 Shanshan Huang
Shanshan Huang Jing Che
Jing Che Qian Chu
Qian Chu Peng Zhang
Peng Zhang- 1Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2College of Life Sciences, Wuhan University, Wuhan, China
The increasing risk of long-term adverse effects from radiotherapy on the cardiovascular structure is receiving increasing attention. However, the mechanisms underlying this increased risk remain poorly understood. Recently, the nucleotide-binding domain and leucine-rich-repeat-containing family pyrin 3 (NLRP3) inflammasome was suggested to play a critical role in radiation-induced cardiovascular injury. However, the relationship between ionizing radiation and the NLRP3 inflammasome in acute and chronic inflammation is complex. We reviewed literature detailing pathological changes and molecular mechanisms associated with radiation-induced damage to the cardiovascular structure, with a specific focus on NLRP3 inflammasome-related cardiovascular diseases. We also summarized possible therapeutic strategies for the prevention of radiation-induced heart disease (RIHD).
Introduction
Chronic health problems in cancer survivors include cardiovascular diseases (CVDs), which are the primary causes of morbidity and mortality in this population. There is a significant correlation between increased risk of CVDs and chest irradiation, which results in short-term and long-term cardiovascular complications in cancer survivors (Abe et al., 2016). Although acute pericarditis can result from high dose radiation, radiation-induced heart injury may take decades to become symptomatic (Nielsen et al., 2017). Common CVDs include accelerated atherosclerosis, myocardial remodeling, fibrosis, and injury to cardiac valves. Recent epidemiological, clinical, and preclinical studies have shown strong evidence that ionizing radiation can cause cardiovascular injury (Stewart et al., 2013; Yusuf et al., 2017; Wang H. et al., 2019). The underlying mechanisms of radiation-induced cardiovascular injury have not been characterized due to the wide spectrum of complex effects of ionizing radiation on the cardiovascular system.
Recent studies showed that NLRP3 inflammasome upregulation and activation were associated with radiation damage (Wei et al., 2019). However, the role of the NLRP3 inflammasome in radiation-induced damage requires further characterization. In this review, we focused on relevant epidemiological and clinical evidence for radiation-induced CVDs. We also reviewed the role of the NLRP3 inflammasome in radiation-induced injury, and highlighted possible therapeutic strategies for preventing radiation-induced heart disease (RIHD).
Overview of Radiation-Induced Cardiovascular Diseases
Clinical Evidence for Radiation-Induced Cardiovascular Diseases
Survivors of childhood cancer and Hodgkin’s lymphoma who experienced conventional fractionated radiotherapy (total tumor dose of 30–40 Gy) are at greater risk of late cardiac death than the general population (Stewart et al., 2013). Long-term follow-up of over 1,400 patients with Hodgkin’s lymphoma with a history of radiotherapy showed a significant increase in standardized incidence ratio of myocardial infarction (MI), angina, valve disease, and congestive heart failure. In addition, patients who received radiotherapy at a young age (<20 years-old) are at significantly higher risk for MI (5.4 standard incidence ratio) than patients irradiated at age 36–40 (2.6 standard incidence ratio). Anthracycline chemotherapy further increased the risk of cardiovascular events in young patients with a history of thoracic irradiation (Aleman et al., 2007). Head and neck cancer patients with a radical radiotherapy history often experience a significant dose of radiation to the carotid arteries. Subsequent vascular injury may result in atherosclerosis, and increased risk of stroke and transient ischemic attack (TIA) (Scott et al., 2009).
Mulrooney et al. (2009) and Tukenova et al. (2010) showed a relationship between cardiovascular death and cardiac radiation dose in childhood cancer survivors. These studies showed that the relative risk of cardiovascular death caused by radiotherapy was significantly associated with mean heart dose, particularly when cardiac doses were ≥ 15 Gy. Mean heart doses over 35 Gy were linked with even greater hazard ratios. Prospective studies showed that the majority of survivors of Hodgkin’s lymphoma exhibited asymptomatic cardiovascular abnormalities (e.g., conduction defects, valve dysfunction, and diastolic dysfunction). These findings suggested that occult RIHD may be underestimated in this patient population. Whether low-dose irradiation damages the cardiovascular system is unknown.
Mean heart doses and dose distribution to certain cardiac structures were estimated in patients with breast cancer who underwent post-operative irradiation (50 Gy in 25 fractions) up to the early 1990s (Taylor et al., 2007). Left-side breast cancer patients, of whom some received internal mammary chain radiation, experienced total average doses of 3–17 Gy across the entire heart. A large-scale investigation suggested that exposure of the internal mammary chain to radiation correlated with a significantly increased risk of cardiovascular abnormalities (average cardiac dose was 6–15 Gy). Patients who underwent breast irradiation alone with a mean heart dose < 7 Gy were not at increased risk for development of CVD (Hooning et al., 2007). However, the effect of low dose irradiation (<2.5 Gy) on the cardiovascular system is unknown. A life-span investigation of Japanese atomic bomb survivors indicated that cardiac abnormalities such as inflammation and fibrosis were increased by exposure to low dose radiation (Shimizu et al., 2010).
Experimental Evidence for Radiation-Induced Cardiovascular Diseases
Use of experimental animals to estimate RIHD is convenient due to shorter life spans and ease of follow-up. Radiation-induced cardiovascular injury depends on timing, dose and fraction, and extent of radiation exposure to the heart and blood vessels. However, most animal studies have applied single high doses or a total high dose across a limited number of fractions (Sylvester et al., 2018). Local single high doses of radiation are more frequently applied to young adult (male) animals. Rodent models are widely used to identify potential biological mechanisms of radiation-induced cardiac injury. Larger animals, such as rats, rabbits, and cynomolgus monkeys, have been used for decades to study RIHD (Boerma, 2012).
Although myocardial changes have been shown to be similar in animals and humans, radiation-induced accelerated atherosclerosis has not been frequently observed in common animal models (Boerma et al., 2016). Several research groups have reported that increased perivascular and interstitial collagen deposition in the myocardium was observed several months after local cardiac radiation in mice (Schultz-Hector, 1992; Unthank et al., 2015). Increased collagen deposition was accompanied by local increased fibrotic cytokine expression. Boerma et al. (2013) found that induction of TGF-β expression further enhanced radiation-induced cardiac fibrosis. Moreover, this radiation-induced fibrosis worsened with loss of microvasculature density and function in the myocardium (Taunk et al., 2015).
To investigate radiation-induced accelerated atherosclerosis, inclusion of additional cardiovascular risk factors in experimental models is necessary. Genetically modified mouse models susceptible to atherogenesis, such as LDL receptor-deficient mice (Ldlr–/–) and apolipoprotein E-deficient mice (Apoe–/–), are commonly used (Schiller et al., 2001; Stewart et al., 2006). High dose radiation to the neck of Apoe–/– mice led to increased atherosclerotic plaque formation and increased macrophage abundance in the carotid artery (Hoving et al., 2008). In addition, local heart radiation resulted in microvascular injury and atherosclerosis in the coronary arteries of Apoe–/– mice that was more extensive (Gabriels et al., 2012). Pathophysiological changes in the cardiovascular systems of small animals exposed to radiation are similar to those in clinical specimens, including increased intima-media thickness and more artery wall lesions (Unthank et al., 2019).
Pathological Features of Radiation-Induced Cardiovascular Diseases
Atherosclerosis and fibrosis are typical pathological changes in RIHD, but the pathophysiology of these changes is poorly understood (Yarnold and Brotons, 2010). Many of the pathological changes in vascular structure secondary to radiation may be much the same as those observed in typical atherosclerosis. However, radiation-accelerated coronary atherosclerosis results in more extensive media damage and adventitia fibrosis compared with ordinary coronary atherosclerosis. In addition, more lipid and calcium deposition has been shown to occur in radiation-induced fibrous tissue of intimal plaques than in non-irradiated atherosclerotic plaques (Virmani et al., 1999). Brosius et al. (1981) found that more than one-fourth of the major coronary arteries were nearly completely narrowed when cross-sections were evaluated. The proximal half of the right and left anterior descending coronary arteries are frequently damaged by radiation (Brosius et al., 1981). Moreover, intramyocardial coronary thickening and myocardial fibrosis of the right ventricle have been observed in response to radiation. The endocardium was significantly thickened in the right ventricle of many patients who underwent radiation (Tadic et al., 2017). Similarly, valve thickening, particularly of the tricuspid valve, occurred in over 80% of patients who underwent irradiation (Gujral et al., 2016a). Nearly all patients experienced pericardial and epicardial fibrosis following radiotherapy, and pericardial tamponade was also common.
Cervical radiotherapy-induced carotid lesions exhibit a greater degree of stenosis than non-typical atherosclerotic lesions. Radiation-induced carotid lesions are frequently observed in the external and common carotid artery. Similar to radiation-accelerated coronary atherosclerosis, ionizing irradiation-induced carotid stenosis presents a more stable phenotype than de novo atherosclerotic plaques (Fokkema et al., 2012). Macrophage infiltration was observed less frequently in response to radiation, but more fibrous plaques were present following cervical irradiation-induced carotid lesions (Gujral et al., 2016b).
Radiation and NLRP3 Inflammasome Activation
Overview of the NLRP3 Inflammasome
A sensor protein (NLRP3), an adaptor protein (apoptosis-associated speck-like protein containing a caspase recruitment domain, ASC), and an effector protein (cysteinyl aspartate specific protease 1, caspase-1) comprise the NLRP3 inflam- masome. Nucleotide-binding domain and leucine-rich-repeat-containing family pyrin 3 is comprised of three domains: an amino-terminal pyrin domain (PYD), a central NATCH domain (an evolutionarily conserved protein domain contains NLP family apoptosis inhibitor protein, MHC class II transcription activator, incompatibility locus protein from Podospora anserina, and telomerase-associated protein), and a carboxy-terminal leucine-rich repeat (LRR) domain (Schroder and Tschopp, 2010). The NATCH domain possesses ATPase activity responsible for NLRP3 self-assembly and function. The LRR domain is responsible for self-inhibition through the NATCH domain. Apoptosis-associated speck-like protein containing CARD (ASC) functions as an adaptor protein with two protein-interacting domains. The amino-terminal domain of the ASC is PYD, and the carboxy-terminal domain is the caspase recruitment domain (CARD) that interacts with inflammatory caspase-1. Upon stimulation, oligomerization of NLRP3 occurs via NATCH–NATCH domain interactions, following by the recruitment of ASC and caspase-1 via homotypic interactions with CARD. Activated caspase-1 then cleaves the pro-inflammatory cytokines IL-1β and IL-18 to their bioactive forms (Swanson et al., 2019). In addition, inflammasome activation results in pyroptosis, a form of cell death characterized by loss of cell integrity, cellular swelling, and lysis (Yuan et al., 2018). Pyroptosis correlates with active IL-1β and IL-18 release by three different mechanisms: secretion through pores formed by active gasdermin D (GSDMD) on the membrane, shedding of microvesicles, and lysosome-mediated exocytosis (Semino et al., 2018). Endogenous danger-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1), APT, and IL-1α produced by stressed cells further activate the NLRP3 inflammasome. Activation of these signaling pathways results in an enhanced inflammatory response (Baldrighi et al., 2017).
In addition to the role of canonical NLRP3 function in activation of caspase-1, non-canonical activation of NLRP3 activated caspase-4/5 in humans and caspase-11 in mice in a TLR-independent manner (Vigano et al., 2015; Yi, 2018). A recent study suggested that NIMA-related kinase 7 (NEK7) might be critical to activation of the NLRP3 inflammasome. Upon inflammasome activation, NEK7 was shown to oligomerize with NLRP3 to form a complex, which indicated that NEK7 was a component of the NLRP3 inflammasome (He Y. et al., 2016).
Radiation Triggers NLRP3 Inflammasome Activation
Inflammasome activation is considered a two-step “priming and activation” process. Priming is characterized by upregulation of the expression of inflammasome core components and induction of post-translational modifications (PTMs) such as ubiquitylation, SUMOylation, and phosphorylation (Swanson et al., 2019). In addition, interfering RNAs modulate the expression of NLRP3 at the transcriptional level (Tezcan et al., 2019). The second step initiates inflammasome formation upon activation. Nucleotide-binding domain and leucine-rich-repeat-containing family pyrin 3 is an effector of the innate immune response that recognizes pathogen-associated molecular patterns (PAMPs) or DAMPs by engaging pattern recognition receptors (PRRs) (Kelley et al., 2019). In addition, NLRP3 can be activated by a number of other stimuli. Furthermore, non-infectious stimuli such as endogenous DAMPs and exposure to environmental irritants can activate the NLRP3 inflammasome. Most activators of NLRP3 are produced by cellular stress responses (Baldrighi et al., 2017). The mechanisms by which NLRP3 senses cellular stress and the signaling pathways involved in NLRP3 activation and inflammasome formation are poorly understood. Activation of NLRP3 may involve multiple upstream pathways, but there is no consensus model. Upstream signals of NLRP3 activation are often interrelated and overlapping, including ion internalization and efflux, lysosomal disruption, mitochondrial disorders, metabolic dysfunction, and sphingolipid metabolic changes (Latz et al., 2013).
Radiation can activate NLRP3 inflammasome via multiple mechanisms. Radiation-related potassium ion efflux and calcium flux have been studied extensively (Merrick and Bruce, 1965; Konings, 1981; Franzen et al., 1991, 1993; Todd and Mikkelsen, 1994). Calcium influx into the cytosol can occur through opening of plasma membrane channel or release from the endoplasmic reticulum (ER) intracellular Ca2+ pool. Potassium efflux functions as a counter regulator of Ca2+ influx. As such, Ca2+ influx and K+ efflux are often found to be coordinated in NLRP3 activation (Yaron et al., 2015). Adenosine triphosphate is another key signaling molecule that mediates radiation-induced biological effects (Kojima et al., 2017). Stimulation of the P2X purinoceptor 7 (P2X7) by ATP induces weak Ca2+ and Na+ influx, with concurrent K+ efflux (Gombault et al., 2012; Karmakar et al., 2016). Low intracellular K+ has been shown to activate the NLRP3 inflammasome in THP1 cells and bone-marrow-derived macrophages in vitro (Petrilli et al., 2007). Radiation can also directly induce cholesterol biosynthesis (Werner et al., 2019). Longitudinal trends in total serum cholesterol levels of atomic bomb survivors also supported the ability of radiation to induce cholesterol biosynthesis (Wong et al., 1999). Phagocytosis of cholesterol crystals results in lysosomal rupture and release of particles into the cytoplasm. Radiation can directly induce lysosomal destabilization (Persson et al., 2005). Cholesterol crystals and radiation have been shown to induce lysosomal rupture, resulting in NLRP3 activation (Duewell et al., 2010). In addition, cathepsins released from ruptured lysosomes have been shown to play a critical role in NLRP3 activation following radiation treatment (Amaral et al., 2018). However, individual knockouts of cathepsin B, X, L, or S had little effect on NLRP3 activation, which demonstrated that cathepsins may have redundant roles in activation of NLRP3 signaling (Orlowski et al., 2015). Recent studies have suggested that radiation-induced up-regulation of cellular ceramide (Kolesnick and Fuks, 2003; Sharma and Czarnota, 2019) acts as a second messenger in initiation of intrinsic apoptosis. Ceramide plays multiple pathophysiological roles during radiation-induced NLRP3 activation. Ceramide-induced cathepsin D activation has been shown to link TNFα-induced acid sphingomyelinase to Bid-related mitochondrial apoptosis (Heinrich et al., 2004). Recent studies identified plasma membrane ion channels as novel targets of ceramide, such as those responsible for Ca2+ influx and K+ efflux (Szabo et al., 1996; Lepple-Wienhues et al., 1999; Church et al., 2005). These findings show that many mechanisms of NLRP3 activation involve either Ca2+ or K+ flux.
