 Sonam Parakh
Sonam Parakh Julie D. Atkin
Julie D. Atkin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 21 May 2015
Sec. Cellular Biochemistry
Volume 3 - 2015 | https://doi.org/10.3389/fcell.2015.00030
This article is part of the Research Topic Recent progress on the functional roles of protein disulfide isomerase View all 6 articles
Protein disulphide isomerase (PDI) is a multifunctional redox chaperone of the endoplasmic reticulum (ER). Since it was first discovered 40 years ago the functions ascribed to PDI have evolved significantly and recent studies have recognized its distinct functions, with adverse as well as protective effects in disease. Furthermore, post translational modifications of PDI abrogate its normal functional roles in specific disease states. This review focusses on recent studies that have identified novel functions for PDI relevant to specific diseases.
Protein disulphide isomerase (PDI) was the first folding catalyst isolated from rat liver (Goldberger et al., 1963) and it is found abundantly in many tissues, accounting for 0.8% of total cellular protein (Freedman et al., 1994). PDI is induced during endoplasmic reticulum (ER) stress (Wilkinson and Gilbert, 2004) and it serves as a vital cellular defense against general protein misfolding via its chaperone activity. It is also responsible for the isomerization, formation, and rearrangement of protein disulphide bonds, thereby providing another mechanism by which native protein conformation is maintained. Disulphide bonds play an important role in the folding and stability of proteins and they are present in more than 30% of all human proteins that traverse the secretory pathway (Fewell et al., 2001). Since most cellular compartments are reducing environments, protein disulphide bonds are usually unstable in the cytosol, although there are exceptions (Frand et al., 2000). PDI assists in redox protein folding, involving oxidation, multiple intramolecular thiol-disulphide exchanges, and isomerization (reduction) activities and it is highly specific in its interaction with different substrates. Whilst PDI is considered to be resident primarily within the ER, nonetheless it has been detected in many other diverse cellular locations, including the cell surface, cytosol, mitochondria, and extracellular matrix (Turano et al., 2002). However, the mechanism by which PDI escapes from the ER is still unclear. PDI is also present in the extracellular medium where it facilitates protein folding and reduces protein aggregation (Delom et al., 2001). Furthermore, specific functions of cell surface PDI have been identified in hepatocytes, platelets, and endothelial cells (Turano et al., 2002). This review focusses on recent advances recognizing the versatile roles of PDI in normal cellular function and also in disease states. These studies highlight novel therapeutic possibilities based on the functional properties of PDI.
PDI is a soluble 55-kDa protein that is the prototype of the PDI family of proteins which all contain the thioredoxin-like βαβαβαββα domain (Kemmink et al., 1997). Thioredoxins are a class of oxidoreductase enzymes containing a dithiol-disulphide active site that are involved in redox signaling (Moran et al., 2001). Besides PDI, 21 more family members have been described (Kozlov et al., 2010). However, the enzymatic properties of these proteins differ in their redox potential and hence substrate specificity (Jessop et al., 2009), the sequence of their active site and the pKa of the active site cysteine residues (Ellgaard and Ruddock, 2005). They are primarily localized in the ER where they maintain an oxidative environment and thereby contribute to ER homeostasis (Anelli et al., 2002).
Full length PDI contains 508 amino acids and consists of four domains namely a, b, b', a' (Figure 1). The homologous a and a' domains share 47% similarity and contain the active site, CGHC (Kemmink et al., 1996). The active site cysteine residues interact with the thiol group of a newly synthesized substrate, thus mediating the formation and isomerization of protein disulphide bonds (Gilbert, 1998). The intermediate b and b' domains are 28% identical and they assist in the binding of protein substrates but they lack the catalytically active cysteine residues (Gruber et al., 2006). PDI also contains a x linker region and an acidic C terminus containing a KDEL-ER retrieval sequence (Darby et al., 1996). Whilst the three dimensional structure of human PDI is still under investigation, the structures of each single thioredoxin domain (Nguyen et al., 2008) and the domain combinations bb'c (Denisov et al., 2009) and bb'cxac (Wang et al., 2012a) have been determined. However, the structure of yeast PDI has been solved (Tian et al., 2006) revealing that it adopts a U shape structure, with the catalytic a and a' domains facing each other. NMR and x-ray crystallography has further demonstrated that the b' domain contains the chaperone activity responsible for binding ligands and protein substrates in its hydrophobic pocket (Denisov et al., 2009).
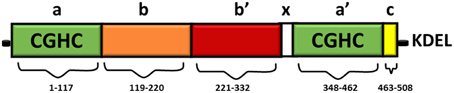
Figure 1. Domain structure of PDI. The thioredoxin-like domains are shown in green, representing the catalytically active domains a and a'. The catalytically inactive b domain and b' domains are illustrated in orange and red respectively. The linker region x (shown in white) is responsible for the U shape structure of PDI. The C terminus is illustrated in yellow, followed by an ER retrieval signal, KDEL.
The CGHC motif modulates the overall reduction potential of PDI and thus it regulates the catalytic ability of the active site cysteines to actively oxidize or reduce disulphide bonds (Chivers et al., 1997). The reduction potential of PDI is −180 mV, higher than other PDI family members, thus making it a strong oxidizing agent. The individual a and a' domains have similar oxidizing ability but conversely they have low isomerase activity (Darby et al., 1998). The b' domain is the main site for binding misfolded protein substrates but the other domains also assist in this process (Klappa et al., 1998). The catalytic domains can only catalyze basic disulphide exchange and all the domains are required to isomerize a protein substrate that has undergone conformational changes (Darby et al., 1996). Deletion of the C-terminal residues of PDI results in deactivation of its chaperone-like activity and its peptide binding ability, but this does not affect its catalytic activity in disulphide bond formation (Dai and Wang, 1997).
Although it is implied that all PDI family members possess the ability to rearrange disulphide bonds, only a few members have actually been demonstrated to perform these activities and the rest are linked to the family through evolution rather than function (Galligan and Petersen, 2012). The most commonly studied members of the PDI family after PDI are ERp57, ERp72, ERp29, ERp44, and PDIA2 (Appenzeller-Herzog and Ellgaard, 2008). There appears to be an interplay of functions amongst the PDI family and some family members are able to recompense for each other. For example, ERp72 is known to compensate for ERp57 deficiency, where it can assist in folding specific proteins (Solda et al., 2006). Certain protein substrates also appear to interact simultaneously with PDI and its family members. ERp57 and PDI engage simultaneously in forming mixed disulphides with thyroglobulin during the production and isomerization of new disulphide bonds. In addition both ERp57 and PDI are released from thyroglobulin when it dissociates from the ER (Di Jeso et al., 2005). Transferrin also requires both PDI and ERp57 for optimal folding. Furthermore, depletion of both PDI and ERp57 leads to generalized protein misfolding, impaired export from the ER, and degradation in human hepatoma cells, implying that these proteins function together (Rutkevich et al., 2010). Functional analysis in yeast revealed that ERp46 substitutes for PDI-mediated disulphide bond formation in vivo (Knoblach et al., 2003). However, PDI itself plays a key role in oxidative protein folding and no other family member can entirely compensate for its loss (Rutkevich et al., 2010).
There is also evidence that PDI family members dimerise and that this property is involved in its function. PDI was recently shown to form disulphide-independent dimers in vivo suggesting that dimerization may control efficient protein folding in the ER (Bastos-Aristizabal et al., 2014). This may be achieved by generating a reserve of inactive protein that allows the ER to respond competently to an abrupt increase in substrate availability (Bastos-Aristizabal et al., 2014). PDI family member ERp29 also dimerises, and it acts as an escort protein in the binding of thyroglobulin in the ER (Rainey-Barger et al., 2007). It has been suggested that the formation of a dimer of PDIA2, which is mediated through glycosylation (Walker et al., 2013), is increased under conditions of oxidative stress, and this dimer has increased chaperone activity compared to the monomeric form (Fu and Zhu, 2009). Several excellent recent reviews have discussed the structural aspects of the PDI family in more detail and the reader is directed to these for further information (Hatahet and Ruddock, 2009; Kozlov et al., 2010; Galligan and Petersen, 2012). This review will focus on recent advances made into the functional roles of PDI.
