 Hye M. Hwang
Hye M. Hwang Ray Y. Ku
Ray Y. Ku Kazue Hashimoto-Torii
Kazue Hashimoto-Torii
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 17 July 2019
Sec. Cell Adhesion and Migration
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00138
This article is part of the Research Topic Cell Migration in Neurological Disorders View all 6 articles
Migration of neurons starts in the prenatal period and continues into infancy. This developmental process is crucial for forming a proper neuronal network, and the disturbance of this process results in dysfunction of the brain such as epilepsy. Prenatal exposure to environmental stress, including alcohol, drugs, and inflammation, disrupts neuronal migration and causes neuronal migration disorders (NMDs). In this review, we summarize recent findings on this topic and specifically focusing on two different modes of migration, radial, and tangential migration during cortical development. The shared mechanisms underlying the NMDs are discussed by comparing the molecular changes in impaired neuronal migration under exposure to different types of prenatal environmental stress.
In utero environment critically affects the brain development, thereby modifying neurobehavior of the offspring after birth (Rees and Harding, 2004). Substances that are ingested by a pregnant woman can cross the placenta and adversely affect fetal development. Some are unavoidable medications due to the mother’s medical condition, such as epilepsy or depression, but others are consumed by an expectant mother unbeknown to the risks and effects. In some cases, an excessive amount may be taken by a pregnant woman due to the addiction.
During brain development, newly generated neurons undergo morphological changes followed by migrating from the germinal layer through the intricate network of extracellular matrix to establish connections with other cells in a highly ordered fashion (Rakic, 1972, 1995; Metin et al., 2008). Two major modes of migration in the cerebral cortex are known (Copp and Harding, 1999; Guerrini and Parrini, 2010). The first is radial migration. Excitatory neurons generated in the proliferative zones of the dorsal telencephalic primordium radially migrate toward the pial surface of the cerebral cortex along the radial axis. These neurons use the cellular processes that are elongated from the radial glial cells, which are ascending neural progenitor cells (NPCs), as the scaffold of their migration (Rakic, 1972; Ayala et al., 2007; Figure 1). Once arriving at the superficial layer, the neurons detach from the radial glial processes to settle down in the cortical plate. Another mode of neuronal migration, known as tangential migration, is employed by inhibitory interneurons. Those neurons migrate tangentially from the ganglionic eminences (GE) to the cerebral cortex (Marín and Rubenstein, 2001; Ayala et al., 2007; Figure 1).
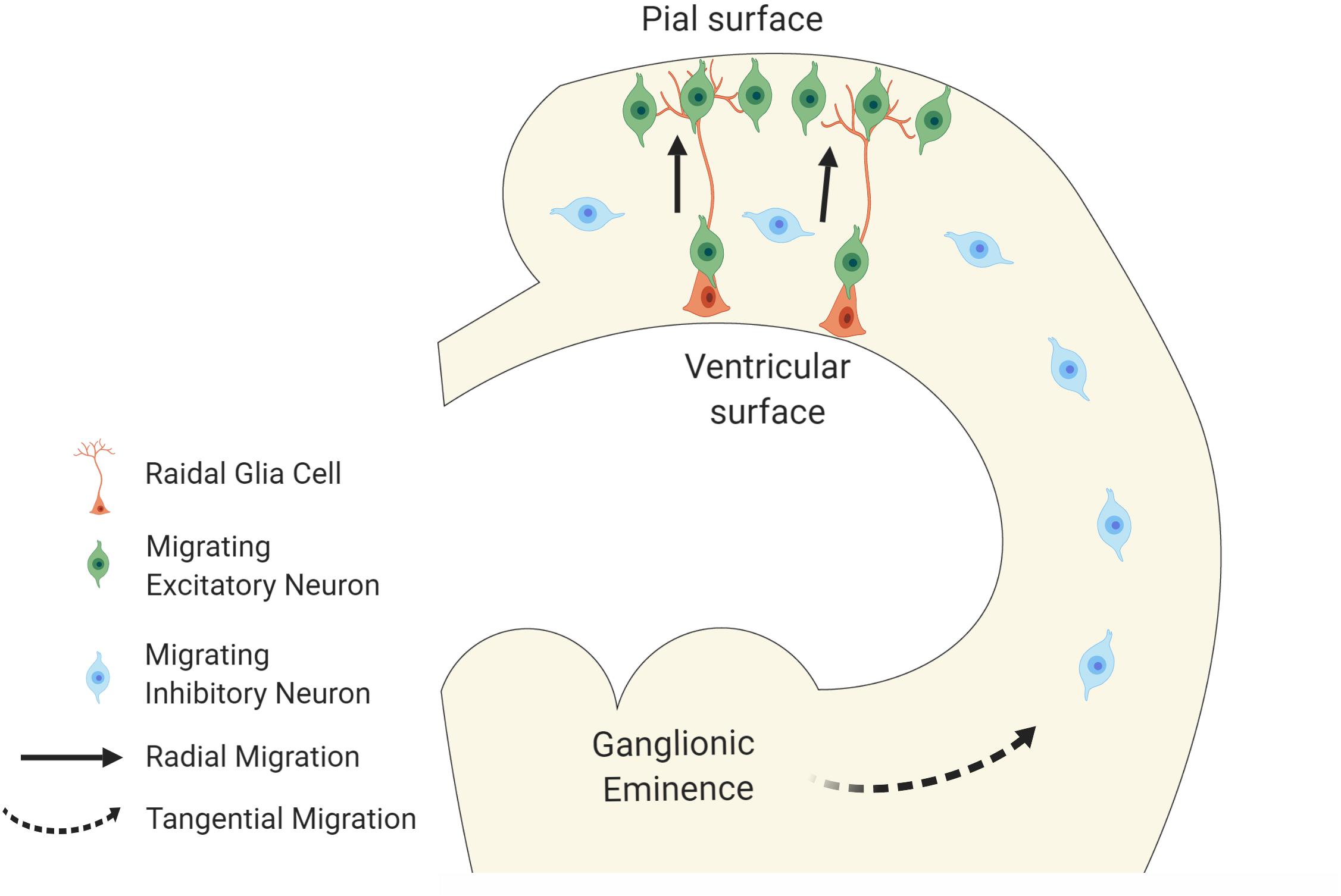
Figure 1. Modes of neuronal migration in cortical development. The schematic diagram illustrates two major modes of migration. Excitatory neurons migrate radially from ventricular surface toward the pial surface of the neocortex using radial glial cells as their scaffolds whereas the inhibitory neurons migrate tangentially from the ganglionic eminences to the neocortex.
Any disturbances of these two modes of migration cause neuronal migration disorders (NMDs), and the consequent malformations are detectable by brain imaging in NMD patients (Roberts, 2018). Profound cases of NMDs include lissencephaly, heterotopia, and focal dysplasia (Guerrini and Parrini, 2010; Roberts, 2018). Precise control of the migration and positioning of both excitatory and inhibitory neurons are particularly important for the formation of synaptic excitation and inhibition (E/I) balanced circuit in the brain. Therefore the E/I imbalance is the main cause of epilepsy in NMD patients (Copp and Harding, 1999). Other neurodevelopmental and psychiatric disorders, including autism spectrum disorders (ASD), and schizophrenia are also associated with NMDs (Canitano and Pallagrosi, 2017).
In addition to the genetic causes, harmful prenatal environment also leads to neuronal migration defects (Metin et al., 2008). When developmental neurotoxicology first emerged, various paradigms of exposure were deployed in many species to recapitulate human pathophysiology, including defects of neuronal migration. Decades later, many recent studies have utilized genetic tools and cutting edge molecular techniques in those preclinical animal models to decipher the underlying mechanisms of neuronal migration defects under the stress exposure. In this review, we summarize such recent findings in animal research and discuss the mechanisms shared by various types of environmental stress as the targets of interventions.
Prenatal alcohol exposure (PAE) is the cause of fetal alcohol spectrum disorders (FASD) (Jones and Smith, 1973; Burd et al., 2007; Riley et al., 2011; Wilhoit et al., 2017). Alcohol consumed by a pregnant woman can easily cross the placenta, and increases fetal blood alcohol concentrations to the levels equivalent to those in maternal side within 2 h of ingestion (Burd et al., 2012). Affected individuals of FASD display a spectrum of defects, including facial malformations and neurobehavior impairments (Subramoney et al., 2018).
