 Yue-Yu Gu
Yue-Yu Gu Xu-Sheng Liu1
Xu-Sheng Liu1 Hui-Yao Lan
Hui-Yao Lan- 1Guangdong Provincial Key Laboratory of Clinical Research on Traditional Chinese Medicine Syndrome, Department of Nephrology, Guangdong Provincial Hospital of Chinese Medicine, The Second Affiliated Hospital, Guangzhou University of Chinese Medicine, Guangzhou, China
- 2Department of Medicine and Therapeutics, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China
- 3Guangdong-Hong Kong Joint Laboratory for Immunity and Genetics of Chronic Kidney Disease, Guangdong Academy of Medical Sciences, Guangdong Provincial People‘s Hospital, Guangzhou, China
Inflammation and fibrosis are two pathological features of chronic kidney disease (CKD). Transforming growth factor-β (TGF-β) has been long considered as a key mediator of renal fibrosis. In addition, TGF-β also acts as a potent anti-inflammatory cytokine that negatively regulates renal inflammation. Thus, blockade of TGF-β inhibits renal fibrosis while promoting inflammation, revealing a diverse role for TGF-β in CKD. It is now well documented that TGF-β1 activates its downstream signaling molecules such as Smad3 and Smad3-dependent non-coding RNAs to transcriptionally and differentially regulate renal inflammation and fibrosis, which is negatively regulated by Smad7. Therefore, treatments by rebalancing Smad3/Smad7 signaling or by specifically targeting Smad3-dependent non-coding RNAs that regulate renal fibrosis or inflammation could be a better therapeutic approach. In this review, the paradoxical functions and underlying mechanisms by which TGF-β1 regulates in renal inflammation and fibrosis are discussed and novel therapeutic strategies for kidney disease by targeting downstream TGF-β/Smad signaling and transcriptomes are highlighted.
Introduction
Increasing evidence shows that chronic kidney disease (CKD) is a global-burden-disease (Romagnani et al., 2017). The prevalence and incidence of CKD have risen by almost 90% over last 30 years (Provenzano et al., 2019). During the progression of CKD, renal function is impaired with a loss of nephrons and the development of renal fibrosis characterized by the excessive accumulation of extracellular matrix (ECM) components, reduction in glomerular filtration rate (GFR), and abnormal albuminuria (Glassock et al., 2017). CKD eventually leads to the development of end-stage renal disease (ESRD) (Eddy and Neilson, 2006; Liu, 2011). Fibrosis and inflammation are the two major features of CKD and prolonged renal inflammation promotes renal fibrosis as well (Meng et al., 2014; Li et al., 2017). Physiologically, fibrosis is a repair and healing process in response to the initial renal insults. However, as the pathological condition prolongs, unresolved renal inflammation turns into a major driving force to promote renal scar formation via a progressive process of renal fibrosis (Meng et al., 2014; Mihai et al., 2018).
Transforming growth factor-β has been long considered as a master cytokine in the pathogenesis of renal inflammation and fibrosis (Meng et al., 2016). The TGF-β superfamily contains members of TGF-βs, activins, inhibins, growth and differentiation factors (GDFs), bone morphogenetic proteins (BMPs), and glial-derived neurotrophic factors (GDNFs) (Zhang and Newfeld, 2013). It is well established that there are three isoforms of TGF-β in mammals, the TGF-β1, 2 and 3 (Roberts et al., 1991). Of these, TGF-β1 has been considered as a profibrotic mediator in various kidney diseases (Sureshbabu et al., 2016). Newly synthesized TGF-β1 releases and binds to the latency-associated peptide (LAP) to form a latent complex which later binds to the TGF-β binding protein (LTBP) to form a larger complex (Ando et al., 1995; Kusakabe et al., 2008). The latent complex is inactive and stored in the ECM until it is released by reactive oxygen species (ROS) and plasmin or acid. Once TGF-β1 is released from LAP and LTBP, it becomes active (Saharinen et al., 1999; Annes et al., 2003). Active TGF-β1 binds to Type II TGF-β receptor (TβRII), which recruits and activates Type I TGF-β receptor (TβRI) and downstream receptor-associated Smads (R-Smads), Smad2, and Smad3. The phosphorylated Smad2/3 then form an oligomeric complex with Smad4 (Derynck and Zhang, 2003; Lan and Chung, 2012). Subsequently, the Smad2/3/4 complex translocate into the nucleus to regulate transcription of target genes, inducing α-smooth muscle actin (α-SMA), collagens, and inhibitory Smad7 (Nakao et al., 1997; Miyazawa and Miyazono, 2017). Interestingly, Smad7 can antagonize TGF-β-mediated fibrosis, carcinogenesis and inflammation in various diseases (Yan et al., 2009; Troncone et al., 2018; Zhou G. et al., 2018). Smad7 negatively regulates TGF-β/Smad signaling by competing with the R-Smad binding to the TβRI (Yan et al., 2016; Figure 1). Moreover, Smad7 also induces the IκBα, a NF-κB inhibitor, to suppress NF-kB-driven inflammatory response (Bitzer et al., 2000; Wang et al., 2005a; Chen et al., 2018).
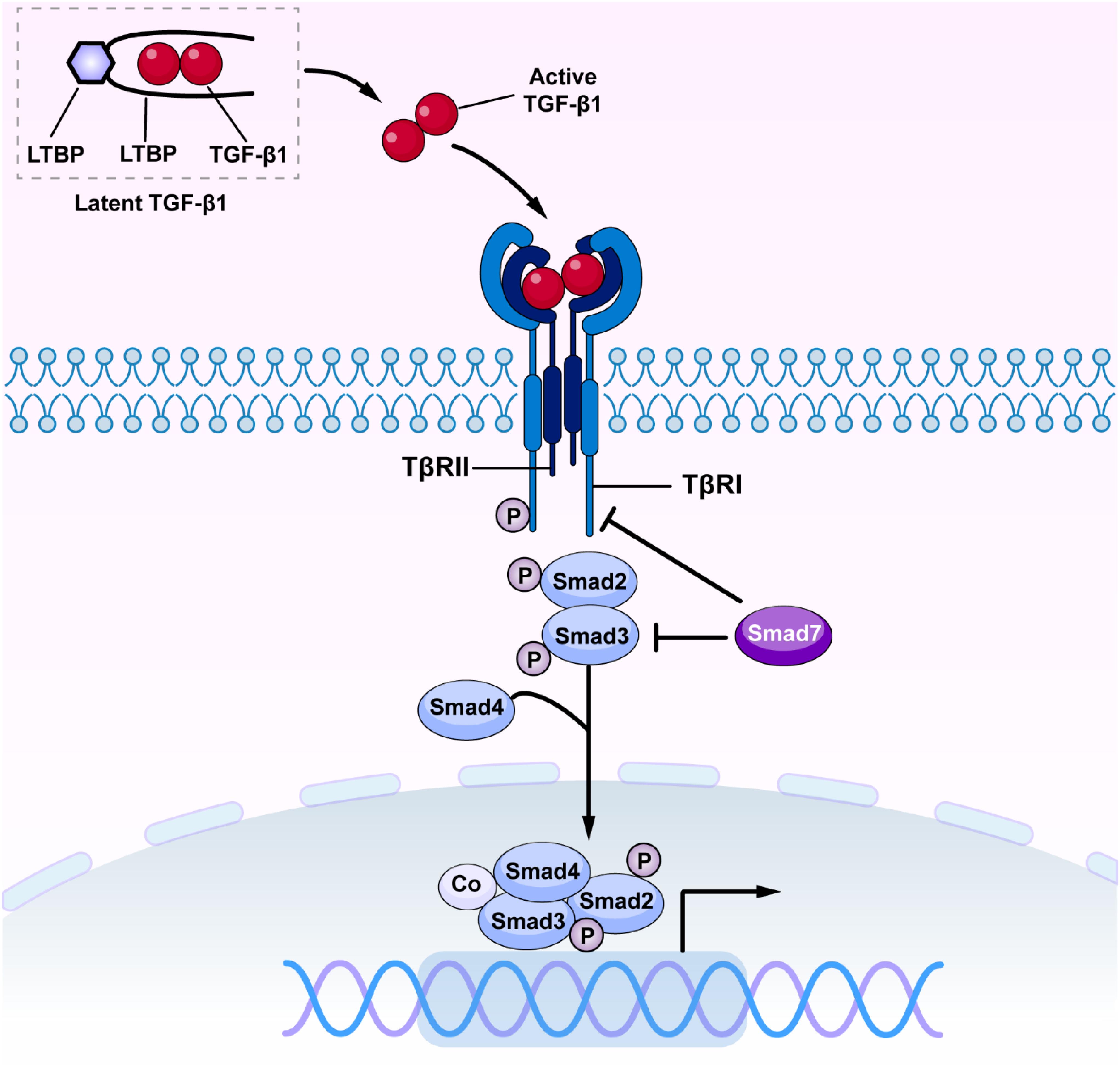
Figure 1. The canonical TGF-β/Smad signaling in fibrosis. Once released, active TGF-β1 binds TβRII and activates TβRI and R-Smads (Smad2 and Smad3), resulting in formation of a complex with Smad4. The Smad2/3/4 complex then translates into the nucleus and binds to the target genes to induce fibrosis and inflammation. TGF-β, transforming growth factor β; TβRI, TGF-β receptor type I; TβRII, TGF-β receptor type II.
In this review, the diverse roles of canonical TGF-β signaling, the distinct roles of downstream Smad proteins, and the potential therapeutic strategies for renal fibrosis and inflammation by targeting downstream TGF-β/Smad signaling are discussed.
Diverse Roles of TGF-β1 in Renal Fibrosis and Inflammation
It is well accepted that TGF-β is a master regulator in renal inflammation and fibrosis (Meng et al., 2016). TGF-β exerts multifunctional effects on cell proliferation, apoptosis, migration, differentiation, and ECM production (Massagué, 2012). TGF-β1 induces tubular and glomerular epithelial cell-to-mesenchymal transition (EMT) and excessive ECM production and deposition in glomeruli and tubulointerstitium (Fan et al., 1999; Ng et al., 1999). TGF-β1 is highly expressed in a wide range of kidney diseases associated with fibrosis (Lopez-Hernandez and Lopez-Novoa, 2012; Wang et al., 2017; Isaka, 2018). The functions of TGF-β1 on renal fibrosis and EMT were further confirmed by the findings that overexpression of active TGF-β1 in liver causes the development of severe renal fibrosis in mice (Bottinger et al., 1996; Kopp et al., 1996). Whereas, anti-TGF-β treatments by using neutralizing antibodies (Border et al., 1990), inhibitors against the TβRII (Sutaria et al., 1998; Liu et al., 2018), or antisense oligonucleotides to TGF-β1 (Akagi et al., 1996; Miyajima et al., 2000; Ziyadeh et al., 2000; Chen et al., 2003) halt the progression of renal fibrosis, suggesting a vital pathological role of TGF-β in CKD.
Renal inflammation is driven by NF-κB-dependent mechanism (Sanz et al., 2010; Ernandez and Mayadas, 2016). TGF-β is considered to be one of anti-inflammatory cytokines during the renal repair process in response to the injuries (Meng et al., 2014; Nikolic-Paterson et al., 2014; Meng, 2019; Tang et al., 2019). A number of studies have reported that mice deficient TGF-β1 suffer from the lethal inflammation and the early death (Kulkarni et al., 1993; Yaswen et al., 1996), suggesting a protective role for TGF-β in renal inflammation. Consistently, conditional deletion of TβRII from mice results in protection against TGF-β/Smad3-mediated renal fibrosis while enhancing NF-κB-driven renal inflammation (Meng et al., 2012a). More importantly, TGF-β is also a master regulator of T cell immune responses in a variety of immune diseases (Li and Flavell, 2008), which makes TGF-β as a key regulator in renal inflammation.
