 Fei Guo
Fei Guo Chunlin Shao
Chunlin Shao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 15 October 2019
Sec. Molecular and Cellular Oncology
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00238
This article is part of the Research Topic The Role of Epigenetic Modifications in Cancer Progression View all 18 articles
Recently, a growing number of ADP ribosylation factor (ARF) family members has been suggested to be critical in tumorigenesis. However, the effects of most ARF members on lung adenocarcinoma pathogenesis are still not well disclosed yet. In this study, ARF-like GTPase 14 (ARL14) was screened as an important prognostic factor of lung adenocarcinoma from The Cancer Genome Atlas (TCGA) database and validated by our in vitro experiments. It was found that silencing of ARL14 gene inhibited cell proliferation and the abilities of cell migration and invasion, and it also attenuated radiation damage of lung adenocarcinoma cells but had no effect on the proliferation of normal lung cells. Notably, ARL14 siRNA blocked the extracellular signal-regulated kinase (ERK)/p38 signaling pathway and induced cell cycle arrest in G0 phase, ultimately leading to cell dormancy. Moreover, ARL14 siRNA enhanced the expression of cell death activator DFFA-like effector (CIDEC) that had opposite roles in cell proliferation and migration to ALR14. Collectively, our results suggest that ARL14 has an important role in the pathogenesis of lung adenocarcinoma through CIDEC/ERK/p38 signaling pathway, and thus it could be applied as a new candidate of prognosis indicator and/or therapeutic target of lung adenocarcinoma.
With the development of diagnostic techniques and treatment strategies, significant improvements have been made in the quality of life of patients with lung cancer; however, malignant lung tumors still show the highest morbidity and mortality rates among cancer types (Siegel et al., 2017), with only 16.8% of patients surviving for 5 years after diagnosis (Ridge et al., 2013). Given the variability between pathology and etiology, lung cancer can be subdivided into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) (Sharma et al., 2007; Cheng et al., 2015). Approximately 40% of all lung cancer cases are lung adenocarcinoma, which is the most common type of NSCLC (Xue et al., 2018). The prognosis of patients with lung adenocarcinoma is extremely poor because of the lack of effective treatment measures against metastatic lung cancer. Therefore, studies to determine the molecular mechanisms governing the oncogenesis and metastasis process of lung adenocarcinoma are urgently needed.
ADP ribosylation factor-like GTPase 14 (ARL14), also known as ARF7, belongs to the ADP ribosylation factor (ARF) family of GTP-binding proteins of the Ras superfamily and it is the closest homolog of ARL11 (Yendamuri et al., 2007). The ARL11 polymorphisms Trp149Stop and Cys148Arg have been shown to be associated with a high risk of familial cancers, such as breast, ovarian, colorectal, and hematological malignancies, among others (Calin et al., 2005; Frank et al., 2006; Masojc et al., 2006; Siltanen et al., 2008; Yang et al., 2009; Hamadou et al., 2017). ARL11 was also reported as a novel tumor suppressor gene in lung and prostate cancer (Yendamuri et al., 2007, 2008; Siltanen et al., 2013). However, the function of ARL14 in the formation and progression of human cancer is unknown.
This study was conducted to determine the function and possible underlying mechanisms of ARL14 in lung adenocarcinoma tumorigenesis. Our results revealed the contribution of ARL14 in lung adenocarcinoma tumorigenesis and suggested that ARL14 might have potential implication as a diagnostic biomarker and therapeutic target for lung adenocarcinoma.
Human lung bronchial epithelial BEAS-2B cells and human lung cancer PC9 cells were obtained as gifts from the Nanjing Medical University and School of Life Sciences of Fudan University, respectively. They were cultured in Dulbecco’s Modified Eagle Medium (DMEM). Human non-small-cell lung cancer A549 cells and human lung fibroblast MRC-5 cells were purchased from Shanghai Cell Bank (Shanghai, China) and cultured in DMEM and α-modified Eagle medium (MEM), respectively. All cells were cultured with suitable medium contained 10% fetal bovine serum (FBS, Gibco, Invitrogen, United States), 100 U/ml penicillin and 100 μg/ml streptomycin, and incubated at 37°C and 5% CO2 atmosphere. For irradiation treatment, cells were exposed to different doses of γ-rays as described previously (He et al., 2014).
Short interfering RNAs (siRNAs) against ARL14, cell death activator DFFA-like effector (CIDEC) and their negative controls (RiboBio Biotechnology, Guangzhou, China) were transfected into cells with lipofectamine 2000 (Invitrogen, Carlsbad, CA, United States) following the manufacturer’s protocol. The target sequences of these transiently transfected siRNAs are listed in Supplementary Table S1.
Following siRNA transfection for 24 h, 1000 cells/well were plated into a 96-well plate and incubated for 24, 48, 72, 96, or 120 h and then measured by Cell Counting Kit-8 assay (CCK-8, Dojindo Laboratories, Kumamoto, Japan) at an absorbance of 450 nm. For the cloning efficiency assay, 200–300 cells/well were seeded into six-well culture plates and grown for 10–14 days. The number of colonies containing more than 50 cells was counted and normalized to corresponding control.
After siRNA transfection for 24 h, 1500–2500 cells/well from all groups were plated onto a 96-well plate and incubated for 24 h. After irradiation with γ-ray at 0, 2, 4, and 8 Gy, the cells were further cultured for 96 h and then issued for cell proliferation assay described above.
In vitro transwell assays were performed to assess cell migration and invasion abilities as previously described (Pan et al., 2016). Briefly, for the migration assays, 5–7 × 104 serum-starved cells were cultured with serum-free medium in a upper insert dish containing enormous 8-μm-diameter pores in its bottom membrane (Corning Inc., Corning, NY, United States) companied with a 6-well plate chamber filled with DMEM containing 10% FBS. For the invasion assays, the above insert dish was replaced with one coated with 1 μg/mL Matrigel (Corning). After 24 h of culture, the cells were fixed with 100% methanol for 30 min and stained with crystal violet staining solution (Beyotime, Shanghai, China) for 25 min. Cells on the upper surface of the insert dish bottom were carefully removed using a wet cotton swab and those that had migrated through the membrane were photographed and counted in five random fields (×10) using an inverted microscope.
Western blot analysis for specific protein expression was performed as previously described (Wang et al., 2017). The antibodies used in this study are listed in Supplementary Table S2.