Depending on the timing of exposure to alcohol and the mother’s drinking pattern, alcohol may have different effects on a child (Foltran et al., 2011). A comprehensive transcriptome study of mice that were exposed to two acute doses (5 g/kg in total) of alcohol at various periods of brain development demonstrated that ethanol exposure at different developmental periods impacts differential biological processes (Kleiber et al., 2013). Notably, the genes that are associated with cell migration and differentiation were altered only in the brains exposed to alcohol on E14 and E16 which is equivalent to second trimester in humans (Kleiber et al., 2013).
Neuronal migration defects have been reported by many groups in animal models of PAE. In a binge-drinking model in which the mice were exposed to 5% w/w alcohol at E13.5–E16.5, defects of radial migration particularly affecting the populations of excitatory neurons were found in the somatosensory cortex (Delatour et al., 2018; Table 1). These animals also showed a decrease of dendritic complexity in excitatory neurons and reduced tactile sensitivity (Delatour et al., 2018). In another study using the same binge drinking paradigm (5% w/w E13.5–E16.5 exposure), the increase of interneurons derived from medial ganglionic eminence (MGE) in the medial prefrontal cortex (mPFC) was observed by using Nkx2.1Cre-Ai14 transgenic mice. This increase of interneurons in the mPFC is due to the increase of proliferation of the progenitor cells in MGE as well as earlier entrance of those neurons into the mPFC (Skorput et al., 2015). Associated E/I imbalance that favors synaptic inhibition was also reported (Skorput et al., 2015; Table 1). Chronic liquid administration of ethanol at lower dosage (1 or 2% w/v) from early to mid-gestational stages (E0.5–E14.5) increased the number of inhibitory interneurons in the neocortex(Cuzon et al., 2008; Table 1).
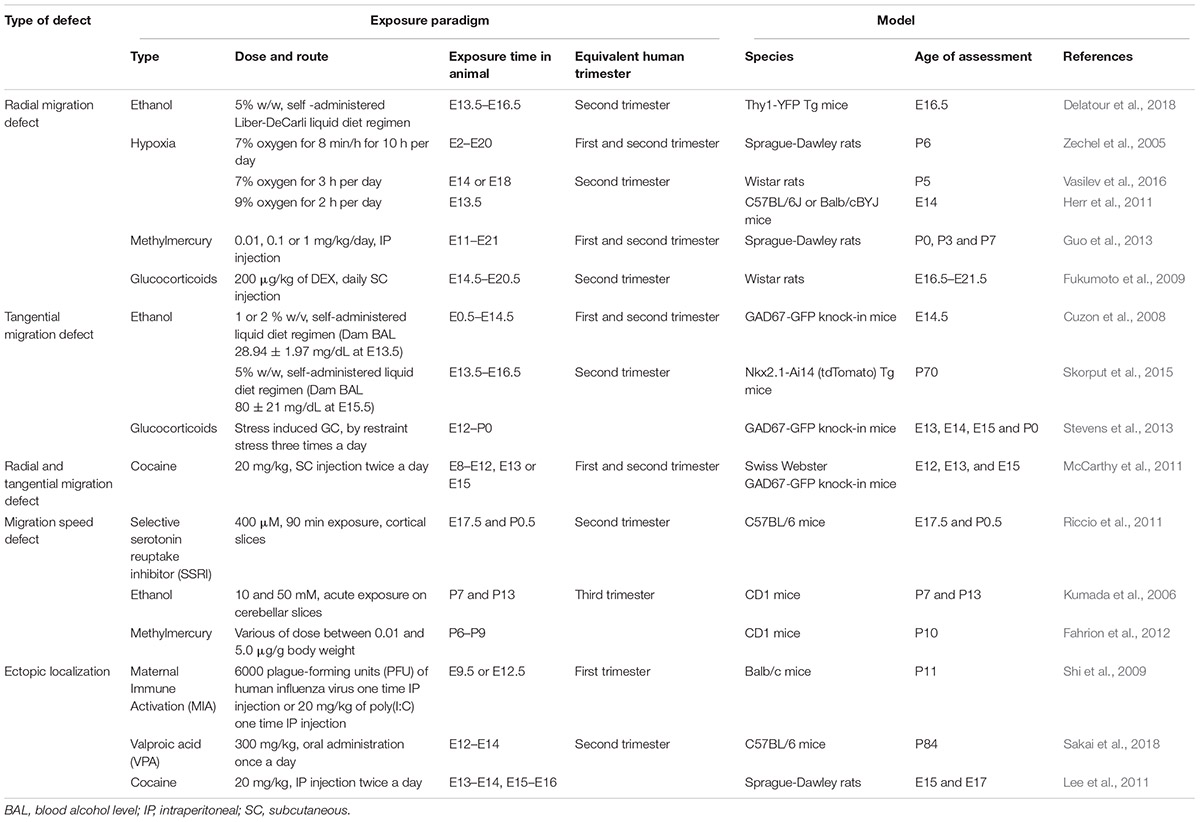
Table 1. Summary of different environmental exposure paradigm studies described in this review.
Intracellular Ca2+ serves as a second messenger of various signaling, and dynamic oscillation of the intracellular Ca2+ is associated with neuronal migration (Komuro and Rakic, 1996). Interestingly, Kumada et al. reported the migration defect observed in cerebellar granule cells following ethanol exposure ex vivo were rescued by modifying the Ca2+ signaling (Table 1). The authors demonstrated that exposure to ethanol reduces the cGMP level, but increases the cAMP level in the tissue, resulting in changes of Ca2+ transients in the migrating neuron. Importantly, the neuronal migration defects were reversed by stimulating the brain with Ca2+ and cGMP or inhibiting cAMP signaling both in vitro and in vivo (Kumada et al., 2006). Rescuing neuronal migration through changing Ca2+ and cyclic nucleotide signaling is mediated by activating the downstream targets that are essential for neuronal migration, such as protein kinase C (PKC), Ca2+/calmodulin-dependent protein kinase II (CaMKII), protein phosphatase 1 (PP1), Rho GTPase, mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) (Huang et al., 2004; Shmueli et al., 2006; Zhao et al., 2009; Khodosevich and Monyer, 2011).
As mentioned earlier, alcohol exposure changes the expressions of many genes that are associated with neuronal migration. Transcriptome analyses of human and mouse fetal cerebral cortices that were acutely exposed to alcohol demonstrated altered expressions of genes that are related to neuronal migration (Hashimoto-Torii et al., 2011; Kleiber et al., 2013; Kawasawa et al., 2017). Of note, a study on the expressions of splicing isoforms in the human brain revealed global exon skipping following alcohol exposure (Kawasawa et al., 2017). Consequently, the functions of the encoded proteins may be different before and after alcohol exposure. However, how the changes in expressing protein isoforms lead to migration deficiency, and the mechanism of alcohol leading to global changes in RNA splicing remains to be investigated.
In human, prenatal cocaine exposure (PCE) is associated with changes in cortical structure, physical and neurocognitive development in the offspring (Bellini et al., 2000; Richardson et al., 2015). The association between cocaine exposure in second trimester and poor motor development in the infants based on psychomotor developmental index (PDI) was reported in a cohort study (Richardson et al., 2008). However, given that cocaine users frequently abuse other drugs and substances, the consequences of PCE at different stages of the trimester is not clearly defined (Martin et al., 2016).
Although animal studies clearly show that PCE in early developmental stages induces a more severe effects in neuronal migration than that in late developmental stages (Lee et al., 2011; McCarthy et al., 2011). The animals subjected to cocaine at 20 mg/kg twice a day during early/mid gestation periods (E13–E14 or E15–E16) showed similar levels of accumulation of postmitotic excitatory neurons in the ventricular zone of the cerebral cortex (Lee et al., 2011; Table 1). This indicates that the migration deficits can be explained by indirect effects due to changes in the proliferation and differentiation of NPCs in the germinal zone.
Disruption of C-X-C motif chemokine ligand 12 (CXCL12)/C-X-C chemokine receptor type 4 (CXCR4) pathway is another potential mechanism of the neuronal migration defects that are elicited by cocaine exposure. Reduction of CXCR4 protein expression following exposure to cocaine was reported by Hu et al. (2006). They exposed human fetal brain-derived NPCs to cocaine in vitro and observed inhibition of cellular proliferation and directional migration toward CXCL12 as well as reduction of CXCR4 expression (Hu et al., 2006). Similarly, the positioning of the interneurons that express CXCR4 in upper cortical layers was disturbed in Cxcr4 knockout mice, providing additional evidence that CXCR4 plays a role in neuronal migration (Stumm et al., 2003). However, neither study replenishes CXCR4 to ameliorate the neuronal migration defects. This piece of data will strengthen the crucial role of CXCR4/CXCL12 signaling in neuronal migration, and indicate the potential of the signaling as a target of therapeutic intervention for NMDs.
