 Brian Lam
Brian Lam Emilie Roudier
Emilie Roudier
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 10 December 2019
Sec. Molecular and Cellular Pathology
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00320
This article is part of the Research Topic Multitasking Biomolecules in Human Pathologies: Known Players on Their Unexpected Journeys View all 6 articles
The E3 ubiquitin ligase Murine double minute 2 (MDM2) is the main negative regulator of the tumor protein p53 (TP53). Extensive studies over more than two decades have confirmed MDM2 oncogenic role through mechanisms both TP53-dependent and TP53-independent oncogenic function. These studies have contributed to designate MDM2 as a therapeutic target of choice for cancer treatment and the number of patents for MDM2 antagonists has increased immensely over the last years. However, the question of the physiological functions of MDM2 has not been fully resolved yet, particularly when expressed and regulated physiologically in healthy tissue. Cardiovascular complications are almost an inescapable side-effect of anti-cancer therapies. While several MDM2 antagonists are entering phase I, II and even III of clinical trials, this review proposes to bring awareness on the physiological role of MDM2 in the cardiovascular system.
Murine double minute 2 was identified in 1993 as a gene that is amplified in about one-third of human soft tissue sarcomas (Leach et al., 1993). This E3 ubiquitin ligase is one of the main negative regulators of the transcription factor tumor protein p53 (TP53). MDM2 regulates TP53 activity through direct interaction and ubiquitination, repressing its transcriptional activity (Levine and Oren, 2009; Vousden and Prives, 2009; Bieging et al., 2014). MDM2-TP53 interaction is highly regulated by multiple PTMs of both proteins (Meek and Anderson, 2009; Meek and Hupp, 2010), and through interactions with other binding partners (Naski et al., 2009; Fåhraeus and Olivares-Illana, 2014). MDM2 also has TP53-independent oncogenic properties (Bohlman and Manfredi, 2014, 2016). Thus, using antagonists that would limit MDM2 action represents a valuable anti-cancer approach, independently of the TP53 status of cancer cells (Toledo and Wahl, 2006; Bohlman and Manfredi, 2014, 2016). Most patents for MDM2 antagonists concern small molecule inhibitors, e.g., Nutlin-3a and RG7112. Some of these molecules are currently under phases I, II and III of clinical trials (Li and Lozano, 2013; Bohlman and Manfredi, 2016; Burgess et al., 2016). Since cardiovascular side effects of anti-cancer therapies are a recurrent problem (Jain et al., 2017), characterizing the cardiovascular function of MDM2 has become a pressing issue. Here, we propose to review the current literature regarding the cardiovascular function of MDM2. This review is divided into three parts. The first part analyzes whether the network of MDM2-interacting proteins plays a role in the cardiovascular system. The second part summarizes the knowledge gained from MDM2-transgenic mice. The last part examines the role of MDM2 in endothelial cells, in vascular smooth muscle cells (VSMCs) and in cardiomyocytes, some of the most important cells of the cardiovascular system.
MDM2 is ubiquitous and regulates key cellular processes, such as differentiation, DNA repair and synthesis, transcription, intracellular trafficking, cell cycle, hypoxia signaling, apoptosis and oxidative stress response. To achieve these functions, MDM2 acts on TP53 and a myriad of other downstream proteins (Fåhraeus and Olivares-Illana, 2014). The activity of MDM2 is tightly regulated through PTMs that include ubiquitination, SUMOylation, putative acetylation and phosphorylation (Kubbutat et al., 1997; Iwakuma and Lozano, 2003; Meek and Anderson, 2009; Marine and Lozano, 2009; Naski et al., 2009; Meek and Hupp, 2010; Nag et al., 2013; Fåhraeus and Olivares-Illana, 2014). As a proof of concept that the MDM2 network plays a role in the cardiovascular system, we discuss here some evidence supporting the idea that upstream regulators (see Table 1 for summary) and downstream targets (see Table 2 for summary) of MDM2 can regulate the homeostasis of vascular beds and the cardiac tissue.

Table 1. Summary of the putative cardiovascular functions of upstream regulatory kinases for MDM2 discussed in this review.
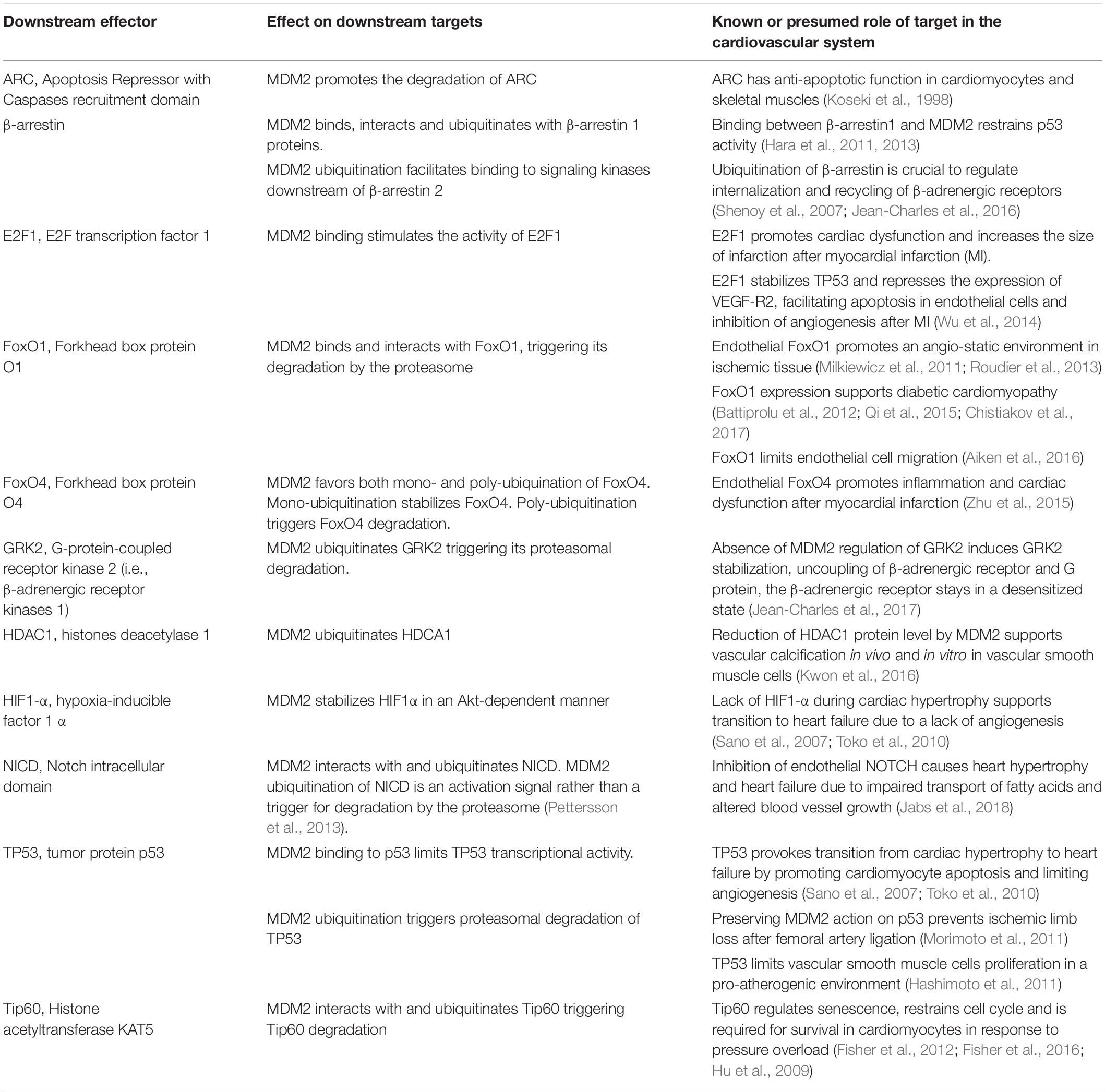
Table 2. Summary of the putative cardiovascular functions of downstream effectors of MDM2 discussed in this review.
Several upstream regulators of MDM2 are important actors of the cardiovascular homeostasis. To illustrate the idea, we have deliberately chosen to discuss the role of a few receptors and kinases (Figure 1).
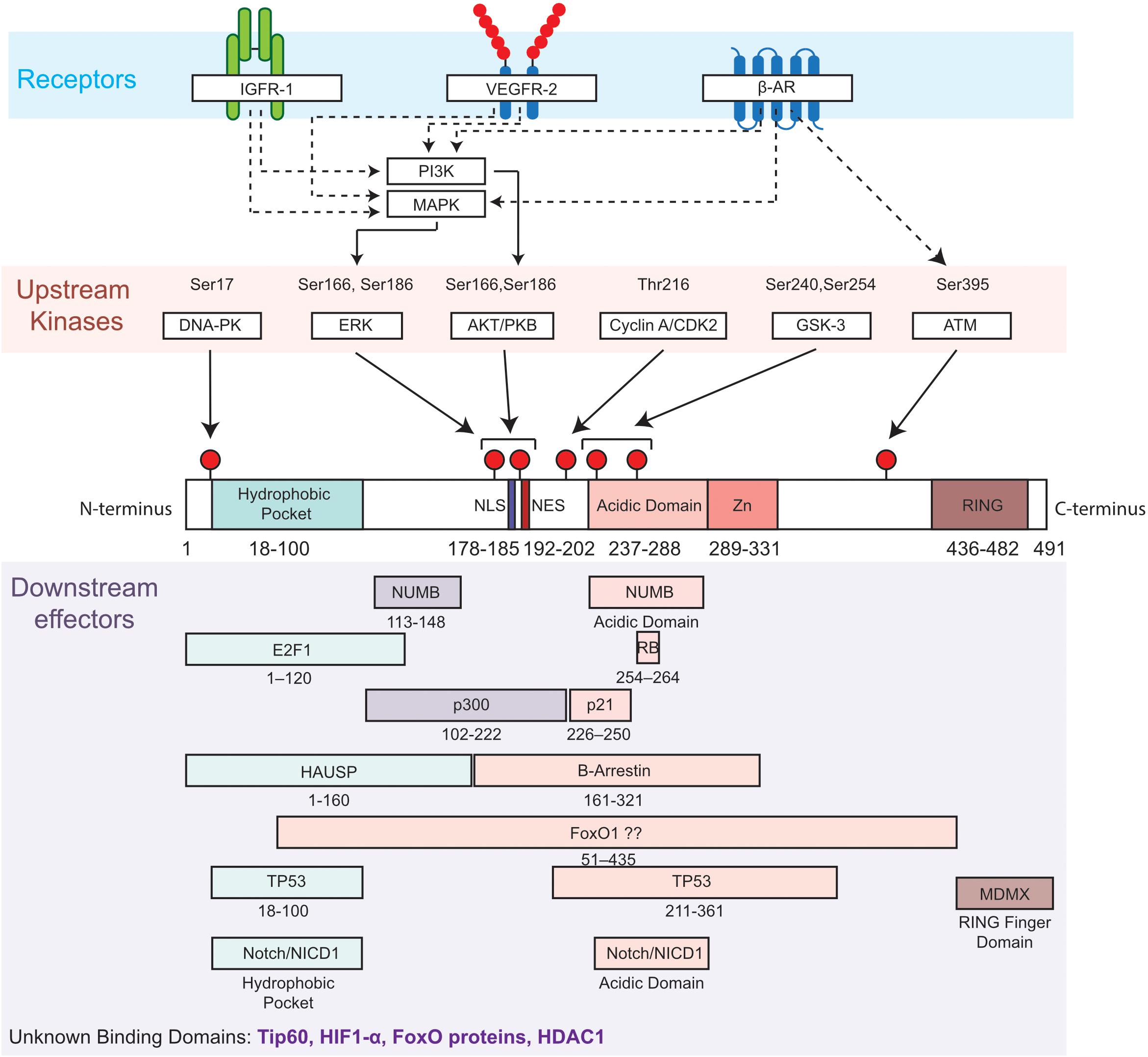
Figure 1. Diagram of MDM2 domains showing phosphorylation sites and upstream regulators (kinases and related receptors) of MDM2 functions, as well as putative binding sites with downstream effectors. Top, most common phosphorylation sites on Serine (Ser) and Threonine (Thr) are indicated by a red pin. Residues are numbered based on amino acid sequence from N-terminus to C-terminus. Middle, the functional domains of MDM2 include the hydrophobic pocket, the nuclear localization sequence (NLS), the nuclear export sequence (NES), the acidic and central domain, the Zinc domain (Zn) and the ring domain (RING). Bottom, selected interactions between MDM2 and its downstream effectors of MDM2 are illustrated. β-Adrenergic receptor, β-AR; DNA-dependent protein kinase, DNA-PK; E2F transcription factor 1, E2F1; extracellular-signal-regulated kinase, ERK; Forkhead box protein, FoxO; herpes virus-associated ubiquitin specific protease, HAUSP; Insulin-like growth factor receptor 1, IGFR1; Mitogen-activated protein kinases, MAPK; Mouse double Minute 4, MDM4; NUMB; Notch intracellular domain, NICD; Phosphotidylinositol 3-kinase, PI3K; Protein kinase B, PKB; protein numb homolog, Retinoblastoma protein, RB; Vascular Endothelial Growth Factor Receptor 2, VEGFR2.