Most radiation damage results from water radiolysis, as 80% or more of total cell mass or tissue is comprised of water. Ionizing irradiation-induced water radiolysis generates reactive oxygen species (ROS), the main source of radiation-induced tissue damage. NLPR3 protein contains a highly conserved disulfide bond connecting the PYD domain and the nucleotide-binding site domain that is highly sensitive to altered redox state (Bae and Park, 2011). Radiation-induced mitochondrial dysfunction, and release of mitochondrial ROS and DNA into the cytoplasm, are essential upstream regulators of NLRP3 activation (Chen et al., 2016). Ionizing radiation can also effectively generate ozone, which has been suggested to have a strong association with cardiovascular injury (Srebot et al., 2009; Wang M. et al., 2019). Ozone induced oxidative stress causes inflammasome activation with the release of IL-1 and inflammatory cytokines (Michaudel et al., 2016). ROS may serve as both “kindling” and “bonfire” for NLRP3 inflammasome activation. ROS can function as a redox signaling messenger to trigger NLRP3 inflammasome activation (Abais et al., 2015). When NLRP3 inflammasome is activated, “bonfire” and inflammatory cytokines are generated. Excess ROS results in intracellular and extracellular oxidative stress, and damage of nucleotides, lipids, proteins eventually and inevitably. Although there is a plethora of evidence supporting radiation-induced ROS activation of NLRP3 inflammasomes, the exact mechanisms are still largely unknown. Two distinct proteins, thioredoxin-interacting protein (TXNIP) and mitochondrial antiviral signaling protein (MAVS), are demonstrated as possible mediators of ROS to activate NLRP3 inflammasomes (Zhou et al., 2010; Subramanian et al., 2013). Moreover, irradiation damage induces mitosis and pyroptosis, resulting in generation of DAMPs and activation of the NLRP3 inflammasome. Reports have shown persistent expression of NF-κB, which promotes NLRP3 transcription, in patient neck vessels over 10 years post-radiation (Halle et al., 2010). This finding further indicated a possible link between NF-κB-dependent NLRP3 activation and chronic fibrosis.
The Role of the NLRP3 Inflammasome in Radiation-Induced Cardiovascular Diseases
Recent studies have indicated that upregulation of the NLRP3 inflammasome may play an essential role in RIHD (Wei et al., 2019). NLRP3 inflammasome has been shown as the primary generator responsible for IL-1 production in atherosclerosis and other CVDs (Grebe et al., 2018). Significant increase in secretion of inflammatory cytokines IL-1β and IL-18 was reported in irradiated-animal models (Hong et al., 1999; Ha et al., 2014). IL-1 was suggested to participate in the development of radiation-induced cardiomyopathy (Mezzaroma et al., 2015). Gene expression of IL-1β was significantly elevated during the first month in rat heart after local ionizing irradiation (Kruse et al., 2001). A markedly increased IL-1 expression was observed in irradiated human arteries and veins when compared to non-irradiated (Christersdottir Bjorklund et al., 2013). Increased level of pro-caspase-1 and caspase 1 in human arteries with chronic radiation injury indicated the involvement of NLRP3 inflammasome in RIHD (Christersdottir et al., 2019). However, the outcomes of vascular damage at organ subsites have not been fully characterized, for example, the underlying mechanisms of NLRP3 in radiation-induced CVDs have not yet been established. In this section, we summarized current knowledge of the consequences and molecular mechanisms of RIHD, with a particular focus on the NLRP3 inflammasome in RIHD.
NLRP3 Inflammasome in Radiation-Induced Atherosclerosis
Activation of the NLRP3 inflammasome has been extensively investigated in macrophages and atheroma cells, including smooth muscle cells (SMC), endothelial cells (ECs), and T cells, each of which play significant roles in onset and development of atherosclerosis (Galkina and Ley, 2009). Recent studies suggested that NLRP3 may have additional functions beyond a regulatory role in inflammasome signaling and the innate immune response (Henao-Mejia et al., 2012). These findings indicated that NLRP3 may be involved in radiation-induced atherosclerosis.
Many endogenous danger signals are present in radiation-induced atherosclerosis, which indicates that the NLRP3 inflammasome may be linked to metabolic disturbances, irradiation damage, and inflammation. Limited studies have evaluated the role of the NLRP3 inflammasome in the pathogenesis of irradiation-induced atherosclerosis, but the role of the NLRP3 inflammasome has been extensively studied in non-irradiation-induced atherosclerosis.
Several epidemiological studies have shown a correlation between aortic NLRP3 expression and CVD (Grebe et al., 2018). Components of the NLRP3 inflammasome are expressed in atheromatous cells (Baldrighi et al., 2017). High aortic expression of NLRP3 has been observed in patients with coronary atherosclerosis. The expression of NLRP3 was correlated with the severity of narrowing or blockage of the coronary artery (Wang et al., 2014). Moreover, aortic NLRP3 expression has been shown to be directly associated with CVD severity and multiple clinical risk factors (Zheng et al., 2013). Analysis of human carotid atherosclerotic plaques showed significantly elevated expression of NLRP3, ASC, caspase-1, IL-1β, and IL-18 compared to those in control mesenteric or iliac arteries (Shi et al., 2015). Furthermore, symptomatic patients with CVD have markedly higher NLRP3 expression than asymptomatic patients. Active atherosclerotic plaques exhibit higher expression of NLRP3 inflammasome components than stable atherosclerotic plaques (Paramel Varghese et al., 2016). In addition, patients with acute coronary syndrome showed significantly increased NLRP3 expression in peripheral blood monocytes, which was directly associated with the severity of the disease (Zhu et al., 2019).
Two murine models developed on the C57BL/6 background, Apoe–/– and Ldlr–/–, have been widely used to investigate the mechanisms of atherogenesis (Emini Veseli et al., 2017). However, these models leverage distinctly different mechanisms to induce diet-induced atherosclerosis (Getz and Reardon, 2016). The importance of activation of the NLRP3 inflammasome in irradiation-induced atherogenesis was first detailed by Duewell et al. (2010), who found that bone marrow transplanted from Nlpr3–/–, Asc–/–, or Il1α–/–/Il1β–/– mice into irradiated-Ldlr–/– mice resulted in impaired progression of atherogenesis. Hendrikx et al. (2015) recently showed that activation of caspase-1 and caspase-11 were important in diet-induced atherosclerosis using irradiated Ldlr–/– mice transplanted with bone marrow from caspase-1 or caspase-11 deficient mice.
Ionizing radiation can also accelerate the development of atherosclerotic lesions in Apoe–/– mice (Stewart et al., 2006). Depletion of inflammasome components has been shown to suppress atherogenesis in Apoe–/– mice. Zheng et al. (2014) showed that NLRP3 inflammasome activation was involved in atherogenesis by silencing NLRP3 in Apoe–/– mice, which resulted in decreased atherogenesis. Two other groups showed reduced atherogenic plaque progression in caspase-1–/–/Apoe–/– dual knockout mice fed a low cholesterol diet (Gage et al., 2012; Usui et al., 2012). In addition, spontaneous development of atherosclerosis was reduced in chow-fed caspase-1–/–/Apoe–/– mice. Inhibition of IL-18 activity or IL-18 deletion in Apoe–/– mice markedly attenuated atherosclerotic plaque progression compared to control mice (Mallat et al., 2001). In addition, IL-1β–/–/Apoe–/– knockout mice showed a 30% reduction in plaque development compared to Apoe–/– control mice (Kirii et al., 2003). Another study showed a prominent reduction in plaque size in both Il1β–/–/Apoe–/– and Il1α–/–/Apoe–/– double knockout mice compared to that in Apoe–/– control mice. Similar findings were observed in radiation-treated Apoe–/– mice transplanted with bone marrow from Il1α–/– or Il1β–/– mice (Kamari et al., 2011). However, Nlpr3–/–/Apoe–/–, Asc–/–/Apoe–/–, and caspase-1–/–/Apoe–/– mice did not exhibit decreased progression of atherosclerosis, which indicated that atherosclerosis can proceed independently of NLRP3 inflammasome activation (Menu et al., 2011). Although Apoe–/– mice exhibit more rapid and more severe progression of diet-induced atherosclerosis than Ldlr–/– mice, spontaneous atherosclerosis, even when fed a normal chow diet, is frequently observed in Apoe–/– mice (Getz and Reardon, 2016). The suitability of the Apoe–/– mouse model for study of atherogenesis remains controversial. A possible explanation is that compensatory inflammatory signaling may drive progression of atherogenesis at later stages of the disease (Grebe et al., 2018). This hypothesis was supported by findings that both IL-1β and IL-1α signaling are required for atherogenesis (Freigang et al., 2013). IL-1α immediately presents its bioactive form after transcription in a NLRP3 inflammasome-independent manner (Hansson, 2017). Prolonged atherogenic stimulation in Ldlr–/– mice transplanted with bone marrow of Il1α–/– mice resulted in less significant atherosclerotic lesions than those observed in mice transplanted with bone marrow from Il1β–/– mice (Freigang et al., 2013). These findings highlighted the involvement of IL-1α, but not IL-1β, in atherogenic progression following prolonged administration of an atherogenic diet.
Though the long-term adverse effects of radiation on organs, and the involvement of activation of the NLRP3 inflammasome in radiation-induced injury, have been extensively demonstrated (Wei et al., 2019), the underlying mechanisms responsible for RIHD are poorly understood. Whether the NLRP3 inflammasome plays a role in onset or development of radiation-accelerated atherogenesis, or both, has not been determined.
Multiple atherogenic factors, which can be induced by ionizing radiation, have been shown to activate the NLRP3 inflammasome, followed by endothelial damage and subsequent atherogenesis. Activation of the NLPR3 inflammasome has been reported to directly induce endothelial dysfunction (Zhang et al., 2015; Erdei et al., 2018; Koka et al., 2019). A recent study showed that inhibition of radiation-induced NLRP3 inflammasome activation by LGM2605, an antioxidant and free radical scavenger, alleviated vascular damage (Chatterjee et al., 2019). Inhibitors of HMGB1 or caspase-1 significantly ameliorated endothelium-associated vasodilation in poloxamer 407 (P-407)-treated Nlrp3+/+ mice (Zhang et al., 2015). In addition, activation of endothelial NLRP3 inflammasome was partially correlated with lysosomal disruption in coronary arteritis, which resulted in endothelial dysfunction (Chen et al., 2015). Induction of cold-inducible RNA-binding protein by irradiation causes endothelial dysfunction via activating NLRP3 inflammasome (Yang et al., 2016). Heme, released from damaged RBCs (possibly by radiation under certain conditions), can function as a DAMP to induce IL-1β secretion through NLRP3 activation (Erdei et al., 2018). Heme-induced elevation of IL-1β levels was intensified by LPS priming (Erdei et al., 2018). Acute hypercholesterolemia has been shown to significantly impair endothelium-associated vasodilation in wide-type mice. However, this effect was markedly attenuated in Nlrp3–/– mice (Zhang et al., 2015). Li and colleagues recently found that acid sphingomyelinase (ASMase, encoded by gene Smpd1) and ceramide-associated membrane raft (MR) signaling platforms were involved in endothelial NLRP3 inflammasome activation and atherogenesis resulting from hypercholesterolemia (Koka et al., 2017). Cholesterol crystal administration significantly promoted NLRP3 inflammasome formation and activation in murine carotid arterial endothelial cells, as evidenced by augmented colocalization of NLRP3 with other inflammasome components and enhanced caspase-1 activity and IL-1β production. These effects were significantly reduced by silencing or deletion of mouse Smpd1 or treatment with an ASM inhibitor. Neointimal formation that is more severe, significant intimal inflammasome formation, and increased IL-1β expression were associated with elevated NLRP3 in Smpd1+/+ mice compared with those in Smpd1-deficient mice. These data showed that ASMase and ceramide-related MR clustering are critical for activation of inflammasomes and subsequent endothelial dysfunction in the carotid arteries, resulting in progression of atherosclerosis (Koka et al., 2017).
Radiation can directly damage cardiovascular endothelial cells through multiple mechanisms, which can further enhance the pathophysiological effects of the NLRP3 inflammasome. Single and fractionated ionizing radiation significantly affect vasculature by altering endothelial expression of connexins and increase release of ATP (Ramadan et al., 2019). Conventional fractionated ionizing radiation induces mitotic catastrophe (mitotic cell death distinct from apoptosis) in ECs (Galluzzi et al., 2018). Radiation (≥10 Gy) can induce cell cycle arrest and Bax-mediated apoptosis of endothelial progenitor cells (EPCs), and cause cellular senescence in well-differentiated ECs similar to that observed in premature atherosclerosis (Venkatesulu et al., 2018). Radiation-induced cell death, which is characterized by mitotic catastrophe, apoptosis, and senescence, could stimulate an acute reduction in capillary density or induce chronic inflammation, resulting in disruption of vascular homeostasis (Venkatesulu et al., 2018).