McCarthy et al. (2011) proposed that the reduction of brain-derived neurotropic factor (BDNF) post exposure to cocaine may also explain impaired migration of the interneurons (Table 1). The animals that were injected with 20 mg/kg of cocaine twice a day show defects not only in radial migration of excitatory neurons but also in tangential migration of inhibitory neurons (McCarthy et al., 2011). These defects were associated with a transient decrease in BDNF protein expression (McCarthy et al., 2011). BDNF plays a pivotal role in neurogenesis in the developing cerebral cortex, and its interaction with CXCR4 has been reported (Ohmiya et al., 2002; Bachis et al., 2003). In fact, Xu and Heilshorn (2013) demonstrated that BDNF significantly enhances the directional migration of human NPCs toward CXCL12. Furthermore, the presence of BAMD3100, a blocker of CXCR4, completely abolishes the migration, suggesting that BDNF-mediated chemotaxis toward CXCL12 is through CXCR4 activation (Xu and Heilshorn, 2013). Although how BDNF and CXCR4 interact during neuronal migration is still unclear, BDNF-mediated CXCL12/CXCR4 signaling may critically contribute to the impairment of neuronal migration under cocaine exposure.
Intrauterine hypoxia occurs when the fetus is deprived of an adequate supply of oxygen. The inadequate oxygen level may be caused by high altitude and pre-existing maternal cardiovascular diseases, including heart failure and pulmonary hypertension. Maternal anemia, infections, chronic inflammation, and smoking also limit the amount of oxygen delivered to the fetus (Hutter et al., 2010). Chronic hypoxia during brain development affects radial and tangential migrations of excitatory and inhibitory neurons, respectively, thereby leading to the thinner cortical plate in newborn rats (Zechel et al., 2005; Table 1). Vasilev et al. (2016) demonstrated a short period of hypoxia at E14 or E18 impairs radial migration. Notably, hypoxia at E14, but not at E18, disturbed lateral dispersion of excitatory neurons in cortical minicolumns (Table 1). This finding indicates that the occurrence of neuronal migration defects due to prenatal hypoxia is timing-dependent. The fetal brain in earlier gestation period may be more susceptible to hypoxia-induced disturbance of neuronal migration. However, the relationship between the timing and severity of neuronal migration defect is still unclear, and thus more investigations are required.
One of the potential mechanisms underlying the impaired neuronal migration under prenatal hypoxia is the activation of lysophosphatidic acid (LPA) signaling. Herr et al. (2011) demonstrated that LPA signaling is activated by acute prenatal hypoxia in mice (Table 1). LPA is a lysophospholipid that activates LPA receptors and affects neurogenesis, neuronal migration, neuritogenesis, and myelination (Ishii et al., 2004). Notably, hypoxia-induced impaired neuronal migration was mitigated in E13 brain slices of Lpa1 receptor knockout mice exposed to hypoxia for 17 h. This observation suggests that defects of neuronal migration under hypoxia is mediated by Lpa1 receptor (Herr et al., 2011).
Maternal use of valproic acid (VPA), an anti-epileptic drug, during pregnancy is closely associated with increased frequency of congenital malformation and neuropsychiatric disorders in human (Meador et al., 2013). A cohort study comparing the various anti-epileptic drugs that are prescribed to pregnant women found that the risks of fetal malformation increase in a dose-dependent manner. VPA exposure especially presents a greater risk of major congenital malformation compared to other antiepileptic drugs such as carbamazepine, lamotrigine, and phenobarbital (Tomson et al., 2011). VPA at daily doses of 1000 mg or higher is clearly associated with developmental abnormalities, thus frequent consumption of the VPA at reduced dose is recommended control epilepsy during pregnancy (Ornoy, 2009). Of note, the manifestation of autism is also strongly associated with prenatal exposure to VPA. Children who are exposed to VPA prenatally have a three-fold higher risk of developing autism (Christensen et al., 2013). Therefore, the mouse model of prenatal exposure to VPA has been widely used as one of non-genetic ASD models. In fact, these animals mimic many of ASD-like behaviors, such as social interaction deficits, repetitive self-grooming, digging, and deficiency in sensory processing (Roullet et al., 2010; Nicolini and Fahnestock, 2018).
The exact mechanism of how VPA treats epilepsy is not fully understood, yet proposed mechanisms include alteration of GABA levels, blockade of voltage-gated sodium channels (VGSCs) and inhibition of histone deacetylases (Rosenberg, 2007). All of which may contribute to abnormal brain development. For example, blocking or enhancing GABAA receptor diminished or promoted the migration of MGE-derived interneurons into the cerebral cortex, respectively (Cuzon et al., 2006), demonstrating ambient level of GABA is important for stimulating the migrating neurons. Similarly, the activation of VGSCs induced Ca2+ signaling in GABA-mediated migration of oligodendrocyte precursor cells, and the knockdown of VGSCs by siRNA diminished intracellular Ca2+ level and reduced the migration (Tong et al., 2009). Thus, the blockage of VGSCs in neurons by VPA can lead to neuronal migration impairment via similar mechanism.
In addition, prenatal exposure to VPA during the peak of cortical neurogenesis in mice changes expressions of the genes that are associated with the cell migration, including BDNF and Cxcr4. This results in the mislocalization of excitatory neurons in the cerebral cortex in these animals (Almeida et al., 2014; Sakai et al., 2018; Table 1). It indicates that disturbance of BDNF/CXCL12/CXCR4 signaling is another potential mechanism underlying the deficiency of the neuronal migration by prenatal VPA exposure.
Selective serotonin reuptake inhibitors (SSRIs) are commonly used to treat depression during pregnancy. The human imaging study demonstrated a significant increase of the gray matter volume in amygdala and insula along with increased connectivity in children exposed to SSRI prenatally (Lugo-Candelas et al., 2018). The expression of the genes involved in serotonin signaling starts during the early stage of fetal brain development, and the disruption of serotonin signaling pathway affects cell proliferation, differentiation, neuronal migration and network formation in various regions of the brain (Sodhi and Sanders-Bush, 2004).
The differential effects of SSRI depending upon the given dosage and/or timing is obscure. A study has shown that the children exposed to SSRIs in late pregnancy (>29 weeks) is strongly associated with increased risk of anxious or depressed behaviors (Lupattelli et al., 2018). Whereas another study has shown that the length of SSRI exposure rather than the timing crucially contributes to the outcome of the fetal development (Oberlander et al., 2008). Similarly, another study shows that extended SSRI exposure in prenatal period increases the risks of lower scores in psychomotor developmental index and behavioral rating scale in infancy (Casper et al., 2011).
Given that the polymorphisms of the genes encoding the serotonin transporters and receptors are strongly associated with neurodevelopmental disorders such as ASD (Anderson et al., 2002), both reduced and increased levels of serotonin signaling are proposed to cause devastating consequence in brain development. Consistent with this hypothesis, excessive activation of serotonin signaling disturbed the migration of pyramidal neurons in the cortex of the serotonin transporter (Sert) knockout (KO) mouse, in which serotonin accumulated extracellular and the activation of serotonin receptors was extended (Riccio et al., 2011; Table 1). The authors also demonstrated that excess activation of a subtype of serotonin receptor 5-HT6 that is expressed in the developing excitatory neurons disturbed the migration in the Sert KO cerebral cortex. In line with the hypothesis above, downregulation of 5-HT6 receptor also resulted in mislocalization of pyramidal neurons in the mouse cortex (Jacobshagen et al., 2014). Given that serotonin signaling also interacts with BDNF signaling, the migration may be disrupted by, at least in part, the similar mechanism observed in the cases of prenatal exposure to cocaine and VPA as described above (Sodhi and Sanders-Bush, 2004).
Exposure to a high level of heavy metal, such as methylmercury, is associated with neuropsychological problems. Methylmercury ingested by a pregnant woman is absorbed in the gastrointestinal tract and forms a placenta-crossing complex with L-cysteine (Broussard et al., 2002). Children exposed to methylmercury in the prenatal period show deficits in finger tapping speed, reaction time on a continuous performance task, and cued naming (Debes et al., 2006). A classic example of neurotoxicity due to prenatal methylmercury exposure is patients of Minamata disease. The brain development of these patients is severely compromised, resulting in ataxia, dysarthria, and tremor (Harada, 1978).
