 John P. Dowling
John P. Dowling Anirudh Nair
Anirudh Nair Jianke Zhang
Jianke Zhang- Department of Microbiology and Immunology, Sidney Kimmel Cancer Center, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, USA
RIP1 is an adaptor kinase originally identified as being able to associate with TNFR1 and Fas, and is later shown to be involved in signaling induced by TLRs. Major signaling pathways regulated by RIP1 include necroptosis, apoptosis, and pro-survival/inflammation NF-κB activation. Previous studies show that RIP1 deficiency has no effect on mouse embryogenesis, but blocks postnatal development. This phenotype could not readily be explained, since mice lacking TNFR1, Fas, or TLRs show no apparent developmental defect. Certain types of RIP1-deficient cells are hypersensitive to TNF-induced apoptosis. However, in our previous study, deletion of the apoptotic adaptor protein, FADD, provides marginal improvement of postnatal development of rip1−/− mice. Remarkably, the current data shows that haploid insufficiency of RIP3, a known mediator of necroptosis, allowed survival of rip1−/−fadd−/− mice beyond weaning age, although the resulting rip1−/−fadd−/− rip3+/− mice were significant smaller in size and weight. Moreover, complete absence of RIP3 further improved postnatal development of the resulting rip1−/−fadd−/−rip3−/− mice, which display normal size and weight. In such triple knockout (TKO) mice, lymphocytes underwent normal development, but progressively accumulated as mice age. This lymphoproliferative (lpr) disease in TKO mice is, however, less severe than that of fadd−/−rip3−/− double knockout mice. In total, the data show that the postnatal developmental defect in rip1−/− mice is due in part to FADD-mediated apoptosis as well as RIP3-dependent necroptosis. Moreover, the function of RIP1 contributes to development of lpr diseases.
Introduction
Apoptosis and necroptosis are programmed cell death (PCD) pathways that play an essential role during animal development and immune responses (Moriwaki and Chan, 2013; Walsh, 2014). Although these two PCD pathways have distinct mechanisms and outcomes, they are intricately connected and, therefore, must be tightly regulated. PCD can be initiated through the death receptors (DRs) like Fas and tumor necrosis factor receptor-1 (TNFR1) (Ashkenazi and Dixit, 1998). Apoptotic death induced by DRs is mediated by the Fas-associated death domain (FADD or MORT1) adaptor protein (Boldin et al., 1995; Chinnaiyan et al., 1995; Zhang and Winoto, 1996). FADD binds to and activates the apical Caspase 8, which then cleaves and activates the effector Caspases, and triggers the apoptotic program (Boldin et al., 1996; Muzio et al., 1996). When apoptotic pathway is blocked, such as through viral defense mechanisms or Caspase inhibitory conditions, DRs signaling could lead to necroptosis which is mediated through the receptor interacting protein (RIP)1-RIP3-mixed lineage kinase domain-like (MLKL) protein axis (Holler et al., 2000; Degterev and Yuan, 2008; Cho et al., 2009; Sun et al., 2012; Zhao et al., 2012).
One striking example of the importance of these PCD pathways in immune homeostasis is illustrated by a mutant mouse strain with lymphoproliferative (lpr) disease caused by mutations in the fas gene (Strasser et al., 2009). In lpr mice, lymphocytes are unable to undergo Fas-mediated cell death, leading to accumulation of a unique subset of T cells that lose expression of CD4 and CD8 and gain expression of B220, a traditional B cell marker (Strasser et al., 2009). Moreover, in certain genetic backgrounds, the lpr Fas mutation facilitates autoimmunity including glomerulonephritis, arthritis, and autoantibody production, and diseases sharing symptoms of systemic lupus erythematosus (SLE) (Cohen and Eisenberg, 1992; Shi et al., 2002). In human patients containing mutations in the fas gene, a similar disease has been identified as autoimmune-lymphoproliferative symdrome (ALPS) (Fisher et al., 1995). Deletion of TNFR1 alone does not affect lymphoid homeostasis (Pfeffer et al., 1993; Rothe et al., 1993), but exacerbates the lpr disease of Fas mutant mice (Zhou et al., 1996).
Besides abnormality in certain immune functions, mice lacking each individual DR undergo normal embryogenesis and postnatal development in mice (Pfeffer et al., 1993; Rothe et al., 1993; Adachi et al., 1996; Diehl et al., 2004). Surprisingly, inactivation of FADD, the common signaling adaptor of the DRs, results in death around embryonic day (E)11.5 (Yeh et al., 1998; Zhang et al., 1998). It has been demonstrated that fadd−/− embryos contain elevated levels of RIP1 and undergo massive necrosis (Zhang et al., 2011). Lack of RIP1 completely corrects the embryonic defect in fadd−/− embryos. Moreover, RIP3 deletion prevents T cell death induced by expression of a FADD dominant-negative (DN) mutant (Lu et al., 2011). Unlike RIP1, RIP3 does not play a role in NF-κB activation (Newton et al., 2004). Deletion of RIP3 completely restores normal development of mice lacking FADD or Caspase 8 (Ch'en et al., 2011; Kaiser et al., 2011; Oberst et al., 2011), indicating that RIP1/RIP3-dependent necroptosis is unleashed in the absence of FADD or Caspase 8.
Though RIP1 was initially identified as a Fas-binding protein (Stanger et al., 1995), apoptosis through Fas is not affected in rip1−/− cells (Ting et al., 1996; Kelliher et al., 1998). However, RIP1 deficiency leads to hypersensitivity to TNF-induced apoptosis, which has been attributed in part to a defect in NF-κB activation in certain cell types. When signaling through receptors like TNFR1 or toll like receptors (TLRs), the ubiquitination status of RIP1 can determine whether a cell lives or dies (Vucic et al., 2011; Schmukle and Walczak, 2012). For example, upon activation of TNFR1, RIP1 promotes NF-κB activation when ubiquitinated, but mediates apoptosis when deubiquitinated and associated with FADD and Caspase 8. Furthermore, if the function of FADD or Caspase 8 is interrupted, RIP1 kinase activity signals through RIP3, leading to necroptosis (Moriwaki and Chan, 2013; Vanden Berghe et al., 2014). These multiple functions of RIP1 participate in regulating inflammatory signaling and have been a target of interest for inflammatory disease (Moriwaki and Chan, 2013; Vanden Berghe et al., 2014; Walsh, 2014).
The importance of RIP1 during development is manifested in rip1−/− mice, which die at birth with extensive cell death in the lymphoid and adipose tissues (Kelliher et al., 1998) Since without RIP1, TNFR1 initiates FADD-mediated apoptosis (Cusson et al., 2002; O'Donnell and Ting, 2011), it was surprising that deletion of FADD does not rescue rip1−/− mice from perinatal lethality (Zhang et al., 2011). This suggests that RIP1 could play different roles during embryonic development, where it may initiate necroptosis in the absence of FADD, and in perinatal mice, where RIP1 is essential for survival. Unlike perinatal lethality in rip1−/− or rip1−/−fadd−/− mice, normal embryonic and postnatal development is observed in fadd−/−rip3−/− mice (Dillon et al., 2012). Moreover, under certain circumstances, RIP3 is able to signal necroptosis in the absence of RIP1 (Upton et al., 2010; Moujalled et al., 2013). In this study, we performed mouse genetic studies and show that lack of RIP3 is insufficient to correct the defect in RIP1 mice. However, the resulting rip1−/−rip3−/− mice could survive to adulthood if FADD is also inactivated. Therefore, our data indicate that RIP1 has a novel function, which appears to provide protection against RIP3-dependent necroptosis and FADD-mediated apoptosis during postnatal development.