It should be pointed out that TGF-β signaling is not the sole pathway mediating the fibrotic process (Luo, 2017). Increasing evidence shows that TGF-β signaling can interact with other signaling pathways to mediate fibrosis. Among TGF-β signaling, both canonical and non-canonical TGF-β/Smad signaling pathways play a role in the renal fibrosis (Figure 2). Importantly, under disease conditions, Smad signaling can also be activated independently TGF-β1 by many stress molecules such as angiotensin II, and advanced glycation end products (AGE) via the ERK/p38/MAPK-Smad crosstalk pathway (Wang et al., 2005b, 2006; Yang et al., 2009; Meng et al., 2016). TGF-β/Smad can also interact with other signaling pathways such as Wnt/β-catenin, Jagged1/Notch, and Hedgehog to regulate epithelial dedifferentiation, myofibroblast transformation and proliferation (Edeling et al., 2016). In addition, TGF-β can induce renal fibrosis by transactivating epidermal growth factor receptor (EGFR) and p53 via proto-oncogene tyrosine-protein kinase Src (c-Src) and ROS-dependent mechanisms (Samarakoon et al., 2013; Harskamp et al., 2016). TGF-β1 also induces phosphorylation and acetylation of p53 and promote formation of p53/Smad3 complexes during renal fibrosis (Higgins et al., 2018; Rane et al., 2019). By contrast, BMP signaling via Smad1/5/8 complex is able to counter regulate TGF-β/Smad-mediated renal fibrosis (Weiskirchen et al., 2009; Meng et al., 2013; Munoz-Felix et al., 2015). Thus, TGF-β may exert its diverse role in renal inflammation and fibrosis by interacting with many other signaling pathways and molecules.
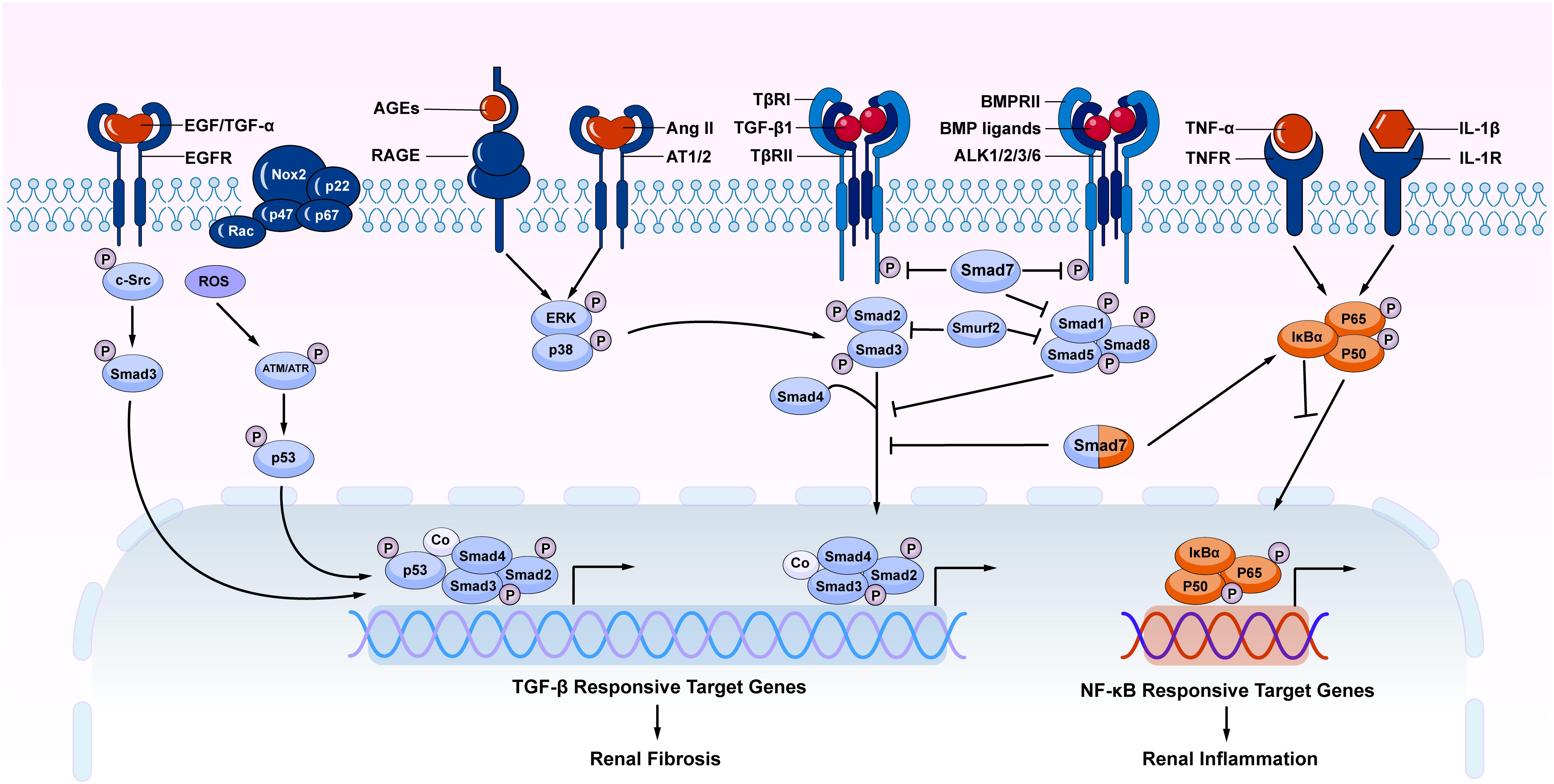
Figure 2. The overview of crosstalk pathways associated with renal fibrosis and inflammation. Many stress molecules such as TGF-β1, EGF, TGF-α, ROS, AGEs, and Ang II can activate individual pathways and interact with TGF-β/Smad signaling pathway to regulate renal fibrosis and inflammation. Among TGF-β super family, the BMP signaling negatively regulates TGF-β/Smad signaling. In TGF-β/Smad signaling, Smad7 inhibits the phosphorylation of TβRI and R-Smads via ubiquitin degradation mechanism. Meanwhile, Smad7 also alleviates renal inflammatory by inducing IκBα, therefore inhibiting NF-κB-driven inflammation. AGEs, advanced glycation end products; RAGE, receptor for AGE; Ang II, angiotensin II; AT1/2, Ang II receptor 1 and 2; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; EGF, epidermal growth factor; EGFR, EGF receptor; c-Src, proto-oncogene tyrosine-protein kinase Src; ROS, reactive oxygen species; BMP, bone morphogenic protein; ALK, activin receptor-like kinases; TNF-α, tumor necrosis factor α; TNFR, TNF receptor; IL-1, Interleukin 1; IL-1R, IL-1 receptor; Nox, NADPH oxidase.
Distinct Roles of Smad2 and Smad3 in Renal Fibrosis
In canonical TGF-β signaling, Smad2, and Smad3 are two key downstream mediators that are highly activated in the fibrotic kidney (Wang et al., 2006; Chung et al., 2010b; Zhou et al., 2010; Loeffler et al., 2018). Although Smad2 and Smad3 bind together, their functional roles are distinct. In the context of fibrosis, Smad3 is pathogenic while Smad2 is protective (Meng et al., 2010, 2016; Duan et al., 2014). Smad3 can induce matrix deposition by directly binding to the promoter region of collagen-producing genes and tissue inhibitor of matrix metalloproteinases (TIMP) while reducing the activity of MMP-1 to inhibit ECM degradation (Hall et al., 2003). By contrast, role of Smad2 in fibrosis is not fully elucidated due to a lack of Smad2 knockout (KO) mice which is embryonic lethal (Ju et al., 2006). However, a recent finding that conditional deletion of Smad2 from TECs accelerates renal fibrosis reveals a protective role of Smad2 in renal fibrosis (Meng et al., 2010). In addition, FSP1-specific Smad2 knockout in renal tubular, endothelial, and interstitial cells is also reported to reduce renal fibrosis and epithelial-to-mesenchymal transition in murine streptozotocin (STZ)-induced diabetic nephropathy (Loeffler et al., 2018).
Diverse Role of Smad4 in Renal Fibrosis and Inflammation
Smad4 is a common Smad associated with nuclear translocation of Smad2/3 and Smad1/5/8 complexes in response to TGF-β and BMP signaling (Gomez-Puerto et al., 2019). Limited evidence has shown a direct role of Smad4 in renal fibrosis due to the lethality of Smad4 knockout mice. However, conditional deletion of Smad4 from TECs significantly reduces renal fibrosis in the obstructive kidney (Meng et al., 2012b). Mechanistically, deletion of Smad4 inhibits renal fibrosis by suppressing Smad3 promoter activity and blocking the binding of Smad3 to the collagen promoter without affecting its phosphorylation and nuclear translocation (Meng et al., 2012b). This finding is consistent with studies in Smad4 knockout mesangial cells and in the folic acid-induced rodent model (Tsuchida et al., 2003; Morishita et al., 2014). It is also reported that the formation of Smad3/Smad4/CDK9 complex drives renal fibrosis during ureteral obstruction (Qu et al., 2015). In contrast, conditional deletion of Smad4 promotes renal inflammation by impairing Smad7-mediated inhibition of NF-κB activation (Meng et al., 2012b). Thus, Smad4 may play a diverse role in renal fibrosis and inflammation and may not be a specific therapeutic target for CKD.
Smad7 as an Inhibitory Protein of Renal Fibrosis and Inflammation
Smad7 is a vital negative regulator of both TGF-β/Smad and NF-κB signaling pathways (Lan, 2008, 2011; Yan and Chen, 2011; Meng et al., 2016). Indeed, although TGF-β1 induces Smad7 transcriptionally, Smad7 inhibits TGF-β signaling by directly binding to the TβRI and blocking the activation of R-Smads (Hayashi et al., 1997). Mechanistically, Smad7 interacts with E3 ubiquitin ligases, such as arkadia, Smurf1 or Smurf2 (Smad ubiquitination regulatory factors), and recruit them to the TRβI to cause its degradation, hence resulting in the inhibition of TGF-β/Smad signaling (Ebisawa et al., 2001; Chong et al., 2006; Liu et al., 2008). Under fibrosis conditions, Smad7 is reduced while Smad3 is highly activated as seen in diabetic nephropathy, hypertensive nephropathy, and aristolochic acid-induced nephropathy (Chen et al., 2011; Liu et al., 2012; Chung et al., 2013a; Tian et al., 2015). Thus, the imbalance between Smad3 and Smad7 signaling may be a key mechanism in fibrogenesis and rebalancing this pathway by overexpressing Smad7 and inactivating Smad3 may represent as a better therapeutic strategy for CKD.
Smad7 can also induce expression of IκBα, an inhibitor of NF-κB, to negatively regulate NF-κB-driven renal inflammation (Wang et al., 2005a, b; Lan, 2008, 2011). Furthermore, Smad7 can interact with NF-κB directly as Smad7 promoter contains a putative NF-κB regulatory site (Nagarajan et al., 2000). Under CKD conditions, loss of renal Smad7 is associated with activation of NF-κB signaling and severe renal inflammation as reported in hypertensive nephropathy (Liu et al., 2013, 2014) and aristolochic acid-induced nephropathy (Dai et al., 2015). In contrast, overexpression of Smad7 suppresses both renal fibrosis and inflammatory in these disease models, making Smad7 as an promising therapeutic strategy for CKD (Lan, 2008).