Similar to the cases of fetal exposure to alcohol described in previous section, mice that were exposed to methylmercury from postnatal day 6 to 9, the peak of neuronal migration in the cerebellum, showed a decrease in the frequency of spontaneous Ca2+ dynamics as well as impaired migration of cerebellar granular cells (CGCs) (Fahrion et al., 2012; Table 1). Notably, the migration defects were ameliorated by caffeine supplementation, which induces Ca2+ release by binding to ryanodine receptors in the brain and stimulating internal Ca2+ release and Ca2+ influx (Usachev et al., 1993; Fahrion et al., 2012). Additionally, a proteomic study of mouse embryo exposed to methylmercury reported a decrease in the phosphorylated form of cofilin in CGCs (Vendrell et al., 2010). LIM-kinase 1 (LMK-1)-mediated phosphorylation inactivates cofilin, by which prevents disassembly of actin filaments. Therefore, the change of phosphorylation ratio of cofilin would disrupt the actin dynamics in the filopodia of migrating CGCs (Yang et al., 1998; Fujimura et al., 2009; Fujimura and Usuki, 2012).
Chronic exposure to methylmercury, even at a very low dose of 0.1 mg/kg/day, affects neuronal migration in the developing rat cerebral cortex. Methylmercury suppresses protein expressions of Rac family small GTPase 1 (RAC1), Cell division cycle 42 (CDC42), and Ras homolog gene family, member A (RHOA), all of which are key players of radial migration in the developing brain (Govek et al., 2011; Guo et al., 2013; Table 1). Altogether, similar to alcohol, methylmercury changes the Ca2+ signaling and the expressions of genes or proteins that are closely linked to the control of cortical neuronal migration.
Another potential mechanism of methylmercury affecting the neuronal migration may be the serotonin signaling. Placental serotonin, which is synthesized from maternal tryptophan precursor, is required for neuronal migration during the forebrain development (Bonnin et al., 2011). Methylmercury not only inhibits cellular proliferation and migration, but also alters the expressions of oxidative stress-related genes such as superoxide dismutase 1 (SOD1) in human placenta-derived trophoblasts (Tucker and Nowak, 2018). The vulnerability of the trophoblasts to methylmercury suggests the potential effects on placental development (Hsiao and Patterson, 2012). Therefore, the maldevelopment of the placenta under exposure to methylmercury leads not only to the loss of protection for the fetus but also to disrupted production of the neurotransmitters required for proper neuronal migration.
Glucocorticoids (GCs) are a class of corticosteroids that are prescribed to pregnant women to promote lung maturation in fetuses when they are at risk of preterm delivery (Marciniak et al., 2011). Synthetic GC binds to the endogenous GC receptors in the fetal lung and promotes the maturation by increasing a production of the surfactant-associated proteins and phospholipids, both of which are crucial for stable lung function (Bolt et al., 2001).
The benefits of GC treatment for fetal lung maturation may come with a cost as studies show that the excess prenatal steroid exposure is associated with a wide spectrum of deficits in brain development, such as loss of hippocampal neurons, delayed myelination, and increased risk of neurodevelopmental disability (Velísek, 2005). Structural abnormalities including reduced cortical thickness in frontal cortex and rostral anterior cingulate cortex (rACC) were also evident in children who had been exposed to GC prenatally (Modi et al., 2001; Davis et al., 2013). Different dosages and timing of GC exposure may have differential effects on the developing brain. Treatment prior to mid-gestation may be well tolerated by the fetus without adverse effects due to the relatively high expression of a GC degrading enzyme, 11β-hydroxysteroid dehydrogenase (11β-HSD2), in the placenta and the fetal brain before mid-gestation (Diaz et al., 1998). In addition, the expressions of GC receptor transcripts start to arise in the pons, medulla, anterior hypothalamus, and spinal cord at mid-gestational stage of pregnancy (E12.5). Then gradually spread to the rest of brain regions such as neocortex in rats (Diaz et al., 1998) which may increase the fetal susceptibility to GC.
Animal studies demonstrate that neuronal migration defect by GC exposure is mediated by alterations in the expression of key neuronal migration genes. The administration of GC retarded the radial migration of excitatory neurons in fetal cerebral cortex. These neurons also showed increase in Cald1 expression. Consistent with this, overexpression of Cald1 led to impaired neuronal migration in vitro (Fukumoto et al., 2009; Table 1). Cald1 encodes Caldesmon that negatively regulates the function of myosin II, an essential protein for the actin dynamics in the leading process of neuron. The upregulation of Cald1 inhibits interactions between actin and myosin II in the radially migrating neurons, thereby disrupting the migration (Sobue and Fukumoto, 2010; Morita et al., 2012).
Interneurons in offspring of dams that underwent daily physical stress (restraint stress) exposure starting from E12 until birth show delayed migration without changes in the survival or proliferation (Table 1; Stevens et al., 2013). The expressions of transcription factors dlx2 and nkx2.1 in the fetal brains are significantly decreased as early as 24 h after the first stress exposure (Stevens et al., 2013). These transcription factors are important in determining cell fate and migration of interneuron (Nóbrega-Pereira et al., 2008). Overall, excess GC affects neuronal migration altering the expressions of the genes that directly and indirectly modulate the neuronal migration.
Maternal immune activation (MIA) is caused by an infection during pregnancy, and it is linked to increased risk of ASD and schizophrenia (Patterson, 2009). Maternal infection to human influenza virus during pregnancy resulted in ectopic cluster formation of Purkinje cells (PC) in the white matter of cerebellar lobules VI and VII in mouse, while the number of PC decreased in lobule VII (Shi et al., 2009; Table 1). A similar reduction in the number of PC in lobule VII was observed in mice born to mothers inoculated with an immunostimulant, poly(I:C) (Shi et al., 2009). These reports suggest that MIA disrupts the migration of the PC during cerebellar development. Given a subclass of small cytokines functions as migrational cues for newly generated neurons, the excess production of maternal cytokines is likely to trigger misguidance of neurons in the fetal brain under MIA (Du et al., 2014). MIA also altered the expressions of genes that are involved in the regulation of cell proliferation, migration, and axon guidance in the fetal brain (Oskvig et al., 2012; Lombardo et al., 2018). However, the contributions of each of such genes to the neuronal migration remain elusive.
As described in previous sections, different types of prenatal environmental stress have common effects on the neuronal migration at the molecular level (Summarized in Figure 2). An example described above is the BDNF/CXCL12/CXCR4 signaling. Many of the chemicals and stress listed in this review alter the expressions of genes involved in this pathway. Interestingly, both excessive and insufficient expressions and activations of the genes in this signaling disturb the neuronal migration. Similarly, as described above, both up- and down-regulation of serotonin signaling disturb neuronal migration.
