
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 28 November 2019
Sec. Cell Death and Survival
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00308
This article is part of the Research TopicAutophagy and Ageing: Ideas, Methods, MoleculesView all 9 articles
 Eleanor R. Stead1†
Eleanor R. Stead1† Jorge I. Castillo-Quan2,3†
Jorge I. Castillo-Quan2,3† Victoria Eugenia Martinez Miguel1
Victoria Eugenia Martinez Miguel1 Celia Lujan1
Celia Lujan1 Robin Ketteler4
Robin Ketteler4 Kerri J. Kinghorn5,6,7
Kerri J. Kinghorn5,6,7 Ivana Bjedov1*
Ivana Bjedov1*Autophagy is a major cellular recycling process that delivers cellular material and entire organelles to lysosomes for degradation, in a selective or non-selective manner. This process is essential for the maintenance of cellular energy levels, components, and metabolites, as well as the elimination of cellular molecular damage, thereby playing an important role in numerous cellular activities. An important function of autophagy is to enable survival under starvation conditions and other stresses. The majority of factors implicated in aging are modifiable through the process of autophagy, including the accumulation of oxidative damage and loss of proteostasis, genomic instability and epigenetic alteration. These primary causes of damage could lead to mitochondrial dysfunction, deregulation of nutrient sensing pathways and cellular senescence, finally causing a variety of aging phenotypes. Remarkably, advances in the biology of aging have revealed that aging is a malleable process: a mild decrease in signaling through nutrient-sensing pathways can improve health and extend lifespan in all model organisms tested. Consequently, autophagy is implicated in both aging and age-related disease. Enhancement of the autophagy process is a common characteristic of all principal, evolutionary conserved anti-aging interventions, including dietary restriction, as well as inhibition of target of rapamycin (TOR) and insulin/IGF-1 signaling (IIS). As an emerging and critical process in aging, this review will highlight how autophagy can be modulated for health improvement.
Aging is characterized by progressive deterioration of tissues and organs, leading to loss of physiological function and increased risk of death. In developed societies, we are witnessing a constant increase in the size of elderly populations, and an ensuing increase in people suffering from age-related diseases, making health improvement during aging an important challenge and a priority (Rae et al., 2010). Over the last few decades, outstanding progress has been made toward understanding the aging process. Specifically, we now know that, despite all of the complexities of aging, a single mutation in just one of a few genes in nutrient-sensing pathways is sufficient to extend lifespan in all model organisms tested (Kenyon, 2010; Partridge, 2010). Moreover, the effect of anti-aging mutations is evolutionary conserved from yeast to mammals, and importantly, the long-lived mutants in all model organisms are healthier (Fontana et al., 2010; Selman and Withers, 2011; Singh et al., 2019). Thus, an improved understanding of the underlying mechanisms of aging based on genetic findings, with translation into pharmacological interventions, has the potential to improve health in the continuously growing elderly populations of modern societies. The hope is that such strategies will at the same time prevent age-related diseases (Niccoli and Partridge, 2012; Kennedy et al., 2014; Partridge et al., 2018). Examples of this approach include the anti-diabetic and the anti-aging drug metformin, which is the first drug to be tested for improvement of various health parameters in elderly people (Barzilai et al., 2016), and rapamycin, which has been shown to improve the efficacy of the flu vaccination in aged individuals (Mannick et al., 2018). Such approaches, if successful in slowing the aging process and age-related diseases, would be expected to have a significant impact on the quality of life of elderly individuals, as well as an important socio-economical benefit (Rae et al., 2010; Partridge et al., 2018; Campisi et al., 2019).
Currently there are nine proposed and well-defined primary hallmarks of aging (Lopez-Otin et al., 2013) that contribute to cellular injury and damage. These comprise genomic instability, telomere attrition, and epigenetic alteration, loss of proteostasis, deregulated nutrient-sensing, mitochondrial dysfunction and cellular senescence. In addition, two integrative hallmarks, stem cell exhaustion and altered intercellular communication, lead to functional deterioration and aging phenotypes.
In this review we focus on the autophagy process, whose upregulation is a common denominator of all major pro-longevity interventions (Hansen et al., 2018), including dietary restriction and mild down-regulation of the nutrient-sensing pathways – insulin (IIS) and mechanistic target-of-rapamycin signalling (mTOR) (Lopez-Otin et al., 2013). We first consider the regulators and effectors of autophagy, and examine the role of autophagy in the aging process. We next focus on how autophagy regulates those processes affected by aging, such as proteostasis, the maintenance of genomic integrity and organelle degradation. Finally, we explore the potential therapeutic role of autophagy modulation in preventing the aging process and age-related diseases.
Under normal physiological conditions autophagic is indispensable for cellular homeostasis, and is upregulated under stress conditions. One of the most common examples of a stress condition is starvation, where active autophagy enables survival by degrading cellular components (Mizushima, 2018; Lahiri et al., 2019). There are three different types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA), and they differ on how cargo is delivered to lysosomes for degradation. Macroautophagy is the principal and most commonly studied type of autophagy that is described below, and is commonly referred to as autophagy (Ktistakis and Tooze, 2016). Microautophagy involves the cytosolic sequesteration of cellular debris by a small invagination of the lysosomal membrane, thereby accessing lysosomal enzymes for degradation. Lastly, CMA relies on the cytosolic heat shock cognate 70 (hsc70) chaperone to recognize a KFERQ motif in target cytosolic proteins, facilitating lysosomal degradation (Kaushik and Cuervo, 2018; Scrivo et al., 2018).
A critical feature of autophagy is the ability to degrade cellular components not only randomly, but also selectively, and the number of autophagy receptor proteins that deliver certain cargo for autophagic degradation is continually growing (Kirkin, 2019). For selective autophagy to occur, LIR (LC3-interacting region)-containing receptors are essential and link specific cargo with LC3-II proteins on autophagosomes. The number of identified selective autophagy cargos are expanding; examples include degradation of glycogen by glycophagy, ferritin by ferrinophagy, protein aggregates by aggrephagy, lipids by lipophagy, and ribosomes by ribophagy. Organelle degradation is referred to as pexophagy for peroxisomes, mitophagy for mitochondria, and reticulophagy for endoplasmic reticulum (Dikic, 2017). This selective degradation is critical for ridding the cell of damaged constituents (Figure 1).
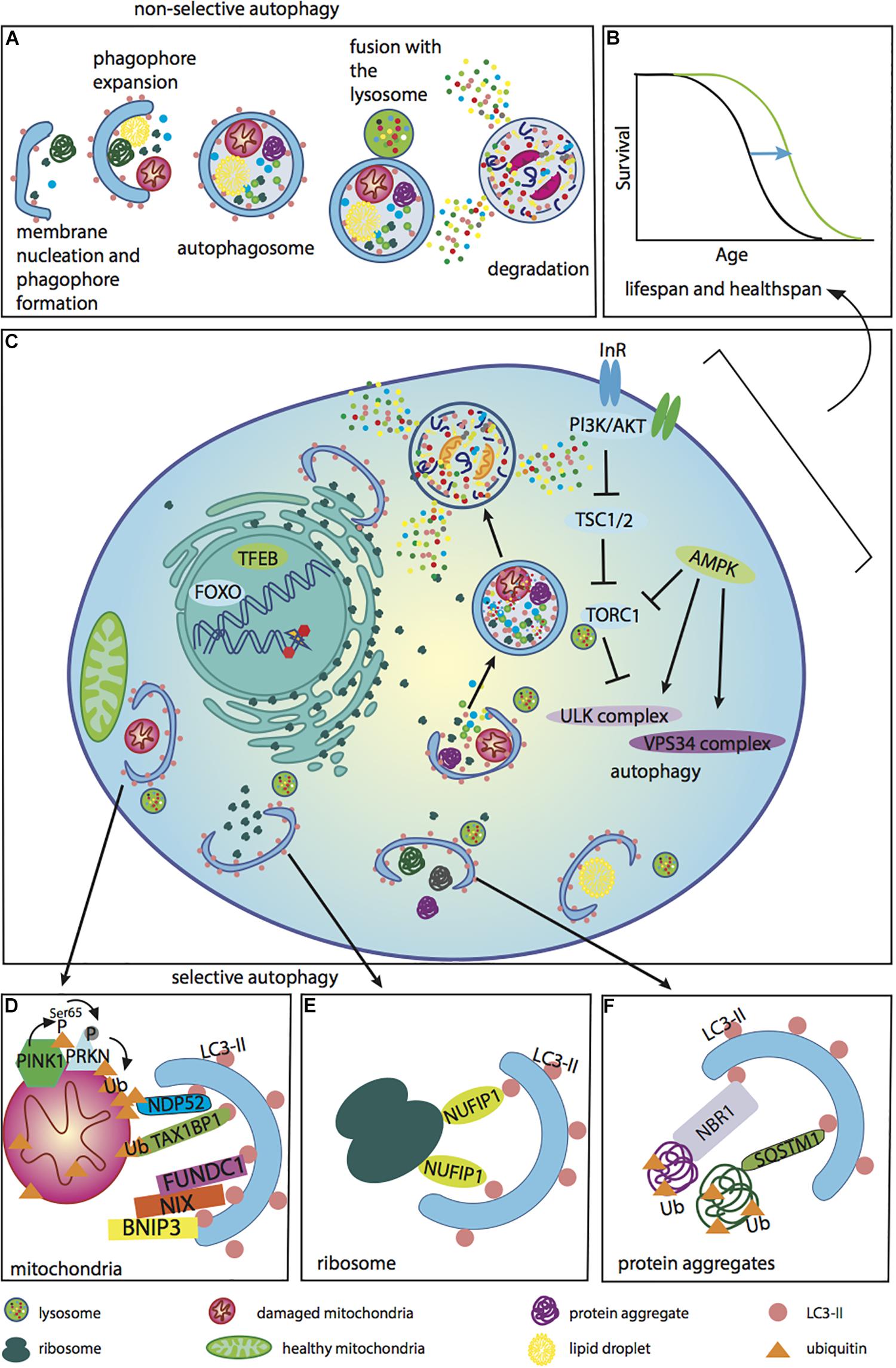
Figure 1. Autophagy regulators and functions associated with anti-aging effects. Different cellular components can be degraded by either non-selective or selective autophagy. These forms of autophagy are regulated by PI3K, mTOR, and AMPK, as well as the transcription factors TFEB and FOXO, which are accountable for the transcription of many genes involved in the autophagy process. (A) The different steps of non-selective autophagy. Upon autophagy initiation, a phagophore is formed and expanded, thereby producing an autophagosome. The matured phagosome fuses with the lysosome, initiating degradation of the autophagosome’s inner membrane. Cellular components captured within are subsequently degraded and released into the cytoplasm. Autophagy occurs under basal conditions in the cell, but can be up-regulated during stress. The autophagy process is inefficient during aging. (B) A lifespan curve illustrating the positive effects of enhancing autophagy during aging to improve the recycling of different cellular components, extending lifespan and healthspan. Anti-aging effects of autophagy up-regulation have been demonstrated in several model organisms. It has been shown in a number of studies that the lifespan of long-lived mutants is (reduced upon down-regulation of autophagy. In addition, there are a few critical studies showing that up-regulation of autophagy by overexpression of one of the autophagy genes extends lifespan. (C) An illustrative diagram demonstrating the negative regulation of autophagy by PI3K/AKT and mTORC1, and the positive modulation of autophagy by AMPK via effects on the autophagy complexes ULK and VPS34. Phagophore expansion leads to the engulfment of various cellular components, such as ribosomes, protein aggregates and lipids, via selective as well as non-selective autophagy. In addition, autophagy plays an important role in the DNA damage response and the nuclear-associated genes, TFEB and FOXO, play important roles in regulating autophagy. (D) Defective mitochondria are cleared by the cell through mitophagy. This is the process whereby damaged depolarized mitochondria can be degraded by the PINK1-Parkin pathway, which is ubiquitin-dependent. In cells with healthy mitochondria, PINK1 is continuously degraded, while Parkin is in the cytoplasm. Upon stress, PINK1 is stabilized on the outer mitochondrial membrane, where it phosphorylates ubiquitin and E3 ubiquitin ligase Parkin. Once Parkin is recruited to the mitochondria, it then ubiquitinates some of the outer membrane mitochondrial proteins. These polyubiquitin K63-linked chains are phosphorylated, creating a degradative signal for autophagy. Receptor proteins involved in this pathway include NDP52 and TAX1BP1. These proteins recognize phosphorylated polyubiquitin chains and link damaged mitochondria to LC3-II. Another mechanism for mitochondrial degradation involves receptor-mediated autophagy by BNIP3, NIX and FUNDC1. These receptors also interact with LC3 via the LIR domain and target depolarized mitochondria for degradation. For mitophagy to occur, damaged mitochondria must be separated by fission. Healthy mitochondria are essential for cellular ATP production and the maintenance of cellular energy homeostasis. (E) The ribophagy receptor NUFIP1 mediates degradation of ribosomes (Wyant et al., 2018). (F) Protein aggregates are ubiquitinated and degraded by autophagy with the help of the autophagy receptors NBR1 and SQSTM1.)
The autophagy process depends on the formation of different complexes on the membrane and can be divided into a number of steps: initiation, nucleation, elongation, autophagosome-lysosome fusion and degradation of sequestered material (Ktistakis and Tooze, 2016; Galluzzi et al., 2017; Lahiri et al., 2019). The Atg1/ULK1 complex, which is regulated by mTOR and AMPK, initiates the formation of a complex that regulates phagophore membrane nucleation at the endoplasmic reticulum and possibly other membrane enclosed organelles (Carlsson and Simonsen, 2015). The ATG1/ULK1 complex further activates the phosphatidylinositol 3-kinase (PI3K) complex through a series of phosphorylation events, which incorporates phosphatidylinositol 3-phosphate into the phagophore to form an autophagosome (Ktistakis et al., 2014). Two conjugation reactions are critical for the elongation of the autophagosome and closure. First, the conjugation of ATG12 to ATG5, and its interacting partner ATG16L1, via the ubiquitin-conjugation like enzymes ATG7 and ATG10, generates an ATG5-12/16L1 complex that is required for mediating the linkage of ATG8/LC3/GABARAP protein family to the autophagosome membrane (Nakatogawa, 2013; Lahiri et al., 2019). Following cleavage of pro-ATG8/LC3/GABARAP by the ATG4 family of proteases, and a two-step conjugation reaction requiring ATG3 and ATG7, the cleaved form of ATG8/LC3/GABARAP is anchored to phosphatidylethanolamine (PE) in the inner and outer autophagosome membrane. ATG8-PE/LC3-GABARAP can also be removed from the autophagosome membrane by the ATG4 family of proteins, although the relevance of this removal in mammalian cells is not clear (Kauffman et al., 2018; Agrotis et al., 2019). Recent data seems to suggest that ATG8-PE/LC3/GABARAP-II is required for late stages of autophagy (fusion and degradation of inner autophagosomal membrane) (Nguyen et al., 2016; Tsuboyama et al., 2016). In addition, ATG8/LC3/GABARAP can bind to selective autophagy receptors, such as SQSTM1/p62, that bind to ubiquitinated proteins and organelles to mediate selective cargo degradation (Kirkin, 2019). Membrane specific LC3-II and p62 are the most commonly used autophagy markers (Klionsky et al., 2012). Once formed, autophagosomes fuse with lysosomes to form autophagolysosomes (Nakamura and Yoshimori, 2017). It is in this latter structure that unwanted cellular components are degraded by acidic lysosomal hydrolases. Autophagy is a highly coordinated and dynamic process that is tightly controlled by post-translational regulation in a spatio-temporal manner (Botti-Millet et al., 2016; Pengo et al., 2017; Delorme-Axford and Klionsky, 2018), and perturbation in any step can lead to the accumulation of undigested material and aggregates.
