 James B. McCarthy
James B. McCarthy Dorraya El-Ashry
Dorraya El-Ashry Eva A. Turley
Eva A. Turley- 1Department of Laboratory Medicine and Pathology, Masonic Comprehensive Cancer Center, Minneapolis, MN, United States
- 2London Regional Cancer Program, Department of Oncology, Biochemistry and Surgery, Schulich School of Medicine and Dentistry, Lawson Health Research Institute, Western University, London, ON, Canada
This review summarizes the roles of CAFs in forming a “cancerized” fibrotic stroma favorable to tumor initiation and dissemination, in particular highlighting the functions of the extracellular matrix component hyaluronan (HA) in these processes. The structural complexity of the tumor and its host microenvironment is now well appreciated to be an important contributing factor to malignant progression and resistance-to-therapy. There are multiple components of this complexity, which include an extensive remodeling of the extracellular matrix (ECM) and associated biomechanical changes in tumor stroma. Tumor stroma is often fibrotic and rich in fibrillar type I collagen and hyaluronan (HA). Cancer-associated fibroblasts (CAFs) are a major source of this fibrotic ECM. CAFs organize collagen fibrils and these biomechanical alterations provide highways for invading carcinoma cells either under the guidance of CAFs or following their epithelial to mesenchymal transition (EMT). The increased HA metabolism of a tumor microenvironment instructs carcinoma initiation and dissemination by performing multiple functions. The key effects of HA reviewed here are its role in activating CAFs in pre-malignant and malignant stroma, and facilitating invasion by promoting motility of both CAFs and tumor cells, thus facilitating their invasion. Circulating CAFs (cCAFs) also form heterotypic clusters with circulating tumor cells (CTC), which are considered to be pre-cursors of metastatic colonies. cCAFs are likely required for extravasation of tumors cells and to form a metastatic niche suitable for new tumor colony growth. Therapeutic interventions designed to target both HA and CAFs in order to limit tumor spread and increase response to current therapies are discussed.
Introduction
Historically, cancers have been studied as diseases whose initiation and progression are caused by the mutation of key oncogenic “driver” genes, loss of suppressor genes and increasing mutational load resulting in genomic instability, immortalization, unrestrained growth and acquisition of colonizing potential (Hanahan and Weinberg, 2011; Garraway and Lander, 2013; Tomasetti et al., 2013; Vogelstein et al., 2013). More recent studies predict this concept of cancer initiation and progression is incomplete. Most genetic changes that are hallmarks of epithelial cancer are already present in pre-malignant lesions that rarely progress to frank cancer. For example, ultra-deep sequencing of 74 cancer genes in small biopsies of normal aged and sun-exposed human skin reveal a high mutation burden in most key drivers of cutaneous squamous cell carcinoma (Martincorena et al., 2015). These were estimated to be present in over a quarter of the keratinocytes in an epidermis that maintained its normal tissue architecture and physiological functions. A similar paradigm has been observed in other tissues. Endometriosis is a benign inflammatory lesion that is cancer-like in its local invasion and resistance to apoptosis but rarely transforms. Exome sequencing shows that over a quarter of these benign lesions harbor oncogenic driver gene mutations confined to the epithelial compartment that do not result in tumors (Anglesio et al., 2017). These clinical findings are remarkably consistent with experimental studies showing that the tumor phenotype is plastic. Tumor cells can be reverted into a normal growth state while retaining a highly mutated genome by blocking signaling pathways commonly activated by tumor microenvironment (Illmensee and Mintz, 1976; Hall et al., 1995; Wang et al., 2002; Kenny and Bissell, 2003; Postovit et al., 2008; Bizzarri et al., 2011; Northey et al., 2017).
Clues as to the factors required for a mutant genome to either manifest as a transformed phenotype or be restrained into apparent normalcy were initially provided by pioneering studies. The classic studies of B. Mintz brought initial attention to the plasticity of the mutant tumor phenotype and the key role of microenvironments in maintaining transformation (Illmensee and Mintz, 1976). Teratocarcinoma cells, injected into blastocysts, unexpectedly participated in normal tissue development rather than forming tumors. In another key report, chick embryos injected with an oncogenic virus only developed tumors at wound sites even though the viral genome was expressed in unwounded tissues (Dolberg et al., 1985). These original results predicted that while oncogenic insults (e.g., mutations, oncogenic viruses) are a first step toward initiation of cancer, the status of host microenvironment is critical and rate-limiting for disease initiation and progression. These predictions have fueled a synergistic interest in characterizing the properties of “cancerized” host tissue that collaborate with mutant epithelial cells to produce tumors, and drive progression and metastasis, as well as targeting these properties with novel therapeutics designed to manage this aspect of the disease (Radisky et al., 2007; Karn et al., 2015; Werb and Lu, 2015; Luo et al., 2016; Turley et al., 2016; Bridelance et al., 2017; Ghosh et al., 2017; Hutchenreuther and Leask, 2017; Zhan et al., 2017).
Host stroma is a complex mixture of phenotypically heterogeneous endothelial cells, pericytes, immune cells and fibroblasts. Normally, each of these cell types are required for tissue homeostasis, and contribute to the maintenance of tissue architecture and physiologically appropriate tissue functions. The collective paracrine signaling networks that sustain these functions have highly effective tumor-suppressor activity. Gene expression analyses have shown that the stroma surrounding tumors is altered from normal stroma, has lost its tumor suppressing activity and participates in rather than limits tumor initiation, growth and spread (Campisi, 1998; Dumont and Arteaga, 2002; Barsky and Karlin, 2006; Coppé et al., 2010; Bissell and Hines, 2011; Hinds and Pietruska, 2017). Expression differences in normal vs. cancer stroma have been mined to identify signatures that add independent prognostic information to classical epithelial biomarkers (Berdiel-Acer et al., 2014; Bedognetti et al., 2015; Nannini et al., 2015; Winslow et al., 2015; Colangelo et al., 2017; Petitprez et al., 2017). These unbiased analyses together with experimental evidence predict the critical importance of neovascularization, inflammation, immune tolerance and fibroblast activation in creating a “cancerized” microenvironment. In this review, we focus upon the roles of carcinoma-associated fibroblasts (CAFs), also known as tumor-associated fibroblasts (TAFs), in creating a remodeling extracellular matrix that drives tumor initiation and mediates tumor cell spread. We concentrate on the tissue polysaccharide, hyaluronan (HA), as a key contributing ECM component in stromal fibrosis and tumor progression. We conclude by reviewing current experimental interventions targeting both stroma ECM and/or CAF functions that may ultimately limit tumor spread and improve current therapies.
Stromal Extracellular Matrix in Carcinoma Initiation and Progression
It is now well-accepted that carcinomas behave like wounds, which force the host tumor microenvironment into a constant state of fibrotic repair (Dvorak, 1986). As with wound repair, carcinoma-associated stromal tissues undergo dynamic changes in cellular composition and extensive remodeling of extracellular matrix (ECM) as they progress. A particular feature of stromal ECM in cancers particularly pancreatic, prostate, lung and esophageal is its highly fibrotic structure that significantly impacts on progression, metastasis and response-to-therapy (Keely, 2011; Tung et al., 2015; Werb and Lu, 2015; Jiang et al., 2017). Although less well-studied, evidence suggests that chronic inflammation and pro-fibrotic changes in host stroma precede and instruct primary tumor initiation or formation of metastatic colonies by creating a microenvironment or niche favorable for transformation and growth. As examples, in healthy individuals with BRCA1 mutations that are at risk for breast cancer, stromal fibroblasts exhibit a CAF-like activation state (Etzold et al., 2016). Similarly healthy individuals with Li Fraumeni syndrome who bear germ line mutations in TP53 and are at an elevated risk of cancer exhibit “cancerization” of their stromal tissues (Pantziarka, 2015). In a mouse model of colon tumor initiation, both a chronically inflamed and fibrotic stroma are an essential pre-requisite for tumor initiation (Sasaki et al., 2014; Tanabe et al., 2016). There has therefore been an intense effort to understand the dynamic changes in stromal ECM composition to identify the changes that impact on cancer progression, metastasis and resistance to therapies.
Biomechanical Properties of Tumor-associated Stroma
A major ECM component of all fibrotic stroma is type I collagen, which provides structural and biochemical cues to cells within the stroma (Keely, 2011; Tung et al., 2015; Werb and Lu, 2015; Jiang et al., 2017) (Figure 1). A notable property of “cancerized” stroma is the accumulation of type I collagen fibrils in the stroma that are extensively crosslinked by lysyl oxidase (LOX) and tissue transglutaminase (TG2) (Perryman and Erler, 2014; Lee et al., 2016). Collagen crosslinking confers proteolytic resistance to the fibrils and increases stroma stiffness, which promotes tumor cell migration, invasion and proliferation. Tumor-associated collagen signatures categorized by increased collagen density and orientation of mature collagen fibers parallel to or perpendicular to the tumor boundary offer prognostic information (Mellone et al., 2016). Orientation of the fibrils is the result of a process of prolonged mechano-signaling mediated by integrin/cytoskeletal linkages, activation of downstream adhesion pathway signaling components particularly focal adhesion kinase (FAK), phosphorylation of myosin light chain kinase and activation of Rho-Kinase (Schedin and Keely, 2011; Boyle and Samuel, 2016). Oriented collagen fibrils are an ominous biomarker of tumor cell invasion, metastasis and poor outcome (Schedin and Keely, 2011; Tung et al., 2015). In experimental models, non-transformed epithelial cell adhesion to stiff collagen matrices results in elevated activation of oncogenic pathways and increased expression of growth-promoting genes, emphasizing that the mechanical property of stiffness contributes to carcinoma progression (Paszek et al., 2005; Provenzano and Keely, 2011; Ray et al., 2017). Carcinoma cells cultured on stiff collagen gels grow as colonies with discrete boundaries, whereas the same cells cultured in oriented collagen gels of equal stiffness invade along these collagen fibers (Provenzano and Keely, 2011). These in vitro observations have been confirmed in vivo using multiphoton laser scanning microscopy and second harmonic generation imaging of live ex-vivo tumors (Provenzano and Keely, 2011). While fibrillar collagen is a major component of fibrotic stroma, many additional prognostic ECM factors impact the biological and biomechanical properties of tumor-associated stroma. One of these is HA, whose elevated accumulation in the tumor microenvironment contributes to cancer initiation, progression and therapy resistance (Karousou et al., 2014; Chanmee et al., 2016; Sato et al., 2016; Turley et al., 2016; Binder et al., 2017; Bourguignon et al., 2017; Safdar et al., 2017). These properties as they relate to tumor initiation and dissemination are discussed in the following sections below.