Materials and Methods
Mice
fadd+/− mice have been previously described (Zhang et al., 1998). rip1+/− mice were provided by Dr. Michelle Kelliher (Kelliher et al., 1998). rip3−/− mice were provided by Drs. Kim Newton and Vishva Dixit (Genentech) (Newton et al., 2004). All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Thomas Jefferson University.
Mouse Genetic Analysis
rip1+/−fadd+/− mice were crossed with rip3−/− mice. The resulting rip1+/−fadd+/−rip3+/− triple heterozygous (THZ) mice were intercrossed. From the offspring, rip1+/−fadd−/−rip3−/− mice were mated with THZ mice, producing viable and fertile rip1−/−fadd−/−rip3−/− triple knockout (TKO) mice, which were mated with rip1+/−fadd−/−rip3−/− mice. For genotyping, genomic DNA was isolated from the tails of newborns to weaning-age mice using proteinase K and sodium dodecyl sulfate (SDS)-containing lysis buffer followed by phenol extraction and ethanol precipitation. Allele-specific primers were used to detect the presence of wild type and knockout alleles of rip1 and fadd as previously described (Zhang et al., 2011). rip3−/− mice were typed using the forward primers 5′-GCCTGCCCATCAGCAACT-3′ and 5′-CCAGAGGCCACTTGTGTAGCG-3′ and reverse primer 5′-CGCTTTAGAAGCCTTCAGGTTGAC-3′ (Newton et al., 2004). PCR produces a 320 bp wild type band and a 485 bp knockout band. For postnatal survival analysis, neonates were monitored for survival from birth to weaning age; deceased pups were immediately collected and genotyped. Survival curves were generated using Prism software (Graphpad Software, Inc.). For genetic analysis, the numbers of mice containing the most numbers of wild type alleles of the genes of concern were used to predict the numbers of other genotypes among the offspring of the indicated crosses, based on Mendelian ratios.
Western Blotting Analysis
The presence or absence of FADD, RIP3, and RIP1 protein was confirmed by western blotting. Total splenocytes were isolated from mice of the indicated genotypes. Cell lysates were prepared in cold RIPA lysis buffer containing 50 mM Tris pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 1 mM phenylmethyl sulphonyl fluoride (PMSF), and a proteinase inhibitor cocktail (Roche). Proteins (30 μg) were separated on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. Proteins on the membrane were stained with Ponceau S (Sigma-Aldrich) as a loading/transfer control. Antibodies specific for FADD (generated in house), RIP3 (ProSci, Catalog #2283), and RIP1 (BD Biosciences, Catalog #610459) were incubated with the membrane overnight at 4°C followed by HRP-conjugated goat anti-rabbit antibodies (1/10,000). Chemiluminescent signals were detected on the ProteinSimple FluorChem M Western Blot Imaging machine.
Flow Cytometric Analysis
Spleen, lymph nodes, and thymus of mice were isolated during mouse dissection. Red blood cell lysis of the spleen was performed using ACK buffer and single cell suspensions were prepared. Cells (106) were stained with fluorochrome-conjugated antibodies in PBS containing 3% BSA, 0.5 mM EDTA, and 0.05% sodium azide, and samples were incubated on ice for 30 min. Antibodies used for flow cytometry were anti-CD3 (BD Biosciences), anti-CD4 (Caltag), anti-CD8 (Invitrogen), and anti-B220 (BD Biosciences). Data were acquired on an LSR II cytometric analyzer (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Cell Culture
Thymocytes and peripheral T cells purified from the spleen and lymph nodes were cultured in complete RPMI media (cRPMI) containing 10% FBS, 2 mM L-Glutamine, penicillin (100 U/mL), streptomycin (100 μg/mL), and β-mercaptoethanol (50 μM).
Total Cellularity Calculations
Total cell number was obtained using single cell suspensions spleen and lymph nodes counted using a Countess Automated Cell Counter (Invitrogen). Cell number of various lymphocyte subsets was obtained by multiplying the total cell number with percentages of subsets of CD3+ and B220+ cells obtained by flow cytometry.
Cell Death Assays
Thymocytes (105) were seeded in a 96-well flat-bottom plate in 100 μL of cRPMI and incubated at 37°C with 5% CO2for 16 h with indicated concentrations of anti-Fas antibodies, TNFα, or dexamethasone. Thymocytes were also incubated with no treatment, staurosporine (1 μg/mL), and ionomycin (1 μg/mL) for indicated time points. Cells were collected, each well was washed with PBS, and 1 μg/mL propidium iodide was added to the sample. Cells were analyzed by flow cytometry on an LSR II cytometric analyzer (BD Biosciences). In some cases, Percent Specific Death was calculated by: (percent cell killing with treatment)—(percent cell killing without treatment).
Proliferation Assays
T cells were purified by pooling total spleen and lymph node cells of five-week-old mice and using antibodies against Thy1.2 and B220. Thy1.2+B220− T cells were sorting using a FACS Aria (BD Biosciences). Purified T cells were labeled with CellTrace Violet (Life Technologies), as per the manufacturer's instructions. Cells were placed in a 96 well round-bottom plate (2 × 105 cells/well) and stimulated with 1 μg/mL anti-CD3 antibody and 0.2 μg/mL anti-CD28 antibody in 100 μL cRPMI for various timepoints. Cells were stained with 1 μg/mL propidium iodide and run on an LSR II cytometric analyzer (BD Biosciences). For 3H-thymidine experiments, cells were placed in a 96 well round-bottom plate (105 cells/well) and stimulated with 1 μg/mL anti-CD3 antibody and 0.2 μg/mL anti-CD28 antibody for 40 h. Two microcuries of tridiated thymidine were added to each well and cells were incubated for an additional 8 h. Samples were placed onto a glass fiber filter using a Mach Harvester 96 (Tomtec) and analyzed using a Wallac 1205 Betaplate Counter (Perkin Elmer).
Statistical Analysis
Data is expressed as mean ± standard deviation. Student's t-tests were performed using Prism software (Graphpad Software, Inc).
Results
Postnatal Development of rip1−/− Mice in the Absence of RIP3 and/or FADD
To determine whether RIP3 plays a role in the postnatal defect of rip1−/− mice, rip1+/−rip3+/− or rip1+/−rip3−/− mice were intercrossed, which produced no viable rip1−/−rip3−/− double knockout (DKO) or rip1−/−rip3+/− mice at weaning age (data not shown). This observation was confirmed when rip1+/−fadd+/−rip3+/− triple heterozygous (THZ) mutant mice were intercrossed, which produced no viable rip1−/−rip3−/− mice containing one or both alleles of FADD, though Mendelian genetics predicts 12 mice of such genotypes (column 5, Figure 1A). Nonetheless, rip1−/−rip3−/− newborns were detected, and showed slightly improved survival up to postpartum day (p)6, as compared to rip1−/− newborns which died at p1 (Figure 1D). In contrast, if one or both alleles of the rip1 gene were present, fadd−/−rip3−/− DKO mice were viable from birth to adulthood (column 9, Figure 1A).