For all groups, 2–4 × 104 cells plated on culture slides were incubated for 48 h at 37°C in 5% CO2, and then the exponentially growing cells were fixed with immune staining fix solution and treated with enhanced immunostaining permeabilization buffer for 15 min at room temperature. Next, non-specific antibody binding sites were blocked with QuickBlockTM blocking buffer for immunological staining for 1 h. Ki67 primary antibody at appropriate dilutions was added and incubated at 4°C overnight followed by further incubation for 1 h at room temperature in the dark with Alexa Fluor® 594 goat anti-mouse IgG (H + L) (Thermo Fisher Scientific, Waltham, MA, United States). Finally, the cell nuclei were counterstained with DAPI Fluromount-GTM (Southern Biotech, Birmingham, AL, United States) for 5 min. The Ki67 positive cells were examined using a Zeiss Axioplan fluorescence microscope (Oberkochen, Germany).
Total RNA was isolated from cells using a MiniBEST Universal RNA Extraction Kit (Takara, Shiga, Japan). Reverse transcription and real-time PCR (qRT-PCR) were performed with PrimeScriptTM RT Master Mix (Perfect Real Time, Takara) and SYBR® Premix Ex TaqTM II (Tli RNaseH Plus, Takara) following the manufacturer’s instructions. The gene-specific primers are shown in Supplementary Table S3. The relative expression level of mRNA was examined as the inverse log of the delta CT and normalized to the reference gene, β-actin.
Following siRNA transfection and 8 Gy γ-ray radiation treatment, the cells were grown in an incubator with 5% CO2 at 37°C for 24 h and then collected, fixed in 70% cold ethanol, and stored at −20°C overnight. The cell pellets were washed twice with 1 × phosphate-buffered saline and centrifuged at 1000 × g and 4°C for 10 min, following by staining with a cell cycle kit (BD Biosciences, Franklin Lakes, NJ, United States) according to the manufacturer’s instructions. The total cellular DNA content was analyzed with a flow cytometer (Beckman, Brea, CA, United States) by acquiring data for at least 10,000 events.
Gene set enrichment analysis (GSEA 3.0)1 was used to explore potential KEGG pathway and GO analysis within the Molecular Signatures Database (MSigDB 6.0) of c2 (curated gene sets) and c5 (GO gene sets). Setting ARL14 gene expression levels as population phenotypes in GSEA, we analyzed gene expression omics predictions and assessed related pathways in lung adenocarcinoma. A nominal P-value <0.05 and false discovery rate (FDR) <0.25 of the enrichment gene sets in the analysis were considered statistically significant. The theory and process of GSEA have been described previously (Subramanian et al., 2005).
All data were obtained from 3 to 5 independent experiments and presented as the means ± SE. Statistical analyses were analyzed with Student’s t-tests using SPSS 19.0 software (SPSS, Inc., Chicago, IL, United States). P < 0.05 was considered to be statistically significant difference between treatment groups.
Analyzing the data of lung adenocarcinoma samples and matched normal control tissues obtained from The Cancer Genome Atlas (TCGA) online resource, we found that the mRNA expression of ARL14 in lung adenocarcinoma samples was significantly higher than that in the matched normal tissues (Figure 1A, N = 57, fold change = 2.3, P = 1.23E-06), and the expression of ARL14 in adenocarcinoma had a very strong negative correlation with the overall survival of those lung cancer patients (Figure 1B, N = 482, P = 4.12E-04). In comparison with the mean level, the lower level of ARL14 mRNA corresponds to the higher survival. Our measurement further confirmed that the expression levels of ARL14 in lung adenocarcinoma cells (A549 and PC9) were higher than that in normal lung cells (BEAS-2B and MRC-5) (Figure 1C), which is consistent with the results obtained from TCGA cohort. Therefore, ARL14 might become a candidate prognostic factor of lung adenocarcinoma development and is worthy of further investigation of its function.