PDI is found in all eukaryotic organisms, whereas in prokaryotes a related homolog, Dsb, performs similar functions in facilitating protein folding (Inaba, 2009). The importance of PDI in cellular function was first realized in yeast, where PDI was found to be essential for cellular viability (LaMantia et al., 1991). To date, no viable PDI knockout strain has been reported in rodents, further emphasizing the importance of PDI in normal cellular physiology (Hatahet and Ruddock, 2009). The disulphide interchange and chaperone functions of PDI are well documented and will be summarized briefly below. Emerging evidence describing novel functions for PDI will then be described.
PDI has the ability to distinguish between partially folded, unfolded, and properly folded protein substrates, and it has a higher affinity to bind to misfolded proteins rather than native proteins through hydrophobic interactions (Klappa et al., 1997). These properties, together with its conformational flexibility, make PDI a highly effective chaperone (Irvine et al., 2014). PDI binds to a large number of protein substrates in the ER, although it is difficult to isolate and identify the individual substrates in vivo. Several methods are used to measure the chaperone activity of PDI in vitro. The rate of protein aggregation is assessed using protein substrates that do not possess cysteine residues, including GAPDH (Cai et al., 1994), rhodanese (Song and Wang, 1995), citrate synthase, alcohol dehydrogenase (Primm et al., 1996), or GFP, which on interaction with PDI causes increased fluorescent intensity as it folds into its native conformation (Mares et al., 2011).
A major function of PDI is a chaperone upregulated during ER stress. Accumulation of misfolded proteins within the ER activates the unfolded protein response (UPR). The UPR aims to reduce the load of unfolded proteins by increasing the curvature of ER, reducing protein synthesis, and by the induction of PDI and other chaperones to further increase the protein folding capacity (Hetz and Mollereau, 2014). This is achieved by activation of sensor ER proteins inositol requiring enzyme-1(IRE-1), protein kinase RNA like ER kinase (PERK), and activating transcription factor kinase 6 (ATF6), which subsequently activate UPR signaling pathways [detailed in (Sovolyova et al., 2014)]. While initially protective, prolonged UPR causes apoptosis (Schroder and Kaufman, 2005).
PDI facilitates the degradation of misfolded proteins via ER association degradation (ERAD) by translocation of these proteins from the ER to the cytoplasm, for subsequent degradation by the ubiquitin protease system. (Molinari et al., 2002; Lee et al., 2010b). It also helps in protein quality control by retaining unassembled procollagen until the correct native structure is achieved (Bottomley et al., 2001).
Other specific functions involving the chaperone activity of PDI have been described, including maintenance of the active conformation of the β subunit of collagen prolyl 4-hydroxylase (Vuori et al., 1992) and stabilization of the major histocompatibility complex's (MHC) class 1 peptide loading complex (PLC) that mediates MHC class 1 folding. Interestingly, PDI exhibits both chaperone and anti-chaperone activity depending upon its initial concentration. When PDI's chaperone activity is dominant, virtually all of the substrate protein is correctly folded. However, at low concentrations, PD1 promotes intermolecular disulphide crosslinking of substrates into large inactive aggregates via anti-chaperone activity (Puig and Gilbert, 1994).
Multiple studies have suggested that the disulphide interchange enzymatic activity of PDI is more important for its function than its chaperone activity. When its catalytic cysteines are reduced, PDI is able to react with non-native disulphides of substrate proteins to form a mixed disulphide complex. PDI then catalyzes the rearrangement of incorrectly formed disulphide bonds via isomerization reactions. This takes place with cycles of reduction (breaking of non-native disulphide bonds) and oxidation (to introduce correct pairing of cysteines) to eventually form the native disulphide bonds (Schwaller et al., 2003). The tripeptide glutathione constitutes the cellular redox buffer that maintains the redox environment of the ER (Hwang et al., 1992). After PDI has oxidized substrate proteins, it then has to be oxidized itself to complete the catalytic cycle. This function is carried out by a number of proteins including FAD binding oxidases, Ero1α, oxidized glutathione, glutathione peroxidase 7, glutathione peroxidase 8 or quiescin sulfhydryl oxidase (Wilkinson and Gilbert, 2004) (Figure 2). Interestingly, the chaperone activity of PDI is regulated by the redox state of its oxidized and reduced forms (Wang et al., 2013), suggesting a link between the two separate functions of PDI. Redox regulation of PDI can be examined experimentally in vitro using scrambled RNAse, ribonuclease and bovine pancreatic trypsin inhibitor (Darby et al., 1998; Xiao et al., 2001).
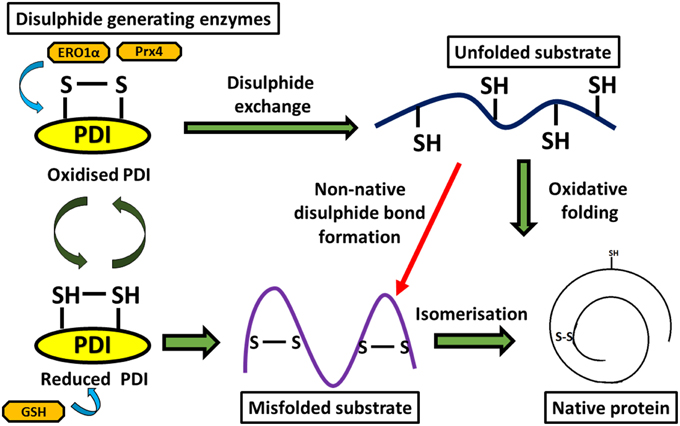
Figure 2. Diagram representing disulphide bond formation in the eukaryotic ER and redox reactions involving PDI. Oxidative folding of PDI leads to disulphide bond formation in native protein substrates. Reduced PDI facilitates isomerization of non-native bonds in protein substrates.
The in vivo redox state of PDI is complex and determined by numerous factors including the reduction potential of PDI, the glutathione redox state in the ER, and the potential reductase activity of the substrate and its availability. However, redox conditions can have a major impact on the functions of PDI. For example, PDI regulates the organization of the cytoskeleton by forming a disulphide bond to Cys 374 of β-actin via a redox dependent mechanism (Sobierajska et al., 2014).
Recent studies provide compelling evidence for a role for PDI in both the physiology and pathophysiology of disease states including diabetes (Grek and Townsend, 2014), cardiovascular diseases (Khan and Mutus, 2014), cancer (Xu et al., 2014), neurodegenerative conditions (Andreu et al., 2012) and the entry of pathogens in infectious diseases (Benham, 2012). However, precise roles for PDI in these diseases have not yet been elucidated. PDI is upregulated in various tissues during disease and surprisingly, both protective and detrimental effects have been described. These effects relate to either a loss of its normal protective function in some situations, or gain of toxic function in others. While the association between the PDI family and human disease states still requires further validation, current improvements in our understanding of the functional roles of PDI provide new insights into the physiological contribution of PDI in vivo.
PDI is highly expressed and up-regulated in numerous cancer cell types, including kidney, lung, brain, ovarian, melanoma, prostrate, and male germ cell tumors (Xu et al., 2014). Also, lower levels of PDI are associated with a higher survival rate in patients with breast cancer and glioblastoma (Thongwatchara et al., 2011), suggesting that PDI promotes the survival of cancer cells. Consistent with this notion, knockdown of PDI induces cytotoxicity in human breast cancer and neuroblastoma cell lines due to caspase activation (Hashida et al., 2011). Suppression of apoptosis by PDI has been proposed as mechanism for tumor growth and metastasis. Over-expression of PDI may therefore serve as a diagnostic marker for cancer, as suggested for glial cell cancer (Goplen et al., 2006), colorectal cancer (Ataman-Onal et al., 2013), and breast cancer (Thongwatchara et al., 2011). Cell surface PDI is also associated with cancer progression and administering of anti-PDI monoclonal antibodies inhibits the invasion of glioma cells (Goplen et al., 2006).