We will first present how ligands relay their signals to MDM2 by discussing the role of two tyrosine kinases receptors, the Vascular Endothelial Growth Factor Receptor (VEGFR2) and the Insulin-like Growth Factor 1 Receptor (IGFR1), as well as one G-protein coupled receptor, the β-Adrenergic Receptor (β-AR).
Kinases sensing DNA damage inhibit MDM2, while growth factors activate MDM2 through the modulation of phosphorylation on multiple sites: the nuclear localization signal, the nuclear export signal, the acidic and central domain (Figure 1). For example, growth factors stimulate MDM2 functions by limiting glycogen synthase kinase-3 (GSK3)-dependent phosphorylation on the acidic domain (Serines 240 and 254) and by enhancing Akt-dependent phosphorylation (Serines 166 and 186). These actions of growth factors counterbalance the effects of DNA damage kinases, such as ATM which phosphorylates MDM2 on Serine 395 (Meek and Anderson, 2009; Meek and Hupp, 2010). Therefore, we will analyze the specific role of the ataxia telangiectasia mutated (ATM) kinase, a DNA damage kinase, and the role of GSK3 and Akt in the cardiovascular system.
The Vascular Endothelial Growth Factor-A (VEGF-A) is a very potent stimulator of angiogenesis, crucial to maintaining vascular homeostasis. VEGF-A binds to VEGFR2, a tyrosine kinase receptor, leading to its activation by auto-phosphorylation. Once activated, VEGFR2 relays its signal to downstream kinases including the Phosphotidylinositol 3-Kinase/Akt (PI3K/Akt) and the Mitogen-activated protein kinases (MAPKs) (Claesson-Welsh, 2016). Both the PI3K/Akt and MAPK pathways can phosphorylate MDM2 on serine residues 166 and 186 (Mayo and Donner, 2001; Feng et al., 2004; Hogan et al., 2008) to promote the oncogenic function of MDM2 in cancer cells. In a physiological context, we reported that VEGF-A promotes MDM2 phosphorylation. Indeed, VEGF-A elicits MDM2 phosphorylation in the skeletal muscle during physical exercise, as well as in primary endothelial cells (Aiken et al., 2016) (for more detailed information see Part 3 ‘Role of the MDM2 pathway in primary endothelial cells’).
IGFR1 and β-AR are important for cellular processes in the heart such as hypertrophy, contractility and cell survival (Stiles et al., 1984; Weeks et al., 2017). Binding of IGF1 to its receptor, IGFR1, promotes the phosphorylation of MDM2 through the Akt and MAPK pathways. This leads to MDM2 nuclear localization and enhances its stability and activity (Ogawara et al., 2002; Feng et al., 2004; Worrall et al., 2017). B -adrenergic stimulation is well known to activate both Akt and MAPK pathways (Morisco et al., 2000; Vidal et al., 2012). Most recently, chronic exposure to β-adrenergic agonist, isoproterenol, was reported to increase the expression of ATM, a kinase upstream of MDM2 (Foster et al., 2011). This suggests that MDM2 can sense the β-adrenergic and IGF1 signals. Interestingly, several publications highlight a more complex relationship. Indeed, MDM2 can regulate the trafficking of both IGFR1 and β-AR, modulating the sensitivity of cells to the IGF1 and catecholamines, respectively (Girnita et al., 2007; Jean-Charles et al., 2017). The relationship between MDM2 and β-AR is discussed in further detail in Part 3, ‘MDM2 and the regulation of the β-adrenergic signal.’
The few studies discussed above support the idea that MDM2 can relay the signals sensed by tyrosine kinase receptors and G-protein coupled receptors within the cardiovascular system. Yet, further investigations are warranted to fully elucidate how these receptors regulate MDM2 functions.
Patients with Ataxia telangiectasia syndrome not only present growth retardation, premature aging and a high risk of cancer, they also exhibit insulin resistance, glucose intolerance and a higher risk of ischemic heart disease (Espach et al., 2015). This suggests that ATM has cardiovascular functions. Interestingly, ATM has emerged as a regulator of atherosclerosis and potentially angiogenesis.
In the atherogenic environment of the Apo null background, haplodeficient ATM± mice present the earliest onset of atherosclerosis (Mercer et al., 2012). Haplodeficient ATM± mice also have worse cardiac remodeling and an impaired ventricular function post-myocardial infarction compared to their wild-type littermate, suggesting that ATM depletion facilitates the development of heart failure (Daniel et al., 2014, 2016; Jia et al., 2017). This suggests a potential role for ATM in maintaining cardiovascular health through the protection against atherosclerosis and pathological cardiac remodeling. Beyond these protective effects, ATM might regulate angiogenesis. It is well known that in response to DNA damage, ATM phosphorylates MDM2 on the Ser395 (de Toledo et al., 2000; Gannon et al., 2012). After hypoxia-reoxygenation, reactive oxygen species (ROS) activate ATM in immature blood vessels. The activation of ATM represses the p38α MAPK. Usually, p38α is activated by the VEGFR2 to support endothelial cell migration; but when overactivated p38α can favor endothelial cell apoptosis. ATM activity can limit p38α proapoptotic functions, thus enhancing endothelial survival (Okuno et al., 2012). Through this mechanism, ATM supports pathological angiogenesis in ischemic retinopathy and tumor growth. Since ATM knock-out in the whole body and specifically in the endothelium protects mice from these pathological forms of angiogenesis, it appears that ATM is a key regulator of angiogenesis driven by hypoxia and oxidative stress. We have reported a central role for MDM2 in muscle angiogenesis during endurance exercise (Roudier et al., 2012; Aiken et al., 2016), a stress-associated oxidative stress and potentially tissue hypoxia. Yet, it remains unknown whether the angiogenic function of MDM2 is regulated by ATM.
A lot remains to be discovered regarding the cardiovascular functions of ATM, however, this DNA damage kinase might modulate how the cardiovascular system adapts to micro-environmental stressors, such as oxidative stress, proatherogenic factors and ischemia.
Growth factors stimulate MDM2 in multiple ways, including the downregulation of GSK3 and the activation of Akt. Interestingly, both GSK3 and Akt play an important role in controlling cardiac hypertrophy (Weeks et al., 2017; Takahashi-Yanaga, 2018).
GSK3 is ubiquitous and normally active in the absence of growth factors. GSK3 phosphorylates the central domain of MDM2, decreasing MDM2 actions on its targets, e.g., TP53 (Kulikov et al., 2005; Lin et al., 2012). To support cardiac function during hypertrophy, it is crucial that angiogenesis occurs to maintain proper blood supply to the cardiac muscle. Chemical inhibition of GSK3 by IM-12 improves myocardial blood flow recovery in pigs fed with a high-fat diet then subjected to myocardial infarction induced by left circumflex coronary artery constriction (Potz et al., 2016). Overall, the increased capillary density suggests improved tissue perfusion. Unfortunately, the capillary-to-cardiomyocytes ratio was not reported in this study. The observed increase in myocardial capillary density could be a result of reduced hypertrophy, since GSK3 is a negative regulator of cardiac hypertrophy (Takahashi-Yanaga, 2018). Indeed, reduced cardiomyocyte size would increase capillary density without any gain of capillaries. Therefore, reporting a capillary-to-cardiomyocytes ratio is crucial to demonstrate whether angiogenesis occurred.
Growth factors induce Akt-dependent phosphorylation of MDM2. This promotes MDM2 interaction with the HIF1-α, stabilizing HIF1-α and enhancing its transcriptional activity on pro-angiogenic genes (Mayo and Donner, 2001; Bárdos et al., 2004). Akt-dependent phosphorylation of MDM2 facilitates MDM2 interaction with TP53, leading to TP53 ubiquitination and degradation. The resulting decrease in TP53 restrains apoptosis and can favor angiogenesis by limiting angiostatic gene expression, such as Thrombospondin-1 (Dameron et al., 1994). It is worth noting that TP53 and HIF1-α can compete for binding to MDM2, which means that under hypoxia HIF1-α could restrain the capacity of MDM2 to inhibit TP53 (Robertson et al., 2014).
In the heart, Akt signaling coordinates the growth of cardiomyocytes with angiogenesis to ensure the cardiac tissue remains well perfused during physiological hypertrophy (Weeks et al., 2017). Conversely, pathological hypertrophy promotes TP53 expression. Cardiac hypertrophy induced by pressure overload leads to TP53 accumulation, restriction of HIF1-α functions and a lack of angiogenesis (Sano et al., 2007); a similar outcome was observed in the failing heart (Toko et al., 2010). While it remains unknown whether Akt-dependent phosphorylation of MDM2 plays a role in these pathological changes in TP53 and HIF1-α, a study suggests that increased MDM2 activity could be beneficial. Indeed, overexpression of IGF1 in vitro and in vivo enhanced MDM2 activity toward TP53 in cardiomyocytes preventing apoptosis and the activation of the renin-angiotensin system, a hallmark of heart failure (Leri et al., 1999a, b).
In the KK-Ay diabetic mice, atorvastatin, a cholesterol-lowering drug, preserved the insulin sensitivity of Akt in the lower limb after femoral artery ligation. Atorvastatin increased the levels of Serine 166 phosphorylation and prevented TP53 accumulation in the ischemic lower limb. Since atorvastatin rescued limb loss after femoral artery ligation. These data suggest that activation of MDM2 supports limb recovery (Morimoto et al., 2011).
GSK3 seems to support adverse remodeling after myocardial infarction, while Akt facilitates beneficial adaptations. Additional investigations are required to fully comprehend how GSK3 and Akt impact MDM2 function in the context of these cardiovascular diseases. Together these studies, however, clearly support that kinases upstream of MDM2 play a role in controlling the balance between microvascular content and striated muscle hypertrophy during pathological remodeling.
MDM2’s functions go beyond the old paradigm that MDM2 is a key regulator of transcriptional factor TP53. Indeed, this E3 ubiquitin ligase has numerous targets in all cellular compartments. Therefore, we have chosen to discuss several examples of MDM2 targets, including some nuclear proteins involved in the regulation of gene expression and several cytoplasmic proteins involved in key intracellular signaling and trafficking pathways.
We and others have reported that MDM2 is key regulator of FoxO transcription factors (Horst et al., 2006; Brenkman et al., 2008; Yang J.-Y. et al., 2008; Yang W. et al., 2008; Fu et al., 2009; Aiken et al., 2016). The original observation that FoxOs are important for longevity (Salih and Brunet, 2008), led researchers to explore the cardiovascular functions of FoxO. FoxO proteins are well-known regulators of antioxidant defense. With no exception, FoxO1 and FoxO3 have been reported to be important to maintain antioxidant molecules (i.e., catalase and SOD2) in response to cardiac ischemia-reperfusion (Sengupta et al., 2011; Puthanveetil et al., 2013). Due to this function, FoxO proteins were thought to have protective properties in the cardiovascular system. However, this might be context- and tissue-dependent.