Diverse Role of TGF-β/Smad Signaling in Regulation of Non-Coding RNAs Expression and Functions During Renal Fibrosis and Inflammation
MicroRNAs (miRNAs) are small (approximately 20–22 nucleotides in length) non-coding single stranded RNAs. More than 200 miRNAs have been identified in renal cells and tissues so far (Jelencsics and Oberbauer, 2015). These miRNAs regulate a wide range of biological processes, including fibrosis and inflammation. Increasing evidence has demonstrated that TGF-β1/Smad3 signaling regulates various miRNAs during the renal pathological processes (Meng et al., 2016; Tang et al., 2018). As a transcriptional factor, Smad3 can bind and upregulate or downregulate miRNAs to promote renal inflammation and fibrosis. It is now clear that Smad3, but not Smad2, regulates these miRNAs by physically interacting with Smad binding site (SBE) located in their promoters to either increase (such as miR-21 and miR-192) or inhibit their transcription (such as miR-29 and miR-200 families) (Chung and Lan, 2015). In addition, Smad7 may inactivate Smad3 to protect kidneys from fibrosis by upregulating renal miR-29b but suppressing miR-192 and miR-21 (Chung and Lan, 2015). Among these miRNAs, miR-21 is well characterized as a profibrotic miRNA. miR-21 is upregulated in renal fibrosis in the patients with CKD as well as AKI (Zarjou et al., 2011; Chau et al., 2012; Glowacki et al., 2013). Mice deficient miR-21 or administration of anti-miR-21 oligonucleotides are able to protect against renal fibrosis (Zhong et al., 2011, 2013). Expression of miR-21 is positively regulated by Smad3 but negatively by Smad7 (Chung et al., 2013a). Overexpression of miRNA-21 promotes renal fibrosis by targeting PTEN and Smad7 (Zhou et al., 2013; McClelland et al., 2015). Thus, knockdown of miR-21 restores renal Smad7 levels and blocks both TGF-β/Smad3 and NF-κB signaling, thereby inhibiting progressive renal fibrosis and inflammation in mouse models of obstructive and diabetic nephropathy (Zhong et al., 2013). However, miR-21 may be also protective in kidney disease as miR-21-deficient TGF-β(1)-transgenic mice show increased proteinuria and glomerular injury in streptozotocin-induced diabetic mice, suggesting a diverse role of miR-21 as a feedback inhibitor of TGF-β/Smad3 signaling (Lai et al., 2015).
MiR-29 family is another well-documented miRNA in fibrotic diseases (He et al., 2013). The miR-29 family consists of miR-29a, b, c. All family members are encoded by two distinct genomic loci in both human and rodent genomes. As all members have the same seed binding sequence, they all bind to the same set of target genes (Kriegel et al., 2012). Renal miR-29b is decreased in association with activation of TGF-β/Smad3 signaling and progressive renal fibrosis in kidney diseases (Qin et al., 2011; Wang et al., 2012; Chen et al., 2014; Meng et al., 2016). miR-29b is negatively regulated by Smad3, but not Smad2, in response to TGF-β1, AGE, and angiotensin II (Qin et al., 2011; Wang et al., 2012; Chen et al., 2014; Yu et al., 2014; Zhang et al., 2014). Overexpression of miR-29 inhibits renal fibrosis and inflammation by targeting TGF-β and Sp1/NF-κB signaling (Chen et al., 2014; Zhang et al., 2014). Interestingly, miR-29b can also target T-bet, a master transcriptional factor for Th-1 T cell immune response. Therefore, overexpression of miR-29b is also capable of inhibiting T cell-mediated type-2 diabetic nephropathy in db/db mice (Chen et al., 2014). Notably, miR-29 also acts as a urinary exosome biomarker of renal fibrosis (Lv et al., 2013). Intramuscular injection of exosome-encapsulated miR-29 has been shown to inhibit renal fibrosis and muscle atrophy (Wang et al., 2019).
Moreover, miR-93, miR-216a, miR-217, miR-377, miR-382, miR-491-5p, miR-433 and miR-17-5p are also demonstrated to be TGF-β1/Smad3-regulated profibrotic miRNAs (Chung and Lan, 2015), whereas miR-let-7, miR-15b, miR-101, and miR-130b exert their antifibrotic effects by inhibiting the expression and activity of TβRI, thus limiting transduction of downstream TGF-β-mediated signals (Wang et al., 2014; Tang et al., 2018). Other miRNAs such as miR-19b, miR-26a, miR-29, and miR-30 inhibit the TGF-β1/Smad signaling by targeting Smads or fibrotic transcriptional factors (Tang et al., 2018). All these findings imply that TGF-β may regulate miRNAs to exert its diverse roles in renal inflammation and fibrosis as shown in Table 1 and Figure 3.
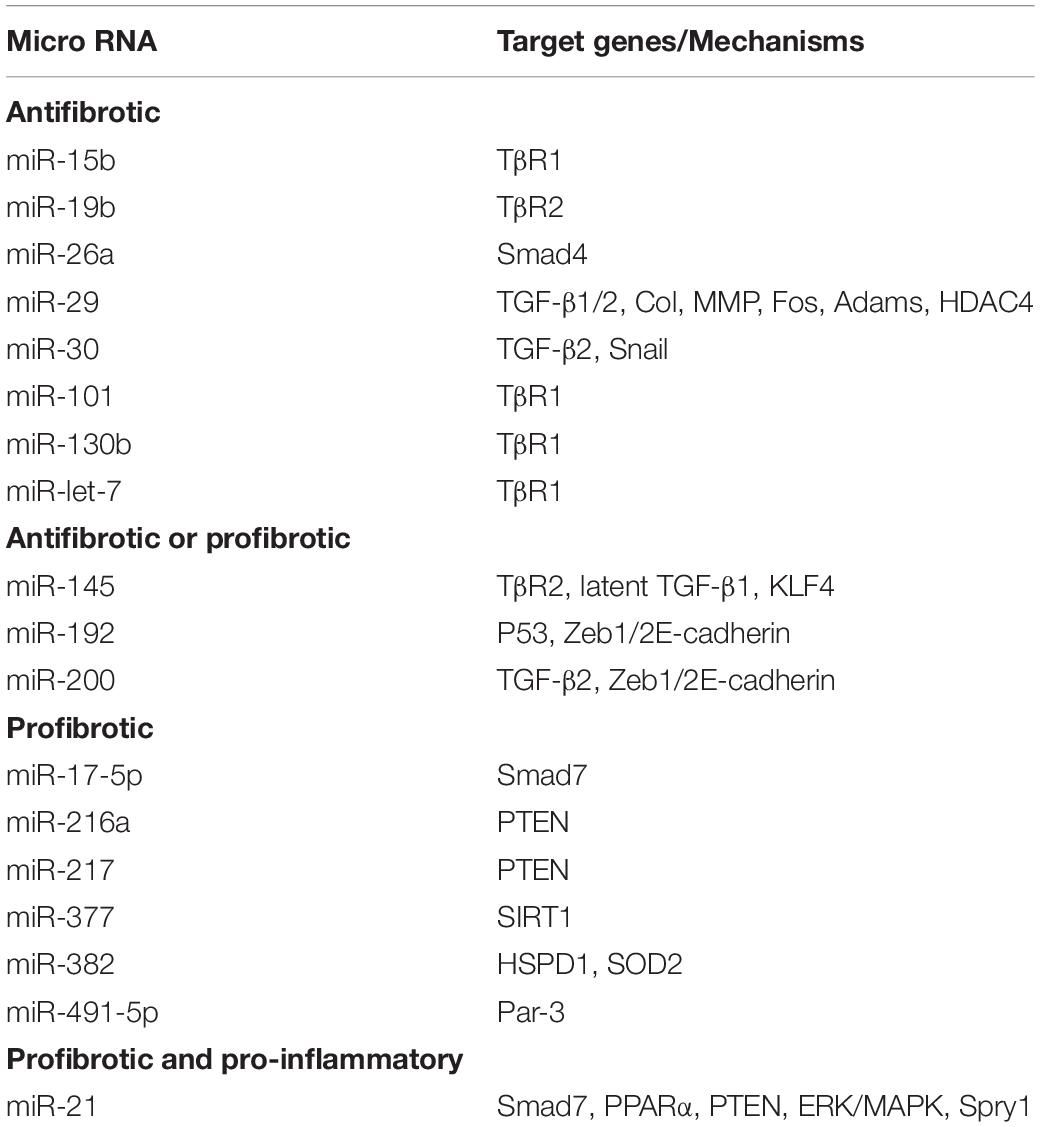
Table 1. MicroRNAs regulated by TGF-β/Smad signaling in renal fibrosis.
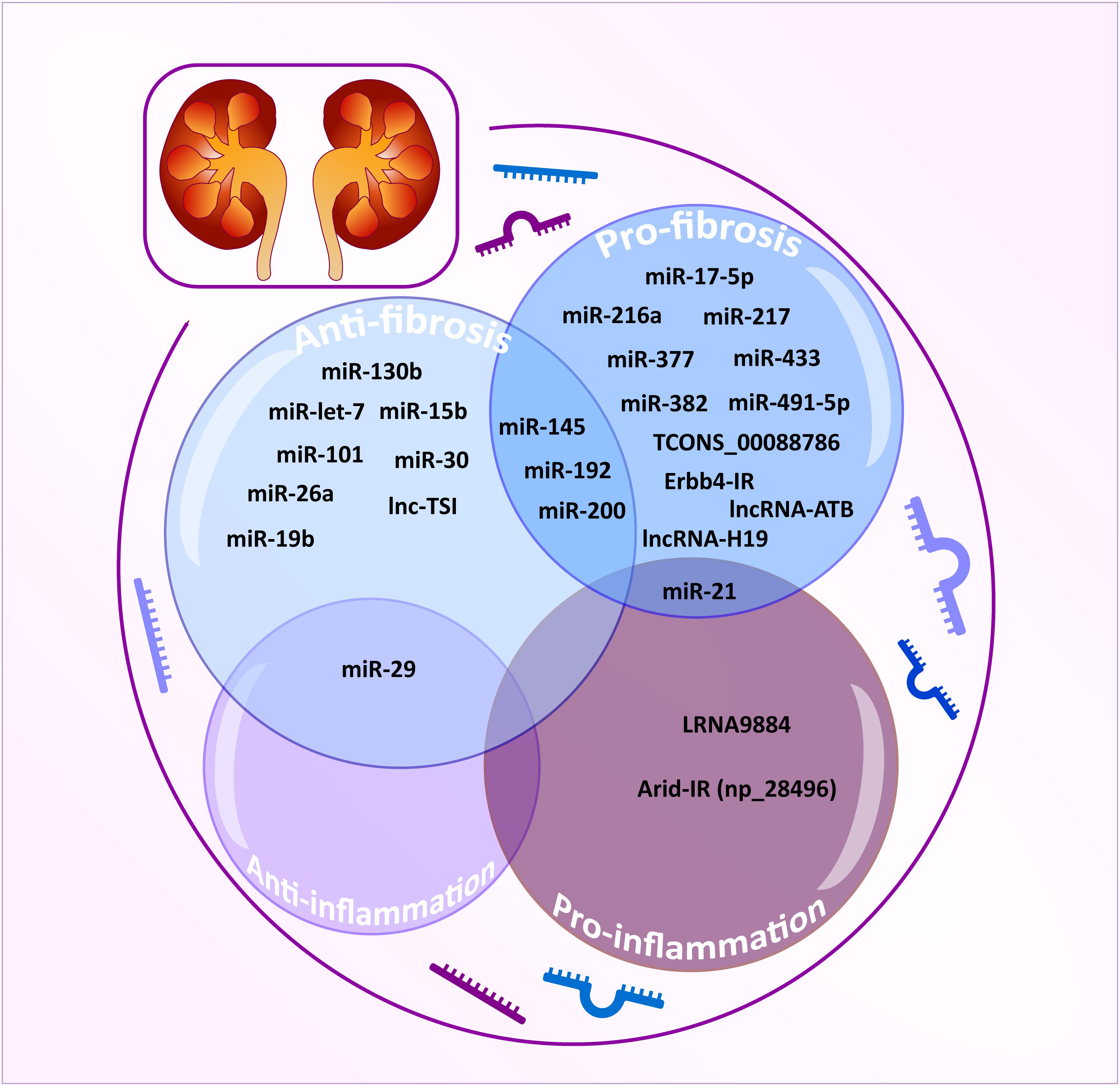
Figure 3. TGF-β/Smad3-dependent miRNAs and lncRNAs related to renal fibrosis and inflammation. TGF-β/Smad3-dependent miRNAs and lncRNAs are classified as anti-fibrotic (powder blue), pro-fibrotic (sky blue), anti-inflammatory (lavender), and pro-inflammatory effect (plum). The integrated area indicates multiple functions for each miRNA/lncRNA.