As increasing evidence suggests that PDI supports the survival and progression of various cancers, inhibitors of PDI may therefore have a therapeutic role against cancer progression (Xu et al., 2014). A synthesized series of PACMA (propynoic acid carbamoyl methyl amides) compounds demonstrated anticancer activity in human ovarian cancer in vitro and in vivo by a mechanism involving inhibition of PDI (Xu et al., 2012). Bacitracin, a pharmacological inhibitor of PDI, reduced the in vitro migration and invasion of human brain glial cells (Goplen et al., 2006). However, the specificity of bacitracin as an inhibitor of PDI has recently been questioned (Karala and Ruddock, 2010). Small-molecule inhibitors of PDI which bind to the CGHC active site may also have potential for improving the efficacy of chemotherapy in melanoma, as inhibition of PDI function proliferates apoptosis (Lovat et al., 2008). However, the effect of PDI in supporting tumor survival is based on the specific type of cancer and may be cell type dependent. Hence it is important to recognize the specific type of cancer cell for future applications in cancer therapy.
Neurodegenerative diseases are also known as protein misfolding disorders due to their characteristic property of accumulating insoluble ubiquitinated aggregated proteins within affected tissues. Protein misfolding within the ER triggers ER stress, and hence up-regulation of PDI, and ER stress is increasingly implicated in these diseases (Hetz and Mollereau, 2014). Most studies suggest that the induction of PDI during ER stress in neurodegenerative diseases reduces the load of misfolded proteins, and is therefore protective thus restoring proteostasis and increasing neuronal viability.
PDI is upregulated in dopaminergic neurons and Lewy bodies of patients with Parkinson's disease. Similarly PDI reduces aggregation of the Parkinson's disease-associated protein synphilin-1 in neuroblastoma cells, an activity which relies on the presence of the CGHC active site motif (Uehara et al., 2006). Similarly, PDI also prevents aggregation of another Parkinson's associated protein, α-synuclein, in cell-free in vitro systems (Cheng et al., 2010). PDI also co-localizes with neurofibrillary tangles in Alzheimer's disease patient brain tissue, and is upregulated in brains of Alzheimer's rodent models (Lee et al., 2010a) implying a role in refolding misfolded proteins in these conditions. Consistent with this notion, ERp57 is present in CSF, where it binds and reduces aggregation of β-amyloid peptides (Erickson et al., 2005). Furthermore, PDI is upregulated in response to hypoxia in the brain and PDI prevents neuronal and cardiomyocyte apoptosis, triggered by hypoxia-ischaemia in cell culture and in rodent models, by decreasing protein misfolding (Tanaka et al., 2000). In prion disorders, Wang and group suggested a pleiotropic role of PDI in the cellular management of misfolded prion protein (Wang et al., 2012b) because PDI and ERp57 are protective against prion induced toxicity in vitro (Hetz et al., 2005) and inhibition of PDI increases the production of misfolded prion proteins (Watts et al., 2009).
An important role for PDI has been implicated in amyotrophic lateral sclerosis (ALS). PDI is up-regulated and recruited to misfolded protein aggregates in sporadic human ALS (Atkin et al., 2008). PDI is also up-regulated in lumbar spinal cords from transgenic SOD1G93A mice, the most widely used animal disease model (Atkin et al., 2006). Furthermore, over-expression of PDI is protective against the formation of mutant SOD1 inclusions and ER stress, whereas knockdown of PDI using siRNA increases mutant SOD1 aggregation and inclusion formation (Walker et al., 2010). Similarly, a small molecule mimic of PDI reduces mutant SOD1 aggregation in vitro (Walker et al., 2010). Endogenous PDI co-localizes with mutant superoxide dismutase 1 (SOD1) (Atkin et al., 2006), TAR DNA-binding protein-43 (TDP-43) (Honjo et al., 2011), vesicle associated protein B (VAPB) (Tsuda et al., 2008), and Fused in Sarcoma (FUS) (Farg et al., 2012) in neuronal cells. PDI and ERp57 were identified as potential biomarkers for ALS using peripheral blood mononuclear cells (Nardo et al., 2011) and mutations in intronic variants of PDI are predicted to be a risk factor in ALS (Kwok et al., 2013).
There is also evidence that the cellular location of PDI is linked to disease outcomes in ALS. PDI is redistributed away from the ER via a reticulon-dependent process in transgenic SOD1G93A mice (Yang et al., 2009). The reticulon family of proteins function in maintaining the curvature of ER and several members of this family modulate the re-distribution of PDI away from the ER when overexpressed (Bernardoni et al., 2013). Furthermore, deletion of reticulon 4a,b enhances disease progression in SOD1G93A mice (Yang et al., 2009), highlighting the importance of a non-ER location of PDI in ALS.
Both beneficial and harmful roles for PDI in cardiovascular disease have been proposed. PDI prevents protein misfolding in the myocardium during ischemic myocardial injury (Toldo et al., 2011). PDI is also up-regulated in hypoxia induced in myocardial capillary endothelial cells (Tian et al., 2009) and this is linked to significant decreases in the rate of cardiomyocyte apoptosis in murine models (Severino et al., 2007). Similarly, PDI is also involved in endothelial cell endurance (Severino et al., 2007) and it is required for platelet derived growth factor (PDGF)-induced vascular smooth muscle cell migration (Primm and Gilbert, 2001) which is an important therapeutic target in atherosclerosis (Pescatore et al., 2012). Furthermore, increased expression of PDI is protective against endothelial cellular migration, adhesion, and tubular formation in mice suggesting an important role for PDI in angiogenesis (Tian et al., 2009). Hence these studies raise the possibility that upregulating PDI has possible future pharmacological applications in treating ischemic cardiomyopathy (Severino et al., 2007).
Diabetes is associated with coronary artery disease and an increased risk of heart failure, and PDI function is impaired in mouse models of diabetes. This may be due to alterations in its oxidoreductive state (Toldo et al., 2011). Reduced PDI has been detected in the diabetic heart after ischemia, which could explain why PDI is not protective in diabetes (Toldo et al., 2011).
However, in contrast to these protective functions, PDI has also been implicated in detrimental activities in cardiovascular diseases. Over-expression of PDI in myocytes attenuates the levels of misfolded pro-insulin while decreasing glucose stimulated insulin secretion, thereby inducing ER stress and apoptosis (Zhang et al., 2009). PDI on the surface of platelets plays an important role in thrombus formation and it is vital for the aggregation of platelets (Kim et al., 2013). Similarly, PDI is also present on at the surface of human B-lymphocytes where it has a putative role in regulating leukocyte adhesion (Bennett et al., 2000). PDI has also been implicated in platelet integrin function, tissue-factor activation, and in mice, it accumulates during fibrin and thrombus formation at sites of vascular injury (Jurk et al., 2011). PDI inhibition prevents both platelet accumulation and fibrin generation during thrombus formation (Jasuja et al., 2012). Therefore, inhibition of PDI could prevent thrombosis in coronary artery disease, suggesting that PDI inhibitors have potential as antithrombotic agents (Jasuja et al., 2012).
PDI is also implicated in mediating the entry of pathogens during infectious disease. Over-expression of PDI increases the fusion of viral membranes, leading to internalization of HIV-1 (Auwerx et al., 2009). Similarly, cell surface PDI facilitates infection of HeLa cells by mouse polyoma virus (Gilbert et al., 2006), and it also mediates the entry of cholera toxin (Stolf et al., 2011). The chaperone activity of PDI is important for folding cholera toxin subunit A1 and reducing its aggregation (Taylor et al., 2011). However, cholera intoxication is a redox dependent process. The oxidized form of PDI mediates translocation of cholera toxin into the host cell cytoplasm (Tsai et al., 2001) whereas the reduced form of PDI leads to its unfolding.