In the vasculature, endothelial knock-down of FoxO1, FoxO3 and FoxO4 appears to be protective against atherosclerosis in LDLr–/– mice, a transgenic mouse model that creates a pro-atherogenic environment due to a lack of low-density lipoprotein receptors (LDLr) (Tsuchiya et al., 2012). This supports the detrimental role of FoxO protein in arteries exposed to an atherogenic environment. Mouse models of type 2 diabetes present increased levels of cardiac FoxO1 (Battiprolu et al., 2012; Qi et al., 2015; Chistiakov et al., 2017). This increase in FoxO1 downregulates insulin receptor substrate 1 (IRS-1) impairing Akt activity and insulin sensitivity. Cardiac-specific knock-down of FoxO1 prevented the development of diabetic cardiomyopathy, making a strong case for a detrimental role of cardiac FoxO1 during diabetic cardiomyopathy. FoxO1, β-arrestin, IRS and IGF1R are all crucial to maintaining an efficient insulin signal (Usui et al., 2004; Girnita et al., 2007). Interestingly, these proteins might compete for binding to MDM2 (Balaji et al., 2018). Yet, the role of cardiac MDM2 in diabetic cardiomyopathy remains unclear. In the context of post-myocardial infarction, endothelial-specific depletion of FoxO4 improved cardiac function by preventing loss of nitric oxide production. Maintenance of nitric oxide restrained inflammation by suppressing monocyte adhesion (Zhu et al., 2015). Interestingly, MDM2 is upregulated in the cardiac tissue 24 h post-myocardial infarction and remains elevated up to 4 weeks post-infarction (Zhao et al., 2016). MDM2 inhibition by Nutlin-3a improved survival and repressed the inflammatory response in this context. MDM2 has been reported to prime the mono-ubiquitination of FoxO4 in response to oxidative stress (Brenkman et al., 2008) leading to FoxO4 poly-ubiquitination and degradation when MDM2 levels are high (Hauck et al., 2017). It is then appealing to hypothesize that the interaction between MDM2 and FoxO4 in the endothelium controls the inflammatory response after myocardial infarction; a hypothesis that remains unverified. Finally, we reported an upregulation of FoxO1 in ischemic skeletal muscle during peripheral artery disease (Milkiewicz et al., 2011; Roudier et al., 2013) that correlated with a restrained binding between MDM2 and FoxO1. Endothelial-directed depletion of FoxO1 enhanced angiogenesis (Milkiewicz et al., 2011). Since constitutive active MDM2 reduces FoxO1 transcriptional activity in endothelial cells (Aiken et al., 2016), it is appealing to hypothesize that MDM2 impedes the angiostatic function of FoxO1 in the endothelium.
Based on the above studies it appears that an in-depth analysis of the FoxO-MDM2 interaction in the cardiovascular tissues has the potential to help fully comprehend how the FoxO functions (i.e., antioxidant defense, angiostasis, inflammation, etc.) are regulated during cardiovascular diseases.
E2 promoter binding Factor 1 (E2F1) is a transcriptional activator mainly known to regulate the cell cycle, apoptosis and angiogenesis to support tumor progression (Schaal et al., 2014). MDM2 was shown to stimulate E2F1 transcriptional activity in tumors (Martin et al., 2014), potentially shifting E2F1 properties from antiproliferative to proliferative (Loughran and Thangue, 2000). Interestingly, E2F1-deficient mice show improved cardiac function, reduced size of myocardial infarction, and enhanced post-infarction cardiac angiogenesis after ligation of the left anterior descending coronary artery. These beneficial effects of E2F1 deficiency in the ischemic heart were due to an upregulation of pro-angiogenic factors (i.e., vascular endothelial growth factor A, VEGF-A and placental growth factor, PIGF (Wu et al., 2014). In cardiac fibroblasts subjected to hypoxia, the overexpression of E2F1 increased the expression of TP53, which was required to repress the expression VEGF-A; while a decrease in PIGF occurred independently of the TP53 status of these cells. These results suggested that E2F1 promotes the expression of pro-angiogenic factors through both TP53-dependent and TP53 independent mechanisms (Wu et al., 2014). Conversely to previous observations made in tumor cells, MDM2-deficiency in cardiac cells was reported to enhance E2F1 transcriptional activity and to have detrimental effects on the heart homeostasis (Hauck et al., 2017) (see below Part ‘Insight from MDM2 transgenic mice’). Further investigations are thus warranted to better understand how the interplay between MDM2 and E2F1 impacts myocardial physiology and recovery post-infarction.
Tip60 is an acetyltransferase that acetylates histones and other proteins, including TP53 and ATM. When TP53 is acetylated on lysine 120 by Tip60, it triggers TP53-dependent apoptosis (Tang et al., 2006). MDM2 can interact with Tip60. MDM2 promotes the proteasomal degradation of Tip60 (Legube et al., 2002), whereas Tip60 binding to MDM2 restrains the neddylation capacity of the E3 ligase (Dohmesen et al., 2008). Tip60 expression in cardiac tissue is enriched during early cardiac development and adult hearts are reported to express Tip60 (Lough, 2002; Hu et al., 2009; Fisher et al., 2012, 2016). Tip60± heterozygous mice (12–14 week-old) present an enhanced cell cycle activity and reduced apoptosis, that could support better cardiac regeneration in the heart subjected to pressure-overload (Fisher et al., 2012).
Specific depletion of Tip60 in cardiomyocytes in mice increased cardiomyocyte cell cycling in vivo in 2–4 week-old hearts (Fisher et al., 2016). However, this was followed by a significant loss of cardiomyocytes after 8 weeks, a reduction of heart mass leading to death between 8 and 12 weeks of age due to cardiac dysfunction. Together these results suggest that Tip60 is crucial to control the balance between cell cycle and apoptosis in the myocardial cells. Yet, the role of MDM2 on the cardiac functions of Tip60 remains to be studied.
The NOTCH pathway acts conjunctly with Vascular Endothelial Growth Factor-A (VEGF-A) to allow patterning of blood vessels during sprouting angiogenesis (Blanco and Gerhardt, 2013). This is essential for vascular growth. An elegant study by Jabs et al. (2018) recently demonstrated the role of the NOTCH pathway in the adult endothelium of the heart. Endothelial-specific downregulation of NOTCH1 signal was induced by conditional ablation of the Rbp-jκ complex in adult mice. In this model, restriction of the endothelial NOTCH pathway was highly associated with cardiac dysfunction, cardiac hypertrophy and impairment of fatty acid transport through the endothelium. The source of energy shifted in favor of glucose instead of fatty acids, likely limiting the capacity to supply ATP for cardiac function. All these changes are hallmarks of heart failure. Down-regulation of the NOTCH pathway was associated with an increase of cardiac microvascular density, supporting the idea that NOTCH imposes a brake on cardiac angiogenesis (Jabs et al., 2018). Usually improved cardiac angiogenesis is associated with physiological cardiac hypertrophy. The observation that enhanced angiogenesis led to heart failure supports the idea that the growth of an abnormal vascular bed occurs when NOTCH is inhibited. The main negative regulator NOTCH1 is NUMB. Indeed, NUMB facilitates NOTCH1 ubiquitination and degradation. MDM2 positively regulates the NOTCH signaling pathways through its capacity to bind to both NUMB and to the NICD1. MDM2 promotes the degradation of NUMB, an event that could enhance NOTCH1 signaling. Conversely, MDM2 binds to the NICD1. This binding does not promote the ubiquitination nor the degradation of NOTCH1, but rather it activates the NOTCH signaling pathway, in cancer cell lines (Pettersson et al., 2013). Since we and others have reported pro-angiogenic functions of MDM2 (Secchiero et al., 2007; Roudier et al., 2012), further investigations are required to decrypt the mechanisms controlling endothelial NOTCH activity in the adult heart, studying the putative involvement of MDM2 might bring new insight on how a normal cardiac microvasculature is established.
MDM2 is associated with key regulators of the beta-adrenergic pathway through its capacity to interact with β-arrestin, a protein adapter of G protein-coupled receptor and G Protein-Coupled Receptor Kinase 2 (GRK2) (Jean-Charles et al., 2017). The pivotal work done by Hara et al. (2011, 2013) clearly established that chronic exposure to β-adrenergic catecholamines facilitates MDM2-β-arrestin1 interaction, restraining TP53 activity. One consequence could be an impairment of the DNA damage response with a potential impact on genomic integrity (Hara et al., 2013). Additional works also support the idea that MDM2 might function to recycle G protein-coupled receptors; playing a key role in the desensitization and re-sensitization of these receptors, including the β-AR (Wang et al., 2003; Shenoy et al., 2009; Jean-Charles et al., 2016, 2017). Ultimately, this is supportive of a role for MDM2 in the cardiovascular system through the modulation of the cardiac response to β-adrenergic stimulation (Cannavo et al., 2013).
The studies presented above, while not exhaustive, serve as examples to highlight the diverse role of MDM2 as a central hub in the cardiovascular system. Most human cardiovascular diseases are associated with DNA damage due to oxidative stress (Jackson and Bartek, 2009; Shimizu et al., 2014; Uryga et al., 2016) and alteration of hormonal signals (Higashi et al., 2019). The DNA damage kinase ATM is an upstream regulator of MDM2, and it does modulate the endothelial function, playing a role in atherosclerosis and angiogenesis. Growth factor responsive kinases known to phosphorylate MDM2 appears as crucial for a proper balance between vascularization and hypertrophy during cardiac remodeling. MDM2 cardiovascular functions are likely TP53- and HIF1-α-dependent, however, numerous TP53-independent MDM2 binding partners also emerge as key actors of cardiac and vascular homeostasis. Yet, a lot remains to be done to fully understand how the “MDM2 social network” participates in cardiovascular homeostasis over the lifespan (Fåhraeus and Olivares-Illana, 2014).
The phenotypic analyses of MDM2 transgenic mouse models further demonstrate a crucial role for MDM2 in the cardiovascular system (Table 3).
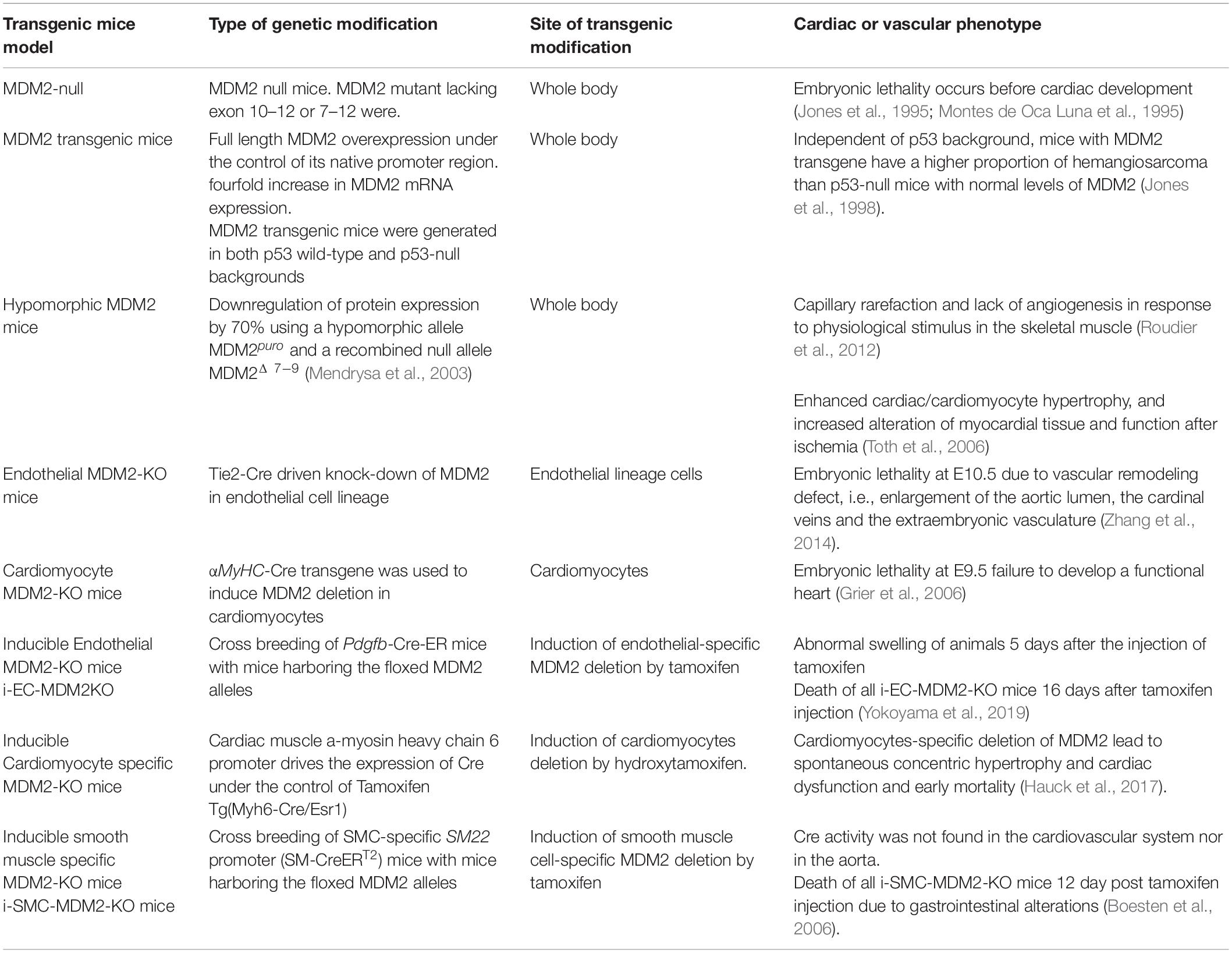
Table 3. Description and summary of the cardiovascular phenotypes observed in MDM2 transgenic mouse model.