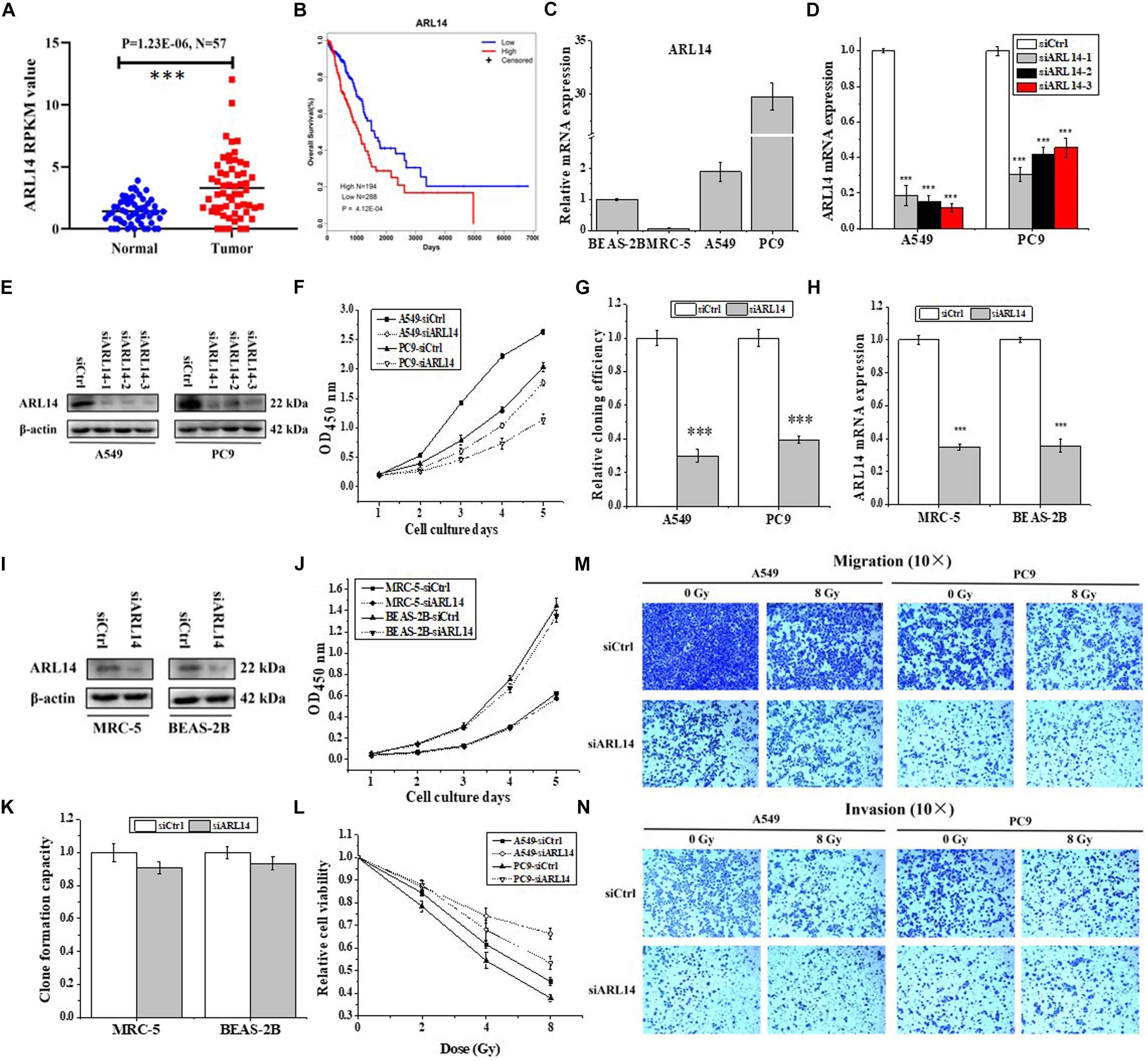
Figure 1. ARL14 is a potential prognostic biomarker for lung adenocarcinoma and contributes to proliferation, radiosensitivity, migration, and invasion of lung adenocarcinoma cells. (A) The expression levels of ARL14 mRNA in lung adenocarcinoma samples and matched normal control tissues (N = 57) in the TCGA cohort. (B) Kaplan–Meier analysis of the correlation between ARL14 level and overall survival of lung adenocarcinoma patients with high (N = 194) and low (N = 288) ARL14 expression in the TCGA cohort. Cut-off value for evaluation of ARL14 mRNA level was the mean expression of 482 lung adenocarcinoma samples. (C) The expression levels of ARL14 mRNA in BEAS-2B, MRC-5, A549, and PC9 cells. (D,E) The expressions of ARL14 mRNA (D) and protein (E) were reduced in A549 and PC9 cells transfected with ARL14 siRNAs for 48 h. (F,G) Silencing of ARL14 decreased proliferation (F) and cloning efficiency (G) of A549 and PC9 cells significantly. (H,I) The expressions of ARL14 mRNA (H) and protein (I) in MRC-5 and BEAS-2B cells transfected with ARL14 siRNAs for 48 h. (J,K) Silencing of ARL14 had no significant influence on proliferation (J) and cloning efficiency (K) of MRC-5 and BEAS-2B cells. (L) The dose responses of the viability of A549 and PC9 cells with or without siARL14 transfection. (M,N) The migration and invasion activities of A549 and PC9 cells with or without ARL14 siRNA transfection. After siRNA transfection, cells were irradiated with 8 Gy γ-rays. ∗∗∗P < 0.001 compared with the corresponding control.
To investigate the functions of ARL14 in lung adenocarcinoma cells, ARL14 gene expression in A549 and PC9 cells was interfered with siRNA and the silencing effect was detected by qRT-PCR and Western blot assays. As shown in Figures 1D,E, siARL14-1 (hereafter called siARL14) had the most effective efficiency in knock-down ARL14 expression and thus was applied for the following studies. It was found that siARL14 significantly inhibited cell proliferation (Figure 1F) and cell colony formation ability (Figure 1G) for both A549 and PC9 tumor cells. In addition, siARL14 also effectively suppressed the expression of ARL14 in normal lung cells MRC-5 and BEAS-2B (Figures 1H,I), however, it had no influence on the proliferation and cloning efficiency of both normal lung cells (Figures 1J,K). These results suggest that ARL14 may have some special function in cancer cells, which could influence the outcome of radiotherapy. To confirm this hypothesis, we studied the influence of siARL14 on radiation responses of lung adenocarcinoma cells. Results showed that cell proliferations of A549 and PC9 were decreased by γ-ray irradiation in a dose dependent manner, but this decrease was effectively weakened when ARL14 was knocked down in these cells (Figure 1L). In addition, radiation reduced the abilities of migration and invasion of A549 and PC9 cells but it had no obvious influence on the migration and invasion of siARL14-transfected cells although siARL14 itself suppressed cell migration and invasiveness (Figures 1M,N).
Uncontrolled proliferation is a well-established hallmark of cancer cells. Nearly all human cancers have deregulated control in cell-cycle progress. G0 phase is an important check-point when cells decide to begin proliferation or remain quiescence. We monitored the cell cycle distribution by flow cytometer at 24 h after irradiation and found that A549 and PC9 cells were arrested in G2/M phase in accompany with a reduction of cells in G1 phase (Figure 2A and Supplementary Table S4). When the cells were transfected with siARL14, radiation-induced G2/M arrest was effectively released, especially for A549 cells. In fact, silencing of ARL14 expression resulted in G0/G1 phase accumulation of A549 cells (Figure 2A).
