 Elizabeth A. Comen
Elizabeth A. Comen Robert L. Bowman2,3
Robert L. Bowman2,3 Maria Kleppe
Maria Kleppe- 1Breast Cancer Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 2Center for Hematopoietic Malignancies, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 3Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, NY, United States
Historically, the link between chronic inflammation and cancer has long been speculated. Only more recently, pre-clinical and epidemiologic data as well as clinical evidence all point to the role of the tumor microenvironment as inextricably connected to the neoplastic process. The tumor microenvironment (TME), a complex mix of vasculature, inflammatory cells, and stromal cells is the essential “soil” helping to modulate tumor potential. Increasingly, evidence suggests that chronic inflammation modifies the tumor microenvironment, via a host of mechanisms, including the production of cytokines, pro-inflammatory mediators, angiogenesis, and tissue remodeling. Inflammation can be triggered by a variety of different pressures, such as carcinogen exposure, immune dysfunction, dietary habits, and obesity, as well as genetic alterations leading to oncogene activation or loss of tumor suppressors. In this review, we examine the concept of the tumor microenvironment as related to both extrinsic and intrinsic stimuli that promote chronic inflammation and in turn tumorigenesis. Understanding the common pathways inherent in an inflammatory response and the tumor microenvironment may shed light on new therapies for both primary and metastatic disease. The concept of personalized medicine has pushed the field of oncology to drill down on the genetic changes of a cancer, in the hopes of identifying individually targeted agents. Given the complexities of the tumor microenvironment, it is clear that effective oncologic therapies will necessitate targeting not only the cancer cells, but their dynamic relationship to the tumor microenvironment as well.
Introduction
Chronic inflammation is a hallmark of cancer and many factors can trigger an inflammatory response in the microenvironment, including infectious pathogens, imbalanced immune regulation, carcinogen exposure, dietary habits and obesity, and genetic alterations leading to oncogene activation or loss of tumor suppressors (Elinav et al., 2013). A growing understanding of the relationship between chronic inflammation and the tumor microenvironment (TME) has dramatically altered our understanding of cancer. Evidence suggests that chronic inflammation creates a pro-tumorigenic environment via the production of pro-inflammatory mediators, angiogenesis, and tissue remodeling (Coussens and Werb, 2002). Some of the most critical external factors that can promote chronic inflammation and increase cancer risk include tobacco, obesity, a sedentary lifestyle, and select infectious agents. Evolving insight into the mechanisms by which chronic inflammation supports a pro-tumorigenic environment has led to new (immuno)-therapies for cancer as well as lifestyle recommendations which may decrease cancer incidence.
In this review, we discuss the current knowledge of how epithelium cancer-initiating events cross talk to inflammatory cells during cancer initiation and progression. Specifically, we review the concept of the TME and both the extrinsic and intrinsic mechanisms that tie an inflammatory response to pro-tumorigenic events (Figure 1).
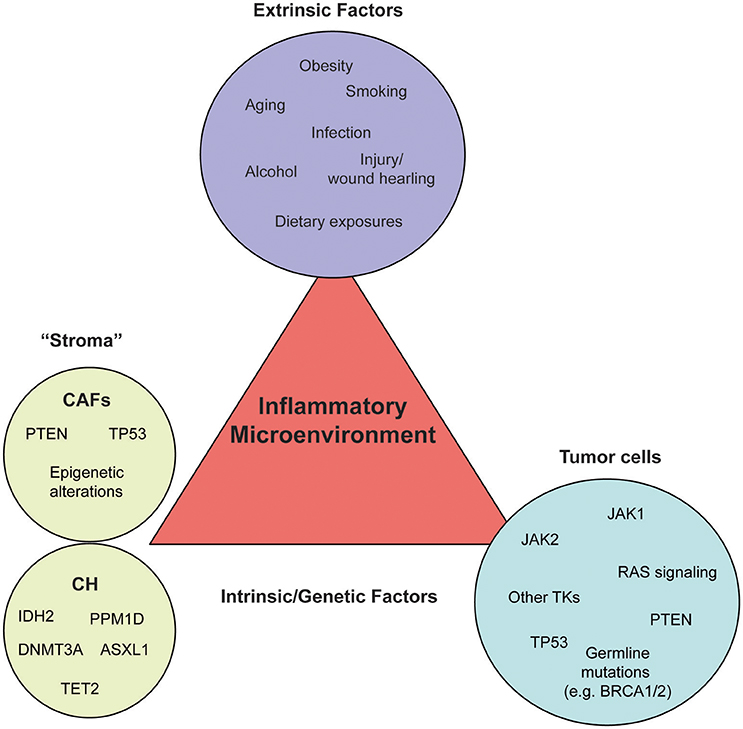
Figure 1. External and intrinsic factors fueling an inflammatory microenvironment. Inflammation can be triggered by a variety of different pressures, such as carcinogen exposure, immune dysfunction, dietary habits, and obesity (extrinsic factors, purple circle), as well as genetic alterations (intrinsic factors) leading to oncogene activation or loss of tumor suppressors in the microenvironment (stroma, green circle) or the tumor cells themselves (tumor, blue circle). Each circle contains a number of examples which belong to the respective category. Most genetic and epigenetic alterations of the stroma have been identified in cancer-associated fibroblasts (CAFs) and white blood cells of healthy individuals or solid cancer patients without signs of hematological malignancies (CH).
Cellular Constituents of an Inflammatory Microenvironment
While the exact composition of the TME will differ from tissue to tissue, as well as between various tumors of the same tissue, key cellular players are recurrently found. In addition to malignant tumor cells, the TME is composed of a number of different cell types including fibroblasts, endothelial cells, pericytes, and various cells associated with the immune system (Quail and Joyce, 2013). In addition to B cells, natural killer (NK) cells and T lymphocytes, the myeloid cells within the TME include tumor-associated macrophages (TAMs), dendritic cells, neutrophils, and monocytes, the latter two of which have often been misclassified as myeloid-derived suppressor cells (MDSCs) on the basis of immunophenotypic markers. In mice, these myeloid cells are identified by immunophenotype (Cd11b+, Gr.1+) with Ly6C and Ly6G to further differentiate a monocytic-MDSC from a granulocytic-MDSC respectively (reviewed here Talmadge and Gabrilovich, 2013). Human MDSCs are defined through a combination of CD11B, CD14, CD15, and CD66 (Bronte et al., 2016). While studies have identified bona fide T cell suppressor function within this compartment, the label of MDSC is often applied even without functional demonstration leading to murky interpretations of their role within the TME. Understanding the molecular functions of these cells within the tumor may provide avenues for disrupting pro-tumorigenic signaling and tailored TME-targeted therapy. Furthermore, identifying markers capable of distinguishing functional MDSC from the immunophenotypically similar monocyte and neutrophil counterparts will be critical to elucidating their role in the TME (Bronte et al., 2016). In this section, we will review the functions of three of the most well studied constituents of the TME, TAMs, cancer-associated fibroblasts (CAFs) and endothelial cells comprising the tumor vasculature.
Tumor-Associated Macrophages
TAMs have emerged as one of the most well studied components of the TME. These cells have been shown to interact with nearly every feature of tumor progression including mediating tumor cell proliferation, migration, invasion, angiogenesis, metastasis, and chemotherapeutic resistance (Noy and Pollard, 2014). The multifaceted role of TAMs in the TME is a direct reflection of the diverse responsibilities of macrophages in normal development, tissue homeostasis, and tissue repair (Okabe and Medzhitov, 2016). Indeed many of the developmental processes mediated by macrophages, including extracellular matrix remodeling, phagocytosis of apoptotic debris and angiogenesis, are critical hallmarks of the TME. As such, in some instances the inflammatory state of the tumor resembles an unresolved wound healing response (Dvorak, 1986; Schäfer and Werner, 2008). Perhaps the most well described function of TAMs involves their role in tumor cell migration and invasion. TAMs have been shown to engage in a CSF1-EGF paracrine signaling loop capable of leading tumor cells to the invasive edge of a tumor (Wyckoff et al., 2004). Additionally, TAMs have been shown to engage in a multicellular interaction with endothelial cells through the release of VEGFA to facilitate the intravasation of tumor cells from the primary site into circulation (Harney et al., 2015). In addition to the production of these growth factors, TAMs are the major source of matrix metalloproteinases and cathepsin proteases (Olson and Joyce, 2015; Varol and Sagi, 2017). In order to execute the diverse functions described above, macrophages employ immense transcriptional plasticity that falls along a continuum of activation (Xue et al., 2014; Glass and Natoli, 2016). On one extreme end of the spectrum lays classically activated macrophages (often termed M1-macrophages mirroring that of Th1 immunity). This activation state is canonically associated with IFNγ stimulation resulting in a STAT1 transcriptional program. Functionally, these cells are capable of perpetuating type I inflammatory responses through the secretion of chemokines such as CCL3, CCL4, and CCL5, as well as the production of nitric oxide and TNFα. Furthermore, IFNγ stimulation can boost antigen presentation capacity through the upregulation of Ciita a master transcriptional regulator of MHC II molecules. On the other extreme, alternatively activated macrophages (or M2 macrophages) are associated with an anti-inflammatory state. Here, the critical molecular regulators of alternative activation are the Th2 cytokines IL-4, IL-13, IL-6, and IL-10. Together these cytokines lead to the activation of STAT6 (IL-4 and IL-13) as well as STAT3 (IL-6 and IL-10). These programs, as well as other transcriptional regulators, drive the upregulation of Arginase 1 leading to decreased nitric oxide signaling, and increased expression of the wound healing associated chemokines CCL17 and CCL22. Functionally, this activation state is associated with inflammation resolution and extracellular matrix remodeling. In addition to the cytokines described above, TAM activation can be influenced by hypoxia and local metabolite concentrations such as lactate (Casazza et al., 2013; Colegio et al., 2014; Carmona-Fontaine et al., 2017). Despite the widespread description of TAMs as either M1 or M2, this dichotomy is clearly an oversimplification of the diverse states in which these cells are capable of existing (Murray et al., 2014). Elucidating the functional capacities of TAMs within the TME and the mechanism that regulate these processes will provide a clearer picture of these cells. Another more recently appreciated factor influencing TAM activation involves the origin of the cells. While TAMs were long thought to derive from circulating monocytes (Qian et al., 2011; Franklin et al., 2014), recent work suggests that TAMs are also derived from local tissue-resident macrophages (Bowman et al., 2016; Zhu et al., 2017). These are important to consider in the setting of therapeutic strategies aimed at reducing TAM accumulation through recruitment blockade, either through CCR2 or CXCR4 inhibition (Kioi et al., 2010; Qian et al., 2011). Further, in most cases, tissue-resident macrophages possess distinct developmental origins from their monocyte-derived counterparts, as they seed the tissue during embryogenesis developing via an erythro-myeloid precursor as opposed to a hematopoietic stem cell (Gomez Perdiguero et al., 2015). This distinct ontogeny appears to imprint a sort of epigenetic memory on the subsequent TAM, eliciting distinct gene expression profiles within the TME (Bowman et al., 2016; Zhu et al., 2017). While clodronate liposome-based depletion strategies have been used to preferentially deplete tissue resident macrophages in the pancreas (Zhu et al., 2017), more selective genetic ablation strategies will be of interest to translate the differences seen in gene expression studies into functional capacities. Translation of these studies from the mouse to human disease will require identification of markers capable of distinguishing the ontogenetically defined TAM populations. One such marker, CD49D, has been found to be absent on brain-resident microglia and present on recruited bone marrow-derived macrophages in multiple brain malignancies (Bowman et al., 2016). Markers such as CD49D will likely be found in many distinct tissues, and may serve as biomarkers for future TME-targeted therapy.
Cancer-Associated Fibroblasts (CAFs)
In addition to the immune components of the TME described above, CAFs are an abundant, heterogeneous pool of cells that play multifactorial roles in cancer progression. CAFs are sometimes referred to as mesenchymal stem cells or tumor-associated fibroblasts (Paunescu et al., 2011). Regardless of the nomenclature, these cells are non-hematopoietic, non-epithelial cells resident to a tissue. These cells can be identified microscopically based on the spindle-like shape and large singular presence within the stroma of a tissue. During tissue homeostasis, these cells are responsible for deposition of type I collagen, laminin, perlecan, nidogen, and fibronectin, but are generally considered quiescent with limited migration and proliferation (Kalluri, 2016). Much like macrophage activation paradigms described above, fibroblasts undergo a similar activation process upon stimulation with factors such as TGFβ, PDGF, and FGF2 (Elenbaas and Weinberg, 2001). Like the TAMs described above, CAFs are distinct from their non tumor-associated counter parts and possess a unique activation state. Upon activation these cells change morphologically, increasing in size and with additional spindle-like processes. Functionally these cells possess increased capacity for migration, collagen crosslinking and secretion of cytokines and chemokines such as VEGFA, TGFβ, HGF, FGF, EGF, CXCL10, CCL5, IL-6, TNFα, and IFNγ (Kalluri, 2016). Through these phenotypic alterations, activated fibroblasts can orchestrate a wound healing response concomitant with extracellular matrix repair, recruitment of immune cells to eliminate pathogens, and regrowth of damaged epithelial tissue (Öhlund et al., 2014). Critically, fibroblast activation is a reversible process and as such wound healing responses are able to resolve and quiescence can be restored. If however, this process is not resolved tissue fibrosis can occur. In cancer, this fibrotic phenotype is widespread with even premalignant lesions are often associated with fibrosis or desmoplastic reactions (Rønnov-Jessen et al., 1996), however the causality of desmoplasia and malignant transformation remain an open discussion in human disease. In developed tumors, pancreatic adenocarcinoma (PDAC) presents an extreme example of unrestrained desmoplasia, driven in part through sonic hedgehog signaling in the stroma (Tian et al., 2009). The fibrotic stroma of PDAC presents a challenge for effective delivery of chemotherapy into tumors, and as such reducing the stromal component of the TME presents an interesting chemo sensitizing therapeutic option (Olive et al., 2009). A more full understanding of how CAFs are activated, and the results of inhibiting these activation states are necessary. While most studies have been completed in vitro, one study utilized genetic mouse models of squamous cell carcinoma. CAFs isolated from these early neosplasms possess a pro-inflammatory gene expression signature driven by NF-κB (Erez et al., 2010). Interestingly, the authors demonstrated that normal dermal fibroblasts could be “educated” to resemble CAFs through co-culture with carcinoma cells. This activation state has since been shown to be regulated through promoter hypermethylation (Zeisberg and Zeisberg, 2013; Li et al., 2015; Xiao et al., 2016). Upon activation CAFs interface with the tumor through many of the same mechanisms as an activated fibroblast during wound healing, yet many differences remain. In vitro proteomic studies identified an altered secretory phenotype in CAFs compared to non-malignant activated fibroblasts (De Boeck et al., 2013). In this study, CAFs were found to secrete higher levels of tenascin and the CXCR4 ligand SDF-1, both of which have been shown to be important in different stages of the metastatic cascade (Oskarsson et al., 2011; Vanharanta et al., 2013). Additionally, while one of the primary functions of fibroblasts in wound healing is to recruit immune cells, CAFs have been shown to negatively regulate immune responses in a TNFα and IFNγ dependent manner (Kraman et al., 2010). As such, targeting CAF-derived cytokines has been shown to enhance CSF1R targeted therapy (Kumar et al., 2017) as well as immune checkpoint blockade (Feig et al., 2013). Clinical efficacy of these combinations remains to be determined.
Angiogenesis and Tumor Vasculature
The formation of new blood vessels, termed angiogenesis, is one of the hallmarks of the tumor microenvironment (Hanahan and Weinberg, 2011). The newly formed vascular network serves as a means to deliver nutrients, cytokines, and oxygen into the tumor. Thus engaging in angiogenesis is a major step in disease development, with earlier stage, smaller tumors possessing fewer vessels than later, more aggressive tumors, which can be highly vascularized (Bergers and Benjamin, 2003). This is, of course, a broad generalization as the vascular content also varies by tissue. For example, while glioblastoma multiforme is one of the most vascularized tumor types (Das and Marsden, 2013), pancreatic ductal adenocarcinoma has low microvascular density and is instead entrenched with dense desmoplastic stroma (Longo et al., 2016). Unlike its' normal tissue counterpart, tumor vasculature often possesses aberrant morphology associated with increased branching and an overall disrupted network of endothelial cells. These abnormal structural findings are often caused by poor pericyte coverage and disruption of a supportive basement membrane (De Palma et al., 2017). Such a distorted network can lead to poor diffusion of oxygen and other small molecules in the tumor resulting in spatial heterogeneity.
While a critical process in malignant development, angiogenesis is not unique to tumors, but rather reminiscent of a wound healing response initiated by inflammation. Indeed, recruited monocytes, eosinophils, and neutrophils are capable of secreting pro-angiogenic factors (De Palma et al., 2017). One of the most potent inducers of angiogenesis is hypoxia, which activates hypoxia inducible factor 1 (HIF1) in both tumor cells and surrounding stromal cells leading to the production of vascular endothelial growth factor (VEGF), a potent mediator of new vessel growth (Krock et al., 2011). In addition, TAMs and CAFs are also capable of stimulating angiogenesis through the secretion of VEGFA as well as the lymphangiogenic factor VEGFC and VEGFD (Quail and Joyce, 2013). Given its critical role in angiogenesis, VEGF-targeted agents have emerged as a major class of therapeutics with broad applicability in cancer (Ferrara and Adamis, 2016). Despite success in some tumor types (renal cell carcinoma), understanding which patients are most likely to benefit from anti-angiogenic therapy remains a challenge. Angiogenic signaling is further complicated in the context of inflammation where the paralogous factors angiopoietin-1 and angiopoietin-2 play a role in dampening and amplifying sensitivity to the inflammatory factor TNFα during wound healing (Fiedler et al., 2006). Two recent studies demonstrated that dual targeting of angiopoietin-2 and VEGF resulted in vascular normalization and extended survival in murine models of glioblastoma (Kloepper et al., 2016; Peterson et al., 2016). This combination was also found to potentiate immunotherapy via PD-1 blockage (Schmittnaegel et al., 2017). Understanding the interplay between these molecules and the tumor's dependency will be critical for maximizing future anti-angiogenic therapeutic approaches. While the vasculature plays a clear role in delivering nutrients to a tumor, several reports have provided evidence for an additional role in promoting cancer stem cells. In colorectal cancer (CRC) endothelial cells have been shown to support cancer stem cells through soluble Jagged-1 mediated activation of the Notch signaling (Lu et al., 2013). Similar results were found for Jagged-1, as well as DLL4, in glioblastoma models (Zhu et al., 2011). These studies suggest that targeting of endothelial cells may provide an avenue for modulating cancer stem cells; however, previous reports have demonstrated that anti-angiogenic therapies can actually lead to an increase in cancer stem cells due to increased hypoxia (Conley et al., 2012). Given the multifaceted role of the tumor vasculature, careful preclinical studies will be necessary to understand the consequences of endothelial-targeted therapy.