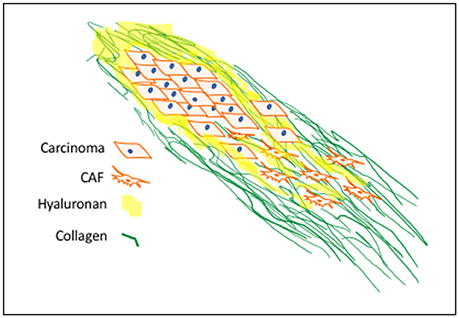
Figure 1. Progression-Associated Fibrosis in Cancerized Stroma: Deregulated synthesis and deposition of ECM components, including HA and type I collagen, leads to tumor-associated fibrosis. HA, a major polysaccharide of provisional wound matrices, contributes to cancer initiation, progression and resistance-to-therapy. CAF activation sustains increased collagen synthesis, structurally oriented by fibroblast contractile forces. These provide structural and biochemical cues to enhance mechano-signaling for carcinoma motility and invasion.
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAF) are the primary cell type in “cancerized” stroma and are a major source of ECM as well as cytokines/growth factors that impact upon both tumor susceptibility/initiation and progression (Kalluri, 2016; Liu et al., 2017; Santi et al., 2017; Yamauchi et al., 2018). CAFs are a heterogeneous mixture of multiple resident fibroblast subtypes and infiltrated circulating mesenchymal cells. Understanding the origin and nature of the fibroblasts that drive oncogenic initiation and progression has been hampered by a paucity of CAF-specific markers and thus their origin remains controversial. Mesenchymal stem cells (MSCs) and resident fibroblast progenitors of CAFs are recruited by chemokines/cytokines and growth factors to specific sites and ECM components at these sites activate these cells into CAFs (Mishra et al., 2008; Shinagawa et al., 2010; Mi et al., 2011). For example, knockdown of the HA receptor CD44 in MSCs blocks both their ability to be recruited to the tumor site, and their tumor promoting functions (Spaeth et al., 2013) Recent studies have identified CAF properties that are distinct from activation of normal fibroblasts responding-to-wounding. For example, CAFs activation status appears to be irreversible while wound repair fibroblasts activation is both reversible and dependent on wound-induced signaling. The secretome, ECM remodeling and tumor promoting properties of CAFs and injury-activated fibroblasts also differ (Kalluri, 2016).
CAFs are most commonly identified by their expression of fibroblast activation protein (FAP) and alpha smooth muscle actin (∂-SMA), however, additional markers including platelet derived growth factor receptor b (PDGFRB), fibroblast specific protein (FSP) and vimentin (VIM), all of whose expression in tumor stroma have, like ∂-SMA, been linked to poor outcome of many cancers, can also be expressed in CAFs (Jacob et al., 2012; Folgueira et al., 2013; Paulsson and Micke, 2014; Han et al., 2015; Peiris-Pagès et al., 2015; Corvigno et al., 2016; Gascard and Tlsty, 2016; Kuzet and Gaggioli, 2016; Hammer et al., 2017; Tao et al., 2017; von Ahrens et al., 2017). The roles of CAFs as promoters of tumor initiation, progression, epithelial to mesenchymal transition, stemness, tumor invasion, angiogenesis, metastasis and drug resistance are well established (Kalluri and Zeisberg, 2006; Shekhar et al., 2007; Straussman et al., 2012) Experimentally, CAFs exhibit activity in all Hallmarks of Cancer categories (Salo et al., 2014; Tommelein et al., 2015; Attieh and Vignjevic, 2016; Mezawa and Orimo, 2016). Many studies of CAF participation in tumorigenesis have viewed their role as a reactive process that is a consequence of signals originating in the epithelial tumor, which results in a permissive environment for tumor cells to grow. A number of studies have demonstrated a more instructive role for CAFs in the initiation and dissemination of tumors. These studies have stimulated interest in the development of therapies that target CAFs and other stromal components of the tumor stroma. These CAF properties are reviewed here.
CAFs and Tumor Initiation
In general, fibroblasts in normal stroma have tumor-suppressing properties (Bhowmick et al., 2004; Augsten, 2014; Klein, 2014; Rhee et al., 2015; Kubo et al., 2016; Lin and Lin, 2017; Mangge et al., 2017). However, when normal fibroblasts are activated (e.g., into myofibroblasts) or become senescent they lose these tumor-suppressing functions and under appropriate conditions convert into tumor-promoting and/or initiating CAFs. Experimentally, such cells can facilitate conversion of pre-malignant epithelial cells into tumors. An early example of this was provided by evidence that irradiated fibroblasts increase the incidence of tumors arising from pre-malignant mammary epithelial cells (Bhowmick et al., 2004; Ji et al., 2017). A number of more recent studies using experimental models provide direct evidence for the ability of CAFs to drive the initiation of cancer (Sasaki et al., 2014). Thus, loss or reduction of a notch effector (CSL) in stromal fibroblasts is sufficient for CAF activation and induction of keratinocyte tumors. Conversely, CCR5 blockade of fibroblast activation in colon tissue of a mouse model of colitis-associated carcinogenesis strongly reduces tumor initiation even though inflammation/colitis is still present. In experimental models, senescent fibroblasts have also been shown to enhance cancers including ovarian and keratinocyte transformation (Lawrenson et al., 2010).
Clinically, CAF-like fibroblast-induced stromal ECM changes have been reported to precede tumor formation and these early changes in ECM provide prognostic information that permit risk stratification. For example, high mammographic density is a strong risk factor in breast cancer (DeFilippis et al., 2012; Ghosh et al., 2017; Vinnicombe, 2017). Clinical features of this condition, which precede detectable tumor formation, include adipocyte loss and high ECM production. This condition has been linked to expression loss of the mesenchymal differentiation regulator CD36 in stromal fibroblasts, which phenocopies the clinical features of high mammographic density breast tissue. In clinical samples, CAFs exhibit loss of CD36 expression. (DeFilippis et al., 2012) and this in breast cancer tissue is strongly associated with poor outcome. Other examples include evidence that primary dermal fibroblasts exhibit a CAF-like state with a germ-line BRCA1 epi-mutation (Etzold et al., 2016). These fibroblasts stimulate rather than suppress epithelial proliferation and migration, express CAF markers including ACTA2, FAP, PDPN, and TNC, and are highly proliferative and migratory relative to normal counterparts from other patients. In early stage breast cancer, high stromal Heat Shock Factor 1 (HSF1) activation is associated with poor outcome and experimental data show that HSF1 expression is elevated/activated and results in potent enabling of malignancy (Scherz-Shouval et al., 2014). Genetic loci have been also identified that affect stromal properties and control mammary tumor susceptibility. These include genes that affect TGFß signaling (Zhang P. et al., 2015). Consistent with these findings, fibroblast-specific deletion of TGFßIIR in a transgenic mouse model results in repression of tumor suppressing functions of fibroblasts and a rapid development of aggressive prostate cancer (Li et al., 2012). HA is one ECM factor that is regulated by TGFß (Heldin et al., 2014) that is linked to tumor susceptibility, initiation and progression of many cancers and will be focused upon here.
Stromal Hyaluronan Is Linked to Tumor Susceptibility and CAF Activation
HA is a simple extracellular matrix polysaccharide that a wealth of experimental approaches has demonstrated is an instructive factor in cancer initiation and progression (Heldin et al., 2014; Tolg et al., 2014; Zhang C. et al., 2015; Chanmee et al., 2016; Turley et al., 2016; Bohaumilitzky et al., 2017; Senbanjo and Chellaiah, 2017; Shih et al., 2017; Wight, 2017; Wong et al., 2017). For example, blocking HA synthesis (Itano et al., 2008; Hamada et al., 2017; Ikuta et al., 2017) or ablating the HA-binding function of one of its receptors RHAMM (gene name HMMR) (Hall et al., 1995), which has been strongly linked to tumorigenesis (Tolg et al., 2014; Turley et al., 2016), attenuates the transformed phenotype. Clinical analyses show that elevated HA accumulation in either the stroma or tumor parenchyma of many cancers is linked to tumor aggression and poor outcome (Sironen et al., 2011; McAtee et al., 2014; Chanmee et al., 2016; Sato et al., 2016; Turley et al., 2016; Bourguignon et al., 2017; Wu et al., 2017). Unexpectedly, HA has also recently been implicated as a stromal tumor-suppressing factor (Tian et al., 2013; Fisher, 2015; Triggs-Raine and Natowicz, 2015; Bohaumilitzky et al., 2017). These opposing effects are not well-understood but have been linked to differences in its metabolism and in particular the regulation of HA polymer size (Simpson and Lokeshwar, 2008; Tian et al., 2013; Khaldoyanidi et al., 2014; Tolg et al., 2014; Litwiniuk et al., 2016; Turley et al., 2016; Fouladi-Nashta et al., 2017).
HA is composed of repeating disaccharide units of N-acetylglucosamine and ß-glucuronic acid linked together by three highly homologous synthases (HAS1,2,3). These are most frequently located at the plasma membrane and the growing HA polymer is extruded directly into the extracellular space through pores in the plasma membrane formed by synthase oligomerization (Weigel, 2015) (e.g., Figure 2). Evolving evidence indicates that the biological effects of HA are primarily determined by size rather than conformational changes typically required for protein activation. In general, large HA polymers, which are mainly present in homeostatic tissues, are immunologically quiescent and contribute to enforcing cell survival and homeostasis. HA fragments (e.g., < 100–200 kDa), which are generated by reactive oxygen/nitrogen species (ROS/RNS) and hyaluronidases produced during tissue stress, repair and chronic disease, are pro-inflammatory and pro-fibrotic (Simpson and Lokeshwar, 2008; Gaudet and Popovich, 2014; Cyphert et al., 2015; Sherman et al., 2015; Gaggar and Weathington, 2016; Maytin, 2016; Turley et al., 2016; Bohaumilitzky et al., 2017; Cowman, 2017; Frevert et al., 2017; Kavasi et al., 2017; Wight et al., 2017; Wu et al., 2017; Avenoso et al., 2018a,b) (Figure 3). The precise effect of specific sizes of HA fragments on immune and mesenchymal cells on such functions as gene expression appears to be cell-context and stimulus-specific, and is currently controversial (Cowman, 2017; Weigel, 2017; Weigel and Baggenstoss, 2017). HA fragment accumulation in quiescent homeostatic tissues is low. In contrast remodeling and diseased tissues such as cancers often contain an elevated level of HA (e.g., Teder et al., 2002; Koyama et al., 2007; Li et al., 2011; Tolg et al., 2017), clear evidence of fragmentation, and overexpression of HAS, hyaluronidases and HA receptors.