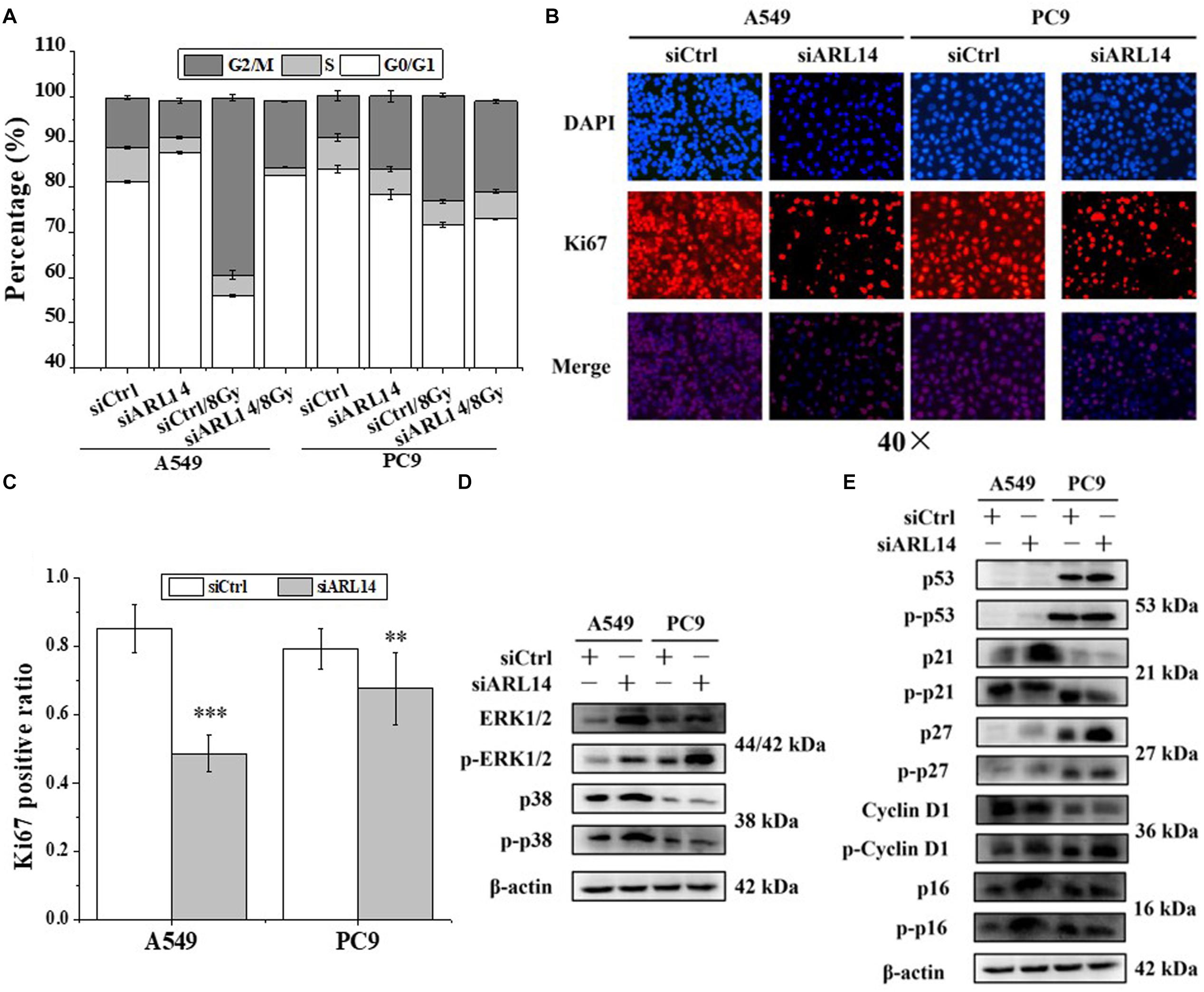
Figure 2. Down-regulation of ARL14 expression induced quiescence of lung adenocarcinoma cells in G0/G1 phase and inhibited cell proliferation. (A) Effects of ARL14 siRNA on cell cycle distribution and radiation-induced G2/M phase arrest of A549 and PC9 cells. (B,C) Ki67 positive ratio in A549 and PC9 cell population transfected with ARL14 siRNA. (D,E) The proteins and their phosphorylation levels of ERK and p38 (D), p16, cyclin D1, p27, p21, and p53 (E) in A549 and PC9 cell population transfected with ARL14 siRNA and its negative control. ∗∗P < 0.01 and ∗∗∗P < 0.001 compared with the corresponding control.
Ki67 protein is an indicator of cell proliferation (Bruno and Darzynkiewicz, 1992). Our immunofluorescence experimental results showed that about 80% cells in the logarithmic growth population of A549 and PC9 had positive Ki67 expression, but when the cells were interfered with siARL14, the ratios of Ki67-positive cells were obviously decreased in both cell lines (Figures 2B,C), indicating cell proliferation ability was attenuated by siARL14.
Imbalance of extracellular signal-regulated kinase (ERK)/p38 signaling activities has also been suggested to determine carcinoma cell proliferation or dormancy (Aguirre-Ghiso et al., 2001, 2003; Ranganathan et al., 2006). We wonder whether ERK and p38 signaling pathways are regulated by ARL14 in lung adenocarcinoma cells. It was found that when ARL14 in A549 and PC9 cells was silenced, both total protein and its phosphorylation level of ERK1/2 were increased and p-p38 protein was activated (Figure 2D). A high level of ERK1/2 activity contributes to the promotion of cell proliferation (Aguirre-Ghiso et al., 2001, 2003; Lents et al., 2002; Ranganathan et al., 2006; Chambard et al., 2007). But here silencing ARL14 caused a distinct decreased cell proliferation, indicating that the ERK signaling pathway is blocked by knockdown of ARL14 expression, which resulted in the accumulation of ERK and its phosphorylation. To verifying this, we examined whether p21 and cyclin D1 were affected by siARL14. Results showed that when ARL14 expressions in A549 and PC9 cells were silenced, the expressions of cyclin D1 protein and its phosphorylation level were both increased. However, the p21 protein and its phosphorylation level were upregulated in A549 cells but downregulated in PC9 cells (Figure 2E). Because the cell dormancy after siARL14 transfection was observed in both A549 and PC9 cells according to above Ki67 immunofluorescence assay, we predicted that other cell cycle associated factors may also play important roles in siARL14-induced cell cycle arrest and cell dormancy, such as p16 (Barkan et al., 2008; Sosa et al., 2011), p27 (Barkan et al., 2008; Cackowski et al., 2017; Fluegen et al., 2017; Liu et al., 2018), and p53 (Nagayama et al., 2000; Sosa et al., 2011; Dai et al., 2016). Indeed, our further assay demonstrated that, after siARL14 transfection, both p16, p27 and p53 protein and their phosphorylation levels were upregulated in PC9 cells, and p16 and p27 were upregulated in A549 cells that had abnormal status of p53 (Figure 2E).
Seldom study has evaluated the transcriptional regulation of ARL14 in human cancers. GSEA is a useful tool to reveal the corresponding pathway and regulation mechanism of specific genes (Li et al., 2018), particularly those with unknown functions (Liu et al., 2016). Therefore, the GSEA of 594 RNA-seq data of lung adenocarcinoma from TCGA was performed to gain the insights of interaction networks of ARL14. The results suggested that 5850 genes are co-expressed with ARL14 (Supplementary Data Sheet S1) and the pathways of both positively and negatively correlated with ARL14 are mostly related to metabolism and immune system (Supplementary Tables S5, S6). Because the absolute magnitude of the correlation index of genes involved in the pathways negatively correlated with ARL14 were much lower than 0.2 (P < 0.05), we focused only on the pathways positively correlated with ARL14 and examined the mRNA expressions of 22 top-ranking protein encoding genes (Supplementary Data Sheet S1). It was found that only the expressions of AXDND1, CIDEC, IL1R2, EPS8L3, and INSC genes were significant increased (fold-change > 1.5) in both A549 and PC9 cells after ARL14 silencing (Figure 3A), indicating that these gene may be downstream of ARL14. Figure 3A also showed that CIDEC and EPS8L3 had the highest expressions and the biggest changes in both cell lines, since the function of EPS8L3 protein is still unknown, we focused on the relationship between ARL14 and CIDEC, which has a correlation coefficient of 0.7756 (Supplementary Data Sheet S1).