Over the last two decades, several transgenic mouse models have been generated to study the function of MDM2, revealing a crucial role of MDM2 in supporting survival, as well as vascular function.
Full Mdm2 deletion results in embryonic lethality at E5 (Jones et al., 1995; Montes de Oca Luna et al., 1995). Embryonic lethality was completely reversed in double-null mice, harboring both MDM2 and TP53 deficiency (Mdm2−/− and Trp53−/−). Due to the early stage of lethality, these initial studies did not allow for the examination of MDM2 function in adult cardiac tissue. To override the lethality observed in mice with full MDM2 knock-down, Zhang et al. (2012) created a transgenic mouse with a selective knock-down of MDM2 in the endothelial cell lineage using the Tie2-Cre transgene. The loss of endothelial MDM2 also resulted in embryonic lethality, however, at a later stage (E10.5). This lethality was due to defective vascular patterning, including an enlargement of the aorta, the cardinal veins, and the extraembryonic vasculature (Zhang et al., 2014). The expression of TP53 targets genes, p21, Nuxa and Puma, was increased at E8.5 and E9.0 in heart tube and apoptosis was enhanced. Most recently, to study the role of MDM2 in the adult endothelium, Yokoyama et al. (2019) generated a transgenic mouse model in which the inducible endothelial cell-specific depletion of MDM2 was achieved by tamoxifen injection using the Pdgfb-Cre-ER transgene in 8-week old mice. Five days after the injection of tamoxifen, some abnormal swelling was observed in mice with endothelial-specific deletion of Mdm2. All mice died 16 days post-injection. This phenotype was associated with severe increases in TP53 expression and apoptosis in the vascular bed, including in the skeletal muscle and the heart. MDM2 appeared as crucial to maintain the vascular barrier and permeability function of the endothelium. Taken together, these studies present a strong case that in the embryo and in adult mice, MDM2 acts in a TP53-dependent manner to limit excessive TP53 overactivation and apoptosis in tissues, including in the endothelium.
While the primary function of MDM2 might be to restrain TP53, additional evidence supports the idea that MDM2 is important for vascular homeostasis due to TP53-independent functions. MDM2 overexpression in mice elicits tumor formation, bringing irrefutable evidence that MDM2 promotes cancer development. A close phenotypic analysis of these mice also reveals an endothelial function of MDM2. The mice overexpressing MDM2 in a Trp53 null background presented a unique subset of tumors compared to mice overexpressing MDM2 in a Trp53 wild-type background. Under a Trp53 null background, a higher rate of hemangiosarcoma was observed. This type of tumor is due to an uncontrolled proliferation of endothelial cells (Jones et al., 1998). These observations suggest that MDM2 can support endothelial cell proliferation independently of simply controlling TP53 activity.
Another very interesting transgenic rodent model is the hypomorphic Mdm2puro/Δ 7–9 mouse. These mice harbor one hypomorphic allele (puro) and one null allele (Δ7-9), thus leading to a reduction of MDM2 expression by 70% of the normal level of expression in tissue (Mendrysa et al., 2003). Under basal conditions, these mice do not present TP53 overactivation. Mdm2 hypomorphic mice, however, have a reduction in MDM2 activity. We reported a decrease of the capillary to fiber ratio by 20% in the skeletal muscle of the hypomorphic Mdm2puro/Δ 7–9 mice (Ogawara et al., 2002) independently of TP53 status (Ogawara et al., 2002). In response to endurance training, a well-established powerful physiological stimulus for muscle angiogenesis, the development of new blood vessels was blunted in exercising Mdm2puro/Δ 7–9 mice. This shows that normal expression of MDM2 is required both for the maintenance and the expansion of the vascular bed. These studies support the notion that beyond its primary function to keep TP53 under control, MDM2 has TP53-independent vascular functions that impact the capacity of endothelial cells to proliferate and to initiate angiogenesis.
The smooth muscle tissue is another key component of blood vessels. Mice with smooth muscle-specific MDM2 deficiency have been generated using a tamoxifen-inducible Cre under the control of the SM22 promoter (Boesten et al., 2006). These mice died shortly (9–13 days) after the induction of MDM2 depletion in the smooth muscle. However, the depletion of MDM2 in the aorta was not be achieved with this approach, the author did not investigate potential depletion in other vascular structures (i.e., arterioles). So, it remains impossible to conclude regarding the in vivo role of MDM2 in the vascular smooth muscle. The putative role of MDM2 in cultivated VSMCs is discussed in Part 3 of this review.
MDM2 appears to be crucial for the embryonic development of the heart. Tissue-specific loss of MDM2 in cardiomyocytes induced by the αMyHC-Cre transgene results in embryonic lethality. At E9.5, embryos with cardiomyocyte-specific Mdm2 deletion failed to develop a functional heart (Grier et al., 2006). Immunohistochemistry staining of cardiomyocytes revealed a significant thinning of the myocardial layer of the ventricles and overall a much smaller heart weight, probably due to an increased level of apoptosis. By E13.5, no embryos survived, and most of them lacked visible signs of heart development (Grier et al., 2006).
Interestingly, hypomorphic Mdm2puro/Δ7–9 mice present a lower heart weight, supporting the notion that MDM2 is required to maintain heart mass. However, the heart-to-body mass ratio, as well as the cardiomyocyte cross-sectional area were increased in the hypomorphic mice (Toth et al., 2006). These mice were also more sensitive to ischemia-induced damage presenting an increased infarction area and an impaired cardiac function (Toth et al., 2006). To study specifically the function of MDM2 in the adult heart, Hauck et al. (2017) generated mice with a cardiomyocyte-specific inducible depletion of Mdm2. This ablation of Mdm2 in the cardiomyocytes led to left ventricular dysfunction and concentric remodeling of the heart, as shown by a 98% increase in the heart-to-body mass and a 1.9-fold increase in cardiomyocyte size 18 days after 4-hydroxytamoxifen injection (Hauck et al., 2017). Mitochondrial dysfunctions were also observed due to a lack of proper mitochondria biogenesis. These mitochondrial effects were a consequence of alterations in the transcription of TP53-dependent genes involved in mitochondria energy metabolism (Pgc1-α, Ppar-α/γ and Esrr-β/γ) (Hauck et al., 2017). Interestingly, Mdm2 mRNA levels were found to be reduced in patients with ischemic cardiomyopathy, idiopathic dilated cardiomyopathy and chemotherapy-induced cardiomyopathy (Toth et al., 2006). Together these data support the idea that reduced expression of MDM2 may facilitate cardiomyocyte hypertrophy and cardiac hypertrophy.
In the embryo, MDM2 is essential to both the cardiac and endothelial tissue development with a primary role as a negative regulator of TP53-induced apoptosis. In adult mice, MDM2 appears essential for controlling cardiac hypertrophy, potentially through TP53 dependent mechanisms (Toth et al., 2006; Hauck et al., 2017). Loss of MDM2 in the endothelium is lethal in adult mice due to TP53 overactivation. However, MDM2 appears to have TP53-independent functions that regulate vascular homeostasis. Too little expression of MDM2 supports vascular rarefaction and lack of angiogenesis, while excessive expression leads to an uncontrolled expansion of the vascular bed.
Aside from the phenotypic observations in transgenic mouse models, evidence for a cardiovascular role of MDM2 is also supported by studies that have investigated MDM2 and its signaling pathways in endothelial cells, VSMCs and cardiomyocytes.
Endothelial cells form the inner lining of blood vessels. In these cells and MDM2 acts as a negative regulator of TP53, limiting apoptosis (Dai et al., 2012; Brockhaus et al., 2018; Si et al., 2018; Yokoyama et al., 2019). MDM2 could elicit other endothelial functions. Originally, the interest in studying MDM2 in the endothelial cells was initiated by the observation that MDM2 could enhance the production of pro-angiogenic factors in tumor cells. Through its interaction with HIF-1α, MDM2 facilitates angiogenesis by stimulating the expression of VEGF-A in response to hypoxia and growth factors (Skinner et al., 2004; Alam et al., 2009; Park et al., 2011; Patterson et al., 2011). MDM2 might have a similar role in endothelial cells (Song et al., 2012). Since autocrine VEGF-A is crucial to endothelial cell survival (Lee et al., 2007), it is plausible that MDM2 also supports endothelial cell survival through a VEGF-dependent pathway. During angiogenesis, endothelial cells switch rapidly from a quiescent state to a migratory and proliferative state. The observation made by Seccherio and colleagues (2007) that MDM2 antagonist Nutlin-3 inhibits cell cycle progression and cell migration in HUVEC was one of the first lines of evidence that MDM2 could facilitate the activation of endothelial cells to support tumor angiogenesis. Interestingly in this study, the author reported that tumor angiogenesis was inhibited by nutlin-3 independently of TP53. This is an additional piece of evidence that MDM2 angiogenic function might be independent of TP53. We have observed in primary human and mouse endothelial cells that the MDM2 signaling pathway is highly sensitive to pro-angiogenic stimuli. Shear stress, VEGF-A and serum stimulation increased the phosphorylation of MDM2 on Serine 166 (Milkiewicz et al., 2011; Aiken et al., 2016). By expressing the phospho-mimetic form of MDM2 in primary endothelial cells, we showed that Ser166 phosphorylation restrained the expression of FoxO1 target genes known to promote angiostasis, such as Sprouty-2 and Thrombospondin-1 and stimulated endothelial migration (Aiken et al., 2016). In primary endothelial cells, we have found that FoxO1 directly binds to MDM2, an interaction that is increased by shear stress and serum stimulation and correlates with decreased levels of FoxO1 protein (Milkiewicz et al., 2011).
Together these studies support an important role of MDM2 in endothelial cells to maintain survival and to support endothelial responsiveness to pro-angiogenic stimuli.
The interest in studying the role of MDM2 in vascular smooth cells has emerged from the observation that TP53 inactivation stimulates atherosclerosis (Ihling et al., 1998; Mercer et al., 2005; Mercer and Bennett, 2006). Atherosclerosis is characterized by inflammation and excessive proliferation of VSMCs into the tunica intima. Hashimoto et al. (2011) have clearly demonstrated that disruption of MDM2-TP53 interaction restrained neointimal hyperplasia following vascular injury. Treatment with the small molecule MDM2 antagonist Nutlin-3 suppresses VSMC proliferation and migration. Nutlin-3 promoted cell cycle arrest in G1 in a TP53-dependent manner. In vivo Nutlin-3 treatment significantly reduced the intima area and the intimal/media area ratio after vascular injury compared to untreated animals. Nutlin-3 treatment also led to lower levels of inflammation in response to Tumor Necrosis Factor-α (TNF-α). This suggests that inhibition of MDM2 prevented inflammation by restoring the capacity of TP53 to inactivate the NF-κB (Hashimoto et al., 2011).
This pivotal work has been followed by other studies that confirmed that inhibition of MDM2-TP53 interaction can prevent the proliferation of VSMCs. Wu et al. (2014) have demonstrated that the Long intervening non-coding-RNA p21 (LincRNA-p21) and TP53 compete for binding to MDM2 in VSMCs. LincRNA-p21 binding to the RING domain of MDM2 facilitates TP53-p300 interaction, increasing TP53 transcriptional activity in VSMCs. LincRNA-p21 expression appeared to be significantly reduced in atherosclerotic plaque of ApoE–/– mice and in coronary arteries of patients with coronary artery disease. In vitro, silencing lincRNA-p21 using siRNAs significantly increased cell viability and proliferation. And in vivo, knockdown of lincRNA-p21 in ApoE–/– mice favored the interaction between MDM2 and TP53 and facilitated neointimal hyperplasia. Additionally, Li et al. (2015) have shown that p55γ, a regulatory subunit of PI3K, inhibited the ubiquitination of TP53 by MDM2 in smooth muscle cells. Overexpression of p55γ dramatically reduced the proliferation of VSMCs in culture, increased TP53 protein levels and significantly decreased TP53 ubiquitination. This demonstrates that p55γ activates and stabilizes TP53 by attenuating the capacity of MDM2 to bind to TP53. In vivo, delivery of p55γ by adenoviral infection reduced the production of neointima formation post artery injury (Li et al., 2015). Together these studies indicate that enhancement of MDM2-TP53 interaction supports vascular smooth muscle hypertrophy during atherosclerosis.
