito, T., Nah, J., Oka, S. I., Mukai, R., Monden, Y., Maejima, Y., et al. (2019). An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Invest. 129, 802–819. doi: 10.1172/jci122035
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). Nix is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 19500–19505. doi: 10.1073/pnas.0708818104
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). Pink1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2:1002.
Sin, J., Andres, A. M., Taylor, D. J., Weston, T., Hiraumi, Y., Stotland, A., et al. (2016). Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 12, 369–380. doi: 10.1080/15548627.2015.1115172
Smith, M. D., Harley, M. E., Kemp, A. J., Wills, J., Lee, M., Arends, M., et al. (2018). Ccpg1 is a non-canonical autophagy cargo receptor essential for Er-Phagy and pancreatic Er proteostasis. Dev. Cell 44, 217–232.e11. doi: 10.1016/j.devcel.2017.11.024
Sun, N., Malide, D., Liu, J., Rovira, I. I., Combs, C. A., and Finkel, T. (2017). A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 12, 1576–1587. doi: 10.1038/nprot.2017.060
Sun, N., Yun, J., Liu, J., Malide, D., Liu, C., Rovira, I. I., et al. (2015). Measuring in vivo mitophagy. Mol. Cell 60, 685–696. doi: 10.1016/j.molcel.2015.10.009
Takahashi, D., Moriyama, J., Nakamura, T., Miki, E., Takahashi, E., Sato, A., et al. (2019). Autacs: cargo-specific degraders using selective autophagy. Mol. Cell. 76, 797–810.10.
Tang, C., Han, H., Liu, Z., Liu, Y., Yin, L., Cai, J., et al. (2019). Activation of Bnip3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 10:677.
Tang, Y. C., Tian, H. X., Yi, T., and Chen, H. B. (2016). The critical roles of mitophagy in cerebral ischemia. Protein Cell 7, 699–713. doi: 10.1007/s13238-016-0307-0
Tong, M., Saito, T., Zhai, P., Oka, S. I., Mizushima, W., Nakamura, M., et al. (2019). Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 124, 1360–1371. doi: 10.1161/circresaha.118.314607
Turco, E., Witt, M., Abert, C., Bock-Bierbaum, T., Su, M. Y., Trapannone, R., et al. (2019). Fip200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol. Cell 74, 330–346.e11. doi: 10.1016/j.molcel.2019.01.035
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Um, J. H., and Yun, J. (2017). Emerging role of mitophagy in human diseases and physiology. BMB Rep. 50, 299–307. doi: 10.5483/bmbrep.2017.50.6.056
Vadysirisack, D. D., and Ellisen, L. W. (2012). mtor activity under hypoxia. Methods Mol. Biol. 821, 45–58. doi: 10.1007/978-1-61779-430-8_4
Vargas, J. N. S., Wang, C., Bunker, E., Hao, L., Maric, D., Schiavo, G., et al. (2019). Spatiotemporal control of Ulk1 activation by Ndp52 and Tbk1 during selective autophagy. Mol. Cell 74:e6.
Walker, S. A., and Ktistakis, N. T. (2019). Autophagosome biogenesis machinery. J. Mol. Biol. [Epub ahead of print].
Wall, C. E., Rose, C. M., Adrian, M., Zeng, Y. J., Kirkpatrick, D. S., and Bingol, B. (2019). Ppef2 opposes Pink1-mediated mitochondrial quality control by dephosphorylating ubiquitin. Cell Rep. 29, 3280–3292.e7. doi: 10.1016/j.celrep.2019.10.130
Wauer, T., Simicek, M., Schubert, A., and Komander, D. (2015). Mechanism of phospho-ubiquitin-induced Parkin activation. Nature 524, 370–374. doi: 10.1038/nature14879
Wei, Y., Chiang, W. C., Sumpter, R. Jr., Mishra, P., and Levine, B. (2017). Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168, 224–238.e10. doi: 10.1016/j.cell.2016.11.042
Wei, Y., Liu, M., Li, X., Liu, J., and Li, H. (2018). Origin of the autophagosome membrane in mammals. Biomed. Res. Int. 2018:1012789.