Various cellular energy and metabolic sensors act as major regulators of autophagy, with their coordinated action leading to different autophagy activities, thereby maintaining cellular homeostasis (Table 1). Activated AMP kinase (AMPK) induces autophagy in response to low ATP/AMP cellular energy status. Several underlying mechanisms have been described for AMPK-mediated autophagy up-regulation, such as phosphorylation of ACSS2 (acetyl-CoA synthetase short-chain family member 2). AMPK can also alter autophagy via induction of TFEB (transcription factor EB)-mediated transcription of lysosomal and autophagy genes (Li X. et al., 2017) and via inhibition of the TOR pathway (Saxton and Sabatini, 2017). Furthermore, AMPK phosphorylates BECN1 and PIK3C3/VPC34, as well ULK1, to stimulate autophagic function (Hardie et al., 2016). Overexpression of AMPK in the Drosophila nervous system induces autophagy in the brain and cell non-autonomously in the intestine, resulting in lifespan extension (Ulgherait et al., 2014). AMPK is a complex sensor of cellular energy status and can be activated genetically, or pharmacologically by metformin, leading to lifespan extension in organisms ranging from yeast to mice (Pryor and Cabreiro, 2015; Salminen et al., 2016). Detailed worm studies uncovered metformin-induced alterations of microbial metabolism (Cabreiro et al., 2013), specifically the production of bacterial agmatine that regulates lipid metabolism and promotes lifespan extension (Pryor et al., 2019).
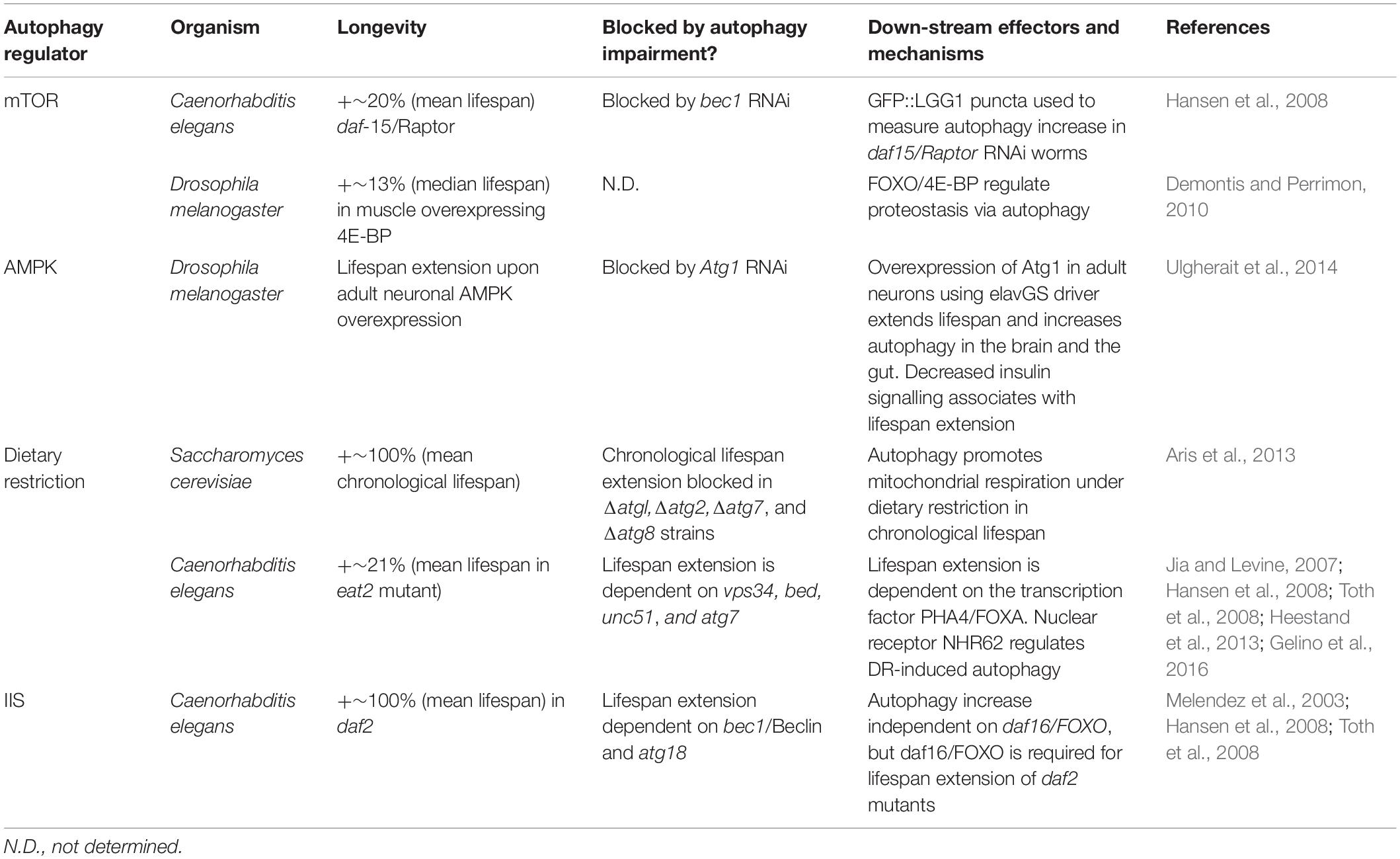
Table 1. Autophagy regulators and down-stream effectors.
Another major autophagy regulator is mTORC1 (mTOR complex 1), which under nutrient-rich conditions, promotes growth and inhibits autophagy. This occurs through inhibitory phosphorylation of ULK1, and of the PIK3C3/VPS34 kinase complex, as well as by phosphorylation of TFEB (the lysosomal biogenesis transcription factor), leading to its cytosolic localization and block in transcriptional activity (Kim and Guan, 2019). Moderate inhibition of the mTOR pathway and its downstream effectors, such as S6K, either genetically or pharmacologically by rapamycin, is one of the most well studied anti-aging interventions. However, not all of its effects are mediated exclusively by increased autophagy, and lower protein synthesis is another pro-longevity effect of decreased mTOR signaling (Bjedov et al., 2010; Castillo-Quan et al., 2015; Saxton and Sabatini, 2017; Hansen et al., 2018).
In addition to amino acid sensing, saturated and unsaturated free fatty acids have also been shown to activate the autophagy process. This is most likely because circulating free fatty acids are abundant during starvation, which is the major autophagy trigger (Papsdorf and Brunet, 2019). Supplementation of food with ω-3 PUFA arachidonic acid (AA) or dihomo-γ-linolenic acid (DGLA) induces autophagy and extends lifespan in Caenorhabditis elegans (O’Rourke et al., 2013; Bustos and Partridge, 2017). In addition, α-linolenic acid induces autophagy in mammalian cells (O’Rourke et al., 2013). There is currently an increasing interest in the role of lipids in aging. Lipids that extend lifespan are mono-unsaturated fatty acids (MUFAs), such as oleic acid, cis-vaccenic acid and palmitoleic acid; albeit the longevity mechanism is linked specifically to modifiers of trimethylated lysine 4 on histone H3 (H3K4me3). This demonstrates an interesting link between lipids and chromatin (Han et al., 2017). However, the specific contribution of autophagy induction to the effects of these lipids is still unknown. Another important anti-aging observation is that in germline-less glp-1 mutant worms, lacking the human ortholog of NOTCH1, longevity is mediated through the intricate interaction of increased lysosomal lipase LIPL-4 and up-regulation of autophagy (Wang et al., 2008; Lapierre et al., 2011; Hansen et al., 2018). LIPL-4 lipase liberates a MUFA, oleoylethanolamide, which mediates longevity effects by binding to lysosomal lipid chaperone LBP-8. This in turn leads to its nuclear localization and activation of the nuclear hormone receptors NHR-80 and NHR-49 (Folick et al., 2015). Taken together, these findings draw attention to the connection between autophagy, lipids and aging. They also demonstrate that autophagy can be regulated by certain fatty acid species, and vice versa. Indeed, active autophagy can alter cellular lipid composition through the activity of lysosomal lipases (Bustos and Partridge, 2017; Papsdorf and Brunet, 2019). Because autophagy is a degradative process, it must occur in moderation and in a coordinated manner. For instance, excessive lipophagy can liberate too many fatty acids from membrane degradation, which if not stored in lipid droplets, can lead to acylcarnitine accumulation and mitochondrial uncoupling and dysfunction (Nguyen and Olzmann, 2017). In addition, lipid alterations can also affect autophagic vesicular fusion (Koga et al., 2010). Clearly, more research on the interaction between autophagy, various lipid species, and aging is required. Particularly because of the pleiotropic role of lipids, this interaction can impact energy storage, signalling and transcription, as well as membrane composition and fluidity. Increasing proportions of unsaturated lipids in the membrane correlate with enhanced fluidity, but also increase the risk of lipid peroxidation, which is associated with reduced lifespan in worms and in several mammals (Shmookler Reis et al., 2011; Jove et al., 2013).
Overall, there is a clear link between energy sensors, nutrients, and nutrient signaling pathways and autophagy. Conversely, autophagy can influence nutrient signaling through liberation of degraded molecules, thus influencing cellular health status (Lahiri et al., 2019). Given that dietary restriction and modulation of nutrient signaling pathways are major pro-longevity interventions (Fontana et al., 2010; Fontana and Partridge, 2015), understanding how autophagy can be modified specifically to alter the metabolic profile of an individual for anti-aging purposes is an essential goal.
Several transcription factors are regulators of autophagy, TFEB (transcription factor EB) being one of the principal ones responsible for the coordination and expression of autophagy genes and lysosomal hydrolases. Overexpression of the TFEB ortholog HLH-30 was shown to increase lifespan in worms, with the extension being dependent on its nuclear localization (Lapierre et al., 2013). Another key transcription factor that regulates autophagy is FOXO (forkhead box class O), which is implicated in nearly all anti-aging interventions (Kenyon, 2010; Tullet, 2015; Martins et al., 2016). It hence became a prime pharmacological anti-aging target (Cheng, 2019). When unphosphorylated in the nucleus, FOXO activates the transcription of several autophagy genes (Mammucari et al., 2007; Zhao et al., 2007). Conversely, when phosphorylated in the cytoplasm it interacts with autophagy proteins (Cheng, 2019). In addition, autophagy directly and indirectly mediates FOXO degradation, providing a negative feedback loop, limiting dangerous excessive autophagic degradation (Webb and Brunet, 2014; Fitzwalter et al., 2018; Cheng, 2019). Interestingly, the FOXO3A variant is one of the few genes associated with longevity in human centenarians (Flachsbart et al., 2009). However, the precise biological effects associated with these FOXO variants are unknown. Another interesting transcriptional regulator of autophagy is p53. When in the cytosol, p53 promotes autophagy suppression, while in the nucleus it induces transcription of autophagy genes (Yee et al., 2009; Kenzelmann Broz et al., 2013). This tumor suppressor gene is one of the most commonly mutated cancer genes and can protect against cancer in mice when carefully overexpressed under endogenous control in a p53 overexpressor strain called “super p53” (Garcia-Cao et al., 2002). It also extends lifespan if combined with Ink4/Arf overexpression (Matheu et al., 2007). The effect of this “super p53” is in stark contrast to overexpression of constitutively active p53 under a heterologous promoter. This triggers excessive apoptosis, and although protecting mice from cancer, it accelerates the aging phenotype, most likely owing to depletion of the stem cell pools (Tyner et al., 2002; Finkel et al., 2007).
Given that autophagy is induced by these very different transcription factors strongly suggests that autophagy is a critical tool for dealing with most forms of stress. However, any excessive autophagy may be detrimental and cause cell death, given that it is a degradative process requiring moderate induction (Kang et al., 2007; Scott et al., 2007).
As described earlier, autophagy is initially triggered by post-translational protein modifications in the cytosol, and can also be regulated at the transcriptional level by various transcription factors. However, it can additionally be regulated at the epigenetic level (Fullgrabe et al., 2014; Lapierre et al., 2015; Baek and Kim, 2017). It is proposed that under prolonged autophagy induction, transcription factors help sustain autophagic flux, while further prolongation entails epigenetic changes that ensure that autophagy does not become lethal (Fullgrabe et al., 2013, 2014).
In addition to potentially preventing autophagy from going awry, it was proposed that an epigenetic regulation of the autophagic process could perhaps also lead to a memory effect enabling a quicker response to subsequent starvation events (Fullgrabe et al., 2014). “Memory effects” for longevity, transmitted across three generations, have been shown in C. elegans having lower H3K4me3 activity, owing to mutations in the corresponding methylation complex. Transgenerational inheritance is not a common feature of long-lived mutants and it does not occur in long-lived daf-2 mutant worms, which have decreased IIS pathway owing to a mutation in the insulin receptor homolog daf-2 (Greer et al., 2011). There is an association between autophagy induction and downregulation of H3K4me3 and H4K16ac (Fullgrabe et al., 2013). Increased autophagy is often linked to deacetylation, particularly of H4K16, which is mediated by Sirt1, and accompanied by the transcription of autophagy genes (Fullgrabe et al., 2014). Overexpression of Sirt1 is a well-studied putative anti-aging intervention (Lopez-Otin et al., 2013), again highlighting the intriguing link between autophagy and longevity. In general, different autophagy stimuli lead to deacetylation of H4K16 (Baek and Kim, 2017). Also LC3/Atg8 is deacetylated by Sirt1 and translocated to the cytosol upon autophagy initiation (Huang et al., 2015).
Another epigenetic alteration linked to autophagy is H3R17, resulting from the coactivation of TFEB by CARM1 (Shin et al., 2016). Autophagy can be suppressed by some of the common epigenetic changes such as G9a-mediated H3K9 dimethylation (Artal-Martinez de Narvajas et al., 2013) and silencing histone mark H3K27 trimethylation via EZH2 (Enhancer of Zest Homologue2) (Wei et al., 2015). Interestingly, H3K27 is linked to aging, albeit evolutionary conservation of this remains to be confirmed. Loss of chromatin repression was observed during aging in species from C. elegans to humans (Bennett-Baker et al., 2003). However, while an increase in H3K27me3 by RNAi against UTX-1 demethylase extends lifespan in worms (Jin et al., 2011; Maures et al., 2011), it is a decrease in H3K27me3 in flies by mutations in PRC2 components E(z) and ESC, that promotes longevity (Siebold et al., 2010). Thus, the intriguing relationship between autophagy and epigenetics has the potential to provide invaluable insights into the aging process itself.