MDM2 is also suggested to play an important role in smooth muscle biology independently of TP53. In fact, MDM2 could mediate vascular calcification through a mechanism involving the ubiquitination of HDAC1 (Kwon et al., 2016). Vascular calcification, the deposition of calcium phosphate, is often associated with the development of atherosclerosis (Giachelli, 2003). Treatment of SMCs with Pi induces calcification. Kwon and colleagues showed that Pi treatment induced the proteasomal degradation of HDAC1, an event that preceded vascular calcification. Pi treatment also upregulated MDM2 and triggered the ubiquitination of HDAC1 by MDM2. Chemical inhibition of MDM2 prevented vascular calcification. The silencing of TP53 did not modulate vascular calcification in this study. This suggests that vascular calcification induced by Pi-treatment requires an MDM2/HDAC1 pathway.
Disturbances of the renin-angiotensin-aldosterone system are part of the physiopathology of atherosclerosis, supporting VSMC hypertrophy, proliferation and migration (Nehme and Zibara, 2017). Several pieces of evidence converge to suggest that crosstalk between MDM2 and elements of the renin-angiotensin-aldosterone system could support atherosclerosis by modulating smooth muscle hypertrophy. First, angiotensin II can regulate upstream kinases of MDM2, particularly PI3K and the ERK (Saward and Zahradka, 1997; Li and Malik, 2005; Cooper et al., 2007; Bunni et al., 2011). While the effect of angiotensin II on MDM2 activity in VSMCs has not been directly examined, it is plausible that phosphorylation of MDM2 by Akt or ERK promotes VSMC proliferation. Aldosterone stimulation of MR significantly increased the expression of MDM2 protein in VSMCs (Nakamura et al., 2006; Yan et al., 2018). In vivo, chronic treatment of rats with aldosterone increased MDM2 protein level as well as smooth muscle cell proliferation and collagen deposition in the aorta. The silencing of MDM2 significantly reduced smooth muscle cell proliferation. These effects were prevented by eplerenone, an inhibitor of MR. This supports the idea that MDM2 expression is upregulated by aldosterone, then contributing to VSMC hypertrophy (Yan et al., 2018).
The CaM pathway is another strong regulator of smooth muscle hypertrophy, mediating neointimal proliferation after vascular injury (Li and Malik, 2005; Bouallegue et al., 2009; Li et al., 2015). After carotid injury, Akt-dependent phosphorylation of MDM2 on residue serine 166 was significantly reduced in mice null for CaMKII-δ compared to wild-type mice (Li et al., 2011). Less binding between TP53 and MDM2 was observed in VSMC lacking the CaMKII-δ protein. Thus, CaMKII-δ appears to function as an upstream regulator of MDM2 activity in smooth muscle cells. Since CaMKII-δ-KO mice presented with less neointima thickening than wild-type mice, this incriminates CaMKII-δ in provoking VSMC proliferation and neointimal formation, potentially involving activation of the Akt/MDM2 pathway.
MDM2 plays an important role in maintaining cardiac function by controlling cellular processes and the stress response in cardiomyocytes.
Toth et al. (2006) demonstrated that cardiomyocytes overexpressing MDM2 acquired resistance to hypoxia/reoxygenation-induced apoptosis. Overexpression of MDM2 by adenoviral infection protected primary cardiomyocytes in culture from apoptosis following 12 h of hypoxia and following hypoxia/reoxygenation (12 h of hypoxia followed by 16 h of re-oxygenation). Chemical inhibition of MDM2 by PNC-28, a peptide that mimics the MDM2-binding domain of TP53, resulted in elevated TP53 levels and increased apoptosis under normoxia, hypoxia, and hypoxia/reoxygenation conditions. This suggests that MDM2 promotes cardiomyocyte survival in vitro after hypoxia/reoxygenation. Toth et al. (2006) also showed that MDM2 overexpression prevented hypertrophy of primary cardiomyocytes following endothelin-1 or phenylephrine treatment. These in vitro results are well aligned with the observations these authors made in vivo that the heart of MDM2 hypomorphic mice had cardiomyocytes with increased cross-sectional areas and an increased area of infarction after ischemia/reperfusion (Toth et al., 2006).
During oxidative stress, MDM2 might exert both pro-apoptotic and anti-apoptotic functions in cardiomyocytes. Pikkarainen et al. (2009) reported an increased MDM2 expression after oxidative stress. They found that knockdown of MDM2 with an antisense strategy resulted in an increased level of caspase 3, supporting the notion that MDM2 protects against oxidative stress in cardiomyocytes (Pikkarainen et al., 2009). Unexpectedly, Foo et al. (2007) colleagues demonstrated that MDM2 induced ARC protein degradation directly through its role as an E3 ubiquitin ligase. ARC is a 22 kDa anti-apoptotic protein that is found abundantly in cardiomyocytes and skeletal myocytes (Koseki et al., 1998). Overexpression of MDM2 led to a decrease in ARC levels. H2O2 significantly upregulated MDM2 protein levels and induced significant apoptotic (50–60%) cell death in cardiomyocytes. This was associated with a time-dependent decrease in ARC protein levels. This was due to an MDM2-dependent degradation of ARC. ARC expression was maintained in MDM2/TP53 double knockout cardiomyocytes, while the reintroduction of MDM2 restored ARC degradation, suggesting a TP53-independent function of MDM2. Foo et al. (2007) study supports a pro-apoptotic role for MDM2 in embryonic cardiomyocytes through its regulation of ARC. It is plausible that MDM2 regulates both TP53-dependent and independent pathways to determine the fate of cardiomyocytes after oxidative stress. Further investigations are required to fully elucidate the role of MDM2 in cardiomyocytes during oxidative stress.
MDM2 can regulate the β-AR signal in cardiomyocytes in a TP53-independent manner that involves G Protein-Coupled Receptor Kinase 2 (GRK2) and β-arrestin2 (Li et al., 2009; Jean-Charles et al., 2016, 2017). The β-arrestin and GRK protein families are multi-functional proteins well known for their roles in desensitizing β-AR signaling by blocking G protein coupling, ultimately controlling contractility. In cardiomyocytes, MDM2 can bind and ubiquitinate both GRK2 and β-arrestin2 (Li et al., 2009; Shenoy et al., 2009). While ubiquitination of GRK2 marks it for degradation by the proteasomes, ubiquitination of β-arrestin2 facilitates its binding to signaling kinases rather than promoting its degradation (Shenoy et al., 2007; Jean-Charles et al., 2016). Cardiomyocytes harboring a double knock-down for Mdm2 and Trp53 have an impairment of Ca2+ handling after stimulation with the β-agonist isoproterenol (Jean-Charles et al., 2017). The polyubiquitination of GRK2 was decreased in the hearts of mice harboring a cardiomyocyte-specific double knockdown of Mdm2 and Trp53, leading to an increase in GRK2 protein levels. These effects were associated with impaired contractility. Since the delivery of the Mdm2 gene rescued the cardiac contractility, it appears that MDM2 is required for proper signaling of the β-adrenergic receptor in cardiomyocytes (Jean-Charles et al., 2017). Therefore, in cardiomyocytes, proper control of MDM2 interaction with TP53 and other binding partners might be important to fine-tune survival, hypertrophy, and sensitivity to β-adrenergic signals.
Due to the role of MDM2 as a negative regulator of TP53, it is not surprising that in vitro data support a pro-survival role for MDM2 in endothelial cells, VSMCs and cardiomyocytes. However, MDM2 has functions that are independent of TP53 in these cells.
In smooth muscle cells, pro-atherogenic stimuli appear to upregulate MDM2. This upregulation of MDM2 might lead to unwanted proliferation and calcification through the inhibition of TP53 and HDAC1 activities. These actions of MDM2 would ultimately facilitate atherosclerosis. In endothelial cells, MDM2 is crucial to survival and migration, potentially determining the capacity of endothelial cells to initiate angiogenesis. Not only does MDM2 reduce TP53 function to support survival, but it also has a dichotomous action on VEGF-A signals. MDM2 supports VEGF-A expression and is also able to sense this proangiogenic signal to modulate endothelial cell gene expression. Finally, in cardiomyocytes, MDM2 is a crucial regulator of the stress response through mechanisms both dependent and independent of TP53. More importantly, MDM2 also controls hypertrophy and the sensitivity to β-adrenergic signals.
Considering the number of small MDM2 inhibitors that are currently under clinical trials, it is crucial to broaden our knowledge of the physiological functions of MDM2 in the cardiovascular system. Many upstream regulators and downstream effectors of MDM2 have cardiovascular functions. The phenotypes of MDM2 transgenic mouse models clearly highlights how important MDM2 is in the cardiovascular system. Studies performed on the three main cell types of the cardiovascular system, i.e., endothelial cells, smooth muscle cells and cardiomyocytes, further support a key role for MDM2. It is highly plausible that MDM2 acts as a central hub for the cardiovascular stress response. How this multitasking E3 ubiquitin ligase operates to coordinate this stress response in the different cells present in the cardiovascular system remains largely unclear. Further investigations are warranted to elucidate how MDM2 are differentially regulated in the cardiovascular system in the context of human health and disease. By delving more into the study of the cardiovascular functions of MDM2, researchers could open new avenues for the treatment and the prevention of cardiovascular diseases, even for the development of cancer treatments where cardiovascular toxicity is curtailed.
BL and ER wrote and edited the manuscript. ER proposed the concept of the review to present the most up-to-date evidence regarding the role of MDM2 in the cardiovascular system.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Birot and Dr. Haas for their fruitful discussion and feedback on the manuscript.
α MyHC, α-Myosin Heavy Chain; Akt, Protein Kinase B; ARC, apoptosis repressor with caspase recruitment domain; ATM, ataxia telangiectasia mutated kinase; β-AR, β-adrenergic receptors; CaM, Ca2+/calmodulin; CaMKII- δ, CaM-dependent protein kinase type II delta chain- δ; E2F1, E2 promoter binding Factor 1; ERK, extracellular signal-regulated kinase; ESRR- β / γ, estrogen related receptor β / γ; FoxO, forkhead box O protein; GRK2, G protein-couple receptor kinase 2; GSK3, Glycogen synthase Kinase 3; HDAC1, histone deacetylase 1; HIF1- α, hypoxia-inducible factor 1- α; HUVEC, human umbilical vein endothelial cells; IGF1, Insulin-like growth factor 1; IGFR1, Insulin-like growth factor receptor 1; IRS1, Insulin-receptor substrate 1; KK-Ay mice, crossbred of diabetic KK and lethal yellow mice carrying a heterozygous mutation of the agouti gene; LDLr, low lipoprotein receptor; lincRNA-p21, Long intergenic non-coding RNA-p21; MAPK, Mitogen-activated protein kinases; MDM2, Murine double minute 2; MR, mineralocorticoid receptors (MR); NF- κ B, Nuclear Factor κ B; NICD1, intracellular domain of Notch1; NOTCH, neurogenic locus notch homolog protein; p55 γ, PI3k regulatory subunit p55 γ; Pdgf β-Cre -ER, tamoxifen-inducible Cre recombinase under the control of the murine platelet derived growth factor receptor β polypeptide promoter/enhancer regions; Pi, inorganic phosphate; PI3K, Phosphotidylinositol 3-Kinase; PGC1- α, Peroxisome proliferator-activated receptor gamma coactivator 1- α; PIGF, placenta growth factor; PPAR- α / γ, peroxisome proliferator-activated receptor α / γ; PTMs, Post-translational modifications; ROS, reactive oxygen species; RSK, Ribosomal S6 Kinase; SM22 α, Smooth Muscle Protein 22- α; Tip60, Histone acetyltransferase KAT5; TP53, Tumor protein p53; VEGF-A, vascular endothelial growth factor A; VSMC, vascular smooth muscle cell.