However, the off-target effects, non-specificity, and toxicity of miRNAs are unavoidable. Thus, research into long non-coding RNAs (lncRNAs) is more promising for a better understanding of the pathogenic mechanisms of kidney diseases (Moghaddas Sani et al., 2018). Compared to miRNAs, lncRNAs are transcripts with lengths exceeding 200 nucleotides without protein-coding functions and are highly tissue-and-cell-type-specific. lncRNA regulates both target DNAs/RNAs and proteins transcriptionally or post-transcriptionally (Dykes and Emanueli, 2017). By using the high-throughput RNA sequencing, 21 TGF-β/Smad3-dependent lncRNAs have been identified in an immunologically induced anti-glomerular basement membranous glomerulonephritis (anti-GBM GN) and obstructive nephropathy (Zhou et al., 2014). Of these, the Arid-IR is a novel and Smad3-related lncRNA as a Smad3 binding site is found in its promoter region. It has been proven that knockdown of Arid2-IR in TECs improves renal inflammation in vivo and in vitro by inhibiting NF-κB-dependent inflammatory transduction without affecting Smad3-mediated fibrosis (Zhou et al., 2015). In contrast, Erbb4-IR is another novel Smad3-dependent lncRNA capable of inhibiting renal fibrosis by targeting miR-29b and Smad7 in both obstructive nephropathy and type II diabetic nephropathy, respectively (Feng et al., 2018; Sun et al., 2018). A recent study also reveals the pathogenic role and mechanism of LRNA9884 in type II diabetic nephropathy (Zhang et al., 2019). LRNA9884 is tightly regulated by Smad3 in response to TGF-β and AGEs and functions to trigger MCP-1 production by directly binding to the MCP-1 promoter, thereby promoting inflammation-driven type II diabetic nephropathy (Zhang et al., 2019). In addition, several TGF-β/Smad3-associated lncRNAs are found to be associated with renal fibrosis. TCONS_00088786 and TCONS_01496394 are TGF-β/Smad3-associated lncRNAs as they contain potential binding sites for Smad3 and silencing TCONS_00088786 inhibits renal interstitial fibrosis by targeting miR-132 (Sun et al., 2017; Zhou S.G. et al., 2018). lncRNA-ATB is highly upregulated in patients with acute renal allograft rejection and renal carcinoma and is able to promote EMT (Qi et al., 2017; Qiu et al., 2017; Zhou and Jiang, 2019). lncRNA uc.412 is able to induce mesangial cell proliferation in vitro although the underlying mechanisms are unclear (Yu et al., 2019). Lnc RNA-H19 is associated with TGF-β2-induced fibrosis in vivo and in vitro (Xie et al., 2016). lncRNA ENST00000453774.1 (LncRNA 74.1) is significantly down-regulated in TGF-β-treated TECs and in fibrotic kidney (Xiao et al., 2019). Interestingly, a recent study also revealed that decreased human lnc-TSI (TGF-β/Smad3-interacting long non-coding RNA) correlates with the degree of renal fibrosis in patients with IgA nephropathy and treatment with lnc-TSI inhibits renal fibrosis by blocking its binding to the MH2 domain of Smad3 (Wang et al., 2018).
Taken together, TGF-β may diversely regulate renal fibrosis and inflammation via Smad3-dependent miRNAs/lncRNAs as shown in Table 2 and Figure 3.

Table 2. Long non-coding RNAs regulated by TGF-β/Smad signaling in renal fibrosis.
Clinical Trials of Anti-TGF-β Therapy
Theoretically, TGF-β is a key mediator for renal fibrosis and thus targeting TGF-β signaling could be a good therapeutic strategy for CKD. There are many approaches to develop anti-TGF-β treatment for CKD clinically (Table 3). It has been shown that treatment with Pirfenidone, a non-specific antifibrotic effect of TGF-β, can improve eGFR in the trials of DN and focal segmental glomerulosclerosis (FSGS) (Lancaster et al., 2017). Disappointingly, a recent clinical trial study using a humanized monoclonal neutralizing antibody against TGF-β1 (LY2382770) for treatment of patients with diabetic nephropathy has been proven no efficacy on the improvements of serum creatinine, estimated GFR (eGFR), and proteinuria (Voelker et al., 2017). In addition, the use of another humanized monoclonal antibody, Fresolimumab that inhibits all three isoforms of TGF-β, also fails to achieve the endpoints of proteinuria reduction in patients with FSGS (Trachtman et al., 2011; Vincenti et al., 2017), demonstrating targeting on the upstream of TGF-β signaling may not be a good therapeutic strategy for CKD. It is possible that blockade of the general effect of TGF-β1, including latent form of TGF-β1, may attribute to the failure of these clinical trials. Our previous studies in latent TGF-β transgenic mice explain this notion since mice overexpressing latent TGF-β1 are protected against renal inflammatory and fibrosis in unilateral ureteral obstructive (UUO) nephropathy and anti-GBM glomerulonephritis model (Huang et al., 2008a, b). Thus, the latent form of TGF-β1 is renal protective while its active form is pathogenic. As most circulating TGF-β1 is latent form, thus, the use of anti-TGF-β1 antibodies may largely block the protective effect of latent TGF-β1, resulting in progressive renal injury as seen in these clinical trials. Results from these studies also suggest that treatment against renal fibrosis in patients with CKD should specifically target the downstream TGF-β signaling molecules, rather than to block the general effect of TGF-β1.
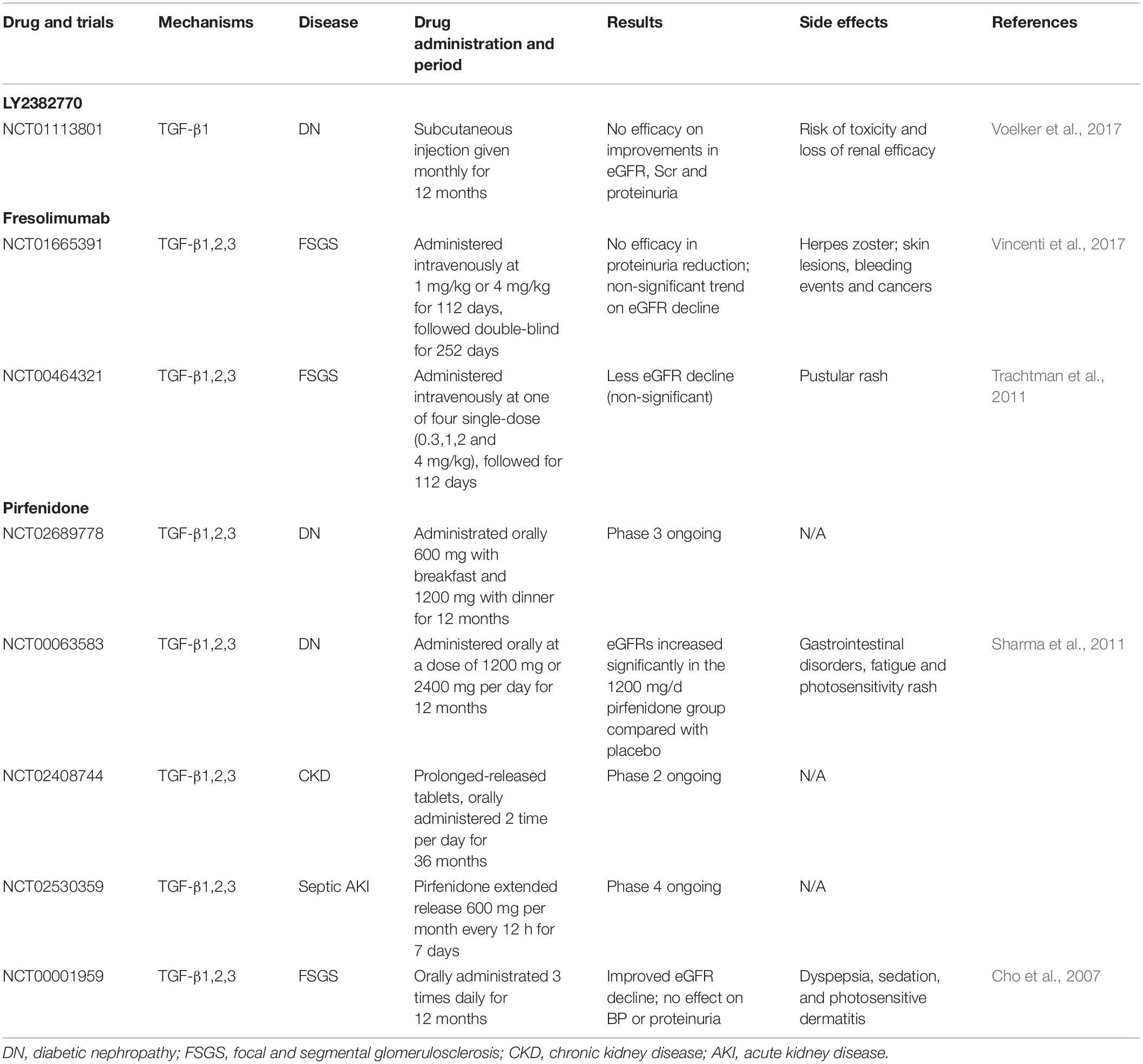
Table 3. Therapeutic drugs and clinical trials for treatment of CKD by targeting TGF-β.
Treatment of CKD by Targeting Downstream TGF-β/Smad Signaling Molecules and Non-Coding RNAs
Given the diversity and the complexity of TGF-β in renal fibrosis and inflammation, direct targeting TGF-β or receptors may not be an ideal tactic due to its involvement in various vital biological processes (Trachtman et al., 2011; Vincenti et al., 2017; Voelker et al., 2017). Although general blockade of the upstream TGF-β signaling may reduce fibrosis, it can also promote renal inflammation and cause unexpected renal injuries (Figure 4a). Because the imbalance of TGF-β/Smad3 signaling with overreactive Smad3 and reduced Smad7 is a key mechanism leading to renal fibrosis and inflammation, rebalancing Smad3/Smad7 signaling may serve as effective strategies to treat renal fibrosis and inflammation (Figure 4b). SIS3, a specific Smad3-inhibitor, has been shown to inhibit renal fibrosis in STZ-induced diabetic nephropathy (Li et al., 2010) and in obstructive nephropathy (Zhang et al., 2018). Overexpression of renal Smad7 is also capable of inhibiting Smad3-mediated renal fibrosis and NF-κB-driven renal inflammation in various kidney diseases, including diabetic and hypertensive nephropathy (Chen et al., 2011; Lan, 2011; Ka et al., 2012; Liu et al., 2014), obstructive nephropathy (Li et al., 2002; Lan et al., 2003; Lan, 2008; Chung et al., 2013a), remnant kidney disease (Hou et al., 2005; Ng et al., 2005), crescentic glomerulonephritis (Ka et al., 2007), and chronic aristolochic acid nephropathy (Dai et al., 2015). Interestingly, treatment of CKD with two Traditional Chinese Medicine compounds, Naringenin from fruits as a Smad3 inhibitor and Asiatic acid derived from Centella asiatica as a Smad7 agonist, is capable of restoring the balance of Smad3/Smad7 signaling and thus additively inhibits renal fibrosis in rodent obstructive nephropathy (Meng et al., 2015). Similarly, the combination of Ginsenoside Rg1 from Panax ginseng C. A. Mey and Astragaloside IV from Radix astragali have also improved fibrosis and inflammation in STZ-induced diabetic nephropathy by inhibiting TGF-β/Smad2/3 while enhancing Smad7 signaling (Du et al., 2018). Asperulosidic acid, a bioactive iridoid glycoside, can also exert renal protective effects by inactivating both TGF-β/Smad and NF-κB signaling pathways (Xianyuan et al., 2019). Similar therapeutic effects are also found in other studies with herbal medicines (Nie et al., 2014; Wan et al., 2014; Zhao et al., 2016).