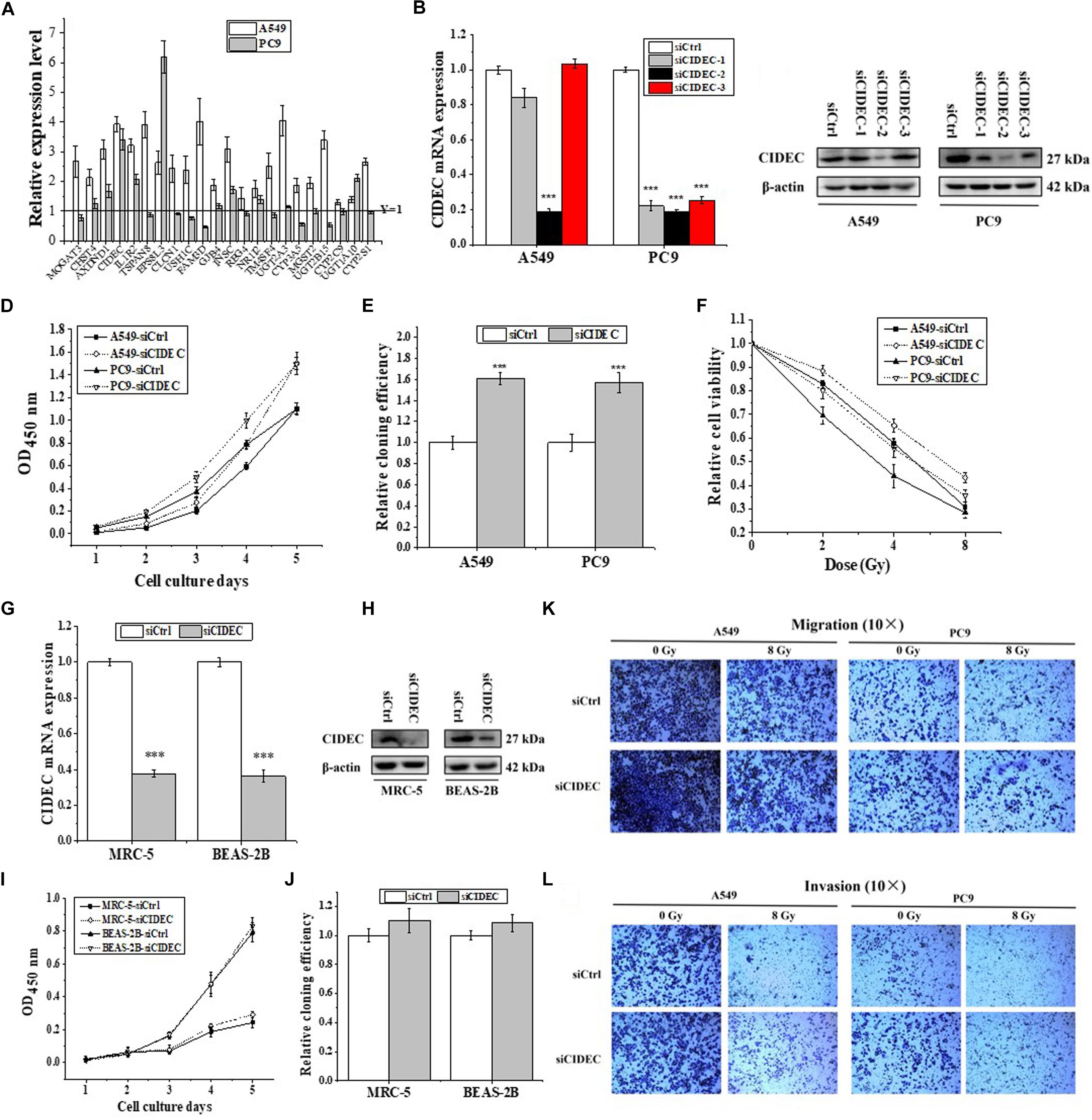
Figure 3. CIDEC is a downstream gene of ARL14 and contributes to proliferation, radiosensitivity, migration and invasion of lung adenocarcinoma cells. (A) The candidate genes negatively correlated with ARL14 analyzed from GESA database and their relative expression levels of mRNA were measured in A549 and PC9 cells transfected with ARL14 siRNA. (B,C) The expressions of CIDEC mRNA (B) and protein (C) in A549 and PC9 cells transfected with CIDEC siRNAs for 48 h. (D–F) Silencing of CIDEC increased proliferation (D), cloning efficiency (E), and radioresistance (F) of A549 and PC9 cells. (G,H) The expressions of CIDEC mRNA (G) and protein (H) were reduced in MRC-5 and BEASA-2B cells transfected with CIDEC siRNAs for 48 h. (I,J) Silencing of CIDEC had no significant influence on proliferation (I) and cloning efficiency (J) of MRC-5 and BEAS-2B cells. (K,L) The migration and invasion activities of A549 and PC9 cells with or without CIDEC siRNA transfection. After siRNA transfection, cells were irradiated with 8 Gy γ-rays. ∗∗P < 0.01 and ∗∗∗P < 0.001 compared with the corresponding control.
To further determine the relationship between ARL14 and CIDEC, the function of CIDEC in tumor proliferation was then detected. We transfected A549 and PC9 cells with siRNAs against CIDEC and the silencing effect were evaluated by qRT-PCR and Western blot assays (Figures 3B,C). The sequence siCIDEC2 (hereafter named siCIDEC) had the most effective efficiency in silencing CIDEC gene and thus applied for further experiments. It was found that siCIDEC significantly increased cell proliferation, clone formation and radiation resistance of A549 and PC9 cells (Figures 3D–F). Interestingly, although siCIDEC also reduced the expressions of CIDEC gene and protein in both MRC-5 and BEAS-2B cells (Figures 3G,H), it had no significant influence in cell proliferation and clone formation of these normal cells (Figures 3I,J). In addition, silencing of CIDEC expression could increase cell migration and invasion and further partly recovered radiation-reduced metastasis ability of both A549 and PC9 cells (Figures 3K,L). Accordingly, CIDEC has opposite roles in cell proliferation and migration to ALR14 in lung adenocarcinoma cells.
In addition, cell cycle analysis revealed that silencing of CIDEC attenuated the accumulation of cell cycle arrested at G2/M phase of irradiated A549 cells but increased the accumulation of PC9 cells arrested at S-phase (Figure 4A and Supplementary Table S4). Western blot assay shows that the protein and phosphorylation levels of p38 and ERK1/2 and their downstream proteins p16, p21, p27, p53, and cyclin D1 were all down-regulated in A549 and PC9 cells after siCIDEC transfection (Figures 4B,C), which also had a conversed pattern in comparison with that in ARL14 silencing cells.