In addition to supporting primary tumor growth, endothelial cells are also involved in the metastatic cascade from the earliest step of intravasation to vascular cooption at the metastatic site. Histological interrogation identified a tri-cellular signaling hub known as TMEM structure, composed of a tumor cell, a Tie2high TAM and an endothelial cell (Robinson and Jones, 2009). Further intravital imaging studies revealed that at this site local VEGF signaling leads to increased vascular permeability followed by a release of tumor cells into the blood stream (Harney et al., 2015). Later in the metastatic cascade, during extravasation, endothelial cells can serve as a barrier between the circulating tumor cells and host tissue. In brain metastasis for instance, cleavage of the endothelial adhesion molecule, JAM-B, is necessary for tumor cells to efficiently extravasate (Sevenich et al., 2014). Upon extravasation, metastatic cells once again rely upon close association with endothelial cells to support their survival in a process known as vascular cooption. These studies collectively highlight that endothelial cells play critical roles in both tumor development through increased vascularization, but are also intricately involved in many steps of disease progression.
Causes of an Inflammatory Microenvironment
External Factors and Exposures Cause Acute and Chronic Inflammation
Evidence indicates that chronic inflammation not only increases the risk of a multitude of cancers, including colon, liver, pancreatic, lung, bladder, gastric, and breast, but may also increase the risk of tumor progression and metastasis (Iyengar et al., 2015). Moreover, it is well known that a variety of external factors and exposures can cause both acute and chronic inflammation. Because many of these factors/exposures, such as diet, are modifiable, there is a growing scientific and public health interest in understanding the relationship between extrinsic pressures that promote chronic inflammation and the TME.
Tobacco
Historically, tobacco has been the most notorious carcinogen. At present, tobacco exposure is the leading preventable cause of death. Roughly 85% of all lung cancers are secondary to smoking with additional cancers attributable to secondary smoke (Warren and Cummings, 2013). In developed countries, tobacco is associated with roughly 30% of all malignancies (McGuire, 2016). Tobacco use is associated with numerous other cancers as well, including but not limited to, head and neck, pancreatic, gastric, esophageal, acute myeloid leukemia (AML), bladder and renal cell cancers (United States Public Health Service, and Office of the Surgeon General, 2010). For example, actively smoking triples the risk of bladder cancer (Freedman et al., 2011; McGuire, 2016). Tobacco increases cancer risk not only by causing direct genetic changes to the epithelium (particularly the lung), but also through altering epigenetic events, eliciting epithelial to mesenchymal cell transition, and inducing a chronic inflammatory and hypoxic microenvironment. Ongoing inflammation promotes apoptotic arrest, angiogenesis and in turn cell proliferation (Milara and Cortijo, 2012). Tobacco inherently contains a multitude of direct carcinogens, including polycyclic aromatic hydrocarbons, N-nitrosamines, aromatic amines, aldehydes, volatile organic hydrocarbons, and metals among others (Pfeifer et al., 2002). The activation of these carcinogens in the host can result in the formation of DNA adducts, a form of DNA damage and source of mutagenesis. In addition to directly affecting DNA function of the cell, tobacco can modify the immune function of the host (Sopori, 2002). For example, tobacco can increase the abundance of alveolar macrophages in the lung, which may increase oxidative stress and oxygen radicals, thereby promoting tumor growth. In a study of 1,819 individuals, systemic levels of 78 makers of inflammation and immunity were measured. The study included 548 never smokers, 857 former smokers, and 414 current smokers. Significant differences in several immune markers were noted between active and never smokers. These include but are not limited to differences in CCL17/TARC, CCL11/EOTAXIN, IL-15, IL-1B, IL-1Rα, CRP, SVEGFR3, IL-16, sIL-6R, and SCF. Interestingly, the authors note that many of these makers are critical to immune function as well as cell growth. Overall, they report that on average smokers appear to have an immune profile consistent with an overall immunosuppressive function of tobacco (Shiels et al., 2014). Indeed multiple studies have demonstrated that tobacco has immunosuppressive functions (Cui and Li, 2010). One mechanism by which nicotine may be immunosuppressive is by impairing macrophage and neutrophil function (Milara and Cortijo, 2012). In tobacco smokers with chronic obstructive airway disease (COPD) the lung epithelium undergoes repeated injury and repair thereby inducing transformation of the normal epithelium to a more malignant phenotype. Tobacco smokers who continue to smoke also have poorer outcomes from a multitude of cancers, perhaps as a result of suppression of NK cell activation (Lu et al., 2007). Despite the significant link between tobacco and lung cancer, some non-smokers still develop lung cancer. Toward this end, understanding the mechanisms by which smokers as well as non-smokers may share common markers related to chronic airway inflammation may shed important light into molecular targets for the diagnosis and treatment of lung cancer. Alternatively, sequencing of non-small cell lung cancers in tobacco smokers vs. non-smokers identifies distinct genetic signatures. Tobacco induced tumors have a greater mutational burden than those found in non-smokers, which may in turn predict response to immunotherapy (Godwin et al., 2013; Hellmann et al., 2016).
Alcohol
Alcohol can act in concert with tobacco to significantly increase the risk of cancer. Historically, alcohol was long associated with liver as well as head and neck cancers (LoConte et al., 2018). However, alcohol alone is now classified as a carcinogen, increasing the risk of several cancers in a linear dose dependent fashion, including breast, pancreas, liver, colon, esophagus and head and neck cancers. In hepatocellular cancers specifically, alcohol is known to induce chronic liver inflammation and fibrosis, which is itself a pre-cursor to malignancy (also discussed above). In addition, there is also evidence suggesting that patients with hepatocellular cancers and increased alcohol consumption show poorer outcomes once diagnosed with cancer (Barbara et al., 1992). More recently, alcohol has been associated with a host of other cancers. According to the American Society of Clinical Oncology (ASCO), between 5 and 6% of new cancers and cancer deaths globally can be “directly attributable to alcohol” (LoConte et al., 2018). Alcohol can cause direct DNA damage as a result of ethanol conversion to acetaldehyde as well as disrupt folate metabolic pathways (Seitz and Becker, 2007). With respect to the TME, evolving preclinical data suggests that ethanol can directly disrupt immune surveillance and innate immune response as well as induce reactive oxygen species (ROS) production and oxidative stress in CAFs (Sanchez-Alvarez et al., 2013). Alcohol may also disrupt the vascular endothelium thereby creating a microenvironment more permissive to metastasis and tumor migration (Xu et al., 2012). In breast cancer patients, particularly those with estrogen receptor (ER) positive breast cancers, Sanchez-Alvarez and colleagues suggest that ethanol induces ketone production in CAFs (Sanchez-Alvarez et al., 2013). In preclinical models, CAFs have also been shown to fuel tumor growth via oxidative mitochondrial metabolism and promote a more aggressive breast cancer phenotype (Donnarumma et al., 2017).
Obesity
Historically, public health campaigns for cancer prevention have focused on tobacco's obvious link to cancer risk. More recently, however, ASCO suggests that obesity may soon outweigh tobacco as the leading modifiable risk factor for cancer (Ligibel et al., 2014). Not only does obesity increase the risk for a multitude of cancers but it may also decrease treatment delivery and worsen outcomes for those newly diagnosed. Frighteningly, over two thirds of the adult US population are overweight or obese and the number is growing (Flegal et al., 2002). Evolving data suggests that excess fat or “hyperadiposity” drives chronic inflammation, which in turn engenders a pro-tumorigenic milieu (Iyengar et al., 2016). White adipose tissue from obese patients is infiltrated by leukocytes, specifically macrophages, and T lymphoctyes (Underhill and Goodridge, 2012). When enlarged fat cells die, they release cytokines that recruit additional macrophages (Cinti et al., 2005). These macrophages then form a “crown” around the dying adipocytes, setting off an inflammatory cascade. The occurrence of these crown- like structures are commonly observed in obese patients with both breast and tongue cancers (Morris et al., 2011). Notably, Iyengar and colleagues have shown that even patients with a normal body mass index (BMI) but a relatively increased amount of body fat can also have inflammation in their breast tissue, indicating that BMI alone is not sufficient to predict for the influence of fat on body composition (Iyengar et al., 2017). Specifically, BMI is the ratio of a person's weight to height. This weight/height measurement alone does not reflect a more nuanced understanding of a person's body composition, or their relative percentages of fat and muscle. A seemingly lean person may be in fact “skinny fat,” wherein they have a relatively low BMI but their body composition is predominantly fat. Much of the work connecting obesity to cancer has been studied in the breast cancer population, because both fat and normal tissue in the breast can be readily evaluated as well as because of the relationship between fat and steroid/hormonal production. In breast cancer studies, inflamed fat tissue within the breast itself can increase local cytokine production, as well as expression of aromatase and ER gene expression (Cleary and Grossmann, 2009). Not surprisingly, obesity increases the risk of death among postmenopausal women with ER-positive breast cancer (Fuentes-Mattei et al., 2014). Obese mouse models suggest that activation of the AKT-mTOR pathway in the breast itself may specifically promote worse outcomes. Obesity has also been linked to poor outcomes for other types of cancers (Ligibel et al., 2014). In squamous cell cancer of the tongue for example, a diagnosis of obesity prior to tongue cancer diagnosis was associated with a five-fold increase of death (Iyengar et al., 2014).
Dietary Exposures and Exercise
Given the relationship of obesity and cancer, diet and exercise modifications have gained increasing interest. Much of the work to date on the role of exercise, cancer, and inflammation has been done in the preclinical setting. Preclinical studies suggest that exercise can decrease inflammatory markers and modulate the TME (Koelwyn et al., 2015). Though the mechanisms by which exercise directly reduces inflammation are not entirely clear, possible hypotheses include decreasing IL-6, reduction in adipose tissue, and inhibition of TNFα (Koelwyn et al., 2015). Exercise may also increase the cytotoxicity and number of NK cells (Bigley and Simpson, 2015). Much like pharmacologic interventions, ongoing clinical trials are evaluating whether exercise as part of ongoing therapy may modulate tumorigenesis. Early data suggests that exercise leads to a reduction in visceral fat mass, decreased low grade chronic inflammation, and a subsequent reduction in pro-inflammatory adipokine secretion, as well as a reduction in macrophage infiltration into adipose tissue (Klionsky et al., 2016). In preclinical cancer models, exercise may decrease TAM and neutrophil infiltration and increase intratumoral cytotoxic T cell infiltration (Koelwyn et al., 2017). While the majority of the work in this space has been preclinical, a more recent randomized controlled trial evaluated the relationship of exercise and outcomes in breast cancer survivors. Specifically, this randomized controlled trial assessed the effects of a 16-week combined aerobic and resistance exercise intervention on metabolic syndrome, sarcopenic obesity, and serum biomarkers among sedentary, overweight, or obese survivors of breast cancer (Dieli-Conwright et al., 2018). Serum biomarkers included IGH1, insulin, IL-6, IL-8, TNFα, and steroid hormones (estrogen and testosterone). Sarcopenic obesity, BMI, and circulating biomarkers, including insulin, IGF-1, leptin, and adiponectin were significantly improved after exercise intervention (Dieli-Conwright et al., 2018). In addition to exercise, although data have not been entirely conclusive, some epidemiology studies have linked a higher fiber, lower fat diet to a decrease in some cancers. In breast cancer patients, for example, a high fiber, low fat diet results in lower circulating levels of estradiol among patients with a history of breast cancer, even in the absence of weight loss (Rock et al., 2004). Traditionally, designing and interpreting clinical trials to assess diet and cancer intervention have been challenging.
Gut Microbiota
More recently, gut bacteria have emerged as a possible link between metabolites in food and the TME. The immune system is dependent in part on exposure via the gut to microbiota as part of immunosurveillance (Kroemer and Zitvogel, 2018). Preclinical data suggests that diet can modify gut bacteria, specifically altering toll-like receptors on macrophages and dendritic cells as well as adipose inflammation (Garrett, 2015). Interestingly, in animal models, modification of gut bacteria affects CRC incidence and natural history (Song and Chan, 2017). Moreover, as reviewed by Song and Chan, a more “Western” diet, high in processed foods, red meat, processed sugars, and refined grains can lead to a dysregulated immune response and increased levels of inflammatory markers that is associated with a higher risk of colon cancer. Multiple, epidemiologic cross-cultural studies of diet and fecal bacteria indicate that diets high in fiber and low in fat alter gut bacteria and in turn are associated with a lower colon cancer risk. Alternatively, a diet high in vegetables, fruits, and whole grains lowers colon cancer risk. It is also well known that antibiotics can modify gut flora and recent data suggests that the use of repeated antibiotics may increase the incidence of lung, prostate, bladder and breast cancer possibly by altering gut flora (Velicer et al., 2004; Iida et al., 2013; Boursi et al., 2015). Evolving evidence suggests that gut bacteria may even influence response to immunotherapy. In melanoma patients, analysis of patient fecal microbiome samples indicated a higher diversity in bacteria and amount of Ruminococcaceae bacteria among those who responded to anti-PD-1 immunotherapy (Gopalakrishnan et al., 2018). Chaput and colleagues also demonstrated that gut microbiota, and in particular Faecalibacterium and other Firmicutes improved response to the CTLA-4 blockade by ipilimumab (Chaput et al., 2017). It is also hypothesized that some chemotherapies may uniquely alter gut bacteria and modulate immune response (Viaud et al., 2013).
Infectious Agents
In addition to modifiable behaviors associated with an inflammatory pro-tumorigenic cascade, several infectious agents have also been linked to cancer incidence (Moore and Chang, 2010). Globally, it is estimated that roughly 15% of all cancers are associated with infections (Parkin, 2006). Microbes (including bacteria and viruses) have been implicated in cancer in a number of different mechanisms, sometimes in combination. Possible mechanisms include direct DNA damage, via oncogenes or tumor suppressor inhibition, as well as via the promotion of chronic inflammation and in some instances, such as HIV, immunosuppression (Kuper et al., 2000). Common microbial associations with cancer include helicobacter pylori (gastric cancer and gastric MALT lymphoma), schistosoma haematobium (bladder cancer), HPV (human papillomavirus) (cervical and oral cancers), clonorchis sinensis (cholangiocarcinoma) and hepatitis B and C viruses (hepatobilliary cancers) to name a few (Kuper et al., 2000). However, the mechanisms by which each infection promotes cancer are not entirely linear. Hepatitis B and C viruses for example do not neatly fit into a category of direct or indirect carcinogens. Rather, it is likely that they contribute to carcinogenesis via a host of mechanisms including the introduction of viral products to the cancer cell as well as by inducing chronic inflammation (Tsai and Chung, 2010). As it is beyond the scope of this article, Moore and Chang provide an extensive review of the relationship between viruses and cancer (Moore and Chang, 2010). Similarly, the relationship between H. Pylori and gastric cancer is also multifactorial. H. Pylori both promotes inflammation of gastric epithelial cells and also induces specific protein changes and gene mutations (Chiba et al., 2008). Notably, not all patients with select infections go on to develop cancer. The ways in which infections promote cancer either by directly inducing changes to host cells or inducing a chronic inflammatory response varies significantly. In addition to efforts to eradicate infections with known cancer associations, such as with the HPV vaccine or H. Pylori treatment, it will be equally important to understand why some people are protected while others progress to infection-associated cancers.
Intrinsic Mechanisms Leading to an Inflammatory Microenvironment
The mechanisms by which extrinsic factors can promote a pro-tumorigenic environment are inextricably linked to the ways in which genetic and epigenetic alterations can aide a cancer cell escape host defense mechanisms. Importantly, oncogenic mechanisms require a tight bidirectional cross talk of cancer cells with their microenvironment mediated by the production of chemokines, cytokines, growth factors, prostaglandins, ROS and nitrogen oxygen species (NOS), as well as recruitment of inflammatory cells into the tumor tissue. Many of the powerful oncogenes possess the ability to initiate a signaling cascade resulting in an inflammatory response in the proximity of the cells that harbor those oncogenes. The discovery that many oncogenic drivers are deeply involved in the modulation of a pro-oncogenic microenvironment and inflammatory processes suggested possible paracrine effects where altered expression or activity of the same genes in a stromal and/or immune cell may dictate epithelial fate and vice versa.
Tumor-Elicited Inflammation, Oncogenes and Tumor Suppressors
The advent of high-throughput sequencing techniques has led to the identification of hundreds of genetic and epigenetic alterations in genes associated with signaling pathways involved in cancer. Historically, genetic and functional studies have focused on a better understanding of the consequences of oncogenic activation in the context of the tumor cell itself and thus far only a limited number of studies has assessed their contribution beyond the cell-intrinsic effects. Here, we review common oncogenic and tumor suppressor pathways that contribute to tumor-associated inflammation (Figure 1). These include receptor and non-receptor tyrosine kinases (TKs), RAS signaling, TP53, APC, and PTEN.
Tyrosine Kinases
The activity of (receptor) TKs is central to many cellular processes. TKs also play cardinal roles in cytokine function, and are crucial for the signal transduction of various pro- and anti-inflammatory cytokines such as TNFα, IL-6, and IL-10. Due to their central status, TKs have received heightened attention as therapeutic targets, partially due to their potential to combat the chronic inflammatory state associated with many malignancies such as rheumatoid arthritis (RA), cardiovascular diseases, and cancer. In solid and blood cancers, TKs are frequently mutated leading to ligand-independent constitutive activation of downstream signaling pathways. For example, more than 90% of the non-leukemic classical myeloproliferative neoplasms (MPNs) are clearly driven by abnormal JAK2 activation, especially the cytokine receptor/JAK2 pathways and their downstream effectors (Passamonti et al., 2011). In line with a crucial role of JAK2 in cytokine signal transduction, MPN patients are characterized by high levels of pro-inflammatory cytokines in their circulation, which can be reduced by JAK inhibitor therapy (Verstovsek et al., 2010; Geyer et al., 2015; Mondet et al., 2015). Indeed, it is believed that the impressive clinical activity of ruxolitinib is a result of its anti-inflammatory effects. This suggests that aberrant JAK-STAT pathway activation is important for the induction and maintenance of the inflammatory state in MPN patients. Notably, whether JAK2 is mutated or not, the efficacy of ruxolitinib is comparable (Deininger et al., 2015). We have recently shown that both mutant and non-mutant hematopoietic cells are the source of pro-inflammatory cytokines in MPN mouse models and patients and that JAK-STAT signaling in mutant and non-mutant cells has to be inhibited in order to achieve therapeutic response (Kleppe et al., 2015b). This data suggests that sequential, interlinked, and selective steps, which bear clear resemblance to tumor-cell-organ microenvironment interactions commonly found in solid cancer and metastasis, also drive aberrant cytokine production in hematological malignancies. Other TKs which may be involved in the induction of an inflammatory state include c-Kit, EGFR, PDGFR, RET, VEGFR, c-Fms, and FGF (extensively reviewed in Yang and Karin, 2014).