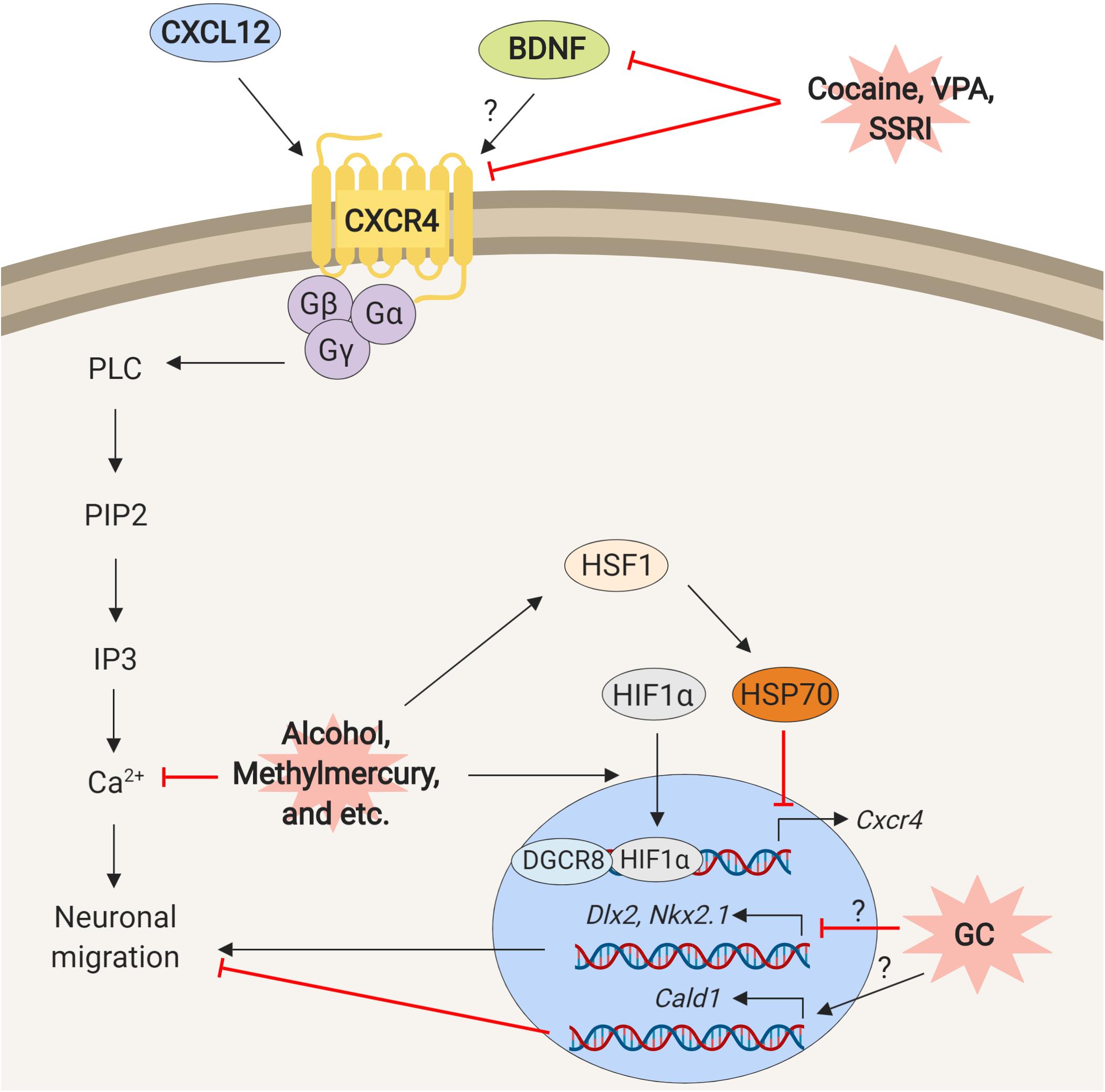
Figure 2. Schematic representation of mechanisms underlying neuronal migration defects elicited by exposure to environmental stress. The prenatal environmental stressors such as cocaine, VPA, SSRI, ethanol, methylmercury similarly, disturb interactions and functions of the molecules that are involved in the control of neuronal migration.
How does CXCR4/CXCL12 signaling control the neuronal migration? The clues may be found in the genetic disorder, 22q11.2 deletion syndrome (22q11.2DS). The mouse model of 22q11.2DS shows neuronal migration defects due to decrease of Cxcr4 (Meechan et al., 2012). This phenotype is rescued by overexpression of the Dgcr8 (Toritsuka et al., 2013). DGCR8 forms a complex with Drosha to process primary microRNA (pri-miRNA) and facilitates microRNA (miRNA) maturation (Gregory et al., 2004; Han et al., 2004). This finding suggests that the disturbance of the CXCL12/CXCR4 pathway may change miRNA-mediated regulation of the transcriptions that control neuronal migration (Sathyan et al., 2007).
The environmental stress may alter the expressions of genes in the CXCL12/CXCR4 pathway via a different route. Heat shock transcription factor 1 – hypoxia inducible factor-1 alpha (HSF1-HIF1α) pathway is a stress response signaling that is activated by various types of environmental stress in fetal brain (Hashimoto-Torii et al., 2014; Ishii et al., 2017; Torii et al., 2017), while HIF1α also directly transcribes CXCR4 (Schioppa et al., 2003; Gabai et al., 2012). Thus the HIF1α may transcribe CXCR4 in response to environmental stress (Ishikawa et al., 2009). On the other hand, overexpression of heat shock protein 70 (hsp70) that is transcriptionally regulated by HSF1 reduces Cxcr4 expression (Sakurai et al., 2015).
Another common molecular trait shared by different types of environmental stress is the changes in expressions of genes that regulate the dynamics of the cytoskeleton. These changes happen partially through the activation of stress response signaling and the Ca2+ signaling (Kumada et al., 2006; Fahrion et al., 2012; Lombardo et al., 2018).
Although animal models convey the mechanisms responding to environmental stress, what happens in the large human brain remains speculative. As rodents and humans do not share the identical mechanism of neuronal migration during the development of brain, studying brain development in a model that closely mimics human brain development is imperative (Semple et al., 2013). Therefore, the human pluripotent stem cell-derived three-dimensional organoid culture system, in which the cortical lamination and thick germinal zone are reproduced, may be a versatile tool to understand the molecular basis of neuronal migration defects in the human brain (Mason and Price, 2016; Wang, 2018). However, current protocols of the brain organoid system often require culture conditions that increase the levels of reactive oxidative stress (ROS). Thereby an optimal protocol that allows testing the effects of environmental stress in organoids without high ROS level is still needed. An especially intriguing question to be answered by using the organoid system will be whether the pathways commonly disrupted during neuronal migration by different types of environmental stressors are similarly, or differently regulated in human brain organoid in comparison to animal models.
HH and RK contributed to the writing and editing of the manuscript, and KH-T reviewed and edited the manuscript. All authors reviewed, discussed, and commented on the manuscript.
We thank NIH/NIAAA F31AA027693 (HH), 5R01AA025215-02 (KH-T) and NARSAD/Scott-Gentle Foundation for their respective support. This work was also supported by National Institute of Health (NIH) grant UH2AA026106 as part of the Collaborative Initiative on Fetal Alcohol Spectrum Disorders (CIFASD) to KH-T.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Almeida, L. E. F., Roby, C. D., and Krueger, B. K. (2014). Increased BDNF expression in fetal brain in the valproic acid model of autism. Mol. Cell. Neurosci. 59, 57–62. doi: 10.1016/j.mcn.2014.01.007
Anderson, G. M., Gutknecht, L., Cohen, D. J., Brailly-Tabard, S., Cohen, J. H. M., Ferrari, P., et al. (2002). Serotonin transporter promoter variants in autism: functional effects and relationship to platelet hyperserotonemia. Mol. Psychiatry 7, 831–836. doi: 10.1038/sj.mp.4001099
Ayala, R., Shu, T., and Tsai, L.-H. (2007). Trekking across the brain: the journey of neuronal migration. Cell 128, 29–43. doi: 10.1016/j.cell.2006.12.021
Bachis, A., Major, E. O., and Mocchetti, I. (2003). Brain-derived neurotrophic factor inhibits human immunodeficiency virus-1/gp120-mediated cerebellar granule cell death by preventing gp120 internalization. J. Neurosci. 23,5715–5722. doi: 10.1523/jneurosci.23-13-05715.2003
Bellini, C., Massocco, D., and Serra, G. (2000). Prenatal cocaine exposure and the expanding spectrum of brain malformations. Arch. Intern. Med. 160, 2393. doi: 10.1001/archinte.160.15.2393
Bolt, R. J., van Weissenbruch, M. M., Lafeber, H. N., and Delemarre-van de Waal, H. A. (2001). Glucocorticoids and lung development in the fetus and preterm infant. Pediatr. Pulmonol. 32, 76–91. doi: 10.1002/ppul.1092
Bonnin, A., Goeden, N., Chen, K., Wilson, M. L., King, J., Shih, J. C., et al. (2011). A transient placental source of serotonin for the fetal forebrain. Nature 472, 347–350. doi: 10.1038/nature09972
Broussard, L. A., Hammett-Stabler, C. A., Winecker, R. E., and Ropero-Miller, J. D. (2002). The Toxicology of Mercury. Lab. Med. 33, 614–625.