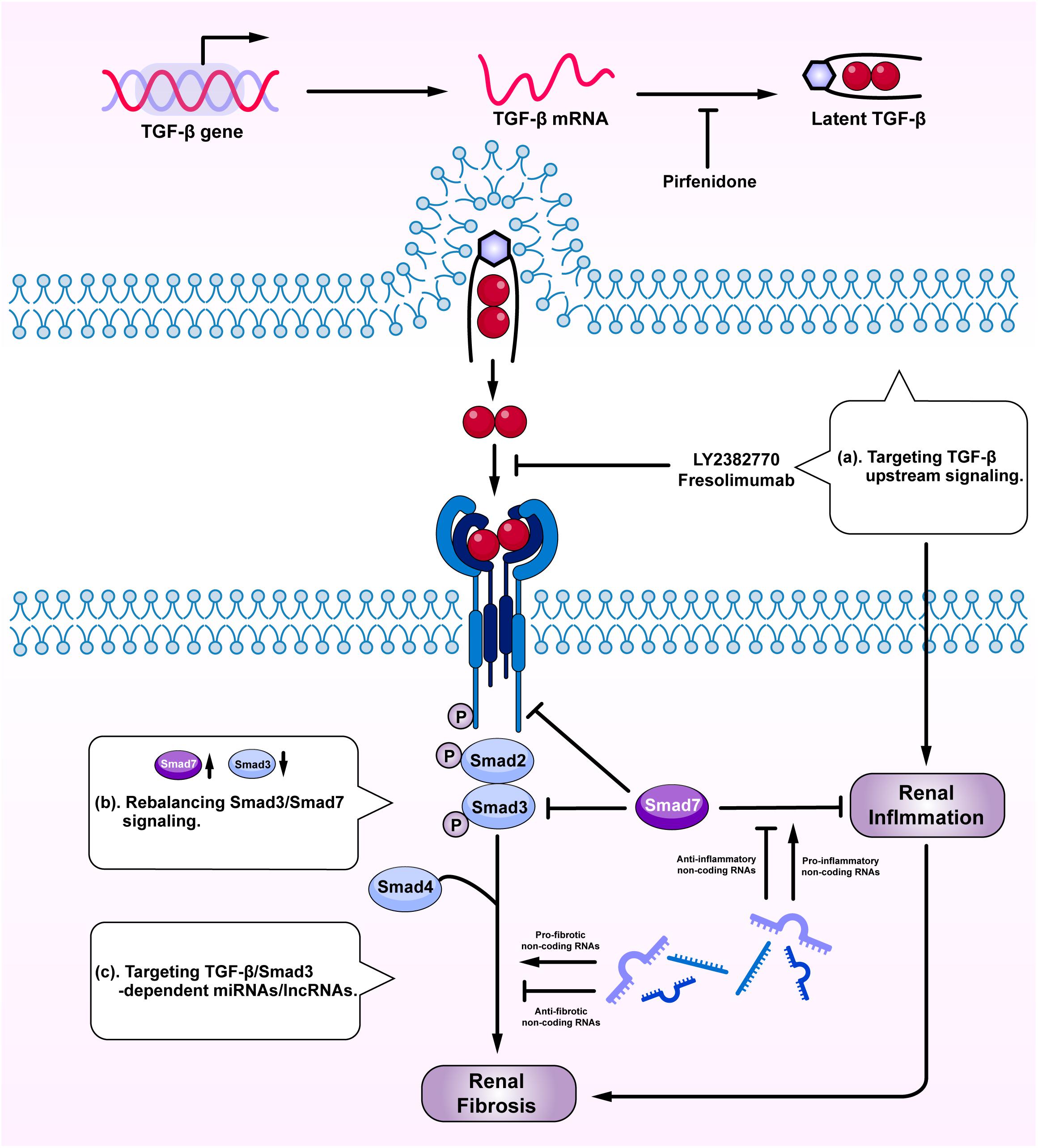
Figure 4. Therapeutic potentials by targeting TGF-β signaling. Anti-TGF-β treatment by: (a) targeting upstream signaling; (b) rebalancing Smad3/Smad7 signaling; and (c) targeting Smad3-dependent miRNAs/lncRNAs.
Targeting Smad3-dependent non-coding RNAs could be another therapeutic approach to treat renal fibrosis and inflammation (Figure 4c). Of Smad3-dependent miRNAs (Figure 3), inhibition of miR-21, miR-192, miR-433, and overexpression of miR-29 and miR-200 have been shown to have therapeutic effects on obstructive nephropathy (Chung et al., 2010a, 2013b; Oba et al., 2010; Qin et al., 2011; Zhong et al., 2011; Li et al., 2013) and diabetic nephropathy (Zhong et al., 2013; Chen et al., 2014). However, the off-target effect of anti-miRNA therapies raises concern and new therapeutic approach by targeting Smad3-dependent lncRNAs is sought. Targeting Arid2-IR and LRNA9884 can specifically inhibit renal inflammation while targeting Erbb4-IR can specifically inhibit renal fibrosis in obstructive and diabetic nephropathy (Zhou et al., 2015; Feng et al., 2018; Sun et al., 2018; Zhang et al., 2019). Furthermore, delivery of a human lncRNA lnc-TSI into the UUO kidney also inhibits Smad3-mediated renal fibrosis (Wang et al., 2018). All these findings highlight the therapeutic potentials by targeting downstream TGF-β signaling molecules including Smad3, Smad7, and non-coding RNAs in renal fibrosis and inflammation.
Conclusion
Transforming growth factor-β plays diverse roles in renal fibrosis and inflammation. Blockade of upstream TGF-β signaling may not be a good therapeutic strategy, which has been proved by unsatisfied clinical trials. TGF-β may specifically regulate renal fibrosis and inflammation via downstream Smad-dependent mechanisms involving Smad3, Smad4, Smad7, and particularly Smad3-dependent non-coding RNAs. Targeting downstream TGF-β/Smad signaling by rebalancing Smad3/Smad7 or by specifically inhibiting or overexpressing Smad3-dependent non-coding RNAs related to fibrosis or inflammation may be a better therapeutic approach. Further studies to understand the diverse role of TGF-β signaling in kidney diseases may promote the translation from bench into clinical settings.
Author Contributions
Y-YG, X-SL, and X-RH wrote and revised the manuscript. X-QY and H-YL revised and edited the manuscript. All authors contributed to the manuscript conception development, data collection and analysis, and discussion on the manuscript writing and revising.
Funding
This work was supported by the Research Grants Council of Hong Kong (Grants GRF 14163317, 14117418, 14104019, R4012-18F, C7018-16G, and T12-402/13N), the Health and Medical Research Fund of Hong Kong (Grants HMRF 05161326, TMP 09094, and 14152321), the Science and Technology Planning Project of Guangdong Province (No. 2017B030314166), the National Natural Science Foundation of China (Nos. 81873261 and 81903956), the Project of Guangdong Province Administration of Traditional Chinese Medicine (No. 20201133), and the Guangdong-Hong Kong-Macao-Joint Labs Program from Guangdong Science and Technology (2019B121205005).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akagi, Y., Isaka, Y., Arai, M., Kaneko, T., Takenaka, M., Moriyama, T., et al. (1996). Inhibition of TGF-beta 1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 50, 148–155. doi: 10.1038/ki.1996.297
Ando, T., Okuda, S., Tamaki, K., Yoshitomi, K., and Fujishima, M. (1995). Localization of transforming growth factor-beta and latent transforming growth factor-beta binding protein in rat kidney. Kidney Int. 47, 733–739. doi: 10.1038/ki.1995.112
Annes, J. P., Munger, J. S., and Rifkin, D. B. (2003). Making sense of latent TGFbeta activation. J. Cell Sci. 116, 217–224. doi: 10.1242/jcs.00229
Bitzer, M., Von Gersdorff, G., Liang, D., Dominguez-Rosales, A., Beg, A. A., Rojkind, M., et al. (2000). A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 14, 187–197.
Border, W. A., Okuda, S., Languino, L. R., Sporn, M. B., and Ruoslahti, E. (1990). Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature 346, 371–374. doi: 10.1038/346371a0
Bottinger, E. P., Factor, V. M., Tsang, M. L., Weatherbee, J. A., Kopp, J. B., Qian, S. W., et al. (1996). The recombinant proregion of transforming growth factor beta1 (latency-associated peptide) inhibits active transforming growth factor beta1 in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 5877–5882. doi: 10.1073/pnas.93.12.5877
Chau, B. N., Xin, C., Hartner, J., Ren, S., Castano, A. P., Linn, G., et al. (2012). MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 4:121ra118. doi: 10.1126/scitranslmed.3003205
Chen, H. Y., Huang, X. R., Wang, W., Li, J. H., Heuchel, R. L., Chung, A. C., et al. (2011). The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes 60, 590–601. doi: 10.2337/db10-0403
Chen, H. Y., Zhong, X., Huang, X. R., Meng, X. M., You, Y., Chung, A. C., et al. (2014). MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. 22, 842–853. doi: 10.1038/mt.2013.235
Chen, L., Yang, T., Lu, D. W., Zhao, H., Feng, Y. L., Chen, H., et al. (2018). Central role of dysregulation of TGF-beta/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 101, 670–681. doi: 10.1016/j.biopha.2018.02.090
Chen, S., Carmen Iglesias-De La Cruz, M., Jim, B., Hong, S. W., Isono, M., and Ziyadeh, F. N. (2003). Reversibility of established diabetic glomerulopathy by anti-TGF-β antibodies in db/db mice. Biochem. Biophys. Res. Commun. 300, 16–22. doi: 10.1016/s0006-291x(02)02708-0
Cho, M. E., Smith, D. C., Branton, M. H., Penzak, S. R., and Kopp, J. B. (2007). Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2, 906–913. doi: 10.2215/cjn.01050207
Chong, P. A., Lin, H., Wrana, J. L., and Forman-Kay, J. D. (2006). An expanded WW domain recognition motif revealed by the interaction between Smad7 and the E3 ubiquitin ligase Smurf2. J. Biol. Chem. 281, 17069–17075. doi: 10.1074/jbc.m601493200
Chung, A. C., Dong, Y., Yang, W., Zhong, X., Li, R., and Lan, H. Y. (2013a). Smad7 suppresses renal fibrosis via altering expression of TGF-beta/Smad3-regulated microRNAs. Mol. Ther. 21, 388–398. doi: 10.1038/mt.2012.251
Chung, A. C., Huang, X. R., Meng, X., and Lan, H. Y. (2010a). miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 21, 1317–1325. doi: 10.1681/ASN.2010020134
Chung, A. C., and Lan, H. Y. (2015). MicroRNAs in renal fibrosis. Front. Physiol. 6:50. doi: 10.3389/fphys.2015.00050
Chung, A. C., Yu, X., and Lan, H. Y. (2013b). MicroRNA and nephropathy: emerging concepts. Int. J. Nephrol. Renovasc. Dis. 6, 169–179. doi: 10.2147/IJNRD.S37885
Chung, A. C., Zhang, H., Kong, Y. Z., Tan, J. J., Huang, X. R., Kopp, J. B., et al. (2010b). Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J. Am. Soc. Nephrol. 21, 249–260. doi: 10.1681/ASN.2009010018
Dai, X. Y., Zhou, L., Huang, X. R., Fu, P., and Lan, H. Y. (2015). Smad7 protects against chronic aristolochic acid nephropathy in mice. Oncotarget 6, 11930–11944.