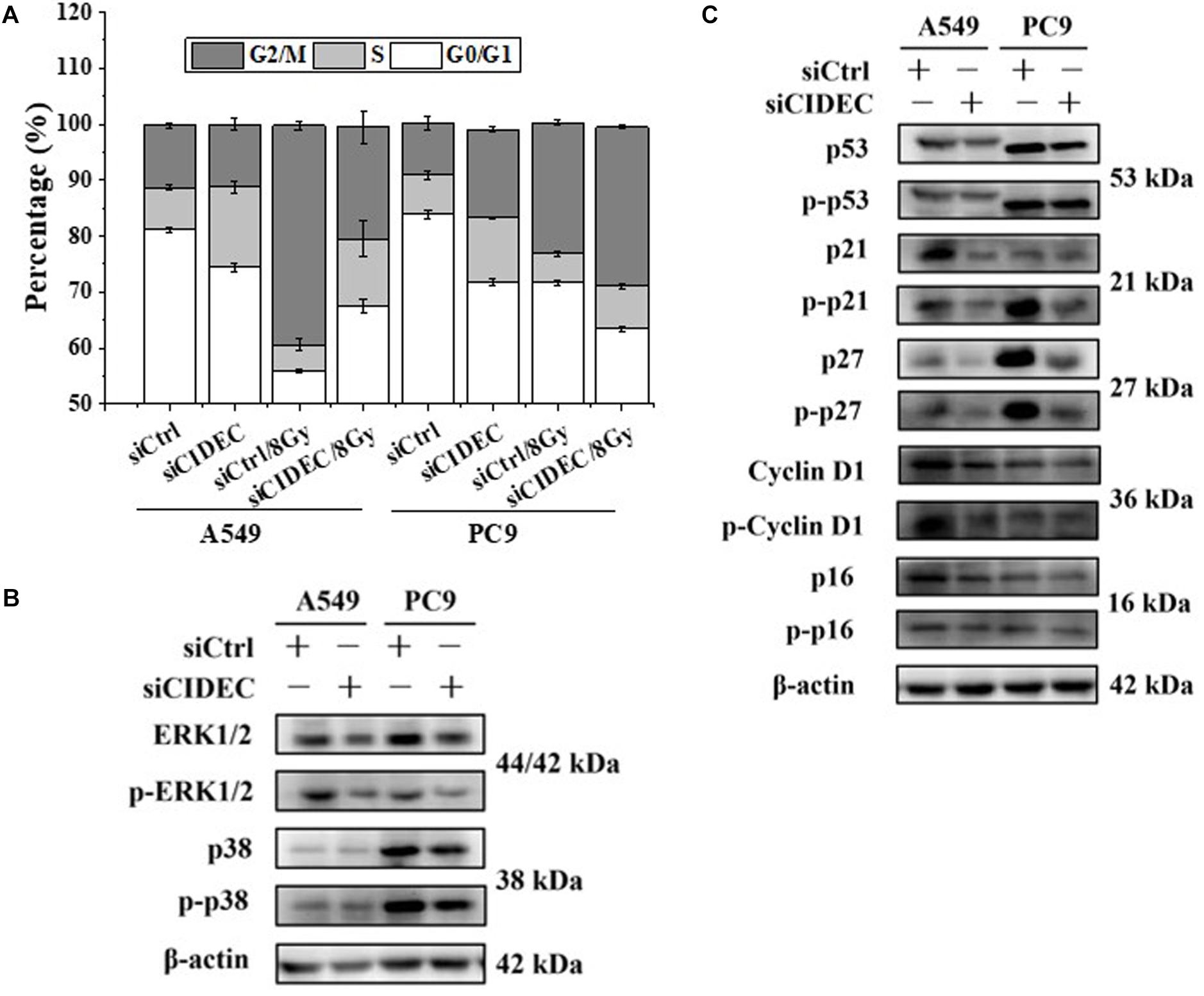
Figure 4. Down-regulation of CIDEC expression contributes to the distribution of cell cycle and the expression of cell cycle-related proteins in A549 and PC9 cells. (A) Effects of CIDEC siRNA on the cell cycle distribution and radiation-induced G2/M phase arrest of A549 and PC9 cells. (B,C) The proteins and their phosphorylation levels of ERK and p38 (B), p16, cyclin D1, p27, p21, and p53 (C) in A549 and PC9 cell population transfected with CIDEC siRNA and its negative control. After siRNA transfection, cells were irradiated with 8 Gy γ-rays.
ARL14 is located on human chromosome 13q14.2, a region closely related to several cancers including lung cancer (Ninomiya et al., 2013; Du et al., 2018) and involved in multidrug resistance in cancer treatment (Litviakov et al., 2016). The ARL14 gene encodes a protein member of the Ras super-family composed of 196 amino acids which is involved in apoptotic signaling and several regulatory pathways (Kahn et al., 1992), indicating its usefulness as a marker for predicting tumor progression and prognosis. We found that ARL14 had significantly different levels between lung adenocarcinoma samples and matched normal control tissues as well between lung adenocarcinoma cells and normal lung cells, and ARL14 level is associated with the prognosis of lung adenocarcinoma. Moreover, silencing ARL14 inhibited the proliferation, migration and invasion of lung adenocarcinoma cells but it had no influence on the proliferation of normal lung cells. Therefore, ARL14 may be applied as an ideal prognostic biomarker and therapeutic target of lung adenocarcinoma.
Our pilot study showed that, when the lung adenocarcinoma A549 cells were transferred with a lentiviral expression vector of ARL14 gene, the cell cloning efficiency became very low (<5%) and the cell proliferation was almost total inhibited, indicating that the silencing ARL14 by lentivirus causes growth arrest and dormancy of lung adenocarcinoma cells. Therefore, this study just transiently transferred siARL14 into lung adenocarcinoma cells and normal lung cells to knock-down ARL14 expression and found that the percentage of G0/G1-phase cells was increased and the protein level of Ki67 was downregulated in ARL14 knockdown lung adenocarcinoma cells.