Collectively, there are numerous genetic and epigenetic autophagy regulators, most of which have already been implicated in aging. Targeting these offers numerous possibilities for modulating autophagy to improve health in older age. However, many of these autophagy regulators have pleiotropic effects, implying that any potential treatments need to be carefully evaluated.
Aging is accompanied by progressive decline of autophagy in many organisms (Hansen et al., 2018). A reduction in autophagy during aging was demonstrated in a study that carefully examined autophagy in different tissues throughout adulthood of long-lived daf-2 and glp-1 C. elegans mutants, and showed that intestinal autophagy inhibition abolishes longevity only in glp-1 mutants (Chang et al., 2017). Mice deficient for essential autophagy genes, such as atg5, atg7 or atg12, are neonatal lethal and die within 1 day due to failure to adapt to starvation and from a suckling defect (Kuma et al., 2017). Moreover, this lethality can be rescued by the neuron-specific expression of ATG5 (Yoshii et al., 2016). Neuronal and glial specific deletion of either atg5 or atg7 results in viable and short-lived mice displaying neuronal protein accumulation and neurodegeneration (Hara et al., 2006; Komatsu et al., 2006). This highlights the importance of autophagy in removing damaged proteins in non-dividing neuronal tissue, and the potential of therapeutic autophagy enhancement in neurodegenerative disease (Menzies et al., 2017; Scrivo et al., 2018).
Evidence for the role of autophagy in aging was first shown in daf-2 long-lived worms, where RNAi-mediated downregulation of the autophagy gene bec-1 completely abrogated their pronounced longevity (Melendez et al., 2003). Since this discovery, dependence on autophagy enhancement has been demonstrated in nearly all longevity-promoting interventions. For instance, lifespan extension by dietary restriction, mTOR inhibition, AMPK up-regulation, mitochondrial mutations, and the above mentioned germline glp-1 mutation, all require functional autophagy for lifespan extension (Toth et al., 2008; Hansen et al., 2018). In all these long-lived mutants, lessening autophagy by RNAi returns lifespan toward wild type levels. However, controls treated with similar autophagy-reducing RNAi interventions do not display altered longevity, suggesting that the residual autophagy levels are sufficient to maintain normal lifespan. It is worth noting that the nutrient-sensing pathways implicated in longevity have pleiotropic effects on metabolism, and often, under conditions when autophagy is up-regulated, this also impacts on other anti-aging processes such translation. It is thus challenging to fully evaluate exact contributions of different down-stream effectors on overall longevity. Another intriguing finding is that if autophagy is inhibited in neurons of post-reproductive adult worms, by targeting genes involved in the early stage of the autophagy process such as bec-1, this results in an extension in lifespan (Wilhelm et al., 2017). One plausible explanation for this is that if aged worms have impaired autophagy flux in the neuronal tissues, then inhibiting autophagy initiation stops clogging the system even further, preventing neuronal decline (Wilhelm and Richly, 2018). Another example where inhibiting autophagy is beneficial is in the intestine of the worm, where this approach limits conversion of intestinal biomass to yolk, thereby preventing age-related visceral pathologies (Ezcurra et al., 2018). This suggests that, rather than being a result of age-associated “wear and tear,” some pathologies may result from a precise genetically-driven biological program (Ezcurra et al., 2018).
This detrimental effect of autophagy late in life has only been observed in worms. It remains to be investigated if autophagy behaves similarly as an antagonistic pleiotropic process in mammals, being beneficial in the young, but detrimental in the aged organism. In this case, interventions to enhance autophagy to improve healthspan may need to be initiated early to prevent tissue deterioration, while later in life once the host’s autophagic machinery has already begun to fail, it should be carefully considered whether autophagy needs to be enhanced or inhibited. However, it should be noted that in mice, the autophagy activating drug rapamycin is beneficial once administered later in life (Harrison et al., 2009), suggesting that either in mammals autophagy activation is beneficial irrespective of age, unlike in worms (Wilhelm et al., 2017; Wilhelm and Richly, 2018), or that rapamycin-mediated downregulation of translation is an important effect for healthspan benefits in the aged mice.
In order to improve health during aging, understanding those alterations that increase lifespan are essential, as they directly demonstrate the importance of a given paradigm in aging. Manipulations that increase autophagy directly are valuable but sparse, and complicated by the fact that that numerous autophagy genes are involved in different stages of this multistep process. Moreover, overexpression of only one autophagy gene does not necessarily trigger autophagy. Nevertheless, there are some very valuable exceptions that directly show how important this process is in aging (Hansen et al., 2018; Maruzs et al., 2019). For instance, overexpression of Atg8a in neurons (Simonsen et al., 2008), as well as Atg1 overexpression in neuronal tissue (Ulgherait et al., 2014) or muscle (Bai et al., 2013), extends lifespan in Drosophila. In addition, mammalian lifespan was extended by an ubiquitous increase of Atg5 in mice, and was accompanied by improved motor function (Pyo et al., 2013). Moreover, knock-in of Becn1 bearing the Phe121Ala mutation, which interrupts its interaction with its negative regulator BCL2, leads to increased autophagy flux and longevity in both male and female mice (Fernandez et al., 2018). These observations are very important to elucidate how an upregulation in autophagy leads directly to benefits at the organismal level. Further studies of autophagy manipulation in different tissues will help to elucidate further tissue-specific effects and the impact of these on organismal aging (Hansen et al., 2018). In particular, combining longevity experiments with healthspan parameters, such as motor function, cardiovascular deterioration, neuronal loss and insulin sensitivity, will facilitate the discovery of pharmacological targets for disease prevention and treatment.
The role of autophagy in delaying aging is commonly attributed to its capacity to degrade damaged proteins and contribute to cellular proteostasis (Hansen et al., 2018). Loss of proteostasis is a prominent feature of aging and a major risk-factor for age-related neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases (Lopez-Otin et al., 2013; Kaushik and Cuervo, 2015; Sands et al., 2017; Hipp et al., 2019). A recent detailed proteomic profiling in worms revealed that a third of the proteome changes in abundance by at least two or threefold with aging (Walther et al., 2015). Interestingly, long-lived daf-2 mutant worms have increased proteasome subunits, but not autophagy proteins, and they maintain proteostasis by sequestering problematic proteins into less toxic chaperone-containing aggregates (Walther et al., 2015). This is in accordance with findings in the neurodegeneration field suggesting that, if unfolded proteins cannot be re-folded, or degraded, then the resulting unfolded soluble oligomeric proteins are far more toxic to the cell than insoluble aggregates (Cohen et al., 2006; Hipp et al., 2019). Consistent with this, reduced insulin signaling in long-lived insulin-like growth factor 1 (IGF-1) heterozygous mice are protected against amyloid β-peptide (Aβ) proteotoxicity by favoring a shift from toxic oligomers toward less toxic aggregates (Cohen et al., 2009). It should be noted, however, that the relationship between insulin signaling and neurodegenerative disease is very complex and not well understood (Ribe and Lovestone, 2016).
Proteostastic mechanisms consist of protein synthesis, protein folding and protein degradation, all of which are interconnected in order to achieve a balanced proteome and robust stress responses when required (Kaushik and Cuervo, 2015; Hipp et al., 2019). The protein synthesis pathway, mTOR, has a well described effect on autophagy inhibition, and has recently also been linked to the proteasome. It has been shown to both activate it via the nuclear factor erythroid 2-related factor 1 (Nrf1) to balance active translation (Zhang et al., 2014), and inhibit the proteasome in a different experimental set up (Zhao et al., 2015). mTOR inhibition can relieve proteotoxic stress by decreasing translation, and specifically, by allowing translation of additional chaperones (Su et al., 2016). Decreasing translation through mutations in mTOR pathway genes, such as S6K (Selman et al., 2009), or by or overexpression of the mTOR suppressor TSC1/2 (Kapahi et al., 2004), are well-described anti-aging interventions for which several underlying mechanisms are suggested. For example, reduced translation extends longevity by reducing the protein load for the proteostatic machinery. Furthermore, the reduction in energy expenditure associated with reduced translation can be invested into cell maintenance processes. Moreover, differential translation may lead to stress resilience and delayed aging (Mehta et al., 2010; Steffen and Dillin, 2016). In addition, it appears that enhancement of proteostasis, either by improved protein folding or by degradation, is also a potentially successful anti-aging strategy (Lopez-Otin et al., 2013). For example, overexpression of the heat shock factor HSF-1 has repeatedly been shown to extend lifespan in worms (Hsu et al., 2003; Li J. et al., 2017). Additionally, overexpression of the proteasomal subunit Rpn6, which stabilizes the 20S core particle and 19S regulatory particle, can induce proteasome activity and extend lifespan in C. elegans. This is an exciting finding as the proteasome has numerous subunits and this suggests that targeting only one key subunit is sufficient to intensify proteasomal degradation (Vilchez et al., 2012). In Drosophila, overexpressing the β5 subunit of the 20S in the whole organism (Nguyen et al., 2019), or just in neurons, extends lifespan (Munkacsy et al., 2019). While whole-body β5 subunit overexpression increases proteostasis in muscle (Nguyen et al., 2019), neuron-specific overexpression prevents age-related decline in learning and memory (Munkacsy et al., 2019). Enhancement of autophagy by overexpression of FOXO transcription factor in muscle tissues in Drosophila is beneficial as well (Demontis and Perrimon, 2010). This tissue-specific upregulation of autophagy in muscle tissues mediates a reduction in aggregate formation during aging, with an improvement in proteostasis in the whole organism, as well as increased longevity (Demontis and Perrimon, 2010).
Autophagy function is highly complementary to that of the proteasome, which targets small short-lived proteins. Autophagy degrades large and long-lived proteins, protein aggregates, entire defective organelles, and essentially any cellular material that is too bulky for proteasomal degradation (Dikic, 2017; Kocaturk and Gozuacik, 2018). Interestingly, results in mammalian cells suggest that there is significant crosstalk and a degree of compensation between these two degradative processes. If the proteasome is inhibited, then autophagy enhances to compensate (Kocaturk and Gozuacik, 2018). However, there is less evidence to support a role for the proteasome in compensating for a block in autophagy. This is likely a result of the fact that the proteasome cannot accomplish all autophagic functions and cannot degrade organelles (Dikic, 2017; Kirkin, 2019). A common characteristic of these two degradative pathways is that they both recognize ubiquitinated substrates. Autophagy often degrades aggregates linked to K63-based polyubiquitin chains, while substrates for the proteasome mainly have K48-polyubiquitin chains (Kocaturk and Gozuacik, 2018). However, it should be noted that the specificity of autophagy toward K63 ubiquitinated substrates is not that clear. For example, in autophagy-deficient mice all types of ubiquitin chains accumulate (Riley et al., 2010).
Aging is accompanied by accumulation of damage and damaged organelles, among which damaged DNA is the most strongly linked to aging. Various syndromes originating from deficiency in DNA repair enzymes recapitulate some of the aging phenotypes and are models of accelerated aging. Examples include Werner and Bloom syndrome, which are caused by mutations in the WRN and BLM genes, respectively, both of which are RecQ-like helicases (Carrero et al., 2016). Cockayne syndrome is another progeria model deficient in ERCC6 or ERCC8, while Hutchinson-Gilford progeria bears a mutation in laminin A gene (LMNA), whose product is a key structural component of nuclear lamina. In addition, defective DNA repair associated with mutations in genes such as Ku70, Ku80, Ercc1, and Xpd leads to shorter lifespan in mice (Vermeij et al., 2016). This strongly suggests a role for DNA damage in aging (Vermeij et al., 2016).
Interestingly, autophagy affects DNA repair, a finding that came as a surprise given that autophagy is mainly a cytoplasmic process. Initially it was reported that mammalian cells lacking beclin1 or atg5 genes have increased genomic instability (Karantza-Wadsworth et al., 2007; Mathew et al., 2007). Since, autophagy was linked to different components of the DNA Damage Response (DDR), as we discuss below, this strengthens the impact of autophagy on genomic integrity and thus aging (Eliopoulos et al., 2016; Hewitt and Korolchuk, 2017).
Maintenance of genomic stability is key for survival. Therefore a complex DDR, with the ability to detect various types of DNA aberrations and engage appropriate DNA repair systems, has evolved (Ciccia and Elledge, 2010). DNA lesions, through post-translational histone modifications, trigger relaxation of the chromatin, and inhibit replication and transcription, in order to promote DNA repair instead (Jeggo et al., 2017). For instance, removal of HP1α (heterochromatin protein 1 alpha) from DSB sites allows formation of Rad51 nucleoprotein filaments and successful homologous recombination (HR). Interestingly, HP1α is ubiquitinated by RAD6, which then triggers autophagic degradation of HP1α and loosening of chromatin compaction for successful double strand break (DSB) repair in mammalian cells (Chen et al., 2015). DSBs are bound by MRN (Mre11-Rad50-Nbs1) and then ATM (ataxia-telangiectasia mutated) kinase, which becomes autophosphorylated and activated, ultimately leading to either homologous recombination (HR) repair, that can occur exclusively in G2/S phase of the cell cycle, or non-homologous end joining (NHEJ). The latter functions in all phases of the cell cycle (Ciccia and Elledge, 2010). Although NHEJ is traditionally considered more erroneous than HR, it has been shown that the increased DNA resection required in different types of NHEJ and HR correlates well with the level of mutations (Rodgers and McVey, 2016). For instance, HR can be mutagenic when hyper-resection by Rad52 mediates single-strand annealing-type of HR (Ochs et al., 2016). Single strand breaks are recognized by RPA (replication protein A), which recruits ATR (ataxia-telangiectasia and Rad3 related) kinase, promoting phosphorylation of p53 and Chk1 as part of the irradiation response. In both ATM-Chk2 and ATR-Chk1 axes of repair, among which cross talk exists, one of the key initial events for DDR is phosphorylation of γH2AX by ATM, ATR or DNA-PKcs (Ciccia and Elledge, 2010; Blackford and Jackson, 2017). In addition to DSB and single strand break (SSB) repair, base excision repair (BER), and nucleotide excision repair (NER) are scanning DNA for different types of damage. Mismatches occurring during replication are fixed by mismatch repair (MMR), while specialized translesion synthesis (TLS) polymerases tolerate damage and avoid replication fork stalling (Ciccia and Elledge, 2010; Jeggo et al., 2017). As well as these DNA damage sensors, mediators and effectors, a number of other cellular responses are essential when timely DNA repair is not achieved (Ciccia and Elledge, 2010). These include cell cycle arrest, changes in energy and metabolism and initiation of cell death. Interestingly, autophagic cell death in cells undergoing a replicative crisis is critical for eliminating cells with genomic instability (Nassour et al., 2019).