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi: 10.1038/nature02006
Du, N., Xu, Z., Gao, M., Liu, P., Sun, B., and Cao, X. (2018). Combination of Ginsenoside Rg1 and Astragaloside IV reduces oxidative stress and inhibits TGF-beta1/Smads signaling cascade on renal fibrosis in rats with diabetic nephropathy. Drug Des. Devel. Ther. 12, 3517–3524. doi: 10.2147/DDDT.S171286
Duan, W. J., Yu, X., Huang, X. R., Yu, J. W., and Lan, H. Y. (2014). Opposing roles for Smad2 and Smad3 in peritoneal fibrosis in vivo and in vitro. Am. J. Pathol. 184, 2275–2284. doi: 10.1016/j.ajpath.2014.04.014
Dykes, I. M., and Emanueli, C. (2017). Transcriptional and post-transcriptional gene regulation by long non-coding RNA. Genomics Proteomics Bioinformatics 15, 177–186. doi: 10.1016/j.gpb.2016.12.005
Ebisawa, T., Fukuchi, M., Murakami, G., Chiba, T., Tanaka, K., Imamura, T., et al. (2001). Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 276, 12477–12480. doi: 10.1074/jbc.c100008200
Eddy, A. A., and Neilson, E. G. (2006). Chronic kidney disease progression. J. Am. Soc. Nephrol. 17, 2964–2966.
Edeling, M., Ragi, G., Huang, S., Pavenstadt, H., and Susztak, K. (2016). Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 12, 426–439. doi: 10.1038/nrneph.2016.54
Ernandez, T., and Mayadas, T. N. (2016). The changing landscape of renal inflammation. Trends Mol. Med. 22, 151–163. doi: 10.1016/j.molmed.2015.12.002
Fan, J. M., Ng, Y. Y., Hill, P. A., Nikolic-Paterson, D. J., Mu, W., Atkins, R. C., et al. (1999). Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 56, 1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x
Feng, M., Tang, P. M., Huang, X. R., Sun, S. F., You, Y. K., Xiao, J., et al. (2018). TGF-beta mediates renal fibrosis via the Smad3-Erbb4-IR long noncoding RNA axis. Mol. Ther. 26, 148–161. doi: 10.1016/j.ymthe.2017.09.024
Glassock, R. J., Warnock, D. G., and Delanaye, P. (2017). The global burden of chronic kidney disease: estimates, variability and pitfalls. Nat. Rev. Nephrol. 13, 104–114. doi: 10.1038/nrneph.2016.163
Glowacki, F., Savary, G., Gnemmi, V., Buob, D., Van Der Hauwaert, C., Lo-Guidice, J. M., et al. (2013). Increased circulating miR-21 levels are associated with kidney fibrosis. PLoS One 8:e58014. doi: 10.1371/journal.pone.0058014
Gomez-Puerto, M. C., Iyengar, P. V., Garcia De Vinuesa, A., Ten Dijke, P., and Sanchez-Duffhues, G. (2019). Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 247, 9–20. doi: 10.1002/path.5170
Hall, M. C., Young, D. A., Waters, J. G., Rowan, A. D., Chantry, A., Edwards, D. R., et al. (2003). The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-beta 1. J. Biol. Chem. 278, 10304–10313. doi: 10.1074/jbc.m212334200
Harskamp, L. R., Gansevoort, R. T., Van Goor, H., and Meijer, E. (2016). The epidermal growth factor receptor pathway in chronic kidney diseases. Nat. Rev. Nephrol. 12, 496–506. doi: 10.1038/nrneph.2016.91
Hayashi, H., Abdollah, S., Qiu, Y., Cai, J., Xu, Y. Y., Grinnell, B. W., et al. (1997). The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89, 1165–1173. doi: 10.1016/s0092-8674(00)80303-7
He, Y., Huang, C., Lin, X., and Li, J. (2013). MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie 95, 1355–1359. doi: 10.1016/j.biochi.2013.03.010
Higgins, S. P., Tang, Y., Higgins, C. E., Mian, B., Zhang, W., Czekay, R. P., et al. (2018). TGF-beta1/p53 signaling in renal fibrogenesis. Cell. Signal. 43, 1–10. doi: 10.1016/j.cellsig.2017.11.005
Hou, C. C., Wang, W., Huang, X. R., Fu, P., Chen, T. H., Sheikh-Hamad, D., et al. (2005). Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am. J. Pathol. 166, 761–771. doi: 10.1016/s0002-9440(10)62297-3
Huang, X. R., Chung, A. C., Wang, X. J., Lai, K. N., and Lan, H. Y. (2008a). Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am. J. Physiol. Renal Physiol. 295, F118–F127. doi: 10.1152/ajprenal.00021.2008
Huang, X. R., Chung, A. C., Zhou, L., Wang, X. J., and Lan, H. Y. (2008b). Latent TGF-beta1 protects against crescentic glomerulonephritis. J. Am. Soc. Nephrol. 19, 233–242. doi: 10.1681/ASN.2007040484
Jelencsics, K., and Oberbauer, R. (2015). microRNA and kidney transplantation. Adv. Exp. Med. Biol. 888, 271–290. doi: 10.1007/978-3-319-22671-2_14
Ju, W., Ogawa, A., Heyer, J., Nierhof, D., Yu, L., Kucherlapati, R., et al. (2006). Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol. Cell. Biol. 26, 654–667. doi: 10.1128/mcb.26.2.654-667.2006
Ka, S. M., Huang, X. R., Lan, H. Y., Tsai, P. Y., Yang, S. M., Shui, H. A., et al. (2007). Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J. Am. Soc. Nephrol. 18, 1777–1788. doi: 10.1681/asn.2006080901
Ka, S. M., Yeh, Y. C., Huang, X. R., Chao, T. K., Hung, Y. J., Yu, C. P., et al. (2012). Kidney-targeting Smad7 gene transfer inhibits renal TGF-beta/MAD homologue (SMAD) and nuclear factor kappaB (NF-kappaB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia 55, 509–519. doi: 10.1007/s00125-011-2364-5
Kopp, J. B., Factor, V. M., Mozes, M., Nagy, P., Sanderson, N., Bottinger, E. P., et al. (1996). Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. 74, 991–1003.
Kriegel, A. J., Liu, Y., Fang, Y., Ding, X., and Liang, M. (2012). The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol. Genomics 44, 237–244. doi: 10.1152/physiolgenomics.00141.2011
Kulkarni, A. B., Huh, C. G., Becker, D., Geiser, A., Lyght, M., Flanders, K. C., et al. (1993). Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U.S.A. 90, 770–774. doi: 10.1073/pnas.90.2.770
Kusakabe, M., Cheong, P. L., Nikfar, R., Mclennan, I. S., and Koishi, K. (2008). The structure of the TGF-beta latency associated peptide region determines the ability of the proprotein convertase furin to cleave TGF-betas. J. Cell. Biochem. 103, 311–320. doi: 10.1002/jcb.21407
Lai, J. Y., Luo, J., O’connor, C., Jing, X., Nair, V., Ju, W., et al. (2015). MicroRNA-21 in glomerular injury. J. Am. Soc. Nephrol. 26, 805–816. doi: 10.1681/ASN.2013121274
Lan, H. Y. (2008). Smad7 as a therapeutic agent for chronic kidney diseases. Front. Biosci. 13, 4984–4992. doi: 10.2741/3057
Lan, H. Y. (2011). Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 7, 1056–1067. doi: 10.7150/ijbs.7.1056
Lan, H. Y., and Chung, A. C. (2012). TGF-beta/Smad signaling in kidney disease. Semin. Nephrol. 32, 236–243. doi: 10.1016/j.semnephrol.2012.04.002
Lan, H. Y., Mu, W., Tomita, N., Huang, X. R., Li, J. H., Zhu, H. J., et al. (2003). Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J. Am. Soc. Nephrol. 14, 1535–1548. doi: 10.1097/01.asn.0000067632.04658.b8
Lancaster, L. H., De Andrade, J. A., Zibrak, J. D., Padilla, M. L., Albera, C., Nathan, S. D., et al. (2017). Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 26:170057. doi: 10.1183/16000617.0057-2017
Li, B., Haridas, B., Jackson, A. R., Cortado, H., Mayne, N., Kohnken, R., et al. (2017). Inflammation drives renal scarring in experimental pyelonephritis. Am. J. Physiol. Renal Physiol. 312, F43–F53. doi: 10.1152/ajprenal.00471.2016
Li, J., Qu, X., Yao, J., Caruana, G., Ricardo, S. D., Yamamoto, Y., et al. (2010). Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59, 2612–2624. doi: 10.2337/db09-1631
Li, J. H., Zhu, H. J., Huang, X. R., Lai, K. N., Johnson, R. J., and Lan, H. Y. (2002). Smad7 inhibits fibrotic effect of TGF-Beta on renal tubular epithelial cells by blocking Smad2 activation. J. Am. Soc. Nephrol. 13, 1464–1472. doi: 10.1097/01.asn.0000014252.37680.e4
Li, M. O., and Flavell, R. A. (2008). TGF-beta: a master of all T cell trades. Cell 134, 392–404. doi: 10.1016/j.cell.2008.07.025
Li, R., Chung, A. C., Dong, Y., Yang, W., Zhong, X., and Lan, H. Y. (2013). The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-beta/Smad3-Azin1 pathway. Kidney Int. 84, 1129–1144. doi: 10.1038/ki.2013.272
Liu, F. Y., Li, X. Z., Peng, Y. M., Liu, H., and Liu, Y. H. (2008). Arkadia regulates TGF-beta signaling during renal tubular epithelial to mesenchymal cell transition. Kidney Int. 73, 588–594. doi: 10.1038/sj.ki.5002713
Liu, G. X., Li, Y. Q., Huang, X. R., Wei, L., Chen, H. Y., Shi, Y. J., et al. (2013). Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PLoS One 8:e53573. doi: 10.1371/journal.pone.0053573
Liu, G. X., Li, Y. Q., Huang, X. R., Wei, L. H., Zhang, Y., Feng, M., et al. (2014). Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin. Sci. 127, 195–208. doi: 10.1042/CS20130706
Liu, H., Zhang, Z., Li, Y., Wang, X., Zhang, Y., Chu, Y., et al. (2018). Preparation and evaluation of anti-renal fibrosis activity of novel truncated TGF-beta receptor type II. Biotechnol. Appl. Biochem. 65, 834–840. doi: 10.1002/bab.1667
Liu, Y. (2011). Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7, 684–696. doi: 10.1038/nrneph.2011.149
Liu, Z., Huang, X. R., and Lan, H. Y. (2012). Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am. J. Physiol. Renal Physiol. 302, F986–F997. doi: 10.1152/ajprenal.00595.2011
Loeffler, I., Liebisch, M., Allert, S., Kunisch, E., Kinne, R. W., and Wolf, G. (2018). FSP1-specific SMAD2 knockout in renal tubular, endothelial, and interstitial cells reduces fibrosis and epithelial-to-mesenchymal transition in murine STZ-induced diabetic nephropathy. Cell Tissue Res. 372, 115–133. doi: 10.1007/s00441-017-2754-1