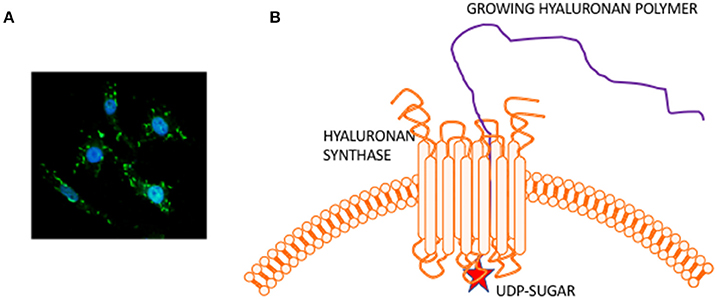
Figure 2. Hyaluronan is a simple polysaccharide produced by cell membrane synthases. (A) Micrograph showing membrane localized hyaluronan synthase 2 in fibroblasts. (B) Hyaluronan synthase has multiple transmembrane domains that cluster to form pores in the cell membrane. UDP-sugars bind to the protein cytoplasmic face and growing polymer is extruded through the pore to the extracellular space.
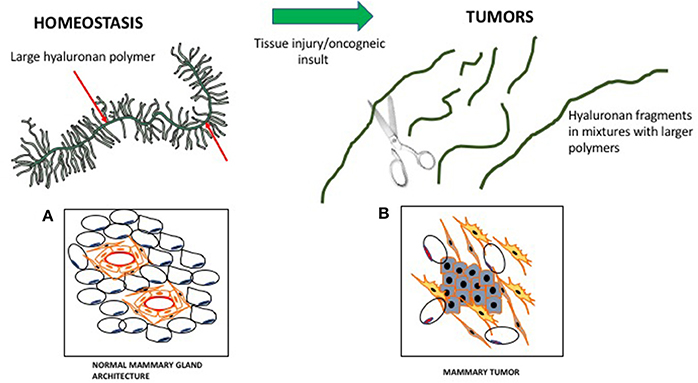
Figure 3. Hyaluronan is a large polymer during tissue homeostasis and fragmented in wounds and tumors. In homeostasis, large hyaluronan polymers are decorated by proteins/proteoglycans, which contribute to normal tissue architecture. (A) Cartoon of a virgin mouse mammary gland. (B) Cartoon of a mouse mammary tumor (Blue, mammary tumor cells). During wounding, fragmented hyaluronan polymers produced by ROS/hyaluronidases activate fibroblasts and attract immune cells, which contribute to the loss of tissue architecture.
The tumor-resistance properties of high molecular weight HA were originally identified in the tumor resistant naked mole rat and resistance of fibroblasts to oncogenic transformation was shown to depend upon production of large HA polymers (Tian et al., 2013). Naked mole rat tissues contain larger HA polymers and less detectable fragmentation than tissues of the more tumor-susceptible mouse. HA-mediated tumor resistance of the naked mole rat is attributed to the ability of high molecular weight HA to hyper-sensitize cells to contact inhibition and induce p16 (ink4a) locus expression with consequent cell cycle arrest (Tian et al., 2015). Consistent with this explanation, HA overproduction has also been shown by other groups to regulate contact inhibition and adhesion in cultured non-malignant cells (Itano et al., 2008). Others have shown that excess production of HA by itself does not promote an aggressive tumor phenotype and can even be tumor-suppressing by blocking G1-S transition in the cell cycle (Bharadwaj et al., 2011). Similarly, exposure of tumor cells to hyaluronidases alone (e.g., HYAL1 or PH-20) can be growth-suppressing (Simpson and Lokeshwar, 2008) and increase response of tumor cells to therapy (Wong et al., 2017). Thus, high HA production combined with an increased capacity for polymer fragmentation appears to be responsible for oncogenic effects of this polysaccharide.
A number of studies using mouse models also predict that elevated HA production, primarily by fibroblasts, pre-disposes epithelial cells to tumor initiation. Examples include evidence that an HA-rich stroma precedes increased mammary tumor formation in transgenic mice expressing both MMTV-driven HAS2 and a c-neu proto-oncogene. HAS2/c-neu mice tumors notably produce higher levels of both high molecular weight and fragmented HA than the c-neu mice (Koyama et al., 2007). Using p38MAPK knock-in mice and tumor xenografts, others have shown that MAPK-driven HAS2 expression and consequent HA production by fibroblasts is required for their activation into CAFs and for loss of their tumor suppressing properties resulting in a pro-tumor niche and increased lung colonization (Brichkina et al., 2016). These studies suggest that the tumor suppressing effects of either HA or processing enzymes alone are converted into a pro-tumor stimulus when HA processing into fragments is enhanced and sustained by elevated expression of one or more HAS genes, and hyaluronidases, often HYAL1. Additional studies predict that the pro-tumor functions of HA also depend upon the display of specific receptors, notably the injury-related HA receptor, RHAMM (gene name HMMR), which activates oncogenic signaling pathways (Tolg et al., 2014; Misra et al., 2015; Nikitovic et al., 2015; Schwertfeger et al., 2015). In a pre-malignant stroma, these genes are expressed by CAF-like fibroblasts.
CAFs and Tumor Dissemnation
CAFs play a significant role in tumor dissemination by inducing an invasive phenotype in tumor cells, promoting motile phenotypes and remodeling the ECM. Invasion is achieved in part by CAF-driven EMT and consequent cell migration driven by factors such as TGF-B, HGF, and CXCL12/SDF-1(Kalluri, 2016). Paladin-expressing CAF create “tunnels” in the ECM which cancer cells migrate through (Brentnall, 2012). Under CAF guidance, tumor cells also migrate and invade as groups in the absence of apparent EMT. This collective migration and invasion is driven by heterotypic E-cadherin/N-cadherin interactions between tumor cells and CAFs (Labernadie et al., 2017) that results in a mechanically active adhesion. CAF-mediated ECM remodeling occurs as a result of secretion of collagen, proteases, and in particular, matrix metalloproteinases. ECM remodeling provides a microenvironment that further supports tumor cell migration and dissemination. Interestingly, CAFs from different breast cancer molecular subtypes including Luminal A, Her2-like, and triple negative/basal-like exhibit subtype-specific differences in stromal gene expression (Tchou et al., 2012), microRNA expression and secretory profiles (Shah et al., 2015). Furthermore, CAFs from more aggressive cancers induce more aggressive breast cancer cell phenotypes than CAFs from more indolent cancers (Shah et al., 2015).
Circulating tumor cell (CTC) clusters were originally described in the 1970's and are now considered to be pre-cursors of metastatic colonies. In mouse breast cancer models, circulating tumor cell clusters exhibit higher metastatic capacity compared with individual or single CTCs (Aceto et al., 2014). Additionally, polyclonal breast cancer metastases have been suggested to arise from circulating tumor cell clusters composed of Keratin 14+ cells (Cheung et al., 2016). Quantification of these CTC clusters in breast cancer patients show that their presence correlates with reduced progression-free survival and poor outcome (Cheung et al., 2016; Jansson et al., 2016; Mu et al., 2016; Wang et al., 2017). Collective migration of tumor cell clusters into the circulation appears to offer a tumor cell survival advantage compared to entry of single tumor cells into the vasculature. CAFs are not only present in primary and metastatic tumor stroma but have recently been shown to occur in the circulation either as individual CAFs, part of CTC clusters or as CAF clusters. Circulating CAFs (cCAFs) likely contribute to CAFs found in pre-metastatic and metastatic niches. Mouse metastasis models suggest that circulating CAFs can exit either with groups of cancer cells or by themselves. In these models, the presence of CAFs from the primary TME promotes metastatic seeding and growth (Duda et al., 2010), likely by helping to create a suitable growth and survival microenvironmental niche for tumor cells and to aid in avoidance of immune surveillance. Additionally, since CAFs are present in pre-metastatic niches prior to the appearance of tumor cells, circulating CAFs likely also play a role in establishing or preparing a niche suitable for future tumor cell colonization. In a pilot study, cCAFs were detected in the blood from patient with Stage IV (metastatic) breast cancer but not from patients with Stage I disease with no evidence of relapse, while CTCs were detected in both patient samples (Ao et al., 2015). Furthermore, CTCs and cCAFs circulate in co-clusters in patient blood, and like CTCs, cCAFs can also cluster with each other (Figure 4). Jones and colleagues also found circulating CK-/CD45/VIM+ fibroblast-like cells in metastatic prostate cancer patient blood (Jones et al., 2013). The development of techniques for isolating circulating CAFs from mouse models of human breast cancer xenografts and mammary tumor susceptibility will greatly aid in characterizing both the origin and contribution of circulating CAFs to successful metastasis. Recent evidence suggests that at least a portion of CTCs are tumor cells transitioning between the epithelial and mesenchymal state (Yu et al., 2013) that possess stem cell-like properties and phenotypically plasticity May et al., 2011. Functional characterization of these circulating cells/clusters will clarify the mechanisms of tumor cell dissemination and likely identify potential therapeutic targets for metastatic disease.
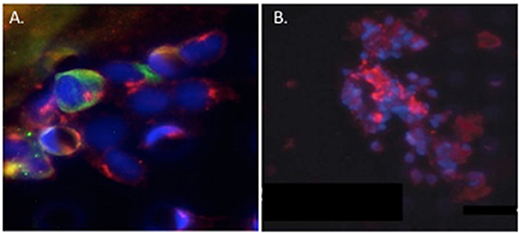
Figure 4. Circulating cCAF/circulating tumor cell (CTC) clusters and cCAF clusters in breast cancer patient blood. (A) cCAF/CTC co-cluster and (B) cCAF cluster. Red: FAP, Green: CK. From Ao et al. (2015).
