 Casper J. E. Wahlund
Casper J. E. Wahlund Anders Eklund2
Anders Eklund2 Susanne Gabrielsson
Susanne Gabrielsson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 26 April 2017
Sec. Membrane Traffic and Organelle Dynamics
Volume 5 - 2017 | https://doi.org/10.3389/fcell.2017.00039
This article is part of the Research Topic Extracellular microvesicles in immune cell communication View all 10 articles
Cells of the airways are constantly exposed to environmental hazards including cigarette smoke, irritants, pathogens, and mechanical insults. Maintaining barrier integrity is vital, and mounting responses to threats depends on intercellular communication. Extracellular vesicles (EVs), including exosomes and microvesicles, are major signal mediators between cells, shuttling cargo in health and disease. Depending on the state of the originating cells, EVs are capable of inducing proinflammatory effects including antigen presentation, cellular migration, apoptosis induction, and inflammatory cytokine release. Cells of the airways release EVs, which can be found in bronchoalveolar lavage fluid. EVs of the airways can support inflammation in the lung, but may also exit into the circulation and carry a cocktail of pro-inflammatory molecules to recipient cells in distant organs. In this review, we discuss the possibility that EVs originating from the airways contribute to dissemination of inflammation in both lung disorders and systemic inflammatory conditions.
Intercellular communication is key in inducing and resolving inflammation. Extracellular vesicles (EV) are intercellular messengers present in all body fluids, transporting proteins, lipids, and nucleic acids(Raposo and Stoorvogel, 2013; Lo Cicero et al., 2015). They are involved in physiological processes at steady state and in pathological conditions, from sperm motility and coagulation to modulating diabetes, cancer and antigen-specific immunogenicity, or tolerance(Raposo and Stoorvogel, 2013; Robbins and Morelli, 2014). EVs are also implicated in inflammatory lung disorders including sarcoidosis, asthma and chronic obstructive pulmonary disorder (COPD), and may be a universal disseminator of inflammation. The two most studied EV species are endosome-originating exosomes (Kowal et al., 2014), and cell surface-shed microvesicles (MV; Cocucci and Meldolesi, 2015), and in this review we discuss how pulmonary EVs support inflammation in the lungs, but also how they may exit the lungs and contribute to dissemination of inflammation.
Gas exchange depends on large air-to-blood contact surface, inevitably with exposure to environmental hazards including particles, pathogens, and chemical irritants. Protective measures of the airways include anatomical barriers, ciliated epithelia, and mucus to trap and transport invaders (Ganesan et al., 2013), and a plethora of innate and adaptive immune cells throughout the airways. Barrier compromise leads to tissue injury, infection or multifactorial inflammatory disorders including asthma, COPD, and sarcoidosis. We first identified exosomes in healthy human bronchoalveolar lavage fluid (BALF), and found that they express the costimulatory molecule CD86, intercellular adhesion molecule 1 (ICAM-1, CD54) and MHC class II, and for most subjects also MHC Class I (Admyre et al., 2003). CD54 adheres to lymphocyte function associated antigen 1 (LFA-1) on antigen presenting cells (APC; Marlin and Springer, 1987), which together with CD86 suggested that pulmonary exosomes have immunomodulatory roles. Further, findings by us and others have linked lung inflammatory disorders to alterations in EV concentrations, cargo or function, and a picture is emerging of a role for EVs in inflammatory diseases.
The pulmonary EV cell sources are likely different during health and disease, but bronchial epithelial cells have been suggested as the main source of lung EVs (Kulshreshtha et al., 2013). This was based on the expression of CD63 or CD81 in epithelial cells, but for full certainty of the cellular origin the analyses should include detection of cell-specific markers in the exosomes. However, epithelial cells are at the front of environmental exposure, and bronchial epithelial cells have long been suggested to orchestrate pulmonary inflammation upon noxious stimuli (Cromwell et al., 1992), and it is likely that they respond to e.g., cigarette smoke with increased EV release. In vitro, interleukin (IL)-13 stimulation of epithelial cells released exosomes inducing proliferation of monocytic cells (Kulshreshtha et al., 2013), and decreasing EV release in vivo by GW4869, an inhibitor known to reduce EV release (Trajkovic et al., 2008), reduced disease burden in a murine asthma model (Kulshreshtha et al., 2013). Conversely, epithelial EVs may play protective roles as they carry mucins, glycoproteins vital for maintaining mucus barriers, and can bind to and neutralize influenza virus via mucin-contained alpha 2,6-sialic acids (Kesimer et al., 2009).
Macrophages are strongly increased in numbers in the lung in both COPD (Barnes, 2016) and Sarcoidosis (Zissel and Müller-Quernheim, 2015), and likely contribute to the pool of EVs. Alveolar macrophage-EVs transport suppressor of cytokine signaling (SOCS) proteins, and are taken up by alveolar epithelial cells, and inhibit IFNγ–induced activation of signal transducer and activator of transcription (STAT; Bourdonnay et al., 2015). This suggests a role for EVs in regulating pulmonary inflammatory processes (Bourdonnay et al., 2015). Exosomes from Mycobacterium-infected macrophages have inflammatory capacity (Bhatnagar and Schorey, 2007), and induce memory CD4+ and CD8+ T cell responses in vivo (Giri and Schorey, 2008). The authors reason that macrophage-released EV even represents a method for macrophages with low antigen presenting capacity to convey antigen-specific immune responses.