Abeyrathna, P., and Su, Y. (2015). The critical role of Akt in cardiovascular function. Vasc. Pharmacol. 74, 38–48. doi: 10.1016/j.vph.2015.05.008
Aiken, J., Roudier, E., Ciccone, J., Drouin, G., Stromberg, A., Vojnovic, J., et al. (2016). Phosphorylation of murine double minute-2 on Ser166 is downstream of VEGF-A in exercised skeletal muscle and regulates primary endothelial cell migration and FoxO gene expression. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 30, 1120–1134. doi: 10.1096/fj.15-276964
Alam, H., Weck, J., Maizels, E., Park, Y., Lee, E. J., Ashcroft, M., et al. (2009). Role of the Phosphatidylinositol-3-Kinase and extracellular regulated kinase pathways in the induction of hypoxia-inducible factor (HIF)-1 activity and the HIF-1 target vascular endothelial growth factor in ovarian granulosa cells in response to follicle-stimulating hormone. Endocrinology 150, 915–928. doi: 10.1210/en.2008-0850
Balaji, V., Pokrzywa, W., and Hoppe, T. (2018). Ubiquitylation pathways in insulin signaling and organismal homeostasis. BioEssays 40:1700223. doi: 10.1002/bies.201700223
Bárdos, J. I., Chau, N.-M., and Ashcroft, M. (2004). Growth factor-mediated induction of HDM2 positively regulates hypoxia-inducible factor 1alpha expression. Mol. Cell Biol. 24, 2905–2914. doi: 10.1128/mcb.24.7.2905-2914.2004
Bartunek, J., Vanderheyden, M., Knaapen, M. W. N., Tack, W., Kockx, M. M., and Goethals, M. (2002). Deoxyribonucleic acid damage/repair proteins are elevated in the failing human myocardium due to idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 40, 1097–103; discussion 1104–1105.
Battiprolu, P. K., Hojayev, B., Jiang, N., Wang, Z. V., Luo, X., Iglewski, M., et al. (2012). Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Invest. 122, 1109–1118. doi: 10.1172/JCI60329
Bieging, K. T., Mello, S. S., and Attardi, L. D. (2014). Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370. doi: 10.1038/nrc3711
Blanco, R., and Gerhardt, H. (2013). VEGF and notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 3:a006569. doi: 10.1101/cshperspect.a006569
Boesten, L. S. M., Zadelaar, S. M., Clercq, S. D., Francoz, S., van Nieuwkoop, A., Biessen, E. A. L., et al. (2006). Mdm2, but not Mdm4, protects terminally differentiated smooth muscle cells from p53-mediated caspase-3-independent cell death. Cell Death Differ. 13, 2089–2098. doi: 10.1038/sj.cdd.4401973
Bohlman, S., and Manfredi, J. J. (2014). p53-independent effects of Mdm2. Subcell Biochem. 85, 235–246. doi: 10.1007/978-94-017-9211-0_13
Bohlman, S., and Manfredi, J. J. (2016). Mdm2-RNA interactions as a target for cancer therapy: it’s not all about p53. Cancer Cell 30, 513–514. doi: 10.1016/j.ccell.2016.09.017
Bouallegue, A., Pandey, N. R., and Srivastava, A. K. (2009). CaMKII knockdown attenuates H2O2-induced phosphorylation of ERK1/2, PKB/Akt, and IGF-1R in vascular smooth muscle cells. Free Radic. Biol. Med. 47, 858–866. doi: 10.1016/j.freeradbiomed.2009.06.022
Brenkman, A. B., de Keizer, P. L. J., Broek, N. J. F., van den, Jochemsen, A. G., Boudewijn, M., et al. (2008). Mdm2 induces mono-ubiquitination of FOXO4. PLoS One 3:e2819. doi: 10.1371/journal.pone.0002819
Brockhaus, K., Böhm, M. R. R., Melkonyan, H., and Thanos, S. (2018). Age-related beta-synuclein alters the p53/Mdm2 pathway and induces the apoptosis of brain microvascular endothelial cells in vitro. Cell Transplant. 27, 796–813. doi: 10.1177/0963689718755706
Bunni, M. A., Kramarenko, I. I., Walker, L., Raymond, J. R., and Garnovskaya, M. N. (2011). Role of integrins in angiotensin II-induced proliferation of vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 300, C647–C656. doi: 10.1152/ajpcell.00179.2010
Burgess, A., Chia, K. M., Haupt, S., Thomas, D., Haupt, Y., and Lim, E. (2016). Clinical overview of MDM2/X-targeted therapies. Front. Oncol. 6:7. doi: 10.3389/fonc.2016.00007/full
Cannavo, A., Liccardo, D., and Koch, W. J. (2013). Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front. Physiol. 4:264. doi: 10.3389/fphys.2013.00264
Chistiakov, D. A., Orekhov, A. N., and Bobryshev, Y. V. (2017). The impact of FOXO-1 to cardiac pathology in diabetes mellitus and diabetes-related metabolic abnormalities. Int. J. Cardiol. 245, 236–244. doi: 10.1016/j.ijcard.2017.07.096
Claesson-Welsh, L. (2016). VEGF receptor signal transduction - A brief update. Vasc. Pharmacol. 86, 14–17. doi: 10.1016/j.vph.2016.05.011
Cooper, S. A., Whaley-Connell, A., Habibi, J., Wei, Y., Lastra, G., Manrique, C., et al. (2007). Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 293, H2009–H2023.
Dai, F., Chen, Y., Song, Y., Huang, L., Zhai, D., Dong, Y., et al. (2012). A natural small molecule harmine inhibits angiogenesis and suppresses tumour growth through activation of p53 in endothelial cells. PLoS One 7:e52162. doi: 10.1371/journal.pone.0052162
Dameron, K. M., Volpert, O. V., Tainsky, M. A., and Bouck, N. (1994). Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 265, 1582–1584. doi: 10.1126/science.7521539
Daniel, L. L., Daniels, C. R., Harirforoosh, S., Foster, C. R., Singh, M., and Singh, K. (2014). Deficiency of ataxia telangiectasia mutated kinase delays inflammatory response in the heart following myocardial infarction. J. Am. Heart Assoc. 3:e001286. doi: 10.1161/JAHA.114.001286
Daniel, L. L., Scofield, S. L. C., Thrasher, P., Dalal, S., Daniels, C. R., Foster, C. R., et al. (2016). Ataxia telangiectasia-mutated kinase deficiency exacerbates left ventricular dysfunction and remodeling late after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 311, H445–H452. doi: 10.1152/ajpheart.00338.2016
de Toledo, S. M., Azzam, E. I., Dahlberg, W. K., Gooding, T. B., and Little, J. B. (2000). ATM complexes with HDM2 and promotes its rapid phosphorylation in a p53-independent manner in normal and tumor human cells exposed to ionizing radiation. Oncogene 19, 6185–6193. doi: 10.1038/sj.onc.1204020
Dohmesen, C., Koeppel, M., and Dobbelstein, M. (2008). Specific inhibition of Mdm2-mediated neddylation by Tip60. Cell Cycle 15, 222–231. doi: 10.4161/cc.7.2.5185
Espach, Y., Lochner, A., Strijdom, H., and Huisamen, B. (2015). ATM protein kinase signaling, type 2 diabetes and cardiovascular disease. Cardiovasc. Drugs Ther. 29, 51–58. doi: 10.1007/s10557-015-6571-z
Fåhraeus, R., and Olivares-Illana, V. (2014). MDM2’s social network. Oncogene 33, 4365–4376. doi: 10.1038/onc.2013.410
Feng, J., Tamaskovic, R., Yang, Z., Brazil, D. P., Merlo, A., Hess, D., et al. (2004). Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 279, 35510–35517. doi: 10.1074/jbc.m404936200
Fisher, J. B., Horst, A., Wan, T., Kim, M.-S., Auchampach, J., and Lough, J. (2016). Depletion of Tip60 from in vivo cardiomyocytes increases myocyte density, followed by cardiac dysfunction, myocyte fallout and lethality. PLoS One 11:e0164855. doi: 10.1371/journal.pone.0164855
Fisher, J. B., Kim, M.-S., Blinka, S., Ge, Z.-D., Wan, T., Duris, C., et al. (2012). Stress-induced cell-cycle activation in Tip60 haploinsufficient adult cardiomyocytes. PLoS One 7:e31569. doi: 10.1371/journal.pone.0031569
Foo, R. S.-Y., Chan, L. K. W., Kitsis, R. N., and Bennett, M. R. (2007). Ubiquitination and degradation of the anti-apoptotic protein ARC by MDM2. J. Biol. Chem. 282, 5529–5535. doi: 10.1074/jbc.m609046200
Foster, C. R., Singh, M., Subramanian, V., and Singh, K. (2011). Ataxia telangiectasia mutated kinase plays a protective role in β-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Mol. Cell Biochem. 353, 13–22. doi: 10.1007/s11010-011-0769-6
Fu, W., Ma, Q., Chen, L., Li, P., Zhang, M., Ramamoorthy, S., et al. (2009). MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J. Biol. Chem. 284, 13987–14000. doi: 10.1074/jbc.M901758200
Gannon, H. S., Woda, B. A., and Jones, S. N. (2012). ATM phosphorylation of Mdm2 Ser394 regulates the amplitude and duration of the DNA damage response in mice. Cancer Cell 21, 668–679. doi: 10.1016/j.ccr.2012.04.011
Giachelli, C. M. (2003). Vascular calcification: in vitro evidence for the role of inorganic phosphate. J. Am. Soc. Nephrol. JASN 14(9 Suppl. 4), S300–S304.