Hyaluronan and Tumor Dissemination
A CAF property that appears to be critical to cancer cell invasion is their active motility and tropism toward tumor cells (e.g., Costea et al., 2013; Berdiel-Acer et al., 2014). These properties culminate in close physical heterotypic contact (Marusyk et al., 2016; Labernadie et al., 2017). Clinically, close proximity of CAFs to tumor cells is linked to poor outcome and resistance to therapy and supports migration and invasion of tumor cells by several mechanisms (Marusyk et al., 2016). HA is one CAF-produced ECM factor that appears to play a key role in these critical autocrine and paracrine migratory interactions of CAFs and tumor cells. Thus, highly motile CAF subtypes produce and rely upon HA for their motogenic properties (Costea et al., 2013) and ability to promote migration of tumor cells. We and others (e.g., Hamilton et al., 2007; Mele et al., 2017; Shigeeda et al., 2017) have also reported that highly aggressive breast cancer cells that have undergone EMT develop a CAF-like autocrine production of HA to sustain their high motility rates. Such tumor cells are able to invade independently from CAFs (Turley et al., 2016). Intriguingly, we have shown using fluorescent HA-probes that HA-binding to breast cancer cells and to activated fibroblasts is heterogeneous (Veiseh et al., 2014, 2015). FACS-sorted tumor cell subsets that bind high levels of HA are more motile, invasive and metastatic than subsets that bind low or no probe. A concept that emerges from these studies is that CAF subsets not only utilize HA to migrate close to tumor cells but that their autocrine production of HA also stimulates the migration of the HA binding tumor subpopulation (Figure 5). Expression of HA receptors CD44 and RHAMM is required for migration of these tumor cells, and we predict that these receptors also mediate HA-dependent, highly motile CAF subsets.
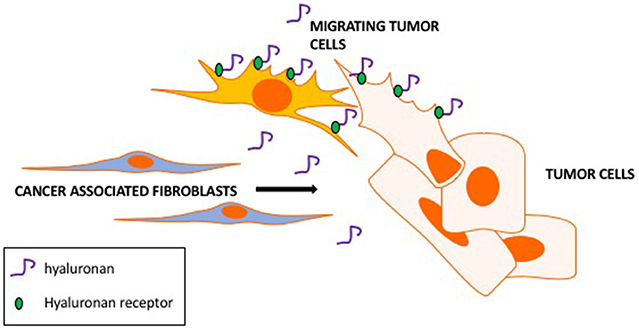
Figure 5. Hyaluronan promotes CAFs motility toward tumor cells and tumor cell motility. CAF subsets produce hyaluronan as a motogenic stimulus for migrating toward tumor cells. Hyaluronan binds to tumor cell subsets via hyaluronan receptors (RHAMM and CD44) contributing to the migration and invasion of CAF-guided tumor cells.
The role of HA and its receptors in circulating CAFs and tumor cells is currently understudied. However, several studies have reported that circulating tumor cells from cancer patients express the HA receptor CD44 (Grillet et al., 2017) and can be captured from circulation by adhering to HA, a process that is mediated by HA receptors (Xu et al., 2017). Interestingly, circulating cells with this dual phenotype are EpCAM- and are therefore distinct form the more commonly studied EpCAM+ circulating tumor cells (Mirza et al., 2017). EpCAM-/CD44+ cells may represent tumor cells that have undergone EMT and/or are circulating cancer stem cells (cCSCs). Circulating cells isolated from lung adenocarcinoma patients that had higher levels of markers such as RHAMM (HMMR) had shorter survival times (Man et al., 2014). CAFs also express CD44 and this CD44 plays important roles in CAF function. These collective results predict a critical importance of HA production and HA receptor display in cCAFs and CTCs to successful metastases.
Targeting Stroma and CAFs
Targeting key genetic or epigenetic alterations in tumors and/or the use of immune checkpoint inhibitors has significantly improved cancer therapy (Jiang et al., 2017). While these advances are encouraging, they are currently either effective in a minority of cancer patients, have significant pro-tumor side-effects or lack long-term durability. Thus, new approaches are necessary to expand the number of patients who will benefit clinically from chemotherapy and targeted therapy. Targeting the fibrotic stroma is emerging as a potentially key approach necessary to achieving therapeutic efficacy. This is particularly true for pancreatic cancer, which typically progresses with an extensive fibrotic stroma that can account for over 80% of the tumor volume (Yu and Tannock, 2012; Tan et al., 2015). Therapies that target the fibrotic stroma, including HA, are being developed and entering clinical trials (Provenzano and Hingorani, 2013; Jiang et al., 2017; Kumari et al., 2017).
High interstitial pressures in the fibrotic stroma of pancreatic cancers, which results from high production of collagen and HA, causes the collapse of the stromal vasculature in pancreatic cancers and impedes exposure of tumor cells to chemo- and immune therapies (Yu and Tannock, 2012). Multiple approaches to target fibrotic stroma are therefore being tested to overcome these delivery issues. One successful strategy is targeting HA. Systemic administration of a recombinant sperm hyaluronidase (PEGPH20), degrades hyaluronan in pancreatic cancer stroma (Provenzano and Hingorani, 2013). This destruction decreases interstitial fluid pressure, increases vasculature patency and improves the delivery of gemcitabine. Importantly, these hyaluronidase-mediated changes both decrease tumor volume and increase animal survival in experimental models of pancreatic cancer. PEGPH20 is now in phase III clinical trials for pancreatic cancer (Doherty et al., 2018). An alternative to the use of recombinant hyaluronidase has been of HA synthesis inhibitors (e.g., 4-methylumbelliferone), which also inhibits tumor growth and could be used in alone or in combination with hyaluronidase to improve therapeutic response (Kudo et al., 2017).
CAF-targeted therapies are also being developed to blunt their fibrosis-activated signaling. For example, a selective FAK inhibitor (VS-4718) targets hyperactive FAK in stromal CAFs. This inhibitor reduces fibrosis, decreases the number of tumor-infiltrating immuno-suppressive cells and results in survival doubling in mouse models of pancreatic ductal adenocarcinoma (Jiang et al., 2016, 2017). Inhibiting FAK activation also increases responsiveness to chemotherapy and immune checkpoint inhibitors with resulting improved outcome. These pre-clinical successes have led to phase 1 clinical trials using this FAK inhibitor in combination with immune checkpoint inhibitors (Jiang et al., 2017). While FAK hyper-activation is a key feature of mechano-signaling in CAFs and provides a proof-of-concept for targeting the microenvironment, stromal immune cells also utilize FAK or the related PYK-2 for survival (Jiang et al., 2017). Off target effects of VS-4718 could contribute to immune-suppression and therefore compromise its effective utility in humans (Jiang et al., 2016).
Active investigations are also underway to target CAF survival in the fibrotic stroma. In contrast to carcinoma cells, CAFs are genetically normal cells that have been co-opted and modified by cancer cells into a state of constitutive activation. CAFs therefore have a less plastic genome than tumor cells limiting their ability to rapidly modify their genome but making them an attractive candidate for stable responses to targeted therapy. CAFs uniquely express FAP, which plays important roles in CAF function (Lai et al., 2012; Koczorowska et al., 2016). In vivo administration of a FAP enzyme inhibitor, Talabostat, in tumor-bearing mice results in tumor regression and upregulation of specific chemokines and cytokines that induce an anti-tumor immune response (Cunningham, 2007). Talabostat is well tolerated in healthy volunteers in both Phase I and II clinical trials but does not result in therapeutic benefit. A CAF-directed, anti-human FAP antibody, sibrotuzumab (Fischer et al., 2012), exhibits specificity and activity in preclinical mouse models (Fischer et al., 2012), and was well tolerated in early Phase I/II clinical trials (Hofheinz et al., 2003; Scott et al., 2003) but has failed to show therapeutic activity in patients with metastatic disease. FAP-targeted chimeric antigen receptor (CAR) T cells reduce ECM, vessel density, and growth of several types of human cancer xenografts and murine pancreatic cancers when introduced into mice by adoptive transfer (Wang et al., 2014; Lo et al., 2015). This technology has not yet entered clinical trials. FAP may be useful for targeting therapies to CAFs. Potentially the development of therapies that impede CAF survival/function in the circulation or their ability to migrate/enter the circulation (e.g., HA/RHAMM) may be a more promising approach.
In conclusion, despite recent advances in targeted therapies, metastases, recurrence and relapse remain as major clinical obstacles to successful cancer treatment. Carcinoma cell epigenetic and genetic heterogeneity are important factors that limit therapeutic efficacy. However, a wealth of studies has now demonstrated that tumor-associated fibrotic stroma is also a major contributing factor to therapeutic failure. The success of new approaches to targeting tumor cells will in the future likely have to include agents that compromise the pro-tumorigenic fibrotic ECM.
Author Contributions
ET: Organized, referenced, edited contributions and wrote introduction, and sections on CAF-mediated tumor initiation, relationship of microenvironment and hyaluronan, prepared model Figures 1, 2, 3. JM: Edited contributions, wrote abstract and wrote/referenced sections on tumor dissembination, biomechanical properties of tumor stroma and therapeutic approaches to targeting CAFs and fibrotic stroma. DE-A: Edited contributions wrote/referenced section on circulating tumor cells and CAFs, tumor dissemination and prepared Figure 4.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JM is supported by Chairman's Fund Professor in Cancer Research and funds from the Pardee Endowed Chair in Cancer Biology. DE-A is supported by start-up funds from the Masonic Cancer Center/Department of Laboratory Medicine and Pathology, University of Minnesota and previously by funds from the Prevent Cancer Foundation and Florida Breast Cancer Foundation. ET is supported by the Breast Cancer Society of Canada and Cancer Research Society.