Radiation-related DNA damage-induced EC senescence often results in secretion of cytokines, proteins, and extracellular vesicles (Alique et al., 2018). The senescence-related secretory phenotype of ECs results in dysfunction of adjacent cells or chronic inflammation. A study showed that irradiation increased the expression of the aging-related atherosclerotic senescence marker CD44 (Mun and Boo, 2010) in human coronary ECs and increased monocyte adhesion to ECs (Lowe and Raj, 2014). Radiation can also activate NEMO-dependent NF-κB signaling, which is involved in transcription and upregulation of NLRP3 (Wang S. et al., 2018), cytokines (Meeren et al., 1997), and adhesion molecules (Linard et al., 2004). Furthermore, irradiation also activates TNFα, resulting in ceramide generation via hydrolysis of sphingomyelin. Ceramides can induce the expression or activation of NF-κB, an important proinflammatory transcription factor (Kitajima et al., 1996; Gill and Windebank, 2000), Moreover, ceramides can directly trigger NLRP3 inflammasome activation (Vandanmagsar et al., 2011; Kolliputi et al., 2012; Scheiblich et al., 2017). In addition, radiation disrupts inflammatory homeostasis in the vascular microenvironment by increasing the expression of selectins, integrins, and adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) (Handschel et al., 1999; Prabhakarpandian et al., 2001). However, the effect of radiation on initiation of NF-κB/NLRP3 signaling, upregulation of adhesion molecules, and recruitment of monocytes/macrophages depends on the types and fractions of radiation, and the types of endothelial cell exposed to radiation (Mavragani et al., 2016). Irradiation-induced EC dysfunction may occur in NLRP3 inflammasome-dependent and -independent manners.
Vascular smooth muscle cells (VSMCs), which are major components of medium and large arteries, participate in arterial wall remodeling and atherogenesis throughout all stages of atherosclerosis (Hu et al., 2019). Although mechanisms of atherogenesis remain poorly understood, studies have shown that crosstalk between inflammatory cells and ECs and VSMCs contributes to CVD progression. Some of these interactions accelerate the genesis and stability of atherosclerotic plaques (Li et al., 2018). Tracing of SMC lineage in Apoe–/– mice fed a long-term western-diet has contributed to understanding of the role of VSMC in atherosclerosis (Shankman et al., 2015). Vascular smooth muscle cells in atherosclerotic lesions have been shown to be derived from a subset of medial Myh11+ VSMCs that propagate the lesions (Chappell et al., 2016). Most VSMCs within atherosclerotic sites lack characteristic SMC marker expression profiles, but they express markers of mesenchymal stem cells, myofibroblast-like cells, or macrophages (Bennett et al., 2016). Milliat et al. (2006) demonstrated the role of proliferation, migration, and fibrogenic phenotype transition of VSMCs in irradiation-induced vascular damage. Co-culture of VSMCs with irradiated ECs resulted in significantly increased proliferation and migration. Further study showed that TGF-β/Smad3 signaling played a role in irradiated EC-induced fibroblastic phenotype transition of VSMCs. However, the role of the NLPR3 inflammasome in VSMC-mediated atherosclerosis has not been characterized. Wang and colleagues found that NLRP3 inflammasome activation promoted foam cell formation in human VSMCs and induced atherogenesis in Apoe–/– mice via release of HMGB1 (Wang R. et al., 2018). Moreover, NLRP3 inflammasome activation has been shown to induce VSMC synthetic phenotype transition and proliferation (Sun et al., 2017). Deficiency of NLRP3 has been shown to reduce Ang II-induced NLRP3 inflammasome activation, synthetic phenotypic transition, and proliferation in murine VSMCs (Ren et al., 2017). However, Duewell et al. (2010) found that lethal irradiation (12 Gy) of Apoe–/– mice resulted in loss of VSMC accumulation in the brachiocephalic and carotid artery, but VSMC levels in the aortic root and in abdominal aortic lesions were normal (Newman et al., 2018). These studies demonstrated differences in the sensitivity to radiation of VSMCs in different vascular beds.
Priming and activation signals are required for NLRP3 inflammasome activation in macrophages. This two-step mechanism tightly regulates activation of NLRP3 inflammasome in macrophages. Nucleotide-binding domain and leucine-rich-repeat-containing family pyrin 3 is easily activated by a variety of DAMPs generated by ionizing radiation. Radiation can induce inflammatory cytokine production in macrophages (Teresa Pinto et al., 2016) and can trigger NLRP3 inflammasome activation, resulting in macrophages pyroptosis (Liu et al., 2017). Pyroptosis is a rapid and highly inflammatory form of programmed cell death that can be initiated by DAMP-activated NLRP3 inflammasome (Guo et al., 2015). Moreover, NLRP3 inflammasome activation has been shown to augment lipid-deposition and macrophage migration (Li et al., 2014). Macrophage recruitment is the primary response to radiation-induced damage (Ahn et al., 2010), and macrophage polarization is affected during the early and late stages of radiation-induced tissue damage (Duru et al., 2016; Meziani et al., 2018). Radiation promotes pro-inflammatory M1 macrophage polarization, which results in enhanced production of M1 cytokines such as TNF-α, interleukins, and IFN-γ (Farooque et al., 2016). Mature M1 macrophages accelerate extracellular matrix breakdown, tissue damage, and fibrosis (Braga et al., 2015). In fact, ionizing radiation can activate NLRP3 inflammasome in multiple immune cells, including T and B lymphocytes (Stoecklein et al., 2015). Both myeloid and adaptive response are involved in atherogenesis (Wolf and Ley, 2019).
NLRP3 Inflammasome Regulation of Radiation-Induced Cardiovascular Fibrosis
The endpoint of RIHD is cardiovascular fibrosis, which is a severe complication associated with a high rate of mortality. Radiation-induced fibrotic diseases of the cardiovascular system include radiation-related coronary artery disease with manifest fibrous tissue, myocardial fibrosis, cardiomyopathy, valvular fibrosis, and fibrosis of the pericardium. Vascular endothelial damage is widely considered to be the primary pathogenic signal that precedes fibrosis (Yarnold and Brotons, 2010).
Radiation damage induces early and delayed cardiac tissue injury. Ionizing radiation induces early endothelial damage, which results in vasodilation and increased blood vessel permeability (Soloviev et al., 2003). Injured ECs secrete inflammatory cytokines (TNFα, IL-1, IL-6, and IL-8), resulting in upregulation of adhesion molecules and activation of the inflammatory response. Inflammatory cells recruited to lesions produce profibrotic cytokines including transforming growth factor-beta (TGF-β), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and connective tissue growth factor (Cuomo et al., 2016). Radiation-induced cardiovascular injury triggers a complex cascade of cellular and molecular responses that result in tissue fibrosis. Fibrogenesis consists of four major phases: (1) The initiation stage is driven by primary vascular endothelial injury. The central event during this phase is recruitment of inflammatory cells, which produce a number of mediators, cytokines, and other factors that strengthen the inflammatory response; (2) Activation of fibrogenic effector cells, include fibroblasts, fibrocytes, tissue-specific pericytes, and myofibroblasts. These types of cells can proliferate and differentiation into mature myofibroblasts in response to injury; (3) Elaboration of the extracellular matrix occurs when effector cells produce a large amount of extracellular matrix proteins, primarily in an autocrine manner. In addition, cell–cell interactions lead to further activation of effector cells, resulting in a positive feedback loop. This process is driven by TGF-β and PDGF; (4) The final stage is fibrosis, which results in organ dysfunction and failure (Rockey et al., 2015).
Myocardial fibrosis commonly features reactive fibrosis and replacement fibrosis (Travers et al., 2016). Reactive fibrosis often occurs in the perivascular space, and replacement fibrosis occurs at locations where myocytes are lost. Radiation-induced myocardial fibrosis may be induced by cardiac fibroblasts, which are the most abundant non-myocyte cell type in the myocardium (Krenning et al., 2010). A recent study indicated that NLRP3 inflammasome in fibroblast linked inflammation with tissue injury (Ershaid et al., 2019). Cardiac fibroblasts might originate from myocardial mesenchymal cells, bone marrow-derived circulating fibroblasts, and epithelial/endothelial–mesenchymal cell transition (EndoMT/EMT) (Moore-Morris et al., 2014). EndoMT is a transition process where ECs lose their endothelial characteristic features and obtain mesenchymal properties. Mounting evidence indicated that EndoMT contributed to cardiac fibrosis (Kovacic et al., 2019). Nearly one-third of all cardiac fibroblasts originated from ECs (Zeisberg et al., 2007). A recent study showed that NLRP3 inflammasome activation during the EMT linked inflammatory injury and endothelial dysfunction (Cho et al., 2018). In addition, studies have shown that myofibroblasts can also originate from resident VSMCs during atherogenesis, as evidenced by expression of smooth muscle myosin heavy chain (SMMHC) and alpha-smooth actin (αSMA). Skin biopsy samples from patients who underwent breast radiotherapy showed collagen accumulation and fibroblast expression of αSMA, which indicated a myofibroblast phenotype. Benderitter and colleagues reported prominent ECM deregulation and VSMC proliferation in hypertrophic blood vessels in patients who received preoperational radiotherapy, as evidenced by the presence of αSMA-expressing cells in the condensed collagen matrix region of intimal hyperplasia. In vitro co-culture experiments showed that VSMCs co-cultured with irradiated human endothelial cells underwent myofibroblastic transition (Milliat et al., 2006). Zhang and colleagues recently reported that PDGF-BB stimulation of Spmd1-deficient VSMCs induced differentiation to a myofibroblast-like phenotype, as evidenced by marked upregulation of fibroblast-specific protein (FSP-1) expression, massive collagen type I deposition, increased IL-6 release, and upregulation of adhesion molecule expression (Zhang et al., 2018). The underlying mechanism of these effects may be due to prolonged Akt activation, and p62/SQSTM1-dependent NRF2 upregulation and nuclear translocation. These findings suggested that myofibroblasts originated from resident VSMCs (Figure 1).
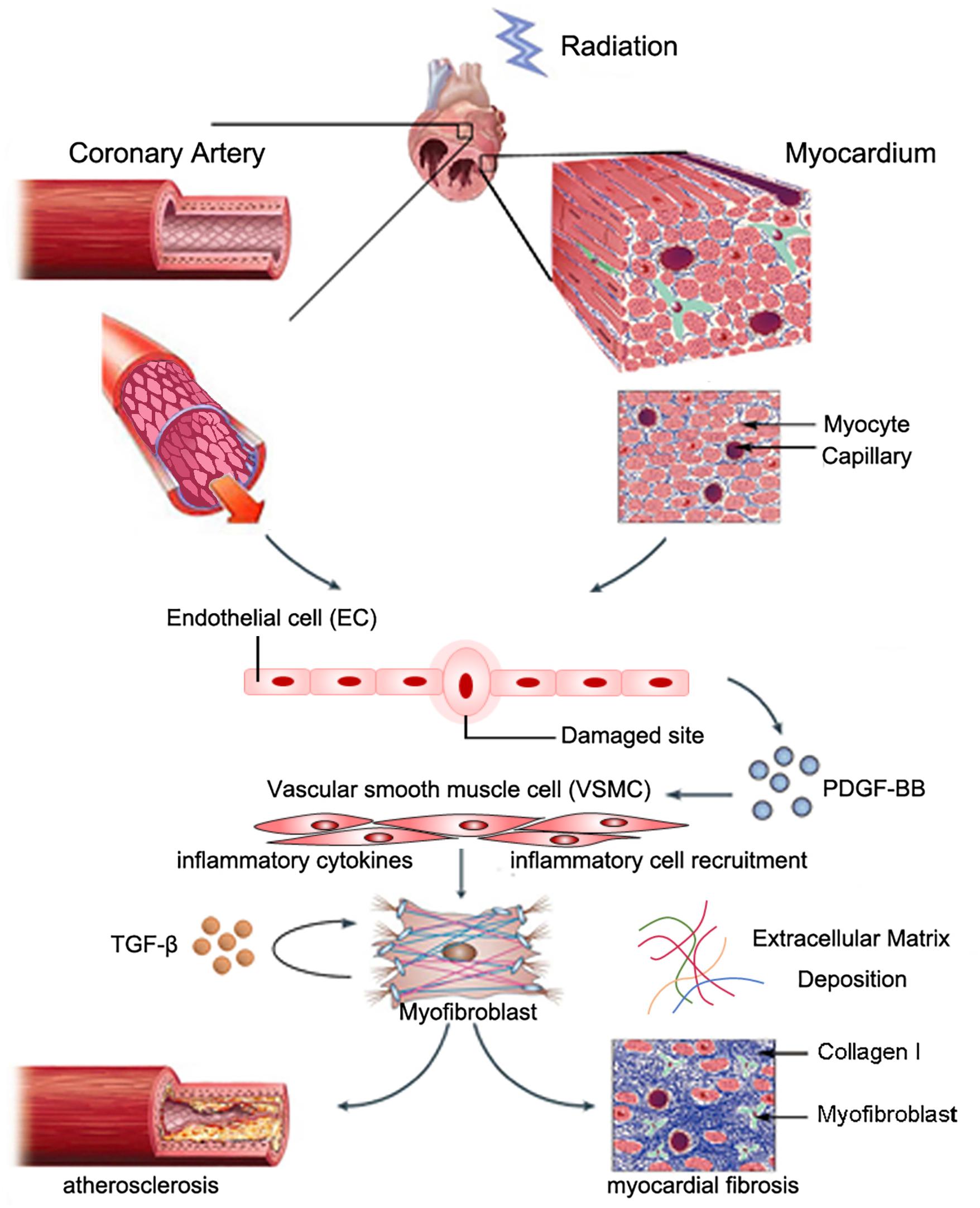
Figure 1. Possible role of VSMCs in radiation-induced cardiac fibrosis. Partially reproduced with permission from Weintraub et al. (2010).
The NLRP3 inflammasome is also involved in regulation of the expression of profibrotic cytokines (e.g., TGF-β, PDGF, and IL-6) (Anders et al., 2018). Wang et al. (2013) showed that NLRP3 inflammasome activation augmented TGF-β signaling in the kidney epithelium. TGF-β1-induced EMT, and induction of MMP-9 and αSMA were significantly impaired in Nlrp3-deficient murine renal tubular epithelial cells (TECs). Mitochondrial NLRP3 protein has been shown to directly induce ROS production, resulting in increased TGF-β/Smad signaling and fibrosis without inflammasome formation (Bracey et al., 2014). The crosstalk between inflammasome-independent NLRP3 and TGFR signaling suggests the essential role of NLRP3 in myofibroblast differentiation and ECM accumulation (Anders et al., 2018). A recent study indicated that triptolide (TP) attenuated cardiac fibrosis by interrupting NLRP3-TGFβ1-Smad signaling (Pan et al., 2019). Coriolus versicolor (CV) was also shown to alleviate cardiac fibrosis through suppression of the TGF-β/Smad pathway and NLRP inflammasome activation (Wang Y. et al., 2019). Similarly, NLRP3 deficiency resulted in reduced TGF-β- and PDGF-BB-induced proliferation of Nlrp3-deficient renal fibroblasts (Anders et al., 2018). Kawaguchi et al. (2011) showed that hypoxia-reoxygenation-induced ROS production and K+ efflux stimulated inflammasome activation in cardiac fibroblasts, but not cardiomyocytes. Radiation has been shown to induce robust production of ROS, which results in differentiation of CD4+ T cells into Th2 lymphocytes that secrete potent profibrotic cytokines such as IL-4 and IL-13 (Besnard et al., 2011; Braga et al., 2019). Nucleotide-binding domain and leucine-rich-repeat-containing family pyrin 3 can directly promote Th2 programming without inflammasome formation (Bruchard et al., 2015), NLRP3 can also modulate M2 macrophage polarization via upregulation of IL-4 (Liu Y. et al., 2018). Taken together, these findings indicate that NLRP3 inflammasome exhibits a complex function in radiation-induced cardiovascular fibrosis.