Endothelial MVs are affected during lung disease as a consequence of damage to lung capillaries. Circulating endothelial-derived MVs increase in smokers with signs of early emphysema (Gordon et al., 2011), and COPD patients have elevated endothelial MV numbers with increased expressions of endothelial markers (Takahashi et al., 2012). Other BAL fluid cells include eosinophils and neutrophils, which release cytokines and granules of strongly proinflammatory contents upon activation via pattern recognition receptors, Fc receptors, or cytokines (Barnes, 2008; Lambrecht and Hammad, 2015). Fernvik et al found that eosinophils have an intracellular pool enriched in the tetraspanin CD9, which was significantly reduced on activation (Fernvik et al., 1995), likely due to shedding of EVs which are enriched in CD9 (Kowal et al., 2014, 2016). Eosinophil-exosomes increase on stimulation with IFNγ, and asthma patient eosinophils release elevated exosome numbers with higher amounts of granule proteins (Mazzeo et al., 2015). Activated neutrophils release EVs carrying the granule protein myeloperoxidase (Hess et al., 1999), and can transport arachidonic acid to recipient platelets (Rossaint et al., 2016). Neutrophils communicate intensely with platelets via lipid mediators during inflammation, and mortality in Escherichia coli-induced murine lung inflammation was reduced by platelet uptake of neutrophilic EVs (Rossaint et al., 2016), speaking for an EV-based crosstalk between neutrophils and platelets in pulmonary inflammation.
Mast cell EVs can induce B and T cell proliferation (Skokos et al., 2001), dendritic cell (DC) maturation (Skokos et al., 2003), and production of anti-coagulant plasminogen activator inhibitor-1 (PAI-1) in endothelial cells (Al-Nedawi et al., 2005). Moreover, proteomic overlap of exosomes from lung patient tracheal aspirations and of exosomes from the human mastocytoma cell line-1 suggest that mast cell-EVs contribute to the pool of lung EVs (Veerappan et al., 2016).
Platelets regulate coagulation, and are also implicated in inflammation by interacting with leukocytes, and releasing proinflammatory granule contents (Herter et al., 2014). Platelet MVs aggravate LPS-induced damage of pulmonary endothelial cells (Xie et al., 2015), increase the adhesion of monocytes to endothelial cells (Barry et al., 1998), and induce IL-6 and IL-8 in an IL-1β dependent fashion in synovial joints of rheumatoid arthritis (RA) patients (Boilard et al., 2010).
APC-derived EVs can present antigen in a fashion that can induce antigen-specific immunogenicity (Raposo and Stoorvogel, 2013; Robbins and Morelli, 2014), and DC exosomes can display TLR ligands to other DCs leading to elevated tumor necrosis factor (TNF) secretion, increased DC-natural killer (NK) cell communication and elevated interferon (IFN)γ (Sobo-Vujanovic et al., 2014).
Asthma can be allergic and induced by antigen-mediated hyperreactivity, or intrinsic and associated with repeated airway epithelial insults such as through cigarette smoke or infections, both leading to breathing difficulties due to bronchoconstriction, mucus overproduction, and airway remodeling (Lambrecht and Hammad, 2015). Asthma is orchestrated by IL-4-induced IgE-production and IL-5-induced expansion of eosinophils releasing proinflammatory and bronchoconstricting granula contents (Lambrecht and Hammad, 2015). We have shown that asthma BAL fluid exosomes display altered miRNA cargo including members of Let-7 miRNAs and the miR-200 family (Levänen et al., 2013), and we also found B-cell derived exosomes to carry allergenic peptides in an MHC context (Admyre et al., 2007). In contrast, BALF exosomes from allergen-exposed mice induced antigen-specific tolerance in recipient mice (Prado et al., 2008), demonstrating the flexible nature of EVs depending on the state of the producing cell. Leukotrienes (LT) are lipid mediators promoting mucus secretion, smooth muscle contraction and airway inflammation (Schauberger et al., 2016). Platelet MVs can transfer the LT-precursor arachidonic acid, and induce LT production in recipient platelets and endothelial cells (Barry et al., 1997). We found that human DC and macrophage exosomes with functional LT-forming enzymes induce granulocyte migration (Esser et al., 2010), and asthmatic patient exosomes induce inflammatory effects in bronchial epithelial cells including elevated LT and IL-8 production (Torregrosa Paredes et al., 2012). Importantly, this IL-8 production was blocked by a Cys-LT1 inhibitor.
COPD is initiated by long-term exposure to cigarette smoke or toxic irritants, followed by a response to tissue damage by DCs and macrophages releasing factors leading to airway remodeling, with a resulting reduction in lung capacity (Barnes, 2016). A small, but increasing, group of COPD patients are individuals associated with prenatal insults such as maternal cigarette smoke during pregnancy, or low birth weight caused by impaired development or pre-term birth (Stocks and Sonnappa, 2013). Cigarette smoke has profound effects on EV release. Mononuclear cells exposed to cigarette smoke extract (CSE) release MVs inducing production of IL-8, monocyte chemoattractant protein-1 and upregulation of CD54 in bronchial epithelial cells (Cordazzo et al., 2014). CSE-exposed epithelial cells release EVs enriched in the COPD-associated protein cysteine-rich angiogenic protein 61 (CCN1), and this increase contributes to the release of vascular endothelial growth factor and IL-8 (Moon et al., 2014). EVs from smoke-exposed bronchial epithelial cells induced differentiation of lung fibroblasts into myofibroblasts, suggesting that EVs may contribute to fibrotic development in COPD (Fujita et al., 2015). The glycoprotein alpha 1 anti-trypsin (A1AT) prevents excessive inflammation by inhibiting neutrophilic and eosinophilic enzymes, and A1AT deficiency leads to emphysema development (Stockley and Turner, 2014), a driver of COPD. EVs from pulmonary endothelial cells carry A1AT and may be involved in shuttling A1AT across the alveolar membrane to recipient epithelial cells (Lockett et al., 2014), possibly to prevent excessive pulmonary inflammation. In our recent investigations currently under review for publication, our preliminary findings showed alterations in the exosomal miRNA content due to both cigarette smoking and COPD, with significant gender difference due to disease. In COPD, dysregulation of exosomal miRNA was primarily observed in men. The observed down-regulations of a subset of miRNA occurred independently of current smoking, with significant correlations to the severity of disease in male, but not female COPD patients (the presentation from the ERS international congress 2016, London, is available online1).