There is an increasing number of conceptually interesting links between autophagy and DDR (Eliopoulos et al., 2016; Hewitt and Korolchuk, 2017). For instance, valproic acid, which is an autophagy stimulator and histone deacetylase inhibitor, targeting HDACs Hda1 and Rpd3, limits DDR by stimulating autophagic degradation of acetylated recombination protein Sea2/CtIP. This confers DNA damage sensitivity – an interesting finding linking acetylation, autophagy and DSB repair (Robert et al., 2011). Genomic instability of autophagy-deficient cells (Karantza-Wadsworth et al., 2007; Mathew et al., 2007) is linked to compensatory upregulation of proteasomal degradation, resulting in less phospho-Chk1 in response to damage. Consequently HR is impaired, making these cells more reliant on NHEJ (Liu et al., 2015). Chk1 is also a substrate for CMA, and high levels of Chk1 accumulating in the nucleus are equally detrimental to genome integrity, as shown when CMA is inhibited and the MRN complex is destabilized (Park et al., 2015). This highlights the fact that in DNA repair, alterations in enzyme activity need to be subtle, and possibly coordinated with other interacting proteins, otherwise repair outcome is perturbed. Furthermore, it has been shown in the hematopoietic system that increasing autophagy using rapamycin is protective against radiation and induced HR and NHEJ (Lin et al., 2015). Rapamycin-independent autophagy is also described in yeast, where DNA damaging agents, acting through DDR kinases Mec1/ATR, Tel1/ATM, and Rad53/CHEK2, induce a novel type of autophagy, named genotoxin-induced targeted autophagy, which relies on Atg11 (Eapen et al., 2017). Another interesting DNA repair link involves the p62/SQSTM1 autophagy receptor protein, which plays a role in proteasomal degradation of ubiquitinated proteins (Liu et al., 2016). p62/SQSTM1-dependent proteasomal degradation of recombination proteins filamin A (FLNA) and RAD51 in the nucleus is excessive under high levels of P62/SQSTM1. This occurs when the autophagic degradation of p62 is impaired, leading to defective DNA repair (Hewitt et al., 2016). A nuclear increase of p62 in autophagy-deficient mammalian cells also inhibits the E3 ligase RNF168, leading to deficient H2A ubiquitination, thereby thwarting recruitment of both NHEJ and HR DNA repair enzymes to the damaged sites (Wang et al., 2016). Inefficient p62-mediated degradation of GATA4 transcription factor leads to induction of cellular senescence, linking autophagy and senescence (Kang et al., 2015). In summary, these findings demonstrate intriguing connections between DNA and protein damage control, and highlight the effect of low levels of nuclear p62 for preventing genomic instability (Mathew et al., 2009). Above, we focused on the links between DSB and SSB repair and the autophagy process. However, BER, NER, and MMR are also associated with autophagy, albeit to a lesser extent. Indeed, it was recently reported that DNA damage, induced by 5-FU, is accompanied by BER and MMR activation and repair, resulting in autophagy-mediated cell death (SenGupta et al., 2013). In the presence of a different cytotoxic chemical, 6-TG, MMR induces autophagy via p53 (Zeng et al., 2007). Finally, in addition to the above mentioned DDR proteins that are modulated by the autophagic status of the cell, autophagy can result in the degradation of entire micronuclei, which are chromosomal fragments that are not incorporated into daughter cells, and which are common markers of genotoxicity (Rello-Varona et al., 2012).
In conclusion, despite being a cytoplasmic process, autophagy can affect numerous aspects of DNA repair. Moderate autophagy enhancement appears to mainly exert positive effects on DDR. It would therefore be interesting to examine whether lifespan in long-lived autophagy mutants depends on DDR. Owing to the complexity of DNA repair systems, overexpression of a single enzyme does not necessarily enhance the entire type of repair, similar to autophagy, making lifespan extension by genetic up-regulation of DDR difficult to investigate. However, these types of findings are particularly valuable for understanding aging, and a few examples do exist. A large overexpression screen in Drosophila uncovered that ubiquitous enhancement of mei-9/XPF resulted in a consistent increase in longevity among tested candidates genes (Shaposhnikov et al., 2015). Remarkably, an extensive study of 18 rodent species revealed that DSB repair co-evolves with longevity, not NER; the latter correlating with sunlight exposure of different species, not their lifespan (Tian et al., 2019). More specifically, it is five amino acids in SIRT6 that dictate the different activities of both HR and NHEJ-types of DSB repair, and which are accountable for longevity (Tian et al., 2019). Thus, it would be interesting to develop drugs that enhance DNA repair, without inflicting DNA damage, as they may prove to be effective anti-aging drugs.
The precise role of mitochondria in aging is an interesting and heavily debated topic. It is increasingly becoming apparent that their degradation by mitophagy, whereby mitochondrial number is regulated and damaged mitochondria recycled, is important for aging and disease. Mitochondrial fission and fusion are vital for the maintenance of mitochondrial shape and health, and are regulated by MFN1/2, FIS1, DRP1, and OPA1. For instance, under starvation conditions mitochondria are protected from mitophagy; mitochondrial fusion results in mitochondria predominantly in the elongated form, thwarting their degradation (Gomes et al., 2011). On the other hand, it is key that damaged parts of the mitochondria are separated through a fission process. These non-functional mitochondria are then degraded by ubiquitin-dependent pathways. Depolarization of the mitochondrial membrane leads to accumulation of PINK1 on the outer membrane, where it phosphorylates ubiquitin and the E3 ubiquitin ligase PARKIN, leading to activation of PARKIN and the ubiquitination of mitochondrial membrane proteins (Kane et al., 2014; Koyano et al., 2014; Gladkova et al., 2018; Harper et al., 2018; Palikaras et al., 2018). It is thought that selective autophagy adaptors, NDP52 and TAX1BP1 bind to ubiquitinated mitochondria, enabling their degradation by selective autophagy (Lazarou et al., 2015). A recent paradigm shift has shown that for various forms of selective autophagy, the autophagy adaptors lead to recruitment of the autophagy initiation machinery, initiating a cascade of autophagosome formation locally (Ravenhill et al., 2019; Vargas et al., 2019; Zachari et al., 2019). Additionally, in ubiquitin-independent mitophagy, mitochondria are linked to the autophagosomes by BNIP3, FUNDC1 and NIX, which are specific autophagy receptors. Enhancing mitophagy has emerged as a promising therapeutic strategy in Parkinson’s disease and other age-related disorders (Padmanabhan et al., 2019).
Mitochondria malfunction with age, leading to perturbations in metabolic and energy homeostasis, releasing reactive oxygen species (ROS) (Lopez-Otin et al., 2013; Munkacsy and Rea, 2014). Increases in ROS, causing molecular damage, have long been considered important culprits in aging. However, careful reassessment suggests that mutants displaying subtle increases in ROS can be long-lived, while highly elevated ROS levels are detrimental (Lopez-Otin et al., 2013; Ristow and Schmeisser, 2014). This is a hormesis effect, where low dose of a substance has a positive stimulatory effect but, contrarily, high dose is toxic (Gems and Partridge, 2008). ROS can be protective until a certain threshold of damage is reached, owing to their role in signaling and ability to trigger defense responses that result in increased robustness and longevity. If this threshold is surpassed, then the amount of DNA damage that occurs becomes toxic for the organism and lifespan is shortened (Gems and Partridge, 2008; Ristow and Schmeisser, 2014; Schieber and Chandel, 2014).
Unexpectedly, given the essential role of mitochondria in physiological functions, it has been observed that mutations in some mitochondrial subunits extend lifespan in worms (Feng et al., 2001; Dillin et al., 2002), flies (Copeland et al., 2009) and mice (Liu et al., 2005). Mutations in all mitochondrial respiratory chain complex genes, except complex II, can extend lifespan in C. elegans (Munkacsy and Rea, 2014). It was suggested that this effect occurs as a result of an imbalance between nuclear and mitochondrial encoded subunits, inducing the mitochondrial unfolded protein (UPRmt) response. Furthermore, it is thought that this does not occur in complex II mutants, since it is the only complex that is exclusively encoded by the nuclear genome (Houtkooper et al., 2013). Remarkably, the triggering of the UPRmt in one tissue can be communicated to another tissue, and this communication leads to enhanced protection against organismal stress (Durieux et al., 2011; Zhang et al., 2018). To fully explain the increased longevity of mitochondrial mutants, in addition to induction of the UPRmt, it was proposed that these mutants display enhanced mitophagy, removing defective mitochondria and improving cellular homeostasis. Indeed, an imbalance between mitophagy and mitochondrial biogenesis plays an important role in aging in C. elegans (Palikaras et al., 2015). Also, increasing mitochondrial fission by overexpression of Drp1 in Drosophila enhances mitophagy, maintains mitochondrial respiratory function during aging and extends healthspan (Rana et al., 2017). Another interesting observation linking autophagy and mitochondria is that mitochondrial permeability determines the effect of autophagy on lifespan (Zhou et al., 2019). More precisely, low mitochondrial permeability is required for various autophagy-mediated lifespan extension effects, while increased permeability is detrimental to the organism (Zhou et al., 2019). In addition, there is evidence to support ROS as secondary messengers, not only as damaging agents. For example, mitochondrial ROS produced by reversing electron transport leads to lifespan extension in Drosophila (Scialò et al., 2016). Overall, it seems that mild induction of stress responses, when damage appears to not be overwhelming, is beneficial for aging, possibly by inducing defense mechanisms and preparing the cell for any subsequent damaging insults that may incur. However, as soon as the stress threshold is exceeded, then either excessive damage, or the stress-response pathway itself, may become detrimental and lead to life-shortening consequences.
One of the critical challenges in modern societies is to improve health during aging. The steep increase in life expectancy seen in populations over recent decades is recognized as a remarkable accomplishment due to advancements in medicine, public health and technology. However, this has been accompanied by a greater number of individuals suffering from age-related diseases, such as cancer and neurodegenerative disorders. Enhanced autophagy is a common mechanism among putative anti-aging interventions, and a number of such drugs exist that are capable of increasing the autophagic process (Table 2). This in turn opens up promising clinical avenues to improve health in the elderly, and to prevent the onset of diseases of old age through pharmacologically-modulated autophagy (Partridge et al., 2018; Campisi et al., 2019; Singh et al., 2019).
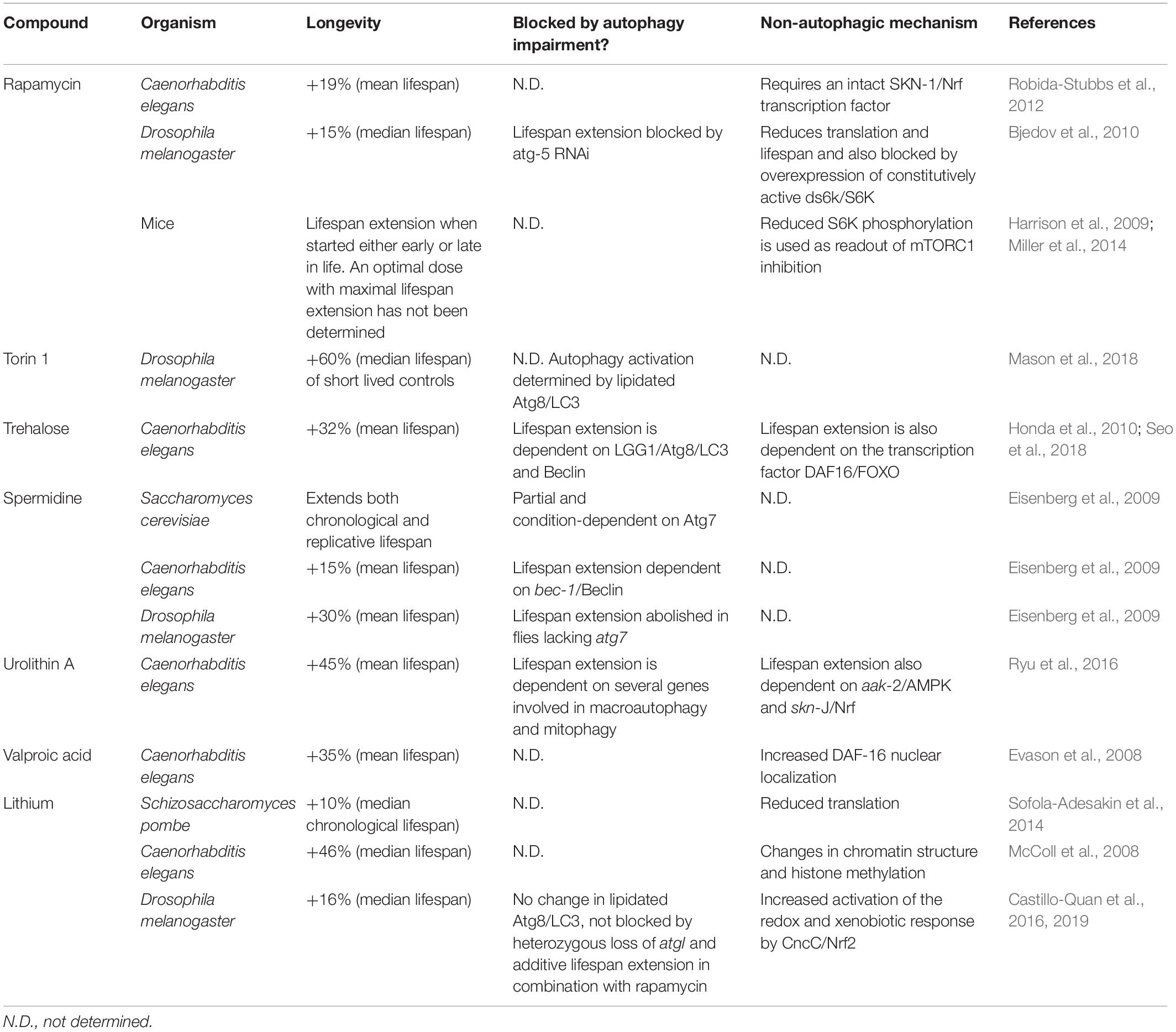
Table 2. Compounds that increase autophagy with potential anti-ageing properties.
Autophagy has a complex relationship with cancer and neurodegeneration (Jiang and Mizushima, 2014; Leidal et al., 2018). Our understanding of the role of autophagy has improved significantly from the study of a variety of genetically engineered mouse models displaying a lack of autophagy, either from Atg7 or Atg5 deletion, in combination with an oncogene mutation KrasG12D, causing either pancreatic intraepithelial neoplasia (PANIN) (Rosenfeldt et al., 2013) or liver adenoma (Takamura et al., 2011). In accordance with the above-mentioned findings, the autophagy-deficient cells suffer from genomic instability (Karantza-Wadsworth et al., 2007; Mathew et al., 2007). Critically, these benign early stage tumors never progress into more malignant ones. This suggests that active autophagy is required for further tumor transformation, most likely to sustain tumor cell survival under stressful conditions owing to lack of nutrients and hypoxia (Kimmelman and White, 2017; Poillet-Perez and White, 2019). It thus appears that human cancers preserve autophagy function. This also points to the fact that complete autophagy ablation in mouse models, while providing a conceptually important insight into the role of autophagy in cancer, may not represent the pathological landscape of the majority of advanced malignant tumors in humans (Poillet-Perez and White, 2019).