Therapeutic Targeting of NLRP3 Against Rihd
Characterization of the underlying mechanisms of activation of the NLRP3 inflammasome may allow for development of effective inhibitors or regulators as therapeutic agents for RIHD. A number of candidate NLRP3 inhibitors have exhibited promising therapeutic effects against inflammatory disorders. These inhibitors primarily target NLRP3, other inflammasome components, or related signaling events.
Three therapeutics agents against IL-1/IL-1R have been approved by the US FDA for treatment of multiple inflammatory diseases: anakinra (recombinant antagonist against the IL-1 receptor), rilonacept (decoy receptor that binds IL-1β), and as canakinumab (a specific antibody against IL-1β) (Dinarello et al., 2012). Two other inhibitory antibodies, MAPp1 for IL-1α and GSK1070806 for IL-18, are under development (Swanson et al., 2019). The success of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) demonstrated the clinical translational potential of inhibitors against NLRP3-driven immunopathological diseases. This study evaluated the impact of canakinumab on occurrence and recurrence of CVDs (e.g., MI, stroke). Standard canakinumab administration resulted in a 15% decrease in MI and cardiovascular mortality compared to placebo. The CANTOS trial was the first study to demonstrate the translational significance of the proatherogenic effect of IL-1β, as evidenced by the finding that clinical outcomes of CVD were improved by interfering with IL-1β production or signaling (Ridker et al., 2012). A recent gene expression analysis of both irradiated and non-irradiated human blood vessels from the Biobank of Radiated Tissues at Karolinska (BiRKa) was conducted, suggesting the involvement of NLRP3-inflammasome and potential treatment using IL-1 inhibitors. According to their findings, treatment with anakinra in irradiated Apoe–/– mice significantly alleviated arterial inflammation (Christersdottir et al., 2019).
Development of oral small molecules specific for NLRP3 inflammasome inhibition might be a more promising, less invasive, and more cost-effective strategy (Zahid et al., 2019). Among the inhibitors summarized in Table 1, compound MCC950 is the most promising NLRP3 inhibitor candidate because it selectively suppresses NLRP3 inflammasome signaling in human and mouse macrophages. In addition, MCC950 does not affect non-NLRP3 inflammasomes, TLR-mediated priming signals, or IL-1α releasing. MCC950 exhibits striking therapeutic effects against a variety of preclinical immunopathological models such as atherosclerosis, cardiac arrhythmia, MI, and colitis (Liston and Masters, 2017; Swanson et al., 2019). Another NLRP3 specific inhibitor, 16673-34-0 exerted protective effects against myocardial ischemia-reperfusion injury and acute peritonitis (Lipinski and Frias, 2014). Pharmacological interventions targeting alternative inflammasome constituents are being actively investigated (Zahid et al., 2019). Of note, only a small number of inhibitory therapeutics that target NLRP3 or its downstream effectors and pathways show potent efficacy for treatment of NLRP3-associated diseases. Other inhibitors such as AZD9059 and GSK1482169 for P2 × 7, and VX VX-740 and VX-756 for caspase-1 also did not induce therapeutic effects against NLRP3-related diseases due to ineffectiveness or severe hepatic toxicity (Ozaki et al., 2015). Interestingly, non-specific pharmacological interventions can also modulate NLRP3 inflammasome activity. Atorvastatin was found to effectively suppress NLRP3 inflammasome level in CAD (Satoh et al., 2014). Certain antidiabetic medicines such as biguanides, thiazolidinediones, and inhibitors of dipeptidyl peptidase 4 have the potential to regulate the activation of NLRP3 inflammasome (Zhang and Wang, 2020). An antioxidant, melatonin, could also function as an anti-inflammatory agent by inhibiting the activity of NLRP3 inflammasome (Garcia et al., 2015).
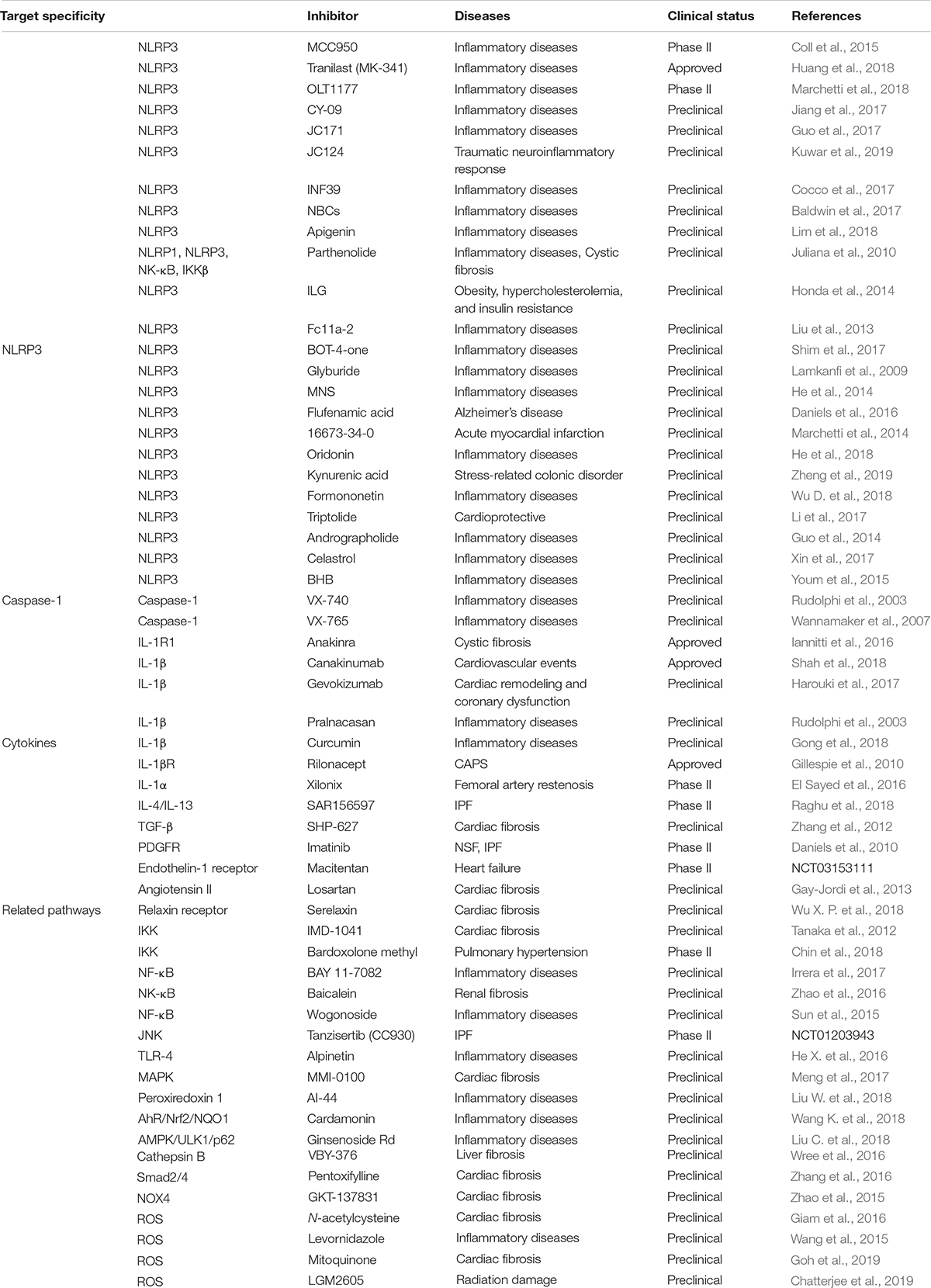
Table 1. Inhibitors against NLRP3 inflammasome.
Conclusion and Perspectives
Radiation-induced cardiovascular injury has received increased attention due to the growing population of cancer survivors exposed to chest radiation. Although great improvements have been made in radiotherapy techniques, asymptomatic patients are at increased risk for development of RIHD. Exposure to low-dose irradiation may result in adverse cardiovascular effects. Studies have shown that acute and chronic inflammatory responses are essential in development of RIHD. Increasing evidence has indicated that the NLRP3 inflammasome is involved in radiation damage. In addition, multiple cellular effects of radiation, such as ROS production and generation of DAMPs resulting from cell death, can further strengthen NLRP3 inflammasome activation (Figure 2). However, the mechanisms by which the NLRP3 inflammasome functions in RIHD requires further characterization. In addition, previous studies showed that two evolutionarily conserved degradation pathways, ubiquitin-proteasome and autophagy, negatively regulate NLRP3 inflammasome activation (Song et al., 2016; Zhong et al., 2016). Small molecule has been developed to inhibit NLRP3 inflammasome activity by interplaying between ubiquitination and autophagy (Han et al., 2019). MicroRNA was also identified to respond to chronic inflammation in RIHD (Eken et al., 2019). Future translational researches are needed to determine whether NLRP3 inflammasome inhibition and whether these pharmacological candidate inhibitors mentioned above are promising to prevent radiation-induced cardiovascular injury, inflammation, and late fibrosis. Additionally, it seems to be rational to start with an early and short-term IL-1β blockage treatment directly after radiotherapy exposure. It is based on the concept that the intervention might not work effectively in already established RIHD, where the chronic inflammatory response is advanced (Christersdottir et al., 2019). Further studies are required to evaluate the optimal timing and treatment-duration of NLRP3 inflammasome signaling inhibition to prevent RIHD. Experimental animal models for RIHD are also needed to be optimized to accelerate the investigation of the underlying mechanism and development of novel treatment strategies.
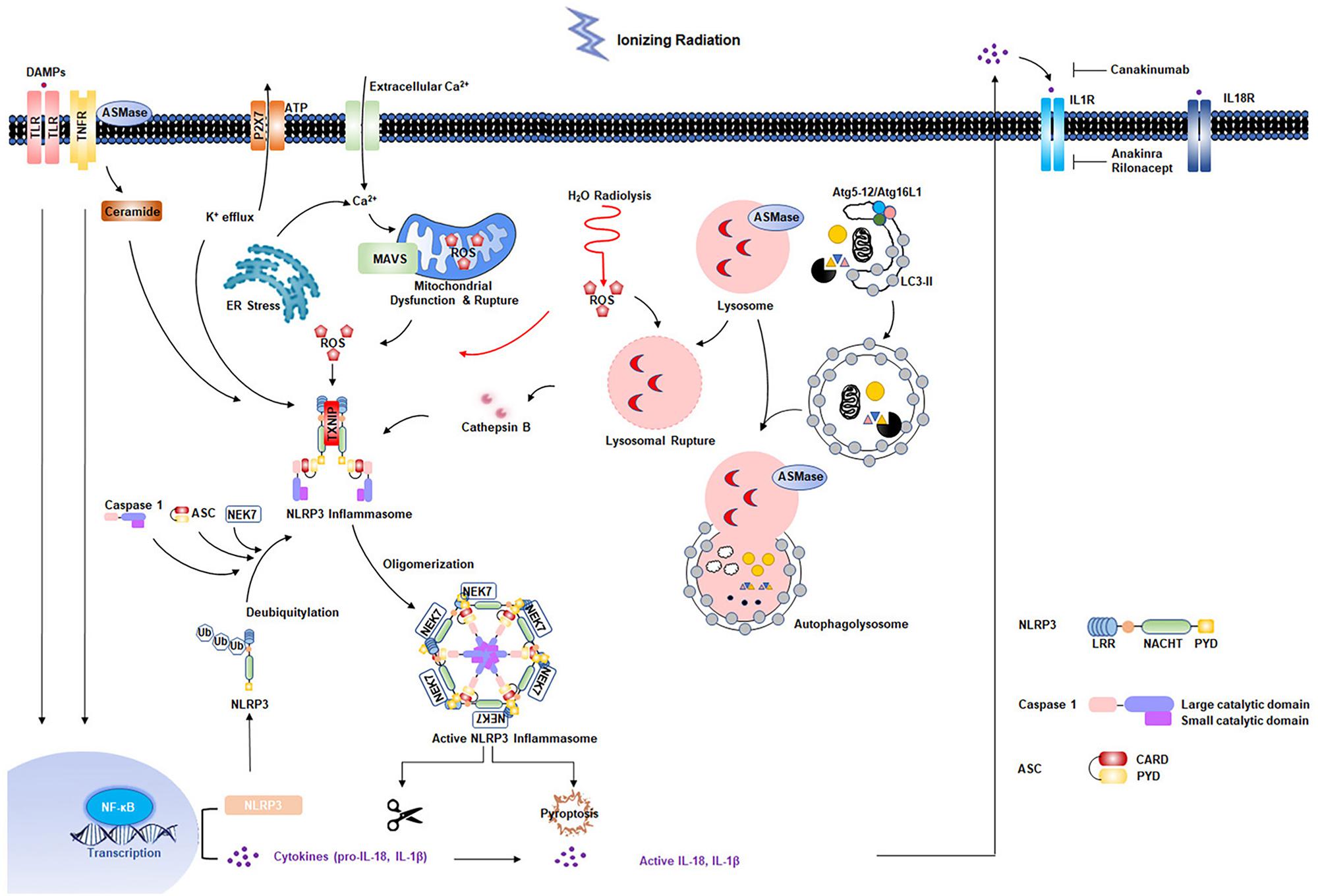
Figure 2. Multiple effects of radiation on NLRP3 inflammasome activation. Radiation-induced NF-κB activation is responsible for the upregulation of NLRP3 and pro-interleukin-1β. Second signals come from multiple pathways: water radiolysis, K+ efflux, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, ceramide pathway, and lysosomal rupture pathways, most of which appear to converge in the production of reactive oxygen species (ROS). The primary and secondary signals triggers the NLRP3 inflammasome activation, resulting in the maturation of caspase 1 and IL-1β. Two evolutionarily conserved degradation pathways, ubiquitin-proteasome and autophagy, negatively regulate NLRP3 activation.