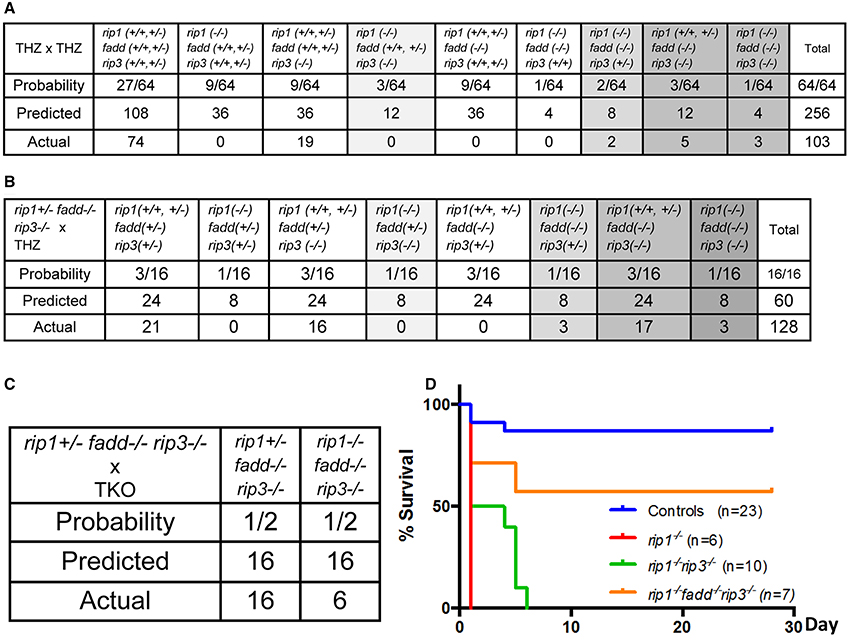
Figure 1. rip1−/− perinatal lethality is mediated by FADD and RIP3. (A–C) Predicted and actual frequency of indicated genotypes of weaning age mice resulting from intercrosses of rip1+/−fadd+/−rip3+/− THZ mice (A), crosses of rip1+/−fadd+/−rip3+/− and rip1+/−fadd−/−rip3−/− mice (B), and crosses of rip1+/−fadd−/−rip3−/− and rip1−/−fadd−/−rip3−/− mice (C). (D) Survival of newborn mice from various intercrosses. For this and following figures, control mice are littermates that have at least one wild type allele of FADD and RIP1. The fadd−/−rip3−/− mice shown contain at least one wild type allele of rip1.
Similar to the previous study (Zhang et al., 2011), no viable rip1−/−fadd−/− mice were present at weaning age among the offspring of THZ intercrosses (column 7, Figure 1A). Strikingly, however, absence of one allele of RIP3 allowed survival of rip1−/−fadd−/− mice beyond weaning ages up to adulthood (column 8, Figures 1A, 2A). Presence or absence of the FADD, RIP1, and RIP3 proteins was analyzed by western blotting (Figure 2B). These observations were also confirmed by analysis of the offspring from crosses of rip1+/−fadd−/−rip3−/− mice with rip1+/−fadd+/−rip3+/− THZ mice (Figure 1B), which produce higher ratios of rip1−/−fadd−/−rip3+/− mice.
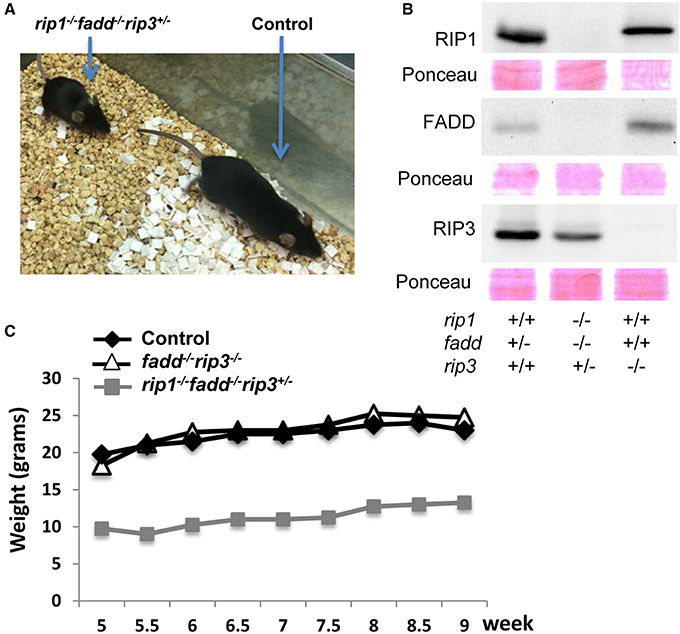
Figure 2. Haploid insufficiency of rip3 improves survival of rip1−/−fadd−/− mice, which are viable but weigh less than wild type controls. (A) Adult rip1−/−fadd−/−rip3+/− mouse and littermate control. (B) Western blot analysis of total splenocytes confirming presence or absence of FADD, RIP3, and RIP1 protein. (C) Weight curves showing the rip1−/−fadd−/−rip3+/− mouse with age-matched controls. Data shown is representative of three mice of each genotype.
Further examination revealed that although rip1−/−fadd−/−rip3+/− mice were viable, they were much smaller in size (Figure 2A), and also weighed less than wild type controls or fadd−/−rip3−/− mice during the indicated period of analysis (Figure 2C). Moreover, rip1−/−fadd−/−rip3+/− mice appeared to be present at lower numbers (2) at weaning age than the predicted Mendelian frequencies (8) (column 8, Figure 1A). Western blotting analysis indicated that ablation of one allele of RIP3 resulted in lower RIP3 protein levels (Figure 2B), which helped improve survival of rip1−/−fadd−/− mice.
Interestingly, when both alleles of RIP3 were absent, the resulting rip1−/−fadd−/−rip3−/− triple knockout (TKO) mice gained weight normally, as compared to wild type control or fadd−/−rip3−/− DKO mice (Figures 1, 3A,C,D). Nonetheless, the numbers of the TKO mice appeared to be lower than that predicted by Mendelian ratios, particularly among the offspring of the mouse cross in Figure 1B (column 10). This defect is more pronounced through analyzing the offspring of crosses between rip1+/−fadd−/−rip3−/− mice and TKO mice (Figure 1C). This correlates with decreased survival of rip1−/−fadd−/−rip3−/−mice from birth to weaning age (Figure 1D). Knockout of these genes was confirmed by western blotting (Figure 3B). In total, these data showed that while rip1−/−rip3−/− mice did survive several days longer than rip1−/−, knockout of both FADD and RIP3 was necessary for survival of rip1−/− mice to adulthood.
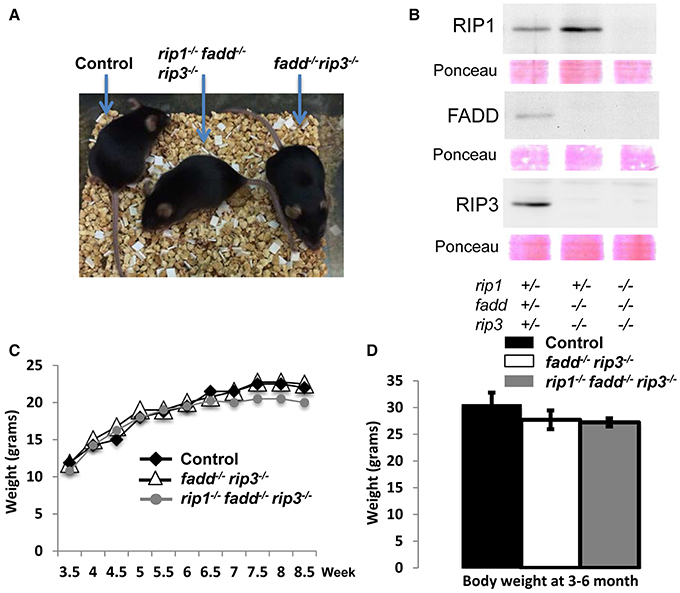
Figure 3. Complete ablation of both RIP3 and FADD rescued rip1−/− perinatal lethality. (A) rip1−/−fadd−/−rip3−/− mouse with fadd−/−rip3−/− and littermate control. (B) Western blot of total splenocytes confirming presence or absence of FADD, RIP3, and RIP1 protein. (C) Weight curves of rip1−/−fadd−/−rip3−/− with age-matched controls, representative of at least three mice of each genotype. (D) Body weight of 3- to 6-month-old mice at time of analysis. Data shown is representative of four mice of each genotype.