Tumors require autophagy to provide the cell with key metabolic intermediates, such as nucleotides and TCA (tricarboxylic acid) cycle metabolites (Guo et al., 2016). This highlights an aspect of tumor vulnerability, whereby autophagy inhibition may be more harmful to tumor tissue than normal healthy tissue. It should be noted that autophagy in the surrounding tissue contributes to tumor growth. One of the key metabolites provided by the host that promotes tumor growth in the pancreas is arginine, which is degraded in autophagy-deficient mice by liver secreted arginase 1 (ARG1) (Poillet-Perez et al., 2018). However, not all tumors tested are arginine auxotrophs, and therefore sensitive to the autophagy status of the host. Another example of the role of autophagy in extra-tumoural tissue can be seen in stroma-associated pancreatic stellate cells, which support pancreatic ductal adenocarcinoma through autophagic alanine secretion (Sousa et al., 2016). Overall, although autophagy can have both tumor-promoting or -reducing effects, depending on the stage of the tumor and its genetic makeup, it is autophagy inhibitors such as hydroxychloroquine that have been primarily tested as anti-cancer therapies, as they are expected to hinder growth in advanced tumors (Poillet-Perez and White, 2019). However, many cancer drugs, such as rapamycin, are autophagy activators. Thus, careful evaluation of the role of autophagy in different tumor types is required. In particular, the effect of autophagy modulation in different drug combinations and regimes needs to be studied in the context of tumor growth.
In the case of neurodegenerative disease, drugs enhancing autophagy, rather than inhibiting, are the most studied. Most misfolded proteins that are deposited in the brains of people affected by neurodegenerative disorders, such as α-synuclein, tau and huntingtin, are autophagy substrates (Menzies et al., 2017). There is evidence that there is insufficient autophagy in many neurodegenerative disorders, and that enhanced autophagy may offer promising therapeutic benefits (Menzies et al., 2017). However, if during aging or in disease, the autophagy process is impaired at any stage, then further up-regulation of autophagy pharmacologically will not aid in aggregate clearance. In keeping with this, an in vitro study demonstrated that autophagy enhancement by rapamycin, or due to starvation, surprisingly can lead to increased toxicity (Tanik et al., 2013). We therefore need to fully understand the role of autophagy, and how perturbations in autophagy flux affect protein aggregation and clearance in neurodegenerative disease. It also highlights the need for more refined autophagy-modulating drugs that can target different stages of the autophagy process. When using autophagy altering drugs that affect other cellular processes, such as in the case of rapamycin, which down-regulates translation, then this additional effect of altered translation on the disease progression should also be evaluated. In aging, the differential rapamycin effects, including the inhibition of translation, and activation of autophagy, are beneficial for longevity (Bjedov et al., 2010), but this may not be the case in disease-related situations. Enhancement of autophagy flux thus offers a potential promising strategy for prevention of neurodegenerative disorders, by improving neuronal proteostasis and preventing cellular toxicity. However, different personalized approaches may be required in terms of optimizing the effects of autophagy modulation and minimizing negative pleiotropic effects of pharmacological interventions.
Similar to neurodegeneration, enhanced autophagy has proven to be an important pharmacological intervention in models of aging. Autophagy can be increased upon inhibition of growth pathways, which are also principal targets of cancer therapy. Interestingly, some of the anti-cancer drugs, such as rapamycin and the MEK/ERK inhibitor trametinib, have been shown to exert pro-longevity effects (Castillo-Quan et al., 2015). The very same nutrient-sensing/growth pathways that are highly up-regulated in cancer, enabling uncontrollable growth of tumor cells, can also promote health if mildly down-regulated to inhibit nutrient-sensing in normal cells (Bjedov and Partridge, 2011; Campisi et al., 2019). Anti-cancer drugs are used in very high concentrations for anti-cancer therapy, often causing side effects. However, in stark contrast, when administered in very low doses to normal non-transformed tissue, they promote anti-aging and disease preventative effects (Bjedov et al., 2010; Slack et al., 2015). It is important to note, however, that not all anti-cancer drugs in small concentrations are expected to improve health. Indeed, many are DNA-damaging agents, such as temozolomide and carboplatin, with negative effects on genome stability (Cheung-Ong et al., 2013). Moreover, as we refine the tools and screening procedures to monitor the autophagic process in healthy and diseased-tissue, it is likely that we will uncover many more autophagy enhancers and inhibitors (Pengo et al., 2018; Panda et al., 2019).
Autophagy activation can be achieved using two licensed drugs that are used to treat epilepsy and mood disorders, namely valproic acid and carbamazepine, as well as the mood-stabilizing drug lithium (Sarkar et al., 2005; Schiebler et al., 2015; Kerr et al., 2018). Valproic acid and lithium have proven anti-aging effects in model organisms. All of these drugs have been linked to mTOR-independent autophagy activation via reducing the recycling of inositol, which in turn reduces inositol 1,4,5-trisphosphate (IP3), disrupting the Beclin-Bcl-2 complex (Ravikumar et al., 2010). Indeed, mTOR-independent autophagy was recently shown to promote longevity and healthspan in mice (Fernandez et al., 2018). Valproic acid has been shown to extend lifespan and to reduce age-related locomotor decline in C. elegans (Evason et al., 2008). Interestingly, while a clear epistatic mechanism of action was not studied, it was observed that valproic acid induces DAF-16/FOXO nuclear accumulation, which may in turn regulate the transcription of autophagy genes (see above). Additionally, the combination of optimal concentrations of valproic acid and trimethadione, another anticonvulsant, had additive effects in extending worm lifespan (Evason et al., 2008). In mice, combinations of valproic acid and lithium retarded the onset and severity of symptomatology, and prolonged the lifespan of a mouse model of amyotrophic lateral sclerosis (ALS) (Feng et al., 2008). Although the main lithium trial in ALS patients was negative (Group et al., 2013), ALS is linked to several different gene mutations, and therefore analysis of genetically defined sub-groups of patients at earlier stages of disease may be necessary. Recent post-mortem analyses of the cerebral cortex transcriptome of ALS patients revealed at least three distinct molecular signatures in which patients could be classified. Intriguingly, the most common molecular subtype included aberrant expression of oxidative and proteostasis stress responses, which included autophagy-related genes (Tam et al., 2019). Therefore, in addition to genetic testing for common gene mutations, a better understanding of the molecular basis of ALS may lead to treatment stratification based upon disease molecular signatures (Group et al., 2013).
The lifespan extending effects of lithium were first shown in C. elegans (McColl et al., 2008) and have been confirmed in yeast and flies (Sofola-Adesakin et al., 2014; Castillo-Quan et al., 2016). In addition, increasing concentrations of lithium in the drinking water have been linked to a reduction in all-cause mortality in a Japanese population (Zarse et al., 2011). Interestingly, none of these studies have tied the longevity properties of lithium to increased autophagy. In yeast, the effects of lithium were linked to reduced translation (Sofola-Adesakin et al., 2014), in C. elegans it was associated with histone methylation and chromatin structure, while in Drosophila it was shown to inhibit glycogen synthase kinase-3 (GSK-3) activity and the transcriptional activation of the cap’n’collar C (CncC)/Nrf2. The latter regulates redox and xenobiotic metabolism (Castillo-Quan et al., 2016). Furthermore, patients suffering from bipolar disorder treated with lithium have longer telomeres, not only in comparison to non-lithium treated patients with bipolar disorder, but also with non-affected relatives (Squassina et al., 2016; Powell et al., 2018). These results suggest that lithium may have many non-autophagic related and pleiotropic anti-aging mechanisms.
Three non-licensed nutraceutical compounds, trehalose, spermidine, and urolithin A have been shown to increase autophagy. Trehalose is a disaccharide used by some species as a mechanism for storing excess sugar to protect against environmental stressors (Sarkar et al., 2007a; Seo et al., 2018). It activates autophagy via a poorly defined mTOR-independent mechanism, and it also acts as chemical chaperone (Ravikumar et al., 2010). It increases clearance of Huntington’s disease-associated polyglutamine expansion aggregates in an atg-5-dependent manner in mammalian cell lines (Sarkar et al., 2007b). When fed to C. elegans, or when trehalose production is genetically enhanced by shifting storage away from glycogen, it increases lifespan (Honda et al., 2010; Seo et al., 2018) with an upregulation of several well-defined markers of autophagy induction in worms (Seo et al., 2018). Lifespan extension and upregulation of atg-9 mRNA levels are DAF-16/FOXO-dependent (Seo et al., 2018). Moreover, the longevity of worms with higher trehalose circulation requires LGG-1/Atg8 and Beclin (Seo et al., 2018), thus demonstrating by genetic epistasis that autophagy is required for trehalose to extend lifespan.
Spermidine is a polyamine that extends the lifespan of yeast, worms, flies and mice (Eisenberg et al., 2009, 2016). In worms, the lifespan-extending effects are dependent on intact Beclin, and in flies on Atg7, demonstrating that autophagy is required for the longevity benefits of spermidine (Eisenberg et al., 2009). In mice, the cardioprotective functions of spermidine were genetically shown to be dependent on intact autophagy. Furthermore, in humans, higher spermidine intake by questionnaire-assessment was associated with lower levels of heart failure (Eisenberg et al., 2016) and a reduction in all-cause mortality in an Italian population (Kiechl et al., 2018).
Urolithin A is one of the three end-products (the other two being Urothilin B and C) of microflora-mediated processing of ellagic acid in the gut from ellagitannins. It is contained in berries, acorns, nuts and tree leaves. When fed to C. elegans, urolithin A extends lifespan and improves healthspan (Ryu et al., 2016), and reduces Aβ-associated memory loss (Fang et al., 2019). The lifespan-extending properties of urolithin A are at least partially dependent on AMPK, and completely dependent on intact mitochondria, as it induces mitophagy (Ryu et al., 2016). The worm lifespan extension produced by urolithins was completely dependent on Pink1, dct-1/BNIP3, Beclin, pdr-1/Parkin, sqst-1, vps-34 and skn-1/Nrf1/2. In mice, urolithin A treatment has also been linked to mitophagy induction and improved muscle function (Ryu et al., 2016).
Among the autophagy-activating drugs with anti-aging effects, rapamycin is the best characterized. It is licensed for clinical use in humans as a co-immunosuppressant in renal transplantation. It is also used to coat coronary stents in the prevention of restenosis following coronary angioplasty, and to treat lymphangioleimyomatosis, a rare lung disease of smooth muscle cell growth. Rapamycin is the flagship mTORC1 inhibitor and acts by allosterically binding to FK506-binding protein12 (FKBP12) to form a complex, which in turn binds and inhibits mTOR (Grolleau et al., 2002; Thoreen et al., 2009). Rapamycin has been shown to extend lifespan in yeast, worms, flies and mice (Powers et al., 2006; Medvedik et al., 2007; Harrison et al., 2009; Bjedov et al., 2010; Robida-Stubbs et al., 2012; Miller et al., 2014), and a randomized controlled preclinical trial in dogs is in the pipeline to study its anti-aging effects (Urfer et al., 2017). In addition, rapamycin treatment in mice and elderly people has been linked to an improved response to vaccination against influenza virus (Chen et al., 2009; Mannick et al., 2014, 2018). The mechanism of action of rapamycin-mediated lifespan extension has been shown to be independent of dietary restriction in flies (Bjedov et al., 2010), and metabolically and transcriptionally different in mice (Miller et al., 2014). While autophagy is assumed to be upregulated when mTOR is inhibited (either genetically or pharmacologically), so far and to the best of our knowledge, the only epistatic investigation showing that autophagy is required for lifespan extension has been performed in flies (Bjedov et al., 2010). However, rapamycin-mediated lifespan extension also requires S6K (Bjedov et al., 2010), another downstream effector important in translation and ribosomal physiology (Pende et al., 2004; Chauvin et al., 2014). In addition, in C. elegans and Drosophila, rapamycin-mediated lifespan extension does not require DAF-16/FOXO (Bjedov et al., 2010; Robida-Stubbs et al., 2012), but instead requires the redox regulator SKN-1/Nrf1/2 in worms (Robida-Stubbs et al., 2012). The effects of rapamycin are mostly specific to mTORC1. However, under certain conditions of prolonged exposure, it can also inhibit mTORC2 (Sarbassov et al., 2006). In rodents and patients, rapamycin leads to insulin resistance and hyperlipidemia (Morrisett et al., 2002; Houde et al., 2010), and this has been attributed to its effects on mTORC2 inhibition (Lamming et al., 2012). Interestingly, in Drosophila higher triacylglycerol content associated with rapamycin treatment is abolished in combination with lithium, and pronounced lifespan extension is achieved when a combination of three drugs, rapamycin, lithium, and trametinib is fed to flies (Castillo-Quan et al., 2019). Efforts continue in developing dual mTORC1/mTORC2 catalytic inhibitors like the Torin1 and Torin2 (Thoreen et al., 2009), which have already been shown to reduce cellular senescence in mammalian cells and extend lifespan in Drosophila (Leontieva and Blagosklonny, 2016; Mason et al., 2018). However, the reality is that we need to identify new compounds that specifically target the autophagy process. Otherwise, the non-autophagy-related effects of inhibiting mTORC1, mTORC2 or other pathways will not allow a clear examination of the potentially beneficial clinical effects of autophagy activation.
In conclusion, we have described the intricate relationship between aging and autophagy, and discussed major anti-aging interventions that depend on autophagy enhancement. There is increasing evidence that boosting autophagy flux and the recycling of damaged cellular components may prove to be an effective anti-aging strategy. However, we need to be mindful of the fact that autophagy is a degradative process, and that strong upregulation may therefore be detrimental to the organism. The more evidence we gather for improvement of health during aging by targeting autophagy, the more complexities we are uncovering. Numerous questions are continually arising concerning the optimal manipulation of autophagy that is required to benefit a given organism. These include factors relating to the intensity of autophagy increase, its timing and effect in the young versus old individuals, whether non-selective autophagy should be targeted or if specific cargoes should be selectively degraded. Finally, the question of tissue specificity needs to be addressed, to determine whether autophagy offers greater benefits when augmented in a specific single tissue or in a combination of tissues. Furthermore, despite tremendous progress in the fields of autophagy and aging, in order for successful therapies to be developed, we need to improve our measurements of autophagy flux. By determining which parts of this multistep process are failing in aging and disease, we can tailor our interventions accordingly. One of the main challenges will be to develop specific strategies that either alter selective autophagy, or restore rate-limiting steps in the autophagy process. Many of the current autophagy activators that have been characterized also affect other intracellular processes, such as the inhibition of translation by rapamycin. It is currently unclear whether autophagy-specific drugs, or those targeting multiple cellular pathways, will provide the most potent health benefits. In order to optimize the benefits of autophagy, short temporary treatment may offer the greatest advantages, cleansing the cell of damaged components, while minimizing toxic side-effects. Similar approaches also apply to senolytic drugs that remove senescent cells from an organism. In summary, autophagy has been implicated in a plethora of essential cellular processes, including those involved in the DNA damage response, immunity, cell death and senescence, and thus has critical importance in the identification of new drugs and strategies to improve healthy aging.
ES, JC-Q, VM, CL, RK, KK, and IB wrote the manuscript. IB prepared the figure.