Burd, L., Blair, J., and Dropps, K. (2012). Prenatal alcohol exposure, blood alcohol concentrations and alcohol elimination rates for the mother, fetus and newborn. J. Perinatol. 32, 652–659. doi: 10.1038/jp.2012.57
Burd, L., Roberts, D., Olson, M., and Odendaal, H. (2007). Ethanol and the placenta: a review. J. Matern. Neonatal Med. 20, 361–375. doi: 10.1080/14767050701298365
Canitano, R., and Pallagrosi, M. (2017). Autism spectrum disorders and schizophrenia spectrum disorders: excitation/inhibition imbalance and developmental trajectories. Front. Psychiatry 8:69. doi: 10.3389/fpsyt.2017.00069
Casper, R. C., Gilles, A. A., Fleisher, B. E., Baran, J., Enns, G., and Lazzeroni, L. C. (2011). Length of prenatal exposure to selective serotonin reuptake inhibitor (SSRI) antidepressants: effects on neonatal adaptation and psychomotor development. Psychopharmacology 217, 211–219. doi: 10.1007/s00213-011-2270-z
Christensen, J., Grønborg, T. K., Sørensen, M. J., Schendel, D., Parner, E. T., Pedersen, L. H., et al. (2013). Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309, 1696–1703. doi: 10.1001/jama.2013.2270
Copp, A. J., and Harding, B. N. (1999). Neuronal migration disorders in humans and in mouse models–an overview. Epilepsy Res. 36, 133–141. doi: 10.1016/s0920-1211(99)00047-9
Cuzon, V. C., Yeh, P. W., Cheng, Q., and Yeh, H. H. (2006). Ambient GABA promotes cortical entry of tangentially migrating cells derived from the medial ganglionic eminence. Cereb. Cortex 16, 1377–1388. doi: 10.1093/cercor/bhj084
Cuzon, V. C., Yeh, P. W. L., Yanagawa, Y., Obata, K., and Yeh, H. H. (2008). Ethanol consumption during early pregnancy alters the disposition of tangentially migrating GABAergic interneurons in the fetal cortex. J. Neurosci. 28,1854–1864. doi: 10.1523/JNEUROSCI.5110-07.2008
Davis, E. P., Sandman, C. A., Buss, C., Wing, D. A., and Head, K. (2013). Fetal glucocorticoid exposure is associated with preadolescent brain development. Biol. Psychiatry 74, 647–655. doi: 10.1016/j.biopsych.2013.03.009
Debes, F., Budtz-Jørgensen, E., Weihe, P., White, R. F., and Grandjean, P. (2006). Impact of prenatal methylmercury exposure on neurobehavioral function at age 14 years. Neurotoxicol. Teratol. 28, 363–375. doi: 10.1016/j.ntt.2006.02.004
Delatour, L. C., Yeh, P. W., and Yeh, H. H. (2018). Ethanol exposure in utero disrupts radial migration and pyramidal cell development in the somatosensory cortex. Cereb. Cortex 29, 2125–2139. doi: 10.1093/cercor/bhy094
Diaz, R., Brown, R. W., and Seckl, J. R. (1998). Distinct ontogeny of glucocorticoid and mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase types I and II mRNAs in the fetal rat brain suggest a complex control of glucocorticoid actions. J. Neurosci. 18, 2570–2580. doi: 10.1523/jneurosci.18-07-02570.1998
Du, M.-R., Wang, S.-C., and Li, D.-J. (2014). The integrative roles of chemokines at the maternal-fetal interface in early pregnancy. Cell. Mol. Immunol. 11, 438–448. doi: 10.1038/cmi.2014.68
Fahrion, J. K., Komuro, Y., Li, Y., Ohno, N., Littner, Y., Raoult, E., et al. (2012). Rescue of neuronal migration deficits in a mouse model of fetal Minamata disease by increasing neuronal Ca2(spike frequency. Proc. Natl. Acad. Sci. U.S.A. 109, 5057–5062. doi: 10.1073/pnas.1120747109
Foltran, F., Gregori, D., Franchin, L., Verduci, E., and Giovannini, M. (2011). Effect of alcohol consumption in prenatal life, childhood, and adolescence on child development. Nutr. Rev. 69, 642–659. doi: 10.1111/j.1753-4887.2011.00417.x
Fujimura, M., and Usuki, F. (2012). Differing effects of toxicants (Methylmercury, Inorganic Mercury, Lead, Amyloid, and Rotenone) on cultured rat cerebrocortical neurons: differential expression of rho proteins associated with neurotoxicity. Toxicol. Sci. 126, 506–514. doi: 10.1093/toxsci/kfr352
Fujimura, M., Usuki, F., Sawada, M., Rostene, W., Godefroy, D., and Takashima, A. (2009). Methylmercury exposure downregulates the expression of Racl and leads to neuritic degeneration and ultimately apoptosis in cerebrocortical neurons. Neurotoxicology 30, 16–22. doi: 10.1016/j.neuro.2008.10.002
Fukumoto, K., Morita, T., Mayanagi, T., Tanokashira, D., Yoshida, T., Sakai, A., et al. (2009). Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol. Psychiatry 14, 1119–1131. doi: 10.1038/mp.2009.60
Gabai, V. L., Meng, L., Kim, G., Mills, T. A., Benjamin, I. J., and Sherman, M. Y. (2012). Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol. Cell. Biol. 32, 929–940. doi: 10.1128/MCB.05921-11
Govek, E.-E., Hatten, M. E., and Van Aelst, L. (2011). The role of Rho GTPase proteins in CNS neuronal migration. Dev. Neurobiol. 71, 528–553. doi: 10.1002/dneu.20850
Gregory, R. I., Yan, K., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., et al. (2004). The microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. doi: 10.1038/nature03120
Guerrini, R., and Parrini, E. (2010). Neuronal migration disorders. Neurobiol. Dis. 38, 154–166. doi: 10.1016/J.NBD.2009.02.008
Guo, B.-Q., Yan, C.-H., Cai, S.-Z., Yuan, X.-B., and Shen, X.-M. (2013). Low level prenatal exposure to methylmercury disrupts neuronal migration in the developing rat cerebral cortex. Toxicology 304, 57–68. doi: 10.1016/j.tox.2012.11.019
Han, J., Lee, Y., Yeom, K.-H., Kim, Y.-K., Jin, H., and Kim, V. N. (2004). The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18, 3016–3027. doi: 10.1101/gad.1262504
Harada, M. (1978). Congenital minamata disease: intrauterine methylmercury poisoning. Teratology 18, 285–288. doi: 10.1002/tera.1420180216
Hashimoto-Torii, K., Kawasawa, Y. I., Kuhn, A., and Rakic, P. (2011). Combined transcriptome analysis of fetal human and mouse cerebral cortex exposed to alcohol. Proc. Natl. Acad. Sci. U.S.A. 108, 4212–4217. doi: 10.1073/pnas.1100903108
Hashimoto-Torii, K., Torii, M., Fujimoto, M., Nakai, A., ElFatimy, R., Mezger, V., et al. (2014). Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron 82, 560–572. doi: 10.1016/j.neuron.2014.03.002
Herr, K. J., Herr, D. R., Lee, C.-W., Noguchi, K., and Chun, J. (2011). Stereotyped fetal brain disorganization is induced by hypoxia and requires lysophosphatidic acid receptor 1 (LPA1) signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 15444–15449. doi: 10.1073/pnas.1106129108
Hsiao, E. Y., and Patterson, P. H. (2012). Placental regulation of maternal-fetal interactions and brain development. Dev. Neurobiol. 72, 1317–1326. doi: 10.1002/dneu.22045
Hu, S., Cheeran, M. C. J., Sheng, W. S., Ni, H. T., Lokensgard, J. R., and Peterson, P. K. (2006). Cocaine alters proliferation, migration, and differentiation of human fetal brain-derived neural precursor cells. J. Pharmacol. Exp. Ther. 318, 1280–1286. doi: 10.1002/esp.1333
Huang, C., Jacobson, K., and Schaller, M. D. (2004). MAP kinases and cell migration. J. Cell Sci. 117, 4619–4628. doi: 10.1242/jcs.01481
Hutter, D., Kingdom, J., and Jaeggi, E. (2010). Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int. J. Pediatr. 2010:401323. doi: 10.1155/2010/401323
Ishii, I., Fukushima, N., Ye, X., and Chun, J. (2004). Lysophospholipid receptors: signaling and biology. Annu. Rev. Biochem. 73, 321–354. doi: 10.1146/annurev.biochem.73.011303.073731
Ishii, S., Torii, M., Son, A. I., Rajendraprasad, M., Morozov, Y. M., Kawasawa, Y. I., et al. (2017). Variations in brain defects result from cellular mosaicism in the activation of heat shock signalling. Nat. Commun. 8:15157. doi: 10.1038/ncomms15157
Ishikawa, T., Nakashiro, K., Klosek, S. K., Goda, H., Hara, S., Uchida, D., et al. (2009). Hypoxia enhances CXCR4 expression by activating HIF-1 in oral squamous cell carcinoma. Oncol. Rep. 21, 707–712.