RAS Signaling
The RAS superfamily of small GTPases comprises a group of more than 150 small G proteins. RAS proteins are signal transduction molecules central to many cellular processes. Mutations in one of the three canonical RAS genes, H-RAS, N-RAS, and K-RAS, are among the most common genetic abnormalities in human cancers. It is well established that aberrant RAS activation drives neoplastic transformation by influencing diverse aspects of the malignant phenotype in a cell autonomous manner, most importantly cell proliferation, survival, and mobility. Interestingly, more recent reports suggest that the role of oncogenic RAS extends beyond the effects on the tumor cell itself. Oncogenic Ras causes genotoxic stress and senescence in cells (Coppé et al., 2008). Intriguingly, senescent cells are known to secrete a myriad of inflammatory factors. In line, oncogenic Ras has been shown to accelerate and amplify a senescence-associated secretory phenotype that largely depends on IL-8 and IL-6 secretion thereby promoting tumorigenesis through effects on non-transformed cells during the process of inducing senescence (Coppé et al., 2008). Interestingly, high Ras activity in pancreatic acinar cells leads to cellular senescence and is sufficient to induce an inflammatory phenotype that is similar to the histological features of chronic pancreatitis suggesting that mutant K-ras is a cause rather than a secondary effect of chronic pancreatitis (Ji et al., 2009). Notably, patients with chronic pancreatitis have an increased risk of developing pancreatic cancer and K-RAS mutations are commonly found in chronic pancreatitis (Lüttges et al., 2000), but also observed in hyperplastic ducts within normal pancreas (Tada et al., 1996). Indeed, a large proportion of the adult human population possesses RAS mutations in tissues besides the pancreas, including colon and lung. Ras-mediated cytokine production has been repeatedly linked to activation of the inflammatory regulator NF-κB. It has been shown that in presence of mutant Ras, inflammatory stimuli initiate a NF-κB-dependent positive feedback loop involving Cox-2 resulting in prolonged Ras signaling and chronic inflammation and precancerous lesions in mice (Daniluk et al., 2012). In keratinocytes, expression of oncogenic Ras instigates an autocrine loop through IL-1α, IL-1R, and MyD88 leading to phosphorylation of IκBα and NF-κB activation (Cataisson et al., 2012). Moreover, activation of oncogenic Ras has been shown to enhance expression of squamous cell carcinoma antigens 1 and 2 and IL-6 via the NF-κB pathway (Catanzaro et al., 2014). K-RAS mutations mediate therapeutic resistance and are associated with poor prognosis, and until now, no effective anti-RAS inhibitor has reached the clinic (Cox et al., 2014). Given the growing body of evidence linking aberrant RAS and NF-κB it is intriguing to speculate that the NF-κB pathway could be exploited as potential preventive and therapeutic target in cancers harboring mutant RAS.
TP53
TP53 is a stress-responsive transcription factor (TF) and acts as a major tumor suppressor inhibiting neoplastic transformation by preventing the escalation of chronic tissue imbalance (Cooks et al., 2014). TP53 is a central hub for diverse stress signals, including ROS and NOS, cytokines, and infectious reagents (Cooks et al., 2014). TP53 also participates in the control of multiple cell cycle checkpoints. Mutations disabling TP53 tumor suppressor functions are the most frequent events in human cancer. For example, molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinomas (Cancer Genome Atlas Research, 2011; Vang et al., 2016). In addition to the cell autonomous effects of TP53 inactivation/dysfunction, compelling evidence suggests that TP53 missense mutants may not merely lose their tumor suppressive functions, but can also acquire new oncogenic properties through the activation of cell non-autonomous pathways. Specifically, multiple studies have linked mutant TP53 (such as TP53 p.R273H) and chronic inflammation to tumorigenic progression through different molecular interactions, including NF-κB (Cooks et al., 2013; Di Minin et al., 2014; Cui and Guo, 2016). In line, it has been demonstrated that TP53 mutants interact directly with NF-κB and that both factors impact the other's binding at diverse sets of active enhancers thus promoting a unique enhancer landscape of cancer cells in response to chronic inflammation (Rahnamoun et al., 2017). Moreover, clinical studies in primary breast carcinoma, head and neck squamous cell carcinoma, and CRC suggest that TP53 inactivation or deletion induces inflammation (Yin et al., 1993; Brentnall et al., 1994; Hussain et al., 2000; Linderholm et al., 2000; Lee et al., 2007). The effects of Tp53 loss have also been studied in diverse mouse models. For example, in a mouse model of prostate cancer, Tp53 loss resulted in enhanced transcription of cytokines and chemokines, accumulation of ROS and protein oxidation products, enhanced macrophage activation and neutrophil clearance, hypersensitivity to LPS, and high expression of metabolic markers (Komarova et al., 2005). Further, Lujambio and colleagues showed that Tp53-deficient hepatic stellate cells secrete factors that stimulate polarization of macrophages into a tumor-promoting M2 state leading to increased liver fibrosis and accelerated transformation of adjacent hepatocytes (Lujambio et al., 2013). Collectively, inhibition of tumor-associated inflammation is likely another important tumor suppressive function of TP53.
APC
CRC represents a paradigm for the link between inflammation and cancer (Lasry et al., 2016). Patients with inflammatory bowel disease, such as ulcerative colitis, are more likely to develop CRC and non-steroidal anti-inflammatory drugs show strong preventive effects (Chia et al., 2012). In mouse models of colitis, genetic, and functional studies have shown that inflammation alone suffices for tumor development and that inflammation-induced DNA damage can link chronic colitis and tumor initiation. Moreover, colorectal tumors exhibit tumor-elicited inflammation and upregulation of inflammatory signature genes (Wang and Karin, 2015). In fact, the type, density, and location of immune cells within human colorectal tumors have proven to be a reliable measure of patient outcome (Galon et al., 2006; Grivennikov et al., 2010; Norton et al., 2015). Inactivating mutations in APC, resulting in aberrant β-catenin activation, are found in 80% of all human colon cancers. In addition, APC loss predisposes humans to familial adenomatous polyposis, an autosomal dominant syndrome, in which patients develop numerous colorectal polyps (Groden et al., 1991). The tumor suppressor activity of APC has been extensively studied in the setting of epithelial transformation. In mice, the presence of an autosomal dominant Apc mutation in intestinal epithelial cells (IECs) leads to tumor development upon inactivation of the other allele due to spontaneous loss of heterozygosity (LOH) (Moser et al., 1990, 1993; Jackstadt and Sansom, 2016). Colon-specific deletion of Apc leads to formation of colorectal tumors with upregulation of pro-inflammatory cytokines. Interestingly, this work suggests that epithelial barrier defects and microbial invasion into the TME leads to an activation of IL-23 producing myeloid cells, which, in turn, drive IL-17 mediated tumor growth (Grivennikov et al., 2012). Chronic NF-κB activation in IECs has been shown to lead to the development of intestinal adenomas linking inflammation and tumorigenesis (Greten and Karin, 2004; Vlantis et al., 2011). In line with a pro-tumorigenic function of NF-κB in CRC, crossing transgenic mice with chronic epithelial NF-κB activation to ApcMin/+ mice leads to accelerated LOH and intestinal tumor initiation through iNOS up-regulation (Shaked et al., 2012). Intriguingly, little is known about the consequences of APC inactivation in immune cells. While it has been shown that Apc mutant mice are characterized by an altered intestinal immune homeostasis and impaired control of inflammation by regulatory T lymphocytes (Gounaris et al., 2009; Akeus et al., 2014; Chae and Bothwell, 2015), only a recent study demonstrated that Apc inactivation in T-cells renders the immune system unable to tackle gut inflammation due to deficient T-cell activation (Agüera-González et al., 2017).
PTEN
The phosphatase PTEN (phosphatase and tensin homolog deleted on chromosome 10) functions as tumor suppressor by inhibiting PI3K-dependent cellular proliferation, survival, growth, and motility. PTEN function is frequently disrupted in human cancer, but has also been shown to play a role in other diseases (Leslie and Downes, 2004). The phosphatase has originally been identified as tumor suppressor through its mutation leading to abolished or greatly decreased phosphatase activity. More recently, it has been shown that PTEN protein expression is lost in a greater number of patients as originally expected based on mutational frequency. Both genetic and epigenetic mechanisms are discussed as possible mechanisms causing loss of PTEN protein expression in the absence of coding sequence mutations (Leslie and Downes, 2004). PTEN represents another tumor suppressor that has been shown to exhibit its oncogenic functions, at least in part, through manipulation of the microenvironment by triggering the release of inflammatory mediators from tumor cells. For example, PTEN dysfunction has been shown to increase the expression and signaling of pro-inflammatory chemokine CXCL8 in prostate cancer cells that resulted in a coordinated response of both tumor and stromal cells. Increased release of Cxcl8 from Pten-deleted tumor cells augmented the sensitivity and responsiveness of tumor cells to stromal chemokines by concurrently inducing the upregulating of chemokine receptors on tumor cells and inducing stromal chemokine production (Maxwell et al., 2014). In one study, progression of Kras mutant PDAC was associated with deletion and loss of expression of Pten. Interestingly, Pten loss and activation of K-Ras cooperated and accelerated pancreatic cancer development by promoting NF-κB activation and its cytokine network, which in turn promoted stromal activation and immune cell infiltration (Ying et al., 2011). Similarly, Kim and colleagues showed that knockdown of tumor suppressor Pten and Tp53 in breast cancer cells synergized to activate a pro-inflammatory Il-6/Stat3/NF-κB signaling axis (Kim et al., 2015). In addition, loss of Pten has been shown to prevent anti-tumor immunity (Spranger et al., 2015; Peng et al., 2016). In conclusion, available data suggests that loss of PTEN leads to activation of an inflammatory loop that contributes to malignant transformation.
Genetic Studies of the Tumor Microenvironment
About two decades ago, Fattaneh Tavassoli and his team were the first to report genetic alterations, specifically LOH at microsatellite markers, in the stroma of mammary carcinomas (Moinfar et al., 2000). Since then a number of human studies have analyzed the mutational spectrum of selected tumor suppressors and LOH/allelic imbalances of specific markers. In 2001, Kurose and colleagues analyzed invasive LCM-procured epithelium and stroma from adenocarcinoma samples of the breast and reported that both epithelial and stromal cells harbor LOH of specific markers including those at 10q23 (in the vicinity of PTEN), 17p13–p15 (in the vicinity of TP53) and 16q24 with a higher frequency in the neoplastic epithelial compartment (Kurose et al., 2001). Around the same time, a different group reported LOH on chromosome 17p13, 3p25-26, and 9q32-33 in the stroma of invasive urothelial carcinoma (Paterson et al., 2003). In 2002, Kurose et al. reported that mutations in the tumor suppressors PTEN and TP53 occur at a high frequency in the neoplastic breast epithelium and/or stroma (Kurose et al., 2002). Charis Eng and her team followed then up on their work with a larger study analyzing the mutational status of TP53 and LOH in 218 invasive breast cancers patients (Patocs et al., 2007). They found a high frequency of TP53 mutations in hereditary (49%) and sporadic tumor (27.4%) stroma. Similarly, 60% (hereditary) and 51% (sporadic) of the patients carried LOH or allelic imbalances in the stroma. In addition to previous studies, the authors related their genetic findings to clinical and pathological features of the disease. Interestingly, in the sporadic group, the presence of TP53 mutations in the stroma was associated with lymph node status and nodal metastasis. Those observations suggest that genetic alteration of TP53 in the stroma may accelerate tumor growth. Overall, numerous independent investigators have described a variety of genetic, epigenetic, and genomic alterations in the stroma of a broad variety of solid tumors and inflammatory conditions (Man et al., 2001; Tuhkanen et al., 2004; Hu et al., 2006; Ishiguro et al., 2006; Kim et al., 2006; Bian et al., 2007; Joshua et al., 2007; Weber et al., 2007; Yagishita et al., 2008). While such findings still remain controversial (Allinen et al., 2004), it challenges the current paradigm that the microenvironment, albeit aberrant, would not be targeted by genetic alterations and highlights the necessity to further our mutational understanding of this crucial component. Importantly, the finding of genetic and epigenetic changes in the stroma raises a number of questions, regarding the mechanisms leading to these genetic lesions, the populations in which they are found, their functional importance to tumor development, and clinical implications for patients with mutations in the stroma. Using a variety of different approaches, multiple groups have tried to model how modulation of known tumor suppressors and/or oncogenes in stromal cell types may affect malignant transformation using mouse models. Early work by the group of Terry van Dyke suggested that oncogenic stress mediated by an initial driver event in the epithelium would create pressure in the microenvironment that leads to loss and selection of a Tp53-deficient stromal compartment (Hill et al., 2005). This is in line with the knowledge that stromal fibroblasts with intact Tp53 can render the microenvironment less supportive of the survival and spread of adjacent tumor cells by secretion of a spectrum of growth inhibitors (Komarova et al., 1998; Moskovits et al., 2006). Intriguingly, Hill and colleagues further showed that loss of Tp53 in the stromal compartment disrupts the homeostasis between the epithelial and stromal tissues ultimately leading to loss of Tp53 also in the tumor suggesting that stromal loss may actually precede epithelial Tp53 loss (Hill et al., 2005; Palumbo et al., 2015). About a decade later, Farmaki and colleagues also showed that the TME induces strong selective pressure onto stromal cells, selecting specific subpopulations of stromal fibroblasts that can survive and expand more efficiently within the TME (Farmaki et al., 2012). Notably, higher numbers of cancer cells were associated with a stronger proliferative advantage of Tp53-deficient fibroblasts as compared to wild-type cells in line with the concept that loss of Tp53 heightens the sensitivity of mutant fibroblasts to epithelial-derived growth factors. Mechanistic studies revealed that the oncogenic effect of Tp53-deficient microenvironment is mediated by enhancing the levels of inflammatory cytokines/chemokines and immunosuppressive molecules, which disturbed immune cell composition and exacerbated immunosuppressive function within the microenvironment (Guo et al., 2013). Further, ablation of Tp53 in fibroblasts has been shown to promote tumor growth in a murine prostate cancer model (Addadi et al., 2010). While TP53 has been the major focus of most mechanistic studies, it is conceivable that mutational inactivation of other tumor suppressors as well as epigenetic alterations such as histone modifications and DNA methylation may be responsible for the generation of stromal cells with pro-tumorigenic properties (Hu et al., 2005; Peng et al., 2005; Bar et al., 2009). It will be important to uncover the identity and regulation of secreted factors that are responsible for the tumor cell-induced inhibition of stromal TP53 induction and other potential tumor suppressors.
Clonal Hematopoiesis and Inflammation
Leukocytes represent a crucial component of the inflammatory TME. The various leukocyte subsets, including macrophages, neutrophils, basophils, and lymphocytes can interact with each other, but also with non-hematopoietic stromal cell types and epithelial tumor cells, thereby orchestrating tumor progression and invasiveness. To date, many groups have studied the prognostic value of infiltrating immune cells in solid tumors (reviewed by Barnes and Amir, 2017; Hammerl et al., 2017). With the increasing interest in harnessing the immune system to treat cancer with checkpoint inhibitors and other novel agents, a better understanding of the composition of the immune infiltrate as prognostic marker is of increasing importance. However, the simple presence of a specific immune cell type in the TME does not predict their function. For example, macrophages cover a continuum of functional states that allows them to fulfill different tasks depending on the microbial and cytokine milieu. Further complicating the scenario, recent data challenge the paradigm that the integrity of the genome of immune cells is intact in solid cancer patients. At first, a large body of genetic data emerged demonstrating that elderly individuals without signs of overt leukemia harbor somatic mutations in hematopoietic cells leading to expansion of mutant blood cells. Most of the mutations were identified in genes encoding for known leukemia drivers such as the chromatin modifiers TET2, ASXL1, and DNMT3A (Busque et al., 2012; Genovese et al., 2014; Jaiswal et al., 2014; Shlush et al., 2014; McKerrell et al., 2015; Young et al., 2016; Buscarlet et al., 2017). Not surprisingly, follow up studies showed that patients with clonal hematopoiesis (CH) are at an increased risk of developing hematological malignancies. However, the same study showed that patients with CH are also at an increased risk of atherosclerotic cardiovascular disease compared to individuals without CH (Jaiswal et al., 2014, 2017a,b). Atherosclerotic cardiovascular disease has long been thought of as an inflammatory disease; however, only recently, data from the CANTOS study was published reporting that selectively targeting inflammation by using a therapeutic monoclonal anti-IL-1β antibody can reduce cardiovascular risk (Ridker et al., 2017a). But is there a mechanistic link between the presence of mutant cells in the blood, the development of atherosclerotic cardiovascular diseases, and inflammation? Intriguingly, a growing body of evidence suggests a causal link between mutations in epigenetic modifiers seen in CH and inflammation. For example, Tet2-deficient macrophages exhibit an increase in inflammasome-mediated IL-1β secretion, which is associated with accelerated development of atherosclerosis in these mice (Fuster et al., 2017). Tet2 also seems to exhibit a suppressive role in the regulation of immunity and inflammation, independent of its role in DNA methylation and hydroxymethylation, but by repression of transcription via histone deacetylation (Zhang et al., 2015). Specifically, loss of Tet2-mediated gene transcription resulted in increased expression of inflammatory mediators upon injection of the mice with the highly potent inflammatory stimulus LPS. Further, Tet2-deficient mice were more susceptible to experimental colitis and endotoxin shock (Zhang et al., 2015). Dnmt3a-deficient mast cells display an increased sensitivity to acute and chronic inflammatory stimulation (Leoni et al., 2017). Yet in another immune cell type, both Dnmt3a and Tet2 seem to be important for the regulation of macrophage activation, polarization and inflammation (Yang et al., 2014; Li et al., 2016). It is likely that this is just the tip of the iceberg and the coming years will provide a greater understanding of the functional and regulatory roles of these important epigenetic regulators in different immune compartments, myeloid malignancies, solid tumors, and non-malignant inflammatory diseases.
The accumulating body of evidence that older individuals have clinically unapparent CH, together with the increasing awareness of the importance of microenvironment in tumor progression, we hypothesized that immune cells infiltrating tumors might be characterized by clonally selected mutations. Targeted sequencing analysis of leukocytes isolated from a small number of tumors from treatment naïve breast cancer patients demonstrated that indeed infiltrating CD45-positive hematopoietic cells harbor somatic mutations in cancer genes, including BCOR, TET2, DNMT3A, in a subset of patients (Kleppe et al., 2015a). These mutations were not found in peripheral blood cells, admixed tumor cells, or epithelial germline samples. This finding suggests that mutant infiltrating leukocytes may interact with cancer cells, which has significant clinical implications for tumor development and response to treatment. Our data was partially corroborated by a recent report that hematopoietic cells are also genetically abnormal in a fraction of patients with advanced solid cancers even at a younger age (Coombs et al., 2017). Coombs and colleagues analyzed paired tumor and blood samples from ~8,000 patients with advanced solid cancers. In total, one fourth of all patients carried at least one CH mutation in the blood sample, with 4.5% of the patients harboring presumptive driver mutations (CH-PD). CH was associated with increasing age, tobacco use, and prior radiation therapy. Further, patients with CH had an increased risk of hematologic cancers and CH-PD was associated with a shorter survival. Notably, the primary cause of death was progression of the primary non-hematopoietic tumor. While both studies support the intriguing idea that immune cells are genetically abnormal which could be of utmost importance from a diagnostic and therapeutic standpoint, there are fundamental differences in the study design and the conclusions. Regardless, neither study data discerns between the different leukocyte subsets. As such, it will be critical to assess the distribution and frequency of CH mutations within the different hematopoietic/immune compartments.