Redox-dependent post translational modifications of PDI are also linked to disease states. Due to cellular conditions, high levels of reactive nitrogen species (RNS), hydrogen peroxide and reactive oxygen species (ROS) can accumulate in cells, inducing nitrosative or oxidative stress. Nitrosative stress can lead to post translation modification of PDI by the addition of NO to active site cysteine residues, resulting in S-nitrosylation. S-nitrosylation of proteins under pathological conditions is an abnormal, irreversible process that is linked to protein misfolding, ER stress and apoptosis. Furthermore, proteins resident in the ER are particularly vulnerable to post translation modification due to the presence of critical redox regulated cysteines. Since PDI is the major enzyme responsible for modification of protein disulphide bonds, the loss of function of PDI could increase cellular protein misfolding and thus increase ER stress. S-nitrosylation of PDI inhibits its normal enzymatic activity and hence the beneficial effects of PDI, and S-nitrosylated PDI has been detected in several neurodegenerative diseases (Nakamura and Lipton, 2011; Chen et al., 2012). S-nitrosylation reduces both its chaperone and isomerase activity (Uehara et al., 2006).
S-nitrosylation of PDI has been detected in postmortem brain tissues of patients with Alzheimer's disease, Parkinson's disease (Uehara et al., 2006) and in lumbar spinal cord tissues of ALS patients and transgenic SOD1G93A mice (Walker et al., 2010). S-nitrosylation has also been reported in prion disease models using brain tissues of scrapie-263K-infected hamsters (Wang et al., 2012b). Exposure of cultured neurons to N-methyl-D-aspartate receptor (NMDA), leading to calcium influx and nitric oxide production, also resulted in the S-nitrosylation of PDI (Forrester et al., 2006). S-nitrosylated PDI (SNO-PDI) increases the levels of polyubiquitinated proteins and triggers cell death, and it is also associated with hyper-activation of NMDA (Forrester et al., 2006) and inhibition of mitochondria, leading to the generation of ROS and nitric oxide (Halloran et al., 2013). SNO-PDI accentuates the misfolding of synphilin in Parkinson disease (Forrester et al., 2006) and S-nitrosylation also increases mutant SOD1 aggregation via incorrect disulphide cross-linking of the immature, misfolded mutant SOD1, leading to neuronal cell death (Jeon et al., 2014).
As well as S-nitrosylation, other aberrant post-translational modifications of PDI have been described, including carbonylation and S-glutathionylation. Oxidized low density lipoproteins induce carbonylation, which disrupts the catalytic activity of PDI, inducing ER stress and apoptosis in vascular cells (Muller et al., 2013). Furthermore, carbonylated PDI detected in the lipid rich atherosclerotic region of human endothelial cells activates CHOP and XBP1 and induces apoptosis (Muller et al., 2013). S-glutathionylation is induced by reactive oxygen or nitrogen species and it results in formation of a disulphide bond between GSH and a cysteine residue of another protein (Xiong et al., 2011). S-glutathionylation, leading to increased protein misfolding and enhancement of the UPR (Townsend et al., 2009), has been detected primarily in relation to cancer. S-glutathionylation of PDI obliterates estrogen receptor α stability in breast cancer cells, which prevents binding of PDI to the receptor. This subsequently leads to dysregulation in ERα signaling (Xiong et al., 2012), and cell death via UPR induction (Xiong et al., 2012). S-glutathionylation also reduces the isomerase activity of PDI in ovarian cancer cells and human leukemia cells and it also decreases chaperone activity. In cultured astrocytes after cerebral ischemic reperfusion, SNO-PDI increases the levels of ubiquitinated aggregates that co-localize with SOD1 (Chen et al., 2012). These modifications can further attenuate UPR and cause neuronal cell death. Hence, aberrant modifications of PDI lead directly to harmful effects as well as loss of the normally protective properties of PDI.
Recent evidence implicates PDI in increasing the levels of ROS, thus directly inducing oxidative stress and apoptosis via its chaperone activity rather than the disulphide interchange activity (Fernandes et al., 2009). Similarly, only oxidized PDI triggers the production of ROS, whereas reduced PDI inhibits the production of ROS (Paes et al., 2011). PDI associates with the NAPDH peroxidase complex (Nox), a major source of ROS, where it stabilizes and associates with the oxidase subunit of Nox in vascular smooth muscle cells (Janiszewski et al., 2005). Similar effects are observed in macrophages and murine microglial cells, where PDI interacts with Nox and increases the levels of ROS (Fernandes et al., 2009). PDI also activates the transcription factors NF-kB and AP-1, thus promoting their binding to DNA (Clive and Greene, 1996). PDI is also a major catalyst of trans-nitrosation reactions, mediating nitric oxide internalization from extracellular S-nitrosothiols (Zai et al., 1999), thus further promoting the production of SNO proteins (Ramachandran et al., 2001).
Whilst SNO-PDI is implicated in triggering apoptosis, recent studies have revealed a direct role for unmodified PDI in apoptosis. In rat models of Huntington and Alzheimer's disease, PDI accumulation at the ER-mitochondrial junction triggers apoptosis via mitochondrial outer membrane permeabilisation (Hoffstrom et al., 2010). This effect is specific for the accumulation of misfolded proteins, but not other triggers of apoptosis, suggesting a specific role for pro-apoptotic PDI in neurodegenerative disease. Inhibitors of PDI including hypotaurine, thiomuscimol, and shRNA that inhibited the activity of PDI, were found to suppress the toxicity associated with misfolded Huntingtin and β-amyloid proteins.
PDI is an important cellular protein given its abundance, multiple biological functions, versatile redox behavior, interaction with other proteins and its implied role in various diseases. However, many issues remain unresolved that warrant further investigation, in particular the role of PDI in non-ER sub-cellular locations, and substrate specificity of the PDI family members. In future studies it will be important to replicate the precise functions of PDI in the ER and other cellular locations, separately from roles ascribed in vitro, before its normal cellular roles are fully understood.
PDI performs an impressive array of cellular functions and the up-regulation of PDI is a cellular defensive mechanism to restore proteostasis. However, despite this up-regulation, the functional properties of PDI can become abrogated due to aberrant post translational modifications. This is of particular relevance in neurodegenerative diseases where disruption to redox regulation is implicated (Parakh et al., 2013). Furthermore, neurons are particularly susceptible to ROS/RNS damage due to their high oxygen demand and a lower availability of antioxidants. Recent evidence implicates PDI as a trigger for apoptosis specifically in relation to the accumulation of misfolded proteins. PDI may therefore act as a regulatory switch, in which PDI is initially is protective against protein misfolding and aggregation. However, in response to an unknown trigger PDI subsequently becomes apoptotic when proteostasis cannot otherwise be resolved (Figure 3). Therefore aberrant post translational modifications together with the pro-apoptotic function of PDI could further accentuate the adverse effects of PDI.
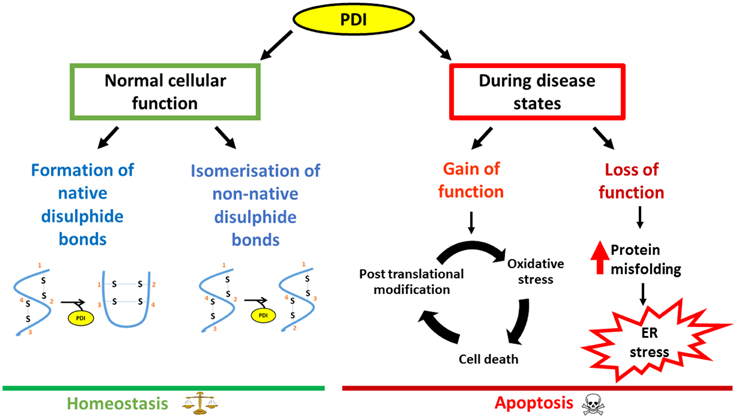
Figure 3. Schematic diagram outlining the dual nature of PDI, focusing on neurodegenerative disorders as an example. Under normal conditions, PDI reduces the load of misfolded proteins either by its chaperone activity or by isomerization of non-native bonds. However, during disease states, loss of the normal protective function of PDI as well as the gain of additional, toxic functions, leads to PDI becoming apoptotic, thus contributing to pathology.