Girnita, L., Shenoy, S. K., Sehat, B., Vasilcanu, R., Vasilcanu, D., Girnita, A., et al. (2007). Beta-arrestin and Mdm2 mediate IGF-1 receptor-stimulated ERK activation and cell cycle progression. J. Biol. Chem. 282, 11329–11338. doi: 10.1074/jbc.m611526200
Grier, J. D., Xiong, S., Elizondo-Fraire, A. C., Parant, J. M., and Lozano, G. (2006). Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol. Cell Biol. 26, 192–198. doi: 10.1128/mcb.26.1.192-198.2006
Hara, M. R., Kovacs, J. J., Whalen, E. J., Rajagopal, S., Strachan, R. T., Grant, W., et al. (2011). A stress response pathway regulates DNA damage through β2-adrenoreceptors and β-arrestin-1. Nature 477, 349–353. doi: 10.1038/nature10368
Hara, M. R., Sachs, B. D., Caron, M. G., and Lefkowitz, R. J. (2013). Pharmacological blockade of a β(2)AR-β-arrestin-1 signaling cascade prevents the accumulation of DNA damage in a behavioral stress model. Cell Cycle Georget Tex. 12, 219–224. doi: 10.4161/cc.23368
Hashimoto, T., Ichiki, T., Ikeda, J., Narabayashi, E., Matsuura, H., Miyazaki, R., et al. (2011). Inhibition of MDM2 attenuates neointimal hyperplasia via suppression of vascular proliferation and inflammation. Cardiovasc. Res. 91, 711–719. doi: 10.1093/cvr/cvr108
Hauck, L., Stanley-Hasnain, S., Fung, A., Grothe, D., Rao, V., Mak, T. W., et al. (2017). Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS One 12:e0189861. doi: 10.1371/journal.pone.0189861
Higashi, Y., Gautam, S., Delafontaine, P., and Sukhanov, S. (2019). IGF-1 and cardiovascular disease. Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc. 45, 6–16. doi: 10.1016/j.ghir.2019.01.002
Hogan, C., Hutchison, C., Marcar, L., Milne, D., Saville, M., Goodlad, J., et al. (2008). Elevated levels of oncogenic protein kinase Pim-1 induce the p53 pathway in cultured cells and correlate with increased Mdm2 in mantle cell lymphoma. J. Biol. Chem. 283, 18012–18023. doi: 10.1074/jbc.M709695200
van der Horst, A., de Vries-Smits, A. M. M., Brenkman, A. B., van Triest, M. H., van den Broek, N., Colland, F., et al. (2006). FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 8, 1064–1073. doi: 10.1038/ncb1469
Hu, Y., Fisher, J. B., Koprowski, S., McAllister, D., Kim, M.-S., and Lough, J. (2009). Homozygous disruption of the Tip60 gene causes early embryonic lethality. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 238, 2912–2921. doi: 10.1002/dvdy.22110
Ihling, C., Haendeler, J., Menzel, G., Hess, R. D., Fraedrich, G., Schaefer, H. E., et al. (1998). Co-expression of p53 and MDM2 in human atherosclerosis: implications for the regulation of cellularity of atherosclerotic lesions. J. Pathol. 185, 303–312. doi: 10.1002/(sici)1096-9896(199807)185:3<303::aid-path106>3.3.co;2-g
Jabs, M., Rose, A. J., Lehmann, L. H., Taylor, J., Moll, I., Sijmonsma, T. P., et al. (2018). Inhibition of endothelial notch signaling impairs fatty acid transport and leads to metabolic and vascular remodeling of the adult heart. Circulation 137, 2592–2608. doi: 10.1161/CIRCULATIONAHA.117.029733
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jain, D., Russell, R. R., Schwartz, R. G., Panjrath, G. S., and Aronow, W. (2017). Cardiac complications of cancer therapy: pathophysiology, identification, prevention, treatment, and future directions. Curr. Cardiol. Rep 19:36. doi: 10.1007/s11886-017-0846-x
Jean-Charles, P.-Y., Rajiv, V., and Shenoy, S. K. (2016). Ubiquitin-related roles of β-Arrestins in endocytic trafficking and signal transduction. J. Cell Physiol. 231, 2071–2080. doi: 10.1002/jcp.25317
Jean-Charles, P.-Y., Yu, S. M.-W., Abraham, D., Kommaddi, R. P., Mao, L., Strachan, R. T., et al. (2017). Mdm2 regulates cardiac contractility by inhibiting GRK2-mediated desensitization of β-adrenergic receptor signaling. JCI Insight 2:95998. doi: 10.1172/jci.insight.95998
Jia, L., Zhang, W., Ma, Y., Chen, B., Liu, Y., Piao, C., et al. (2017). Haplodeficiency of ataxia telangiectasia mutated accelerates heart failure after myocardial infarction. J. Am. Heart Assoc. 6:e006349. doi: 10.1161/JAHA.117.006349
Jones, S. N., Hancock, A. R., Vogel, H., Donehower, L. A., and Bradley, A. (1998). Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 95, 15608–15612. doi: 10.1073/pnas.95.26.15608
Jones, S. N., Roe, A. E., Donehower, L. A., and Bradley, A. (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208. doi: 10.1038/378206a0
Kinoshita, D., Nagasawa, A., Shimizu, I., Ito, T. K., Yoshida, Y., Tsuchida, M., et al. (2017). Progerin impairs vascular smooth muscle cell growth via the DNA damage response pathway. Oncotarget 23, 34045–34056. doi: 10.18632/oncotarget.15973
Koseki, T., Inohara, N., Chen, S., and Núñez, G. (1998). ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proc. Natl. Acad. Sci. U.S.A. 95, 5156–5160. doi: 10.1073/pnas.95.9.5156
Kubbutat, M. H., Jones, S. N., and Vousden, K. H. (1997). Regulation of p53 stability by Mdm2. Nature 387, 299–303. doi: 10.1038/387299a0
Kulikov, R., Boehme, K. A., and Blattner, C. (2005). Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol. Cell Biol. 25, 7170–7180. doi: 10.1128/mcb.25.16.7170-7180.2005
Kwon, D.-H., Eom, G. H., Ko, J. H., Shin, S., Joung, H., Choe, N., et al. (2016). MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 7:10492. doi: 10.1038/ncomms10492
Leach, F. S., Tokino, T., Meltzer, P., Burrell, M., Oliner, J. D., Smith, S., et al. (1993). p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res. 53(10 Suppl.), 2231–2234.
Lee, S., Chen, T. T., Barber, C. L., Jordan, M. C., Murdock, J., Desai, S., et al. (2007). Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703. doi: 10.1016/j.cell.2007.06.054
Legube, G., Linares, L. K., Lemercier, C., Scheffner, M., Khochbin, S., and Trouche, D. (2002). Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. EMBO J. 21, 1704–1712. doi: 10.1093/emboj/21.7.1704
Leri, A., Liu, Y., Claudio, P. P., Kajstura, J., Wang, X., Wang, S., et al. (1999a). Insulin-Like growth factor-1 induces Mdm2 and down-regulates p53, attenuating the myocyte renin-angiotensin system and stretch-mediated apoptosis. Am. J. Pathol. 154, 567–580. doi: 10.1016/s0002-9440(10)65302-3
Leri, A., Liu, Y., Wang, X., Kajstura, J., Malhotra, A., Meggs, L. G., et al. (1999b). Overexpression of insulin-like growth factor-1 attenuates the myocyte renin-angiotensin system in transgenic mice. Circ. Res. 84, 752–762. doi: 10.1161/01.res.84.7.752
Levine, A. J., and Oren, M. (2009). The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758. doi: 10.1038/nrc2723
Li, F., and Malik, K. U. (2005). Angiotensin II-induced Akt activation is mediated by metabolites of arachidonic acid generated by CaMKII-stimulated Ca2+-dependent phospholipase A2. Am. J. Physiol. Heart Circ. Physiol. 288, H2306–H2316.
Li, G., Xie, N., Yao, Y., Zhang, Y., Guo, J., Feng, Y., et al. (2015). Identification of PI3K regulatory subunit p55γ as a novel inhibitor of vascular smooth muscle cell proliferation and neointimal formation. Cardiovasc. Res. 105, 75–85. doi: 10.1093/cvr/cvu235
Li, Q., and Lozano, G. (2013). Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 19, 34–41. doi: 10.1158/1078-0432.ccr-12-0053
Li, W., Li, H., Sanders, P. N., Mohler, P. J., Backs, J., Olson, E. N., et al. (2011). The multifunctional Ca2+/Calmodulin-dependent Kinase II δ (CaMKIIδ) controls neointima formation after carotid ligation and vascular smooth muscle cell proliferation through cell cycle regulation by p21. J. Biol. Chem. 286, 7990–7999. doi: 10.1074/jbc.M110.163006
Li, X., Baillie, G. S., and Houslay, M. D. (2009). Mdm2 directs the ubiquitination of β-Arrestin-sequestered cAMP Phosphodiesterase-4D5. J. Biol. Chem. 284, 16170–16182. doi: 10.1074/jbc.M109.008078
Lin, S.-Y., Li, T. Y., Liu, Q., Zhang, C., Li, X., Chen, Y., et al. (2012). GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336, 477–481. doi: 10.1126/science.1217032
Lough, J. W. (2002). Transient expression of TIP60 protein during early chick heart development. Dev. Dyn. 223, 419–425. doi: 10.1002/dvdy.10058
Loughran, Ö., and Thangue, N. B. L. (2000). Apoptotic and growth-promoting activity of E2F modulated by MDM2. Mol. Cell Biol. 20, 2186–2197. doi: 10.1128/mcb.20.6.2186-2197.2000
Mannell, H., Hammitzsch, A., Mettler, R., Pohl, U., and Krötz, F. (2010). Suppression of DNA-PKcs enhances FGF-2 dependent human endothelial cell proliferation via negative regulation of Akt. Cell Signal. 22, 88–96. doi: 10.1016/j.cellsig.2009.09.015
Marine, J.-C., and Lozano, G. (2009). Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 17, 93–102. doi: 10.1038/cdd.2009.68
Martin, K., Trouche, D., Hagemeier, C., Sorensen, T. S., Thangue, N. B. L., and Kouzarides, T. (2014). Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature 375, 691–694. doi: 10.1038/375691a0
Mayo, L. D., and Donner, D. B. (2001). A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. U.S.A. 98, 11598–11603. doi: 10.1073/pnas.181181198
Medunjanin, S., Daniel, J.-M., Weinert, S., Dutzmann, J., Burgbacher, F., Brecht, S., et al. (2015). DNA-dependent protein kinase (DNA-PK) permits vascular smooth muscle cell proliferation through phosphorylation of the orphan nuclear receptor NOR1. Cardiovasc. Res. 106, 488–497. doi: 10.1093/cvr/cvv126
Meek, D. W., and Anderson, C. W. (2009). Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1:a000950. doi: 10.1101/cshperspect.a000950
Meek, D. W., and Hupp, T. R. (2010). The regulation of MDM2 by multisite phosphorylation–opportunities for molecular-based intervention to target tumours? Semin. Cancer Biol. 20, 19–28. doi: 10.1016/j.semcancer.2009.10.005
Mendrysa, S. M., McElwee, M. K., Michalowski, J., O’Leary, K. A., Young, K. M., and Perry, M. E. (2003). mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol. Cell Biol. 23, 462–472. doi: 10.1128/mcb.23.2.462-473.2003
Mercer, J., and Bennett, M. (2006). The role of p53 in atherosclerosis. Cell Cycle Georget Tex. 5, 1907–1909. doi: 10.4161/cc.5.17.3166
Mercer, J., Figg, N., Stoneman, V., Braganza, D., and Bennett, M. R. (2005). Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ. Res. 96, 667–674. doi: 10.1161/01.res.0000161069.15577.ca
Mercer, J. R., Gray, K., Figg, N., Kumar, S., and Bennett, M. R. (2012). The methyl xanthine caffeine inhibits DNA damage signaling and reactive species and reduces atherosclerosis in ApoE(-/-) mice. Arterioscler. Thromb. Vasc. Biol. 32, 2461–2467. doi: 10.1161/ATVBAHA.112.251322
Milkiewicz, M., Roudier, E., Doyle, J. L., Trifonova, A., Birot, O., and Haas, T. L. (2011). Identification of a mechanism underlying regulation of the anti-angiogenic forkhead transcription factor FoxO1 in cultured endothelial cells and ischemic muscle. Am. J. Pathol. 178, 935–944. doi: 10.1016/j.ajpath.2010.10.042
Montes de Oca Luna, R., Wagner, D. S., and Lozano, G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206. doi: 10.1038/378203a0
Morimoto, Y., Bando, Y. K., Shigeta, T., Monji, A., and Murohara, T. (2011). Atorvastatin prevents ischemic limb loss in type 2 diabetes: role of p53. J. Atheroscler. Thromb. 18, 200–208. doi: 10.5551/jat.6437
Morisco, C., Zebrowski, D., Condorelli, G., Tsichlis, P., Vatner, S. F., and Sadoshima, J. (2000). The Akt-glycogen synthase kinase 3beta pathway regulates transcription of atrial natriuretic factor induced by beta-adrenergic receptor stimulation in cardiac myocytes. J. Biol. Chem. 275, 14466–14475. doi: 10.1074/jbc.275.19.14466
Muslin, A. J. (2008). MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin. Sci. Lond. Engl. 115, 203–218. doi: 10.1042/CS20070430
Nag, S., Qin, J., Srivenugopal, K. S., Wang, M., and Zhang, R. (2013). The MDM2-p53 pathway revisited. J. Biomed. Res. 27, 254–271. doi: 10.7555/JBR.27.20130030
Nakamura, Y., Suzuki, S., Suzuki, T., Ono, K., Miura, I., Satoh, F., et al. (2006). MDM2: a novel mineralocorticoid-responsive gene involved in aldosterone-induced human vascular structural remodeling. Am. J. Pathol. 169, 362–371. doi: 10.2353/ajpath.2006.051351
Naski, N., Gajjar, M., Bourougaa, K., Malbert-Colas, L., Fåhraeus, R., and Candeias, M. M. (2009). The p53 mRNA-Mdm2 interaction. Cell Cycle Georget Tex. 8, 31–34.