Prevention and Therapeutic Intervention
Checkpoint Blockade Immunotherapy
Historically, there has been a longstanding clinical interest in the overlap between the immune system and cancer. Most famously, in the early nineteenth century, William Coley developed a “Coley's Vaccine,” a concoction of bacteria, after noticing tumor regression among patients who developed high fevers from Steptococcus pyogenes infection. His treatments and the research therein largely fell out of favor with the advent of surgical advances, radiation, and chemotherapy. In the last few years, however, the growing understanding of the tumor microenvironment and interplay between the immune system and cancer has dramatically changed the landscape of immunotherapy options. In addition to investigational efforts into vaccines and oncolytic viruses, several immunotherapy treatments have recently been approved. Specifically, an understanding of immune response and activation in cancer, and in particular the role of immune blockade by CTLA-4 (cytotoxic T lymphocyte-associated protein 4) and PD-1/PD-L1 (programmed cell death protein 1/programmed cell death protein ligand 1) has revolutionized the treatment of several types of cancers, including but not limited to melanoma, non-small cell lung cancer, urothelial, head and neck, and renal cancers. Ipilimumab was the first anti- CTLA-4 antibody to be approved in advanced melanoma (Sharma and Allison, 2015). In determining which cancers benefit from checkpoint inhibition, evidence suggests that those with a higher mutational burden (such as in response to tobacco) and in some instances microsatellite instability-high (MSI-h) tumors may respond best (Pleasance et al., 2010; Snyder et al., 2014; Le et al., 2015). Not surprisingly, given the activation of the immune system with these therapies, oncologists have had to become increasingly facile with managing a host of immune related side effects (Postow and Hellmann, 2018; Postow et al., 2018). For a more extensive review of clinically relevant checkpoint inhibitors and immunotherapy please see: (Farkona et al., 2016).
Non-steroidal Anti-inflammatory Drugs (NSAIDs)
COX enzymes (COX1 and COX2) are the primary targets of non-selective non-steroidal anti-inflammatory drugs (NSAIDs), which include aspirin, indomethacin, piroxicam, sulindac, and ibuprofen. Inhibition of COX enzymes results in the inhibition of prostaglandins, which play important roles in many physiological processes. NSAIDs are commonly used for the treatment of fever, pain, and swelling. Slowly, albeit still somewhat controversial and largely based on epidemiologic studies, NSAIDs have emerged as drugs with potential anti-cancer activity which may decrease the incidence and mortality of colon, breast, stomach, and lung cancers (reviewed in Ulrich et al., 2006, Figure 2). For example, chronic use of aspirin has been suggested to reduce the risk of pro-inflammatory conditions such as inflammatory bowel disease and the risk to develop colorectal cancer (Chia et al., 2012). Similarly, different studies suggest that ibuprofen might also stop certain cancers from developing (Harris et al., 2003; Johnson et al., 2010). Overall, a large body of observational data regarding a protective effect of NSAIDs from developing certain cancers, specifically CRC, is strong. A recently performed meta-analysis investigated the relationship between NSAIDs and lymph nodes/distant metastasis. The study suggests that NSAIDs hold potential in the management of cancer metastasis, regardless of whether NSAIDs are used at pre-diagnosis or post-diagnosis (Zhao et al., 2017). At this point, it cannot be denied that anti-inflammatory compounds may represent novel, less toxic, agents for cancer therapy, nonetheless, carefully designed, controlled, blinded, and randomized trials are required to create a benefit-risk assessment and address many outstanding questions such as the lowest effective dose, the age at which to initiate therapy, the duration of treatment, and which population benefits of NSAIDs chemopreventive activity. Toward this end, the AspECT trial, a large phase III, randomized study is designed to assess the long-term chemoprevention effect of esomeprazole in combination with or without aspirin in patients with Barrett's metaplasia (NCT02070757).
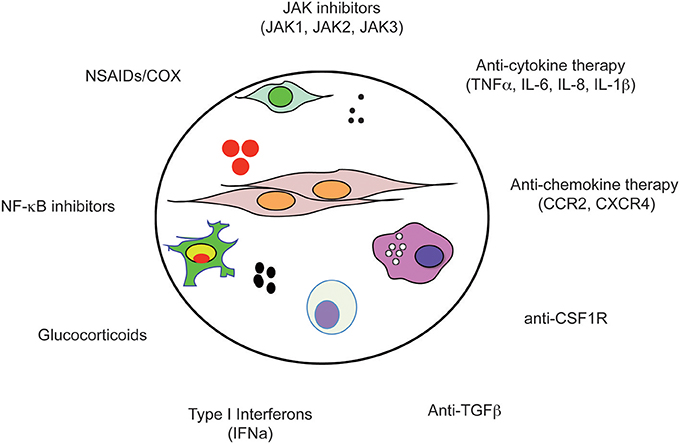
Figure 2. Anti-inflammatory drugs. The tumor microenvironment (circle) is composed of tumor cells, fibroblasts, endothelial cells, pericytes, and various cells associated with the immune system such as macrophages and NK cells. Importantly, within the microenvironment a tight bidirectional cross talk of cancer cells with their microenvironment occurs which is mediated by the production of chemokines, cytokines, growth factors, prostaglandins, ROS, and NOS, as well as recruitment of inflammatory cells into the tumor tissue. Due to the complex nature of the microenvironment, a large number of druggable dependencies have been identified and are currently under investigation as therapeutic targets for anti-inflammatory drugs.
Glucocorticoids
Glucocorticoids (GCs) have been used in the clinic for over half a century. Indeed, due to their strong anti-inflammatory and immunosuppressive properties, GCs are the most prescribed immune suppression medications worldwide. Orally inhaled GCs are commonly used to suppress various allergic, inflammatory, and autoimmune disorders. For example, they are by far the most effective drugs for the treatment of asthma, which is largely due to their efficacy to inhibit inflammatory cytokine gene expression (Barnes, 2011). GCs exhibit multiple modes of action and interfere with the function of basically all immune cell types. For example, GCs suppress cytokine release and cell migration, induce apoptosis, and change cell differentiation fates (Perretti and Ahluwalia, 2000). In oncology, GCs have only shown modest efficacy in breast and prostate cancer and not in other cancer types. In general complexity and controversial observations are associated with GC treatment in non-hematologic cancer types (Lin and Wang, 2016). Nonetheless, GR antagonists are currently tested in several clinic trials in combinations with chemotherapy, including breast, prostate, and lung cancer. In hematological cancers, GCs such as dexamethasone have proven astonishingly effective in the treatment of lymphoid neoplasms including acute lymphoblastic leukemia, chronic lymphocytic leukemia, MM, Hodgkin's lymphoma, and non-Hodgkin's lymphoma where GCs induce growth arrest and apoptosis. GC-induced apoptosis of lymphoid cells is sought to be induced via multiple signaling pathways including Bim, a member of the Bcl2 family, and suppression of cytokines via inhibition of the activity of different TFs such as NF-κB (Lin and Wang, 2016). In contrast, only little is known about the mode of action of GCs in solid cancers.
JAK Inhibitors
The Janus family of kinases (JAKs) comprises four members: JAK1, JAK2, JAK3, and TYK2. JAKs are critical for the signal transduction of about 60 different cytokines that rely on type I and II cytokine receptors. Many of these cytokines are central to the growth of malignant cells and drivers of immune-mediated diseases. Consequently, JAKs have emerged as a new class of pharmacologic agents. The first selective JAK inhibitor to enter clinical trials was tofacitinib. Tofacitinib potently inhibits JAK3 and JAK1 and to a lesser extent JAK2 and shows a high degree of kinome selectivity (O'Shea et al., 2013). Tofacitinib is the first JAK inhibitor approved for the treatment of moderate to severe RA and active psoriatic arthritis. Mechanistically, as a JAK inhibitor, tofacitinib efficiently blocks the biological effects of common γchain cytokines including IL-2, IL-4, IL-15, and IL-21 and consequently suppresses allergic Th2 responses (Fukuyama et al., 2015). For example, in a Th2-dependent asthma mouse model, tofacitinib reduces pulmonary eosinophilia (Kudlacz et al., 2008). Further, tofacitinib has been shown suppress the differentiation of pathogenic Th1 and Th17 cells as well as innate immune signaling by limiting the production of pro-inflammatory cytokines in a LPS-induced sepsis model (Ghoreschi et al., 2011). Interestingly, tofacitinib and ruxolitinib, a JAK1/2 inhibitor, are currently being tested in patients with skin and hair disorders, including the autoimmune disease alopecia areata, and mild to moderate atopic dermatitis (Bissonnette et al., 2016; Mackay-Wiggan et al., 2016; Liu et al., 2017). Also other pharmacologic agents targeting different JAK family members have found increasing attention as anti-inflammatory targets in different disease contexts. Ruxolitinib (Jakafi) was the first FDA approved JAK inhibitor for the treatment of myelofibrosis (MF) and, more recently, for patients with polycythemia vera (PV) who have had an inadequate response to hydroxurea (Verstovsek et al., 2010; Raedler, 2015). Ongoing clinical trials also assess the efficacy of ruxolitinib in patients with post-MPN AML and CML with minimal residual disease, another form of MPN (Eghtedar et al., 2012; Pemmaraju et al., 2015; Assi et al., 2018). Interestingly, on a mechanistic level, recent work from Tarafdar and colleagues suggests that CML stem cells downregulate MHC-II, allowing them to evade the host immune response. They found that this deregulation can be reverted by JAK inhibition and IFNγ (Tarafdar et al., 2017). Besides ruxolitinib, other JAK2 inhibitors are also under clinical development for the treatment of MPNs (Kontzias et al., 2012) and their strong anti-inflammatory potential provides a rationale for repurposing these drugs as solid tumor therapeutics (Quintás-Cardama et al., 2011; Plimack et al., 2013; Buchert et al., 2016). However, clinical studies of different JAK inhibitors in solid tumors have been marked by lack of activity. For example, a phase 1 study of JAK1/2 inhibitor AZD-1480 in solid tumor patients was discontinued due to unusual adverse side effects and lack of clinical activity (Plimack et al., 2013). A comprehensive table listing clinical trials conducted with JAK inhibitors can be found in Buchert et al. (2016). Despite initial excitement, in 2016 Incyte discontinued several clinical trials due to insufficient level of efficacy including: the phase III study (JANUS 2) of ruxolitinib or placebo plus capecitabine in patients with advanced or metastatic pancreatic cancer, the phase II sub-study of ruxolitinib in patients with metastatic colorectal cancer and low CRP, and the phase II studies in breast and lung cancer. In addition, Incyte discontinued the investigation of INCB39110, a selective JAK1 inhibitor as first-line treatment for metastatic pancreatic cancer, however, preclinical studies still continue. The JAK/STAT pathway also presents new potential targets in graft-vs.-host disease (GvHD). Despite major improvements in allogeneic hematopoietic stem cell transplantation (HSCT), GvHD still remains a matter of concern, especially if patients show no adequate response to systemic corticosteroid. Corticosteroid-refractory (SR) acute and chronic GvHD is associated with poor prognosis and therapeutic options for salvage therapy are needed. Due to its strong anti-inflammatory properties, multiple groups have performed retrospective studies to assess the potential of ruxolitinib as salvage therapy in steroid resistant GVHD patients (Zeiser et al., 2015; Mori et al., 2016; Khandelwal et al., 2017). The data suggests that while low-dose ruxolitinib shows potential in this setting, it will be important to determine in prospective studies the optimal dose to achieve the best-tolerated dose and an optimized tapering schedule to avoid withdrawal symptoms which are also observed in MF patients upon discontinuation of the drug. Overall, JAK inhibitors have a wide range of indications due to their central role in the regulation of cytokine signaling. It can be expected that the market for JAK inhibitors will continue to grow in the coming years and, similarly, the field will probably move toward using novel strategies to achieve more specific and versatile inhibition of this important family of kinases.
Anti-Cytokine Therapy
Cytokine and cytokine receptors have been recognized as excellent drug targets for a variety of diseases characterized by chronic inflammation due to their important function as rate-limiting signaling molecules (Feldmann, 2008). TNFα blockade in patients with rheumatoid arthritic was the first major success demonstrating the beneficial effect of anti-cytokine therapy in a disease associated with chronic inflammation (Elliott et al., 1993; Feldmann and Maini, 2001). The role of TNFα and IL-6 as master regulators of tumor-associated inflammation and tumor-promoting functions makes them promising targets for adjuvant anti-cancer therapy (Yan et al., 2006; Grivennikov and Karin, 2011). Clinically, elevated TNFα serum levels have been detected in patients with a wide range of tumor types, although TNFα is not universally detectable. Notably, high levels of TNFα have been correlated with tumor stage, extent of para-neoplastic complications, and worse survival. Due to a prospective study showing that TNFα and IL-6 levels correlate with the degree of disease, and PSA progression in prostate cancer patients, TNFα and IL-6 are currently suggested as additional markers that reflect the activity level of this disease (Michalaki et al., 2004). Three biologics targeting TNFα are currently approved including etanercept, infliximab, and adalimumab and different small molecules that inhibit TNFα signaling or synthesis (for example thalidomide) are under development. TNFα blocking agents may not only find a place as anti-tumor agents, but may have a role in controlling the severe cancer pain associated with metastatic bone lesions. Similarly, anti-IL-6 therapy holds potential to alleviate cancer-related symptoms. Several early phase clinical studies with IL-6 targeting agents currently support the hypothesis that IL-6 may be indeed an effective anti-cancer target. Several humanized monoclonal antibodies against soluble and membrane bound IL-6R (tocilizumab, REGN88) or against IL-6 (siltuximab and sirukumab) are currently in clinical trials at various development stages for several types of cancer including MM, metastatic renal cell carcinoma, B-cell lymphoproliferative diseases (Heo et al., 2016). A fully human anti-IL-8 antibody, ABX-IL8, has shown promising therapeutic efficacy in preclinical models (Yang et al., 1999; Huang et al., 2002) however the therapeutic value of blocking IL-8 has yet to be assessed in cancer patients. Excitingly, recently published data from the CANTOS study, a randomized trial of the role of IL-1β inhibition in atherosclerosis, suggests that anti-inflammatory therapy targeting the IL-1β pathway could significantly reduce incidence of lung cancer and lung cancer mortality (Ridker et al., 2017b). CANTOS was not formally designed as a cancer detection or treatment trial and will need to be replicated; however, there is precedent for other cancer types (MAPp1, NCT01767857; Lust et al., 2009; Hong et al., 2014; Goel et al., 2016). In contrast to the success stories of anti-TNFα and anti-IL-6 targeting studies, little clinical success has been observed with therapeutics targeting single cytokines in other diseases such as COPD or asthma. One can speculate that the lack of clinical success of therapeutic targeting single cytokines or cytokine receptors in other inflammatory diseases may be due to the redundancy of the cytokine network and thus required to target more than one cytokine at a time. This may also be true for the treatment of different types of cancer; however, it is too early to say what place anti-cytokine therapy will claim in the field of oncology.
Interferons
IFNs present another group of cytokines which exhibit anti-tumor activity and can activate the immune system and are leveraged in the clinic (reviewed in Parker et al., 2016). Type I IFNs, IFNα and IFNβ, can be produced and released by tumor cells and by most cell types in the human body (Parker et al., 2016). In contrast, IFNγ, a type II IFN, is primarily expressed by T cells and NK cells. Regardless of their source, all IFNs can act via intrinsic and extrinsic mechanisms thereby exerting direct antitumor effects or indirect effects through antitumor immune responses. Tumor cell intrinsic effects of IFNs are a result of their capability to regulate the expression of genes involved in various cellular processes such as cell growth, proliferation, and survival. For example in patients with PV, IFNα has been shown to inhibit the growth of JAK2V617F-mutant hematopoietic stem and progenitor cells via activation of the p38 MAPK pathway (Lu et al., 2010). Tumor cell extrinsic effects of IFNs are mediated through their ability to influence the activity of nearly every immune cell type including T cells, NK cells, monocytes, and macrophages (Hervas-Stubbs et al., 2011). Recent work by Parker and colleagues showed that intact type I IFN signaling is required to induce anti-metastatic immunity by NK and CD8 T cells. Interestingly, their work further showed that the tumor cells themselves were a source of type I IFNs and that suppression of the tumor cells own IFN production functions as immune-evasion mechanism (Bidwell et al., 2012). IFNs exert their immunoregulatory effects on the cells of the immune system through the regulation of tumor antigens on tumor cells (Greiner et al., 1984) and antigen presentation by MHC complexes and ligands for receptors of immune checkpoints (Propper et al., 2003; Schreiner et al., 2004). Further, IFNs can regulate immunity by triggering the release of “downstream” chemokines, cytokines, and interleukins such as IL-15 (Nguyen et al., 2002; Burkett et al., 2004). Besides their role in immunity, IFNs also promote angiogenesis and osteoclastogenesis which are important to tumor growth (Cheon et al., 2014). Unlike most cytokines which are targeted with antagonists, IFNα is used as therapeutic protein in the treatment of patients with some types of solid cancer, including Kaposi sarcoma, melanoma, and renal cell carcinoma, and different hematologic malignancies such as MPNs, and viral diseases (Kirkwood, 2002). The first type of IFN-based therapy showed striking results on the survival rates of patients with hairy cell leukemia and CML (Platanias, 2013; Stein and Tiu, 2013). While the discovery of the Philadelphia chromosome as genetic alteration in CML and the arrival of BCR-ABL targeting TKIs in the clinic have largely replaced the need for IFNα-based treatment in this disease, some recent clinical studies have shown that combination of imatinib and IFNα is superior to either therapy alone, perhaps due to the fact that IFNα targets preferentially CML stem cells (Talpaz et al., 2013). Notably, due to its success in numerous clinical trials, IFNα is used as first-line treatment choice for patients with high-risk PV (Barbui et al., 2011; Falchi et al., 2015). Regardless of the specific use, IFN-based therapies have their limitations due to dose-limiting side effects. As a direct consequence, various other strategies such as the use of IFNα conjugated with polyethylene glycol moieties or pattern recognition receptor agonists to stimulate type I IFN production in patients are under development aimed at increasing efficacy over toxicity (Parker et al., 2016).