In conclusion, PDI is an efficient catalyst and protein chaperone. It has the ability to restore proteostasis by catalyzing the efficient folding of newly synthesized proteins, and it plays an important role in protein quality control and ERAD by reducing the burden of misfolded proteins, thus inhibiting abnormal protein aggregation. The protective or harmful functions of PDI may be modulated by the subcellular location of PDI, levels of ER stress and the redox environment. While further investigations are clearly needed in this area, PDI has the potential to be exploited therapeutically in a variety of diseases. However, specific approaches depending on the disease in question will be required. In neurodegenerative conditions, elevation of the levels of total PDI, with the aim of restoring PDI function to reduce protein misfolding, could be an effective therapeutic approach. However, in contrast, reduction of the levels of PDI might be an appropriate strategy in cancer or cardiovascular diseases (Figure 4). Similarly, reducing the levels of aberrantly modified PDI might also be necessary in neurodegeneration in order to defend against the pro-apoptotic properties of PDI. At the cellular level there are important unanswered questions that need addressing, before the therapeutic applications of PDI can become realized in the future.
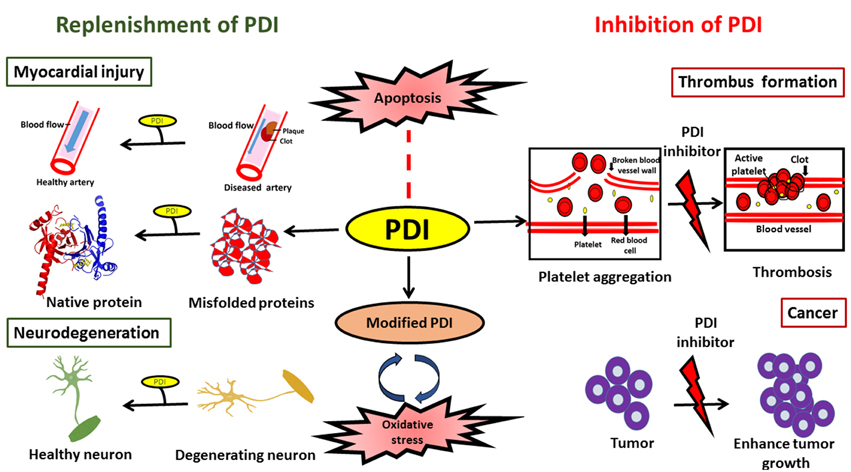
Figure 4. Schematic diagram illustrating possible therapeutic applications to modulate PDI function.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the National Health and Medical Research Council of Australia (NHMRC) Project grants (1006141 and 1030513), Bethlehem Griffiths Research Foundation, and Motor Neurone Disease Research Institute of Australia Angie Cunningham Laugh to Cure MND Grant, Zo-ee Research Grant and Grants in Aid. SP is supported by a Macquarie University Postgraduate Research Scholarship, and previously by a La Trobe Post Graduate Research Scholarship.
Andreu, C. I., Woehlbier, U., Torres, M., and Hetz, C. (2012). Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. 586: 2826–2834. doi: 10.1016/j.febslet.2012.07.023
Anelli, T., Alessio, M., Mezghrani, A., Simmen, T., Talamo, F., Bachi, A., et al. (2002). ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 21, 835–844. doi: 10.1093/emboj/21.4.835
Appenzeller-Herzog, C., and Ellgaard, L. (2008). The human PDI family: versatility packed into a single fold. Biochim. Biophys. Acta 1783, 535–548. doi: 10.1016/j.bbamcr.2007.11.010
Ataman-Onal, Y., Beaulieu, C., Busseret, S., Charrier, J.-P., Choquet-Kastylevsky, G., Rolland, D., et al. (2013). Protein disulfide isomerase assay method for the in vitro diagnosis of colorectal cancer. Patents 1–30.
Atkin, J. D., Farg, M. A., Turner, B. J., Tomas, D., Lysaght, J. A., Cheema, S. S., et al. (2006). Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165. doi: 10.1074/jbc.M603393200
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Auwerx, J., Isacsson, O., Söderlund, J., Balzarini, J., Johansson, M., and Lundberg, M. (2009). Human glutaredoxin-1 catalyzes the reduction of HIV-1 gp120 and CD4 disulfides and its inhibition reduces HIV-1 replication. Int. J. Biochem. Cell Biol. 41, 1269–1275. doi: 10.1016/j.biocel.2008.10.031
Bastos−Aristizabal, S., Kozlov, G., and Gehring, K. (2014). Structural insight into the dimerization of human protein disulfide isomerase. Protein Sci. 23, 618–626. doi: 10.1002/pro.2444
Benham, A. M. (2012). The protein disulfide isomerase family: key players in health and disease. Antioxid. Redox Signal. 16, 781–789. doi: 10.1089/ars.2011.4439
Bennett, T. A., Edwards, B. S., Sklar, L. A., and Rogelj, S. (2000). Sulfhydryl regulation of L-selectin shedding: phenylarsine oxide promotes activation-independent L-selectin shedding from leukocytes. J. Immunol. 164, 4120–4129. doi: 10.4049/jimmunol.164.8.4120
Bernardoni, P., Fazi, B., Costanzi, A., Nardacci, R., Montagna, C., Filomeni, G., et al. (2013). Reticulon1-C modulates protein disulphide isomerase function. Cell Death Dis. 4:e581. doi: 10.1038/cddis.2013.113
Bottomley, M. J., Batten, M. R., Lumb, R. A., and Bulleid, N. J. (2001). Quality control in the endoplasmic reticulum: PDI mediates the ER retention of unassembled procollagen C-propeptides. Curr. Biol. 11, 1114–1118. doi: 10.1016/S0960-9822(01)00317-7
Cai, H., Wang, C.-C., and Tsou, C.-L. (1994). Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J. Biol. Chem. 269, 24550–24552.
Chen, X., Guan, T., Li, C., Shang, H., Cui, L., Lui, X.-M., et al. (2012). SOD1 aggregation in astrocytes following ischemia/reperfusion injury: a role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI). J. Neuroinflammation 9:237. doi: 10.1186/1742-2094-9-237
Cheng, H., Wang, L., and Wang, C.-C. (2010). Domain a'of protein disulfide isomerase plays key role in inhibiting α-synuclein fibril formation. Cell Stress Chaperones. 15, 415–421. doi: 10.1007/s12192-009-0157-2
Chivers, P. T., Prehoda, K. E., and Raines, R. T. (1997). The CXXC motif: a rheostat in the active site. Biochemistry 36, 4061–4066. doi: 10.1021/bi9628580
Clive, D. R., and Greene, J. J. (1996). Cooperation of protein disulfide isomerase and redox environment in the regulation of NF−κB and AP1 binding to DNA. Cell Biochem. Funct. 14, 49–55. doi: 10.1002/cbf.638
Dai, Y., and Wang, C.-C. (1997). A mutant truncated protein disulfide isomerase with no chaperone activity. J. Biol. Chem. 272, 27572–27576. doi: 10.1074/jbc.272.44.27572
Darby, N. J., Kemmink, J., and Creighton, T. E. (1996). Identifying and characterizing a structural domain of protein disulfide isomerase. Biochemistry 35, 10517–10528.