Author Contributions
PZ conceived of the topic for this review. All the authors listed made substantial and intellectual contributions to the work.
Funding
This work was funded by the National Natural Science Foundation of China (No. 81974483).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abais, J. M., Xia, M., Zhang, Y., Boini, K. M., and Li, P. L. (2015). Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 22, 1111–1129. doi: 10.1089/ars.2014.5994
Abe, J., Martin, J. F., and Yeh, E. T. (2016). The future of onco-cardiology: we are not just side effect hunters. Circ. Res. 119, 896–899. doi: 10.1161/circresaha.116.309573
Ahn, G. O., Tseng, D., Liao, C. H., Dorie, M. J., Czechowicz, A., and Brown, J. M. (2010). Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc. Natl. Acad. Sci. U.S.A. 107, 8363–8368. doi: 10.1073/pnas.0911378107
Aleman, B. M., Van Den Belt-Dusebout, A. W., De Bruin, M. L., Van ‘T Veer, M. B., Baaijens, M. H., De Boer, J. P., et al. (2007). Late cardiotoxicity after treatment for Hodgkin lymphoma. Blood 109, 1878–1886. doi: 10.1182/blood-2006-07-034405
Alique, M., Ramirez-Carracedo, R., Bodega, G., Carracedo, J., and Ramirez, R. (2018). Senescent microvesicles: a novel advance in molecular mechanisms of atherosclerotic calcification. Int. J. Mol. Sci. 19:2003. doi: 10.3390/ijms19072003
Amaral, E. P., Riteau, N., Moayeri, M., Maier, N., Mayer-Barber, K. D., Pereira, R. M., et al. (2018). Lysosomal cathepsin release is required for nlrp3-inflammasome activation by mycobacterium tuberculosis in infected macrophages. Front. Immunol. 9:1427. doi: 10.3389/fimmu.2018.01427
Anders, H. J., Suarez-Alvarez, B., Grigorescu, M., Foresto-Neto, O., Steiger, S., Desai, J., et al. (2018). The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int. 93, 656–669. doi: 10.1016/j.kint.2017.09.022
Bae, J. Y., and Park, H. H. (2011). Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 286, 39528–39536. doi: 10.1074/jbc.M111.278812
Baldrighi, M., Mallat, Z., and Li, X. (2017). NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 267, 127–138. doi: 10.1016/j.atherosclerosis.2017.10.027
Baldwin, A. G., Rivers-Auty, J., Daniels, M. J. D., White, C. S., Schwalbe, C. H., Schilling, T., et al. (2017). Boron-based inhibitors of the NLRP3 inflammasome. Cell Chem. Biol. 24:e1325. doi: 10.1016/j.chembiol.2017.08.011
Bennett, M. R., Sinha, S., and Owens, G. K. (2016). Vascular smooth muscle cells in atherosclerosis. Circ. Res. 118, 692–702. doi: 10.1161/CIRCRESAHA.115.306361
Besnard, A. G., Guillou, N., Tschopp, J., Erard, F., Couillin, I., Iwakura, Y., et al. (2011). NLRP3 inflammasome is required in murine asthma in the absence of aluminum adjuvant. Allergy 66, 1047–1057. doi: 10.1111/j.1398-9995.2011.02586.x
Boerma, M. (2012). Experimental radiation-induced heart disease: past, present, and future. Radiat. Res. 178, 1–6. doi: 10.1667/rr2933.1
Boerma, M., Sridharan, V., Mao, X. W., Nelson, G. A., Cheema, A. K., Koturbash, I., et al. (2016). Effects of ionizing radiation on the heart. Mutat. Res. 770, 319–327. doi: 10.1016/j.mrrev.2016.07.003
Boerma, M., Wang, J., Sridharan, V., Herbert, J. M., and Hauer-Jensen, M. (2013). Pharmacological induction of transforming growth factor-beta1 in rat models enhances radiation injury in the intestine and the heart. PLoS One 8:e70479. doi: 10.1371/journal.pone.0070479
Bracey, N. A., Gershkovich, B., Chun, J., Vilaysane, A., Meijndert, H. C., Wright, J. R., et al. (2014). Mitochondrial NLRP3 protein induces reactive oxygen species to promote smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 289, 19571–19584. doi: 10.1074/jbc.M114.550624
Braga, T. T., Agudelo, J. S., and Camara, N. O. (2015). Macrophages during the fibrotic process: M2 as friend and foe. Front. Immunol. 6:602. doi: 10.3389/fimmu.2015.00602
Braga, T. T., Brandao, W. N., Azevedo, H., Terra, F. F., Melo, A. C. L., Pereira, F. V., et al. (2019). NLRP3 gain-of-function in CD4+ T lymphocytes ameliorates experimental autoimmune encephalomyelitis. Clin. Sci. 133, 1901–1916. doi: 10.1042/CS20190506
Brosius, F. C. III, Waller, B. F., and Roberts, W. C. (1981). Radiation heart disease. analysis of 16 young (aged 15 to 33 years) necropsy patients who received over 3,500 rads to the heart. Am. J. Med. 70, 519–530.
Bruchard, M., Boidot, R., Ghiringhelli, F., and Vegran, F. (2015). Transcriptome analysis of TH2 CD4(+) T cells differentiated from wild-type and NLRP3KO mice. Genom. Data 5, 314–315. doi: 10.1016/j.gdata.2015.06.031
Chappell, J., Harman, J. L., Narasimhan, V. M., Yu, H., Foote, K., Simons, B. D., et al. (2016). Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ. Res. 119, 1313–1323. doi: 10.1161/circresaha.116.309799
Chatterjee, S., Pietrofesa, R. A., Park, K., Tao, J. Q., Carabe-Fernandez, A., Berman, A. T., et al. (2019). LGM2605 reduces space radiation-induced NLRP3 inflammasome activation and damage in in vitro lung vascular networks. Int. J. Mol. Sci. 20:176. doi: 10.3390/ijms20010176
Chen, Y., Li, X., Boini, K. M., Pitzer, A. L., Gulbins, E., Zhang, Y., et al. (2015). Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim. Biophys. Acta 1853, 396–408. doi: 10.1016/j.bbamcr.2014.11.012
Chen, Y., Wang, L., Pitzer, A. L., Li, X., Li, P. L., and Zhang, Y. (2016). Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 94, 1335–1347. doi: 10.1007/s00109-016-1481-5
Chin, M. P., Bakris, G. L., Block, G. A., Chertow, G. M., Goldsberry, A., Inker, L. A., et al. (2018). Bardoxolone methyl improves kidney function in patients with chronic kidney disease stage 4 and Type 2 diabetes: post-hoc analyses from bardoxolone methyl evaluation in patients with chronic kidney disease and Type 2 diabetes study. Am. J. Nephrol. 47, 40–47. doi: 10.1159/000486398
Cho, J. G., Lee, A., Chang, W., Lee, M. S., and Kim, J. (2018). Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front. Immunol. 9:294. doi: 10.3389/fimmu.2018.00294
Christersdottir Bjorklund, T., Reilly, S. J., Gahm, C., Bottazzi, B., Mantovani, A., Tornvall, P., et al. (2013). Increased long-term expression of pentraxin 3 in irradiated human arteries and veins compared to internal controls from free tissue transfers. J. Transl. Med. 11:223. doi: 10.1186/1479-5876-11-223
Christersdottir, T., Pirault, J., Gistera, A., Bergman, O., Gallina, A. L., Baumgartner, R., et al. (2019). Prevention of radiotherapy-induced arterial inflammation by interleukin-1 blockade. Eur. Heart J. 40, 2495–2503. doi: 10.1093/eurheartj/ehz206
Church, L. D., Hessler, G., Goodall, J. E., Rider, D. A., Workman, C. J., Vignali, D. A., et al. (2005). TNFR1-induced sphingomyelinase activation modulates TCR signaling by impairing store-operated Ca2+ influx. J. Leukoc. Biol. 78, 266–278. doi: 10.1189/jlb.1003456
Cocco, M., Pellegrini, C., Martinez-Banaclocha, H., Giorgis, M., Marini, E., Costale, A., et al. (2017). Development of an acrylate derivative targeting the nlrp3 inflammasome for the treatment of inflammatory bowel disease. J. Med. Chem. 60, 3656–3671. doi: 10.1021/acs.jmedchem.6b01624
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Munoz-Planillo, R., Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255. doi: 10.1038/nm.3806
Cuomo, J. R., Sharma, G. K., Conger, P. D., and Weintraub, N. L. (2016). Novel concepts in radiation-induced cardiovascular disease. World J. Cardiol. 8, 504–519.
Daniels, C. E., Lasky, J. A., Limper, A. H., Mieras, K., Gabor, E., Schroeder, D. R., et al. (2010). Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am. J. Respir. Crit. Care Med. 181, 604–610. doi: 10.1164/rccm.200906-0964OC
Daniels, M. J., Rivers-Auty, J., Schilling, T., Spencer, N. G., Watremez, W., Fasolino, V., et al. (2016). Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 7:12504. doi: 10.1038/ncomms12504
Dinarello, C. A., Simon, A., and Van Der Meer, J. W. (2012). Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652. doi: 10.1038/nrd3800
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. doi: 10.1038/nature08938
Duru, N., Wolfson, B., and Zhou, Q. (2016). Mechanisms of the alternative activation of macrophages and non-coding RNAs in the development of radiation-induced lung fibrosis. World J. Biol. Chem. 7, 231–239.
Eken, S. M., Christersdottir, T., Winski, G., Sangsuwan, T., Jin, H., Chernogubova, E., et al. (2019). miR-29b mediates the chronic inflammatory response in radiotherapy-induced vascular disease. JACC Transl. Sci. 4, 72–82. doi: 10.1016/j.jacbts.2018.10.006
El Sayed, H., Kerensky, R., Stecher, M., Mohanty, P., and Davies, M. (2016). A randomized phase II study of Xilonix, a targeted therapy against interleukin 1alpha, for the prevention of superficial femoral artery restenosis after percutaneous revascularization. J. Vasc. Surg. 63:e131. doi: 10.1016/j.jvs.2015.08.069
Emini Veseli, B., Perrotta, P., De Meyer, G. R. A., Roth, L., Van Der Donckt, C., Martinet, W., et al. (2017). Animal models of atherosclerosis. Eur. J. Pharmacol. 816, 3–13. doi: 10.1016/j.ejphar.2017.05.010
Erdei, J., Toth, A., Balogh, E., Nyakundi, B. B., Banyai, E., Ryffel, B., et al. (2018). Induction of NLRP3 Inflammasome activation by heme in human endothelial cells. Oxid. Med. Cell Longev. 2018:4310816. doi: 10.1155/2018/4310816
Ershaid, N., Sharon, Y., Doron, H., Raz, Y., Shani, O., Cohen, N., et al. (2019). NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 10:4375. doi: 10.1038/s41467-019-12370-8
Farooque, A., Afrin, F., Adhikari, J. S., and Dwarakanath, B. S. (2016). Polarization of macrophages towards M1 phenotype by a combination of 2-deoxy-d-glucose and radiation: implications for tumor therapy. Immunobiology 221, 269–281. doi: 10.1016/j.imbio.2015.10.009
Fokkema, M., Den Hartog, A. G., Van Lammeren, G. W., Bots, M. L., Pasterkamp, G., Vink, A., et al. (2012). Radiation-induced carotid stenotic lesions have a more stable phenotype than de novo atherosclerotic plaques. Eur. J. Vasc. Endovasc. Surg. 43, 643–648. doi: 10.1016/j.ejvs.2012.02.023
Franzen, L., Funegard, U., Sundstrom, S., Gustafsson, H., Danielsson, A., and Henriksson, R. (1991). Fractionated irradiation and early changes in salivary glands. Different effects on potassium efflux, exocytotic amylase release and gland morphology. Lab. Invest. 64, 279–283.
Franzen, L., Gustafsson, H., Sundstrom, S., Karlsson, M., Littbrand, B., and Henriksson, R. (1993). Fractionated irradiation and late changes in rat parotid gland: effects on the number of acinar cells, potassium efflux, and amylase secretion. Int. J. Radiat. Biol. 64, 93–101. doi: 10.1080/09553009314551141
Freigang, S., Ampenberger, F., Weiss, A., Kanneganti, T. D., Iwakura, Y., Hersberger, M., et al. (2013). Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 14, 1045–1053. doi: 10.1038/ni.2704
Gabriels, K., Hoving, S., Seemann, I., Visser, N. L., Gijbels, M. J., Pol, J. F., et al. (2012). Local heart irradiation of ApoE(-/-) mice induces microvascular and endocardial damage and accelerates coronary atherosclerosis. Radiother. Oncol. 105, 358–364. doi: 10.1016/j.radonc.2012.08.002
Gage, J., Hasu, M., Thabet, M., and Whitman, S. C. (2012). Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol. 28, 222–229. doi: 10.1016/j.cjca.2011.10.013
Galkina, E., and Ley, K. (2009). Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 27, 165–197. doi: 10.1146/annurev.immunol.021908.132620
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Garcia, J. A., Volt, H., Venegas, C., Doerrier, C., Escames, G., Lopez, L. C., et al. (2015). Disruption of the NF-kappaB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-alpha and blocks the septic response in mice. FASEB J. 29, 3863–3875. doi: 10.1096/fj.15-273656
Gay-Jordi, G., Guash, E., Benito, B., Brugada, J., Nattel, S., Mont, L., et al. (2013). Losartan prevents heart fibrosis induced by long-term intensive exercise in an animal model. PLoS One 8:e55427. doi: 10.1371/journal.pone.0055427
Getz, G. S., and Reardon, C. A. (2016). Do the Apoe-/- and Ldlr-/- Mice yield the same insight on atherogenesis? Arterioscler. Thromb. Vasc. Biol. 36, 1734–1741. doi: 10.1161/ATVBAHA.116.306874
Giam, B., Chu, P. Y., Kuruppu, S., Smith, A. I., Horlock, D., Kiriazis, H., et al. (2016). N-acetylcysteine attenuates the development of cardiac fibrosis and remodeling in a mouse model of heart failure. Physiol. Rep. 4:e12757. doi: 10.14814/phy2.12757
Gill, J. S., and Windebank, A. J. (2000). Ceramide initiates NFkappaB-mediated caspase activation in neuronal apoptosis. Neurobiol. Dis. 7, 448–461. doi: 10.1006/nbdi.2000.0312
Gillespie, J., Mathews, R., and Mcdermott, M. F. (2010). Rilonacept in the management of cryopyrin-associated periodic syndromes (CAPS). J. Inflamm. Res. 3, 1–8.
