 Nikolas Capra1†Chloé Lelièvre2†
Nikolas Capra1†Chloé Lelièvre2† Océane Touré2†
Océane Touré2† Aurélie Fossey-Jouenne2
Aurélie Fossey-Jouenne2 Carine Vergne-Vaxelaire2
Carine Vergne-Vaxelaire2 Dick B. Janssen1
Dick B. Janssen1 Andy-Mark W. H. Thunnissen1*
Andy-Mark W. H. Thunnissen1* Anne Zaparucha2*
Anne Zaparucha2*- 1Biotransformation and Biocatalysis, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Groningen, Netherlands
- 2Génomique Métabolique, Genoscope, Institut François Jacob, Commissariat à l’énergie atomique et aux énergies alternatives, Centre National de la Recherche Scientifique, Université Evry Paris-Saclay, Evry, France
The CoA ligase from Metallosphaera sedula (MsACL) can be used for the chemoenzymatic synthesis of amides from carboxylic acids. In this CoA-independent conversion, the enzyme catalyzes the adenylation of a carboxylic acid with the help of ATP, followed by the uncatalyzed cleavage of acyl-AMP by a nucleophilic amine to yield an amide. With ω-amino acids as substrates this reaction may result in formation of lactams, but unfortunately the substrate preference of the wild-type enzyme is rather limited. To allow structure-based protein engineering and expand the substrate scope of the enzyme, crystal structures of MsACL were solved in the thioesterification conformational state with AMP, CoA and with the reaction intermediate acetyl-AMP bound in the active site. Using substrate docking and by comparing the crystals structures and sequence of MsACL to those of related CoA ligases, mutations were predicted which increase the affinity in the carboxylic acid binding pocket for ω-amino acids. The resulting mutations transformed a non-active enzyme into an active enzyme for ε-caprolactam synthesis, highlighting the potential of the thermophilic CoA ligase for this synthetic and biotechnologically relevant reaction.
1 Introduction
Carboxylic acid-CoA ligases (also referred to as acyl/aroyl-CoA ligases or ACLs) catalyze the ATP-dependent conversion of carboxylic acids into acyl/aroyl-CoA thioesters (D’Ambrosio and Derbyshire, 2020). Belonging to the ANL (acyl-CoA synthetase, nonribosomal peptide synthetase, luciferase) enzyme superfamily, ACLs share similar structures and catalytic mechanisms, despite low sequence identities and varying substrate specificities (Gulick, 2009; Schmelz and Naismith, 2009; Clark et al., 2018). Typically, ACLs consist of a monomeric architecture, comprised of a large N-terminal domain and a smaller C-terminal domain connected by a flexible linker, with the active site situated at the domain interface. Catalysis proceeds in two distinct steps: ATP and the carboxylic acid substrate first combine to form a high-energy adenylate intermediate. In the second step, CoA reacts with this intermediate, yielding a CoA-thioester conjugate (Figure 1A). These steps involve a substantial reorientation of the C-terminal domain relative to the N-terminal domain, resulting in two main conformational states of the protein tuned to either catalyze the first reaction step (adenylate-forming state) or the second reaction step (thioester-forming state), the latter corresponding to the protein’s resting state (Wu et al., 2009; Chen et al., 2015; Li and Nair, 2015; Chen et al., 2017). The catalytic mechanism of ACLs is further distinguished by the dynamic creation of binding sites for the carboxylic acid substrate and ATP-Mg2+ in the adenylate-forming state, and for the acyl/aroyl-AMP intermediate and CoA in the thioester-forming state.