Sarcoidosis is a multi-organ disease affecting the lungs in most patients, but also eyes, skin, spleen, nervous system, heart, kidney and liver may be affected. Pulmonary sarcoidosis is characterized by granuloma formation, pulmonary fibrotic development and functional impairment. Sarcoidosis has an unclear etiology, the most plausible explanation being a combination of genetic predisposition and non-resolved inflammation induced by pathogens or irritants leading to granuloma formation (Baughman et al., 2011). The mechanisms for multi-organ involvement are not clear, but dissemination of any pathogen involved, as well as aggravation of inflammation may be associated to EVs which exit pulmonary compartments reaching distant sites. We have identified exosomes in the BALF of sarcoidosis patients, and found a dramatic increase in exosome numbers, with more exosomal MHC Class I and II, tetraspanins as well as Heat shock protein(HSP)70 (Qazi et al., 2010). Compared to healthy subjects, patient exosomes induced more IL-13 and IFNγ production in autologous peripheral blood mononuclear cells (PBMC), and more IL-8 release by bronchial epithelial cells. Recently, our proteomic characterization of BALF exosomes from sarcoidosis patients revealed many proteins associated with inflammation and cellular migration, most strikingly an increase in most complement components (Martinez-Bravo et al., 2017). In addition, Vitamin D-binding protein (VDBP), a transporter of Vitamin D but also a potent chemoattractant for leukocytes, and precursor to a macrophage-activating factor, was significantly increased on both BALF and serum exosomes, which may be the result of pulmonary exosomes exiting into systemic circulation.
Aside from their potential roles in pulmonary disease, we argue that lung EVs may be general disseminators of inflammation, with implications also for non-pulmonary disorders. In essence the airways represent an immunological frontline to pathogens and irritants, and airway cells exposed to e.g., cigarette smoke may respond by releasing EVs which in turn spread inflammation systemically. During inflammation, vascular permeability is dramatically increased (Nagy et al., 2008), with severe consequences for the integrity of the alveolar-capillary barrier in acute lung injury (Herold et al., 2013) and other pulmonary disorders. This will influence the exchange of EVs between the blood and the epithelial lining, with proinflammatory lung EVs reaching systemic circulation and potentially distant organs. In support of systemic spread, EVs have been shown to cross the blood-brain-barrier (BBB), and even seem to be the major transporter of a folate receptor from the choroid plexus (CP) into the cerebrospinal fluid (CSF; Grapp et al., 2013). On systemic LPS administration, the CP releases EVs inducing brain inflammation (Balusu et al., 2016), which is reduced by decreased EV release (Balusu et al., 2016). Exosomes have been used to deliver small interfering RNA across the BBB (Alvarez-Erviti et al., 2011), and to deliver curcumin with anti-inflammatory effects in the brain after nasal administration (Zhuang et al., 2011).
We hypothesize that airway cells thus convey pathogenic EVs to the circulation and thereby play yet undefined roles in other inflammatory diseases such as RA and multiple sclerosis (MS). In fact, smoking is the environmental factor most strongly linked to RA, and the early phase of RA has even been suggested to be initiated in the lungs after exposure to cigarette smoke or other irritants (Klareskog et al., 2009). Indeed, RA patients often present with immunological events preceding joint symptoms, possibly as a consequence of autoimmune events initiated in the lungs (as recently reviewed by Catrina et al. (2017). Furthermore, amongst smokers, the risk for MS is 50% higher and directly correlated to smoking frequency (Ascherio et al., 2012), thus MS is possibly also a disease with early events associated with pulmonary insult. EVs could be vehicles participating in this detrimental spread of inflammation.
Several studies have demonstrated the presence of pro-inflammatory EVs at the site of inflammation in RA (Berckmans et al., 2002; Song et al., 2005), MS (Marcos-Ramiro et al., 2014; Sáenz-Cuesta et al., 2014a,b), and inflammatory bowel disease (IBD; Mitsuhashi et al., 2016). MVs (Lindemann et al., 2001; MacKenzie et al., 2001) and exosomes (Qu et al., 2007) from several different cells are transporters of the strongly pro-inflammatory IL-1β, and membrane-bound TNF (mTNF) was found on synovial fibroblast exosomes from RA patients, but not from (non-autoimmune) osteoarthritis patients (Zhang et al., 2006). Synovial MVs from RA patients also promote coagulation, and bear markers of monocytic and granulocytic origin (Berckmans et al., 2002). In a murine autoimmune arthritis model, platelet-EV internalization by neutrophils was mediated by EVs carrying the lipid mediator enzyme 12-lipoxygenase (12-LO) combined with neutrophilic secreted phospholipase A2 IIA (sPLA2-IIA; Duchez et al., 2015). MS patients have increased serum levels of platelet MVs, which also correlate with clinical subtype (Sáenz-Cuesta et al., 2014a). MVs from MS patients disrupted endothelial barriers in vitro (Marcos-Ramiro et al., 2014), and EVs have been suggested to contribute to MS by degrading the blood brain barrier, increasing endothelial activation and promoting neural inflammation (Sáenz-Cuesta et al., 2014b). Fas ligand (FasL)-induced apoptosis is an immune regulatory mechanism, and DC-derived EVs with TNF, FasL and TNF related apoptosis inducing ligand (TRAIL) can activate natural killer cells, and induce tumor cell apoptosis (Munich et al., 2012), and could also play a role in systemic inflammation. In addition, the transcriptional regulator High mobility group box protein 1 (HMGB1), normally located in the cytosol, is released during necrosis and contributes to inflammation e.g., in acute lung injury (Kim et al., 2005), and one of the main exocytic routes of HMGB1 is via exosomes (Liu et al., 2006). HMGB1-exosomes from the lung could thus also contribute to inflammation systemically or locally at distant sites.