Darby, N. J., Penka, E., and Vincentelli, R. (1998). The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency. J. Mol. Biol. 276, 239–247. doi: 10.1006/jmbi.1997.1504
Delom, F., Mallet, B., Carayon, P., and Lejeune, P.-J. (2001). Role of extracellular molecular chaperones in the folding of oxidized proteins refolding of colloidal thyroglobulin by protein disulfide isomerase and immunoglobulin heavy chain-binding protein. J. Biol. Chem. 276, 21337–21342. doi: 10.1074/jbc.M101086200
Denisov, A. Y., Määttänen, P., Dabrowski, C., Kozlov, G., Thomas, D. Y., Gehring, K., et al. (2009). Solution structure of the bb' domains of human protein disulfide isomerase. FEBS J. 276, 1440–1449. doi: 10.1111/j.1742-4658.2009.06884.x
Di Jeso, B., Park, Y.-N., Ulianich, L., Treglia, A. S., Urbanas, M. L., High, S., et al. (2005). Mixed-disulfide folding intermediates between thyroglobulin and endoplasmic reticulum resident oxidoreductases ERp57 and protein disulfide isomerase. Mol. Cell. Biol. 25, 9793–9805. doi: 10.1128/MCB.25.22.9793-9805.2005
Ellgaard, L., and Ruddock, L. W. (2005). The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32. doi: 10.1038/sj.embor.7400311
Erickson, R. R., Dunning, L. M., Olson, D. A., Cohen, S. J., Davis, A. T., Holtzman, J. L., et al. (2005). In cerebrospinal fluid ER chaperones ERp57 and calreticulin bind β-amyloid. Biochem. Biophys. Res. Commun. 332, 50–57. doi: 10.1016/j.bbrc.2005.04.090
Farg, M. A., Soo, K. Y., Walker, A. K., Pham, H., Orian, J., Atkin, J. D., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
Fernandes, D. C., Manoel, A. H. O., Wosniak, J. Jr., and Laurindo, F. R. (2009). Protein disulfide isomerase overexpression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: effects of nitrosothiol exposure. Arch. Biochem. Biophys. 484, 197–204. doi: 10.1016/j.abb.2009.01.022
Fewell, S. W., Travers, K. J., Weissman, J. S., and Brodsky, J. L. (2001). The action of molecular chaperones in the early secretory pathway. Annu. Rev. Genet. 35, 149–191. doi: 10.1146/annurev.genet.35.102401.090313
Forrester, M. T., Benhar, M., and Stamler, J. S. (2006). Nitrosative stress in the ER: a new role for S-nitrosylation in neurodegenerative diseases. ACS Chem. Biol. 1, 355–358. doi: 10.1021/cb600244c
Frand, A. R., Cuozzo, J. W., and Kaiser, C. A. (2000). Pathways for protein disulphide bond formation. Trends Cell Biol. 10, 203–210. doi: 10.1016/S0962-8924(00)01745-1
Freedman, R. B., Hirst, T. R., and Tuite, M. F. (1994). Protein disulphide isomerase: building bridges in protein folding. Trends Biochem. Sci. 19, 331–336. doi: 10.1016/0968-0004(94)90072-8
Fu, X.-M., and Zhu, B. T. (2009). Human pancreas-specific protein disulfide isomerase homolog (PDIp) is an intracellular estrogen-binding protein that modulates estrogen levels and actions in target cells. J. Steroid Biochem. Mol. Biol. 115, 20–29. doi: 10.1016/j.jsbmb.2009.02.008
Galligan, J. J., and Petersen, D. R. (2012). The human protein disulfide isomerase gene family. Hum. Genomics 6, 1–15. doi: 10.1186/1479-7364-6-6
Gilbert, H. F. (1998). Protein disulfide isomerases. Meth. Enzymol. 290, 26–50. doi: 10.1016/S0076-6879(98)90005-2
Gilbert, J., Ou, W., Silver, J., and Benjamin, T. (2006). Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 80, 10868–10870. doi: 10.1128/JVI.01117-06
Goldberger, R. F., Epstein, C. J., and Anfinsen, C. B. (1963). Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J. Biol. Chem. 238, 628–635.
Goplen, D., Wang, J., Enger, P. Ø., Tysnes, B. B., Terzis, A., Bjerkvig, R., et al. (2006). Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 66, 9895–9902. doi: 10.1158/0008-5472.CAN-05-4589
Grek, C., and Townsend, D. (2014). Protein disulfide isomerase superfamily in disease and the regulation of apoptosis. Endoplasmic Reticulum Stress Dis. 1, 4–17. doi: 10.2478/ersc-2013-0001
Gruber, C. W., Čemažar, M., Heras, B., Martin, J. L., and Craik, D. J. (2006). Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem. Sci. 31, 455–464. doi: 10.1016/j.tibs.2006.06.001
Halloran, M., Parakh, S., and Atkin, J. (2013). The role of S-nitrosylation and S-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013:797914. doi: 10.1155/2013/797914
Hashida, T., Kotake, Y., and Ohta, S. (2011). Protein disulfide isomerase knockdown-induced cell death is cell-line-dependent and involves apoptosis in MCF-7 cells. J. Toxicol. Sci. 36, 1–7. doi: 10.2131/jts.36.1
Hatahet, F., and Ruddock, L. W. (2009). Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850. doi: 10.1089/ars.2009.2466
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hetz, C., Russelakis-Carneiro, M., Wälchli, S., Carboni, S., Vial-Knecht, E., Maundrell, K., et al. (2005). The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 25, 2793–2802. doi: 10.1523/JNEUROSCI.4090-04.2005
Hoffstrom, B. G., Kaplan, A., Letso, R., Schmid, R. S., Turmel, G. J., Stockwell, B. R., et al. (2010). Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 6, 900–906. doi: 10.1038/nchembio.467
Honjo, Y., Kaneko, S., Ito, H., Horibe, T., Nagashima, M., Nakamura, M., et al. (2011). Protein disulfide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12, 444–450. doi: 10.3109/17482968.2011.594055
Hwang, C., Sinskey, A. J., and Lodish, H. F. (1992). Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502. doi: 10.1126/science.1523409
Inaba, K. (2009). Disulfide bond formation system in Escherichia coli. J. Biochem. 146, 591–597. doi: 10.1093/jb/mvp102
Irvine, A. G., Wallis, A. K., Sanghera, N., Rowe, M. L., Ruddock, L. W., Freedman, R. B., et al. (2014). Protein disulfide-isomerase interacts with a substrate protein at all stages along its folding pathway. PLoS ONE 9:e82511. doi: 10.1371/journal.pone.0082511
Janiszewski, M., Lopes, L. R., Carmo, A. O., Pedro, M. A., Brandes, R. P., Laurindo, F. R., et al. (2005). Regulation of NAD (P) H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 280, 40813–40819. doi: 10.1074/jbc.M509255200
Jasuja, R., Passam, F. H., Kennedy, D. R., Kim, S. H., van Hessem, L., Furie, B., et al. (2012). Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J. Clin. Invest. 122, 2104–2113. doi: 10.1172/JCI61228
Jeon, G. S., Nakamura, T., Lee, W.-J., Choi, S.-W., Ahn, K.-W., Lipton, S. A., et al. (2014). Potential effect of S-nitrosylated protein disulfide isomerase on mutant SOD1 aggregation and neuronal cell death in amyotrophic lateral sclerosis. Mol. Neurobiol. 49, 796–807. doi: 10.1007/s12035-013-8562-z
Jessop, C. E., Watkins, R. H., Simmons, J. J., Tasab, M., and Bulleid, N. J. (2009). Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J. Cell Sci. 122, 4287. doi: 10.1242/jcs.059154
Jurk, K., Lahav, J., Van Aken, H., Brodde, M., Nofer, J. R., and Kehrel, B. (2011). Extracellular protein disulfide isomerase regulates feedback activation of platelet thrombin generation via modulation of coagulation factor binding. J. Thromb. Haemost. 9, 2278–2290. doi: 10.1111/j.1538-7836.2011.04509.x
Karala, A. R., and Ruddock, L. W. (2010). Bacitracin is not a specific inhibitor of protein disulfide isomerase. FEBS J. 277, 2454–2462. doi: 10.1111/j.1742-4658.2010.07660.x
Kemmink, J., Darby, N. J., Dijkstra, K., Nilges, M., and Creighton, T. E. (1996). Structure determination of the N-terminal thioredoxin-like domain of protein disulfide isomerase using multidimensional heteronuclear 13C/15N NMR spectroscopy. Biochemistry 35, 7684–7691. doi: 10.1021/bi960335m
Kemmink, J., Darby, N. J., Dijkstra, K., Nilges, M., and Creighton, T. E. (1997). The folding catalyst protein disulfide isomerase is constructed of active and inactive thioredoxin modules. Curr. Biol. 7, 239–245. doi: 10.1016/S0960-9822(06)00119-9
Khan, H. A., and Mutus, B. (2014). Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2:70. doi: 10.3389/fchem.2014.00070
Kim, K., Hahm, E., Li, J., Holbrook, L.-M., Sasikumar, P., Cho, J., et al. (2013). Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood 122, 1052–1061. doi: 10.1182/blood-2013-03-492504
Klappa, P., Hawkins, H. C., and Freedman, R. B. (1997). Interactions between protein disulphide isomerase and peptides. Eur. J. Biochem. 248, 37–42. doi: 10.1111/j.1432-1033.1997.t01-1-00037.x
Klappa, P., Ruddock, L. W., Darby, N. J., and Freedman, R. B. (1998). The b domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927–935.