This work was supported by the American Federation for Aging Research/Glenn Foundation for Medical Research Postdoctoral Fellowship (Grant PD18019 to JC-Q), as well as a Wellcome Trust Clinical Research Career Development Fellowship and a Rosetrees UCL Excellence Fellowship (KK). IB acknowledges funding from ERC StG 311331, ERC PoC 842174, Royal Society Research Grant, The Bill Lyons Foundation, and the CRUK-UCL Centre Award (C416/A25145). RK was supported by the Medical Research Council (MC_U12266B).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agrotis, A., Pengo, N., Burden, J. J., and Ketteler, R. (2019). Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 15, 976–997. doi: 10.1080/15548627.2019.1569925
Aris, J. P., Alvers, A. L., Ferraiuolo, R. A., Fishwick, L. K., Hanvivatpong, A., Hu, D., et al. (2013). Autophagy and leucine promote chronological longevity and respiration proficiency during calorie restriction in yeast. Exp. Gerontol. 48, 1107–1119. doi: 10.1016/j.exger.2013.01.006
Artal-Martinez de Narvajas, A., Gomez, T. S., Zhang, J. S., Mann, A. O., Taoda, Y., Gorman, J. A., et al. (2013). Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell Biol. 33, 3983–3993. doi: 10.1128/MCB.00813-13
Baek, S. H., and Kim, K. I. (2017). Epigenetic control of autophagy: nuclear events gain more attention. Mol. Cell 65, 781–785. doi: 10.1016/j.molcel.2016.12.027
Bai, H., Kang, P., Hernandez, A. M., and Tatar, M. (2013). Activin signaling targeted by insulin/dFOXO regulates aging and muscle proteostasis in Drosophila. PLoS Genet. 9:e1003941. doi: 10.1371/journal.pgen.1003941
Barzilai, N., Crandall, J. P., Kritchevsky, S. B., and Espeland, M. A. (2016). Metformin as a tool to target aging. Cell Metab. 23, 1060–1065. doi: 10.1016/j.cmet.2016.05.011
Bennett-Baker, P. E., Wilkowski, J., and Burke, D. T. (2003). Age-associated activation of epigenetically repressed genes in the mouse. Genetics 165, 2055–2062.
Bjedov, I., and Partridge, L. (2011). A longer and healthier life with TOR down-regulation: genetics and drugs. Biochem. Soc. Trans. 39, 460–465. doi: 10.1042/BST0390460
Bjedov, I., Toivonen, J. M., Kerr, F., Slack, C., Jacobson, J., Foley, A., et al. (2010). Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 11, 35–46. doi: 10.1016/j.cmet.2009.11.010
Blackford, A. N., and Jackson, S. P. (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817. doi: 10.1016/j.molcel.2017.05.015
Botti-Millet, J., Nascimbeni, A. C., Dupont, N., Morel, E., and Codogno, P. (2016). Fine-tuning autophagy: from transcriptional to posttranslational regulation. Am. J. Physiol. Cell Physiol. 311, C351–C362. doi: 10.1152/ajpcell.00129.2016
Bustos, V., and Partridge, L. (2017). Good Ol’ fat: links between lipid signaling and longevity. Trends Biochem. Sci. 42, 812–823. doi: 10.1016/j.tibs.2017.07.001
Cabreiro, F., Au, C., Leung, K. Y., Vergara-Irigaray, N., Cocheme, H. M., Noori, T., et al. (2013). Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 153, 228–239. doi: 10.1016/j.cell.2013.02.035
Campisi, J., Kapahi, P., Lithgow, G. J., Melov, S., Newman, J. C., and Verdin, E. (2019). From discoveries in ageing research to therapeutics for healthy ageing. Nature 571, 183–192. doi: 10.1038/s41586-019-1365-2
Carlsson, S. R., and Simonsen, A. (2015). Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 128, 193–205. doi: 10.1242/jcs.141036
Carrero, D., Soria-Valles, C., and Lopez-Otin, C. (2016). Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells. Dis. Model. Mech. 9, 719–735. doi: 10.1242/dmm.024711
Castillo-Quan, J. I., Kinghorn, K. J., and Bjedov, I. (2015). Genetics and pharmacology of longevity: the road to therapeutics for healthy aging. Adv. Genet. 90, 1–101. doi: 10.1016/bs.adgen.2015.06.002
Castillo-Quan, J. I., Li, L., Kinghorn, K. J., Ivanov, D. K., Tain, L. S., Slack, C., et al. (2016). Lithium promotes longevity through GSK3/NRF2-dependent hormesis. Cell Rep. 15, 638–650. doi: 10.1016/j.celrep.2016.03.041
Castillo-Quan, J. I., Tain, L. S., Kinghorn, K. J., Li, L., Gronke, S., Hinze, Y., et al. (2019). A triple drug combination targeting components of the nutrient-sensing network maximizes longevity. Proc. Natl. Acad. Sci. U.S.A. 116, 20817–20819. doi: 10.1073/pnas.1913212116
Chang, J. T., Kumsta, C., Hellman, A. B., Adams, L. M., and Hansen, M. (2017). Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife 6:e18459. doi: 10.7554/eLife.18459
Chauvin, C., Koka, V., Nouschi, A., Mieulet, V., Hoareau-Aveilla, C., Dreazen, A., et al. (2014). Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 33, 474–483. doi: 10.1038/onc.2012.606
Chen, C., Liu, Y., and Zheng, P. (2009). mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2:ra75. doi: 10.1126/scisignal.2000559
Chen, S., Wang, C., Sun, L., Wang, D. L., Chen, L., Huang, Z., et al. (2015). RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol. Cell Biol. 35, 406–416. doi: 10.1128/MCB.01044-14
Cheng, Z. (2019). The FoxO-autophagy axis in health and disease. Trends Endocrinol. Metab. 30, 658–671. doi: 10.1016/j.tem.2019.07.009
Cheung-Ong, K., Giaever, G., and Nislow, C. (2013). DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem. Biol. 20, 648–659. doi: 10.1016/j.chembiol.2013.04.007
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610. doi: 10.1126/science.1124646
Cohen, E., Paulsson, J. F., Blinder, P., Burstyn-Cohen, T., Du, D., Estepa, G., et al. (2009). Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139, 1157–1169. doi: 10.1016/j.cell.2009.11.014
Copeland, J. M., Cho, J., Lo, T Jr., Hur, J. H., Bahadorani, S., Arabyan, T., et al. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. doi: 10.1016/j.cub.2009.08.016
Delorme-Axford, E., and Klionsky, D. J. (2018). Transcriptional and post-transcriptional regulation of autophagy in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 293, 5396–5403. doi: 10.1074/jbc.R117.804641
Demontis, F., and Perrimon, N. (2010). FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 143, 813–825. doi: 10.1016/j.cell.2010.10.007
Dikic, I. (2017). Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 86, 193–224. doi: 10.1146/annurev-biochem-061516-044908
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. doi: 10.1126/science.1077780
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. doi: 10.1016/j.cell.2010.12.016
Eapen, V. V., Waterman, D. P., Bernard, A., Schiffmann, N., Sayas, E., Kamber, R., et al. (2017). A pathway of targeted autophagy is induced by DNA damage in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 114, E1158–E1167. doi: 10.1073/pnas.1614364114
Eisenberg, T., Abdellatif, M., Schroeder, S., Primessnig, U., Stekovic, S., Pendl, T., et al. (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438. doi: 10.1038/nm.4222
Eisenberg, T., Knauer, H., Schauer, A., Buttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314. doi: 10.1038/ncb1975
Eliopoulos, A. G., Havaki, S., and Gorgoulis, V. G. (2016). DNA damage response and autophagy: a meaningful partnership. Front. Genet. 7:204. doi: 10.3389/fgene.2016.00204
Evason, K., Collins, J. J., Huang, C., Hughes, S., and Kornfeld, K. (2008). Valproic acid extends Caenorhabditis elegans lifespan. Aging Cell 7, 305–317. doi: 10.1111/j.1474-9726.2008.00375.x
Ezcurra, M., Benedetto, A., Sornda, T., Gilliat, A. F., Au, C., Zhang, Q., et al. (2018). C. elegans eats its own intestine to make yolk leading to multiple senescent pathologies. Curr. Biol. 28:2544-2556.e5. doi: 10.1016/j.cub.2018.06.035
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Feng, H. L., Leng, Y., Ma, C. H., Zhang, J., Ren, M., and Chuang, D. M. (2008). Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 155, 567–572. doi: 10.1016/j.neuroscience.2008.06.040
Feng, J., Bussiere, F., and Hekimi, S. (2001). Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644. doi: 10.1016/s1534-5807(01)00071-5
Fernandez, A. F., Sebti, S., Wei, Y., Zou, Z., Shi, M., McMillan, K. L., et al. (2018). Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140. doi: 10.1038/s41586-018-0162-7
Finkel, T., Serrano, M., and Blasco, M. A. (2007). The common biology of cancer and ageing. Nature 448, 767–774. doi: 10.1038/nature05985
Fitzwalter, B. E., Towers, C. G., Sullivan, K. D., Andrysik, Z., Hoh, M., Ludwig, M., et al. (2018). Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev. Cell 44:555-565.e3. doi: 10.1016/j.devcel.2018.02.014
Flachsbart, F., Caliebe, A., Kleindorp, R., Blanche, H., von Eller-Eberstein, H., Nikolaus, S., et al. (2009). Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. U.S.A. 106, 2700–2705. doi: 10.1073/pnas.0809594106
Folick, A., Oakley, H. D., Yu, Y., Armstrong, E. H., Kumari, M., Sanor, L., et al. (2015). Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347, 83–86. doi: 10.1126/science.1258857
Fontana, L., and Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. doi: 10.1016/j.cell.2015.02.020
Fontana, L., Partridge, L., and Longo, V. D. (2010). Extending healthy life span–from yeast to humans. Science 328, 321–326. doi: 10.1126/science.1172539
Fullgrabe, J., Klionsky, D. J., and Joseph, B. (2014). The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 15, 65–74. doi: 10.1038/nrm3716
Fullgrabe, J., Lynch-Day, M. A., Heldring, N., Li, W., Struijk, R. B., Ma, Q., et al. (2013). The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500, 468–471. doi: 10.1038/nature12313
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi: 10.15252/embj.201796697
Garcia-Cao, I., Garcia-Cao, M., Martin-Caballero, J., Criado, L. M., Klatt, P., Flores, J. M., et al. (2002). “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 21, 6225–6235. doi: 10.1093/emboj/cdf595
Gelino, S., Chang, J. T., Kumsta, C., She, X., Davis, A., Nguyen, C., et al. (2016). Intestinal autophagy improves healthspan and longevity in C. elegans during dietary restriction. PLoS Genet. 12:e1006135. doi: 10.1371/journal.pgen.1006135
Gems, D., and Partridge, L. (2008). Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab. 7, 200–203. doi: 10.1016/j.cmet.2008.01.001
Gladkova, C., Maslen, S. L., Skehel, J. M., and Komander, D. (2018). Mechanism of parkin activation by PINK1. Nature 559, 410–414. doi: 10.1038/s41586-018-0224-x
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. doi: 10.1038/ncb2220
Greer, E. L., Maures, T. J., Ucar, D., Hauswirth, A. G., Mancini, E., Lim, J. P., et al. (2011). Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479, 365–371. doi: 10.1038/nature10572
Grolleau, A., Bowman, J., Pradet-Balade, B., Puravs, E., Hanash, S., Garcia-Sanz, J. A., et al. (2002). Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J. Biol. Chem. 277, 22175–22184. doi: 10.1074/jbc.m202014200
Group, U. K.-L. S., Morrison, K. E., Dhariwal, S., Hornabrook, R., Savage, L., Burn, D. J., et al. (2013). Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 12, 339–345. doi: 10.1016/S1474-4422(13)70037-1
Guo, J. Y., Teng, X., Laddha, S. V., Ma, S., Van Nostrand, S. C., Yang, Y., et al. (2016). Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717. doi: 10.1101/gad.283416.116
Han, S., Schroeder, E. A., Silva-Garcia, C. G., Hebestreit, K., Mair, W. B., and Brunet, A. (2017). Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 544, 185–190. doi: 10.1038/nature21686
Hansen, M., Chandra, A., Mitic, L. L., Onken, B., Driscoll, M., and Kenyon, C. (2008). A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 4:e24. doi: 10.1371/journal.pgen.0040024
Hansen, M., Rubinsztein, D. C., and Walker, D. W. (2018). Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol. 19, 579–593. doi: 10.1038/s41580-018-0033-y
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hardie, D. G., Schaffer, B. E., and Brunet, A. (2016). AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201. doi: 10.1016/j.tcb.2015.10.013
Harper, J. W., Ordureau, A., and Heo, J. M. (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108. doi: 10.1038/nrm.2017.129
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi: 10.1038/nature08221
Heestand, B. N., Shen, Y., Liu, W., Magner, D. B., Storm, N., Meharg, C., et al. (2013). Dietary restriction induced longevity is mediated by nuclear receptor NHR-62 in Caenorhabditis elegans. PLoS Genet. 9:e1003651. doi: 10.1371/journal.pgen.1003651
Hewitt, G., and Korolchuk, V. I. (2017). Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol. 27, 340–351. doi: 10.1016/j.tcb.2016.11.011
Hewitt, G., Carroll, B., Sarallah, R., Correia-Melo, C., Ogrodnik, M., Nelson, G., et al. (2016). SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 12, 1917–1930. doi: 10.1080/15548627.2016.1210368
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435. doi: 10.1038/s41580-019-0101-y
Honda, Y., Tanaka, M., and Honda, S. (2010). Trehalose extends longevity in the nematode Caenorhabditis elegans. Aging Cell 9, 558–569. doi: 10.1111/j.1474-9726.2010.00582.x
Houde, V. P., Brule, S., Festuccia, W. T., Blanchard, P. G., Bellmann, K., Deshaies, Y., et al. (2010). Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59, 1338–1348. doi: 10.2337/db09-1324
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. doi: 10.1038/nature12188
Hsu, A. L., Murphy, C. T., and Kenyon, C. (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145. doi: 10.1126/science.1083701
Huang, R., Xu, Y., Wan, W., Shou, X., Qian, J., You, Z., et al. (2015). Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell 57, 456–466. doi: 10.1016/j.molcel.2014.12.013
Jeggo, P. A., Downs, J. A., and Gasser, S. M. (2017). Chromatin modifiers and remodellers in DNA repair and signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 5:372.