References
Aceto, N., Bardia, A., Miyamoto, D. T., Donaldson, M. C., Wittner, B. S., Maheswaran, S., et al. (2014). Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. doi: 10.1016/j.cell.2014.07.013
Anglesio, M. S., Papadopoulos, N., Ayhan, A., Nazeran, T. M., Noe, M., Shih, I. M., et al. (2017). Cancer-associated mutations in endometriosis without cancer. N. Engl. J. Med. 376, 1835–1848. doi: 10.1056/NEJMoa1614814
Ao, Z., Shah, S. H., Machlin, L. M., Parajuli, R., Miller, P. C., El-Ashry, D., et al. (2015). Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Res. 75, 4681–4687. doi: 10.1158/0008-5472.CAN-15-1633
Attieh, Y., and Vignjevic, D. M. (2016). The hallmarks of CAFs in cancer invasion. Eur. J. Cell Biol. 95, 493–502. doi: 10.1016/j.ejcb.2016.07.004
Augsten, M. (2014). Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 4:62. doi: 10.3389/fonc.2014.00062
Avenoso, A., D'Ascola, A., Scuruchi, M., Mandraffino, G., Calatroni, A., Saitta, A., et al. (2018a). Hyaluronan in experimental injured/inflamed cartilage: in vivo studies. Life Sci. 193, 132–140. doi: 10.1016/j.lfs.2017.11.006
Avenoso, A., D'Ascola, A., Scuruchi, M., Mandraffino, G., Calatroni, A., Saitta, A., et al. (2018b). Hyaluronan in the experimental injury of the cartilage: biochemical action and protective effects. Inflamm. Res. 67, 5–20. doi: 10.1007/s00011-017-1084-9
Barsky, S. H., and Karlin, N. J. (2006). Mechanisms of disease: breast tumor pathogenesis and the role of the myoepithelial cell. Nat. Clin. Pract. Oncol. 3. 138–151. doi: 10.1038/ncponc0450
Bedognetti, D., Hendrickx, W., Marincola, F. M., and Miller, L. D. (2015). Prognostic and predictive immune gene signatures in breast cancer. Curr. Opin. Oncol. 27, 433–444. doi: 10.1097/CCO.0000000000000234
Berdiel-Acer, M., Berenguer, A., Sanz-Pamplona, R., Cuadras, D., Sanjuan, X., Paules, M., et al. (2014). A 5-gene classifier from the carcinoma-associated fibroblast transcriptomic profile and clinical outcome in colorectal cancer. Oncotarget 5, 6437–6452. doi: 10.18632/oncotarget.2237
Bharadwaj, A. G., Goodrich, N. P., McAtee, C. O., Haferbier, K., Oakley, G. G., et al. (2011). Hyaluronan suppresses prostate tumor cell proliferation through diminished expression of N-cadherin and aberrant growth factor receptor signaling. Exp. Cell Res. 317, 1214–1225. doi: 10.1016/j.yexcr.2011.01.026
Bhowmick, N. A., Neilson, E. G., and Moses, H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337. doi: 10.1038/nature03096
Binder, M. J., McCoombe, S., Williams, E. D., McCulloch, D. R., and Ward, A. C. (2017). The extracellular matrix in cancer progression: Role of hyalectan proteoglycans and ADAMTS enzymes. Cancer Lett. 385:55–64. doi: 10.1016/j.canlet.2016.11.001
Bissell, M. J., and Hines, W. C. (2011). Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 17, 320–329. doi: 10.1038/nm.2328
Bizzarri, M., Cucina, A., Biava, P. M., Proietti, S., D'Anselmi, F., Lisi, E., et al. (2011). Embryonic morphogenetic field induces phenotypic reversion in cancer cells. Review article. Curr. Pharm. Biotechnol. 12, 243–253. doi: 10.2174/138920111794295701
Bohaumilitzky, L., Huber, A. K., Stork, E. M., Wengert, S., Woelfl, F., and Boehm, H. (2017). A trickster in disguise: hyaluronan's ambivalent roles in the matrix. Front. Oncol. 7:242. doi: 10.3389/fonc.2017.00242
Bourguignon, L. Y. W., Earle, C., and Shiina, M. (2017). Activation of matrix hyaluronan-mediated CD44 signaling, epigenetic regulation and chemoresistance in head and neck cancer stem cells. Int. J. Mol. Sci. 18:E1849. doi: 10.3390/ijms18091849
Boyle, S. T., and Samuel, M. S. (2016). Mechano-reciprocity is maintained between physiological boundaries by tuning signal flux through the Rho-associated protein kinase. Small GTPases 7, 139–146. doi: 10.1080/21541248.2016.1173771
Brentnall, T. A. (2012). Arousal of cancer-associated stromal fibroblasts: palladin-activated fibroblasts promote tumor invasion. Cell Adh. Migr. 6, 488–494. doi: 10.4161/cam.21453
Brichkina, A., Bertero, T., Loh, H. M., Nguyen, N. T., Emelyanov, A., Bulavin, D. V., et al. (2016). p38MAPK builds a hyaluronan cancer niche to drive lung tumorigenesis. Genes Dev. 30, 2623–2636. doi: 10.1101/gad.290346.116
Bridelance, J., Drebert, Z., De Wever, O., Bracke, M., and Beck, I. M. (2017). When neighbors talk: colon cancer cell invasion and tumor microenvironment myofibroblasts. Curr. Drug Targets 18, 964–982. doi: 10.2174/1389450117666161028142351
Campisi, J. (1998). The role of cellular senescence in skin aging. J. Investig. Dermatol. Symp. Proc. 3, 1–5.
Chanmee, T., Ontong, P., and Itano, N. (2016). Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 375 20–30. doi: 10.1016/j.canlet.2016.02.031
Cheung, K. J., Padmanaban, V., Silvestri, V., Schipper, K., Cohen, J. D., Ewald, A. J., et al. (2016). Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. U.S.A. 113, E854–E863. doi: 10.1073/pnas.1508541113
Colangelo, T., Polcaro, G., Muccillo, L., D'Agostino, G., Rosato, V., Ziccardi, P., et al. (2017). Friend or foe? The tumour microenvironment dilemma in colorectal cancer. Biochim Biophys Acta 1867, 1–18. doi: 10.1016/j.bbcan.2016.11.001
Coppé, J. P., Desprez, P. Y., Krtolica, A., and Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144
Corvigno, S., Wisman, G. B., Mezheyeuski, A., van der Zee, A. G., Nijman, H. W., Dahlstrand, H., et al. (2016). Markers of fibroblast-rich tumor stroma and perivascular cells in serous ovarian cancer: inter- and intra-patient heterogeneity and impact on survival. Oncotarget 7, 18573–18584. doi: 10.18632/oncotarget.7613
Costea, D. E., Hills, A., Osman, A. H., Thurlow, J., Kalna, G., Partridge, M., et al. (2013). Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res. 73, 3888–3901. doi: 10.1158/0008-5472.CAN-12-4150
Cowman, M. K. (2017). Hyaluronan and hyaluronan fragments. Adv. Carbohydr. Chem. Biochem. 74, 1–59. doi: 10.1016/bs.accb.2017.10.001
Cunningham, C. C. (2007). Talabostat. Expert Opin. Investig. Drugs 16, 1459–1465. doi: 10.1517/13543784.16.9.1459
Cyphert, J. M., Trempus, C. S., and Garantziotis, S. (2015). Size matters: molecular weight specificity of hyaluronan effects in cell biology. Int. J. Cell Biol. 2015:563818. doi: 10.1155/2015/563818
DeFilippis, R. A., Chang, H., Dumont, N., Rabban, J. T., Chen, Y. Y., Tlsty, T. D., et al. (2012). CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2, 826–839. doi: 10.1158/2159-8290.CD-12-0107
Doherty, G. J., Tempero, M., and Corrie, P. G. (2018). HALO-109-301: a Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 14, 13–22. doi: 10.2217/fon-2017-0338
Dolberg, D. S., Hollingsworth, R., Hertle, M., and Bissell, M. J. (1985). Wounding and its role in RSV-mediated tumor formation. Science 230, 676–678. doi: 10.1126/science.2996144
Duda, D. G., Duyverman, A. M., Kohno, M., Snuderl, M., Steller, E. J., Jain, R. K., et al. (2010). Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. U.S.A. 107, 21677–21682. doi: 10.1073/pnas.1016234107
Dumont, N., and Arteaga, C. L. (2002). The tumor microenvironment: a potential arbitrator of the tumor suppressive and promoting actions of TGFbeta. Differentiation 70, 574–582. doi: 10.1046/j.1432-0436.2002.700910.x
Dvorak, H. F. (1986). Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659. doi: 10.1056/NEJM198612253152606
Etzold, A., Galetzka, D., Weis, E., Bartsch, O., Haaf, T., Spix, C., et al. (2016). CAF-like state in primary skin fibroblasts with constitutional BRCA1 epimutation sheds new light on tumor suppressor deficiency-related changes in healthy tissue. Epigenetics 11, 120–131. doi: 10.1080/15592294.2016.1140295
Fischer, E., Chaitanya, K., Wüest, T., Wadle, A., Scott, A. M. M., et al. (2012). Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clin. Cancer Res. 18, 6208–6218. doi: 10.1158/1078-0432.CCR-12-0644
Fisher, G. J. (2015). Cancer resistance, high molecular weight hyaluronic acid, and longevity. J. Cell Commun. Signal. 9, 91–92. doi: 10.1007/s12079-015-0278-6
Folgueira, M. A., Maistro, S., Katayama, M. L., Roela, R. A., Mundim, F. G., Brentani, M. M., et al. (2013). Markers of breast cancer stromal fibroblasts in the primary tumour site associated with lymph node metastasis: a systematic review including our case series. Biosci. Rep. 33:e00085. doi: 10.1042/BSR20130060
Fouladi-Nashta, A. A., Raheem, K. A., Marei, W. F., Ghafari, F., and Hartshorne, G. M. (2017). Regulation and roles of the hyaluronan system in mammalian reproduction. Reproduction 153, R43–R58. doi: 10.1530/REP-16-0240
Frevert, C. W., Felgenhauer, J., Wygrecka, M., Nastase, M. V., and Schaefer, L. (2017). Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J. Histochem. Cytochem. 66, 213–227. doi: 10.1369/0022155417740880
Gaggar, A., and Weathington, N. (2016). Bioactive extracellular matrix fragments in lung health and disease. J. Clin. Invest. 126, 3176–3184. doi: 10.1172/JCI83147
Garraway, L. A., and Lander, E. S. (2013). Lessons from the cancer genome. Cell 153, 17–37. doi: 10.1016/j.cell.2013.03.002
Gascard, P., and Tlsty, T. D. (2016). Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 30, 1002–1019. doi: 10.1101/gad.279737.116
Gaudet, A. D., and Popovich, P. G. (2014). Extracellular matrix regulation of inflammation in the healthy and injured spinal cord. Exp. Neurol. 258, 24–34. doi: 10.1016/j.expneurol.2013.11.020
Ghosh, K., Vierkant, R. A., Frank, R. D., Winham, S., Visscher, D. W., Vachon, C. M., et al. (2017). Association between mammographic breast density and histologic features of benign breast disease. Breast Cancer Res. 19:134. doi: 10.1186/s13058-017-0922-6
Grillet, F., Bayet, E., Villeronce, O., Zappia, L., Lagerqvist, E. L., Pannequin, J., et al. (2017). Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 66, 1802–1810. doi: 10.1136/gutjnl-2016-311447
Hall, C. L., Yang, B., Yang, X., Zhang, S., Turley, M., Samuel, S., Turley, E. A., et al. (1995). Overexpression of the hyaluronan receptor RHAMM is transforming and is also required for H-ras transformation. Cell 82, 19–26. doi: 10.1016/0092-8674(95)90048-9
Hamada, S., Nishida, Y., Zhuo, L., Shinomura, T., Ikuta, K., Arai, E., et al. (2017). Suppression of hyaluronan synthesis attenuates the tumorigenicity of low-grade chondrosarcoma. J. Orthop. Res. doi: 10.1002/jor.23794. [Epub ahead of print].