The Role of RIP1, FADD, and RIP3 in Lymphocyte Responses
The role of these proteins in lymphocyte development and function has been previously investigated through gene deletion in germ cells or in specific lineages. The lymphoid system of rip3−/− mice is normal (Newton et al., 2004). Without FADD function, peripheral T cell number decreases (Walsh et al., 1998; Zhang et al., 2005), but the B cell compartment is normal (Imtiyaz et al., 2006). Lastly, inducible deletion of RIP1 results in greatly reduced lymphocyte numbers (Roderick et al., 2014). We initially analyzed young (4 week old) mice, due to concerns of age-dependent diseases (see below). The fadd−/−rip3−/− DKO and rip1−/−fadd−/−rip3−/− TKO mutant mice contained normal T cell populations in the thymus, similar to that in control mice (Figure 4B). This indicates that RIP1 deficiency does not affect T lymphocyte development, as long as FADD and RIP3 are absent. The lymphoid organs of DKO and TKO mice at 5 weeks of age were not significantly larger than control mice, and these mutant mice appeared to contain higher numbers of B cells in the lymph nodes (Figures 4A,C,D).
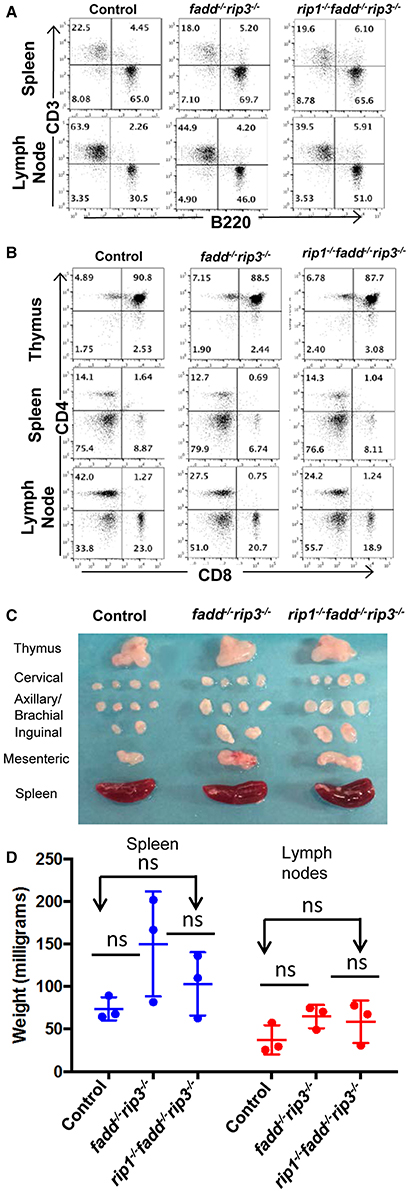
Figure 4. Young rip1−/−fadd−/−rip3−/− mice contain normal T and B cell populations. (A,B) Spleen, lymph node, and thymus of 5 week old mice of the indicated genotypes were stained with antibodies for CD3 and B220 (A) or CD4 and CD8 (B). Numbers indicated are percentages of T cell and B cell subsets. (C) Lymph nodes, spleen, thymus of 5 week-old mice. (D) Weight of spleen and lymph node of mice 4–6 week old mice. ns, not significant. Data shown is representative of at least three independent experiments with one mouse of each genotype.
The extrinsic cell death pathways mediated by the DRs are essential for immune homeostasis, as indicated by the lpr mouse model. Disruption of FADD function blocks Fas-induced apoptosis in T cells (Walsh, 2014). We previously have shown that absence of RIP1 has no effect on Fas-induced death, but leads to hypersensitivity to TNF-induced death in thymocytes (Zhang et al., 2011). In contrast, rip1−/−fadd−/− thymocytes were resistant to TNF, similar to wild type thymocytes (Zhang et al., 2011). In order to determine the status of these pathways in rip1−/−fadd−/−rip3−/− mice, thymocytes were isolated and treated with various stimuli to initiate extrinsic or intrinsic cell death. Similar to fadd−/−rip3−/− DKO thymocytes, TKO thymocytes were highly resistant to cell death responses induced by Fas (Figure 5A). Unlike rip1−/− thymocytes (Zhang et al., 2011), TKO and DKO mutant thymocytes were not sensitive to TNFα-induced cell death (Figure 5B). However, they were able to undergo intrinsic apoptosis in response to dexamethasone, similar to wild type thymocytes (Figure 5C). Furthermore, DKO and TKO thymocytes were sensitive to staurosporine and ionomycin-induced intrinsic apoptosis, as compared with untreated cells (Figures 5D–F). These results imply that resistance to extrinsic cell death is essential for the survival of rip1−/− mice.
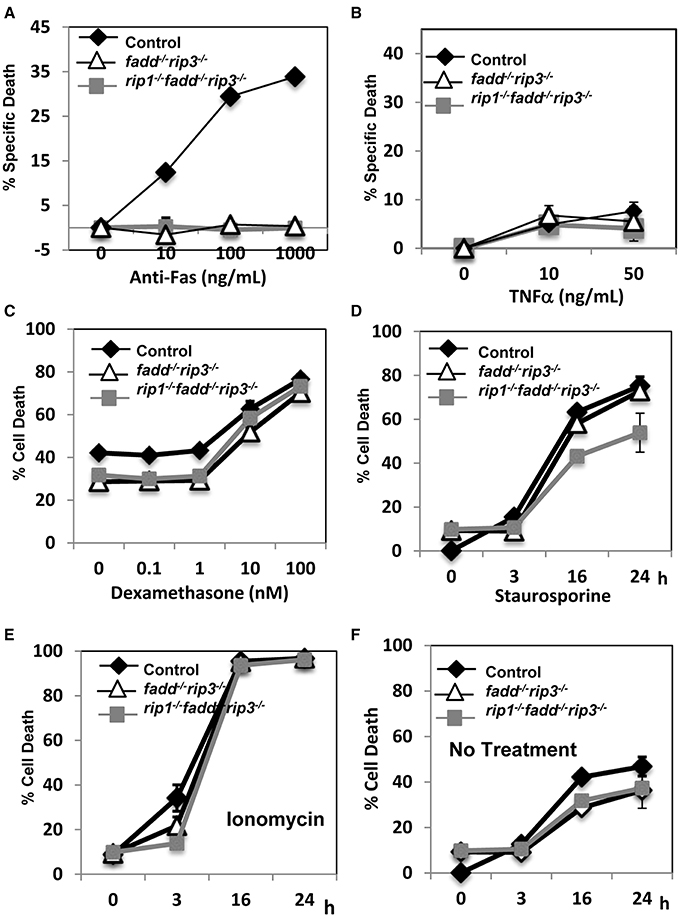
Figure 5. rip1−/−fadd−/−rip3−/− mice had impairment of extrinsic, but not intrinsic, cell death pathways. (A–D) Thymocytes of indicated genotypes were treated with various doses of (A) anti-Fas antibody, (B) TNFα, (C) Dexamethasone and cell death was determined by PI uptake after 16 h incubation. (D–F) Thymocytes were treated with (D) staurosporin (1 μg/ml), (E) ionomycin (1 μg/mL) or (F) no treatment for indicated time and cell death was determined by PI uptake. Experiments were performed in triplicate.
Previous studies indicated that FADD is required during lymphocyte proliferative responses, to sequester RIP1/RIP3-dependent necrosis (Lu et al., 2011; Zhang et al., 2011). To analyze TCR-induced proliferation, peripheral T cells were isolated and stimulated with anti-CD3 antibodies. In tritiated thymidine incorporation assays, there appeared to be more proliferation in fadd−/−rip3−/− T cells and rip1−/−fadd−/−rip3−/− T cells than control T cells (Figure 6A). In further analysis, T cell division kinetics and death responses were analyzed through two-color flow cytometry. T cells were pre-labeled with the CellTrace dye, and then stimulated with anti-CD3/CD28 antibodies. T cell division is indicated by dilution of CellTrace, and cell death measured through propidium iodide dye staining. As shown in Figure 6B, accelerated proliferative responses were seen in both fadd−/−rip3−/− and rip1−/−fadd−/−rip3−/− T cells, when compared to control T cells. Moreover, there was less cell death in DKO and TKO mutant T cells than in control T cells (Figure 6B).