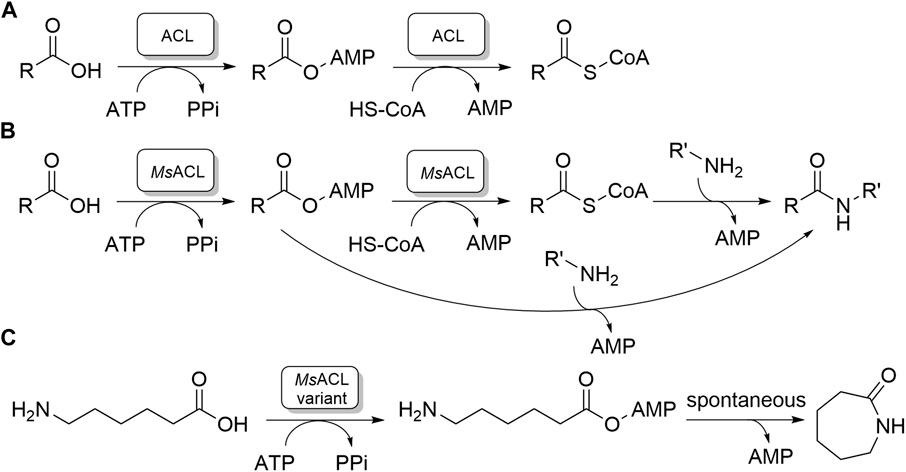
FIGURE 1. Carboxylic acid CoA ligase activities. (A) The native two-step reaction catalyzed by ACLs. (B, C). Amide and lactam formation by chemoenzymatic two-step reaction catalyzed by MsACL; (B): previous work; (C): this work.
A hydrophobic pocket designed for accommodating the acyl/aroyl moiety of the carboxylic acid substrate plays a pivotal role in determining substrate specificity. The shape and chemical nature of this pocket allow ACLs to select substrates based on their length, extent of branching, or presence of an aromatic group (Ingram-Smith et al., 2006; Reger et al., 2007; Shah et al., 2009; Sofeo et al., 2019). Short chain acyl-CoA ligases, whose preferred substrates are acetate and propionate, have a very shallow pocket that is typically enclosed at the base by a conserved tryptophan residue. In aryl and medium chain acyl-CoA ligases this conserved tryptophan residue is replaced by a glycine residue, creating a deeper binding pocket for binding aromatic carboxylic acids, medium sized fatty acids or branched carboxylic acids. Long chain ACLs use an exceptionally spacious hydrophobic acyl binding pocket for binding long fatty acids.
ACLs have garnered significant interest due to their potential in aiding the synthesis of amides and lactams (Caruano et al., 2016; Winnacker and Rieger, 2016; Zhang et al., 2017; Sibikin and Karger-Kocsis, 2018; Stockmann et al., 2020). Amides are crucial precursors for fine chemical and pharmaceutical production, while lactams, such as ε-caprolactam, are essential for nylon polymer manufacturing (Caruano et al., 2016; Winnacker and Rieger, 2016; Zhang et al., 2017; Sibikin and Karger-Kocsis, 2018; Stockmann et al., 2020). Synthesis of amides can be accomplished chemoenzymatically via uncatalyzed amine addition and cleavage of the acyl-CoA product released by ACL, or by amine addition to the acyl-adenylate intermediate, skipping substrate activation by CoA which is an expensive and poorly available substrate (Figure 1B). (Lelièvre et al., 2020) Thus, ACLs may convert ω-amino acids to lactams, by catalyzing the formation of ω-amino acyl adenylates, which then, in the case of five to seven membered chain lengths, undergo spontaneous intramolecular aminolysis of the phosphoester bond (Figure 1C). (Qin et al., 2022) In this way 4-aminobutyric acid, 5-aminopentanoic acid and 6-aminohexanoic acid can be converted into lactams from five to seven membered ring, γ-butyrolactam, δ-valerolactam and ε-caprolactam, respectively. The availability of a chemoenzymatic route towards lactam production would offer clear environmental and economic advantages, as current industrial processes for lactam production rely heavily on petrochemical resources. Unfortunately, the substrate preferences of many native ACLs, particularly for hydrophobic carboxylic acids of specific lengths, as well as their limited thermostability, hinder their direct application in biotechnological lactam production process.