Nehme, A., and Zibara, K. (2017). Cellular distribution and interaction between extended renin-angiotensin-aldosterone system pathways in atheroma. Atherosclerosis 263, 334–342. doi: 10.1016/j.atherosclerosis.2017.05.029
Ogawara, Y., Kishishita, S., Obata, T., Isazawa, Y., Suzuki, T., Tanaka, K., et al. (2002). Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 277, 21843–21850. doi: 10.1074/jbc.m109745200
Okuno, Y., Nakamura-Ishizu, A., Otsu, K., Suda, T., and Kubota, Y. (2012). Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat. Med. 18, 1208–1216. doi: 10.1038/nm.2846
Park, J. H., Lee, J. Y., Shin, D. H., Jang, K. S., Kim, H. J., and Kong, G. (2011). Loss of Mel-18 induces tumor angiogenesis through enhancing the activity and expression of HIF-1α mediated by the PTEN/PI3K/Akt pathway. Oncogene 30, 4578–4589. doi: 10.1038/onc.2011.174
Patterson, D., Gao, D., Trahan, D., Johnson, B., Ludwig, A., Barbieri, E., et al. (2011). Effect of MDM2 and vascular endothelial growth factor inhibition on tumor angiogenesis and metastasis in neuroblastoma. Angiogenesis 14, 255–266. doi: 10.1007/s10456-011-9210-8
Pettersson, S., Sczaniecka, M., McLaren, L., Russell, F., Gladstone, K., Hupp, T., et al. (2013). Non-degradative ubiquitination of the Notch1 receptor by the E3 ligase MDM2 activates the Notch signalling pathway. Biochem. J. 450, 523–536. doi: 10.1042/BJ20121249
Pikkarainen, S., Kennedy, R. A., Marshall, A. K., Tham, E. L., Lay, K., Kriz, T. A., et al. (2009). Regulation of expression of the rat orthologue of mouse double minute 2 (MDM2) by H2O2-induced oxidative stress in neonatal rat cardiac myocytes. J. Biol. Chem. 284, 27195–27210. doi: 10.1074/jbc.m109.037887
Potz, B. A., Sabe, A. A., Elmadhun, N. Y., Clements, R. T., Robich, M. P., Sodha, N. R., et al. (2016). Glycogen synthase kinase 3β inhibition improves myocardial angiogenesis and perfusion in a swine model of metabolic syndrome. J. Am. Heart Assoc. 5:e003694.
Puthanveetil, P., Wan, A., and Rodrigues, B. (2013). FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 97, 393–403. doi: 10.1093/cvr/cvs426
Qi, Y., Zhu, Q., Zhang, K., Thomas, C., Wu, Y., Kumar, R., et al. (2015). Activation of Foxo1 by insulin resistance promotes cardiac dysfunction and β–myosin heavy chain gene expression clinical perspective. Circ. Heart Fail. 8, 198–208. doi: 10.1161/circheartfailure.114.001457
Robertson, E. D., Semenchenko, K., and Wasylyk, B. (2014). Crosstalk between Mdm2, p53 and HIF1-α: distinct responses to oxygen stress and implications for tumour hypoxia. Subcell Biochem. 85, 199–214. doi: 10.1007/978-94-017-9211-0_11
Roudier, E., Forn, P., Perry, M. E., and Birot, O. (2012). Murine double minute-2 expression is required for capillary maintenance and exercise-induced angiogenesis in skeletal muscle. FASEB J. 26, 4530–4539. doi: 10.1096/fj.12-212720
Roudier, E., Milkiewicz, M., Birot, O., Slopack, D., Montelius, A., Gustafsson, T., et al. (2013). Endothelial FoxO1 is an intrinsic regulator of thrombospondin 1 expression that restrains angiogenesis in ischemic muscle. Angiogenesis 16, 759–772. doi: 10.1007/s10456-013-9353-x
Salih, D. A. M., and Brunet, A. (2008). FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 20, 126–136. doi: 10.1016/j.ceb.2008.02.005
Sano, M., Minamino, T., Toko, H., Miyauchi, H., Orimo, M., Qin, Y., et al. (2007). p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446, 444–448. doi: 10.1038/nature05602
Saward, L., and Zahradka, P. (1997). Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ. Res. 81, 249–257. doi: 10.1161/01.res.81.2.249
Schaal, C., Pillai, S., and Chellappan, S. P. (2014). The Rb-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv. Cancer Res. 121, 147–182. doi: 10.1016/B978-0-12-800249-0.00004-4
Secchiero, P., Corallini, F., Gonelli, A., Dell’Eva, R., Vitale, M., Capitani, S., et al. (2007). Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ. Res. 100, 61–69. doi: 10.1161/01.res.0000253975.76198.ff
Sengupta, A., Molkentin, J. D., Paik, J.-H., DePinho, R. A., and Yutzey, K. E. (2011). FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 286, 7468–7478. doi: 10.1074/jbc.M110.179242
Shenoy, S. K., Barak, L. S., Xiao, K., Ahn, S., Berthouze, M., Shukla, A. K., et al. (2007). Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J. Biol. Chem. 282, 29549–29562. doi: 10.1074/jbc.m700852200
Shenoy, S. K., Modi, A. S., Shukla, A. K., Xiao, K., Berthouze, M., Ahn, S., et al. (2009). β-Arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc. Natl. Acad. Sci. U.S.A. 106, 6650–6655. doi: 10.1073/pnas.0901083106
Shimizu, I., Yoshida, Y., Suda, M., and Minamino, T. (2014). DNA damage response and metabolic disease. Cell Metab. 20, 967–977. doi: 10.1016/j.cmet.2014.10.008
Si, S., Nakajima-Takagi, Y., Iga, T., Tsuji, M., Hou, L., Oshima, M., et al. (2018). Hematopoietic insults damage bone marrow niche by activating p53 in vascular endothelial cells. Exp. Hematol. 63, 41.e–51.e. doi: 10.1016/j.exphem.2018.04.006
Skinner, H. D., Zheng, J. Z., Fang, J., Agani, F., and Jiang, B.-H. (2004). Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J. Biol. Chem. 279, 45643–45651. doi: 10.1074/jbc.m404097200
Song, H., Yin, D., and Liu, Z. (2012). GDF-15 promotes angiogenesis through modulating p53/HIF-1α signaling pathway in hypoxic human umbilical vein endothelial cells. Mol. Biol. Rep. 39, 4017–4022. doi: 10.1007/s11033-011-1182-7
Stanley-Hasnain, S., Hauck, L., Grothe, D., Aschar-Sobbi, R., Beca, S., Butany, J., et al. (2017). p53 and Mdm2 act synergistically to maintain cardiac homeostasis and mediate cardiomyocyte cell cycle arrest through a network of microRNAs. Cell Cycle 16, 1585–1600. doi: 10.1080/15384101.2017.1346758
Stiles, G. L., Caron, M. G., and Lefkowitz, R. J. (1984). Beta-adrenergic receptors: biochemical mechanisms of physiological regulation. Physiol. Rev. 64, 661–743. doi: 10.1152/physrev.1984.64.2.661
Takahashi-Yanaga, F. (2018). Roles of glycogen synthase kinase-3 (GSK-3) in cardiac development and heart disease. J. UOEH. 40, 147–156. doi: 10.7888/juoeh.40.147
Tang, Y., Luo, J., Zhang, W., and Gu, W. (2006). Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell. 24, 827–839. doi: 10.1016/j.molcel.2006.11.021
Toko, H., Takahashi, H., Kayama, Y., Oka, T., Minamino, T., Okada, S., et al. (2010). Ca2+/Calmodulin-dependent kinase iiδ causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation 122, 891–899. doi: 10.1161/circulationaha.109.935296
Toledo, F., and Wahl, G. M. (2006). Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 6, 909–923. doi: 10.1038/nrc2012
Toth, A., Nickson, P., Qin, L. L., and Erhardt, P. (2006). Differential regulation of cardiomyocyte survival and hypertrophy by MDM2, an E3 ubiquitin ligase. J. Biol. Chem. 281, 3679–3689. doi: 10.1074/jbc.m509630200
Tsuchiya, K., Tanaka, J., Shuiqing, Y., Welch, C. L., DePinho, R. A., Tabas, I., et al. (2012). FoxOs integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell Metab. 15, 372–381. doi: 10.1016/j.cmet.2012.01.018
Uryga, A., Gray, K., and Bennett, M. (2016). DNA damage and repair in vascular disease. Annu. Rev. Physiol. 78, 45–66. doi: 10.1146/annurev-physiol-021115-105127
Usui, I., Imamura, T., Huang, J., Satoh, H., Shenoy, S. K., Lefkowitz, R. J., et al. (2004). beta-arrestin-1 competitively inhibits insulin-induced ubiquitination and degradation of insulin receptor substrate 1. Mol. Cell Biol. 24, 8929–8937. doi: 10.1128/mcb.24.20.8929-8937.2004
Vidal, M., Wieland, T., and Lohse, M. J. (2012). Lorenz K. β-Adrenergic receptor stimulation causes cardiac hypertrophy via a Gβγ/Erk-dependent pathway. Cardiovasc. Res. 96, 255–264. doi: 10.1093/cvr/cvs249
Vousden, K. H., and Prives, C. (2009). Blinded by the light: the growing complexity of p53. Cell 137, 413–431. doi: 10.1016/j.cell.2009.04.037
Wang, P., Gao, H., Ni, Y., Wang, B., Wu, Y., Ji, L., et al. (2003). Beta-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J. Biol. Chem. 278, 6363–6370. doi: 10.1074/jbc.m210350200
Weeks, K. L., Bernardo, B. C., Ooi, J. Y. Y., Patterson, N. L., and McMullen, J. R. (2017). The IGF1-PI3K-Akt signaling pathway in mediating exercise-induced cardiac hypertrophy and protection. Adv. Exp. Med. Biol. 1000, 187–210. doi: 10.1007/978-981-10-4304-8_12
Worrall, C., Suleymanova, N., Crudden, C., Trocoli Drakensjö, I., Candrea, E., Nedelcu, D., et al. (2017). Unbalancing p53/Mdm2/IGF-1R axis by Mdm2 activation restrains the IGF-1-dependent invasive phenotype of skin melanoma. Oncogene 36, 3274–3286. doi: 10.1038/onc.2016.472
Wu, M., Zhou, J., Cheng, M., Boriboun, C., Biyashev, D., Wang, H., et al. (2014). E2F1 suppresses cardiac neovascularization by down-regulating VEGF and PlGF expression. Cardiovasc. Res. 104, 412–422. doi: 10.1093/cvr/cvu222
Yan, Y., Wang, C., Lu, Y., Gong, H., Wu, Z., Ma, X., et al. (2018). Mineralocorticoid receptor antagonism protects the aorta from vascular smooth muscle cell proliferation and collagen deposition in a rat model of adrenal aldosterone-producing adenoma. J. Physiol. Biochem. 74, 17–24. doi: 10.1007/s13105-017-0600-2
Yang, J.-Y., Zong, C. S., Xia, W., Yamaguchi, H., Ding, Q., Xie, X., et al. (2008). ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 10, 138–148. doi: 10.1038/ncb1676
Yang, W., Dolloff, N. G., and El-Deiry, W. S. (2008). ERK and MDM2 prey on FOXO3a. Nat. Cell Biol. 10, 125–126. doi: 10.1038/ncb0208-125
Yokoyama, M., Shimizu, I., Nagasawa, A., Yoshida, Y., Katsuumi, G., Wakasugi, T., et al. (2019). p53 plays a crucial role in endothelial dysfunction associated with hyperglycemia and ischemia. J. Mol. Cell Cardiol. 129, 105–117. doi: 10.1016/j.yjmcc.2019.02.010
Zhang, Q., He, X., Chen, L., Zhang, C., Gao, X., Yang, Z., et al. (2012). Synergistic regulation of p53 by Mdm2 and Mdm4 is critical in cardiac endocardial cushion morphogenesis during heart development. J. Pathol. 228, 416–428. doi: 10.1002/path.4077
Zhang, Y., Xiong, S., Li, Q., Hu, S., Tashakori, M., Van Pelt, C., et al. (2014). Tissue-specific and age-dependent effects of global Mdm2 loss. J. Pathol. 233, 380–391. doi: 10.1002/path.4368
Zhao, H., Shen, R., Dong, X., and Shen, Y. (2016). Murine double minute-2 inhibition attenuates cardiac dysfunction and fibrosis by modulating NF-κB pathway after experimental myocardial infarction. Inflammation 40, 232–239. doi: 10.1007/s10753-016-0473-5
Keywords: murine double minute 2 (MDM2), cardiovascular health, intracellular signaling, cardiac, endothelial, vascular smooth muscle cells
Citation: Lam B and Roudier E (2019) Considering the Role of Murine Double Minute 2 in the Cardiovascular System? Front. Cell Dev. Biol. 7:320. doi: 10.3389/fcell.2019.00320
Received: 08 August 2019; Accepted: 21 November 2019;
Published: 10 December 2019.
Edited by:
Grazyna Kwapiszewska, Ludwig Boltzmann Institute for Lung Vascular Research, AustriaReviewed by:
Rossella Rota, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2019 Lam and Roudier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emilie Roudier, ZXJvdWRpZXJAeW9ya3UuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.