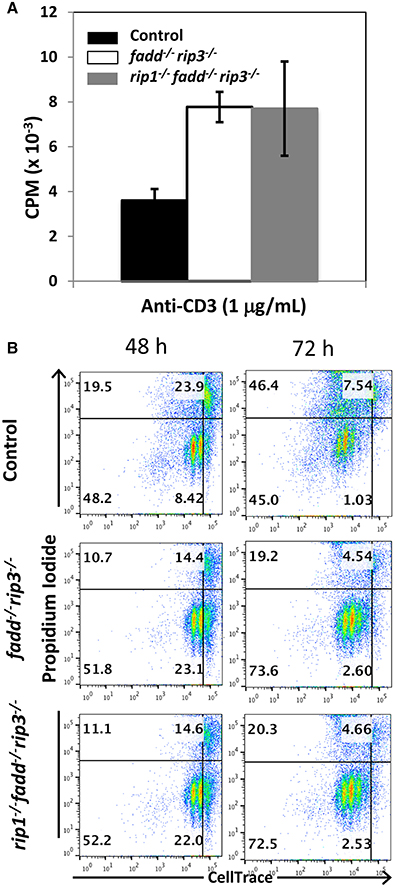
Figure 6. Proliferative responses of TKO and DKO T cells are more robust than control T cells. (A) T cells of indicated genotypes were stimulated with anti-CD3, anti-CD28 antibodies. Proliferation was measured by [3H]-thymidine incorporation (cpm, counts per minute). (B) T cells labeled with CellTrace dye were stimulated with anti-CD3, anti-CD28 antibodies. Dilution of CellTrace dye and uptake of propidium iodide was monitored by flow cytometry at various time points. Data shown is representative of at least three independent experiments with one mouse of each genotype.
The Role of RIP1 in Age-Dependent lpr Diseases
Previous studies have shown that disruption of the function of FADD by using a FADD-DN mutant results in an lpr-like symptom in aged rip3−/− mice (Lu et al., 2011). Similarly, fadd−/−rip3−/− DKO mutant mice developed lpr-like symptoms including enlarged lymph nodes and spleen in as early as 5-week old mice (Figure 7 and data not shown). As mice age, the lymphadenopathy and splenomegaly phenotype become more pronounced, as indicated by greatly increased size, weight, and total cellularity of the secondary lymphoid organs of the fadd−/−rip3−/− DKO mice (Figures 7A–C). Interestingly, these signature lpr symptoms were diminished in TKO mice, as compared to DKO mice (Figure 7).
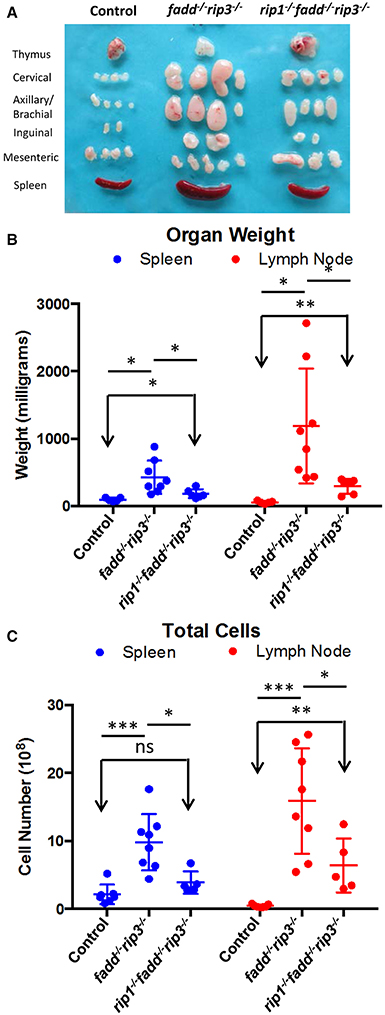
Figure 7. Lymphadenopathy and splenomegaly is less severe in TKO mice, compared to DKO mice. (A) Thymus, lymph nodes, and spleen of 6-month-old mice (B) Weight of spleen and lymph nodes of mice >3 months old (C) Total cell number of spleen and lymph node of mice 3–6.5 months old. At least five mice from each group were used to generate given data. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
Flow cytometric analyses showed that DKO mice contained the unique subset of CD3+ T cells, which express the B cell marker B220+, a signature phenotype of the Fas mutant mice (Figure 8A). Furthermore, these T cells lose the expression of CD4 and CD8 (Figure 8B). When absolute numbers of distinct lymphocyte populations were determined, DKO mice aged 3–6 months were shown to accumulate dramatically higher numbers of CD3+B220+ T cells (Figure 8C). Moreover, B cells and conventional T cells (CD3+B220−) number also increased in DKO mice (Figures 8D,E). Such lpr-like phenotypes were significantly less severe when RIP1 was deleted (Figures 7, 8).
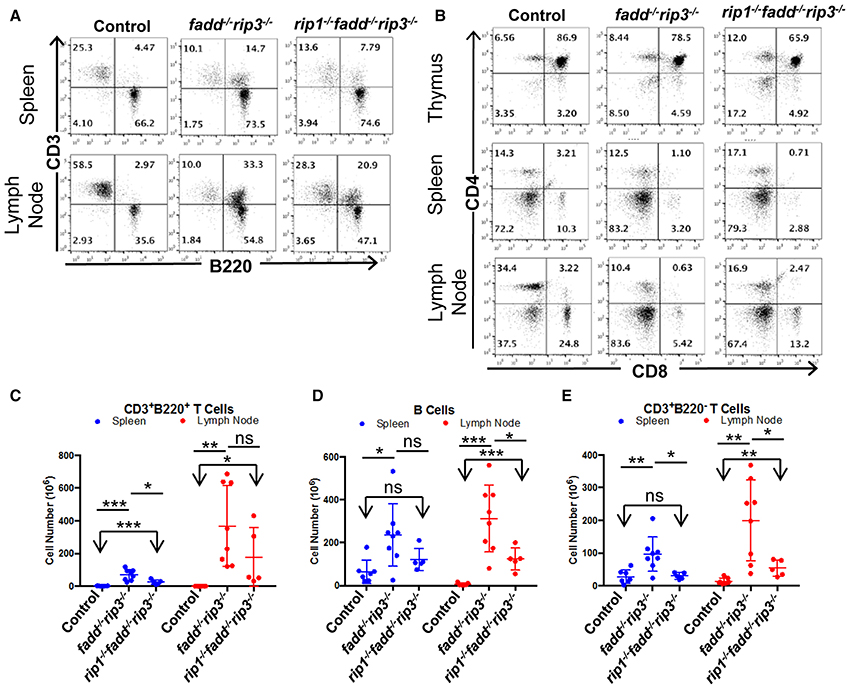
Figure 8. Knockout of RIP1 reduced severity of the lpr symptoms of lymphadenopathy and splenomegaly in fadd−/−rip3−/−. (A,B) Spleen, lymph node, and thymus of six-month-old mice of the indicated genotypes were stained with antibodies for CD3 and B220 (A) or for CD4 and CD8 (B). Numbers indicated are percentages of T cell and B cell subsets. (C–E) Scatter plots of total number of (C) CD3+B220+ T Cells, (D) B cells, and (E) conventional CD3+B220− T cells. At least five mice from each group were used to generate given data. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
Discussion
PCD is tightly controlled in order to ensure proper embryonic and postnatal development as well as to maintain tissue homeostasis. Two major PCD pathways, intrinsic and extrinsic apoptosis, have been well-studied. Intrinsic apoptosis is mediated by pro-apoptotic members of the Bcl-2 family proteins including Bax and Bak, which are counteracted by Bcl-2 and Bcl-XS/L (Marsden and Strasser, 2003; Danial and Korsmeyer, 2004). Absence of Bcl-XS/L blocks embryogenesis at early-midgestation stages due to massive cell death, and deletion of Bcl-2 results in lymphopenic conditions due to greatly increased apoptosis in lymphocytes. In contrast, when proapoptotic Bim is inactivated, mice develop autoimmune and lymphoproliferative diseases (Bouillet et al., 2002). The significance of extrinsic PCD pathways during animal development mediated by DRs or TLRs has been underappreciated, mainly because mice lacking these receptors undergo normal embryonic and postnatal development. As such, developmental defects in mice without the downstream signaling proteins of the DRs, including RIP1, FADD, Caspase 8, or cFLIP, remained largely a conundrum for over a decade, until a latent, RIP1/RIP3-dependent necrosis-like death pathway has been revealed recently (Moriwaki and Chan, 2013; Walsh, 2014).