Jacobshagen, M., Niquille, M., Chaumont-Dubel, S., Marin, P., and Dayer, A. (2014). The serotonin 6 receptor controls neuronal migration during corticogenesis via a ligand-independent Cdk5-dependent mechanism. Development 141, 3370–3377. doi: 10.1242/dev.108043
Jones, K. L., and Smith, D. W. (1973). Recognition of the fetal alcohol syndrome in early infancy. Lancet 302, 999–1001. doi: 10.1016/S0140-6736(73)91092-1
Kawasawa, Y. I., Mohammad, S., Son, A. I., Morizono, H., Basha, A., Salzberg, A. C., et al. (2017). Genome-wide profiling of differentially spliced mRNAs in human fetal cortical tissue exposed to alcohol. Alcohol 62, 1–9. doi: 10.1016/j.alcohol.2017.05.001
Khodosevich, K., and Monyer, H. (2011). Signaling in migrating neurons: from molecules to networks. Front. Neurosci. 5:28. doi: 10.3389/fnins.2011.00028
Kleiber, M. L., Mantha, K., Stringer, R. L., and Singh, S. M. (2013). Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure. J. Neurodev. Disord. 5:6. doi: 10.1186/1866-1955-5-6
Komuro, H., and Rakic, P. (1996). Intracellular Ca2(fluctuations modulate the rate of neuronal migration. Neuron 17, 275–285. doi: 10.1016/S0896-6273(00)80159-2
Kumada, T., Lakshmana, M. K. and Komuro, H. (2006). Reversal of neuronal migration in a mouse model of fetal alcohol syndrome by controlling second-messenger signalings. J. Neurosci. 26, 742–756. doi: 10.1523/JNEUROSCI.4478-05.2006
Lee, C.-T., Chen, J., Worden, L. T., and Freed, W. J. (2011). Cocaine causes deficits in radial migration and alters the distribution of glutamate and GABA neurons in the developing rat cerebral cortex. Synapse 65, 21–34. doi: 10.1002/syn.20814
Lombardo, M. V., Moon, H. M., Su, J., Palmer, T. D., Courchesne, E., and Pramparo, T. (2018). Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 23, 1001–1013. doi: 10.1038/mp.2017.15
Lugo-Candelas, C., Cha, J., Hong, S., Bastidas, V., Weissman, M., Fifer, W. P., et al. (2018). Associations between brain structure and connectivity in infants and exposure to selective serotonin reuptake inhibitors during pregnancy. JAMA Pediatr. 172:525. doi: 10.1001/jamapediatrics.2017.5227
Lupattelli, A., Wood, M., Ystrom, E., Skurtveit, S., Handal, M., and Nordeng, H. (2018). Effect of time-dependent selective serotonin reuptake inhibitor antidepressants during pregnancy on behavioral, emotional, and social development in preschool-aged children. J. Am. Acad. Child Adolesc. Psychiatry 57, 200–208. doi: 10.1016/j.jaac.2017.12.010
Marciniak, B., Patro-Małysza, J., Poniedziałek-Czajkowska, E., Kimber-Trojnar, Z., Leszczyńska-Gorzelak, B., and Oleszczuk, J. (2011). Glucocorticoids in pregnancy. Curr. Pharm. Biotechnol. 12, 750–757.
Marín, O., and Rubenstein, J. L. R. (2001). A long, remarkable journey: tangential migration in the telencephalon. Nat. Rev. Neurosci. 2, 780–790. doi: 10.1038/35097509
Martin, M. M., Graham, D. L., McCarthy, D. M., Bhide, P. G., and Stanwood, G. D. (2016). Cocaine-induced neurodevelopmental deficits and underlying mechanisms. Birth Defects Res. C Embryo Today 108, 147–173. doi: 10.1002/bdrc.21132
Mason, J. O., and Price, D. J. (2016). Building brains in a dish: prospects for growing cerebral organoids from stem cells. Neuroscience 334, 105–118. doi: 10.1016/j.neuroscience.2016.07.048
McCarthy, D. M., Zhang, X., Darnell, S. B., Sangrey, G. R., Yanagawa, Y., Sadri-Vakili, G., et al. (2011). Cocaine alters BDNF expression and neuronal migration in the embryonic mouse forebrain. J. Neurosci. 31, 13400–13411. doi: 10.1523/JNEUROSCI.2944-11.2011
Meador, K. J., Baker, G. A., Browning, N., Cohen, M. J., Bromley, R. L., Clayton-Smith, J., et al. (2013). Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurol. 12, 244–252. doi: 10.1016/S1474-4422(12)70323-X
Meechan, D. W., Tucker, E. S., Maynard, T. M., and LaMantia, A.-S. (2012). Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome. Proc. Natl. Acad. Sci. U.S.A. 109, 18601–18606. doi: 10.1073/pnas.1211507109
Metin, C., Vallee, R. B., Rakic, P., and Bhide, P. G. (2008). Modes and mishaps of neuronal migration in the mammalian brain. J. Neurosci. 28, 11746–11752. doi: 10.1523/jneurosci.3860-08.2008
Modi, N., Lewis, H., Al-Naqeeb, N., Ajayi-Obe, M., Doré, C. J., and Rutherford, M. (2001). The effects of repeated antenatal glucocorticoid therapy on the developing brain. Pediatr. Res. 50, 581–585. doi: 10.1203/00006450-200111000-00008
Morita, T., Mayanagi, T., and Sobue, K. (2012). Caldesmon regulates axon extension through interaction with myosin II. J. Biol. Chem. 287, 3349–3356. doi: 10.1074/jbc.M111.295618
Nicolini, C., and Fahnestock, M. (2018). The valproic acid-induced rodent model of autism. Exp. Neurol. 299, 217–227. doi: 10.1016/j.expneurol.2017.04.017
Nóbrega-Pereira, S., Kessaris, N., Du, T., Kimura, S., Anderson, S. A., and Marín, O. (2008). Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron 59, 733–745. doi: 10.1016/j.neuron.2008.07.024
Oberlander, T. F., Warburton, W., Misri, S., Aghajanian, J., and Hertzman, C. (2008). Effects of timing and duration of gestational exposure to serotonin reuptake inhibitor antidepressants: population-based study. Br. J. Psychiatry 192, 338–343. doi: 10.1192/bjp.bp.107.037101
Ohmiya, M., Shudai, T., Nitta, A., Nomoto, H., Furukawa, Y., and Furukawa, S. (2002). Brain-derived neurotrophic factor alters cell migration of particular progenitors in the developing mouse cerebral cortex. Neurosci. Lett. 317, 21–24. doi: 10.1016/s0304-3940(01)02412-0
Ornoy, A. (2009). Valproic acid in pregnancy: how much are we endangering the embryo and fetus? Reprod. Toxicol. 28, 1–10. doi: 10.1016/j.reprotox.2009.02.014
Oskvig, D. B., Elkahloun, A. G., Johnson, K. R., Phillips, T. M., and Herkenham, M. (2012). Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain. Behav. Immun. 26, 623–634. doi: 10.1016/j.bbi.2012.01.015
Patterson, P. H. (2009). Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav. Brain Res. 204, 313–321. doi: 10.1016/j.bbr.2008.12.016
Rakic, P. (1972). Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 145, 61–83. doi: 10.1002/cne.901450105
Rakic, P. (1995). Radial versus tangential migration of neuronal clones in the developing cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 92, 11323–11327. doi: 10.1073/pnas.92.25.11323
Rees, S., and Harding, R. (2004). Brain development during fetal life: influences of the intra-uterine environment. Neurosci. Lett. 361, 111–114. doi: 10.1016/j.neulet.2004.02.002
Riccio, O., Jacobshagen, M., Golding, B., Vutskits, L., Jabaudon, D., Hornung, J. P., et al. (2011). Excess of serotonin affects neocortical pyramidal neuron migration. Transl. Psychiatry 1:e47. doi: 10.1038/tp.2011.49
Richardson, G. A., Goldschmidt, L., Larkby, C., and Day, N. L. (2015). Effects of prenatal cocaine exposure on adolescent development. Neurotoxicol. Teratol. 49, 41–48. doi: 10.1016/j.ntt.2015.03.002
Richardson, G. A., Goldschmidt, L., and Willford, J. (2008). The effects of prenatal cocaine use on infant development. Neurotoxicol. Teratol. 30, 96–106. doi: 10.1016/j.ntt.2007.12.006