Lopez-Hernandez, F. J., and Lopez-Novoa, J. M. (2012). Role of TGF-beta in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell Tissue Res. 347, 141–154. doi: 10.1007/s00441-011-1275-6
Luo, K. (2017). Signaling cross talk between TGF-beta/Smad and other signaling pathways. Cold Spring Harb. Perspect. Biol. 9:a022137. doi: 10.1101/cshperspect.a022137
Lv, L. L., Cao, Y. H., Ni, H. F., Xu, M., Liu, D., Liu, H., et al. (2013). MicroRNA-29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am. J. Physiol. Renal Physiol. 305, F1220–F1227. doi: 10.1152/ajprenal.00148.2013
Massagué, J. (2012). TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630. doi: 10.1038/nrm3434
McClelland, A. D., Herman-Edelstein, M., Komers, R., Jha, J. C., Winbanks, C. E., Hagiwara, S., et al. (2015). miR-21 promotes renal fibrosis in diabetic nephropathy by targeting PTEN and SMAD7. Clin. Sci. 129, 1237–1249. doi: 10.1042/CS20150427
Meng, X. M. (2019). Inflammatory mediators and renal fibrosis. Adv. Exp. Med. Biol. 1165, 381–406. doi: 10.1007/978-981-13-8871-2_18
Meng, X. M., Chung, A. C., and Lan, H. Y. (2013). Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin. Sci. 124, 243–254. doi: 10.1042/CS20120252
Meng, X. M., Huang, X. R., Chung, A. C., Qin, W., Shao, X., Igarashi, P., et al. (2010). Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 21, 1477–1487. doi: 10.1681/ASN.2009121244
Meng, X. M., Huang, X. R., Xiao, J., Chen, H. Y., Zhong, X., Chung, A. C., et al. (2012a). Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 227, 175–188. doi: 10.1002/path.3976
Meng, X. M., Huang, X. R., Xiao, J., Chung, A. C., Qin, W., Chen, H. Y., et al. (2012b). Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 81, 266–279. doi: 10.1038/ki.2011.327
Meng, X. M., Nikolic-Paterson, D. J., and Lan, H. Y. (2014). Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 10, 493–503. doi: 10.1038/nrneph.2014.114
Meng, X. M., Nikolic-Paterson, D. J., and Lan, H. Y. (2016). TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol 12, 325–338. doi: 10.1038/nrneph.2016.48
Meng, X. M., Zhang, Y., Huang, X. R., Ren, G. L., Li, J., and Lan, H. Y. (2015). Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget 6, 36984–36997. doi: 10.18632/oncotarget.6100
Mihai, S., Codrici, E., Popescu, I. D., Enciu, A. M., Albulescu, L., Necula, L. G., et al. (2018). Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J. Immunol. Res. 2018:2180373. doi: 10.1155/2018/2180373
Miyajima, A., Chen, J., Lawrence, C., Ledbetter, S., Soslow, R. A., Stern, J., et al. (2000). Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 58, 2301–2313. doi: 10.1046/j.1523-1755.2000.00414.x
Miyazawa, K., and Miyazono, K. (2017). Regulation of TGF-beta family signaling by inhibitory smads. Cold Spring Harb. Perspect. Biol. 9:a022095. doi: 10.1101/cshperspect.a022095
Moghaddas Sani, H., Hejazian, M., Hosseinian Khatibi, S. M., Ardalan, M., and Zununi Vahed, S. (2018). Long non-coding RNAs: an essential emerging field in kidney pathogenesis. Biomed. Pharmacother. 99, 755–765. doi: 10.1016/j.biopha.2018.01.122
Morishita, Y., Yoshizawa, H., Watanabe, M., Ishibashi, K., Muto, S., Kusano, E., et al. (2014). siRNAs targeted to Smad4 prevent renal fibrosis in vivo. Sci. Rep. 4:6424. doi: 10.1038/srep06424
Munoz-Felix, J. M., Gonzalez-Nunez, M., Martinez-Salgado, C., and Lopez-Novoa, J. M. (2015). TGF-beta/BMP proteins as therapeutic targets in renal fibrosis. Where have we arrived after 25 years of trials and tribulations? Pharmacol. Ther. 156, 44–58. doi: 10.1016/j.pharmthera.2015.10.003
Nagarajan, R. P., Chen, F., Li, W., Vig, E., Harrington, M. A., Nakshatri, H., et al. (2000). Repression of transforming-growth-factor-beta-mediated transcription by nuclear factor kappaB. Biochem. J. 348(Pt 3), 591–596. doi: 10.1042/bj3480591
Nakao, A., Afrakhte, M., Moren, A., Nakayama, T., Christian, J. L., Heuchel, R., et al. (1997). Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 389, 631–635. doi: 10.1038/39369
Ng, Y. Y., Fan, J. M., Mu, W., Nikolic-Paterson, D. J., Yang, W. C., Huang, T. P., et al. (1999). Glomerular epithelial-myofibroblast transdifferentiation in the evolution of glomerular crescent formation. Nephrol. Dial. Transplant. 14, 2860–2872. doi: 10.1093/ndt/14.12.2860
Ng, Y. Y., Hou, C. C., Wang, W., Huang, X. R., and Lan, H. Y. (2005). Blockade of NFkappaB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int. 94, S83–S91.
Nie, Y., Li, S., Yi, Y., Su, W., Chai, X., Jia, D., et al. (2014). Effects of astragalus injection on the TGFbeta/Smad pathway in the kidney in type 2 diabetic mice. BMC Complement. Altern. Med. 14:148. doi: 10.1186/1472-6882-14-148
Nikolic-Paterson, D. J., Wang, S., and Lan, H. Y. (2014). Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. 4, 34–38. doi: 10.1038/kisup.2014.7
Oba, S., Kumano, S., Suzuki, E., Nishimatsu, H., Takahashi, M., Takamori, H., et al. (2010). miR-200b precursor can ameliorate renal tubulointerstitial fibrosis. PLoS One 5:e13614. doi: 10.1371/journal.pone.0013614
Provenzano, M., Coppolino, G., De Nicola, L., Serra, R., Garofalo, C., Andreucci, M., et al. (2019). Unraveling cardiovascular risk in renal patients: a new take on old tale. Front. Cell Dev. Biol. 7:314. doi: 10.3389/fcell.2019.00314
Qi, J. J., Liu, Y. X., and Lin, L. (2017). High expression of long non-coding RNA ATB is associated with poor prognosis in patients with renal cell carcinoma. Eur. Rev. Med. Pharmacol. Sci. 21, 2835–2839.
Qin, W., Chung, A. C., Huang, X. R., Meng, X. M., Hui, D. S., Yu, C. M., et al. (2011). TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 22, 1462–1474. doi: 10.1681/ASN.2010121308
Qiu, J., Chen, Y., Huang, G., Zhang, Z., Chen, L., and Na, N. (2017). Transforming growth factor-beta activated long non-coding RNA ATB plays an important role in acute rejection of renal allografts and may impacts the postoperative pharmaceutical immunosuppression therapy. Nephrology 22, 796–803. doi: 10.1111/nep.12851
Qu, X., Jiang, M., Sun, Y. B., Jiang, X., Fu, P., Ren, Y., et al. (2015). The Smad3/Smad4/CDK9 complex promotes renal fibrosis in mice with unilateral ureteral obstruction. Kidney Int. 88, 1323–1335. doi: 10.1038/ki.2015.235
Rane, M. J., Zhao, Y., and Cai, L. (2019). Krupsilonppel-like factors (KLFs) in renal physiology and disease. EBioMedicine 40, 743–750. doi: 10.1016/j.ebiom.2019.01.021
Roberts, A. B., Kim, S. J., Noma, T., Glick, A. B., Lafyatis, R., Lechleider, R., et al. (1991). Multiple forms of TGF-beta: distinct promoters and differential expression. Ciba Found Symp. 157, 7–15.; discussion 15–28,
Romagnani, P., Remuzzi, G., Glassock, R., Levin, A., Jager, K. J., Tonelli, M., et al. (2017). Chronic kidney disease. Nat. Rev. Dis. Primers 3:17088. doi: 10.1038/nrdp.2017.88
Saharinen, J., Hyytiainen, M., Taipale, J., and Keski-Oja, J. (1999). Latent transforming growth factor-beta binding proteins (LTBPs)–structural extracellular matrix proteins for targeting TGF-beta action. Cytokine Growth Factor Rev. 10, 99–117. doi: 10.1016/s1359-6101(99)00010-6
Samarakoon, R., Dobberfuhl, A. D., Cooley, C., Overstreet, J. M., Patel, S., Goldschmeding, R., et al. (2013). Induction of renal fibrotic genes by TGF-beta1 requires EGFR activation, p53 and reactive oxygen species. Cell. Signal. 25, 2198–2209. doi: 10.1016/j.cellsig.2013.07.007
Sanz, A. B., Sanchez-Nino, M. D., Ramos, A. M., Moreno, J. A., Santamaria, B., Ruiz-Ortega, M., et al. (2010). NF-kappaB in renal inflammation. J. Am. Soc. Nephrol. 21, 1254–1262. doi: 10.1681/ASN.2010020218
Sharma, K., Ix, J. H., Mathew, A. V., Cho, M., Pflueger, A., and Dunn, S. R. (2011). Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 22, 1144–1151. doi: 10.1681/ASN.2010101049
Sun, J., Zhang, S., Shi, B., Zheng, D., and Shi, J. (2017). Transcriptome identified lncRNAs associated with renal fibrosis in UUO rat model. Front. Physiol. 8:658. doi: 10.3389/fphys.2017.00658
Sun, S. F., Tang, P. M. K., Feng, M., Xiao, J., Huang, X. R., Li, P., et al. (2018). Novel lncRNA Erbb4-IR promotes diabetic kidney injury in db/db mice by targeting miR-29b. Diabetes 67, 731–744. doi: 10.2337/db17-0816
Sureshbabu, A., Muhsin, S. A., and Choi, M. E. (2016). TGF-beta signaling in the kidney: profibrotic and protective effects. Am. J. Physiol. Renal Physiol. 310, F596–F606. doi: 10.1152/ajprenal.00365.2015
Sutaria, P. M., Ohebshalom, M., Mccaffrey, T. A., Vaughan, E. D. Jr., and Felsen, D. (1998). Transforming growth factor-beta receptor types I and II are expressed in renal tubules and are increased after chronic unilateral ureteral obstruction. Life Sci. 62, 1965–1972. doi: 10.1016/s0024-3205(98)00166-0
Tang, P. M., Zhang, Y. Y., Mak, T. S., Tang, P. C., Huang, X. R., and Lan, H. Y. (2018). Transforming growth factor-beta signalling in renal fibrosis: from Smads to non-coding RNAs. J. Physiol. 596, 3493–3503. doi: 10.1113/JP274492
Tang, P. M., Nikolic-Paterson, D. J., and Lan, H. Y. (2019). Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 15, 144–158. doi: 10.1038/s41581-019-0110-2
Tian, Y., Liao, F., Wu, G., Chang, D., Yang, Y., Dong, X., et al. (2015). Ubiquitination and regulation of Smad7 in the TGF-beta1/Smad signaling of aristolochic acid nephropathy. Toxicol. Mech. Methods 25, 645–652. doi: 10.3109/15376516.2015.1061082
Trachtman, H., Fervenza, F. C., Gipson, D. S., Heering, P., Jayne, D. R., Peters, H., et al. (2011). A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 79, 1236–1243. doi: 10.1038/ki.2011.33