Goh, K. Y., He, L., Song, J., Jinno, M., Rogers, A. J., Sethu, P., et al. (2019). Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol. 21:101100. doi: 10.1016/j.redox.2019.101100
Gombault, A., Baron, L., and Couillin, I. (2012). ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 3:414. doi: 10.3389/fimmu.2012.00414
Gong, Z., Zhao, S., Zhou, J., Yan, J., Wang, L., Du, X., et al. (2018). Curcumin alleviates DSS-induced colitis via inhibiting NLRP3 inflammsome activation and IL-1beta production. Mol. Immunol. 104, 11–19. doi: 10.1016/j.molimm.2018.09.004
Grebe, A., Hoss, F., and Latz, E. (2018). NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 122, 1722–1740. doi: 10.1161/CIRCRESAHA.118.311362
Gujral, D. M., Lloyd, G., and Bhattacharyya, S. (2016a). Radiation-induced valvular heart disease. Heart 102, 269–276. doi: 10.1136/heartjnl-2015-308765
Gujral, D. M., Shah, B. N., Chahal, N. S., Bhattacharyya, S., Hooper, J., Senior, R., et al. (2016b). Carotid intima-medial thickness as a marker of radiation-induced carotid atherosclerosis. Radiother. Oncol. 118, 323–329. doi: 10.1016/j.radonc.2015.11.025
Guo, C., Fulp, J. W., Jiang, Y., Li, X., Chojnacki, J. E., Wu, J., et al. (2017). Development and characterization of a Hydroxyl-Sulfonamide analogue, 5-chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem. Neurosci. 8, 2194–2201. doi: 10.1021/acschemneuro.7b00124
Guo, H., Callaway, J. B., and Ting, J. P. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687. doi: 10.1038/nm.3893
Guo, W., Sun, Y., Liu, W., Wu, X., Guo, L., Cai, P., et al. (2014). Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 10, 972–985. doi: 10.4161/auto.28374
Ha, C. T., Li, X. H., Fu, D., Moroni, M., Fisher, C., Arnott, R., et al. (2014). Circulating interleukin-18 as a biomarker of total-body radiation exposure in mice, minipigs, and nonhuman primates (NHP). PLoS One 9:e109249. doi: 10.1371/journal.pone.0109249
Halle, M., Gabrielsen, A., Paulsson-Berne, G., Gahm, C., Agardh, H. E., Farnebo, F., et al. (2010). Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J. Am. Coll. Cardiol. 55, 1227–1236. doi: 10.1016/j.jacc.2009.10.047
Han, X., Sun, S., Sun, Y., Song, Q., Zhu, J., Song, N., et al. (2019). Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease. Autophagy 15, 1860–1881. doi: 10.1080/15548627.2019.1596481
Handschel, J., Prott, F. J., Sunderkotter, C., Metze, D., Meyer, U., and Joos, U. (1999). Irradiation induces increase of adhesion molecules and accumulation of beta2-integrin-expressing cells in humans. Int. J. Radiat. Oncol. Biol. Phys. 45, 475–481. doi: 10.1016/s0360-3016(99)00202-3
Hansson, G. K. (2017). Inflammation and atherosclerosis: the end of a controversy. Circulation 136, 1875–1877. doi: 10.1161/circulationaha.117.030484
Harouki, N., Nicol, L., Remy-Jouet, I., Henry, J. P., Dumesnil, A., Lejeune, A., et al. (2017). The IL-1beta antibody gevokizumab limits cardiac remodeling and coronary dysfunction in rats with heart failure. JACC Transl. Sci. 2, 418–430. doi: 10.1016/j.jacbts.2017.06.005
He, H., Jiang, H., Chen, Y., Ye, J., Wang, A., Wang, C., et al. (2018). Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 9:2550. doi: 10.1038/s41467-018-04947-6
He, X., Wei, Z., Wang, J., Kou, J., Liu, W., Fu, Y., et al. (2016). Alpinetin attenuates inflammatory responses by suppressing TLR4 and NLRP3 signaling pathways in DSS-induced acute colitis. Sci. Rep. 6:28370. doi: 10.1038/srep28370
He, Y., Varadarajan, S., Munoz-Planillo, R., Burberry, A., Nakamura, Y., and Nunez, G. (2014). 3,4-methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 289, 1142–1150. doi: 10.1074/jbc.M113.515080
He, Y., Zeng, M. Y., Yang, D., Motro, B., and Nunez, G. (2016). NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357. doi: 10.1038/nature16959
Heinrich, M., Neumeyer, J., Jakob, M., Hallas, C., Tchikov, V., Winoto-Morbach, S., et al. (2004). Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 11, 550–563. doi: 10.1038/sj.cdd.4401382
Henao-Mejia, J., Elinav, E., Strowig, T., and Flavell, R. A. (2012). Inflammasomes: far beyond inflammation. Nat. Immunol. 13, 321–324. doi: 10.1038/ni.2257
Hendrikx, T., Jeurissen, M. L., Van Gorp, P. J., Gijbels, M. J., Walenbergh, S. M., Houben, T., et al. (2015). Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 282, 2327–2338. doi: 10.1111/febs.13279
Honda, H., Nagai, Y., Matsunaga, T., Okamoto, N., Watanabe, Y., Tsuneyama, K., et al. (2014). Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 96, 1087–1100. doi: 10.1189/jlb.3A0114-005RR
Hong, J. H., Chiang, C. S., Tsao, C. Y., Lin, P. Y., Mcbride, W. H., and Wu, C. J. (1999). Rapid induction of cytokine gene expression in the lung after single and fractionated doses of radiation. Int. J. Radiat. Biol. 75, 1421–1427. doi: 10.1080/095530099139287
Hooning, M. J., Botma, A., Aleman, B. M., Baaijens, M. H., Bartelink, H., Klijn, J. G., et al. (2007). Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J. Natl. Cancer Inst. 99, 365–375. doi: 10.1093/jnci/djk064
Hoving, S., Heeneman, S., Gijbels, M. J., Te Poele, J. A., Russell, N. S., Daemen, M. J., et al. (2008). Single-dose and fractionated irradiation promote initiation and progression of atherosclerosis and induce an inflammatory plaque phenotype in ApoE(-/-) mice. Int. J. Radiat. Oncol. Biol. Phys. 71, 848–857. doi: 10.1016/j.ijrobp.2008.02.031
Hu, D., Yin, C., Luo, S., Habenicht, A. J. R., and Mohanta, S. K. (2019). Vascular smooth muscle cells contribute to atherosclerosis immunity. Front. Immunol. 10:1101. doi: 10.3389/fimmu.2019.01101
Huang, Y., Jiang, H., Chen, Y., Wang, X., Yang, Y., Tao, J., et al. (2018). Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 10:e8689. doi: 10.15252/emmm.201708689
Iannitti, R. G., Napolioni, V., Oikonomou, V., De Luca, A., Galosi, C., Pariano, M., et al. (2016). IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat. Commun. 7:10791. doi: 10.1038/ncomms10791
Irrera, N., Vaccaro, M., Bitto, A., Pallio, G., Pizzino, G., Lentini, M., et al. (2017). BAY 11-7082 inhibits the NF-kappaB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin. Sci. 131, 487–498. doi: 10.1042/CS20160645
Jiang, H., He, H., Chen, Y., Huang, W., Cheng, J., Ye, J., et al. (2017). Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 214, 3219–3238. doi: 10.1084/jem.20171419
Juliana, C., Fernandes-Alnemri, T., Wu, J., Datta, P., Solorzano, L., Yu, J. W., et al. (2010). Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 285, 9792–9802. doi: 10.1074/jbc.M109.082305
Kamari, Y., Shaish, A., Shemesh, S., Vax, E., Grosskopf, I., Dotan, S., et al. (2011). Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1alpha. Biochem. Biophys. Res. Commun. 405, 197–203. doi: 10.1016/j.bbrc.2011.01.008
Karmakar, M., Katsnelson, M. A., Dubyak, G. R., and Pearlman, E. (2016). Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat. Commun. 7:10555. doi: 10.1038/ncomms10555
Kawaguchi, M., Takahashi, M., Hata, T., Kashima, Y., Usui, F., Morimoto, H., et al. (2011). Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123, 594–604. doi: 10.1161/CIRCULATIONAHA.110.982777
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20:3328. doi: 10.3390/ijms20133328
Kirii, H., Niwa, T., Yamada, Y., Wada, H., Saito, K., Iwakura, Y., et al. (2003). Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 656–660. doi: 10.1161/01.atv.0000064374.15232.c3
Kitajima, I., Soejima, Y., Takasaki, I., Beppu, H., Tokioka, T., and Maruyama, I. (1996). Ceramide-induced nuclear translocation of NF-kappa B is a potential mediator of the apoptotic response to TNF-alpha in murine clonal osteoblasts. Bone 19, 263–270. doi: 10.1016/8756-3282(96)00181-0
Kojima, S., Ohshima, Y., Nakatsukasa, H., and Tsukimoto, M. (2017). Role of ATP as a key signaling molecule mediating radiation-induced biological effects. Dose Res. 15:1559325817690638. doi: 10.1177/1559325817690638
Koka, S., Xia, M., Chen, Y., Bhat, O. M., Yuan, X., Boini, K. M., et al. (2017). Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 13, 336–344. doi: 10.1016/j.redox.2017.06.004
Koka, S., Xia, M., Zhang, C., Zhang, Y., Li, P. L., and Boini, K. M. (2019). Podocyte NLRP3 inflammasome activation and formation by adipokine visfatin. Cell Physiol. Biochem. 53, 355–365. doi: 10.33594/000000143
Kolesnick, R., and Fuks, Z. (2003). Radiation and ceramide-induced apoptosis. Oncogene 22, 5897–5906. doi: 10.1038/sj.onc.1206702
Kolliputi, N., Galam, L., Parthasarathy, P. T., Tipparaju, S. M., and Lockey, R. F. (2012). NALP-3 inflammasome silencing attenuates ceramide-induced transepithelial permeability. J. Cell Physiol. 227, 3310–3316. doi: 10.1002/jcp.24026
Konings, A. W. (1981). Radiation-induced efflux of potassium ions and haemoglobin in bovine erythrocytes at low doses and low dose-rates. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 40, 441–444. doi: 10.1080/09553008114551391
Kovacic, J. C., Dimmeler, S., Harvey, R. P., Finkel, T., Aikawa, E., Krenning, G., et al. (2019). Endothelial to Mesenchymal transition in cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 73, 190–209. doi: 10.1016/j.jacc.2018.09.089
Krenning, G., Zeisberg, E. M., and Kalluri, R. (2010). The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell Physiol. 225, 631–637. doi: 10.1002/jcp.22322
Kruse, J. J., Zurcher, C., Strootman, E. G., Bart, C. I., Schlagwein, N., Leer, J. W., et al. (2001). Structural changes in the auricles of the rat heart after local ionizing irradiation. Radiother. Oncol. 58, 303–311. doi: 10.1016/s0167-8140(00)00327-3
Kuwar, R., Rolfe, A., Di, L., Xu, H., He, L., Jiang, Y., et al. (2019). A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflam. 16:81. doi: 10.1186/s12974-019-1471-y
Lamkanfi, M., Mueller, J. L., Vitari, A. C., Misaghi, S., Fedorova, A., Deshayes, K., et al. (2009). Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 187, 61–70. doi: 10.1083/jcb.200903124
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Lepple-Wienhues, A., Belka, C., Laun, T., Jekle, A., Walter, B., Wieland, U., et al. (1999). Stimulation of CD95 (Fas) blocks T lymphocyte calcium channels through sphingomyelinase and sphingolipids. Proc. Natl. Acad. Sci. U.S.A. 96, 13795–13800. doi: 10.1073/pnas.96.24.13795
Li, M., Qian, M., Kyler, K., and Xu, J. (2018). Endothelial-vascular smooth muscle cells interactions in atherosclerosis. Front. Cardiovasc. Med. 5:151. doi: 10.3389/fcvm.2018.00151
Li, R., Lu, K., Wang, Y., Chen, M., Zhang, F., Shen, H., et al. (2017). Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem. Biophys. Res. Commun. 485, 69–75. doi: 10.1016/j.bbrc.2017.02.021
Li, X., Zhang, Y., Xia, M., Gulbins, E., Boini, K. M., and Li, P. L. (2014). Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One 9:e87552. doi: 10.1371/journal.pone.0087552
Lim, H., Min, D. S., Park, H., and Kim, H. P. (2018). Flavonoids interfere with NLRP3 inflammasome activation. Toxicol. Appl. Pharmacol. 355, 93–102. doi: 10.1016/j.taap.2018.06.022
Linard, C., Marquette, C., Mathieu, J., Pennequin, A., Clarencon, D., and Mathe, D. (2004). Acute induction of inflammatory cytokine expression after gamma-irradiation in the rat: effect of an NF-kappaB inhibitor. Int. J. Radiat. Oncol. Biol. Phys. 58, 427–434. doi: 10.1016/j.ijrobp.2003.09.039
Lipinski, M. J., and Frias, J. C. (2014). Molecule 16673-34-0: a new tool in our arsenal against inflammation. J. Cardiovasc. Pharmacol. 63, 314–315. doi: 10.1097/FJC.0000000000000070
Liston, A., and Masters, S. L. (2017). Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 17, 208–214. doi: 10.1038/nri.2016.151
Liu, C., Wang, J., Yang, Y., Liu, X., Zhu, Y., Zou, J., et al. (2018). Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem. Pharmacol. 155, 366–379. doi: 10.1016/j.bcp.2018.07.010
Liu, W., Guo, W., Wu, J., Luo, Q., Tao, F., Gu, Y., et al. (2013). A novel benzo[d]imidazole derivate prevents the development of dextran sulfate sodium-induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem. Pharmacol. 85, 1504–1512. doi: 10.1016/j.bcp.2013.03.008
Liu, W., Guo, W., Zhu, Y., Peng, S., Zheng, W., Zhang, C., et al. (2018). Targeting peroxiredoxin 1 by a curcumin analogue, AI-44, Inhibits NLRP3 inflammasome activation and attenuates lipopolysaccharide-induced sepsis in mice. J. Immunol. 201, 2403–2413. doi: 10.4049/jimmunol.1700796
Liu, Y. G., Chen, J. K., Zhang, Z. T., Ma, X. J., Chen, Y. C., Du, X. M., et al. (2017). NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 8:e2579. doi: 10.1038/cddis.2016.460
Liu, Y., Gao, X., Miao, Y., Wang, Y., Wang, H., Cheng, Z., et al. (2018). NLRP3 regulates macrophage M2 polarization through up-regulation of IL-4 in asthma. Biochem. J. 475, 1995–2008. doi: 10.1042/BCJ20180086
Lowe, D., and Raj, K. (2014). Premature aging induced by radiation exhibits pro-atherosclerotic effects mediated by epigenetic activation of CD44 expression. Aging Cell 13, 900–910. doi: 10.1111/acel.12253
Mallat, Z., Corbaz, A., Scoazec, A., Graber, P., Alouani, S., Esposito, B., et al. (2001). Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 89, E41–E45.