Jia, K., and Levine, B. (2007). Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3, 597–599. doi: 10.4161/auto.4989
Jiang, P., and Mizushima, N. (2014). Autophagy and human diseases. Cell Res. 24, 69–79. doi: 10.1038/cr.2013.161
Jin, C., Li, J., Green, C. D., Yu, X., Tang, X., Han, D., et al. (2011). Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 14, 161–172. doi: 10.1016/j.cmet.2011.07.001
Jove, M., Naudi, A., Aledo, J. C., Cabre, R., Ayala, V., Portero-Otin, M., et al. (2013). Plasma long-chain free fatty acids predict mammalian longevity. Sci. Rep. 3:3346. doi: 10.1038/srep03346
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kang, C., Xu, Q., Martin, T. D., Li, M. Z., Demaria, M., Aron, L., et al. (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349:aaa5612. doi: 10.1126/science.aaa5612
Kang, C., You, Y. J., and Avery, L. (2007). Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 21, 2161–2171. doi: 10.1101/gad.1573107
Kapahi, P., Zid, B. M., Harper, T., Koslover, D., Sapin, V., and Benzer, S. (2004). Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 14, 885–890. doi: 10.1016/j.cub.2004.03.059
Karantza-Wadsworth, V., Patel, S., Kravchuk, O., Chen, G., Mathew, R., Jin, S., et al. (2007). Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 21, 1621–1635. doi: 10.1101/gad.1565707
Kauffman, K. J., Yu, S., Jin, J., Mugo, B., Nguyen, N., O’Brien, A., et al. (2018). Delipidation of mammalian Atg8-family proteins by each of the four ATG4 proteases. Autophagy 14, 992–1010. doi: 10.1080/15548627.2018.1437341
Kaushik, S., and Cuervo, A. M. (2018). The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381. doi: 10.1038/s41580-018-0001-6
Kennedy, B. K., Berger, S. L., Brunet, A., Campisi, J., Cuervo, A. M., Epel, E. S., et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. doi: 10.1016/j.cell.2014.10.039
Kenyon, C. (2010). The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 9–16. doi: 10.1098/rstb.2010.0276
Kenzelmann Broz, D., Spano Mello, S., Bieging, K. T., Jiang, D., Dusek, R. L., Brady, C. A., et al. (2013). Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031. doi: 10.1101/gad.212282.112
Kerr, F., Bjedov, I., and Sofola-Adesakin, O. (2018). Molecular mechanisms of lithium action: switching the light on multiple targets for dementia using animal models. Front. Mol. Neurosci. 11:297. doi: 10.3389/fnmol.2018.00297
Kiechl, S., Pechlaner, R., Willeit, P., Notdurfter, M., Paulweber, B., Willeit, K., et al. (2018). Higher spermidine intake is linked to lower mortality: a prospective population-based study. Am. J. Clin. Nutr. 108, 371–380. doi: 10.1093/ajcn/nqy102
Kim, J., and Guan, K. L. (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell. Biol. 21, 63–71. doi: 10.1038/s41556-018-0205-1
Kimmelman, A. C., and White, E. (2017). Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043. doi: 10.1016/j.cmet.2017.04.004
Kirkin, V. (2019). History of the selective autophagy research: how did it begin and where does it stand today? J. Mol. Biol. doi: 10.1016/j.jmb.2019.05.010 [Epub ahead of print].
Klionsky, D. J., Abdalla, F. C., Abeliovich, H., Abraham, R. T., Acevedo-Arozena, A., Adeli, K., et al. (2012). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544.
Kocaturk, N. M., and Gozuacik, D. (2018). Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 6:128. doi: 10.3389/fcell.2018.00128
Koga, H., Kaushik, S., and Cuervo, A. M. (2010). Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 24, 3052–3065. doi: 10.1096/fj.09-144519
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Ktistakis, N. T., and Tooze, S. A. (2016). Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 26, 624–635. doi: 10.1016/j.tcb.2016.03.006
Ktistakis, N. T., Karanasios, E., and Manifava, M. (2014). Dynamics of autophagosome formation: a pulse and a sequence of waves. Biochem. Soc. Trans. 42, 1389–1395. doi: 10.1042/BST20140183
Kuma, A., Komatsu, M., and Mizushima, N. (2017). Autophagy-monitoring and autophagy-deficient mice. Autophagy 13, 1619–1628. doi: 10.1080/15548627.2017.1343770
Lahiri, V., Hawkins, W. D., and Klionsky, D. J. (2019). Watch what you (Self-) eat: autophagic mechanisms that modulate metabolism. Cell Metab. 29, 803–826. doi: 10.1016/j.cmet.2019.03.003
Lamming, D. W., Ye, L., Katajisto, P., Goncalves, M. D., Saitoh, M., Stevens, D. M., et al. (2012). Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643. doi: 10.1126/science.1215135
Lapierre, L. R., De Magalhaes Filho, C. D., McQuary, P. R., Chu, C. C., Visvikis, O., Chang, J. T., et al. (2013). The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 4:2267. doi: 10.1038/ncomms3267
Lapierre, L. R., Gelino, S., Melendez, A., and Hansen, M. (2011). Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol. 21, 1507–1514. doi: 10.1016/j.cub.2011.07.042
Lapierre, L. R., Kumsta, C., Sandri, M., Ballabio, A., and Hansen, M. (2015). Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11, 867–880. doi: 10.1080/15548627.2015.1034410
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Leidal, A. M., Levine, B., and Debnath, J. (2018). Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 20, 1338–1348. doi: 10.1038/s41556-018-0235-8
Leontieva, O. V., and Blagosklonny, M. V. (2016). Gerosuppression by pan-mTOR inhibitors. Aging 8, 3535–3551. doi: 10.18632/aging.101155
Li, J., Labbadia, J., and Morimoto, R. I. (2017). Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol. 27, 895–905. doi: 10.1016/j.tcb.2017.08.002
Li, X., Yu, W., Qian, X., Xia, Y., Zheng, Y., Lee, J. H., et al. (2017). Nucleus-Translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol. Cell 66:e689. doi: 10.1016/j.molcel.2017.04.026
Lin, W., Yuan, N., Wang, Z., Cao, Y., Fang, Y., Li, X., et al. (2015). Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Sci. Rep. 5:12362. doi: 10.1038/srep12362
Liu, E. Y., Xu, N., O’Prey, J., Lao, L. Y., Joshi, S., Long, J. S., et al. (2015). Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad. Sci. U.S.A. 112, 773–778. doi: 10.1073/pnas.1409563112
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016). p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 21:29. doi: 10.1186/s11658-016-0031-z
Liu, X., Jiang, N., Hughes, B., Bigras, E., Shoubridge, E., and Hekimi, S. (2005). Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 19, 2424–2434. doi: 10.1101/gad.1352905
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi: 10.1016/j.cmet.2007.11.001
Mannick, J. B., Del Giudice, G., Lattanzi, M., Valiante, N. M., Praestgaard, J., Huang, B., et al. (2014). mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 6:268ra179. doi: 10.1126/scitranslmed.3009892
Mannick, J. B., Morris, M., Hockey, H. P., Roma, G., Beibel, M., Kulmatycki, K., et al. (2018). TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl Med. 10:eaaq1564. doi: 10.1126/scitranslmed.aaq1564
Martins, R., Lithgow, G. J., and Link, W. (2016). Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15, 196–207. doi: 10.1111/acel.12427
Maruzs, T., Simon-Vecsei, Z., Kiss, V., Csizmadia, T., and Juhasz, G. (2019). On the fly: recent progress on autophagy and aging in Drosophila. Front. Cell Dev. Biol. 7:140. doi: 10.3389/fcell.2019.00140
Mason, J. S., Wileman, T., and Chapman, T. (2018). Lifespan extension without fertility reduction following dietary addition of the autophagy activator Torin1 in Drosophila melanogaster. PLoS One 13:e0190105. doi: 10.1371/journal.pone.0190105
Matheu, A., Maraver, A., Klatt, P., Flores, I., Garcia-Cao, I., Borras, C., et al. (2007). Delayed ageing through damage protection by the Arf/p53 pathway. Nature 448, 375–379. doi: 10.1038/nature05949
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H. Y., et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075. doi: 10.1016/j.cell.2009.03.048
Mathew, R., Kongara, S., Beaudoin, B., Karp, C. M., Bray, K., Degenhardt, K., et al. (2007). Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21, 1367–1381. doi: 10.1101/gad.1545107
Maures, T. J., Greer, E. L., Hauswirth, A. G., and Brunet, A. (2011). The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10, 980–990. doi: 10.1111/j.1474-9726.2011.00738.x
McColl, G., Killilea, D. W., Hubbard, A. E., Vantipalli, M. C., Melov, S., and Lithgow, G. J. (2008). Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J. Biol. Chem. 283, 350–357. doi: 10.1074/jbc.m705028200
Medvedik, O., Lamming, D. W., Kim, K. D., and Sinclair, D. A. (2007). MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 5:e261. doi: 10.1371/journal.pbio.0050261
Mehta, R., Chandler-Brown, D., Ramos, F. J., Shamieh, L. S., and Kaeberlein, M. (2010). Regulation of mRNA translation as a conserved mechanism of longevity control. Adv. Exp. Med. Biol. 694, 14–29. doi: 10.1007/978-1-4419-7002-2_2
Melendez, A., Talloczy, Z., Seaman, M., Eskelinen, E. L., Hall, D. H., and Levine, B. (2003). Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391. doi: 10.1126/science.1087782
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Miller, R. A., Harrison, D. E., Astle, C. M., Fernandez, E., Flurkey, K., Han, M., et al. (2014). Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13, 468–477. doi: 10.1111/acel.12194
Mizushima, N. (2018). A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527. doi: 10.1038/s41556-018-0092-5
Morrisett, J. D., Abdel-Fattah, G., Hoogeveen, R., Mitchell, E., Ballantyne, C. M., Pownall, H. J., et al. (2002). Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J. Lipid Res. 43, 1170–1180.
Munkacsy, E., and Rea, S. L. (2014). The paradox of mitochondrial dysfunction and extended longevity. Exp. Gerontol. 56C, 221–233. doi: 10.1016/j.exger.2014.03.016
Munkacsy, E., Chocron, E. S., Quintanilla, L., Gendron, C. M., Pletcher, S. D., and Pickering, A. M. (2019). Neuronal-specific proteasome augmentation via Prosbeta5 overexpression extends lifespan and reduces age-related cognitive decline. Aging Cell 18:e13005. doi: 10.1111/acel.13005
Nakamura, S., and Yoshimori, T. (2017). New insights into autophagosome-lysosome fusion. J. Cell Sci. 130, 1209–1216. doi: 10.1242/jcs.196352
Nakatogawa, H. (2013). Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 55, 39–50. doi: 10.1042/bse0550039
Nassour, J., Radford, R., Correia, A., Fuste, J. M., Schoell, B., Jauch, A., et al. (2019). Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663. doi: 10.1038/s41586-019-0885-0
Nguyen, N. N., Rana, A., Goldman, C., Moore, R., Tai, J., Hong, Y., et al. (2019). Proteasome beta5 subunit overexpression improves proteostasis during aging and extends lifespan in Drosophila melanogaster. Sci. Rep. 9:3170. doi: 10.1038/s41598-019-39508-4
Nguyen, T. B., and Olzmann, J. A. (2017). Lipid droplets and lipotoxicity during autophagy. Autophagy 13, 2002–2003. doi: 10.1080/15548627.2017.1359451
Nguyen, T. N., Padman, B. S., Usher, J., Oorschot, V., Ramm, G., and Lazarou, M. (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874. doi: 10.1083/jcb.201607039
Niccoli, T., and Partridge, L. (2012). Ageing as a risk factor for disease. Curr. Biol. 22, R741–R752. doi: 10.1016/j.cub.2012.07.024
Ochs, F., Somyajit, K., Altmeyer, M., Rask, M. B., Lukas, J., and Lukas, C. (2016). 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 23, 714–721. doi: 10.1038/nsmb.3251
O’Rourke, E. J., Kuballa, P., Xavier, R., and Ruvkun, G. (2013). omega-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 27, 429–440. doi: 10.1101/gad.205294.112
Padmanabhan, S., Polinski, N. K., Menalled, L. B., Baptista, M. A. S., and Fiske, B. K. (2019). The Michael J. Fox foundation for Parkinson’s research strategy to advance therapeutic development of PINK1 and parkin. Biomolecules 9:E296. doi: 10.3390/biom9080296
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Panda, P. K., Fahrner, A., Vats, S., Seranova, E., Sharma, V., Chipara, M., et al. (2019). Chemical screening approaches enabling drug discovery of autophagy modulators for biomedical applications in human diseases. Front. Cell Dev. Biol. 7:38. doi: 10.3389/fcell.2019.00038
Papsdorf, K., and Brunet, A. (2019). Linking lipid metabolism to chromatin regulation in aging. Trends Cell Biol. 29, 97–116. doi: 10.1016/j.tcb.2018.09.004
Park, C., Suh, Y., and Cuervo, A. M. (2015). Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 6:6823. doi: 10.1038/ncomms7823
Partridge, L. (2010). The new biology of ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 147–154.
Partridge, L., Deelen, J., and Slagboom, P. E. (2018). Facing up to the global challenges of ageing. Nature 561, 45–56. doi: 10.1038/s41586-018-0457-8
Pende, M., Um, S. H., Mieulet, V., Sticker, M., Goss, V. L., Mestan, J., et al. (2004). S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell Biol. 24, 3112–3124. doi: 10.1128/mcb.24.8.3112-3124.2004
Pengo, N., Agrotis, A., Prak, K., Jones, J., and Ketteler, R. (2017). A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat. Commun. 8:294. doi: 10.1038/s41467-017-00303-2
Pengo, N., Prak, K., Costa, J. R., Luft, C., Agrotis, A., Freeman, J., et al. (2018). Identification of kinases and phosphatases that regulate ATG4B activity by sirna and small molecule screening in cells. Front. Cell Dev. Biol. 6:148. doi: 10.3389/fcell.2018.00148
Poillet-Perez, L., and White, E. (2019). Role of tumor and host autophagy in cancer metabolism. Genes Dev. 33, 610–619. doi: 10.1101/gad.325514.119
Poillet-Perez, L., Xie, X., Zhan, L., Yang, Y., Sharp, D. W., Hu, Z. S., et al. (2018). Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573. doi: 10.1038/s41586-018-0697-7
Powell, T. R., Dima, D., Frangou, S., and Breen, G. (2018). Telomere length and bipolar disorder. Neuropsychopharmacology 43:454. doi: 10.1038/npp.2017.239
Powers, R. W. III, Kaeberlein, M., Caldwell, S. D., Kennedy, B. K., and Fields, S. (2006). Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184. doi: 10.1101/gad.1381406
Pryor, R., and Cabreiro, F. (2015). Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem. J. 471, 307–322. doi: 10.1042/BJ20150497
Pryor, R., Norvaisas, P., Marinos, G., Best, L., Thingholm, L. B., Quintaneiro, L. M., et al. (2019). Host-microbe-drug-nutrient screen identifies bacterial effectors of metformin therapy. Cell 178:e1229. doi: 10.1016/j.cell.2019.08.003
Pyo, J. O., Yoo, S. M., Ahn, H. H., Nah, J., Hong, S. H., Kam, T. I., et al. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4:2300. doi: 10.1038/ncomms3300
Rae, M. J., Butler, R. N., Campisi, J., de Grey, A. D., Finch, C. E., Gough, M., et al. (2010). The demographic and biomedical case for late-life interventions in aging. Sci. Transl. Med. 240cm21. doi: 10.1126/scitranslmed.3000822
Rana, A., Oliveira, M. P., Khamoui, A. V., Aparicio, R., Rera, M., Rossiter, H. B., et al. (2017). Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 8:448. doi: 10.1038/s41467-017-00525-4
Ravenhill, B. J., Boyle, K. B., von Muhlinen, N., Ellison, C. J., Masson, G. R., Otten, E. G., et al. (2019). The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol. Cell 74:e326. doi: 10.1016/j.molcel.2019.01.041
Ravikumar, B., Sarkar, S., Davies, J. E., Futter, M., Garcia-Arencibia, M., Green-Thompson, Z. W., et al. (2010). Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435.