Knoblach, B., Keller, B. O., Groenendyk, J., Aldred, S., Zheng, J., Michalak, M., et al. (2003). ERp19 and ERp46, new members of the thioredoxin family of endoplasmic reticulum proteins. Mol. Cell. Proteomics 2, 1104–1119. doi: 10.1074/mcp.M300053-MCP200
Kozlov, G., Määttänen, P., Thomas, D. Y., and Gehring, K. (2010). A structural overview of the PDI family of proteins. FEBS J. 277, 3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x
Kwok, C. T., Morris, A. G., Frampton, J., Smith, B., Shaw, C. E., de Belleroche, J., et al. (2013). Association studies indicate that protein disulfide isomerase is a risk factor in amyotrophic lateral sclerosis. Free Radic. Biol. Med. 58, 81–86. doi: 10.1016/j.freeradbiomed.2013.01.001
LaMantia, M., Miura, T., Tachikawa, H., Kaplan, H. A., Lennarz, W. J., Mizunaga, T., et al. (1991). Glycosylation site binding protein and protein disulfide isomerase are identical and essential for cell viability in yeast. Proc. Natl. Acad. Sci. U.S.A. 88, 4453–4457. doi: 10.1073/pnas.88.10.4453
Lee, J. H., Won, S. M., Suh, J., Son, S. J., Moon, G. J., Park, U.-J., et al. (2010a). Induction of the unfolded protein response and cell death pathway in Alzheimer's disease, but not in aged Tg2576 mice. Exp. Mol. Med. 42, 386–394. doi: 10.3858/emm.2010.42.5.040
Lee, S. O., Cho, K., Cho, S., Kim, I., Oh, C., and Ahn, K. (2010b). Protein disulphide isomerase is required for signal peptide peptidase−mediated protein degradation. EMBO J. 29, 363–375. doi: 10.1038/emboj.2009.359
Lovat, P. E., Corazzari, M., Armstrong, J. L., Martin, S., Pagliarini, V., Redfern, C. P., et al. (2008). Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 68, 5363–5369. doi: 10.1158/0008-5472.CAN-08-0035
Mares, R. E., Meléndez-López, S. G., and Ramos, M. A. (2011). Acid-denatured Green Fluorescent Protein (GFP) as model substrate to study the chaperone activity of protein disulfide isomerase. Int. J. Mol. Sci. 12, 4625–4636. doi: 10.3390/ijms12074625
Molinari, M., Galli, C., Piccaluga, V., Pieren, M., and Paganetti, P. (2002). Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J. Cell Biol. 158, 247–257. doi: 10.1083/jcb.200204122
Moran, L. K., Gutteridge, J., and Quinlan, G. J. (2001). Thiols in cellular redox signalling and control. Curr. Med. Chem. 8, 763–772. doi: 10.2174/0929867013372904
Muller, C., Bandemer, J., Vindis, C., Camaré, C., Mucher, E., Guéraud, F., et al. (2013). Protein disulfide isomerase modification and inhibition contribute to ER stress and apoptosis induced by oxidized low density lipoproteins. Antioxid. Redox Signal. 18, 731–742.
Nakamura, T., and Lipton, S. A. (2011). S-nitrosylation of critical protein thiols mediates protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Antioxid. Redox Signal. 14, 1479–1492. doi: 10.1089/ars.2010.3570
Nardo, G., Pozzi, S., Pignataro, M., Lauranzano, E., Spano, G., Garbelli, S., et al. (2011). Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 6:e25545. doi: 10.1371/journal.pone.0025545
Nguyen, V. D., Wallis, K., Howard, M. J., Haapalainen, A. M., Salo, K. E., Ruddock, L. W., et al. (2008). Alternative conformations of the x region of human protein disulphide-isomerase modulate exposure of the substrate binding b'domain. J. Mol. Biol. 383, 1144–1155. doi: 10.1016/j.jmb.2008.08.085
Paes, A. M. D. A., Veríssimo-Filho, S., Guimarães, L. L., Silva, A. C. B., Takiuti, J. T., Lopes, L. R., et al. (2011). Protein disulfide isomerase redox-dependent association with p47phox: evidence for an organizer role in leukocyte NADPH oxidase activation. J. Leukoc. Biol. 90, 799–810. doi: 10.1189/jlb.0610324
Parakh, S., Spencer, D. M., Halloran, M. A., Soo, K. Y., and Atkin, J. D. (2013). Redox regulation in amyotrophic lateral sclerosis. Oxid. Med. Cell. Longev. 2013:408681. doi: 10.1155/2013/408681
Pescatore, L. A., Bonatto, D., Forti, F. L., Sadok, A., Kovacic, H., Laurindo, F. R., et al. (2012). Protein disulfide isomerase is required for platelet-derived growth factor-induced vascular smooth muscle cell migration, Nox1 NADPH oxidase expression, and RhoGTPase activation. J. Biol. Chem. 287, 29290–29300. doi: 10.1074/jbc.M112.394551
Primm, T. P., and Gilbert, H. F. (2001). Hormone binding by protein disulfide isomerase, a high capacity hormone reservoir of the endoplasmic reticulum. J. Biol. Chem. 276, 281–286. doi: 10.1074/jbc.M007670200
Primm, T. P., Walker, K. W., and Gilbert, H. F. (1996). Facilitated protein aggregation effects of calcium on the chaperone and anti-chaperone activity of protein disulfide-isomerase. J. Biol. Chem. 271, 33664–33669. doi: 10.1074/jbc.271.52.33664
Puig, A., and Gilbert, H. F. (1994). Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme. J. Biol. Chem. 269, 7764–7771.