CIDEC, a member of the cell-death-inducing DFF45-like effectors family (Liang et al., 2003), is located on human chromosome 3p25, a region associated with a high frequency of loss of heterozygosity in a wide range of tumor tissues. This region plays an important role in the pathogenesis of lung cancer (Graziano et al., 1991). However, few studies of CIDEC expression in cancer cells have been reported. Min et al. (2011) found that CIDEC expression was decreased in hepatocellular carcinoma tissue compared with its adjacent normal tissues, and overexpression of CIDEC inhibited the proliferation of SMMC-7721 cells. In agreement with those studies, our results showed that CIDEC silencing promoted cell proliferation, while ARL14 silencing inhibited proliferation and upregulated CIDEC expression in lung adenocarcinoma cells. Moreover, CIDEC and ARL14 had opposite effects on the expressions of proliferation-related proteins and the migration and invasion capacities of lung adenocarcinoma cells. These results supply an evidence that CIDEC is downstream of ARL14 and has antagonistic effect on the biological function of ARL14.
Overall, our results demonstrate that silencing ARL14 can block ERK1/2 and p38 signaling and stimulates its downstream gene CIDEC expression, which further activates their downstream effectors of p16, p21, p27, p53, and cyclin D1, resulting in cell cycle arrest of the lung cancer cells. These findings should have implication in identifying the predictive biomarker and treatment targets for lung adenocarcinoma.
The datasets generated for this study are available on request to the corresponding author.
FG, DY, and CS: conceptualization. FG, JuZ, HZ, CW, LZ, and JiZ: methodology. FG, DY, and JuZ: formal analysis. FG and DY: investigation. FG: draft writing. YP and CS: review and editing. FG: visualization. CS: project administration. YP and CS: funding acquisition. CS: supervision.
This work was supported by the National Key R&D Program of China (Grant Number 2017YFC0108604) and the National Natural Science Foundation of China (Grant Numbers 31770910, 31570850, and 11775052).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2019.00238/full#supplementary-material
Aguirre-Ghiso, J. A., Estrada, Y., Liu, D., and Ossowski, L. (2003). ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. 63, 1684–1695.
Aguirre-Ghiso, J. A., Liu, D., Mignatti, A., Kovalski, K., and Ossowski, L. (2001). Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 12, 863–879. doi: 10.1091/mbc.12.4.863
Barkan, D., Kleinman, H., Simmons, J. L., Asmussen, H., Kamaraju, A. K., Hoenorhoff, M. J., et al. (2008). Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 68, 6241–6250. doi: 10.1158/0008-5472.can-07-6849
Bruno, S., and Darzynkiewicz, Z. (1992). Cell cycle dependent expression and stability of the nuclear protein detected by Ki-67 antibody in HL-60 cells. Cell Prolif. 25, 31–40. doi: 10.1111/j.1365-2184.1992.tb01435.x
Cackowski, F. C., Eber, M. R., Rhee, J., Decker, A. M., Yumoto, K., Berry, J. E., et al. (2017). Mer tyrosine kinase regulates disseminated prostate cancer cellular dormancy. J. Cell. Biochem. 118, 891–902. doi: 10.1002/jcb.25768
Calin, G. A., Trapasso, F., Shimizu, M., Dumitru, C. D., Yendamuri, S., Godwin, A. K., et al. (2005). Familial cancer associated with a polymorphism in ARLTS1. N. Engl. J. Med. 352, 1667–1676. doi: 10.1056/NEJMoa042280
Chambard, J. C., Lefloch, R., Pouyssegur, J., and Lenormand, P. (2007). ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310. doi: 10.1016/j.bbamcr.2006.11.010
Cheng, H., Shcherba, M., Kandavelou, K., Liang, Y., Liu, H., and Perez-Soler, R. (2015). Emerging drugs for squamous cell lung cancer. Expert Opin. Emerg. Drugs 20, 149–160. doi: 10.1517/14728214.2015.1001365
Dai, Y., Wang, L., Tang, J., Cao, P., Luo, Z., Sun, J., et al. (2016). Activation of anaphase-promoting complex by p53 induces a state of dormancy in cancer cells against chemotherapeutic stress. Oncotarget 7, 25478–25492. doi: 10.18632/oncotarget.8172
Du, M., Thompson, J., Fisher, H., Zhang, P., Huang, C. C., and Wang, L. (2018). Genomic alterations of plasma cell-free DNAs in small cell lung cancer and their clinical relevance. Lung cancer 120, 113–121. doi: 10.1016/j.lungcan.2018.04.008
Fluegen, G., Avivar-Valderas, A., Wang, Y., Padgen, M. R., Williams, J. K., Nobre, A. R., et al. (2017). Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 19, 120–132. doi: 10.1038/ncb3465
Frank, B., Hemminki, K., Brenner, H., Hoffmeister, M., Chang-Claude, J., and Burwinkel, B. (2006). ARLTS1 variants and risk of colorectal cancer. Cancer Lett. 244, 172–175. doi: 10.1016/j.canlet.2005.12.006
Graziano, S. L., Pfeifer, A. M., Testa, J. R., Mark, G. E., Johnson, B. E., Hallinan, E. J., et al. (1991). Involvement of the RAF1 locus, at band 3p25, in the 3p deletion of small-cell lung cancer. Genes Chromosomes Cancer 3, 283–293. doi: 10.1002/gcc.2870030407
Hamadou, W. S., Besbes, S., Mani, R., Bourdon, V., Ben Youssef, Y., Achour, B., et al. (2017). ARLTS1, potential candidate gene in familial aggregation of hematological malignancies. Bull. Cancer 104, 123–127. doi: 10.1016/j.bulcan.2016.10.016
He, M., Dong, C., Konishi, T., Tu, W., Liu, W., Shiomi, N., et al. (2014). Differential effects of p53 on bystander phenotypes induced by gamma ray and high LET heavy ion radiation. Life Sci. Space Res. 1, 53–59. doi: 10.1016/j.lssr.2014.02.003
Kahn, R. A., Der, C. J., and Bokoch, G. M. (1992). The ras superfamily of GTP-binding proteins: guidelines on nomenclature. FASEB J. 6, 2512–2513. doi: 10.1096/fasebj.6.8.1592203