CSF1R Inhibition
In contrast to IFN-based therapy, colony-stimulating factor 1 receptor (CSF1R) has only recently emerged as a cancer drug target. CSF1 is essential for the development and maintenance of macrophages, and as such provides a critical target for depleting these cells within the TME. The efficacy of CSF1R inhibition, and its capacity to deplete TAMs seems to vary depending upon tumor type and tissue. Administration of BLZ945, a small molecule inhibitor of CSF1R, in a murine model of glioma led to regression and long term increases in survival (Pyonteck et al., 2013). Surprisingly, this reduction in tumor burden was not associated with TAM depletion, but rather TAMs underwent “re-education,” an alteration in gene expression profile with decreased expression of M2-associated anti-inflammatory markers. Meanwhile breast cancer models utilizing either BLZ945 or CSF1R neutralizing antibodies led to depletion of TAMs with limited therapeutic efficacy as a monotherapy (DeNardo et al., 2011; Shiao et al., 2015; Olson et al., 2017). Understanding why an environment is permissive to maintaining TAMs in the face of CSF1R inhibition will be critical for clinical implementation. One possibility is the expression of GM-CSF, IFNγ, and CXCL10, factors that were shown to rescue macrophage from BLZ945-mediated killing (Pyonteck et al., 2013). Whether these factors are also causatively involved in “re-education” remains to be determined. While CSF1R inhibition as a monotherapy has shown various model dependent results, combination therapy with cytotoxic agents has shown broader applicability. In murine breast tumor models, combinations of CSF1R inhibitors such as PLX3397 or BLZ945 with radiotherapy or paclitaxel displayed greater efficacy than with either single agent alone (DeNardo et al., 2011; Shiao et al., 2015; Olson et al., 2017). This combined efficacy does not appear to be limited to cytotoxic therapy as in a murine model of pancreatic adenocarcinoma PLX3397 treatment synergized with immune checkpoint blockade (Zhu et al., 2014). In this setting, treatment with PLX3397 led to a reduction in macrophages, where the remaining cells underwent a reprogramming not dissimilar from that seen in gliomas with BLZ945 treatment (Pyonteck et al., 2013). Similar combined efficacy was also seen in glioma models combining CSF1R inhibition with multiple TKIs (Yan et al., 2017). In another study, prolonged CSF1R inhibition led resistance through macrophage-derived IGF1 signaling and PI3K activation in tumor cells (Quail et al., 2016). In this setting, combination CSF1R and PI3K/IGF1R inhibition was capable of preventing relapse. Collectively, these studies demonstrate that CSF1R inhibition shows promise in chemo sensitizing combination therapies and in combination with TKIs. Multiple clinical trials are currently ongoing evaluating CSF1R inhibition with checkpoint immunotherapy, radiation therapy, and targeted small molecules (Cannarile et al., 2017). In addition to identifying the optimal tumor types for CSF1R inhibition, proper dosing schedules will also need to be determined. While the goal of many Phase I trials is to identify the maximum tolerable dose of the drug, care should be taken to understand the different outcomes of high dose and low dose CSF1R inhibition. It is possible, that at high doses, CSF1R inhibitors may deplete macrophages even in “protective” environments, and that lower doses may offer a capacity to “re-educate” macrophages without depleting them in the TME. Proper biomarkers, potentially derived from gene expression studies following CSF1R inhibition, will be necessary to determine the optimal dose based on the desired outcome of depletion vs. reprogramming.
Inflammation and Drug Resistance
Diverse resistance mechanisms to targeted cancer therapy have emerged and present one of the foremost challenges in cancer today. Drug resistance may pre-exist (intrinsic, primary drug resistance) or may be acquired under the strong selective pressure during the course of treatment (acquired resistance) (Zahreddine and Borden, 2013). Importantly, some of these resistance pathways lead to multi-drug resistance (MDR), generating an even more difficult clinical problem to overcome. Alterations in the level of cytokines, chemokines, and growth factors have emerged as yet another mechanism conferring resistance to chemotherapeutic treatments (Jones et al., 2016). For example, mounting evidence suggests a crosstalk between IL-6 and MDR in cancer and potential therapeutic opportunities arising from this role of IL-6 (Ghandadi and Sahebkar, 2016). Since the 1990s, elevated serum levels of IL-6 have been associated with worse survival in breast cancer patients (Zhang and Adachi, 1999; Salgado et al., 2003; Knüpfer and Preiss, 2007). Subsequently, functional studies revealed that autocrine production of IL-6 by tumor cells confers resistance to several chemotherapeutic agents, suggesting that acquisition of the ability of tumor cells to produce IL-6 represents another self-protective mechanism (Conze et al., 2001). Further, increased secretion of IL-6 has been linked to resistance to bortezomib in MM and to etoposide and cisplatin in hormone-independent prostate carcinomas (Borsellino et al., 1995; Frassanito et al., 2001; Voorhees et al., 2007). IL-6-induced STAT3 feedback activation in response to the EGFR inhibitor erlotinib has been associated with the development of resistance and poor prognosis in lung adenocarcinoma (Lee et al., 2014). Work published by Korkaya and colleagues suggests that trastuzumab resistance may be mediated by an IL-6 inflammatory loop (Korkaya et al., 2012). Importantly, soluble factors mediating drug resistance such as IL-6 can be released by the tumor cell itself or the TME. For example, Ara and colleagues demonstrated that in neuroblastoma the bone marrow microenvironment is a source of IL-6 and sIL-6R, thereby allowing cancer cells to escape the cytotoxic effects of multiple chemotherapeutics (Ara et al., 2013). Regardless of the source of the cytokine IL-6, IL-6 targeting strategies promise to be effective in combination with cytotoxic agents, TKIs, or targeted antibody therapy to prevent drug resistance. Besides IL-6, IL-8 has emerged as another resistance cytokine. Similarly to IL-6, clinical studies have linked high serum levels of IL-8 to disease progression of various cancer types (Xie, 2001). In line, by using cytokine antibody arrays to identify cytokines associated with drug resistance, two independent groups concordantly found that IL-6 and IL-8 are key markers for the development of drug resistance (He et al., 2011; Shi et al., 2012). The list of “resistance cytokines” is long including AMF (autocrine mobility factor), AM (adrenomedullin), IL-4, and IL-10 (Jones et al., 2016). In addition, a recent study showed that patients with increased levels of the inflammatory biomarkers ferritin and C-reactive protein (CRP) had a markedly poorer response to trastuzumab-containing therapy (Alkhateeb et al., 2012). Another important factor to consider when discussing the relation between cytokines and cancer drug resistance is the fact that the inflammatory master regulator NF-κB is activated by a variety of cytotoxic chemotherapy agents including cisplatin, paclitaxel, docetaxel, and doxorubicin (Nakanishi and Toi, 2005; Li and Sethi, 2010). Anticancer therapeutic induced NF-κB activation often leads to the activation of pro-survival signaling pathways. In addition, NF-κB is often constitutively active in response to a variety of cancer-promoting agents. Both the constitutive and therapy-induced NF-κB activation eventuates in drug-resistant tumors, at least in part, by the induction of inflammatory cytokine secretion. Consequently, NF-κB signaling has emerged as an attractive molecular target for pharmacological intervention and its inhibitors as potential sensitizer to anticancer drugs; however, despite the clinical success in newly diagnosed and relapsed/refractory multiple myeloma (MM) and mantle cell lymphoma patients, the NF-κB inhibitor bortezomib (Valcade) has fallen short of original expectations (Lin et al., 2010; Godwin et al., 2013). Paradoxically, in some instances bortezomib has been shown to activate NF-κB signaling and stimulate cytokine secretion. For example, exposure to bortezomib of prostate cells led increased expression and release of IL-8. Mechanistically, bortezomib increased the accumulation of IκB kinase β (IKKβ) in the nucleus and increased recruitment of nuclear IKKβ, phosphorylated p65, and transcription factor early growth response-1 (EGR1) to the IL-8 promoter (Singha et al., 2014). Given that IL-8 is another resistance-associated cytokine this may explain why bortezomib is less effective in certain tumor types. Overall, there is a strong link between inflammation and drug resistance and beyond their ability to prevent or decrease chemotoxicity, anti-inflammatory agents may also have therapeutic effects when combined with conventional agents, acting additively or synergistically, or sensitizing cancer cells to treatment with conventional cancer therapies. In order to optimize the therapeutic window and regimens of anti-inflammatory cancer therapy, clinical oncologists, and experimental cancer researchers will need to identify for each drug the appropriate molecular target, cancer type, disease stage, and treatment duration. Based on the current state of knowledge, it can be expected that anti-inflammatory agents will be most effective in combination with anti-angiogenic, cytotoxic, and cytostatic agents.
Future Direction
Herein we discuss the constituents of the TME, the causes of inflammation within, and therapeutic strategies aimed at disrupting the signaling pathways critical to bolstering cancer development. Given the evolutionary nature of cancer, it is unlikely that any single therapeutic option will lead to durable cures or eradication of complex tumors. Indeed combinations therapies targeting tumor cell intrinsic oncogenic pathways, and TME-derived support pathways will offer the best means to effectively eradicate disease. These combinations are not trivial, as the TME itself is an evolving system with relatively understudied kinetics in the clinical setting. Effective therapeutic intervention may require new paradigms of dosing and schedule to identify therapeutically optimal schedules that may be non-linear and independent of maximum tolerable doses. These arduous studies may benefit from preclinical animal model development, but will inevitably require clinical testing. To expedite studies, the development of biomarkers sensitive to TME-targeted is of the utmost importance. Without these tools, potentially viable compounds and strategies may be left behind due to suboptimal dosing or rather suboptimal understanding of the correct dosing paradigm. While the molecules and pathways described here may provide sound mechanistic and molecular targets for clinical development, unveiling their full potential will require an integrated understanding of the systems-level effects of inflammation. Instead of strict molecular pathways, inflammatory microenvironments can be thought of as ecological landscapes and causes of inflammation as selective pressures sculpting tumor evolution along these landscapes. This ecological landscape is both dynamic and systemic, extending beyond the simple genetics within malignant tumor cells and beyond invasive tumor edge.
Author Contributions
MK has written most of the part of this review dealing with prevention and therapeutic invention as well as intrinsic mechanisms. RB has written most of the part on cellular constituents of an inflammatory microenvironment. EC has written most of the part of this review dealing with external factors and exposures causing inflammation. All authors have corrected the whole manuscript.
Funding
MK acknowledges financial support through a grant by the National Heart, Lung, Blood Institute (K99 HL122503-01A1). MK is a fellow of the American Society of Hematology and RB is a fellow of the Damon Runyon-Sohn Cancer Research Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AML, acute myeloid leukemia; APC, adenomatous polyposis coli; ASCO, American Society of Clinical Oncology; BMI, body mass index; CAFs, cancer-associated fibroblasts; CANTOS, Canakinumab Anti-inflammatory Thrombosis Outcome Study; CH, clonal hematopoiesis; CH-PD, CH with a presumptive driver mutation; COPD, chronic obstructive airway disease; ER, estrogen receptor; FDA, Food and Drug Administration; GCs, glucocorticoids; GI, gastrointestinal; GvHD, graft-versus-host disease; NOS, nitrogen species; HPV, human papillomavirus; HSCT, allogeneic hematopoietic stem cell transplantation; IECs, intestinal epithelial cells; IFN, interferons; JAKs, janus family of kinases; LOH, loss of heterozygosity; LPS, lipopolysaccharides; MDSCs, macrophage-derives suppressor cells; MDR, multi-drug resistance; MM, multiple myeloma; MPNs, myeloproliferative neoplasms; NSAIDs, non-selective non-steroidal anti-inflammatory drugs; PDAC, pancreatic ductal adenocarcinoma; Pten, phosphatase and tensin homolog; PV, polycythemia vera; RA, rheumatoid arthritis; ROS, reactive oxygen species; TAMs, tumor associated macrophages; TF, transcription factor; TKs, tyrosine kinases; TKIs, tyrosine kinase inhibitors.
References
Addadi, Y., Moskovits, N., Granot, D., Lozano, G., Carmi, Y., Apte, R. N., et al. (2010). p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 70, 9650–9658. doi: 10.1158/0008-5472.CAN-10-1146
Agüera-González, S., Burton, O. T., Vázquez-Chávez, E., Cuche, C., Herit, F., Bouchet, J., et al. (2017). Adenomatous polyposis coli defines treg differentiation and anti-inflammatory function through microtubule-mediated NFAT localization. Cell Rep. 21, 181–194. doi: 10.1016/j.celrep.2017.09.020
Akeus, P., Langenes, V., von Mentzer, A., Yrlid, U., Sjöling, Å., Saksena, P., et al. (2014). Altered chemokine production and accumulation of regulatory T cells in intestinal adenomas of APC(Min/+) mice. Cancer Immunol. Immunother. 63, 807–819. doi: 10.1007/s00262-014-1555-6
Alkhateeb, A. A., Leitzel, K., Ali, S. M., Campbell-Baird, C., Evans, M., Fuchs, E. M., et al. (2012). Elevation in inflammatory serum biomarkers predicts response to trastuzumab-containing therapy. PLoS ONE 7:e51379. doi: 10.1371/journal.pone.0051379
Allinen, M., Beroukhim, R., Cai, L., Brennan, C., Lahti-Domenici, J., Huang, H., et al. (2004). Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6, 17–32. doi: 10.1016/j.ccr.2004.06.010
Ara, T., Nakata, R., Sheard, M. A., Shimada, H., Buettner, R., Groshen, S. G., et al. (2013). Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer Res. 73, 3852–3864. doi: 10.1158/0008-5472.CAN-12-2353
Assi, R., Kantarjian, H. M., Garcia-Manero, G., Cortes, J. E., Pemmaraju, N., Wang, X., et al. (2018). A phase II trial of ruxolitinib in combination with azacytidine in myelodysplastic syndrome/myeloproliferative neoplasms. Am. J. Hematol. 93, 277–285. doi: 10.1002/ajh.24972
Bar, J., Feniger-Barish, R., Lukashchuk, N., Shaham, H., Moskovits, N., Goldfinger, N., et al. (2009). Cancer cells suppress p53 in adjacent fibroblasts. Oncogene 28, 933–936. doi: 10.1038/onc.2008.445
Barbara, L., Benzi, G., Gaiani, S., Fusconi, F., Zironi, G., Siringo, S., et al. (1992). Natural history of small untreated hepatocellular carcinoma in cirrhosis: a multivariate analysis of prognostic factors of tumor growth rate and patient survival. Hepatology 16, 132–137. doi: 10.1002/hep.1840160122
Barbui, T., Barosi, G., Birgegard, G., Cervantes, F., Finazzi, G., Griesshammer, M., et al. (2011). Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J. Clin. Oncol. 29, 761–770. doi: 10.1200/JCO.2010.31.8436
Barnes, P. J. (2011). Glucocorticosteroids: current and future directions. Br. J. Pharmacol. 163, 29–43. doi: 10.1111/j.1476-5381.2010.01199.x
Barnes, T. A., and Amir, E. (2017). HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 117, 451–460. doi: 10.1038/bjc.2017.220
Bergers, G., and Benjamin, L. E. (2003). Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 3, 401–410. doi: 10.1038/nrc1093
Bian, Y., Knobloch, T. J., Sadim, M., Kaklamani, V., Raji, A., Yang, G. Y., et al. (2007). Somatic acquisition of TGFBR1*6A by epithelial and stromal cells during head and neck and colon cancer development. Hum. Mol. Genet. 16, 3128–3135. doi: 10.1093/hmg/ddm274
Bidwell, B. N., Slaney, C. Y., Withana, N. P., Forster, S., Cao, Y., Loi, S., et al. (2012). Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 18, 1224–1231. doi: 10.1038/nm.2830
Bigley, A. B., and Simpson, R. J. (2015). NK cells and exercise: implications for cancer immunotherapy and survivorship. Discov. Med. 19, 433–445.
Bissonnette, R., Papp, K. A., Poulin, Y., Gooderham, M., Raman, M., Mallbris, L., et al. (2016). Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br. J. Dermatol. 175, 902–911. doi: 10.1111/bjd.14871
Borsellino, N., Belldegrun, A., and Bonavida, B. (1995). Endogenous interleukin 6 is a resistance factor for cis-diamminedichloroplatinum and etoposide-mediated cytotoxicity of human prostate carcinoma cell lines. Cancer Res. 55, 4633–4639.
Boursi, B., Haynes, K., Mamtani, R., and Yang, Y. X. (2015). Impact of antibiotic exposure on the risk of colorectal cancer. Pharmacoepidemiol. Drug Saf. 24, 534–542. doi: 10.1002/pds.3765
Bowman, R. L., Klemm, F., Akkari, L., Pyonteck, S. M., Sevenich, L., Quail, D. F., et al. (2016). Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 17, 2445–2459. doi: 10.1016/j.celrep.2016.10.052
Brentnall, T. A., Crispin, D. A., Rabinovitch, P. S., Haggitt, R. C., Rubin, C. E., Stevens, A. C., et al. (1994). Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology 107, 369–378. doi: 10.1016/0016-5085(94)90161-9
Bronte, V., Brandau, S., Chen, S. H., Colombo, M. P., Frey, A. B., Greten, T. F., et al. (2016). Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 7:12150. doi: 10.1038/ncomms12150
Buchert, M., Burns, C. J., and Ernst, M. (2016). Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 35, 939–951. doi: 10.1038/onc.2015.150
Burkett, P. R., Koka, R., Chien, M., Chai, S., Boone, D. L., and Ma, A. (2004). Coordinate expression and trans presentation of interleukin (IL)-15Rα and IL-15 supports natural killer cell and memory CD8+ T cell homeostasis. J. Exp. Med. 200, 825–834. doi: 10.1084/jem.20041389
Buscarlet, M., Provost, S., Zada, Y. F., Barhdadi, A., Bourgoin, V., Lépine, G., et al. (2017). DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 130, 753–762. doi: 10.1182/blood-2017-04-777029
Busque, L., Patel, J. P., Figueroa, M. E., Vasanthakumar, A., Provost, S., Hamilou, Z., et al. (2012). Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 44, 1179–1181. doi: 10.1038/ng.2413
Cancer Genome Atlas Research, N. (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. doi: 10.1038/nature10166
Cannarile, M. A., Weisser, M., Jacob, W., Jegg, A. M., Ries, C. H., and Rüttinger, D. (2017). Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5, 53. doi: 10.1186/s40425-017-0257-y
Carmona-Fontaine, C., Deforet, M., Akkari, L., Thompson, C. B., Joyce, J. A., and Xavier, J. B. (2017). Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. U.S.A. 114, 2934–2939. doi: 10.1073/pnas.1700600114
Casazza, A., Laoui, D., Wenes, M., Rizzolio, S., Bassani, N., Mambretti, M., et al. (2013). Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24, 695–709. doi: 10.1016/j.ccr.2013.11.007
Cataisson, C., Salcedo, R., Hakim, S., Moffitt, B. A., Wright, L., Yi, M., et al. (2012). IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. J. Exp. Med. 209, 1689–1702. doi: 10.1084/jem.20101355
Catanzaro, J. M., Sheshadri, N., Pan, J. A., Sun, Y., Shi, C., Li, J., et al. (2014). Oncogenic ras induces inflammatory cytokine production by upregulating the squamous cell carcinoma antigens SerpinB3/B4. Nat. Commun. 5, 3729. doi: 10.1038/ncomms4729
Chae, W. J., and Bothwell, A. L. (2015). Spontaneous intestinal tumorigenesis in Apc (/Min+) mice requires altered T cell development with IL-17A. J. Immunol. Res. 2015:860106. doi: 10.1155/2015/860106
Chaput, N., Lepage, P., Coutzac, C., Soularue, E., Le Roux, K., Monot, C., et al. (2017). Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 28, 1368–1379. doi: 10.1093/annonc/mdx108
Cheon, H., Borden, E. C., and Stark, G. R. (2014). Interferons and their stimulated genes in the tumor microenvironment. Semin. Oncol. 41, 156–173. doi: 10.1053/j.seminoncol.2014.02.002
Chia, W. K., Ali, R., and Toh, H. C. (2012). Aspirin as adjuvant therapy for colorectal cancer–reinterpreting paradigms. Nat. Rev. Clin. Oncol. 9, 561–570. doi: 10.1038/nrclinonc.2012.137
Chiba, T., Marusawa, H., Seno, H., and Watanabe, N. (2008). Mechanism for gastric cancer development by Helicobacter pylori infection. J. Gastroenterol. Hepatol. 23, 1175–1181. doi: 10.1111/j.1440-1746.2008.05472.x
Cinti, S., Mitchell, G., Barbatelli, G., Murano, I., Ceresi, E., Faloia, E., et al. (2005). Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 46, 2347–2355. doi: 10.1194/jlr.M500294-JLR200
Cleary, M. P., and Grossmann, M. E. (2009). Minireview: obesity and breast cancer: the estrogen connection. Endocrinology 150, 2537–2542. doi: 10.1210/en.2009-0070
Colegio, O. R., Chu, N. Q., Szabo, A. L., Chu, T., Rhebergen, A. M., Jairam, V., et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. doi: 10.1038/nature13490
Conley, S. J., Gheordunescu, E., Kakarala, P., Newman, B., Korkaya, H., Heath, A. N., et al. (2012). Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. U.S.A. 109, 2784–2789. doi: 10.1073/pnas.1018866109
Conze, D., Weiss, L., Regen, P. S., Bhushan, A., Weaver, D., Johnson, P., et al. (2001). Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 61, 8851–8858.