The fact that RIP1, as implicated in studies based on cell systems, is involved in the regulation of NF-κB activation, apoptosis, and necroptosis, poses as a challenge in understanding the perinatal lethality phenotype of rip1−/− mice (Kelliher et al., 1998). One possible mechanism is that rip1−/− cells become hypersensitive to extrinsic cell death, as observed previously(Ting et al., 1996; Kelliher et al., 1998). However, our previous study shows that blocking extrinsic apoptosis pathways alone through deletion of FADD is insufficient, as postnatal survival of rip1−/−fadd−/− mice were only marginally improved (Zhang et al., 2011). Alternatively, evidence exists, which implies that RIP3 mediates necroptosis in the absence of RIP1 (Upton et al., 2010; Moujalled et al., 2013). Indeed, we found that deletion of RIP3 helped improve the survival of rip1−/− mice for several additional days (Figure 1 and data not shown). Importantly, the current study demonstrates that postnatal lethality in rip1−/− mice is due to the functions of not only FADD but also RIP3, because rip1−/− mice could reach adulthood only if both the FADD and RIP3 genes are ablated simultaneously (Figures 1–3). Firstly, decreasing the RIP3 protein level through deletion of just one allele of the RIP3 gene significantly improved survival of rip1−/−fadd−/−, although the resulting rip1−/−fadd−/−rip3+/− mice had trouble gaining weight (Figures 1, 2). This could suggest that the initial pathology of cell death in the adipose tissue rip1−/− mice is partially mediated by RIP3, which may explain the systemic inflammation displayed in rip1−/−fadd−/−rip3+/− mice, as indicated in a previous independent study (Kaiser et al., 2014). Secondly, when both alleles of RIP3 were deleted, the resulting TKO mice show no defect in body sizes and weights as compared to DKO or control littermates (Figure 3). This data implies that during postnatal development, rip1−/− cells may undergo both FADD-mediated apoptosis and RIP3-dependent necroptosis. This new finding is counterintuitive because RIP1 appears to signal through RIP3 in promoting necroptosis in embryonic cells lacking FADD or Caspase 8 or cFLIP (Kaiser et al., 2011; Oberst et al., 2011; Dillon et al., 2012). In particular, our previous study showed that deletion of the RIP1 adaptor kinase is able to correct the embryonic defect in fadd−/− mice (Zhang et al., 2011). Moreover, fadd−/− or caspase 8−/− mice could reach adulthood when RIP3 is deleted (Kaiser et al., 2011; Oberst et al., 2011; Dillon et al., 2012). It appears that RIP1 plays distinct roles at different stages during development. While dispensable in embryos, RIP1 is required to restrict FADD-mediated apoptosis and RIP3-dependent necroptosis in order to ensure proper postnatal development.
The precise mechanism involved in RIP1-mediated sequestration of apoptosis and necroptosis remains to be further defined. One scenario involves cFLIP, an inhibitor of both apoptosis and necroptosis. cFLIP is a target of NF-κB (Kreuz et al., 2001), which could be activated through RIP1 under certain circumstances. Likely, lack of RIP1 impairs the expression of cFLIP. It was previously thought that the defect in rip1−/− mice is due, in part, to TNFR1-triggered apoptosis. Recently, deletion of TNFR1 or caspase 8 in addition to RIP1 was attempted to mitigate uncontrolled apoptosis, but provided no survival advantage (Rickard et al., 2014). A RIP1-independent necroptotic pathway is induced by TLR3 and TLR4 through its adaptor TIR-domain-containing adaptor-inducing interferon-β (TRIF), which can bind RIP3 via the RIP homotypic interaction motif (RHIM) (Kaiser et al., 2013; Dillon et al., 2014). These two TLRs could also trigger activation of NF-κB through RIP1 (Meylan et al., 2004; Zhang et al., 2011). The type I and type II interferon (IFN) receptors are also capable of inducing necroptosis (Balachandran et al., 2004; Kaiser et al., 2014). In concordance with these models, knockout of TRIF or the type I IFN receptor, in addition to knockout of RIP1 and TNFR, modestly prolonged survival, allowing a small number of these mice to survive to weaning age (Dillon et al., 2014). The perinatal lethality of rip1−/− mice was partially rescued when apoptosis or necroptosis mediated by TNFR, TLRs, or IFN receptors was blunted (Dillon et al., 2014; Kaiser et al., 2014; Rickard et al., 2014).
RIP3 and MLKL have been shown to execute necroptosis in a RIP1-independent manner through Toll-like Receptor-3 (TLR-3) (Upton et al., 2010; He et al., 2011; Kaiser et al., 2013). Recent independent study has shown that ablation of either RIP3 or MLKL in addition to RIP1 resulted in lower levels of cleaved caspase 3 in various organs in newborns compared with rip1−/− newborns as well as lower levels of inflammatory cytokines, potentially due to lower levels of damage-associated molecular patterns (DAMPs) released from necrotic cells (Rickard et al., 2014). However, these mice did not survive past day p4. Another study found that a small percentage of rip1−/−rip3−/− mice survived up to 21 days, however, we see no survival of rip1−/−rip3−/− mice beyond day p6 (Figure 1D). Furthermore, some rip1−/−tnfr−/− survived for 18 days (Dillon et al., 2014), whereas such mice die right at birth in a separate study (Rickard et al., 2014). Combined knockout of RIP3 and TNFR1 did allow survival of rip1−/− mice into adulthood, but these mice ultimately succumbed to blood bacteremia, sepsis, and prevalent apoptosis in the intestines (Dillon et al., 2014). This lethal intestinal defect caused by RIP1 ablation that was recently characterized using cell-specific Cre-mediated deletion of RIP1 (Dannappel et al., 2014; Takahashi et al., 2014). In another study, it was noted that rip1−/−rip3−/−tnf−/− mice did not survive past day 28 (Kaiser et al., 2014). The discrepancy in survival of mice in these studies could be due to differences in genetic backgrounds of the mice used. The lethality of rip1−/−rip3−/−tnfr−/− mice also suggests a key role for FADD/caspase 8-mediated apoptosis in RIP1 perinatal lethality. Therefore, lethality of RIP1 knockout mice is due to multiple factors.