Hamilton, S. R., Fard, S. F., Paiwand, F. F., Tolg, C., Veiseh, M., Turley, E. A., et al. (2007). The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J. Biol. Chem. 282, 16667–16680. doi: 10.1074/jbc.M702078200
Hammer, A. M., Sizemore, G. M., Shukla, V. C., Avendano, A., Sizemore, S. T., Ostrowski, M. C., et al. (2017). Stromal PDGFR-alpha activation enhances matrix stiffness, impedes mammary ductal development, and accelerates tumor growth. Neoplasia 19, 496–508. doi: 10.1016/j.neo.2017.04.004
Han, Y., Zhang, Y., Jia, T., and Sun, Y. (2015). Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumour Biol. 36, 1385–1394. doi: 10.1007/s13277-015-3230-8
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Heldin, P., Basu, K., Kozlova, I., and Porsch, H. (2014). HAS2 and CD44 in breast tumorigenesis. Adv. Cancer Res. 123, 211–229. doi: 10.1016/B978-0-12-800092-2.00008-3
Hinds, P., and Pietruska, J. (2017). Senescence and tumor suppression. F1000Res 6:2121. doi: 10.12688/f1000research.11671.1
Hofheinz, R. D., al-Batran, S. E., Hartmann, F., Hartung, G., Jager, D., Renner, C., et al. (2003). Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26, 44–48. doi: 10.1159/000069863
Hutchenreuther, J., and Leask, A. (2017). Why target the tumor stroma in melanoma? J. Cell Commun. Signal. 12, 113–118. doi: 10.1007/s12079-017-0419-1
Ikuta, K., Ota, T., Zhuo, L., Urakawa, H., Kozawa, E., Hamada, S., et al. (2017). Antitumor effects of 4-methylumbelliferone, a hyaluronan synthesis inhibitor, on malignant peripheral nerve sheath tumor. Int. J. Cancer 140, 469–479. doi: 10.1002/ijc.30460
Illmensee, K., and Mintz, B. (1976). Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts Proc. Natl. Acad. Sci. U.S.A. 73, 549–553. doi: 10.1073/pnas.73.2.549
Itano, N., Zhuo, L., and Kimata, K. (2008). Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer Sci. 99, 1720–1725. doi: 10.1111/j.1349-7006.2008.00885.x
Jacob, M., Chang, L., and Pure, E. (2012). Fibroblast activation protein in remodeling tissues. Curr. Mol. Med. 12, 1220–1243. doi: 10.2174/156652412803833607
Jansson, S., Bendahl, P. O., Larsson, A. M., Aaltonen, K. E., and Ryden, L. (2016). Prognostic impact of circulating tumor cell apoptosis and clusters in serial blood samples from patients with metastatic breast cancer in a prospective observational cohort. BMC Cancer 16:433. doi: 10.1186/s12885-016-2406-y
Ji, X., Zhu, X., and Lu, X. (2017). Effect of cancer-associated fibroblasts on radiosensitivity of cancer cells. Future Oncol. 13(17): 1537–1550. doi: 10.2217/fon-2017-0054
Jiang, H., Hegde, S., and DeNardo, D. G. (2017). Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. 66, 1037–1048. doi: 10.1007/s00262-017-2003-1
Jiang, H., Hegde, S., Knolhoff, B. L., Zhu, Y., Herndon, J. M., DeNardo, D. G., et al. (2016). Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 22, 851–860. doi: 10.1038/nm.4123
Jones, M. L., Siddiqui, J., Pienta, K. J., and Getzenberg, R. H. (2013). Circulating fibroblast-like cells in men with metastatic prostate cancer. Prostate 73, 176–181. doi: 10.1002/pros.22553
Kalluri, R. (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. doi: 10.1038/nrc.2016.73
Kalluri, R., and Zeisberg, M. (2006). Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401. doi: 10.1038/nrc1877
Karn, T., Pusztai, L., Rody, A., Holtrich, U., and Becker, S. (2015). The influence of host factors on the prognosis of breast cancer: stroma and immune cell components as cancer biomarkers. Curr. Cancer Drug Targets 15, 652–664. doi: 10.2174/156800961508151001101209
Karousou, E., D'Angelo, M. L., Kouvidi, K., Vigetti, D., Viola, M., Passi, A., et al. (2014). Collagen VI and hyaluronan: the common role in breast cancer. Biomed. Res. Int. 2014:606458. doi: 10.1155/2014/606458
Kavasi, R. M., Berdiaki, A., Spyridaki, I., Corsini, E., Tsatsakis, A., Tzanakakis, G., et al. (2017). HA metabolism in skin homeostasis and inflammatory disease. Food Chem. Toxicol. 101, 128–138. doi: 10.1016/j.fct.2017.01.012
Keely, P. J. (2011). Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 16, 205–219. doi: 10.1007/s10911-011-9226-0
Kenny, P. A., and Bissell, M. J. (2003). Tumor reversion: correction of malignant behavior by microenvironmental cues. Int. J. Cancer 107, 5688–695. doi: 10.1002/ijc.11491
Khaldoyanidi, S. K., Goncharova, V., Mueller, B., and Schraufstatter, I. U. (2014). Hyaluronan in the healthy and malignant hematopoietic microenvironment. Adv. Cancer Res. 123, 149–189. doi: 10.1016/B978-0-12-800092-2.00006-X
Klein, G. (2014). Evolutionary aspects of cancer resistance. Semin. Cancer Biol. 25, 10–14. doi: 10.1016/j.semcancer.2014.01.001
Koczorowska, M. M., Tholen, S., Bucher, F., Lutz, L., Kizhakkedathu, J. N., Schilling, O., et al. (2016). Fibroblast activation protein-alpha, a stromal cell surface protease, shapes key features of cancer associated fibroblasts through proteome and degradome alterations. Mol. Oncol. 10, 40–58. doi: 10.1016/j.molonc.2015.08.001
Koyama, H., Hibi, T., Isogai, Z., Yoneda, M., Fujimori, M., Amano, J., et al. (2007). Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: possible involvement of versican/PG-M. Am. J. Pathol. 170, 1086–1099. doi: 10.2353/ajpath.2007.060793
Kubo, N., Araki, K., Kuwano, H., and Shirabe, K. (2016). Cancer-associated fibroblasts in hepatocellular carcinoma. World J. Gastroenterol. 22, 6841–6850. doi: 10.3748/wjg.v22.i30.6841
Kudo, D., Suto, A., and Hakamada, K. (2017). The Development of a Novel Therapeutic Strategy to Target Hyaluronan in the Extracellular Matrix of Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 18(3). doi: 10.3390/ijms18030600
Kumari, S., Panda, T. K., and Pradhan, T. (2017). Lysyl OXIDASE: its diversity in health and diseases. Indian J. Clin. Biochem. 32, 134–141. doi: 10.1007/s12291-016-0576-7
Kuzet, S. E., and Gaggioli, C. (2016). Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 365, 607–619. doi: 10.1007/s00441-016-2467-x
Labernadie, A., Kato, T., Brugues, A., Serra-Picamal, X., Derzsi, S., Arwert, E., et al. (2017). A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 19, 224–237. doi: 10.1038/ncb3478
Lai, D., Ma, L., and Wang, F. (2012). Fibroblast activation protein regulates tumor-associated fibroblasts and epithelial ovarian cancer cells. Int. J. Oncol. 41, 541–550. doi: 10.3892/ijo.2012.1475
Lawrenson, K., Grun, B., Benjamin, E., Jacobs, I. J., Dafou, D., and Gayther, S. A. (2010). Senescent fibroblasts promote neoplastic transformation of partially transformed ovarian epithelial cells in a three-dimensional model of early stage ovarian cancer. Neoplasia 12, 317–325. doi: 10.1593/neo.91948
Lee, D. E., Ayoub, N., and Agrawal, D. K. (2016). Mesenchymal stem cells and cutaneous wound healing: novel methods to increase cell delivery and therapeutic efficacy. Stem Cell Res. Ther. 7:37. doi: 10.1186/s13287-016-0303-6
Li, X., Sterling, J. A., Fan, K. H., Vessella, R. L., Shyr, Y., Bhowmick, N. A., et al. (2012). Loss of TGF-beta responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol. Cancer Res. 10, 494–503. doi: 10.1158/1541-7786.MCR-11-0506
Lin, H. J., and Lin, J. (2017). Seed-in-soil: pancreatic cancer influenced by tumor microenvironment. Cancers 9:E93. doi: 10.3390/cancers9070093
Li, Y., Jiang, D., Liang, J., Meltzer, E. B., Gray, A., Noble, P. W., et al. (2011). Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 208, 1459–1471. doi: 10.1084/jem.20102510
Litwiniuk, M., Krejner, A., Speyrer, M. S., Gauto, A. R., and Grzela, T. (2016). Hyaluronic acid in inflammation and tissue regeneration. Wounds 28, 78–88.