Cooks, T., Harris, C. C., and Oren, M. (2014). Caught in the cross fire: p53 in inflammation. Carcinogenesis 35, 1680–1690. doi: 10.1093/carcin/bgu134
Cooks, T., Pateras, I. S., Tarcic, O., Solomon, H., Schetter, A. J., Wilder, S., et al. (2013). Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 23, 634–646. doi: 10.1016/j.ccr.2013.03.022
Coombs, C. C., Zehir, A., Devlin, S. M., Kishtagari, A., Syed, A., Jonsson, P., et al. (2017). Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374.e374–382.e374. doi: 10.1016/j.stem.2017.07.010
Coppé, J. P., Patil, C. K., Rodier, F., Sun, Y., Muñoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868. doi: 10.1371/journal.pbio.0060301
Coussens, L. M., and Werb, Z. (2002). Inflammation and cancer. Nature 420, 860–867. doi: 10.1038/nature01322
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J., and Der, C. J. (2014). Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851. doi: 10.1038/nrd4389
Cui, W. Y., and Li, M. D. (2010). Nicotinic modulation of innate immune pathways via alpha7 nicotinic acetylcholine receptor. J. Neuroimmune Pharmacol. 5, 479–488. doi: 10.1007/s11481-010-9210-2
Cui, Y., and Guo, G. (2016). Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int. J. Mol. Sci. 17:E1942. doi: 10.3390/ijms17111942
Daniluk, J., Liu, Y., Deng, D., Chu, J., Huang, H., Gaiser, S., et al. (2012). An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Invest. 122, 1519–1528. doi: 10.1172/JCI59743
Das, S., and Marsden, P. A. (2013). Angiogenesis in glioblastoma. N. Engl. J. Med. 369, 1561–1563. doi: 10.1056/NEJMcibr1309402
De Boeck, A., Hendrix, A., Maynard, D., Van Bockstal, M., Daniëls, A., Pauwels, P., et al. (2013). Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics 13, 379–388. doi: 10.1002/pmic.201200179
De Palma, M., Biziato, D., and Petrova, T. V. (2017). Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 17, 457–474. doi: 10.1038/nrc.2017.51
Deininger, M., Radich, J., Burn, T. C., Huber, R., Paranagama, D., and Verstovsek, S. (2015). The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 126, 1551–1554. doi: 10.1182/blood-2015-03-635235
DeNardo, D. G., Brennan, D. J., Rexhepaj, E., Ruffell, B., Shiao, S. L., Madden, S. F., et al. (2011). Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1, 54–67. doi: 10.1158/2159-8274.CD-10-0028
Di Minin, G., Bellazzo, A., Dal Ferro, M., Chiaruttini, G., Nuzzo, S., Bicciato, S., et al. (2014). Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 56, 617–629. doi: 10.1016/j.molcel.2014.10.013
Dieli-Conwright, C. M., Courneya, K. S., Demark-Wahnefried, W., Sami, N., Lee, K., Buchanan, T. A., et al. (2018). Effects of aerobic and resistance exercise on metabolic syndrome, sarcopenic obesity, and circulating biomarkers in overweight or obese survivors of breast cancer: a randomized controlled Trial. J. Clin. Oncol. 36, 875–883. doi: 10.1200/JCO.2017.75.7526
Donnarumma, E., Fiore, D., Nappa, M., Roscigno, G., Adamo, A., Iaboni, M., et al. (2017). Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 8, 19592–19608. doi: 10.18632/oncotarget.14752
Dvorak, H. F. (1986). Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659. doi: 10.1056/NEJM198612253152606
Eghtedar, A., Verstovsek, S., Estrov, Z., Burger, J., Cortes, J., Bivins, C., et al. (2012). Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood 119, 4614–4618. doi: 10.1182/blood-2011-12-400051
Elenbaas, B., and Weinberg, R. A. (2001). Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 264, 169–184. doi: 10.1006/excr.2000.5133
Elinav, E., Nowarski, R., Thaiss, C. A., Hu, B., Jin, C., and Flavell, R. A. (2013). Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771. doi: 10.1038/nrc3611
Elliott, M. J., Maini, R. N., Feldmann, M., Long-Fox, A., Charles, P., Katsikis, P., et al. (1993). Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 36, 1681–1690. doi: 10.1002/art.1780361206
Erez, N., Truitt, M., Olson, P., Arron, S. T., and Hanahan, D. (2010). Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 17, 135–147. doi: 10.1016/j.ccr.2009.12.041
Falchi, L., Newberry, K. J., and Verstovsek, S. (2015). New therapeutic approaches in polycythemia vera. Clin. Lymphoma Myeloma Leuk. 15(Suppl.), S27–S33. doi: 10.1016/j.clml.2015.02.013
Farkona, S., Diamandis, E. P., and Blasutig, I. M. (2016). Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 14:73. doi: 10.1186/s12916-016-0623-5
Farmaki, E., Chatzistamou, I., Bourlis, P., Santoukou, E., Trimis, G., Papavassiliou, A. G., et al. (2012). Selection of p53-deficient stromal cells in the tumor microenvironment. Genes Cancer 3, 592–598. doi: 10.1177/1947601912474002
Feig, C., Jones, J. O., Kraman, M., Wells, R. J., Deonarine, A., Chan, D. S., et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U.S.A. 110, 20212–20217. doi: 10.1073/pnas.1320318110
Feldmann, M. (2008). Many cytokines are very useful therapeutic targets in disease. J. Clin. Invest. 118, 3533–3536. doi: 10.1172/JCI37346
Feldmann, M., and Maini, R. N. (2001). Anti-TNF α therapy of rheumatoid arthritis: what have we learned? Annu. Rev. Immunol. 19, 163–196. doi: 10.1146/annurev.immunol.19.1.163
Ferrara, N., and Adamis, A. P. (2016). Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 15, 385–403. doi: 10.1038/nrd.2015.17
Fiedler, U., Reiss, Y., Scharpfenecker, M., Grunow, V., Koidl, S., Thurston, G., et al. (2006). Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat. Med. 12, 235–239. doi: 10.1038/nm1351
Flegal, K. M., Carroll, M. D., Ogden, C. L., and Curtin, L. R. (2002). Prevalence and trends in obesity among US adults, 1999-2008. JAMA 303, 235–241. doi: 10.1001/jama.2009.2014
Franklin, R. A., Liao, W., Sarkar, A., Kim, M. V., Bivona, M. R., Liu, K., et al. (2014). The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925. doi: 10.1126/science.1252510
Frassanito, M. A., Cusmai, A., Iodice, G., and Dammacco, F. (2001). Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood 97, 483–489. doi: 10.1182/blood.V97.2.483
Freedman, N. D., Silverman, D. T., Hollenbeck, A. R., Schatzkin, A., and Abnet, C. C. (2011). Association between smoking and risk of bladder cancer among men and women. JAMA 306, 737–745. doi: 10.1001/jama.2011.1142
Fuentes-Mattei, E., Velazquez-Torres, G., Phan, L., Zhang, F., Chou, P. C., Shin, J. H., et al. (2014). Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J. Natl. Cancer Inst. 106:dju158. doi: 10.1093/jnci/dju158
Fukuyama, T., Ehling, S., Cook, E., and Bäumer, W. (2015). Topically administered janus-kinase inhibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatory responses in a model of allergic dermatitis. J. Pharmacol. Exp. Ther. 354, 394–405. doi: 10.1124/jpet.115.223784
Fuster, J. J., MacLauchlan, S., Zuriaga, M. A., Polackal, M. N., Ostriker, A. C., Chakraborty, R., et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847. doi: 10.1126/science.aag1381
Galon, J., Costes, A., Sanchez-Cabo, F., Kirilovsky, A., Mlecnik, B., Lagorce-Pagès, C., et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. doi: 10.1126/science.1129139
Genovese, G., Kähler, A. K., Handsaker, R. E., Lindberg, J., Rose, S. A., Bakhoum, S. F., et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487. doi: 10.1056/NEJMoa1409405
Geyer, H. L., Dueck, A. C., Scherber, R. M., and Mesa, R. A. (2015). Impact of inflammation on myeloproliferative neoplasm symptom development. Mediators Inflamm. 2015, 284706. doi: 10.1155/2015/284706
Ghandadi, M., and Sahebkar, A. (2016). Interleukin-6: a critical cytokine in cancer multidrug resistance. Curr. Pharm. Des. 22, 518–526. doi: 10.2174/1381612822666151124234417
Ghoreschi, K., Jesson, M. I., Li, X., Lee, J. L., Ghosh, S., Alsup, J. W., et al. (2011). Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 186, 4234–4243. doi: 10.4049/jimmunol.1003668
Glass, C. K., and Natoli, G. (2016). Molecular control of activation and priming in macrophages. Nat. Immunol. 17, 26–33. doi: 10.1038/ni.3306
Godwin, P., Baird, A. M., Heavey, S., Barr, M. P., O'Byrne, K. J., and Gately, K. (2013). Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 3:120. doi: 10.3389/fonc.2013.00120
Goel, S., Wyrwicz, L., Choi, M., Coveler, A. L., Ucar, A., Brown, A. W., et al. (2016). Phase III double-blinded, placebo-controlled study of MABp1 for improving survival in metastatic colorectal cancer. J. Clin. Oncol. 34:TPS784. doi: 10.1200/jco.2016.34.4_suppl.tps784
Gomez Perdiguero, E., Klapproth, K., Schulz, C., Busch, K., Azzoni, E., Crozet, L., et al. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. doi: 10.1038/nature13989
Gopalakrishnan, V., Spencer, C. N., Nezi, L., Reuben, A., Andrews, M. C., Karpinets, T. V., et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. doi: 10.1126/science.aan4236
Gounaris, E., Blatner, N. R., Dennis, K., Magnusson, F., Gurish, M. F., Strom, T. B., et al. (2009). T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res. 69, 5490–5497. doi: 10.1158/0008-5472.CAN-09-0304
Greiner, J. W., Hand, P. H., Noguchi, P., Fisher, P. B., Pestka, S., and Schlom, J. (1984). Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte α-interferon treatment. Cancer Res. 44, 3208–3214.
Greten, F. R., and Karin, M. (2004). The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 206, 193–199. doi: 10.1016/j.canlet.2003.08.029
Grivennikov, S. I., and Karin, M. (2011). Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 70(Suppl 1), i104–i108. doi: 10.1136/ard.2010.140145
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. doi: 10.1016/j.cell.2010.01.025
Grivennikov, S. I., Wang, K., Mucida, D., Stewart, C. A., Schnabl, B., Jauch, D., et al. (2012). Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258. doi: 10.1038/nature11465
Groden, J., Thliveris, A., Samowitz, W., Carlson, M., Gelbert, L., Albertsen, H., et al. (1991). Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66, 589–600. doi: 10.1016/0092-8674(81)90021-0
Guo, G., Marrero, L., Rodriguez, P., Del Valle, L., Ochoa, A., and Cui, Y. (2013). Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid-like stromal network. Cancer Res. 73, 1668–1675. doi: 10.1158/0008-5472.CAN-12-3810
Hammerl, D., Smid, M., Timmermans, A. M., Sleijfer, S., Martens, J. W. M., and Debets, R. (2017). Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2017.11.003. [Epub ahead of print].
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Harney, A. S., Arwert, E. N., Entenberg, D., Wang, Y., Guo, P., Qian, B. Z., et al. (2015). Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. 5, 932–943. doi: 10.1158/2159-8290.CD-15-0012
Harris, R. E., Chlebowski, R. T., Jackson, R. D., Frid, D. J., Ascenseo, J. L., Anderson, G., et al. (2003). Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women's Health Initiative. Cancer Res. 63, 6096–6101.
He, W., Luistro, L., Carvajal, D., Smith, M., Nevins, T., Yin, X., et al. (2011). High tumor levels of IL6 and IL8 abrogate preclinical efficacy of the gamma-secretase inhibitor, RO4929097. Mol. Oncol. 5, 292–301. doi: 10.1016/j.molonc.2011.01.001
Hellmann, M., Rizvi, N., Wolchok, J. D., and Chan, T. A. (2016). Genomic profile, smoking, and response to anti-PD-1 therapy in non-small cell lung carcinoma. Mol. Cell Oncol. 3:e1048929. doi: 10.1080/23723556.2015.1048929
Heo, T. H., Wahler, J., and Suh, N. (2016). Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 7, 15460–15473. doi: 10.18632/oncotarget.7102
Hervas-Stubbs, S., Perez-Gracia, J. L., Rouzaut, A., Sanmamed, M. F., Le Bon, A., and Melero, I. (2011). Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 17, 2619–2627. doi: 10.1158/1078-0432.CCR-10-1114
Hill, R., Song, Y., Cardiff, R. D., and Van Dyke, T. (2005). Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 123, 1001–1011. doi: 10.1016/j.cell.2005.09.030
Hong, D. S., Hui, D., Bruera, E., Janku, F., Naing, A., Falchook, G. S., et al. (2014). MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 15, 656–666. doi: 10.1016/S1470-2045(14)70155-X
Hu, M., Yao, J., Cai, L., Bachman, K. E., van den Brûle, F., Velculescu, V., et al. (2005). Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 37, 899–905. doi: 10.1038/ng1596
Hu, N., Wang, C., Hu, Y., Yang, H. H., Kong, L. H., Lu, N., et al. (2006). Genome-wide loss of heterozygosity and copy number alteration in esophageal squamous cell carcinoma using the Affymetrix GeneChip Mapping 10 K array. BMC Genomics 7:299. doi: 10.1186/1471-2164-7-299
Huang, S., Mills, L., Mian, B., Tellez, C., McCarty, M., Yang, X. D., et al. (2002). Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am. J. Pathol. 161, 125–134. doi: 10.1016/S0002-9440(10)64164-8
Hussain, S. P., Amstad, P., Raja, K., Ambs, S., Nagashima, M., Bennett, W. P., et al. (2000). Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 60, 3333–3337.