Riley, E. P., Infante, M. A., and Warren, K. R. (2011). Fetal alcohol spectrum disorders: an overview. Neuropsychol. Rev. 21, 73–80. doi: 10.1007/s11065-011-9166-x
Rosenberg, G. (2007). The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell. Mol. Life Sci. 64, 2090–2103. doi: 10.1007/s00018-007-7079-x
Roullet, F. I., Wollaston, L., deCatanzaro, D., and Foster, J. A. (2010). Behavioral and molecular changes in the mouse in response to prenatal exposure to the anti-epileptic drug valproic acid. Neuroscience 170, 514–522. doi: 10.1016/j.neuroscience.2010.06.069
Sakai, A., Matsuda, T., Doi, H., Nagaishi, Y., Kato, K., and Nakashima, K. (2018). Ectopic neurogenesis induced by prenatal antiepileptic drug exposure augments seizure susceptibility in adult mice. Proc. Natl. Acad. Sci. U.S.A. 115, 4270–4275. doi: 10.1073/PNAS.1716479115
Sakurai, T., Kashida, H., Hagiwara, S., Nishida, N., Watanabe, T., Fujita, J., et al. (2015). Heat shock protein A4 controls cell migration and gastric ulcer healing. Dig. Dis. Sci. 60, 850–857. doi: 10.1007/s10620-015-3561-8
Sathyan, P., Golden, H. B., and Miranda, R. C. (2007). Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 27, 8546–8557. doi: 10.1523/JNEUROSCI.1269-07.2007
Schioppa, T., Uranchimeg, B., Saccani, A., Biswas, S. K., Doni, A., Rapisarda, A., et al. (2003). Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 198, 1391–1402. doi: 10.1084/jem.20030267
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M., and Noble-Haeusslein, L. J. (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol.106–107, 1–16. doi: 10.1016/j.pneurobio.2013.04.001
Shi, L., Smith, S. E. P., Malkova, N., Tse, D., Su, Y., and Patterson, P. H. (2009). Activation of the maternal immune system alters cerebellar development in the offspring. Brain. Behav. Immun. 23, 116–123. doi: 10.1016/j.bbi.2008.07.012
Shmueli, A., Gdalyahu, A., Sapoznik, S., Sapir, T., Tsukada, M., and Reiner, O. (2006). Site-specific dephosphorylation of doublecortin (DCX) by protein phosphatase 1 (PP1). Mol. Cell. Neurosci. 32, 15–26. doi: 10.1016/j.mcn.2006.01.014
Skorput, A. G. J., Gupta, V. P., Yeh, P. W. L., and Yeh, H. H. (2015). Persistent interneuronopathy in the prefrontal cortex of young adult offspring exposed to ethanol in utero. J. Neurosci. 35, 10977–10988. doi: 10.1523/JNEUROSCI.1462-15.2015
Sobue, K., and Fukumoto, K. (2010). Caldesmon, an actin-linked regulatory protein, comes across glucocorticoids. Cell Adh. Migr. 4, 185–189. doi: 10.4161/cam.4.2.10886
Sodhi, M. S., and Sanders-Bush, E. (2004). Serotonin and brain development. Int. Rev. Neurobiol. 59, 111–174. doi: 10.1016/S0074-7742(04)59006-2
Stevens, H. E., Su, T., Yanagawa, Y., and Vaccarino, F. M. (2013). Prenatal stress delays inhibitory neuron progenitor migration in the developing neocortex. Psychoneuroendocrinology 38, 509–521. doi: 10.1016/j.psyneuen.2012.07.011
Stumm, R. K., Zhou, C., Ara, T., Lazarini, F., Dubois-Dalcq, M., Nagasawa, T., et al. (2003). CXCR4 regulates interneuron migration in the developing neocortex. J. Neurosci. 23, 5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003
Subramoney, S., Eastman, E., Adnams, C., Stein, D. J., and Donald, K. A. (2018). The early developmental outcomes of prenatal alcohol exposure: a review. Front. Neurol. 9:1108. doi: 10.3389/fneur.2018.01108
Tomson, T., Battino, D., Bonizzoni, E., Craig, J., Lindhout, D., Sabers, A., et al. (2011). Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 10, 609–617. doi: 10.1016/S1474-4422(11)70107-7
Tong, X., Li, X., Zhou, B., Shen, W., Zhang, Z., Xu, T., et al. (2009). Ca(2() signaling evoked by activation of Na(() channels and Na(()/Ca(2() exchangers is required for GABA-induced NG2 cell migration. J. Cell Biol. 186, 113–128. doi: 10.1083/jcb.200811071
Torii, M., Sasaki, M., Chang, Y.-W., Ishii, S., Waxman, S. G., Kocsis, J. D., et al. (2017). Detection of vulnerable neurons damaged by environmental insults in utero. Proc. Natl. Acad. Sci. U.S.A. 114, 2367–2372. doi: 10.1073/pnas.1620641114
Toritsuka, M., Kimoto, S., Muraki, K., Landek-Salgado, M. A., Yoshida, A., Yamamoto, N., et al. (2013). Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc. Natl. Acad. Sci. U.S.A. 110, 17552–17557. doi: 10.1073/pnas.1312661110
Tucker, E. K., and Nowak, R. A. (2018). Methylmercury alters proliferation, migration, and antioxidant capacity in human HTR8/SV-neo trophoblast cells. Reprod. Toxicol. 78, 60–68. doi: 10.1016/J.REPROTOX.2018.03.008
Usachev, Y., Shmigol, A., Pronchuk, N., Kostyuk, P., and Verkhratsky, A. (1993). Caffeine-induced calcium release from internal stores in cultured rat sensory neurons. Neuroscience 57, 845–859. doi: 10.1016/0306-4522(93)90029-f
Vasilev, D. S., Dubrovskaya, N. M., Tumanova, N. L., and Zhuravin, I. A. (2016). Prenatal hypoxia in different periods of embryogenesis differentially affects cell migration, neuronal plasticity, and rat behavior in postnatal ontogenesis. Front. Neurosci. 10:126. doi: 10.3389/fnins.2016.00126
Velísek, L. (2005). Prenatal corticosteroid impact on hippocampus: implications for postnatal outcomes. Epilepsy Behav. 7, 57–67. doi: 10.1016/j.yebeh.2005.04.008
Vendrell, I., Carrascal, M., Campos, F., Abián, J., and Suñol, C. (2010). Methylmercury disrupts the balance between phosphorylated and non-phosphorylated cofilin in primary cultures of mice cerebellar granule cells A proteomic study. Toxicol. Appl. Pharmacol. 242, 109–118. doi: 10.1016/j.taap.2009.09.022
Wang, H. (2018). Modeling neurological diseases with human brain organoids. Front. Synap. Neurosci. 10:15. doi: 10.3389/fnsyn.2018.00015
Wilhoit, L. F., Scott, D. A., and Simecka, B. A. (2017). Fetal alcohol spectrum disorders: characteristics, complications, and treatment. Commun. Ment. Health J. 53, 711–718. doi: 10.1007/s10597-017-0104-0
Xu, H., and Heilshorn, S. C. (2013). Microfluidic investigation of BDNF-enhanced neural stem cell chemotaxis in CXCL12 gradients. Small 9, 585–595. doi: 10.1002/smll.201202208
Yang, N., Higuchi, O., Ohashi, K., Nagata, K., Wada, A., Kangawa, K., et al. (1998). Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812. doi: 10.1038/31735
Zechel, J. L., Gamboa, J. L., Peterson, A. G., Puchowicz, M. A., Selman, W. R., and Lust, W. D. (2005). Neuronal migration is transiently delayed by prenatal exposure to intermittent hypoxia. Birth Defects Res. Part B Dev. Reprod. Toxicol. 74, 287–299. doi: 10.1002/bdrb.20051
Keywords: prenatal environmental stress, neuronal migration, neuronal migration disorders, fetal brain development, Cxcl12/Cxcr4 signaling, heat shock signaling
Citation: Hwang HM, Ku RY and Hashimoto-Torii K (2019) Prenatal Environment That Affects Neuronal Migration. Front. Cell Dev. Biol. 7:138. doi: 10.3389/fcell.2019.00138
Received: 15 April 2019; Accepted: 08 July 2019;
Published: 17 July 2019.
Edited by:
Kazuhito Toyo-oka, Drexel University, United StatesReviewed by:
Zhizhan Gu, Dana-Farber Cancer Institute, United StatesCopyright © 2019 Hwang, Ku and Hashimoto-Torii. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazue Hashimoto-Torii, S0hUb3JpaUBjaGlsZHJlbnNuYXRpb25hbC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.