Lents, N. H., Keenan, S. M., Bellone, C., and Baldassare, J. J. (2002). Stimulation of the Raf/MEK/ERK cascade is necessary and sufficient for activation and Thr-160 phosphorylation of a nuclear-targeted CDK2. J. Biol. Chem. 277, 47469–47475. doi: 10.1074/jbc.M207425200
Li, Y., Deng, L., Zhao, X., Li, B., Ren, D., Yu, L., et al. (2018). Tripartite motif-containing 37 (TRIM37) promotes the aggressiveness of non-small-cell lung cancer cells by activating the NF-kappaB pathway. J. Pathol. 246, 366–378. doi: 10.1002/path.5144
Liang, L., Zhao, M., Xu, Z., Yokoyama, K. K., and Li, T. (2003). Molecular cloning and characterization of CIDE-3, a novel member of the cell-death-inducing DNA-fragmentation-factor (DFF45)-like effector family. Biochem. J. 370, 195–203. doi: 10.1042/BJ20020656
Litviakov, N. V., Cherdyntseva, N. V., Tsyganov, M. M., Slonimskaya, E. M., Ibragimova, M. K., Kazantseva, P. V., et al. (2016). Deletions of multidrug resistance gene loci in breast cancer leads to the down-regulation of its expression and predict tumor response to neoadjuvant chemotherapy. Oncotarget 7, 7829–7841. doi: 10.18632/oncotarget.6953
Liu, Y., Lv, J., Liu, J., Liang, X., Jin, X., Xie, J., et al. (2018). STAT3/p53 pathway activation disrupts IFN-beta-induced dormancy in tumor-repopulating cells. J. Clin. Invest. 128, 1057–1073. doi: 10.1172/JCI96329
Liu, Y. Z., Yang, H., Cao, J., Jiang, Y. Y., Hao, J. J., Xu, X., et al. (2016). KIAA1522 is a novel prognostic biomarker in patients with non-small cell lung cancer. Sci. Rep. 6:24786. doi: 10.1038/srep24786
Masojc, B., Mierzejewski, M., Cybulski, C., van de Wetering, T., Debniak, T., Gorski, B., et al. (2006). Cancer familial aggregation (CFA) and G446A polymorphism in ARLTS1 gene. Breast Cancer Res. Treat. 99, 59–62. doi: 10.1007/s10549-006-9180-9185
Min, J., Zhang, W., Gu, Y., Hong, L., Yao, L., Li, F., et al. (2011). CIDE-3 interacts with lipopolysaccharide-induced tumor necrosis factor, and overexpression increases apoptosis in hepatocellular carcinoma. Med. Oncol. 28(Suppl. 1), S219–S227. doi: 10.1007/s12032-010-9702-9701
Nagayama, Y., Shigematsu, K., Namba, H., Zeki, K., Yamashita, S., and Niwa, M. (2000). Inhibition of angiogenesis and tumorigenesis, and induction of dormancy by p53 in a p53-null thyroid carcinoma cell line in vivo. Anticancer. Res. 20, 2723–2728.
Ninomiya, H., Kato, M., Sanada, M., Takeuchi, K., Inamura, K., Motoi, N., et al. (2013). Allelotypes of lung adenocarcinomas featuring ALK fusion demonstrate fewer onco- and suppressor gene changes. BMC Cancer 13:8. doi: 10.1186/1471-2407-13-18
Pan, Y., Zhou, C., Yuan, D., Zhang, J., and Shao, C. (2016). Radiation exposure promotes hepatocarcinoma cell invasion through epithelial mesenchymal transition mediated by H2S/CSE pathway. Radiat. Res. 185, 96–105. doi: 10.1667/rr14177.1
Ranganathan, A. C., Adam, A. P., and Aguirre-Ghiso, J. A. (2006). Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle 5, 1799–1807. doi: 10.4161/cc.5.16.3109
Ridge, C. A., McErlean, A. M., and Ginsberg, M. S. (2013). Epidemiology of lung cancer. Semin. Int. Radiol. 30, 93–98. doi: 10.1055/s-0033-1342949
Sharma, S. V., Bell, D. W., Settleman, J., and Haber, D. A. (2007). Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 7, 169–181. doi: 10.1038/nrc2088
Siegel, R. L., Miller, K. D., and Jemal, A. (2017). Cancer statistics, 2017. Cancer J. Clin. 67, 7–30. doi: 10.3322/caac.21387
Siltanen, S., Fischer, D., Rantapero, T., Laitinen, V., Mpindi, J. P., Kallioniemi, O., et al. (2013). ARLTS1 and prostate cancer risk–analysis of expression and regulation. PLoS One 8:e72040. doi: 10.1371/journal.pone.0072040
Siltanen, S., Syrjakoski, K., Fagerholm, R., Ikonen, T., Lipman, P., Mallott, J., et al. (2008). ARLTS1 germline variants and the risk for breast, prostate, and colorectal cancer. Eur. J. Hum. Genet. 16, 983–991. doi: 10.1038/ejhg.2008.43
Sosa, M. S., Avivar-Valderas, A., Bragado, P., Wen, H. C., and Aguirre-Ghiso, J. A. (2011). ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clin. Cancer Res. 17, 5850–5857. doi: 10.1158/1078-0432.ccr-10-2574
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. doi: 10.1073/pnas.0506580102
Wang, P., Yuan, D., Guo, F., Chen, X., Zhu, L., Zhang, H., et al. (2017). Chromatin remodeling modulates radiosensitivity of the daughter cells derived from cell population exposed to low- and high-LET irradiation. Oncotarget 8, 52823–52836. doi: 10.18632/oncotarget.17275
Xue, X., Fei, X., Hou, W., Zhang, Y., Liu, L., and Hu, R. (2018). miR-342-3p suppresses cell proliferation and migration by targeting AGR2 in non-small cell lung cancer. Cancer Lett. 412, 170–178. doi: 10.1016/j.canlet.2017.10.024
Yang, X. Y., Yu, H., Xi, M. R., Yang, K. X., Pan, X. L., Hu, M., et al. (2009). Association of the ARLTS1 variants with familial ovarian cancer risk in China. Int. J. Gynecol. Cancer 19, 585–590. doi: 10.1111/IGC.0b013e3181a39d03
Yendamuri, S., Trapasso, F., and Calin, G. A. (2008). ARLTS1 - a novel tumor suppressor gene. Cancer Lett. 264, 11–20. doi: 10.1016/j.canlet.2008.02.021
Keywords: ARL14, CIDEC, ERK/p38, lung cancer dormancy, radiation
Citation: Guo F, Yuan D, Zhang J, Zhang H, Wang C, Zhu L, Zhang J, Pan Y and Shao C (2019) Silencing of ARL14 Gene Induces Lung Adenocarcinoma Cells to a Dormant State. Front. Cell Dev. Biol. 7:238. doi: 10.3389/fcell.2019.00238
Received: 27 August 2019; Accepted: 01 October 2019;
Published: 15 October 2019.
Edited by:
Lei Chang, Soochow University Medical College (SUMC), ChinaReviewed by:
Guan Jian, Southern Medical University, ChinaCopyright © 2019 Guo, Yuan, Zhang, Zhang, Wang, Zhu, Zhang, Pan and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Pan, c3dhbGxvd3BhbkBmdWRhbi5lZHUuY24=; Chunlin Shao, Y2xzaGFvQHNobXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.