Troncone, E., Marafini, I., Stolfi, C., and Monteleone, G. (2018). Transforming growth factor-beta1/Smad7 in intestinal immunity, inflammation, and cancer. Front. Immunol. 9:1407. doi: 10.3389/fimmu.2018.01407
Tsuchida, K., Zhu, Y., Siva, S., Dunn, S. R., and Sharma, K. (2003). Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int. 63, 2000–2009. doi: 10.1046/j.1523-1755.2003.00009.x
Vincenti, F., Fervenza, F. C., Campbell, K. N., Diaz, M., Gesualdo, L., Nelson, P., et al. (2017). A Phase 2, double-blind, placebo-controlled, randomized study of fresolimumab in patients with steroid-resistant primary focal segmental glomerulosclerosis. Kidney Int. Rep. 2, 800–810. doi: 10.1016/j.ekir.2017.03.011
Voelker, J., Berg, P. H., Sheetz, M., Duffin, K., Shen, T., Moser, B., et al. (2017). Anti-TGF-beta1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 28, 953–962. doi: 10.1681/ASN.2015111230
Wan, Y. G., Che, X. Y., Sun, W., Huang, Y. R., Meng, X. J., Chen, H. L., et al. (2014). Low-dose of multi-glycoside of Tripterygium wilfordii Hook. f., a natural regulator of TGF-beta1/Smad signaling activity improves adriamycin-induced glomerulosclerosis in vivo. J. Ethnopharmacol. 151, 1079–1089. doi: 10.1016/j.jep.2013.12.005
Wang, B., Jha, J. C., Hagiwara, S., Mcclelland, A. D., Jandeleit-Dahm, K., Thomas, M. C., et al. (2014). Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 85, 352–361. doi: 10.1038/ki.2013.372
Wang, B., Komers, R., Carew, R., Winbanks, C. E., Xu, B., Herman-Edelstein, M., et al. (2012). Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol. 23, 252–265. doi: 10.1681/ASN.2011010055
Wang, H., Wang, B., Zhang, A., Hassounah, F., Seow, Y., Wood, M., et al. (2019). Exosome-mediated miR-29 transfer reduces muscle atrophy and kidney fibrosis in mice. Mol. Ther. 27, 571–583. doi: 10.1016/j.ymthe.2019.01.008
Wang, P., Luo, M. L., Song, E., Zhou, Z., Ma, T., Wang, J., et al. (2018). Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-beta/Smad3 pathway. Sci. Transl. Med. 10:eaat2039. doi: 10.1126/scitranslmed.aat2039
Wang, W., Huang, X. R., Canlas, E., Oka, K., Truong, L. D., Deng, C., et al. (2006). Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 98, 1032–1039. doi: 10.1161/01.res.0000218782.52610.dc
Wang, W., Huang, X. R., Li, A. G., Liu, F., Li, J. H., Truong, L. D., et al. (2005a). Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J. Am. Soc. Nephrol. 16, 1371–1383. doi: 10.1681/asn.2004121070
Wang, W., Koka, V., and Lan, H. Y. (2005b). Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology 10, 48–56. doi: 10.1111/j.1440-1797.2005.00334.x
Wang, Z., Han, Z., Tao, J., Wang, J., Liu, X., Zhou, W., et al. (2017). Role of endothelial-to-mesenchymal transition induced by TGF-beta1 in transplant kidney interstitial fibrosis. J. Cell. Mol. Med. 21, 2359–2369. doi: 10.1111/jcmm.13157
Weiskirchen, R., Meurer, S. K., Gressner, O. A., Herrmann, J., Borkham-Kamphorst, E., and Gressner, A. M. (2009). BMP-7 as antagonist of organ fibrosis. Front. Biosci. 14, 4992–5012. doi: 10.2741/3583
Xianyuan, L., Wei, Z., Yaqian, D., Dan, Z., Xueli, T., Zhanglu, D., et al. (2019). Anti-renal fibrosis effect of asperulosidic acid via TGF-beta1/smad2/smad3 and NF-kappaB signaling pathways in a rat model of unilateral ureteral obstruction. Phytomedicine 53, 274–285. doi: 10.1016/j.phymed.2018.09.009
Xiao, X., Yuan, Q., Chen, Y., Huang, Z., Fang, X., Zhang, H., et al. (2019). LncRNA ENST00000453774.1 contributes to oxidative stress defense dependent on autophagy mediation to reduce extracellular matrix and alleviate renal fibrosis. J. Cell. Physiol. 234, 9130–9143. doi: 10.1002/jcp.27590
Xie, H., Xue, J. D., Chao, F., Jin, Y. F., and Fu, Q. (2016). Long non-coding RNA-H19 antagonism protects against renal fibrosis. Oncotarget 7, 51473–51481. doi: 10.18632/oncotarget.10444
Yan, X., and Chen, Y. G. (2011). Smad7: not only a regulator, but also a cross-talk mediator of TGF-beta signalling. Biochem. J. 434, 1–10. doi: 10.1042/BJ20101827
Yan, X., Liao, H., Cheng, M., Shi, X., Lin, X., Feng, X. H., et al. (2016). Smad7 Protein Interacts with Receptor-regulated Smads (R-Smads) to Inhibit Transforming Growth Factor-beta (TGF-beta)/Smad Signaling. J. Biol. Chem. 291, 382–392. doi: 10.1074/jbc.M115.694281
Yan, X., Liu, Z., and Chen, Y. (2009). Regulation of TGF-beta signaling by Smad7. Acta Biochim. Biophys. Sin. 41, 263–272. doi: 10.1093/abbs/gmp018
Yang, F., Chung, A. C., Huang, X. R., and Lan, H. Y. (2009). Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 54, 877–884. doi: 10.1161/HYPERTENSIONAHA.109.136531
Yaswen, L., Kulkarni, A. B., Fredrickson, T., Mittleman, B., Schiffman, R., Payne, S., et al. (1996). Autoimmune manifestations in the transforming growth factor-beta 1 knockout mouse. Blood 87, 1439–1445. doi: 10.1182/blood.v87.4.1439.bloodjournal8741439
Yu, J. W., Duan, W. J., Huang, X. R., Meng, X. M., Yu, X. Q., and Lan, H. Y. (2014). MicroRNA-29b inhibits peritoneal fibrosis in a mouse model of peritoneal dialysis. Lab Invest. 94, 978–990. doi: 10.1038/labinvest.2014.91
Yu, M., Guan, Z., Li, S., Wen, X., Shi, H., Qu, G., et al. (2019). Gene expression profiling analysis reveals that the long non-coding RNA uc.412 is involved in mesangial cell proliferation. Mol. Med. Rep. 20, 5297–5303. doi: 10.3892/mmr.2019.10753
Zarjou, A., Yang, S., Abraham, E., Agarwal, A., and Liu, G. (2011). Identification of a microRNA signature in renal fibrosis: role of miR-21. Am. J. Physiol. Renal Physiol. 301, F793–F801. doi: 10.1152/ajprenal.00273.2011
Zhang, Y., Huang, X. R., Wei, L. H., Chung, A. C., Yu, C. M., and Lan, H. Y. (2014). miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. 22, 974–985. doi: 10.1038/mt.2014.25
Zhang, Y., Meng, X. M., Huang, X. R., and Lan, H. Y. (2018). The preventive and therapeutic implication for renal fibrosis by targetting TGF-beta/Smad3 signaling. Clin. Sci. 132, 1403–1415. doi: 10.1042/CS20180243
Zhang, Y. E., and Newfeld, S. J. (2013). Meeting report - TGF-beta superfamily: signaling in development and disease. J. Cell Sci. 126, 4809–4813. doi: 10.1242/jcs.142398
Zhang, Y. Y., Tang, P. M., Tang, P. C., Xiao, J., Huang, X. R., Yu, C., et al. (2019). LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1-Dependent Renal Inflammation. Diabetes 68, 1485–1498. doi: 10.2337/db18-1075
Zhao, T., Sun, S., Zhang, H., Huang, X., Yan, M., Dong, X., et al. (2016). Therapeutic effects of tangshen formula on diabetic nephropathy in rats. PLoS One 11:e0147693. doi: 10.1371/journal.pone.0147693
Zhong, X., Chung, A. C., Chen, H. Y., Dong, Y., Meng, X. M., Li, R., et al. (2013). miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 56, 663–674. doi: 10.1007/s00125-012-2804-x
Zhong, X., Chung, A. C., Chen, H. Y., Meng, X. M., and Lan, H. Y. (2011). Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 22, 1668–1681. doi: 10.1681/ASN.2010111168
Zhou, G., Sun, X., Qin, Q., Lv, J., Cai, Y., Wang, M., et al. (2018). Loss of Smad7 promotes inflammation in rheumatoid arthritis. Front. Immunol. 9:2537. doi: 10.3389/fimmu.2018.02537
Zhou, J., and Jiang, H. (2019). Livin is involved in TGF-beta1-induced renal tubular epithelial-mesenchymal transition through lncRNA-ATB. Ann. Transl. Med. 7:463. doi: 10.21037/atm.2019.08.29
Zhou, L., Fu, P., Huang, X. R., Liu, F., Chung, A. C., Lai, K. N., et al. (2010). Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am. J. Physiol. Renal Physiol. 298, F1006–F1017. doi: 10.1152/ajprenal.00675.2009
Zhou, Q., Chung, A. C., Huang, X. R., Dong, Y., Yu, X., and Lan, H. Y. (2014). Identification of novel long noncoding RNAs associated with TGF-beta/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol. 184, 409–417. doi: 10.1016/j.ajpath.2013.10.007
Zhou, Q., Huang, X. R., Yu, J., Yu, X., and Lan, H. Y. (2015). Long noncoding RNA Arid2-IR is a novel therapeutic target for renal inflammation. Mol. Ther. 23, 1034–1043. doi: 10.1038/mt.2015.31
Zhou, S. G., Zhang, W., Ma, H. J., Guo, Z. Y., and Xu, Y. (2018). Silencing of LncRNA TCONS_00088786 reduces renal fibrosis through miR-132. Eur. Rev. Med. Pharmacol. Sci. 22, 166–173. doi: 10.26355/eurrev_201801_14114
Zhou, Y., Xiong, M., Fang, L., Jiang, L., Wen, P., Dai, C., et al. (2013). miR-21-containing microvesicles from injured tubular epithelial cells promote tubular phenotype transition by targeting PTEN protein. Am. J. Pathol. 183, 1183–1196. doi: 10.1016/j.ajpath.2013.06.032
Ziyadeh, F. N., Hoffman, B. B., Han, D. C., Iglesias-De La Cruz, M. C., Hong, S. W., Isono, M., et al. (2000). Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 97, 8015–8020. doi: 10.1073/pnas.120055097
Keywords: TGF-β, Smads, fibrosis, inflammation, mechanisms, therapy
Citation: Gu Y-Y, Liu X-S, Huang X-R, Yu X-Q and Lan H-Y (2020) Diverse Role of TGF-β in Kidney Disease. Front. Cell Dev. Biol. 8:123. doi: 10.3389/fcell.2020.00123
Received: 15 October 2019; Accepted: 12 February 2020;
Published: 28 February 2020.
Edited by:
Zhonglin Chai, Monash University, AustraliaReviewed by:
Phillip Kantharidis, Monash University, AustraliaPaul J. Higgins, Albany Medical College, United States
Copyright © 2020 Gu, Liu, Huang, Yu and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui-Yao Lan, aHlsYW5AY3Voay5lZHUuaGs=