Airway EVs directly released from pathogens or APCs that have encountered pathogens, could also contribute to inflammation. Pseudomonas aeruginosa releases EVs with hemolysin and protease, suggested by the authors to play a role in bacterial sepsis (Kadurugamuwa and Beveridge, 1995). Also, Mycobacterium avium is an inhaled opportunistic pathogen infecting macrophages, which in turn release exosomes inducing TLR-dependent inflammation (Bhatnagar and Schorey, 2007). In a similar fashion, also patients with Inflammatory bowel disease (IBD) had intestinal proinflammatory EVs with elevated levels of IL-6, IL-8, and TNF on mRNA as well as protein level compared to healthy controls (Mitsuhashi et al., 2016). Connecting these systemic inflammatory EVs to the lung, pulmonary EVs can induce many of the effects discussed including adhesion, cellular migration and lipid mediator activities, as illustrated in Figure 1 and summarized in Table 1. Taken together, we propose that pulmonary EVs with pro-inflammatory cargo may contribute to spread of inflammation in both pulmonary and extrapulmonary disease.
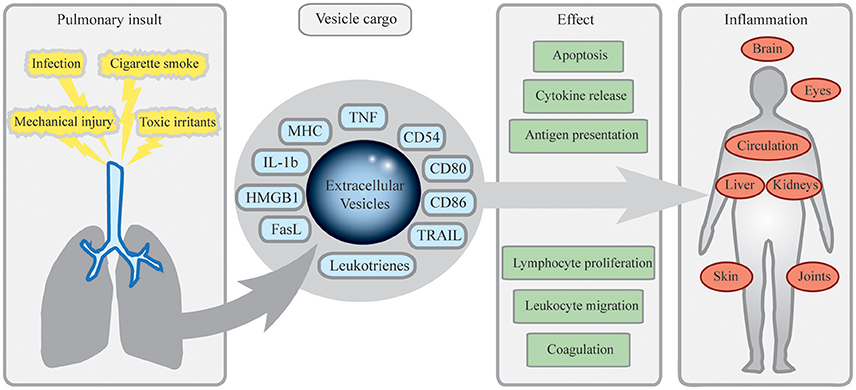
Figure 1. Mechanisms of inflammatory dissemination by pulmonary extracellular vesicles (EV). Pulmonary insults including cigarette smoke, toxic irritants, infection, or mechanical injury such as burns or particle exposure lead to release of EVs from cells of the airways. Equipped with antigen presenting, costimulatory and pro-inflammatory molecules, these EVs may exit pulmonary compartments, spread systemically and induce cellular migration, apoptosis, cytokine release, and other effects contributing to inflammation. In peripheral lymphoid organs, this could induce antigen-specific reactions leading to inflammation and autoimmunity. Pulmonary EVs may also contribute to joint inflammation in rheumatoid arthritis, fever, and systemic effects via the central nervous system, spread of sarcoidosis to the eyes and the heart, and to dissemination of components from pathogens of the airways.
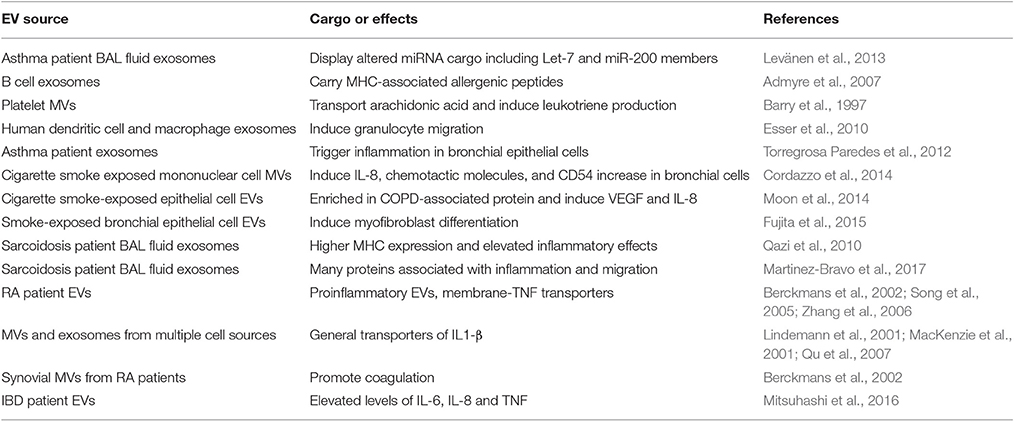
Table 1. Summary of EVs associated with inflammatory disease.
It is of importance to mention that EV research is still facing general challenges including finding the most suitable techniques for isolation and characterization of vesicles. Most commonly, EV isolation is based on differential centrifugations, which can be combined with density gradient separations to refine EV isolations. Alternative isolation methods include ultrafiltration and liquid chromatography, and it is important to carefully evaluate the choice of isolation method. Characterizing EVs involves a combination of methods, commonly including electron microscopy, flow cytometry and western blot detection of EV proteins (Théry et al., 2006). However, several markers previously considered exosome-associated including the tetraspanins CD9, CD63, and CD81 along with HSP70 (Raposo and Stoorvogel, 2013; Colombo et al., 2014) appear to be present in most EV categories (Kowal et al., 2016). Further, studying EV effects by specifically inhibiting their release or production is not possible as no universal EV-specific inhibitor has been reported so far. GW4869, an inhibitor of neutral sphingomyelases, reduces exosome production (Trajkovic et al., 2008), but is likely to also have other profound effects on the cells. Silencing of the small GTPase Rab27, implicated in exosome formation, reduces exosome-associated CD63, Alix, HSP70, and tumor susceptibility gene (TSG)101, but not CD9 or milk-fat globule-EGF factor 8 protein (MFGE8; Bobrie et al., 2012), pointing to a redundancy in exosome biogenesis.