Iida, N., Dzutsev, A., Stewart, C. A., Smith, L., Bouladoux, N., Weingarten, R. A., et al. (2013). Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970. doi: 10.1126/science.1240527
Ishiguro, K., Yoshida, T., Yagishita, H., Numata, Y., and Okayasu, T. (2006). Epithelial and stromal genetic instability contributes to genesis of colorectal adenomas. Gut 55, 695–702. doi: 10.1136/gut.2005.079459
Iyengar, N. M., Brown, K. A., Zhou, X. K., Gucalp, A., Subbaramaiah, K., Giri, D. D., et al. (2017). Metabolic obesity, adipose inflammation and elevated breast aromatase in women with normal body mass index. Cancer Prev. Res. 10, 235–243. doi: 10.1158/1940-6207.CAPR-16-0314
Iyengar, N. M., Gucalp, A., Dannenberg, A. J., and Hudis, C. A. (2016). Obesity and cancer mechanisms: tumor microenvironment and inflammation. J. Clin. Oncol. 34, 4270–4276. doi: 10.1200/JCO.2016.67.4283
Iyengar, N. M., Hudis, C. A., and Dannenberg, A. J. (2015). Obesity and cancer: local and systemic mechanisms. Annu. Rev. Med. 66, 297–309. doi: 10.1146/annurev-med-050913-022228
Iyengar, N. M., Kochhar, A., Morris, P. G., Morris, L. G., Zhou, X. K., Ghossein, R. A. G., et al. (2014). Impact of obesity on the survival of patients with early-stage squamous cell carcinoma of the oral tongue. Cancer 120, 983–991. doi: 10.1002/cncr.28532
Jackstadt, R., and Sansom, O. J. (2016). Mouse models of intestinal cancer. J. Pathol. 238, 141–151. doi: 10.1002/path.4645
Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498. doi: 10.1056/NEJMoa1408617
Jaiswal, S., Natarajan, P., and Ebert, B. L. (2017a). Clonal hematopoiesis and atherosclerosis. N. Engl. J. Med. 377, 1401–1402. doi: 10.1056/NEJMc1710381
Jaiswal, S., Natarajan, P., Silver, A. J., Gibson, C. J., Bick, A. G., Shvartz, E., et al. (2017b). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121. doi: 10.1056/NEJMoa1701719
Ji, B., Tsou, L., Wang, H., Gaiser, S., Chang, D. Z., Daniluk, J., et al. (2009). Ras activity levels control the development of pancreatic diseases. Gastroenterology 137, 1072–1082, 1082.e1–6. doi: 10.1053/j.gastro.2009.05.052
Johnson, C. C., Hayes, R. B., Schoen, R. E., Gunter, M. J., and Huang, W. Y. (2010). Non-steroidal anti-inflammatory drug use and colorectal polyps in the prostate, lung, colorectal, and ovarian cancer screening trial. Am. J. Gastroenterol. 105, 2646–2655. doi: 10.1038/ajg.2010.349
Jones, V. S., Huang, R. Y., Chen, L. P., Chen, Z. S., Fu, L., and Huang, R. P. (2016). Cytokines in cancer drug resistance: cues to new therapeutic strategies. Biochim. Biophys. Acta 1865, 255–265. doi: 10.1016/j.bbcan.2016.03.005
Joshua, A. M., Vukovic, B., Braude, I., Hussein, S., Zielenska, M., Srigley, J., et al. (2007). Telomere attrition in isolated high-grade prostatic intraepithelial neoplasia and surrounding stroma is predictive of prostate cancer. Neoplasia 9, 81–89. doi: 10.1593/neo.06745
Kalluri, R. (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. doi: 10.1038/nrc.2016.73
Khandelwal, P., Teusink-Cross, A., Davies, S. M., Nelson, A. S., Dandoy, C. E., El-Bietar, J., et al. (2017). Ruxolitinib as salvage therapy in steroid-refractory acute graft-versus-host disease in pediatric hematopoietic stem cell transplant patients. Biol. Blood Marrow Transplant. 23, 1122–1127. doi: 10.1016/j.bbmt.2017.03.029
Kim, G., Ouzounova, M., Quraishi, A. A., Davis, A., Tawakkol, N., Clouthier, S. G., et al. (2015). SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene 34, 671–680. doi: 10.1038/onc.2014.4
Kim, H. S., Lim, H. S., Lee, S. H., Lee, J. W., Nam, S. W., Park, W. S., et al. (2006). Mitochondrial microsatellite instability of colorectal cancer stroma. Int. J. Cancer 119, 2607–2611. doi: 10.1002/ijc.22244
Kioi, M., Vogel, H., Schultz, G., Hoffman, R. M., Harsh, G. R., and Brown, J. M. (2010). Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 120, 694–705. doi: 10.1172/JCI40283
Kirkwood, J. (2002). Cancer immunotherapy: the interferon-alpha experience. Semin. Oncol. 29, 18–26. doi: 10.1053/sonc.2002.33078
Kleppe, M., Comen, E., Wen, H. Y., Bastian, L., Blum, B., Rapaport, F. T., et al. (2015a). Somatic mutations in leukocytes infiltrating primary breast cancers. NPJ Breast Cancer 1, 15005. doi: 10.1038/npjbcancer.2015.5
Kleppe, M., Kwak, M., Koppikar, P., Riester, M., Keller, M., Bastian, L., et al. (2015b). JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 5, 316–331. doi: 10.1158/2159-8290.CD-14-0736
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Kloepper, J., Riedemann, L., Amoozgar, Z., Seano, G., Susek, K., Yu, V., et al. (2016). Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. U.S.A. 113, 4476–4481. doi: 10.1073/pnas.1525360113
Knüpfer, H., and Preiss, R. (2007). Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res. Treat. 102, 129–135. doi: 10.1007/s10549-006-9328-3
Koelwyn, G. J., Quail, D. F., Zhang, X., White, R. M., and Jones, L. W. (2017). Exercise-dependent regulation of the tumour microenvironment. Nat. Rev. Cancer 17, 620–632. doi: 10.1038/nrc.2017.78
Koelwyn, G. J., Wennerberg, E., Demaria, S., and Jones, L. W. (2015). Exercise in regulation of inflammation-immune axis function in cancer initiation and progression. Oncology 29, 908–920, 922.
Komarova, E. A., Diatchenko, L., Rokhlin, O. W., Hill, J. E., Wang, Z. J., Krivokrysenko, V. I., et al. (1998). Stress-induced secretion of growth inhibitors: a novel tumor suppressor function of p53. Oncogene 17, 1089–1096. doi: 10.1038/sj.onc.1202303
Komarova, E. A., Krivokrysenko, V., Wang, K., Neznanov, N., Chernov, M. V., Komarov, P. G., et al. (2005). p53 is a suppressor of inflammatory response in mice. FASEB J. 19, 1030–1032. doi: 10.1096/fj.04-3213fje
Kontzias, A., Kotlyar, A., Laurence, A., Changelian, P., and O'Shea, J. J. (2012). Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr. Opin. Pharmacol. 12, 464–470. doi: 10.1016/j.coph.2012.06.008
Korkaya, H., Kim, G. I., Davis, A., Malik, F., Henry, N. L., Ithimakin, S., et al. (2012). Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 47, 570–584. doi: 10.1016/j.molcel.2012.06.014
Kraman, M., Bambrough, P. J., Arnold, J. N., Roberts, E. W., Magiera, L., Jones, J. O., et al. (2010). Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830. doi: 10.1126/science.1195300
Krock, B. L., Skuli, N., and Simon, M. C. (2011). Hypoxia-induced angiogenesis: good and evil. Genes Cancer 2, 1117–1133. doi: 10.1177/1947601911423654
Kroemer, G., and Zitvogel, L. (2018). Cancer immunotherapy in 2017: the breakthrough of the microbiota. Nat. Rev. Immunol. 18, 87–88. doi: 10.1038/nri.2018.4
Kudlacz, E., Conklyn, M., Andresen, C., Whitney-Pickett, C., and Changelian, P. (2008). The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur. J. Pharmacol. 582, 154–161. doi: 10.1016/j.ejphar.2007.12.024
Kumar, V., Donthireddy, L., Marvel, D., Condamine, T., Wang, F., Lavilla-Alonso, S., et al. (2017). Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell 32, 654.e655–668.e655. doi: 10.1016/j.ccell.2017.10.005
Kuper, H., Adami, H. O., and Trichopoulos, D. (2000). Infections as a major preventable cause of human cancer. J. Intern. Med. 248, 171–183. doi: 10.1046/j.1365-2796.2000.00742.x
Kurose, K., Gilley, K., Matsumoto, S., Watson, P. H., Zhou, X. P., and Eng, C. (2002). Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 32, 355–357. doi: 10.1038/ng1013
Kurose, K., Hoshaw-Woodard, S., Adeyinka, A., Lemeshow, S., Watson, P. H., and Eng, C. (2001). Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: clues to tumour-microenvironment interactions. Hum. Mol. Genet. 10, 1907–1913. doi: 10.1093/hmg/10.18.1907
Lasry, A., Zinger, A., and Ben-Neriah, Y. (2016). Inflammatory networks underlying colorectal cancer. Nat. Immunol. 17, 230–240. doi: 10.1038/ni.3384
Le, D. T., Uram, J. N., Wang, H., Bartlett, B. R., Kemberling, H., Eyring, A. D., et al. (2015). PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520. doi: 10.1056/NEJMoa1500596
Lee, H. J., Zhuang, G., Cao, Y., Du, P., Kim, H. J., and Settleman, J. (2014). Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 26, 207–221. doi: 10.1016/j.ccr.2014.05.019
Lee, T. L., Yang, X. P., Yan, B., Friedman, J., Duggal, P., Bagain, L., et al. (2007). A novel nuclear factor-kappaB gene signature is differentially expressed in head and neck squamous cell carcinomas in association with TP53 status. Clin. Cancer Res. 13, 5680–5691. doi: 10.1158/1078-0432.CCR-07-0670
Leoni, C., Montagner, S., Rinaldi, A., Bertoni, F., Polletti, S., Balestrieri, C., et al. (2017). Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. U.S.A. 114, E1490–E1499. doi: 10.1073/pnas.1616420114
Leslie, N. R., and Downes, C. P. (2004). PTEN function: how normal cells control it and tumour cells lose it. Biochem. J. 382, 1–11. doi: 10.1042/BJ20040825
Li, F., and Sethi, G. (2010). Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 1805, 167–180. doi: 10.1016/j.bbcan.2010.01.002
Li, P., Shan, J. X., Chen, X. H., Zhang, D., Su, L. P., Huang, X. Y., et al. (2015). Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res. 25, 588–603. doi: 10.1038/cr.2015.51
Li, X., Zhang, Q., Ding, Y., Liu, Y., Zhao, D., Zhao, K., et al. (2016). Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol. 17, 806–815. doi: 10.1038/ni.3464
Ligibel, J. A., Alfano, C. M., Courneya, K. S., Demark-Wahnefried, W., Burger, R. A., Chlebowski, R. T., et al. (2014). American Society of Clinical Oncology position statement on obesity and cancer. J. Clin. Oncol. 32, 3568–3574. doi: 10.1200/JCO.2014.58.4680
Lin, K. T., and Wang, L. H. (2016). New dimension of glucocorticoids in cancer treatment. Steroids 111, 84–88. doi: 10.1016/j.steroids.2016.02.019
Lin, Y., Bai, L., Chen, W., and Xu, S. (2010). The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 14, 45–55. doi: 10.1517/14728220903431069
Linderholm, B., Lindh, B., Tavelin, B., Grankvist, K., and Henriksson, R. (2000). p53 and vascular-endothelial-growth-factor (VEGF) expression predicts outcome in 833 patients with primary breast carcinoma. Int. J. Cancer 89, 51–62. doi: 10.1002/(SICI)1097-0215(20000120)89:1<51::AID-IJC9>3.0.CO;2-8
Liu, L. Y., Craiglow, B. G., Dai, F., and King, B. A. (2017). Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J. Am. Acad. Dermatol. 76, 22–28. doi: 10.1016/j.jaad.2016.09.007
LoConte, N. K., Brewster, A. M., Kaur, J. S., Merrill, J. K., and Alberg, A. J. (2018). Alcohol and cancer: a statement of the American Society of Clinical Oncology. J. Clin. Oncol. 36, 83–93. doi: 10.1200/JCO.2017.76.1155
Longo, V., Brunetti, O., Gnoni, A., Cascinu, S., Gasparini, G., Lorusso, V., et al. (2016). Angiogenesis in pancreatic ductal adenocarcinoma: a controversial issue. Oncotarget 7, 58649–58658. doi: 10.18632/oncotarget.10765
Lu, J., Ye, X., Fan, F., Xia, L., Bhattacharya, R., Bellister, S., et al. (2013). Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 23, 171–185. doi: 10.1016/j.ccr.2012.12.021
Lu, L. M., Zavitz, C. C., Chen, B., Kianpour, S., Wan, Y., and Stämpfli, M. R. (2007). Cigarette smoke impairs NK cell-dependent tumor immune surveillance. J. Immunol. 178, 936–943. doi: 10.4049/jimmunol.178.2.936
Lu, M., Zhang, W., Li, Y., Berenzon, D., Wang, X., Wang, J., et al. (2010). Interferon-α targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp. Hematol. 38, 472–480. doi: 10.1016/j.exphem.2010.03.005
Lujambio, A., Akkari, L., Simon, J., Grace, D., Tschaharganeh, D. F., Bolden, J. E., et al. (2013). Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460. doi: 10.1016/j.cell.2013.03.020
Lust, J. A., Lacy, M. Q., Zeldenrust, S. R., Dispenzieri, A., Gertz, M. A., Witzig, T. E., et al. (2009). Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{β}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 84, 114–122. doi: 10.4065/84.2.114
Luttges, J., Diederichs, A., Menke, M. A., Vogel, I., Kremer, B., and Klöppel, G. (2000). Ductal lesions in patients with chronic pancreatitis show K-ras mutations in a frequency similar to that in the normal pancreas and lack nuclear immunoreactivity for p53. Cancer 88, 2495–2504. doi: 10.1002/1097-0142(20000601)88:11<2495::AID-CNCR10>3.0.CO;2-B
Mackay-Wiggan, J., Jabbari, A., Nguyen, N., Cerise, J. E., Clark, C., Ulerio, G., et al. (2016). Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight 1:e89790. doi: 10.1172/jci.insight.89790
Man, Y., Mannion, C., Kuhls, E., Moinfar, F., Bratthauer, G. L., Albores-Saavedra, J., et al. (2001). Allelic losses at 3p and 11p are detected in both epithelial and stromal components of cervical small-cell neuroendocrine carcinoma. Appl. Immunohistochem. Mol. Morphol. 9, 340–345. doi: 10.1097/00129039-200112000-00009
Maxwell, P. J., Neisen, J., Messenger, J., and Waugh, D. J. (2014). Tumor-derived CXCL8 signaling augments stroma-derived CCL2-promoted proliferation and CXCL12-mediated invasion of PTEN-deficient prostate cancer cells. Oncotarget 5, 4895–4908. doi: 10.18632/oncotarget.2052
McGuire, S. (2016). World Cancer Report 2014. World Health Organization; International Agency for Research on Cancer, Geneva.
McKerrell, T., Park, N., Moreno, T., Grove, C. S., Ponstingl, H., Stephens, J., et al. (2015). Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 10, 1239–1245. doi: 10.1016/j.celrep.2015.02.005
Michalaki, V., Syrigos, K., Charles, P., and Waxman, J. (2004). Serum levels of IL-6 and TNF-α correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 90, 2312–2316. doi: 10.1038/sj.bjc.6601814
Milara, J., and Cortijo, J. (2012). Tobacco, inflammation, and respiratory tract cancer. Curr. Pharm. Des. 18, 3901–3938. doi: 10.2174/138161212802083743
Moinfar, F., Man, Y. G., Arnould, L., Bratthauer, G. L., Ratschek, M., and Tavassoli, F. A. (2000). Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 60, 2562–2566.
Mondet, J., Hussein, K., and Mossuz, P. (2015). Circulating cytokine levels as markers of inflammation in philadelphia negative myeloproliferative neoplasms: diagnostic and prognostic interest. Mediators Inflamm. 2015:670580. doi: 10.1155/2015/670580
Moore, P. S., and Chang, Y. (2010). Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 10, 878–889. doi: 10.1038/nrc2961
Mori, Y., Ikeda, K., Inomata, T., Yoshimoto, G., Fujii, N., Ago, H., et al. (2016). Ruxolitinib treatment for GvHD in patients with myelofibrosis. Bone Marrow Transplant. 51, 1584–1587. doi: 10.1038/bmt.2016.256
Morris, P. G., Hudis, C. A., Giri, D., Morrow, M., Falcone, D. J., Zhou, X. K., et al. (2011). Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev. Res. 4, 1021–1029. doi: 10.1158/1940-6207.CAPR-11-0110
Moser, A. R., Mattes, E. M., Dove, W. F., Lindstrom, M. J., Haag, J. D., and Gould, M. N. (1993). ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc. Natl. Acad. Sci. U.S.A. 90, 8977–8981. doi: 10.1073/pnas.90.19.8977
Moser, A. R., Pitot, H. C., and Dove, W. F. (1990). A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247, 322–324. doi: 10.1126/science.2296722
Moskovits, N., Kalinkovich, A., Bar, J., Lapidot, T., and Oren, M. (2006). p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 66, 10671–10676. doi: 10.1158/0008-5472.CAN-06-2323
Murray, P. J., Allen, J. E., Biswas, S. K., Fisher, E. A., Gilroy, D. W., Goerdt, S., et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. doi: 10.1016/j.immuni.2014.06.008
Nakanishi, C., and Toi, M. (2005). Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 5, 297–309. doi: 10.1038/nrc1588
Nguyen, K. B., Salazar-Mather, T. P., Dalod, M. Y., Van Deusen, J. B., Wei, X. Q., Liew, F. Y., et al. (2002). Coordinated and distinct roles for IFN-α β, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169, 4279–4287. doi: 10.4049/jimmunol.169.8.4279
Norton, S. E., Ward-Hartstonge, K. A., Taylor, E. S., and Kemp, R. A. (2015). Immune cell interplay in colorectal cancer prognosis. World J. Gastrointest. Oncol. 7, 221–232. doi: 10.4251/wjgo.v7.i10.221
Noy, R., and Pollard, J. W. (2014). Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61. doi: 10.1016/j.immuni.2014.06.010
Öhlund, D., Elyada, E., and Tuveson, D. (2014). Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 211, 1503–1523. doi: 10.1084/jem.20140692
Okabe, Y., and Medzhitov, R. (2016). Tissue biology perspective on macrophages. Nat. Immunol. 17, 9–17. doi: 10.1038/ni.3320
Olive, K. P., Jacobetz, M. A., Davidson, C. J., Gopinathan, A., McIntyre, D., Honess, D., et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. doi: 10.1126/science.1171362
Olson, O. C., and Joyce, J. A. (2015). Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 15, 712–729. doi: 10.1038/nrc4027
Olson, O. C., Kim, H., Quail, D. F., Foley, E. A., and Joyce, J. A. (2017). Tumor-associated macrophages suppress the cytotoxic activity of antimitotic agents. Cell Rep. 19, 101–113. doi: 10.1016/j.celrep.2017.03.038
O'Shea, J. J., Kontzias, A., Yamaoka, K., Tanaka, Y., and Laurence, A. (2013). Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 72(Suppl. 2), ii111–ii115. doi: 10.1136/annrheumdis-2012-202576
Oskarsson, T., Acharyya, S., Zhang, X. H., Vanharanta, S., Tavazoie, S. F., Morris, P. G., et al. (2011). Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874. doi: 10.1038/nm.2379
Palumbo, A. Jr., Da Costa Nde, O., Bonamino, M. H., Pinto, L. F., and Nasciutti, L. E. (2015). Genetic instability in the tumor microenvironment: a new look at an old neighbor. Mol. Cancer 14, 145. doi: 10.1186/s12943-015-0409-y
Parker, B. S., Rautela, J., and Hertzog, P. J. (2016). Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144. doi: 10.1038/nrc.2016.14
Parkin, D. M. (2006). The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 118, 3030–3044. doi: 10.1002/ijc.21731
Passamonti, F., Maffioli, M., Caramazza, D., and Cazzola, M. (2011). Myeloproliferative neoplasms: from JAK2 mutations discovery to JAK2 inhibitor therapies. Oncotarget 2, 485–490. doi: 10.18632/oncotarget.281
Paterson, R. F., Ulbright, T. M., MacLennan, G. T., Zhang, S., Pan, C. X., Sweeney, C. J., et al. (2003). Molecular genetic alterations in the laser-capture-microdissected stroma adjacent to bladder carcinoma. Cancer 98, 1830–1836. doi: 10.1002/cncr.11747
Patocs, A., Zhang, L., Xu, Y., Weber, F., Caldes, T., Mutter, G. L., et al. (2007). Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 357, 2543–2551. doi: 10.1056/NEJMoa071825
Paunescu, V., Bojin, F. M., Tatu, C. A., Gavriliuc, O. I., Rosca, A., Gruia, A. T., et al. (2011). Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J. Cell. Mol. Med. 15, 635–646. doi: 10.1111/j.1582-4934.2010.01044.x
Pemmaraju, N., Kantarjian, H., Kadia, T., Cortes, J., Borthakur, G., Newberry, K., et al. (2015). A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 15, 171–176. doi: 10.1016/j.clml.2014.08.003
Peng, D. F., Kanai, Y., Sawada, M., Ushijima, S., Hiraoka, N., Kosuge, T., et al. (2005). Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci. 96, 403–408. doi: 10.1111/j.1349-7006.2005.00071.x
Peng, W., Chen, J. Q., Liu, C., Malu, S., Creasy, C., Tetzlaff, M. T., et al. (2016). Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 6, 202–216. doi: 10.1158/2159-8290.CD-15-0283
Perretti, M., and Ahluwalia, A. (2000). The microcirculation and inflammation: site of action for glucocorticoids. Microcirculation 7, 147–161. doi: 10.1111/j.1549-8719.2000.tb00117.x
Peterson, T. E., Kirkpatrick, N. D., Huang, Y., Farrar, C. T., Marijt, K. A., Kloepper, J., et al. (2016). Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. U.S.A. 113, 4470–4475. doi: 10.1073/pnas.1525349113
Pfeifer, G. P., Denissenko, M. F., Olivier, M., Tretyakova, N., Hecht, S. S., and Hainaut, P. (2002). Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21, 7435–7451. doi: 10.1038/sj.onc.1205803
Platanias, L. C. (2013). Interferons and their antitumor properties. J. Interferon Cytokine Res. 33, 143–144. doi: 10.1089/jir.2013.0019
Pleasance, E. D., Stephens, P. J., O'Meara, S., McBride, D. J., Meynert, A., Jones, D., et al. (2010). A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463, 184–190. doi: 10.1038/nature08629
Plimack, E. R., Lorusso, P. M., McCoon, P., Tang, W., Krebs, A. D., Curt, G., et al. (2013). AZD1480: a phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist 18, 819–820. doi: 10.1634/theoncologist.2013-0198
Postow, M. A., and Hellmann, M. D. (2018). Adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 378, 1165. doi: 10.1056/NEJMc1801663
Postow, M. A., Sidlow, R., and Hellmann, M. D. (2018). Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 378, 158–168. doi: 10.1056/NEJMra1703481
Propper, D. J., Chao, D., Braybrooke, J. P., Bahl, P., Thavasu, P., Balkwill, F., et al. (2003). Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin. Cancer Res. 9, 84–92.