Williams, J. A., and Ding, W. X. (2018). Mechanisms, pathophysiological roles and methods for analyzing mitophagy - recent insights. Biol. Chem. 399, 147–178. doi: 10.1515/hsz-2017-0228
Wong, Y. C., and Holzbaur, E. L. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an Als-linked mutation. Proc. Natl. Acad. Sci. U.S.A. 111, E4439–E4448.
Wu, W., Lin, C., Wu, K., Jiang, L., Wang, X., Li, W., et al. (2016). Fundc1 regulates mitochondrial dynamics at the Er-mitochondrial contact site under hypoxic conditions. EMBO J. 35, 1368–1384. doi: 10.15252/embj.201593102
Wu, W., Tian, W., Hu, Z., Chen, G., Huang, L., Li, W., et al. (2014). Ulk1 translocates to mitochondria and phosphorylates Fundc1 to regulate mitophagy. EMBO Rep. 15, 566–575. doi: 10.1002/embr.201438501
Yamano, K., Wang, C., Sarraf, S. A., Munch, C., Kikuchi, R., Noda, N. N., et al. (2018). Endosomal Rab cycles regulate parkin-mediated mitophagy. Elife 7:e31326.
Yamano, K., and Youle, R. J. (2013). Pink1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi: 10.4161/auto.24633
Yamashita, S. I., Jin, X., Furukawa, K., Hamasaki, M., Nezu, A., Otera, H., et al. (2016). Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J. Cell Biol. 215, 649–665. doi: 10.1083/jcb.201605093
Yan, C., Gong, L., Chen, L., Xu, M., Abou-Hamdan, H., Tang, M., et al. (2019). Phb2 (prohibitin 2) promotes Pink1-Prkn/Parkin-dependent mitophagy by the Parl-Pgam5-Pink1 axis. Autophagy 16, 419–434. doi: 10.1080/15548627.2019.1628520
Yang, J. Y., and Yang, W. Y. (2013). Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 4:2428.
Yuan, Y., Zheng, Y., Zhang, X., Chen, Y., Wu, X., Wu, J., et al. (2017). Bnip3L/Nix-mediated mitophagy protects against ischemic brain injury independent of Park2. Autophagy 13, 1754–1766. doi: 10.1080/15548627.2017.1357792
Yun, J., Puri, R., Yang, H., Lizzio, M. A., Wu, C., Sheng, Z. H., et al. (2014). Mul1 acts in parallel to the Pink1/parkin pathway in regulating mitofusin and compensates for loss of Pink1/parkin. Elife 3:e01958.
Zachari, M., Gudmundsson, S. R., Li, Z., Manifava, M., Shah, R., Smith, M., et al. (2019). Selective autophagy of mitochondria on a ubiquitin-endoplasmic-reticulum platform. Dev. Cell. 50, 627–643.e5. doi: 10.1016/j.devcel.2019.06.016
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an Hif-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. doi: 10.1074/jbc.m800102200
Zhang, T., Xue, L., Li, L., Tang, C., Wan, Z., Wang, R., et al. (2016). Bnip3 protein suppresses Pink1 kinase proteolytic cleavage to promote mitophagy. J. Biol. Chem. 291, 21616–21629. doi: 10.1074/jbc.m116.733410
Zhang, W., Ren, H., Xu, C., Zhu, C., Wu, H., Liu, D., et al. (2016). Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5:e21407.
Keywords: autophagy, mitophagy, mTOR, endoplasmic reticulum, membrane potential
Citation: Zachari M and Ktistakis NT (2020) Mammalian Mitophagosome Formation: A Focus on the Early Signals and Steps. Front. Cell Dev. Biol. 8:171. doi: 10.3389/fcell.2020.00171
Received: 14 January 2020; Accepted: 02 March 2020;
Published: 18 March 2020.
Edited by:
Konstantinos Palikaras, Foundation for Research and Technology-Hellas, GreeceReviewed by:
Shinji Saiki, Juntendo University, JapanMorten Scheibye-Knudsen, University of Copenhagen, Denmark
Copyright © 2020 Zachari and Ktistakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Zachari, bS56YWNoYXJpQGR1bmRlZS5hYy51aw==; Nicholas T. Ktistakis, bmljaG9sYXMua3Rpc3Rha2lzQGJhYnJhaGFtLmFjLnVr