Thymocytes isolated from DKO and TKO mice were resistant to death induced by the extrinsic pathways triggered through stimulations with anti-Fas antibodies or TNFα (Figures 5A,B). However, these mutant T cells were sensitive to cell death induced by dexamethasone, staurosporine, or ionomycin, similar to that of the wild-type control mice (Figures 5C,D,E). Extrinsic cell death has been shown to play a key role in lymphocyte homeostasis and peripheral tolerance (Strasser et al., 2009). When lymphocytes are not capable of being eliminated in the periphery, progressive accumulation of lymphocytes can occur in the peripheral lymphoid organs. This is the case in lpr mice, displaying splenomegaly and lymphadenopathy as a result of a mutation in the Fas gene. Whereas disruption of the FADD function alone through gene deletion or using FADD-DN results in a lymphopenic condition (Newton et al., 1998; Walsh et al., 1998; Zhang et al., 2005), double deletion of both FADD and RIP3 leads to lpr-like symptoms (Figures 7, 8), which is similar to that in caspase8−/−rip3−/− mice (Kaiser et al., 2011; Oberst et al., 2011), but is more severe that in fas−/− mutant mice. Notably, however, a diminished lpr disease was present in rip1−/−fadd−/−rip3−/− mice (Figures 7, 8). This indicates that RIP1 has additional functions besides blocking FADD-mediated apoptosis and RIP3-dependent necroptosis. Interestingly, rip1−/−fadd−/−rip3+/− mice develop splenomegaly, but not lymphadenopathy, after several months of age (data not shown), indicating a role for RIP3-dependent necroptosis in the clearance of lymphocytes in the lymph nodes, but not the spleen. These rip1−/−fadd−/−rip3+/−, as well as DKO and TKO mice, contain varying numbers of the peculiar CD3+B220+ T cells, which lose CD4 and CD8 expression (Figure 8 and data not shown). These data, in total, illustrate that in the immune system, chronic activation of lymphocytes disrupts homeostasis and leads to autoimmunity, which can be prevented by FADD-mediated apoptosis, a sterile PCD. RIP3-deficient T cells undergo normal activation-induced cell death (AICD), indicating that FADD-mediated apoptosis is sufficient for maintaining homeostasis. Intuitively, apoptosis is a preferred pathway for AICD as necroptosis may potentially lead to inflammation which needs to be suppressed by FADD. Therefore, although pro-inflammatory, RIP1/RIP3-dependent necroptosis serves as a backup, when the FADD pathway is compromised. It is interesting to note that there appeared to be more cell division in activated fadd−/−rip3−/− T cells than in wild type T cells (Figure 6). This may due in part to decreased cell death in activated fadd−/−rip3−/− T cells, and/or a role for FADD in negatively regulating T cell proliferative responses. As such, both of these functions of FADD likely contribute to the greatly accelerated lpr disease in fadd−/−rip3−/− mice.
Clearly, RIP1 plays an essential role in the life of an organism and the absence of this protein can only be compensated for by removing both apoptosis and necroptosis pathways. Due to its essential function of regulating not only cell death, but also cell survival, there may be further important consequences for the loss of such a key protein. Future investigations will drive the discoveries of the essential functions of RIP1 and the consequences for deleting RIP1. These studies will broaden our understanding of RIP1 function and could lead to the development of new strategies to target inflammatory conditions as well as cancer and autoimmune diseases, that may be exacerbated due to unregulated necrosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study is supported in part by NIH grant R21 AI105794 to JZ. We thank Dr. Michelle Kelliher for providing rip1+/− mice and Drs. Kim Newton and Vishva Dixit for providing rip3−/− mice, Jennifer Wilson for critical reading of the manuscript, technical contribution by Ben Davis, the technical staff for their help at the Flow Cytometry Facility and Research Animal Facility of the Sidney Kimmel Cancer Center, which is supported in part by NCI Cancer Center support grant P30CA56036.
References
Adachi, M., Suematsu, S., Suda, T., Watanabe, D., Fukuyama, H., Ogasawara, J., et al. (1996). Enhanced and accelerated lymphoproliferation in Fas-null mice. Proc. Natl. Acad. Sci. U.S.A. 93, 2131–2136. doi: 10.1073/pnas.93.5.2131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ashkenazi, A., and Dixit, V. M. (1998). Death receptors: signaling and modulation. Science 281, 1305–1308. doi: 10.1126/science.281.5381.1305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balachandran, S., Thomas, E., and Barber, G. N. (2004). A FADD-dependent innate immune mechanism in mammalian cells. Nature 432, 401–405. doi: 10.1038/nature03124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boldin, M. P., Goncharov, T. M., Goltsev, Y. V., and Wallach, D. (1996). Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1-and TNF receptor-induced cell death. Cell 85, 803–815. doi: 10.1016/S0092-8674(00)81265-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boldin, M. P., Varfolomeev, E. E., Pancer, Z., Mett, I. L., Camonis, J. H., and Wallach, D. (1995). A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 270, 7795–7798. doi: 10.1074/jbc.270.14.7795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouillet, P., Purton, J. F., Godfrey, D. I., Zhang, L. C., Coultas, L., Puthalakath, H., et al. (2002). BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415, 922–926. doi: 10.1038/415922a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ch'en, I. L., Tsau, J. S., Molkentin, J. D., Komatsu, M., and Hedrick, S. M. (2011). Mechanisms of necroptosis in T cells. J. Exp. Med. 208, 633–641. doi: 10.1084/jem.20110251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chinnaiyan, A. M., O'Rourke, K., Tewari, M., and Dixit, V. M. (1995). FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81, 505–512. doi: 10.1016/0092-8674(95)90071-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. doi: 10.1016/j.cell.2009.05.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, P. L., and Eisenberg, R. A. (1992). The lpr and gld genes in systemic autoimmunity: life and death in the Fas lane. Immunol. Today 13, 427–428. doi: 10.1016/0167-5699(92)90066-G
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cusson, N., Oikemus, S., Kilpatrick, E. D., Cunningham, L., and Kelliher, M. (2002). The death domain kinase RIP protects thymocytes from tumor necrosis factor receptor type 2-induced cell death. J. Exp. Med. 196, 15–26. doi: 10.1084/jem.20011470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Danial, N. N., and Korsmeyer, S. J. (2004). Cell death: critical control points. Cell 116, 205–219. doi: 10.1016/S0092-8674(04)00046-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dannappel, M., Vlantis, K., Kumari, S., Polykratis, A., Kim, C., Wachsmuth, L., et al. (2014). RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94. doi: 10.1038/nature13608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Degterev, A., and Yuan, J. (2008). Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 9, 378–390. doi: 10.1038/nrm2393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Diehl, G. E., Yue, H. H., Hsieh, K., Kuang, A. A., Ho, M., Morici, L. A., et al. (2004). TRAIL-R as a negative regulator of innate immune cell responses. Immunity 21, 877–889. doi: 10.1016/j.immuni.2004.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dillon, C. P., Weinlich, R., Rodriguez, D. A., Cripps, J. G., Quarato, G., Gurung, P., et al. (2014). RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202. doi: 10.1016/j.cell.2014.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dillon, C. P., Oberst, A., Weinlich, R., Janke, L. J., Kang, T. B., Ben-Moshe, T., et al. (2012). Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 1, 401–407. doi: 10.1016/j.celrep.2012.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fisher, G. H., Rosenberg, F. J., Straus, S. E., Dale, J. K., Middleton, L. A., Lin, A. Y., et al. (1995). Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81, 935–946. doi: 10.1016/0092-8674(95)90013-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, S., Liang, Y., Shao, F., and Wang, X. (2011). Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20054–20059. doi: 10.1073/pnas.1116302108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi: 10.1038/82732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imtiyaz, H. Z., Rosenberg, S., Zhang, Y., Rahman, Z. S., Hou, Y. J., Manser, T., et al. (2006). The Fas-associated death domain protein is required in apoptosis and TLR-induced proliferative responses in B cells. J. Immunol. 176, 6852–6861. doi: 10.4049/jimmunol.176.11.6852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaiser, W. J., Daley-Bauer, L. P., Thapa, R. J., Mandal, P., Berger, S. B., Huang, C., et al. (2014). RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. U.S.A. 111, 7753–7758. doi: 10.1073/pnas.1401857111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaiser, W. J., Sridharan, H., Huang, C., Mandal, P., Upton, J. W., Gough, P. J., et al. (2013). Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279. doi: 10.1074/jbc.M113.462341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaiser, W. J., Upton, J. W., Long, A. B., Livingston-Rosanoff, D., Daley-Bauer, L. P., Hakem, R., et al. (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372. doi: 10.1038/nature09857