Liu, J., Xia, J., Zhang, Y., Fu, M., Gong, S., and Guo, Y. (2017). Associations between the expression of MTA1 and VEGF-C in esophageal squamous cell carcinoma with lymph angiogenesis and lymph node metastasis. Oncol. Lett. 14, 3275–3281. doi: 10.3892/ol.2017.6530
Lo, A., Wang, L. C., Scholler, J., Monslow, J., Avery, D., Pure, E., et al. (2015). Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 75, 2800–2810. doi: 10.1158/0008-5472.CAN-14-3041
Luo, Z., Wang, Q., Lau, W. B., Lau, B., Xu, L., Zhou, S., et al. (2016). Tumor microenvironment: the culprit for ovarian cancer metastasis? Cancer Lett. 377, 174–182. doi: 10.1016/j.canlet.2016.04.038
Mangge, H., Niedrist, T., Renner, W., Lyer, S., Alexiou, C., and Haybaeck, J. (2017). New diagnostic and therapeutic aspects of pancreatic ductal adenocarcinoma. Curr. Med. Chem. 24, 3012–3024. doi: 10.2174/0929867324666170510150124
Man, Y., Cao, J., Jin, S., Xu, G., Pan, B., Shang, L., et al. (2014). Newly identified biomarkers for detecting circulating tumor cells in lung adenocarcinoma. Tohoku J. Exp. Med. 234, 29–40. doi: 10.1620/tjem.234.29
Martincorena, I., Roshan, A., Gerstung, M., Ellis, P., Van Loo, P., McLaren, S., et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. doi: 10.1126/science.aaa6806
Marusyk, A., Tabassum, D. P., Janiszewska, M., Place, A. E., Trinh, A., Polyak, K., et al. (2016). Spatial proximity to fibroblasts impacts molecular features and therapeutic sensitivity of breast cancer cells influencing clinical outcomes. Cancer Res. 76, 6495–6506. doi: 10.1158/0008-5472.CAN-16-1457
May, C. D., Sphyris, N., Evans, K. W., Werden, S. J., and Guo, W. S. A. (2011). Mani. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 13:202. doi: 10.1186/bcr2789
Maytin, E. V. (2016). Hyaluronan: more than just a wrinkle filler. Glycobiology 26, 553–559. doi: 10.1093/glycob/cww033
McAtee, C. O., Barycki, J. J., and Simpson, M. A. (2014). Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv. Cancer Res. 123, 1–34. doi: 10.1016/B978-0-12-800092-2.00001-0
Mele, V., Sokol, L., Kolzer, V. H., Pfaff, D., Muraro, M. G., Lugli, A., et al. (2017). The hyaluronan-mediated motility receptor RHAMM promotes growth, invasiveness and dissemination of colorectal cancer. Oncotarget 8, 70617–70629. doi: 10.18632/oncotarget.19904
Mellone, M., Hanley, C. J., Thirdborough, S., Mellows, T., Garcia, E., Thomas, G. J., et al. (2016). Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 9, 114–132 doi: 10.18632/aging.101127
Mezawa, Y., and Orimo, A. (2016). The roles of tumor- and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas. Cell Tissue Res. 365, 675–689. doi: 10.1007/s00441-016-2471-1
Mirza, S., Jain, N., and Rawal, R. (2017). Evidence for circulating cancer stem-like cells and epithelial-mesenchymal transition phenotype in the pleurospheres derived from lung adenocarcinoma using liquid biopsy. Tumour Biol. 39:1010428317695915. doi: 10.1177/1010428317695915
Mishra, P. J., Humeniuk, R., Medina, D. J., Alexe, G., Mesirov, J. P., Banerjee, D., et al. (2008). Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 68, 4331–4339. doi: 10.1158/0008-5472.CAN-08-0943
Misra, S., Hascall, V. C., Markwald, R. R., and Ghatak, S. (2015). Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 6:201. doi: 10.3389/fimmu.2015.00201
Mi, Z., Bhattacharya, S. D., Kim, V. M., Guo, H., Talbot, L. J., and Kuo, P. C. (2011). Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 32, 477–487. doi: 10.1093/carcin/bgr009
Mu, Z., Benali-Furet, N., Uzan, G., Znaty, A., Ye, Z., Paolillo, C., et al. (2016). Detection and characterization of circulating tumor associated cells in metastatic breast cancer. Int J Mol Sci 17:E1665. doi: 10.3390/ijms17101665
Nannini, M., Ravegnini, G., Angelini, S., Astolfi, A., Biasco, G., and Pantaleo, M. A. (2015). miRNA profiling in gastrointestinal stromal tumors: implication as diagnostic and prognostic markers. Epigenomics 7, 1033–1049. doi: 10.2217/epi.15.52
Nikitovic, D., Tzardi, M., Berdiaki, A., Tsatsakis, A., and Tzanakakis, G. N. (2015). Cancer microenvironment and inflammation: role of hyaluronan. Front. Immunol. 6:169. doi: 10.3389/fimmu.2015.00169
Northey, J. J., Przybyla, L., and Weaver, V. M. (2017). Tissue force programs cell fate and tumor aggression. Cancer Discov. 7, 1224–1237. doi: 10.1158/2159-8290.CD-16-0733
Pantziarka, P. (2015). Primed for cancer: Li Fraumeni Syndrome and the pre-cancerous niche. Ecancermedicalscience 9:541. doi: 10.3332/ecancer.2015.541
Paszek, M. J., Zahir, N., Johnson, K. R., Lakins, J. N., Rozenberg, G. I., Weaver, V. M., et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254. doi: 10.1016/j.ccr.2005.08.010
Paulsson, J., and Micke, P. (2014). Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin. Cancer Biol. 25, 61–68. doi: 10.1016/j.semcancer.2014.02.006
Peiris-Pagès, M., Smith, D. L., Gyorffy, B., Sotgia, F., and Lisanti, M. P. (2015). Proteomic identification of prognostic tumour biomarkers, using chemotherapy-induced cancer-associated fibroblasts. Aging 7, 816–838. doi: 10.18632/aging.100808
Perryman, L., and Erler, J. T. (2014). Lysyl oxidase in cancer research. Future Oncol. 10, 1709–1717. doi: 10.2217/fon.14.39
Petitprez, F., Vano, Y. A., Becht, E., Giraldo, N. A., de Reynies, A., Fridman, W. H., et al. (2017). Transcriptomic analysis of the tumor microenvironment to guide prognosis and immunotherapies. Cancer Immunol. Immunother. doi: 10.1007/s00262-017-2058-z. [Epub ahead of print].
Postovit, L. M., Margaryan, N. V., Seftor, E. A., and Hendrix, M. J. (2008). Role of nodal signaling and the microenvironment underlying melanoma plasticity. Pigment Cell Melanoma Res. 21, 348–357. doi: 10.1111/j.1755-148X.2008.00463.x
Provenzano, P. P., and Hingorani, S. R. (2013). Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 108, 1–8. doi: 10.1038/bjc.2012.569
Provenzano, P. P., and Keely, P. J. (2011). Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J Cell Sci 124(Pt 8), 1195–1205. doi: 10.1242/jcs.067009
Radisky, D. C., Kenny, P. A., and Bissell, M. J. (2007). Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J. Cell. Biochem. 101, 830–839. doi: 10.1002/jcb.21186
Ray, A., Slama, Z. M., Morford, R. K., Madden, S. A., and Provenzano, P. P. (2017). Enhanced directional migration of cancer stem cells in 3D aligned collagen matrices. Biophys. J. 112, 1023–1036. doi: 10.1016/j.bpj.2017.01.007
Rhee, K. J., Lee, J. I., and Eom, Y. W. (2015). Mesenchymal stem cell-mediated effects of tumor support or suppression. Int. J. Mol. Sci. 16, 30015–30033. doi: 10.3390/ijms161226215
Safdar, M. H., Hussain, Z., Abourehab, M. A. S., Hasan, H., Afzal, S., and Thu, H. E. (2017). New developments and clinical transition of hyaluronic acid-based nanotherapeutics for treatment of cancer: reversing multidrug resistance, tumour-specific targetability and improved anticancer efficacy. Artif. Cells Nanomed. Biotechnol. doi: 10.1080/21691401.2017.1397001. [Epub ahead of print].
Salo, T., Vered, M., Bello, I. O., Nyberg, P., Bitu, C. C., Dayan, D., et al. (2014). Insights into the role of components of the tumor microenvironment in oral carcinoma call for new therapeutic approaches. Exp. Cell Res. 325, 58–64. doi: 10.1016/j.yexcr.2013.12.029
Santi, A., Kugeratski, F. G., and Zanivan, S. (2017). Cancer associated fibroblasts: the architects of stroma remodelling. Proteomics 18:e1700167. doi: 10.1002/pmic.201700167
Sasaki, S., Baba, T., Shinagawa, K., Matsushima, K., and Mukaida, N. (2014). Crucial involvement of the CCL3-CCR5 axis-mediated fibroblast accumulation in colitis-associated carcinogenesis in mice. Int. J. Cancer 135, 1297–1306. doi: 10.1002/ijc.28779
Sato, N., Cheng, X. B., Kohi, S., Koga, A., and Hirata, K. (2016). Targeting hyaluronan for the treatment of pancreatic ductal adenocarcinoma. Acta Pharm. Sin. B 6, 101–105. doi: 10.1016/j.apsb.2016.01.002
Schedin, P., and Keely, P. J. (2011). Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb. Perspect. Biol. 3:a003228. doi: 10.1101/cshperspect.a003228
Scherz-Shouval, R., Santagata, S., Mendillo, M. L., Sholl, L. M., Ben-Aharon, I., Lindquist, S., et al. (2014). The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158, 564–578. doi: 10.1016/j.cell.2014.05.045
Schwertfeger, K. L., Cowman, M. K., Telmer, P. G., Turley, E. A., and McCarthy, J. B. (2015). Hyaluronan, inflammation, and breast cancer progression. Front. Immunol. 6:236. doi: 10.3389/fimmu.2015.00236
Scott, A. M., Wiseman, G., Welt, S., Adjei, A., Lee, F. T., Old, L. J., et al. (2003). A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 9, 1639–1647.