Marchetti, C., Chojnacki, J., Toldo, S., Mezzaroma, E., Tranchida, N., Rose, S. W., et al. (2014). A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 63, 316–322. doi: 10.1097/fjc.0000000000000053
Marchetti, C., Swartzwelter, B., Gamboni, F., Neff, C. P., Richter, K., Azam, T., et al. (2018). OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. U.S.A. 115, E1530–E1539. doi: 10.1073/pnas.1716095115
Mavragani, I. V., Laskaratou, D. A., Frey, B., Candeias, S. M., Gaipl, U. S., Lumniczky, K., et al. (2016). Key mechanisms involved in ionizing radiation-induced systemic effects. Curr. Rev. Toxicol. Res. 5, 12–33. doi: 10.1039/c5tx00222b
Meeren, A. V., Bertho, J. M., Vandamme, M., and Gaugler, M. H. (1997). Ionizing radiation enhances IL-6 and IL-8 production by human endothelial cells. Med. Inflamm. 6, 185–193. doi: 10.1080/09629359791677
Meng, Q., Bhandary, B., Osinska, H., James, J., Xu, N., Shay-Winkler, K., et al. (2017). MMI-0100 inhibits cardiac fibrosis in a mouse model overexpressing cardiac myosin binding protein C. J. Am. Heart Assoc. 6:e006590. doi: 10.1161/JAHA.117.006590
Menu, P., Pellegrin, M., Aubert, J. F., Bouzourene, K., Tardivel, A., Mazzolai, L., et al. (2011). Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2:e137. doi: 10.1038/cddis.2011.18
Merrick, T. P., and Bruce, A. K. (1965). Radiation response of potassium efflux in Micrococcus Radiodurans and Sarcina Lutea. Radiat. Res. 24, 612–618.
Meziani, L., Deutsch, E., and Mondini, M. (2018). Macrophages in radiation injury: a new therapeutic target. Oncoimmunology 7:e1494488. doi: 10.1080/2162402X.2018.1494488
Mezzaroma, E., Mikkelsen, R. B., Toldo, S., Mauro, A. G., Sharma, K., Marchetti, C., et al. (2015). Role of interleukin-1 in radiation-induced cardiomyopathy. Mol. Med. 21, 210–218. doi: 10.2119/molmed.2014.00243
Michaudel, C., Couturier-Maillard, A., Chenuet, P., Maillet, I., Mura, C., Couillin, I., et al. (2016). Inflammasome, IL-1 and inflammation in ozone-induced lung injury. Am. J. Clin. Exp. Immunol. 5, 33–40.
Milliat, F., Francois, A., Isoir, M., Deutsch, E., Tamarat, R., Tarlet, G., et al. (2006). Influence of endothelial cells on vascular smooth muscle cells phenotype after irradiation: implication in radiation-induced vascular damages. Am. J. Pathol. 169, 1484–1495. doi: 10.2353/ajpath.2006.060116
Moore-Morris, T., Guimaraes-Camboa, N., Banerjee, I., Zambon, A. C., Kisseleva, T., Velayoudon, A., et al. (2014). Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Invest. 124, 2921–2934. doi: 10.1172/JCI74783
Mulrooney, D. A., Yeazel, M. W., Kawashima, T., Mertens, A. C., Mitby, P., Stovall, M., et al. (2009). Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: retrospective analysis of the childhood cancer survivor study cohort. BMJ 339:b4606. doi: 10.1136/bmj.b4606
Mun, G. I., and Boo, Y. C. (2010). Identification of CD44 as a senescence-induced cell adhesion gene responsible for the enhanced monocyte recruitment to senescent endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 298, H2102–H2111. doi: 10.1152/ajpheart.00835.2009
Newman, A. A., Baylis, R. A., Hess, D. L., Griffith, S. D., Shankman, L. S., Cherepanova, O. A., et al. (2018). Irradiation abolishes smooth muscle investment into vascular lesions in specific vascular beds. JCI Insight 3, e121017. doi: 10.1172/jci.insight.121017
Nielsen, K. M., Offersen, B. V., Nielsen, H. M., Vaage-Nilsen, M., and Yusuf, S. W. (2017). Short and long term radiation induced cardiovascular disease in patients with cancer. Clin. Cardiol. 40, 255–261. doi: 10.1002/clc.22634
Orlowski, G. M., Colbert, J. D., Sharma, S., Bogyo, M., Robertson, S. A., and Rock, K. L. (2015). Multiple cathepsins promote pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J. Immunol. 195, 1685–1697. doi: 10.4049/jimmunol.1500509
Ozaki, E., Campbell, M., and Doyle, S. L. (2015). Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J. Inflamm. Res. 8, 15–27. doi: 10.2147/JIR.S51250
Pan, X. C., Liu, Y., Cen, Y. Y., Xiong, Y. L., Li, J. M., Ding, Y. Y., et al. (2019). Dual role of triptolide in interrupting the nlrp3 inflammasome pathway to attenuate cardiac fibrosis. Int. J. Mol. Sci. 20:360. doi: 10.3390/ijms20020360
Paramel Varghese, G., Folkersen, L., Strawbridge, R. J., Halvorsen, B., Yndestad, A., Ranheim, T., et al. (2016). NLRP3 Inflammasome expression and activation in human atherosclerosis. J. Am. Heart Assoc. 5:e003031. doi: 10.1161/JAHA.115.003031
Persson, H. L., Kurz, T., Eaton, J. W., and Brunk, U. T. (2005). Radiation-induced cell death: importance of lysosomal destabilization. Biochem. J. 389, 877–884. doi: 10.1042/bj20050271
Petrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., and Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589. doi: 10.1038/sj.cdd.4402195
Prabhakarpandian, B., Goetz, D. J., Swerlick, R. A., Chen, X., and Kiani, M. F. (2001). Expression and functional significance of adhesion molecules on cultured endothelial cells in response to ionizing radiation. Microcirculation 8, 355–364. doi: 10.1111/j.1549-8719.2001.tb00182.x
Raghu, G., Richeldi, L., Crestani, B., Wung, P., Bejuit, R., Esperet, C., et al. (2018). SAR156597 in idiopathic pulmonary fibrosis: a phase 2 placebo-controlled study (DRI11772). Eur. Respir. J. 52, 1801130. doi: 10.1183/13993003.01130-2018
Ramadan, R., Vromans, E., Anang, D. C., Decrock, E., Mysara, M., Monsieurs, P., et al. (2019). Single and fractionated ionizing radiation induce alterations in endothelial connexin expression and channel function. Sci. Rep. 9:4643. doi: 10.1038/s41598-019-39317-9
Ren, X. S., Tong, Y., Ling, L., Chen, D., Sun, H. J., Zhou, H., et al. (2017). NLRP3 Gene deletion attenuates angiotensin ii-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell Physiol. Biochem. 44, 2269–2280. doi: 10.1159/000486061
Ridker, P. M., Howard, C. P., Walter, V., Everett, B., Libby, P., Hensen, J., et al. (2012). Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation 126, 2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556
Rockey, D. C., Bell, P. D., and Hill, J. A. (2015). Fibrosis–a common pathway to organ injury and failure. N. Engl. J. Med. 372, 1138–1149. doi: 10.1056/nejmra1300575
Rudolphi, K., Gerwin, N., Verzijl, N., Van Der Kraan, P., and Van Den Berg, W. (2003). Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthr. Cartilage 11, 738–746. doi: 10.1016/s1063-4584(03)00153-5
Satoh, M., Tabuchi, T., Itoh, T., and Nakamura, M. (2014). NLRP3 inflammasome activation in coronary artery disease: results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin. Sci. 126, 233–241. doi: 10.1042/cs20130043
Scheiblich, H., Schlutter, A., Golenbock, D. T., Latz, E., Martinez-Martinez, P., and Heneka, M. T. (2017). Activation of the NLRP3 inflammasome in microglia: the role of ceramide. J. Neurochem. 143, 534–550. doi: 10.1111/jnc.14225
Schiller, N. K., Kubo, N., Boisvert, W. A., and Curtiss, L. K. (2001). Effect of gamma-irradiation and bone marrow transplantation on atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 21, 1674–1680. doi: 10.1161/hq1001.096724
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Schultz-Hector, S. (1992). Radiation-induced heart disease: review of experimental data on dose response and pathogenesis. Int. J. Radiat. Biol. 61, 149–160. doi: 10.1080/09553009214550761
Scott, A. S., Parr, L. A., and Johnstone, P. A. (2009). Risk of cerebrovascular events after neck and supraclavicular radiotherapy: a systematic review. Radiother. Oncol. 90, 163–165. doi: 10.1016/j.radonc.2008.12.019
Semino, C., Carta, S., Gattorno, M., Sitia, R., and Rubartelli, A. (2018). Progressive waves of IL-1beta release by primary human monocytes via sequential activation of vesicular and gasdermin D-mediated secretory pathways. Cell Death Dis. 9:1088. doi: 10.1038/s41419-018-1121-9
Shah, S. R., Abbasi, Z., Fatima, M., Ochani, R. K., Shahnawaz, W., Asim Khan, M., et al. (2018). Canakinumab and cardiovascular outcomes: results of the CANTOS trial. J. Commun. Hosp. Intern. Med. Perspect. 8, 21–22. doi: 10.1080/20009666.2018.1428023
Shankman, L. S., Gomez, D., Cherepanova, O. A., Salmon, M., Alencar, G. F., Haskins, R. M., et al. (2015). KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 21, 628–637. doi: 10.1038/nm.3866
Sharma, D., and Czarnota, G. J. (2019). Role of acid sphingomyelinase-induced ceramide generation in response to radiation. Oncotarget 10, 6–7.
Shi, X., Xie, W. L., Kong, W. W., Chen, D., and Qu, P. (2015). Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 24, 2455–2466. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
Shim, D. W., Shin, W. Y., Yu, S. H., Kim, B. H., Ye, S. K., Koppula, S., et al. (2017). BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci. Rep. 7:15020. doi: 10.1038/s41598-017-15314-8
Shimizu, Y., Kodama, K., Nishi, N., Kasagi, F., Suyama, A., Soda, M., et al. (2010). Radiation exposure and circulatory disease risk: Hiroshima and Nagasaki atomic bomb survivor data, 1950-2003. BMJ 340:b5349. doi: 10.1136/bmj.b5349
Soloviev, A. I., Tishkin, S. M., Parshikov, A. V., Ivanova, I. V., Goncharov, E. V., and Gurney, A. M. (2003). Mechanisms of endothelial dysfunction after ionized radiation: selective impairment of the nitric oxide component of endothelium-dependent vasodilation. Br. J. Pharmacol. 138, 837–844. doi: 10.1038/sj.bjp.0705079
Song, H., Liu, B., Huai, W., Yu, Z., Wang, W., Zhao, J., et al. (2016). The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 7:13727. doi: 10.1038/ncomms13727
Srebot, V., Gianicolo, E. A., Rainaldi, G., Trivella, M. G., and Sicari, R. (2009). Ozone and cardiovascular injury. Cardiovasc. Ultrasound 7:30. doi: 10.1186/1476-7120-7-30
Stewart, F. A., Heeneman, S., Te Poele, J., Kruse, J., Russell, N. S., Gijbels, M., et al. (2006). Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE-/- mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am. J. Pathol. 168, 649–658. doi: 10.2353/ajpath.2006.050409
Stewart, F. A., Seemann, I., Hoving, S., and Russell, N. S. (2013). Understanding radiation-induced cardiovascular damage and strategies for intervention. Clin. Oncol. 25, 617–624. doi: 10.1016/j.clon.2013.06.012
Stoecklein, V. M., Osuka, A., Ishikawa, S., Lederer, M. R., Wanke-Jellinek, L., and Lederer, J. A. (2015). Radiation exposure induces inflammasome pathway activation in immune cells. J. Immunol. 194, 1178–1189. doi: 10.4049/jimmunol.1303051
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z., and Germain, R. N. (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. doi: 10.1016/j.cell.2013.02.054
Sun, H. J., Ren, X. S., Xiong, X. Q., Chen, Y. Z., Zhao, M. X., Wang, J. J., et al. (2017). NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 8:e3074. doi: 10.1038/cddis.2017.470
Sun, Y., Zhao, Y., Yao, J., Zhao, L., Wu, Z., Wang, Y., et al. (2015). Wogonoside protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting NF-kappaB and NLRP3 inflammasome activation. Biochem. Pharmacol. 94, 142–154. doi: 10.1016/j.bcp.2015.02.002
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. doi: 10.1038/s41577-019-0165-0
Sylvester, C. B., Abe, J. I., Patel, Z. S., and Grande-Allen, K. J. (2018). Radiation-induced cardiovascular disease: mechanisms and importance of linear energy transfer. Front. Cardiovasc. Med. 5:5. doi: 10.3389/fcvm.2018.00005
Szabo, I., Gulbins, E., Apfel, H., Zhang, X., Barth, P., Busch, A. E., et al. (1996). Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J. Biol. Chem. 271, 20465–20469. doi: 10.1074/jbc.271.34.20465
Tadic, M., Cuspidi, C., Hering, D., Venneri, L., and Grozdic-Milojevic, I. (2017). Radiotherapy-induced right ventricular remodelling: The missing piece of the puzzle. Arch. Cardiovasc. Dis. 110, 116–123. doi: 10.1016/j.acvd.2016.10.003
Tanaka, T., Ogawa, M., Suzuki, J., Sekinishi, A., Itai, A., Hirata, Y., et al. (2012). Inhibition of IkappaB phosphorylation prevents load-induced cardiac dysfunction in mice. Am. J. Physiol. Heart Circ. Physiol. 303, H1435–H1445. doi: 10.1152/ajpheart.00290.2012
Taunk, N. K., Haffty, B. G., Kostis, J. B., and Goyal, S. (2015). Radiation-induced heart disease: pathologic abnormalities and putative mechanisms. Front. Oncol. 5:39. doi: 10.3389/fonc.2015.00039
Taylor, C. W., Nisbet, A., Mcgale, P., and Darby, S. C. (2007). Cardiac exposures in breast cancer radiotherapy: 1950s-1990s. Int. J. Radiat. Oncol. Biol. Phys. 69, 1484–1495. doi: 10.1016/j.ijrobp.2007.05.034
Teresa Pinto, A., Laranjeiro Pinto, M., Patricia Cardoso, A., Monteiro, C., Teixeira Pinto, M., Filipe Maia, A., et al. (2016). Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci. Rep. 6:18765. doi: 10.1038/srep18765
Tezcan, G., Martynova, E. V., Gilazieva, Z. E., Mcintyre, A., Rizvanov, A. A., and Khaiboullina, S. F. (2019). MicroRNA post-transcriptional regulation of the NLRP3 inflammasome in immunopathologies. Front. Pharmacol. 10:451. doi: 10.3389/fphar.2019.00451
Todd, D. G., and Mikkelsen, R. B. (1994). Ionizing radiation induces a transient increase in cytosolic free [Ca2+] in human epithelial tumor cells. Cancer Res. 54, 5224–5230.