Pyonteck, S. M., Akkari, L., Schuhmacher, A. J., Bowman, R. L., Sevenich, L., Quail, D. F., et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272. doi: 10.1038/nm.3337
Qian, B. Z., Li, J., Zhang, H., Kitamura, T., Zhang, J., Campion, L. R., et al. (2011). CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225. doi: 10.1038/nature10138
Quail, D. F., and Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. doi: 10.1038/nm.3394
Quail, D. F., Bowman, R. L., Akkari, L., Quick, M. L., Schuhmacher, A. J., Huse, J. T., et al. (2016). The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352:aad3018. doi: 10.1126/science.aad3018
Quintás-Cardama, A., Kantarjian, H., Cortes, J., and Verstovsek, S. (2011). Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat. Rev. Drug Discov. 10, 127–140. doi: 10.1038/nrd3264
Raedler, L. A. (2015). Jakafi (Ruxolitinib): first FDA-approved medication for the treatment of patients with polycythemia vera. Am. Health Drug Benefits 8, 75–79.
Rahnamoun, H., Lu, H., Duttke, S. H., Benner, C., Glass, C. K., and Lauberth, S. M. (2017). Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat. Commun. 8, 754. doi: 10.1038/s41467-017-01117-y
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017a). Antiinflammatory therapy with canakinumab for Atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131. doi: 10.1056/NEJMoa1707914
Ridker, P. M., MacFadyen, J. G., Thuren, T., Everett, B. M., Libby, P., Glynn, R. J., et al. (2017b). Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842. doi: 10.1016/S0140-6736(17)32247-X
Robinson, B. D., and Jones, J. G. (2009). Tumor microenvironment of metastasis (TMEM): a novel tissue-based assay for metastatic risk in breast cancer. Future Oncol. 5, 919–921. doi: 10.2217/fon.09.79
Rock, C. L., Flatt, S. W., Thomson, C. A., Stefanick, M. L., Newman, V. A., Jones, L. A., et al. (2004). Effects of a high-fiber, low-fat diet intervention on serum concentrations of reproductive steroid hormones in women with a history of breast cancer. J. Clin. Oncol. 22, 2379–2387. doi: 10.1200/JCO.2004.09.025
Rønnov-Jessen, L., Petersen, O. W., and Bissell, M. J. (1996). Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol. Rev. 76, 69–125. doi: 10.1152/physrev.1996.76.1.69
Salgado, R., Junius, S., Benoy, I., Van Dam, P., Vermeulen, P., Van Marck, E., et al. (2003). Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 103, 642–646. doi: 10.1002/ijc.10833
Sanchez-Alvarez, R., Martinez-Outschoorn, U. E., Lin, Z., Lamb, R., Hulit, J., Howell, A., et al. (2013). Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle 12, 289–301. doi: 10.4161/cc.23109
Schäfer, M., and Werner, S. (2008). Cancer as an overhealing wound: an old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 9, 628–638. doi: 10.1038/nrm2455
Schmittnaegel, M., Rigamonti, N., Kadioglu, E., Cassará, A., Wyser Rmili, C., Kiialainen, A., et al. (2017). Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 9:eaak9670. doi: 10.1126/scitranslmed.aak9670
Schreiner, B., Mitsdoerffer, M., Kieseier, B. C., Chen, L., Hartung, H. P., Weller, M., et al. (2004). Interferon-β enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J. Neuroimmunol. 155, 172–182. doi: 10.1016/j.jneuroim.2004.06.013
Seitz, H. K., and Becker, P. (2007). Alcohol metabolism and cancer risk. Alcohol Res. Health 30, 38–41, 44–37.
Sevenich, L., Bowman, R. L., Mason, S. D., Quail, D. F., Rapaport, F., Elie, B. T., et al. (2014). Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol. 16, 876–888. doi: 10.1038/ncb3011
Shaked, H., Hofseth, L. J., Chumanevich, A., Chumanevich, A. A., Wang, J., Wang, Y., et al. (2012). Chronic epithelial NF-kappaB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc. Natl. Acad. Sci. U.S.A. 109, 14007–14012. doi: 10.1073/pnas.1211509109
Sharma, P., and Allison, J. P. (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214. doi: 10.1016/j.cell.2015.03.030
Shi, Z., Yang, W. M., Chen, L. P., Yang, D. H., Zhou, Q., Zhu, J., et al. (2012). Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res. Treat. 135, 737–747. doi: 10.1007/s10549-012-2196-0
Shiao, S. L., Ruffell, B., DeNardo, D. G., Faddegon, B. A., Park, C. C., and Coussens, L. M. (2015). TH2-Polarized CD4(+) T Cells and macrophages limit efficacy of radiotherapy. Cancer Immunol. Res. 3, 518–525. doi: 10.1158/2326-6066.CIR-14-0232
Shiels, M. S., Katki, H. A., Freedman, N. D., Purdue, M. P., Wentzensen, N., Trabert, B., et al. (2014). Cigarette smoking and variations in systemic immune and inflammation markers. J. Natl. Cancer Inst. 106:dju294. doi: 10.1093/jnci/dju294
Shlush, L. I., Zandi, S., Mitchell, A., Chen, W. C., Brandwein, J. M., Gupta, V., et al. (2014). Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–333. doi: 10.1038/nature13038
Singha, B., Gatla, H. R., Manna, S., Chang, T. P., Sanacora, S., Poltoratsky, V., et al. (2014). Proteasome inhibition increases recruitment of IkappaB kinase beta (IKKbeta), S536P-p65, and transcription factor EGR1 to interleukin-8 (IL-8) promoter, resulting in increased IL-8 production in ovarian cancer cells. J. Biol. Chem. 289, 2687–2700. doi: 10.1074/jbc.M113.502641
Snyder, A., Makarov, V., Merghoub, T., Yuan, J., Zaretsky, J. M., Desrichard, A., et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199. doi: 10.1056/NEJMoa1406498
Song, M., and Chan, A. T. (2017). Diet, gut microbiota, and colorectal cancer prevention: a review of potential mechanisms and promising targets for future research. Curr. Colorectal Cancer Rep. 13, 429–439. doi: 10.1007/s11888-017-0389-y
Sopori, M. (2002). Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2, 372–377. doi: 10.1038/nri803
Spranger, S., Bao, R., and Gajewski, T. F. (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. doi: 10.1038/nature14404
Stein, B. L., and Tiu, R. V. (2013). Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J. Interferon Cytokine Res. 33, 145–153. doi: 10.1089/jir.2012.0120
Tada, M., Ohashi, M., Shiratori, Y., Okudaira, T., Komatsu, Y., Kawabe, T., et al. (1996). Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology 110, 227–231. doi: 10.1053/gast.1996.v110.pm8536861
Talmadge, J. E., and Gabrilovich, D. I. (2013). History of myeloid-derived suppressor cells. Nat. Rev. Cancer 13, 739–752. doi: 10.1038/nrc3581
Talpaz, M., Hehlmann, R., Quintas-Cardama, A., Mercer, J., and Cortes, J. (2013). Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia 27, 803–812. doi: 10.1038/leu.2012.313
Tarafdar, A., Hopcroft, L. E., Gallipoli, P., Pellicano, F., Cassels, J., Hair, A., et al. (2017). CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 129, 199–208. doi: 10.1182/blood-2016-09-742049
Tian, H., Callahan, C. A., DuPree, K. J., Darbonne, W. C., Ahn, C. P., Scales, S. J., et al. (2009). Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 4254–4259. doi: 10.1073/pnas.0813203106
Tsai, W. L., and Chung, R. T. (2010). Viral hepatocarcinogenesis. Oncogene 29, 2309–2324. doi: 10.1038/onc.2010.36
Tuhkanen, H., Anttila, M., Kosma, V. M., Ylä-Herttuala, S., Heinonen, S., Kuronen, A., et al. (2004). Genetic alterations in the peritumoral stromal cells of malignant and borderline epithelial ovarian tumors as indicated by allelic imbalance on chromosome 3p. Int. J. Cancer 109, 247–252. doi: 10.1002/ijc.11733
Ulrich, C. M., Bigler, J., and Potter, J. D. (2006). Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat. Rev. Cancer 6, 130–140. doi: 10.1038/nrc1801
Underhill, D. M., and Goodridge, H. S. (2012). Information processing during phagocytosis. Nat. Rev. Immunol. 12, 492–502. doi: 10.1038/nri3244
United States Public Health Service, and Office of the Surgeon General. (2010). How Tobacco Smoke Causes Disease: the Biology and Behavioral Basis for Smoking-Attributable Disease: a Report of the Surgeon General. (Rockville, MD; Washington, DC: U.S. Dept. of Health and Human Services; Public Health Service For sale by the Supt. of Docs U.S. G.P.O.).
Vang, R., Levine, D. A., Soslow, R. A., Zaloudek, C., Shih Ie, M., and Kurman, R. J. (2016). Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a rereview of cases lacking TP53 mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. 35, 48–55. doi: 10.1097/PGP.0000000000000207
Vanharanta, S., Shu, W., Brenet, F., Hakimi, A. A., Heguy, A., Viale, A., et al. (2013). Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat. Med. 19, 50–56. doi: 10.1038/nm.3029
Varol, C., and Sagi, I. (2017). Phagocyte-extracellular matrix crosstalk empowers tumor development and dissemination. FEBS J. 285, 734–751. doi: 10.1111/febs.14317
Velicer, C. M., Heckbert, S. R., Lampe, J. W., Potter, J. D., Robertson, C. A., and Taplin, S. H. (2004). Antibiotic use in relation to the risk of breast cancer. JAMA 291, 827–835. doi: 10.1001/jama.291.7.827
Verstovsek, S., Kantarjian, H., Mesa, R. A., Pardanani, A. D., Cortes-Franco, J., Thomas, D. A., et al. (2010). Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 363, 1117–1127. doi: 10.1056/NEJMoa1002028
Viaud, S., Saccheri, F., Mignot, G., Yamazaki, T., Daillère, R., Hannani, D., et al. (2013). The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976. doi: 10.1126/science.1240537
Vlantis, K., Wullaert, A., Sasaki, Y., Schmidt-Supprian, M., Rajewsky, K., Roskams, T., et al. (2011). Constitutive IKK2 activation in intestinal epithelial cells induces intestinal tumors in mice. J. Clin. Invest. 121, 2781–2793. doi: 10.1172/JCI45349
Voorhees, P. M., Chen, Q., Kuhn, D. J., Small, G. W., Hunsucker, S. A., Strader, J. S., et al. (2007). Inhibition of interleukin-6 signaling with CNTO 328 enhances the activity of bortezomib in preclinical models of multiple myeloma. Clin. Cancer Res. 13, 6469–6478. doi: 10.1158/1078-0432.CCR-07-1293
Wang, K., and Karin, M. (2015). Tumor-elicited inflammation and colorectal cancer. Adv. Cancer Res. 128, 173–196. doi: 10.1016/bs.acr.2015.04.014
Warren, G. W., and Cummings, K. M. (2013). Tobacco and lung cancer: risks, trends, and outcomes in patients with cancer. Am. Soc. Clin. Oncol. 359–364. doi: 10.1200/EdBook_AM.2013.33.359
Weber, F., Xu, Y., Zhang, L., Patocs, A., Shen, L., Platzer, P., et al. (2007). Microenvironmental genomic alterations and clinicopathological behavior in head and neck squamous cell carcinoma. JAMA 297, 187–195. doi: 10.1001/jama.297.2.187
Wyckoff, J., Wang, W., Lin, E. Y., Wang, Y., Pixley, F., Stanley, E. R., et al. (2004). A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64, 7022–7029. doi: 10.1158/0008-5472.CAN-04-1449
Xiao, Q., Zhou, D., Rucki, A. A., Williams, J., Zhou, J., Mo, G., et al. (2016). Cancer-associated fibroblasts in pancreatic cancer are reprogrammed by tumor-induced alterations in genomic DNA methylation. Cancer Res. 76, 5395–5404. doi: 10.1158/0008-5472.CAN-15-3264
Xie, K. (2001). Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 12, 375–391. doi: 10.1016/S1359-6101(01)00016-8
Xu, M., Chen, G., Fu, W., Liao, M., Frank, J. A., Bower, K. A., et al. (2012). Ethanol disrupts vascular endothelial barrier: implication in cancer metastasis. Toxicol. Sci. 127, 42–53. doi: 10.1093/toxsci/kfs087
Xue, J., Schmidt, S. V., Sander, J., Draffehn, A., Krebs, W., Quester, I., et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. doi: 10.1016/j.immuni.2014.01.006
Yagishita, H., Yoshida, T., Ishiguro, K., Numata, Y., and Okayasu, I. (2008). Epithelial and stromal genetic instability linked to tumor suppressor genes in ulcerative colitis-associated tumorigenesis. Scand. J. Gastroenterol. 43, 559–566. doi: 10.1080/00365520701817419
Yan, D., Kowal, J., Akkari, L., Schuhmacher, A. J., Huse, J. T., West, B. L., et al. (2017). Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 36, 6049–6058. doi: 10.1038/onc.2017.261
Yan, L., Anderson, G. M., DeWitte, M., and Nakada, M. T. (2006). Therapeutic potential of cytokine and chemokine antagonists in cancer therapy. Eur. J. Cancer 42, 793–802. doi: 10.1016/j.ejca.2006.01.013
Yang, L., and Karin, M. (2014). Roles of tumor suppressors in regulating tumor-associated inflammation. Cell Death Differ. 21, 1677–1686. doi: 10.1038/cdd.2014.131
Yang, X. D., Corvalan, J. R., Wang, P., Roy, C. M., and Davis, C. G. (1999). Fully human anti-interleukin-8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J. Leukoc. Biol. 66, 401–410. doi: 10.1002/jlb.66.3.401
Yang, X., Wang, X., Liu, D., Yu, L., Xue, B., and Shi, H. (2014). Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 28, 565–574. doi: 10.1210/me.2013-1293
Yin, J., Harpaz, N., Tong, Y., Huang, Y., Laurin, J., Greenwald, B. D., et al. (1993). p53 point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 104, 1633–1639. doi: 10.1016/0016-5085(93)90639-T
Ying, H., Elpek, K. G., Vinjamoori, A., Zimmerman, S. M., Chu, G. C., Yan, H., et al. (2011). PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 1, 158–169. doi: 10.1158/2159-8290.CD-11-0031
Young, A. L., Challen, G. A., Birmann, B. M., and Druley, T. E. (2016). Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 7:12484. doi: 10.1038/ncomms12484
Zahreddine, H., and Borden, K. L. (2013). Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 4:28. doi: 10.3389/fphar.2013.00028
Zeisberg, E. M., and Zeisberg, M. (2013). The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J. Pathol. 229, 264–273. doi: 10.1002/path.4120
Zeiser, R., Burchert, A., Lengerke, C., Verbeek, M., Maas-Bauer, K., Metzelder, S. K., et al. (2015). Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia 29, 2062–2068. doi: 10.1038/leu.2015.212
Zhang, G. J., and Adachi, I. (1999). Serum interleukin-6 levels correlate to tumor progression and prognosis in metastatic breast carcinoma. Anticancer Res. 19, 1427–1432.
Zhang, Q., Zhao, K., Shen, Q., Han, Y., Gu, Y., Li, X., et al. (2015). Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393. doi: 10.1038/nature15252
Zhao, X., Xu, Z., and Li, H. (2017). NSAIDs use and reduced metastasis in cancer patients: results from a meta-analysis. Sci. Rep. 7:1875. doi: 10.1038/s41598-017-01644-0
Zhu, T. S., Costello, M. A., Talsma, C. E., Flack, C. G., Crowley, J. G., Hamm, L. L., et al. (2011). Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 71, 6061–6072. doi: 10.1158/0008-5472.CAN-10-4269
Zhu, Y., Herndon, J. M., Sojka, D. K., Kim, K. W., Knolhoff, B. L., Zuo, C., et al. (2017). Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 597. doi: 10.1016/j.immuni.2017.08.018
Zhu, Y., Knolhoff, B. L., Meyer, M. A., Nywening, T. M., West, B. L., Luo, J., et al. (2014). CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069. doi: 10.1158/0008-5472.CAN-13-3723
Keywords: chronic inflammation, clonal hematopoiesis, stroma, microenvironment, tumor suppressors, oncogenes, anti-inflammatory drugs
Citation: Comen EA, Bowman RL and Kleppe M (2018) Underlying Causes and Therapeutic Targeting of the Inflammatory Tumor Microenvironment. Front. Cell Dev. Biol. 6:56. doi: 10.3389/fcell.2018.00056
Received: 01 February 2018; Accepted: 11 May 2018;
Published: 12 June 2018.
Edited by:
William Curtis Hines, University of New Mexico School of Medicine, United StatesReviewed by:
Paola Rizzo, University of Ferrara, ItalyRonny Drapkin, University of Pennsylvania, United States
Copyright © 2018 Comen, Bowman and Kleppe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Kleppe, a2xlcHBlbUBtc2tjYy5vcmc=