Research based only on cell culture supernatants may oversimplify the in vivo functions of EVs due to the inherently limited milieu of cellular cooperation. Clinical samples provide the biological complexity necessary to study EVs, but are subject to experimental bias due to patient heterogeneity, and sample collection procedures. For BALF EV studies, large enough patient cohorts are necessary to reduce bias by individual variation, and importantly the BAL procedure should be performed prior to any biopsies to avoid blood contamination with plasma EVs in the BALF. Further, the volume and number of aliquots of instilled fluid, and the dwell time should preferably be constant between subjects, to minimize sampling differences. Another question is how representative BALF EVs are of the EVs present in the epithelial lining fluid and/or the tissues. Any differences in vascular permeability might lead to a difference in BALF EV profile, but this profile should still reflect the status of the lung, where more blood-derived EVs are likely to be found during inflammation.
EVs have the potential to initiate, aggravate and propagate inflammation, owing to their ability to ship proinflammatory molecules and to reach distant organs and compartments including the central nervous system. During inflammation there is a general increase in intercellular signaling with release of cytokines and lipid mediators regulating inflammatory activities, and EVs may be particularly potent. Each exosome and MV can transport a combination of inflammatory molecules, densely packed and even in combination with targeting molecules, antigen peptide-loaded MHC and costimulatory molecules. Furthermore, with the abilities to reach distant sites, EVs can expand communicative perimeters from inducing local to remote, or even systemic, effects. It is possible that EVs are central in mediating inflammation as they are major transporters of both TNF and IL1-β, so EV research has the potential of increasing understanding of fundamental inflammatory processes. With increasing evidence of EVs playing roles in pulmonary inflammatory disorders, we believe that they hold great potential in lung research to dissect pathogenesis, but also to identify biomarkers for disease. We further propose that inflammatory diseases affecting other specific organs or with systemic inflammation may be aggravated, or even initiated, by inflammatory EVs originating in and exiting from the lungs. Cigarette smoke, infection, injury and other insult to cells of the airways could lead to release of proinflammatory EVs. With the lungs being highly vascularized and on the border to the environment, they represent gates via which EVs can access systemic circulation and ship proinflammatory cargo. Further efforts are needed to study pulmonary EVs and their role in inflammation.
All authors planned the manuscript, CW drafted it and all authors critically revised it.
The authors are supported by the Swedish Medical Research Council (grant no. K2013-67x-15242-10-5), the Swedish Heart-Lung Foundation (grant no. 20140497), Hesselman's Foundation, the Stockholm County Council, the Cancer and Allergy Research Foundation, the Oscar II Jubilee Foundation, the Mats Kleberg Foundation, the Center for Allergy Research and the ChAMP consortium at the Karolinska Institutet, and the Karolinska Institutet.
SG holds a patent for the use of exosomes in immune therapy.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Åsa Wheelock, Karolinska Institute, for valuable discussions.
1. ^ERS International Congress 2016, London, Session: Extracellular vesicles in lung disease pathogenesis, presentation ID 4771, http://www.ers-education.org/home/browse-all-content.aspx?idParent=156465
Admyre, C., Bohle, B., Johansson, S. M., Focke-Tejkl, M., Valenta, R., Scheynius, A., et al. (2007). B cell-derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J. Allergy Clin. Immunol. 120, 1418–1424. doi: 10.1016/j.jaci.2007.06.040
Admyre, C., Grunewald, J., Thyberg, J., Gripenbäck, S., Tornling, G., Eklund, A., et al. (2003). Exosomes with major histocompatibility complex class II and co-stimulatory molecules are present in human BAL fluid. Eur. Respir. J. 22, 578–583. doi: 10.1183/09031936.03.00041703
Al-Nedawi, K., Szemraj, J., and Cierniewski, C. S. (2005). Mast cell-derived exosomes activate endothelial cells to secrete plasminogen activator inhibitor type 1. Arterioscler. Thromb. Vasc. Biol. 25, 1744–1749. doi: 10.1161/01.ATV.0000172007.86541.76
Alvarez-Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S., and Wood, M. J. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345. doi: 10.1038/nbt.1807
Ascherio, A., Munger, K. L., and Lünemann, J. D. (2012). The initiation and prevention of multiple sclerosis. Nat. Rev. Neurol. 8, 602–612. doi: 10.1038/nrneurol.2012.198
Balusu, S., Van Wonterghem, E., De Rycke, R., Raemdonck, K., Stremersch, S., Gevaert, K., et al. (2016). Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med. 8, 1162–1183. doi: 10.15252/emmm.201606271
Barnes, P. J. (2008). Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 8, 183–192. doi: 10.1038/nri2254