Rainey-Barger, E. K., Mkrtchian, S., and Tsai, B. (2007). Dimerization of ERp29, a PDI-like protein, is essential for its diverse functions. Mol. Biol. Cell. 18, 1253–1260. doi: 10.1091/mbc.E06-11-1004
Ramachandran, N., Root, P., Jiang, X.-M., Hogg, P. J., and Mutus, B. (2001). Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc. Natl. Acad. Sci. U.S.A. 98, 9539–9544. doi: 10.1073/pnas.171180998
Rutkevich, L. A., Cohen-Doyle, M. F., Brockmeier, U., and Williams, D. B. (2010). Functional relationship between protein disulfide isomerase family members during the oxidative folding of human secretory proteins. Mol. Biol. Cell. 21, 3093–3105. doi: 10.1091/mbc.E10-04-0356
Schroder, M., and Kaufman, R. J. (2005). ER stress and the unfolded protein response. mutation research/fundamental and molecular mechanisms of mutagenesis. Mutat. Res. 569, 29–63. doi: 10.1016/j.mrfmmm.2004.06.056
Schwaller, M., Wilkinson, B., and Gilbert, H. F. (2003). Reduction-reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 278, 7154–7159. doi: 10.1074/jbc.M211036200
Severino, A., Campioni, M., Straino, S., Salloum, F. N., Schmidt, N., Bussani, R., et al. (2007). Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J. Am. Coll. Cardiol. 50, 1029–1037. doi: 10.1016/j.jacc.2007.06.006
Sobierajska, K., Skurzynski, S., Stasiak, M., Kryczka, J., Cierniewski, C. S., Swiatkowska, M., et al. (2014). Protein disulfide isomerase directly interacts with β-Actin Cys374 and regulates cytoskeleton reorganization. J. Biol. Chem. 289, 5758–5773. doi: 10.1074/jbc.M113.479477
Solda, T., Garbi, N., Hämmerling, G. J., and Molinari, M. (2006). Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J. Biol. Chem. 281, 6219–6226. doi: 10.1074/jbc.M513595200
Song, J. L., and Wang, C. C. (1995). Chaperone−like activity of protein disulfide−isomerase in the refolding of rhodanese. Eur. J. Biochem. 231, 312–316. doi: 10.1111/j.1432-1033.1995.tb20702.x
Sovolyova, N., Healy, S., Samali, A., and Logue, S. E. (2014). Stressed to death–mechanisms of ER stress-induced cell death. Biol. Chem. 395, 1–13. doi: 10.1515/hsz-2013-0174
Stolf, B. S., Smyrnias, I., Lopes, L. R., Vendramin, A., Goto, H., Santos, C. X., et al. (2011). Protein disulfide isomerase and host-pathogen interaction. ScientificWorldJournal 11, 1749–1761. doi: 10.1100/2011/289182
Tanaka, S., Uehara, T., and Nomura, Y. (2000). Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J. Biol. Chem. 275, 10388–10393. doi: 10.1074/jbc.275.14.10388
Taylor, M., Banerjee, T., Ray, S., Tatulian, S. A., and Teter, K. (2011). Protein-disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J. Biol. Chem. 286, 22090–22100. doi: 10.1074/jbc.M111.237966
Thongwatchara, P., Promwikorn, W., Srisomsap, C., Chokchaichamnankit, D., Boonyaphiphat, P., Thongsuksai, P., et al. (2011). Differential protein expression in primary breast cancer and matched axillary node metastasis. Oncol. Rep. 26, 185–191. doi: 10.3892/or.2011.1266
Tian, F., Zhou, X., Wikström, J., Karlsson, H., Sjöland, L.-M., Gan, J., et al. (2009). Protein disulfide isomerase increases in myocardial endothelial cells in mice exposed to chronic hypoxia: a stimulatory role in angiogenesis. Am. J. Physiol. 297, H1078–H1086. doi: 10.1152/ajpheart.00937.2008
Tian, G., Xiang, S., Noiva, R., Lennarz, W., and Schindelin, H. (2006). The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124, 61–73. doi: 10.1016/j.cell.2005.10.044
Toldo, S., Boccellino, M., Rinaldi, B., Seropian, I. M., Mezzaroma, E., Paolisso, G., et al. (2011). Altered oxido-reductive state in the diabetic heart: loss of cardioprotection due to protein disulfide isomerase. Mol. Med. 17, 1012. doi: 10.2119/molmed.2011.00100
Townsend, D. M., Manevich, Y., He, L., Xiong, Y., Bowers, R., Tew, K. D., et al. (2009). Nitrosative stress–induced S-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 69, 7626–7634. doi: 10.1158/0008-5472.CAN-09-0493
Tsai, B., Rodighiero, C., Lencer, W. I., and Rapoport, T. A. (2001). Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948. doi: 10.1016/S0092-8674(01)00289-6
Tsuda, H., Han, S. M., Yang, Y., Tong, C., Lin, Y. Q., Kwan, J., et al. (2008). The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 133, 963–977. doi: 10.1016/j.cell.2008.04.039
Turano, C., Coppari, S., Altieri, F., and Ferraro, A. (2002). Proteins of the PDI family: unpredicted non−ER locations and functions. J. Cell. Physiol. 193, 154–163. doi: 10.1002/jcp.10172
Uehara, T., Nakamura, T., Yao, D., Shi, Z.-Q., Gu, Z., Lipton, S. A., et al. (2006). S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517. doi: 10.1038/nature04782
Vuori, K., Pihlajaniemi, T., Marttila, M., and Kivirikko, K. I. (1992). Characterization of the human prolyl 4-hydroxylase tetramer and its multifunctional protein disulfide-isomerase subunit synthesized in a baculovirus expression system. Proc. Natl. Acad. Sci. U.S.A. 89, 7467.
Walker, A. K., Farg, M. A., Bye, C. R., McLean, C. A., Horne, M. K., Atkin, J. D., et al. (2010). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116. doi: 10.1093/brain/awp267
Walker, A. K., Soo, K. Y., Levina, V., Talbo, G. H., and Atkin, J., D. (2013). N–linked glycosylation modulates dimerization of protein disulfide isomerase family A member 2 (PDIA2). FEBS J. 280, 233–243. doi: 10.1111/febs.12063
Wang, C., Li, W., Ren, J., Fang, J., Ke, H., Gong, W., et al. (2013). Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 19, 36–45. doi: 10.1089/ars.2012.4630
Wang, C., Yu, J., Huo, L., Wang, L., Feng, W., Wang, C.-C., et al. (2012a). Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a'. J. Biol. Chem. 287, 1139–1149. doi: 10.1074/jbc.M111.303149
Wang, S.-B., Shi, Q., Xu, Y., Xie, W.-L., Zhang, J., Chen, C., et al. (2012b). Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS ONE 7:e38221. doi: 10.1371/journal.pone.0038221
Watts, J. C., Huo, H., Bai, Y., Ehsani, S., Won, A. H., Xu, L., et al. (2009). Interactome Analyses identify ties of PrPC and its mammalian paralogs to oligomannosidic N-Glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 5:e1000608. doi: 10.1371/journal.ppat.1000608
Wilkinson, B., and Gilbert, H. F. (2004). Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44. doi: 10.1016/j.bbapap.2004.02.017
Xiao, R., Solovyov, A., Gilbert, H. F., Holmgren, A., and Lundström-Ljung, J. (2001). Combinations of protein-disulfide isomerase domains show that there is little correlation between isomerase activity and wild-type growth. J. Biol. Chem. 276, 27975–27980. doi: 10.1074/jbc.M104203200
Xiong, Y., Manevich, Y., Tew, K. D., and Townsend, D. M. (2012). S-glutathionylation of protein disulfide isomerase regulates estrogen receptor stability and function. Int. J. Cell Biol. 2012:273549. doi: 10.1155/2012/273549
Xiong, Y., Uys, J. D., Tew, K. D., and Townsend, D. M. (2011). S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid. Redox Signal. 15, 233–270. doi: 10.1089/ars.2010.3540/
Xu, S., Butkevich, A. N., Yamada, R., Zhou, Y., Debnath, B., Neamati, N., et al. (2012). Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc. Natl. Acad. Sci. U.S.A. 109, 16348–16353. doi: 10.1073/pnas.1205226109
Xu, S., Sankar, S., and Neamati, N. (2014). Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov. Today 19, 222–240. doi: 10.1016/j.drudis.2013.10.017
Yang, Y. S., Harel, N. Y., and Strittmatter, S. M. (2009). Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. J. Neurosci. 29, 13850–13859. doi: 10.1523/JNEUROSCI.2312-09.2009
Zai, A., Rudd, M. A., Scribner, A. W., and Loscalzo, J. (1999). Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. J. Clin. Invest. 103, 393–399. doi: 10.1172/JCI4890
Keywords: protein disulfide isomerase family, neurodegnerative diseases, protein chaperones, post-translational modifications, cancer, amyotrophic lateral sclerosis
Citation: Parakh S and Atkin JD (2015) Novel roles for protein disulphide isomerase in disease states: a double edged sword? Front. Cell Dev. Biol. 3:30. doi: 10.3389/fcell.2015.00030
Received: 30 January 2015; Accepted: 28 April 2015;
Published: 21 May 2015.
Edited by:
Bulent Mutus, University of Windsor, CanadaCopyright © 2015 Parakh and Atkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie D. Atkin, Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, 2 Technology Place, Sydney, NSW 2109, Australia,anVsaWUuYXRraW5AbXEuZWR1LmF1; website:www.medicine.mq.edu.au
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.