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelliher, M. A., Grimm, S., Ishida, Y., Kuo, F., Stanger, B. Z., and Leder, P. (1998). The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303. doi: 10.1016/S1074-7613(00)80535-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kreuz, S., Siegmund, D., Scheurich, P., and Wajant, H. (2001). NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 21, 3964–3973. doi: 10.1128/MCB.21.12.3964-3973.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, J. V., Weist, B. M., van Raam, B. J., Marro, B. S., Nguyen, L. V., Srinivas, P., et al. (2011). Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc. Natl. Acad. Sci. U.S.A. 108, 15312–15317. doi: 10.1073/pnas.1102779108
Marsden, V. S., and Strasser, A. (2003). Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 21, 71–105. doi: 10.1146/annurev.immunol.21.120601.141029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meylan, E., Burns, K., Hofmann, K., Blancheteau, V., Martinon, F., Kelliher, M., et al. (2004). RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 5, 503–507. doi: 10.1038/ni1061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moriwaki, K., and Chan, F. K. (2013). RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 27, 1640–1649. doi: 10.1101/gad.223321.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moujalled, D. M., Cook, W. D., Okamoto, T., Murphy, J., Lawlor, K. E., Vince, J. E., et al. (2013). TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 4, e465. doi: 10.1038/cddis.2012.201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muzio, M., Chinnaiyan, A. M., Kischkel, F. C., O'Rourke, K., Shevchenko, A., Ni, J., et al. (1996). FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85, 817–827. doi: 10.1016/S0092-8674(00)81266-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newton, K., Harris, A. W., Bath, M. L., Smith, K. G. C., and Strasser, A. (1998). A dominant interfering mutant of FADD/MORT1 enhance deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J. 17, 706–718. doi: 10.1093/emboj/17.3.706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newton, K., Sun, X., and Dixit, V. M. (2004). Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol. 24, 1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., et al. (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. doi: 10.1038/nature09852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Donnell, M. A., and Ting, A. T. (2011). RIP1 comes back to life as a cell death regulator in TNFR1 signaling. FEBS J. 278, 877–887. doi: 10.1111/j.1742-4658.2011.08016.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeffer, K., Matsuyama, T., Kundig, T. M., Wakeham, A., Kishihara, K., Shahinian, A., et al. (1993). Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73, 457–467. doi: 10.1016/0092-8674(93)90134-C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rickard, J. A., O'Donnell, J. A., Evans, J. M., Lalaoui, N., Poh, A. R., Rogers, T., et al. (2014). RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188. doi: 10.1016/j.cell.2014.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roderick, J. E., Hermance, N., Zelic, M., Simmons, M. J., Polykratis, A., Pasparakis, M., et al. (2014). Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc. Natl. Acad. Sci. U.S.A. 111, 14436–14441. doi: 10.1073/pnas.1409389111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rothe, J., Lesslauer, W., Lotscher, H., Lang, Y., Koebel, P., Kontgen, F., et al. (1993). Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364, 798–802. doi: 10.1038/364798a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmukle, A. C., and Walczak, H. (2012). No one can whistle a symphony alone—how different ubiquitin linkages cooperate to orchestrate NF-kappaB activity. J. Cell Sci. 125, 549–559. doi: 10.1242/jcs.091793
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, X., Xie, C., Kreska, D., Richardson, J. A., and Mohan, C. (2002). Genetic dissection of SLE: SLE1 and FAS impact alternate pathways leading to lymphoproliferative autoimmunity. J. Exp. Med. 196, 281–292. doi: 10.1084/jem.20010955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stanger, B. Z., Leder, P., Lee, T.-H., Kim, E., and Seed, B. (1995). RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81, 513–523. doi: 10.1016/0092-8674(95)90072-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strasser, A., Jost, P. J., and Nagata, S. (2009). The many roles of FAS receptor signaling in the immune system. Immunity 30, 180–192. doi: 10.1016/j.immuni.2009.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takahashi, N., Vereecke, L., Bertrand, M. J. M., Duprez, L., Berger, S. B., Divert, T., et al. (2014). RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513, 95–99. doi: 10.1038/nature13706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ting, A. T., Pimentel-Muinos, F. X., and Seed, B. (1996). RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-intitiated apoptosis. EMBO J. 15, 6189–6196.
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2010). Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313. doi: 10.1016/j.chom.2010.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15, 135–147. doi: 10.1038/nrm3737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vucic, D., Dixit, V. M., and Wertz, I. E. (2011). Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 12, 439–452. doi: 10.1038/nrm3143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walsh, C. M. (2014). Grand challenges in cell death and survival: apoptosis vs. necroptosis. Front. Cell Dev. Biol. 2:3. doi: 10.3389/fcell.2014.00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walsh, C. M., Wen, B. G., Chinnaiyan, A. M., O'Rourke, K., Dixit, V. M., and Hedrick, S. M. (1998). A role for FADD in T cell activation and development. Immunity 8, 439–449. doi: 10.1016/S1074-7613(00)80549-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeh, W.-C., Pompa, J. L., McCurrach, M. E., Shu, H.-B., Elia, A. J., Shahinian, A., et al. (1998). FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279, 1954–1958. doi: 10.1126/science.279.5358.1954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Zhou, X., McQuade, T., Li, J., Chan, F. K., and Zhang, J. (2011). Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471, 373–376. doi: 10.1038/nature09878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, J., Cado, D., Chen, A., Kabra, N. H., and Winoto, A. (1998). Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392, 296–300. doi: 10.1038/32681
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, J., and Winoto, A. (1996). A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol. Cell. Biol. 16, 2756–2763.
Zhang, Y., Rosenberg, S., Wang, H., Imtiyaz, H. Z., Hou, Y. J., and Zhang, J. (2005). Conditional Fas-Associated Death Domain Protein (FADD):GFP Knockout mice reveal FADD Is dispensable in thymic development but essential in peripheral T cell homeostasis. J. Immunol. 175, 3033–3044. doi: 10.4049/jimmunol.175.5.3033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, J., Jitkaew, S., Cai, Z., Choksi, S., Li, Q., Luo, J., et al. (2012). Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. U.S.A. 109, 5322–5327. doi: 10.1073/pnas.1200012109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mouse development, apoptosis, necroptosis, programmed cell death (PCD), immune homeostasis
Citation: Dowling JP, Nair A and Zhang J (2015) A novel function of RIP1 in postnatal development and immune homeostasis by protecting against RIP3-dependent necroptosis and FADD-mediated apoptosis. Front. Cell Dev. Biol. 3:12. doi: 10.3389/fcell.2015.00012
Received: 23 January 2015; Paper pending published: 06 February 2015;
Accepted: 10 February 2015; Published online: 25 February 2015.
Edited by:
Francis Kaming Chan, University of Massachusetts Medical School, USAReviewed by:
Han-Ming Shen, National University of Singapore, SingaporeAdrian T. Ting, Icahn School of Medicine at Mount Sinai, USA
Copyright © 2015 Dowling, Nair and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianke Zhang, Department of Microbiology and Immunology, Sidney Kimmel Cancer Center, Sidney Kimmel Medical College, Thomas Jefferson University, 233 S. 10th St., Room 731 BLSB, Philadelphia, PA 19107, USA e-mail:amlhbmtlLnpoYW5nQGplZmZlcnNvbi5lZHU=