Barnes, P. J. (2016). Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 138, 16–27. doi: 10.1016/j.jaci.2016.05.011
Barry, O. P., Pratico, D., Lawson, J. A., and FitzGerald, G. A. (1997). Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Invest. 99, 2118–2127. doi: 10.1172/JCI119385
Barry, O. P., Praticò, D., Savani, R. C., and FitzGerald, G. A. (1998). Modulation of monocyte-endothelial cell interactions by platelet microparticles. J. Clin. Invest. 102, 136–144. doi: 10.1172/JCI2592
Baughman, R. P., Culver, D. A., and Judson, M. A. (2011). A concise review of pulmonary sarcoidosis. Am. J. Respir. Crit. Care Med. 183, 573–581. doi: 10.1164/rccm.201006-0865CI
Berckmans, R. J., Nieuwland, R., Tak, P. P., Böing, A. N., Romijn, F. P., Kraan, M. C., et al. (2002). Cell-derived microparticles in synovial fluid from inflamed arthritic joints support coagulation exclusively via a factor VII-dependent mechanism. Arthritis Rheum. 46, 2857–2866. doi: 10.1002/art.10587
Bhatnagar, S., and Schorey, J. S. (2007). Exosomes released from infected macrophages contain Mycobacterium avium glycopeptidolipids and are proinflammatory. J. Biol. Chem. 282, 25779–25789. doi: 10.1074/jbc.M702277200
Bobrie, A., Colombo, M., Krumeich, S., Raposo, G., and Théry, C. (2012). Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 1. doi: 10.3402/jev.v1i0.18397
Boilard, E., Nigrovic, P. A., Larabee, K., Watts, G. F., Coblyn, J. S., Weinblatt, M. E., et al. (2010). Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327, 580–583. doi: 10.1126/science.1181928
Bourdonnay, E., Zaslona, Z., Penke, L. R., Speth, J. M., Schneider, D. J., Przybranowski, S., et al. (2015). Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J. Exp. Med. 212, 729–742. doi: 10.1084/jem.20141675
Catrina, A. I., Svensson, C. I., Malmström, V., Schett, G., and Klareskog, L. (2017). Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat. Rev. Rheumatol. 13, 79–86. doi: 10.1038/nrrheum.2016.200
Cocucci, E., and Meldolesi, J. (2015). Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 25, 364–372. doi: 10.1016/j.tcb.2015.01.004
Colombo, M., Raposo, G., and Théry, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Ann. Rev. Cell Dev. Biol. 30, 255–289. doi: 10.1146/annurev-cellbio-101512-122326
Cordazzo, C., Petrini, S., Neri, T., Lombardi, S., Carmazzi, Y., Pedrinelli, R., et al. (2014). Rapid shedding of proinflammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on Ca2+ mobilization. Inflamm. Res. 63, 539–547. doi: 10.1007/s00011-014-0723-7
Cromwell, O., Hamid, Q., Corrigan, C. J., Barkans, J., Meng, Q., Collins, P. D., et al. (1992). Expression and generation of interleukin-8, IL-6 and granulocyte-macrophage colony-stimulating factor by bronchial epithelial cells and enhancement by IL-1 beta and tumour necrosis factor-alpha. Immunology 77, 330–337.
Duchez, A. C., Boudreau, L. H., Naika, G. S., Bollinger, J., Belleannee, C., Cloutier, N., et al. (2015). Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc. Natl. Acad. Sci. U.S.A. 112, E3564–E3573. doi: 10.1073/pnas.1507905112
Esser, J., Gehrmann, U., D'Alexandri, F. L., Hidalgo-Estévez, A. M., Wheelock, C. E., Scheynius, A., et al. (2010). Exosomes from human macrophages and dendritic cells contain enzymes for leukotriene biosynthesis and promote granulocyte migration. J. Allergy Clin. Immunol. 126, 1032–40, 40.e1–4. doi: 10.1016/j.jaci.2010.06.039
Fernvik, E., Halldén, G., Hed, J., and Lundahl, J. (1995). Intracellular and surface distribution of CD9 in human eosinophils. APMIS 103, 699–706. doi: 10.1111/j.1699-0463.1995.tb01426.x
Fujita, Y., Araya, J., Ito, S., Kobayashi, K., Kosaka, N., Yoshioka, Y., et al. (2015). Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J. Extracellular Vesicles 4:28388. doi: 10.3402/jev.v4.28388
Ganesan, S., Comstock, A. T., and Sajjan, U. S. (2013). Barrier function of airway tract epithelium. Tissue Barriers. 1:e24997. doi: 10.4161/tisb.24997
Giri, P. K., and Schorey, J. S. (2008). Exosomes derived from M. Bovis BCG infected macrophages activate antigen-specific CD4+ and CD8+ T cells in vitro and in vivo. PloS ONE 3:e2461. doi: 10.1371/journal.pone.0002461
Gordon, C., Gudi, K., Krause, A., Sackrowitz, R., Harvey, B. G., Strulovici-Barel, Y., et al. (2011). Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am. J. Respir. Critical Care Medicine 184, 224–232. doi: 10.1164/rccm.201012-2061OC
Grapp, M., Wrede, A., Schweizer, M., Huwel, S., Galla, H. J., Snaidero, N., et al. (2013). Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 4, 2123. doi: 10.1038/ncomms3123
Herold, S., Gabrielli, N. M., and Vadász, I. (2013). Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 305, L665–L6681. doi: 10.1152/ajplung.00232.2013
Herter, J. M., Rossaint, J., and Zarbock, A. (2014). Platelets in inflammation and immunity. J. Thromb. Haemost. 12, 1764–1775. doi: 10.1111/jth.12730
Hess, C., Sadallah, S., Hefti, A., Landmann, R., and Schifferli, J. A. (1999). Ectosomes released by human neutrophils are specialized functional units. J. Immunol. 163, 4564–4573.