Travers, J. G., Kamal, F. A., Robbins, J., Yutzey, K. E., and Blaxall, B. C. (2016). Cardiac fibrosis: the fibroblast awakens. Circ. Res. 118, 1021–1040. doi: 10.1161/CIRCRESAHA.115.306565
Tukenova, M., Guibout, C., Oberlin, O., Doyon, F., Mousannif, A., Haddy, N., et al. (2010). Role of cancer treatment in long-term overall and cardiovascular mortality after childhood cancer. J. Clin. Oncol. 28, 1308–1315. doi: 10.1200/JCO.2008.20.2267
Unthank, J. L., Miller, S. J., Quickery, A. K., Ferguson, E. L., Wang, M., Sampson, C. H., et al. (2015). Delayed effects of acute radiation exposure in a murine model of the H-ARS: multiple-organ injury consequent to <10 Gy total body irradiation. Health Phys. 109, 511–521. doi: 10.1097/HP.0000000000000357
Unthank, J. L., Ortiz, M., Trivedi, H., Pelus, L. M., Sampson, C. H., Sellamuthu, R., et al. (2019). Cardiac and Renal delayed effects of acute radiation exposure: organ differences in vasculopathy, inflammation, senescence and oxidative balance. Radiat. Res. 191, 383–397. doi: 10.1667/RR15130.1
Usui, F., Shirasuna, K., Kimura, H., Tatsumi, K., Kawashima, A., Karasawa, T., et al. (2012). Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 425, 162–168. doi: 10.1016/j.bbrc.2012.07.058
Vandanmagsar, B., Youm, Y. H., Ravussin, A., Galgani, J. E., Stadler, K., Mynatt, R. L., et al. (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188. doi: 10.1038/nm.2279
Venkatesulu, B. P., Mahadevan, L. S., Aliru, M. L., Yang, X., Bodd, M. H., Singh, P. K., et al. (2018). Radiation-induced endothelial vascular injury: a review of possible mechanisms. JACC Transl. Sci. 3, 563–572. doi: 10.1016/j.jacbts.2018.01.014
Vigano, E., Diamond, C. E., Spreafico, R., Balachander, A., Sobota, R. M., and Mortellaro, A. (2015). Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 6:8761. doi: 10.1038/ncomms9761
Virmani, R., Farb, A., Carter, A. J., and Jones, R. M. (1999). Pathology of radiation-induced coronary artery disease in human and pig. Cardiovasc. Radiat. Med. 1, 98–101. doi: 10.1016/s1522-1865(98)00010-9
Wang, H., Wei, J., Zheng, Q., Meng, L., Xin, Y., Yin, X., et al. (2019). Radiation-induced heart disease: a review of classification, mechanism and prevention. Int. J. Biol. Sci. 15, 2128–2138. doi: 10.7150/ijbs.35460
Wang, K., Lv, Q., Miao, Y. M., Qiao, S. M., Dai, Y., and Wei, Z. F. (2018). Cardamonin, a natural flavone, alleviates inflammatory bowel disease by the inhibition of NLRP3 inflammasome activation via an AhR/Nrf2/NQO1 pathway. Biochem. Pharmacol. 155, 494–509. doi: 10.1016/j.bcp.2018.07.039
Wang, L., Qu, P., Zhao, J., and Chang, Y. (2014). NLRP3 and downstream cytokine expression elevated in the monocytes of patients with coronary artery disease. Arch. Med. Sci. 10, 791–800. doi: 10.5114/aoms.2014.44871
Wang, M., Sampson, P. D., Sheppard, L. E., Stein, J. H., Vedal, S., and Kaufman, J. D. (2019). Long-term exposure to ambient ozone and progression of subclinical arterial disease: the multi-ethnic study of atherosclerosis and air pollution. Environ. Health Perspect. 127, 57001. doi: 10.1289/EHP3325
Wang, R., Wu, W., Li, W., Huang, S., Li, Z., Liu, R., et al. (2018). Activation of NLRP3 inflammasome promotes foam cell formation in vascular smooth muscle cells and atherogenesis Via HMGB1. J. Am. Heart Assoc. 7:e008596. doi: 10.1161/JAHA.118.008596
Wang, S., Lin, Y., Yuan, X., Li, F., Guo, L., and Wu, B. (2018). REV-ERBalpha integrates colon clock with experimental colitis through regulation of NF-kappaB/NLRP3 axis. Nat. Commun. 9:4246. doi: 10.1038/s41467-018-06568-5
Wang, W., Wang, X., Chun, J., Vilaysane, A., Clark, S., French, G., et al. (2013). Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J. Immunol. 190, 1239–1249. doi: 10.4049/jimmunol.1201959
Wang, X., Wang, S., Hu, C., Chen, W., Shen, Y., Wu, X., et al. (2015). A new pharmacological effect of levornidazole: Inhibition of NLRP3 inflammasome activation. Biochem. Pharmacol. 97, 178–188. doi: 10.1016/j.bcp.2015.06.030
Wang, Y., Li, H., Li, Y., Zhao, Y., Xiong, F., Liu, Y., et al. (2019). Coriolus versicolor alleviates diabetic cardiomyopathy by inhibiting cardiac fibrosis and NLRP3 inflammasome activation. Phytother. Res. 33, 2737–2748. doi: 10.1002/ptr.6448
Wannamaker, W., Davies, R., Namchuk, M., Pollard, J., Ford, P., Ku, G., et al. (2007). (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoy l)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J. Pharmacol. Exp. Ther. 321, 509–516. doi: 10.1124/jpet.106.111344
Wei, J., Wang, H., Wang, H., Wang, B., Meng, L., Xin, Y., et al. (2019). The role of NLRP3 inflammasome activation in radiation damage. Biomed. Pharmacother. 118:109217. doi: 10.1016/j.biopha.2019.109217
Weintraub, N. L., Jones, W. K., and Manka, D. (2010). Understanding radiation-induced vascular disease. J. Am. Coll. Cardiol. 55, 1237–1239. doi: 10.1016/j.jacc.2009.11.053
Werner, E., Alter, A., Deng, Q., Dammer, E. B., Wang, Y., Yu, D. S., et al. (2019). Ionizing radiation induction of cholesterol biosynthesis in Lung tissue. Sci. Rep. 9:12546. doi: 10.1038/s41598-019-48972-x
Wolf, D., and Ley, K. (2019). Immunity and Inflammation in atherosclerosis. Circ. Res. 124, 315–327. doi: 10.1161/CIRCRESAHA.118.313591
Wong, F. L., Yamada, M., Sasaki, H., Kodama, K., and Hosoda, Y. (1999). Effects of radiation on the longitudinal trends of total serum cholesterol levels in the atomic bomb survivors. Radiat. Res. 151, 736–746.
Wree, A., Mehal, W. Z., and Feldstein, A. E. (2016). Targeting cell death and sterile inflammation loop for the treatment of nonalcoholic steatohepatitis. Semin. Liver Dis. 36, 27–36. doi: 10.1055/s-0035-1571272
Wu, D., Wu, K., Zhu, Q., Xiao, W., Shan, Q., Yan, Z., et al. (2018). Formononetin administration ameliorates dextran sulfate sodium-induced acute colitis by inhibiting NLRP3 inflammasome signaling pathway. Mediat. Inflamm. 2018:3048532.
Wu, X. P., Wang, H. J., Wang, Y. L., Shen, H. R., and Tan, Y. Z. (2018). Serelaxin inhibits differentiation and fibrotic behaviors of cardiac fibroblasts by suppressing ALK-5/Smad2/3 signaling pathway. Exp. Cell Res. 362, 17–27. doi: 10.1016/j.yexcr.2017.10.004
Xin, W., Wang, Q., Zhang, D., and Wang, C. (2017). A new mechanism of inhibition of IL-1beta secretion by celastrol through the NLRP3 inflammasome pathway. Eur. J. Pharmacol. 814, 240–247. doi: 10.1016/j.ejphar.2017.08.036
Yang, W. L., Sharma, A., Wang, Z., Li, Z., Fan, J., and Wang, P. (2016). Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci. Rep. 6:26571. doi: 10.1038/srep26571
Yarnold, J., and Brotons, M. C. (2010). Pathogenetic mechanisms in radiation fibrosis. Radiother. Oncol. 97, 149–161. doi: 10.1016/j.radonc.2010.09.002
Yaron, J. R., Gangaraju, S., Rao, M. Y., Kong, X., Zhang, L., Su, F., et al. (2015). K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. 6:e1954. doi: 10.1038/cddis.2015.277
Yi, Y. S. (2018). Regulatory roles of the caspase-11 non-canonical inflammasome in inflammatory diseases. Immun. Netw. 18:e41. doi: 10.4110/in.2018.18.e41
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Yuan, Y. Y., Xie, K. X., Wang, S. L., and Yuan, L. W. (2018). Inflammatory caspase-related pyroptosis: mechanism, regulation and therapeutic potential for inflammatory bowel disease. Gastroenterol. Rep. 6, 167–176. doi: 10.1093/gastro/goy011
Yusuf, S. W., Venkatesulu, B. P., Mahadevan, L. S., and Krishnan, S. (2017). Radiation-induced cardiovascular disease: a clinical perspective. Front. Cardiovasc. Med. 4:66. doi: 10.3389/fcvm.2017.00066
Zahid, A., Li, B., Kombe, A. J. K., Jin, T., and Tao, J. (2019). Pharmacological inhibitors of the NLRP3 inflammasome. Front. Immunol. 10:2538. doi: 10.3389/fimmu.2019.02538
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., Mcmullen, J. R., Gustafsson, E., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961.
Zhang, H., and Wang, Z. C. (2020). Effect and regulation of the NLRP3 inflammasome during renal fibrosis. Front. Cell Dev. Biol. 7:379. doi: 10.3389/fcell.2019.00379
Zhang, P., Guan, Y., Chen, J., Li, X., Mcconnell, B. K., Zhou, W., et al. (2018). Contribution of p62/SQSTM1 to PDGF-BB-induced myofibroblast-like phenotypic transition in vascular smooth muscle cells lacking Smpd1 gene. Cell Death Dis. 9:1145. doi: 10.1038/s41419-018-1197-2
Zhang, X., Meng, F., Song, J., Zhang, L., Wang, J., Li, D., et al. (2016). Pentoxifylline ameliorates cardiac fibrosis, pathological hypertrophy, and cardiac dysfunction in angiotensin ii-induced hypertensive rats. J. Cardiovasc. Pharmacol. 67, 76–85. doi: 10.1097/FJC.0000000000000316
Zhang, Y., Edgley, A. J., Cox, A. J., Powell, A. K., Wang, B., Kompa, A. R., et al. (2012). FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy. Eur. J. Heart Fail. 14, 549–562. doi: 10.1093/eurjhf/hfs011
Zhang, Y., Li, X., Pitzer, A. L., Chen, Y., Wang, L., and Li, P. L. (2015). Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: beyond inflammation. Antioxid. Redox Signal. 22, 1084–1096. doi: 10.1089/ars.2014.5978
Zhao, F., Fu, L., Yang, W., Dong, Y., Yang, J., Sun, S., et al. (2016). Cardioprotective effects of baicalein on heart failure via modulation of Ca(2+) handling proteins in vivo and in vitro. Life Sci. 145, 213–223. doi: 10.1016/j.lfs.2015.12.036
Zhao, Q. D., Viswanadhapalli, S., Williams, P., Shi, Q., Tan, C., Yi, X., et al. (2015). NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation 131, 643–655. doi: 10.1161/CIRCULATIONAHA.114.011079
Zheng, F., Xing, S., Gong, Z., and Xing, Q. (2013). NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 22, 746–750. doi: 10.1016/j.hlc.2013.01.012
Zheng, F., Xing, S., Gong, Z., Mu, W., and Xing, Q. (2014). Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediat. Inflamm. 2014:507208. doi: 10.1155/2014/507208
Zheng, X., Hu, M., Zang, X., Fan, Q., Liu, Y., Che, Y., et al. (2019). Kynurenic acid/GPR35 axis restricts NLRP3 inflammasome activation and exacerbates colitis in mice with social stress. Brain Behav. Immun. 79, 244–255. doi: 10.1016/j.bbi.2019.02.009
Zhong, Z., Sanchez-Lopez, E., and Karin, M. (2016). Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin. Exp. Rheumatol. 34, 12–16.
Zhou, R., Tardivel, A., Thorens, B., Choi, I., and Tschopp, J. (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140. doi: 10.1038/ni.1831
Keywords: NLRP inflammasome, radiation, cardiovascular injury, atherosclerosis, radiation fibrosis
Citation: Huang S, Che J, Chu Q and Zhang P (2020) The Role of NLRP3 Inflammasome in Radiation-Induced Cardiovascular Injury. Front. Cell Dev. Biol. 8:140. doi: 10.3389/fcell.2020.00140
Received: 31 October 2019; Accepted: 19 February 2020;
Published: 12 March 2020.
Edited by:
Zhengchao Wang, Fujian Normal University, ChinaReviewed by:
Yang Chen, Guangzhou University of Chinese Medicine, ChinaGuangbi Li, Virginia Commonwealth University, United States
Eleonora Mezzaroma, Virginia Commonwealth University, United States
Copyright © 2020 Huang, Che, Chu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Zhang, cGVuZ3poYW5nQHRqaC50am11LmVkdS5jbg==