Senbanjo, L. T., and Chellaiah, M. A. (2017). CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 5:18. doi: 10.3389/fcell.2017.00018
Shah, S. H., Miller, P., Garcia-Contreras, M., Ao, Z., Machlin, L., Issa, E., et al. (2015). Hierarchical paracrine interaction of breast cancer associated fibroblasts with cancer cells via hMAPK-microRNAs to drive ER-negative breast cancer phenotype. Cancer Biol. Ther. 16, 1–11. doi: 10.1080/15384047.2015.1071742
Shekhar, M. P., Santner, S., Carolin, K. A., and Tait, L. (2007). Direct involvement of breast tumor fibroblasts in the modulation of tamoxifen sensitivity. Am. J. Pathol. 170, 1546–1560. doi: 10.2353/ajpath.2007.061004
Sherman, L. S., Matsumoto, S., Su, W., Srivastava, T., and Back, S. A. (2015). Hyaluronan synthesis, catabolism, and signaling in neurodegenerative diseases. Int. J. Cell Biol. 2015:368584. doi: 10.1155/2015/368584
Shigeeda, W., Shibazaki, M., Yasuhira, S., Masuda, T., Tanita, T., Kaneko, Y., et al. (2017). Hyaluronic acid enhances cell migration and invasion via the YAP1/TAZ-RHAMM axis in malignant pleural mesothelioma. Oncotarget 8, 93729–93740. doi: 10.18632/oncotarget.20750
Shih, F. Y., Wu, Y. C., Shih, Y. S., Shih, M. C., Wu, T. S., Wang, W. H., et al. (2017). Environment-insensitive and gate-controllable photocurrent enabled by bandgap engineering of MoS2 junctions. Sci. Rep. 7:44768. doi: 10.1038/srep44768
Shinagawa, K., Kitadai, Y., Tanaka, M., Sumida, T., Kodama, M., Higashi, Y., et al. (2010). Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int. J. Cancer 127, 2323–2333. doi: 10.1002/ijc.25440
Simpson, M. A., and Lokeshwar, V. B. (2008). Hyaluronan and hyaluronidase in genitourinary tumors. Front. Biosci. 13, 5664–5680. doi: 10.2741/3108
Sironen, R. K., Tammi, M., Tammi, R., Auvinen, P. K., Anttila, M., and Kosma, V. M. (2011). Hyaluronan in human malignancies. Exp. Cell Res. 317, 383–391. doi: 10.1016/j.yexcr.2010.11.017
Spaeth, E. L., Labaff, A. M., Toole, B. P., Klopp, A., Andreeff, M., and Marini, F. C. (2013). Mesenchymal CD44 expression contributes to the acquisition of an activated fibroblast phenotype via TWIST activation in the tumor microenvironment. Cancer Res. 73, 5347–5359. doi: 10.1158/0008-5472.CAN-13-0087
Straussman, R., Morikawa, T., Shee, K., Barzily-Rokni, M., Qian, Z. R., Golub, T. R., et al. (2012). Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504. doi: 10.1038/nature11183
Tanabe, Y., Sasaki, S., Mukaida, N., and Baba, T. (2016). Blockade of the chemokine receptor, CCR5, reduces the growth of orthotopically injected colon cancer cells via limiting cancer-associated fibroblast accumulation. Oncotarget 7, 48335–48345. doi: 10.18632/oncotarget.10227
Tan, Q., Saggar, J. K., Yu, M., Wang, M., and Tannock, I. F. (2015). Mechanisms of drug resistance related to the microenvironment of solid tumors and possible strategies to inhibit them. Cancer J. 21, 254–262. doi: 10.1097/PPO.0000000000000131
Tao, L., Huang, G., Song, H., Chen, Y., and Chen, L. (2017). Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol. Lett. 14, 2611–2620. doi: 10.3892/ol.2017.6497
Tchou, J., Kossenkov, A. V., Chang, L., Satija, C., Herlyn, M., Pure, E., et al. (2012). Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Med. Genomics 5:39. doi: 10.1186/1755-8794-5-39
Teder, P., Vandivier, R. W., Jiang, D., Liang, J., Cohn, L., Noble, P. W., et al. (2002). Resolution of lung inflammation by CD44. Science 296, 155–58. doi: 10.1126/science.1069659
Tian, X., Azpurua, J., Hine, C., Vaidya, A., Myakishev-Rempel, M., Ablaeva, J., et al. (2013). High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349. doi: 10.1038/nature12234
Tian, X., Azpurua, J., Ke, Z., Augereau, A., Zhang, Z. D., Seluanov, A., et al. (2015). INK4 locus of the tumor-resistant rodent, the naked mole rat, expresses a functional p15/p16 hybrid isoform. Proc. Natl. Acad. Sci. U.S.A. 112, 1053–1058. doi: 10.1073/pnas.1418203112
Tolg, C., McCarthy, J. B., Yazdani, A., and Turley, E. A. (2014). Hyaluronan and RHAMM in wound repair and the “cancerization” of stromal tissues. Biomed Res. Int. 2014:103923. doi: 10.1155/2014/103923
Tolg, C., Yuan, H., Flynn, S. M., Basu, K., Ma, J., Turley, E. A., et al. (2017). Hyaluronan modulates growth factor induced mammary gland branching in a size dependent manner. Matrix Biol. 63:117–132. doi: 10.1016/j.matbio.2017.02.003
Tomasetti, C., Vogelstein, B., and Parmigiani, G. (2013). Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl. Acad. Sci. U.S.A. 110, 1999–2004. doi: 10.1073/pnas.1221068110
Tommelein, J., Verset, L., Boterberg, T., Demetter, P., Bracke, M., and De Wever, O. (2015). Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front. Oncol. 5:63. doi: 10.3389/fonc.2015.00063
Triggs-Raine, B., and Natowicz, M. R. (2015). Biology of hyaluronan: insights from genetic disorders of hyaluronan metabolism. World J. Biol. Chem. 6, 110–120. doi: 10.4331/wjbc.v6.i3.110
Tung, J. C., Barnes, J. M., Desai, S. R., Sistrunk, C., Conklin, M. W., Weaver, V. M., et al. (2015). Tumor mechanics and metabolic dysfunction. Free Radic. Biol. Med. 79, 269–280. doi: 10.1016/j.freeradbiomed.2014.11.020
Turley, E. A., Wood, D. K., and McCarthy, J. B. (2016). Carcinoma cell hyaluronan as a portable cancerized prometastatic microenvironment. Cancer Res. 76, 2507–2512. doi: 10.1158/0008-5472.CAN-15-3114
Veiseh, M., Kwon, D. H., Borowsky, A. D., Tolg, C., Leong, H. S., Bissell, M. J., et al. (2014). Cellular heterogeneity profiling by hyaluronan probes reveals an invasive but slow-growing breast tumor subset. Proc. Natl. Acad. Sci. U.S.A. 111, E1731–E1739. doi: 10.1073/pnas.1402383111
Veiseh, M., Leith, S. J., Tolg, C., Elhayek, S. S., Bahrami, S. B., Turley, E., et al. (2015). Uncovering the dual role of RHAMM as an HA receptor and a regulator of CD44 expression in RHAMM-expressing mesenchymal progenitor cells. Front. Cell Dev. Biol. 3:63. doi: 10.3389/fcell.2015.00063
Vinnicombe, S. J. (2017). Breast density: why all the fuss?. Clin. Radiol. 73, 334–357. doi: 10.1016/j.crad.2017.11.018
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A. W., et al. (2013). Cancer genome landscapes.” Science 339, 1546–1558. doi: 10.1126/science.1235122
von Ahrens, D., Bhagat, T. D., Nagrath, D., Maitra, A., and Verma, A. (2017). The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 10:76. doi: 10.1186/s13045-017-0448-5
Wang, C., Mu, Z., Chervoneva, I., Austin, L., Ye, Z., Rossi, G., Yang, H., et al. (2017). Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 161 83–94. doi: 10.1007/s10549-016-4026-2
Wang, F., Hansen, R. K., Radisky, D., Yoneda, T., Barcellos-Hoff, M. H., Bissell, M. J., et al. (2002). Phenotypic reversion or death of cancer cells by altering signaling pathways in three-dimensional contexts. J. Natl. Cancer Inst. 94, 1494–1503. doi: 10.1093/jnci/94.19.1494
Wang, L. C., Lo, A., Scholler, J., Sun, J., Majumdar, R., S. Albelda, S. M., et al. (2014). Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2, 154–166. doi: 10.1158/2326-6066.CIR-13-0027
Weigel, P. H. (2015). Hyaluronan Synthase: The mechanism of initiation at the reducing end and a pendulum model for polysaccharide translocation to the cell exterior. Int. J. Cell Biol. 2015:367579. doi: 10.1155/2015/367579
Weigel, P. H. (2017). Planning, evaluating and vetting receptor signaling studies to assess hyaluronan size-dependence and specificity. Glycobiology 27, 796–799. doi: 10.1093/glycob/cwx056
Weigel, P. H., and Baggenstoss, B. A. (2017). What is special about 200 kDa hyaluronan that activates hyaluronan receptor signaling? Glycobiology 27, 868–877. doi: 10.1093/glycob/cwx039
Werb, Z., and Lu, P. (2015). The Role of Stroma in Tumor Development. Cancer J. 21, 250–253. doi: 10.1097/PPO.0000000000000127
Wight, T. N. (2017). Provisional matrix: A role for versican and hyaluronan. Matrix Biol. 60–61, 38–56. doi: 10.1016/j.matbio.2016.12.001
Wight, T. N., Frevert, C. W., Debley, J. S., Reeves, S. R., Parks, W. C., and Ziegler, S. F. (2017). Interplay of extracellular matrix and leukocytes in lung inflammation. Cell. Immunol. 312, 1–14. doi: 10.1016/j.cellimm.2016.12.003
Winslow, S., Leandersson, K., Edsjo, A., and Larsson, C. (2015). Prognostic stromal gene signatures in breast cancer. Breast Cancer Res. 17:23. doi: 10.1186/s13058-015-0530-2
Wong, K. M., Horton, K. J., Coveler, A. L., Hingorani, S. R., and Harris, W. P. (2017). Targeting the tumor stroma: the biology and clinical development of pegylated recombinant human hyaluronidase (PEGPH20). Curr. Oncol. Rep. 19:47. doi: 10.1007/s11912-017-0608-3
Wu, R. L., Huang, L., Zhao, H. C., and Geng, X. P. (2017). Hyaluronic acid in digestive cancers. J. Cancer Res. Clin. Oncol. 143, 1–16. doi: 10.1007/s00432-016-2213-5
Xu, G., Tan, Y., Xu, T., Yin, D., Wang, M., Shen, M., et al. (2017). Hyaluronic acid-functionalized electrospun PLGA nanofibers embedded in a microfluidic chip for cancer cell capture and culture. Biomater. Sci. 5, 752–761. doi: 10.1039/C6BM00933F
Yamauchi, M., Barker, T. H., Gibbons, D. L., and Kurie, J. M. (2018). The fibrotic tumor stroma. J. Clin. Invest. 128, 16–25. doi: 10.1172/JCI93554
Yu, M., Bardia, A., Wittner, B. S., Stott, S. L., Smas, M. E., Haber, D. A., et al. (2013). Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584. doi: 10.1126/science.1228522
Yu, M., and Tannock, I. F. (2012). Targeting tumor architecture to favor drug penetration: a new weapon to combat chemoresistance in pancreatic cancer? Cancer Cell 21, 327–329. doi: 10.1016/j.ccr.2012.03.002
Zhang, C., Cao, S., Toole, B. P., and Xu, Y. (2015). Cancer may be a pathway to cell survival under persistent hypoxia and elevated ROS: a model for solid-cancer initiation and early development. Int. J. Cancer 136, 2001–2011. doi: 10.1002/ijc.28975
Zhang, P., Lo, A., Huang, Y., Huang, G., Liang, G., Mott, J., Mao, J. H., et al. (2015). Identification of genetic loci that control mammary tumor susceptibility through the host microenvironment. Sci. Rep. 5:8919. doi: 10.1038/srep08919
Keywords: hyaluronan, cancer-associated fibroblasts, migration, tumor microenvironment, tumor initiation, circulating cancer-associated fibroblasts, metastasis
Citation: McCarthy JB, El-Ashry D and Turley EA (2018) Hyaluronan, Cancer-Associated Fibroblasts and the Tumor Microenvironment in Malignant Progression. Front. Cell Dev. Biol. 6:48. doi: 10.3389/fcell.2018.00048
Received: 17 January 2018; Accepted: 13 April 2018;
Published: 08 May 2018.
Edited by:
Catherine Park, University of California, San Francisco, United StatesReviewed by:
Kandice Tanner, National Institutes of Health (NIH), United StatesAnn E. Walts, Cedars-Sinai Medical Center, United States
Copyright © 2018 McCarthy, El-Ashry and Turley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva A. Turley, ZXZhLnR1cmxleUBsaHNjLm9uLmNh