Kadurugamuwa, J. L., and Beveridge, T. J. (1995). Virulence factors are released from Pseudomonas aeruginosa in association with membrane vesicles during normal growth and exposure to gentamicin: a novel mechanism of enzyme secretion. J. Bacteriol. 177, 3998–4008. doi: 10.1128/jb.177.14.3998-4008.1995
Kesimer, M., Scull, M., Brighton, B., DeMaria, G., Burns, K., O'Neal, W., et al. (2009). Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J. 23, 1858–1868. doi: 10.1096/fj.08-119131
Kim, J. Y., Park, J. S., Strassheim, D., Douglas, I., Diaz del Valle, F., Asehnoune, K., et al. (2005). HMGB1 contributes to the development of acute lung injury after hemorrhage. Am. J. Physiol. Lung Cell. Mol. Physiol. 288, L958–L965. doi: 10.1152/ajplung.00359.2004
Klareskog, L., Catrina, A. I., and Paget, S. (2009). Rheumatoid arthritis. Lancet 373, 659–672. doi: 10.1016/S0140-6736(09)60008-8
Kowal, J., Arras, G., Colombo, M., Jouve, M., Morath, J. P., Primdal-Bengtson, B., et al. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U.S.A. 113, E968–E977. doi: 10.1073/pnas.1521230113
Kowal, J., Tkach, M., and Thery, C. (2014). Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 29C, 116–125. doi: 10.1016/j.ceb.2014.05.004
Kulshreshtha, A., Ahmad, T., Agrawal, A., and Ghosh, B. (2013). Proinflammatory role of epithelial cell-derived exosomes in allergic airway inflammation. J. Allergy Clin. Immunol. 131, 1194–1203, 203.e1–14. doi: 10.1016/j.jaci.2012.12.1565
Lambrecht, B. N., and Hammad, H. (2015). The immunology of asthma. Nat. Immunol. 16, 45–56. doi: 10.1038/ni.3049
Levänen, B., Bhakta, N. R., Torregrosa Paredes, P., Barbeau, R., Hiltbrunner, S., Pollack, J. L., et al. (2013). Altered microRNA profiles in bronchoalveolar lavage fluid exosomes in asthmatic patients. J. Allergy Clin. Immunol. 131, 894–903. doi: 10.1016/j.jaci.2012.11.039
Lindemann, S., Tolley, N. D., Dixon, D. A., McIntyre, T. M., Prescott, S. M., Zimmerman, G. A., et al. (2001). Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 154, 485–490. doi: 10.1083/jcb.200105058
Liu, S., Stolz, D. B., Sappington, P. L., Macias, C. A., Killeen, M. E., Tenhunen, J. J., et al. (2006). HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am. J. Physiol. Cell Physiol. 290, C990–C999. doi: 10.1152/ajpcell.00308.2005
Lo Cicero, A., Stahl, P. D., and Raposo, G. (2015). Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr. Opin. Cell Biol. 35, 69–77. doi: 10.1016/j.ceb.2015.04.013
Lockett, A. D., Brown, M. B., Santos-Falcon, N., Rush, N. I., Oueini, H., Oberle, A. J., et al. (2014). Active trafficking of alpha 1 antitrypsin across the lung endothelium. PloS ONE 9:e93979. doi: 10.1371/journal.pone.0093979
MacKenzie, A., Wilson, H. L., Kiss-Toth, E., Dower, S. K., North, R. A., and Surprenant, A. (2001). Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 15, 825-835. doi: 10.1016/S1074-7613(01)00229-1
Marcos-Ramiro, B., Oliva Nacarino, P., Serrano-Pertierra, E., Blanco-Gelaz, M. A., Weksler, B. B., Romero, I. A., et al. (2014). Microparticles in multiple sclerosis and clinically isolated syndrome: effect on endothelial barrier function. BMC Neurosci. 15:110. doi: 10.1186/1471-2202-15-110
Marlin, S. D., and Springer, T. A. (1987). Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell 51, 813–819. doi: 10.1016/0092-8674(87)90104-8
Martinez-Bravo, M. J., Wahlund, C. J., Qazi, K. R., Moulder, R., Lukic, A., Rådmark, O., et al. (2017). Pulmonary sarcoidosis is associated with exosomal vitamin D-binding protein and inflammatory molecules. J. Allergy Clin. Immunol. 139, 1186–1194. doi: 10.1016/j.jaci.2016.05.051
Mazzeo, C., Cañas, J. A., Zafra, M. P., Marco, A. R., Fernandez-Nieto, M., Sanz, V., et al. (2015). Exosome secretion by eosinophils: A possible role in asthma pathogenesis. J. Allergy Clin. Immunol. 135, 1603–1613. doi: 10.1016/j.jaci.2014.11.026
Mitsuhashi, S., Feldbrugge, L., Csizmadia, E., Mitsuhashi, M., Robson, S. C., and Moss, A. C. (2016). Luminal Extracellular Vesicles (EVs) in Inflammatory Bowel Disease (IBD) exhibit proinflammatory effects on epithelial cells and macrophages. Inflamm. Bowel Dis. 22, 1587–1595. doi: 10.1097/MIB.0000000000000840
Moon, H. G., Kim, S. H., Gao, J., Quan, T., Qin, Z., Osorio, J. C., et al. (2014). CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. Am. J. Physiol. Lung Cell. Mol. Physiol. 2307, L326–L337. doi: 10.1152/ajplung.00102.2014
Munich, S., Sobo-Vujanovic, A., Buchser, W. J., Beer-Stolz, D., and Vujanovic, N. L. (2012). Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology 1, 1074–1083. doi: 10.4161/onci.20897
Nagy, J. A., Benjamin, L., Zeng, H., Dvorak, A. M., and Dvorak, H. F. (2008). Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 11, 109–119. doi: 10.1007/s10456-008-9099-z
Prado, N., Marazuela, E. G., Segura, E., Fernández-García, H., Villalba, M., Théry, C., et al. (2008). Exosomes from bronchoalveolar fluid of tolerized mice prevent allergic reaction. J. Immunol. 181, 1519–1525. doi: 10.4049/jimmunol.181.2.1519
Qazi, K. R., Torregrosa Paredes, P., Dahlberg, B., Grunewald, J., Eklund, A., and Gabrielsson, S. (2010). Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax 65, 1016–1024. doi: 10.1136/thx.2009.132027