Rello-Varona, S., Lissa, D., Shen, S., Niso-Santano, M., Senovilla, L., Marino, G., et al. (2012). Autophagic removal of micronuclei. Cell Cycle 11, 170–176. doi: 10.4161/cc.11.1.18564
Ribe, E. M., and Lovestone, S. (2016). Insulin signalling in Alzheimer’s disease and diabetes: from epidemiology to molecular links. J. Intern. Med. 280, 430–442. doi: 10.1111/joim.12534
Riley, B. E., Kaiser, S. E., Shaler, T. A., Ng, A. C., Hara, T., Hipp, M. S., et al. (2010). Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J. Cell Biol. 191, 537–552. doi: 10.1083/jcb.201005012
Ristow, M., and Schmeisser, K. (2014). Mitohormesis: promoting health and lifespan by increased levels of Reactive Oxygen Species (ROS). Dose Response 12, 288–341.
Robert, T., Vanoli, F., Chiolo, I., Shubassi, G., Bernstein, K. A., Rothstein, R., et al. (2011). HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471, 74–79. doi: 10.1038/nature09803
Robida-Stubbs, S., Glover-Cutter, K., Lamming, D. W., Mizunuma, M., Narasimhan, S. D., Neumann-Haefelin, E., et al. (2012). TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 15, 713–724. doi: 10.1016/j.cmet.2012.04.007
Rodgers, K., and McVey, M. (2016). Error-prone repair of DNA double-strand breaks. J. Cell Physiol. 231, 15–24. doi: 10.1002/jcp.25053
Rosenfeldt, M. T., O’Prey, J., Morton, J. P., Nixon, C., MacKay, G., Mrowinska, A., et al. (2013). p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300. doi: 10.1038/nature12865
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Felix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888. doi: 10.1038/nm.4132
Salminen, A., Kaarniranta, K., and Kauppinen, A. (2016). Age-related changes in AMPK activation: role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 28, 15–26. doi: 10.1016/j.arr.2016.04.003
Sands, W. A., Page, M. M., and Selman, C. (2017). Proteostasis and ageing: insights from long-lived mutant mice. J. Physiol. 595, 6383–6390. doi: 10.1113/JP274334
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. doi: 10.1016/j.molcel.2006.03.029
Sarkar, S., Davies, J. E., Huang, Z., Tunnacliffe, A., and Rubinsztein, D. C. (2007a). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 282, 5641–5652. doi: 10.1074/jbc.m609532200
Sarkar, S., Floto, R. A., Berger, Z., Imarisio, S., Cordenier, A., Pasco, M., et al. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111. doi: 10.1083/jcb.200504035
Sarkar, S., Perlstein, E. O., Imarisio, S., Pineau, S., Cordenier, A., Maglathlin, R. L., et al. (2007b). Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 3, 331–338. doi: 10.1038/nchembio883
Saxton, R. A., and Sabatini, D. M. (2017). mTOR Signaling in Growth. Metabolism, and Disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Schieber, M., and Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462. doi: 10.1016/j.cub.2014.03.034
Schiebler, M., Brown, K., Hegyi, K., Newton, S. M., Renna, M., Hepburn, L., et al. (2015). Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 7, 127–139. doi: 10.15252/emmm.201404137
Scialò, F., Sriram, A., Fernández-Ayala, D., Gubina, N., Lõhmus, M., Nelson, G., et al. (2016). Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734. doi: 10.1016/j.cmet.2016.03.009
Scott, R. C., Juhasz, G., and Neufeld, T. P. (2007). Direct induction of autophagy by atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17, 1–11. doi: 10.1016/j.cub.2006.10.053
Scrivo, A., Bourdenx, M., Pampliega, O., and Cuervo, A. M. (2018). Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 17, 802–815. doi: 10.1016/S1474-4422(18)30238-2
Selman, C., and Withers, D. J. (2011). Mammalian models of extended healthy lifespan. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 99–107. doi: 10.1098/rstb.2010.0243
Selman, C., Tullet, J. M., Wieser, D., Irvine, E., Lingard, S. J., Choudhury, A. I., et al. (2009). Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144. doi: 10.1126/science.1177221
SenGupta, T., Torgersen, M. L., Kassahun, H., Vellai, T., Simonsen, A., and Nilsen, H. (2013). Base excision repair AP endonucleases and mismatch repair act together to induce checkpoint-mediated autophagy. Nat. Commun. 4:2674. doi: 10.1038/ncomms3674
Seo, Y., Kingsley, S., Walker, G., Mondoux, M. A., and Tissenbaum, H. A. (2018). Metabolic shift from glycogen to trehalose promotes lifespan and healthspan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 115, E2791–E2800. doi: 10.1073/pnas.1714178115
Shaposhnikov, M., Proshkina, E., Shilova, L., Zhavoronkov, A., and Moskalev, A. (2015). Lifespan and stress resistance in drosophila with overexpressed DNA repair genes. Sci. Rep. 5:15299. doi: 10.1038/srep15299
Shin, H. J., Kim, H., Oh, S., Lee, J. G., Kee, M., Ko, H. J., et al. (2016). AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534, 553–557. doi: 10.1038/nature18014
Shmookler Reis, R. J., Xu, L., Lee, H., Chae, M., Thaden, J. J., Bharill, P., et al. (2011). Modulation of lipid biosynthesis contributes to stress resistance and longevity of C. elegans mutants. Aging 3, 125–147. doi: 10.18632/aging.100275
Siebold, A. P., Banerjee, R., Tie, F., Kiss, D. L., Moskowitz, J., and Harte, P. J. (2010). Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. U.S.A. 107, 169–174. doi: 10.1073/pnas.0907739107
Simonsen, A., Cumming, R. C., Brech, A., Isakson, P., Schubert, D. R., and Finley, K. D. (2008). Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184. doi: 10.4161/auto.5269
Singh, P. P., Demmitt, B. A., Nath, R. D., and Brunet, A. (2019). The genetics of aging: a vertebrate perspective. Cell 177, 200–220. doi: 10.1016/j.cell.2019.02.038
Slack, C., Alic, N., Foley, A., Cabecinha, M., Hoddinott, M. P., and Partridge, L. (2015). The Ras-Erk-ETS-signaling pathway is a drug target for longevity. Cell 162, 72–83. doi: 10.1016/j.cell.2015.06.023
Sofola-Adesakin, O., Castillo-Quan, J. I., Rallis, C., Tain, L. S., Bjedov, I., Rogers, I., et al. (2014). Lithium suppresses Abeta pathology by inhibiting translation in an adult Drosophila model of Alzheimer’s disease. Front. Aging Neurosci. 6:190. doi: 10.3389/fnagi.2014.00190
Sousa, C. M., Biancur, D. E., Wang, X., Halbrook, C. J., Sherman, M. H., Zhang, L., et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. doi: 10.1038/nature19084
Squassina, A., Pisanu, C., Congiu, D., Caria, P., Frau, D., Niola, P., et al. (2016). Leukocyte telomere length positively correlates with duration of lithium treatment in bipolar disorder patients. Eur. Neuropsychopharmacol. 26, 1241–1247. doi: 10.1016/j.euroneuro.2016.03.020
Steffen, K. K., and Dillin, A. (2016). A ribosomal perspective on proteostasis and aging. Cell Metab. 23, 1004–1012. doi: 10.1016/j.cmet.2016.05.013
Su, K. H., Cao, J., Tang, Z., Dai, S., He, Y., Sampson, S. B., et al. (2016). HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat. Cell Biol. 18, 527–539. doi: 10.1038/ncb3335
Takamura, A., Komatsu, M., Hara, T., Sakamoto, A., Kishi, C., Waguri, S., et al. (2011). Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25, 795–800. doi: 10.1101/gad.2016211
Tam, O. H., Rozhkov, N. V., Shaw, R., Kim, D., Hubbard, I., Fennessey, S., et al. (2019). Postmortem cortex samples identify distinct molecular subtypes of als: retrotransposon activation, oxidative stress, and activated glia. Cell Rep. 29, 1164–1177.e5. doi: 10.1016/j.celrep.2019.09.066
Tanik, S. A., Schultheiss, C. E., Volpicelli-Daley, L. A., Brunden, K. R., and Lee, V. M. (2013). Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 288, 15194–15210. doi: 10.1074/jbc.M113.457408
Thoreen, C. C., Kang, S. A., Chang, J. W., Liu, Q., Zhang, J., Gao, Y., et al. (2009). An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032. doi: 10.1074/jbc.M900301200
Tian, X., Firsanov, D., Zhang, Z., Cheng, Y., Luo, L., Tombline, G., et al. (2019). SIRT6 is responsible for more efficient dna double-strand break repair in long-lived species. Cell 177:e622. doi: 10.1016/j.cell.2019.03.043
Toth, M. L., Sigmond, T., Borsos, E., Barna, J., Erdelyi, P., Takacs-Vellai, K., et al. (2008). Longevity pathways converge on autophagy genes to regulate life span in caenorhabditis elegans. Autophagy 4, 330–338. doi: 10.4161/auto.5618
Tsuboyama, K., Koyama-Honda, I., Sakamaki, Y., Koike, M., Morishita, H., and Mizushima, N. (2016). The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041. doi: 10.1126/science.aaf6136
Tullet, J. M. (2015). DAF-16 target identification in C. elegans: past, present and future. Biogerontology 16, 221–234. doi: 10.1007/s10522-014-9527-y
Tyner, S. D., Venkatachalam, S., Choi, J., Jones, S., Ghebranious, N., Igelmann, H., et al. (2002). p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53. doi: 10.1038/415045a
Ulgherait, M., Rana, A., Rera, M., Graniel, J., and Walker, D. W. (2014). AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep. 8, 1767–1780. doi: 10.1016/j.celrep.2014.08.006
Urfer, S. R., Kaeberlein, T. L., Mailheau, S., Bergman, P. J., Creevy, K. E., Promislow, D. E. L., et al. (2017). A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. Geroscience 39, 117–127. doi: 10.1007/s11357-017-9972-z
Vargas, J. N. S., Wang, C., Bunker, E., Hao, L., Maric, D., Schiavo, G., et al. (2019). Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol. Cell 74, e346. doi: 10.1016/j.molcel.2019.02.010
Vermeij, W. P., Hoeijmakers, J. H., and Pothof, J. (2016). Genome integrity in aging: human syndromes, mouse models, and therapeutic options. Annu. Rev. Pharmacol. Toxicol. 56, 427–445. doi: 10.1146/annurev-pharmtox-010814-124316
Vilchez, D., Morantte, I., Liu, Z., Douglas, P. M., Merkwirth, C., Rodrigues, A. P., et al. (2012). RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 489, 263–268. doi: 10.1038/nature11315
Walther, D. M., Kasturi, P., Zheng, M., Pinkert, S., Vecchi, G., Ciryam, P., et al. (2015). Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932. doi: 10.1016/j.cell.2015.03.032
Wang, M. C., O’Rourke, E. J., and Ruvkun, G. (2008). Fat metabolism links germline stem cells and longevity in C. elegans. Science 322, 957–960. doi: 10.1126/science.1162011
Wang, Y., Zhang, N., Zhang, L., Li, R., Fu, W., Ma, K., et al. (2016). Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol Cell 63, 34–48. doi: 10.1016/j.molcel.2016.05.027
Webb, A. E., and Brunet, A. (2014). FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci. 39, 159–169. doi: 10.1016/j.tibs.2014.02.003
Wei, F. Z., Cao, Z., Wang, X., Wang, H., Cai, M. Y., Li, T., et al. (2015). Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy 11, 2309–2322. doi: 10.1080/15548627.2015.1117734
Wilhelm, T., and Richly, H. (2018). Autophagy during ageing - from Dr Jekyll to Mr Hyde. FEBS J. 285, 2367–2376. doi: 10.1111/febs.14453
Wilhelm, T., Byrne, J., Medina, R., Kolundzic, E., Geisinger, J., Hajduskova, M., et al. (2017). Neuronal inhibition of the autophagy nucleation complex extends life span in post-reproductive C. elegans. Genes Dev. 31, 1561–1572. doi: 10.1101/gad.301648.117
Wyant, G. A., Abu-Remaileh, M., Frenkel, E. M., Laqtom, N. N., Dharamdasani, V., Lewis, C. A., et al. (2018). NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758. doi: 10.1126/science.aar2663
Yee, K. S., Wilkinson, S., James, J., Ryan, K. M., and Vousden, K. H. (2009). PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 16, 1135–1145. doi: 10.1038/cdd.2009.28
Yoshii, S. R., Kuma, A., Akashi, T., Hara, T., Yamamoto, A., Kurikawa, Y., et al. (2016). Systemic Analysis of Atg5-Null Mice Rescued from Neonatal Lethality by Transgenic ATG5 Expression in Neurons. Dev. Cell 39, 116–130. doi: 10.1016/j.devcel.2016.09.001
Zachari, M., Gudmundsson, S. R., Li, Z., Manifava, M., Shah, R., Smith, M., et al. (2019). Selective autophagy of mitochondria on a ubiquitin-endoplasmic-reticulum platform. Dev. Cell 50:e625. doi: 10.1016/j.devcel.2019.06.016
Zarse, K., Terao, T., Tian, J., Iwata, N., Ishii, N., and Ristow, M. (2011). Low-dose lithium uptake promotes longevity in humans and metazoans. Eur. J. Nutr. 50, 387–389. doi: 10.1007/s00394-011-0171-x
Zeng, X., Yan, T., Schupp, J. E., Seo, Y., and Kinsella, T. J. (2007). DNA mismatch repair initiates 6-thioguanine–induced autophagy through p53 activation in human tumor cells. Clin. Cancer Res. 13, 1315–1321. doi: 10.1158/1078-0432.ccr-06-1517
Zhang, Q., Wu, X., Chen, P., Liu, L., Xin, N., Tian, Y., et al. (2018). The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent wnt signaling. Cell 174:870-883.e17. doi: 10.1016/j.cell.2018.06.029
Zhang, Y., Nicholatos, J., Dreier, J. R., Ricoult, S. J., Widenmaier, S. B., Hotamisligil, G. S., et al. (2014). Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513, 440–443. doi: 10.1038/nature13492
Zhao, J., Brault, J. J., Schild, A., Cao, P., Sandri, M., Schiaffino, S., et al. (2007). FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483. doi: 10.1016/j.cmet.2007.11.004
Zhao, J., Zhai, B., Gygi, S. P., and Goldberg, A. L. (2015). mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. PNAS 112, 15790–15797. doi: 10.1073/pnas.1521919112
Keywords: autophagy, aging, target of rapamycin, insulin/IGF-1 signaling, proteostasis, DNA damage, mitophagy, anti-aging drugs
Citation: Stead ER, Castillo-Quan JI, Martinez Miguel VE, Lujan C, Ketteler R, Kinghorn KJ and Bjedov I (2019) Agephagy – Adapting Autophagy for Health During Aging. Front. Cell Dev. Biol. 7:308. doi: 10.3389/fcell.2019.00308
Received: 26 September 2019; Accepted: 12 November 2019;
Published: 28 November 2019.
Edited by:
Nicholas Ktistakis, Babraham Institute (BBSRC), United KingdomReviewed by:
Viktor Korolchuk, Newcastle University, United KingdomCopyright © 2019 Stead, Castillo-Quan, Martinez Miguel, Lujan, Ketteler, Kinghorn and Bjedov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivana Bjedov, aS5iamVkb3ZAdWNsLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.