Qu, Y., Franchi, L., Nunez, G., and Dubyak, G. R. (2007). Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 179, 1913–1925. doi: 10.4049/jimmunol.179.3.1913
Raposo, G., and Stoorvogel, W. (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383. doi: 10.1083/jcb.201211138
Robbins, P. D., and Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208. doi: 10.1038/nri3622
Rossaint, J., Kühne, K., Skupski, J., Van Aken, H., Looney, M. R., Hidalgo, A., et al. (2016). Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 7:13464. doi: 10.1038/ncomms13464
Sáenz-Cuesta, M., Irizar, H., Castillo-Triviño, T., Muñoz-Culla, M., Osorio-Querejeta, I., Prada, A., et al. (2014a). Circulating microparticles reflect treatment effects and clinical status in multiple sclerosis. Biomarkers Med. 8, 653–661. doi: 10.2217/bmm.14.9
Sáenz-Cuesta, M., Osorio-Querejeta, I., and Otaegui, D. (2014b). Extracellular vesicles in multiple sclerosis: what are they telling us? Front. Cell. Neurosci. 8:100. doi: 10.3389/fncel.2014.00100
Schauberger, E., Peinhaupt, M., Cazares, T., and Lindsley, A. W. (2016). Lipid Mediators of allergic disease: pathways, treatments, and emerging therapeutic targets. Curr. Allergy Asthma Rep. 16:48. doi: 10.1007/s11882-016-0628-3
Skokos, D., Botros, H. G., Demeure, C., Morin, J., Peronet, R., Birkenmeier, G., et al. (2003). Mast cell-derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo. J. Immunol. 170, 3037–3045. doi: 10.4049/jimmunol.170.6.3037
Skokos, D., Le Panse, S., Villa, I., Rousselle, J. C., Peronet, R., David, B., et al. (2001). Mast cell-dependent B and T lymphocyte activation is mediated by the secretion of immunologically active exosomes. J. Immunol. 166, 868–876. doi: 10.4049/jimmunol.166.2.868
Sobo-Vujanovic, A., Munich, S., and Vujanovic, N. L. (2014). Dendritic-cell exosomes cross-present Toll-like receptor-ligands and activate bystander dendritic cells. Cell Immunol. 289, 119–127. doi: 10.1016/j.cellimm.2014.03.016
Song, Z., Marzilli, L., Greenlee, B. M., Chen, E. S., Silver, R. F., Askin, F. B., et al. (2005). Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J. Exp. Med. 201, 755–767. doi: 10.1084/jem.20040429
Stockley, R. A., and Turner, A. M. (2014). alpha-1-Antitrypsin deficiency: clinical variability, assessment, and treatment. Trends Mol. Med. 20, 105–115. doi: 10.1016/j.molmed.2013.11.006
Stocks, J., and Sonnappa, S. (2013). Early life influences on the development of chronic obstructive pulmonary disease. Ther. Adv. Respir. Dis. 7, 161–173. doi: 10.1177/1753465813479428
Takahashi, T., Kobayashi, S., Fujino, N., Suzuki, T., Ota, C., He, M., et al. (2012). Increased circulating endothelial microparticles in COPD patients: a potential biomarker for COPD exacerbation susceptibility. Thorax 67, 1067–1074. doi: 10.1136/thoraxjnl-2011-201395
Théry, C., Amigorena, S., Raposo, G., and Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protocols Cell Biol. Chapter 3, Unit 3.22. doi: 10.1002/0471143030.cb0322s30
Torregrosa Paredes, P., Esser, J., Admyre, C., Nord, M., Rahman, Q. K., Lukic, A., et al. (2012). Bronchoalveolar lavage fluid exosomes contribute to cytokine and leukotriene production in allergic asthma. Allergy 67, 911–919. doi: 10.1111/j.1398-9995.2012.02835.x
Trajkovic, K., Hsu, C., Chiantia, S., Rajendran, L., Wenzel, D., Wieland, F., et al. (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 319, 1244–1247. doi: 10.1126/science.1153124
Veerappan, A., Thompson, M., Savage, A. R., Silverman, M. L., Chan, W. S., Sung, B., et al. (2016). Mast cells and exosomes in hyperoxia-induced neonatal lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 310, L1218-L1232. doi: 10.1152/ajplung.00299.2015
Xie, R. F., Hu, P., Wang, Z. C., Yang, J., Yang, Y. M., Gao, L., et al. (2015). Platelet-derived microparticles induce polymorphonuclear leukocyte-mediated damage of human pulmonary microvascular endothelial cells. Transfusion 55, 1051–1057. doi: 10.1111/trf.12952
Zhang, H. G., Liu, C., Su, K., Yu, S., Zhang, L., Zhang, S., et al. (2006). A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J. Immunol. 176, 7385–7393. doi: 10.4049/jimmunol.176.12.7385
Zhuang, X., Xiang, X., Grizzle, W., Sun, D., Zhang, S., Axtell, R. C., et al. (2011). Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 19, 1769–1779. doi: 10.1038/mt.2011.164
Keywords: exosomes, microvesicles, inflammation mediators, Extracellular vesicles (EVs), sarcoidosis, pulmonary, COPD, asthma
Citation: Wahlund CJE, Eklund A, Grunewald J and Gabrielsson S (2017) Pulmonary Extracellular Vesicles as Mediators of Local and Systemic Inflammation. Front. Cell Dev. Biol. 5:39. doi: 10.3389/fcell.2017.00039
Received: 27 January 2017; Accepted: 30 March 2017;
Published: 26 April 2017.
Edited by:
Kaushik Choudhuri, University of Michigan Health System, USAReviewed by:
Geri Kreitzer, CUNY School of Medicine, USACopyright © 2017 Wahlund, Eklund, Grunewald and Gabrielsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanne Gabrielsson, U3VzYW5uZS5HYWJyaWVsc3NvbkBraS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.