Optimizing the acyl-binding pockets of ACLs by structure-guided protein engineering to change their substrate scope, in particular for converting ω-amino acid substrates to lactams, would increase the applicability of these enzymes. A promising candidate for such an approach is the promiscuous propionate-CoA ligase isolated from Metallosphaera sedula (MsACL, Uniprot ID: A4YDT1). This thermophilic enzyme prefers propionate as substrate, but also shows activity towards longer linear carboxylic acids, up to hexanoic acid, and towards 3-hydroxy-propanoic acid and 4-hydroxybutyric acid, which have structural features in common with short ω-amino acids (Hawkins et al., 2013; Hawkins et al., 2014). Importantly, it has been demonstrated that MsACL can be used in chemoenzymatic reactions for converting carboxylic acid substrates into amides (Hawkins et al., 2013; Hawkins et al., 2014; Lelièvre et al., 2020). Its thermoactivity and stability allow these reactions to run at high temperature, which is highly favorable for the intramolecular aminolysis reaction step leading to lactam formation. Unfortunately, activity with substrates larger than butyric acid is low, which renders the wild-type enzyme unsuitable for the synthesis of lactams.
In this work we focused on modifying the acyl binding pocket of MsACL to accommodate larger and more polar carboxylated substrates, including ω-amino acids, to expand its applicability in lactam synthesis. Initially, we employed X-ray crystallography to determine three-dimensional structures of MsACL in a thioesterification state with bound acetyl-AMP and CoA. By comparing these structures with related ACLs and conducting molecular docking simulations with adenylated substrate intermediates, we gained insight into key residues shaping the acyl-binding pocket and determining substrate specificity. This insight guided the rational prediction of mutations to broaden MsACL’s substrate scope. Some of the resultant MsACL variants exhibited increased activity with ω-amino acids, particularly towards 6-aminohexanoic acid, precursor of ε-caprolactam, enabling promising application in lactam synthesis.
2 Materials and methods
2.1 Chemicals and equipment
All reagents were purchased from commercial sources and used without additional purification, except N-methylamides which were synthesized according to a previously described protocol (Sibikin and Karger-Kocsis, 2018). Structures were confirmed by 1H-NMR and 13C-NMR.
UHPLC-MS analyses were performed on a UHPLC U3000 RS 1034 bar instrument (Thermo Fischer Scientific) coupled to an MSQ Plus Single Quadrupole mass spectrometer with electrospray ESI in positive mode for amide detection with the probe temperature at 450 °C and cone voltage at 50 V. For detection of lactams, the UPLC was coupled to an ISQ EM Single Quadrupole mass spectrometer with APCI in positive mode with the probe current at 15 μA, the source CID voltage at 60V, the vaporization temperature set at 200°C and the Ion Transfer Tube (ITT) set at 300°C. Spectrophotometric assays were done on a Safas UVMC2 spectrophotometer thermostated at the indicated temperature with a refrigerated/heating circulator Corio CD-200F (Jubalo) using high-precision quartz microcells with 10-mm light path (Hellma Analytics).
2.2 Protein expression and purification
The CoA ligase was produced as described previously (Vergne-Vaxelaire et al., 2013). It was purified by loading cell-free extract onto a Ni-NTA column (QIAGEN) according to the manufacturer’s instructions. The elution buffer was 50 mM phosphate (pH 7.5), 50 mM NaCl, 250 mM imidazole and 10% glycerol and the desalting buffer was 50 mM sodium potassium phosphate (pH 7.5), 50 mM NaCl and 10% glycerol. Large-scale purification was conducted from a 500 mL culture by nickel affinity chromatography in tandem with a HiPrep 26/10 (Cytiva) desalting column as described elsewhere (Perchat et al., 2018). Protein concentrations were determined by the Bradford method with bovine serum albumin as the standard. Protein purity was analyzed by SDS-PAGE using the Invitrogen NuPage system. The purified protein was stored at −80 °C. Purified MsACL in 50 mM phosphate buffer, pH 7.5, 50 mM NaCl, 10% glycerol was obtained and used for assays and crystallization.
2.3 Generation of mutants and expression
The QuikChange II site-directed mutagenesis kit (Agilent) and the QuikChange multisite-directed mutagenesis kit (Agilent) were used according to manufacturer’s instructions for introducing mutations. Successful cloning and mutagenesis were confirmed by sequencing. The mutants were produced in E. coli BL-21 CodonPlus (DE3) RIPL and purified by nickel affinity chromatography with an Ni-NTA column (Qiagen) according to the manufacturer’s instructions as described above.
2.4 Crystallization
To prepare the protein for crystallization screening, an additional cation-exchange chromatography step was carried out at 4°C using a MonoS 5/55 HR column (Cytiva) equilibrated with 25 mM N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES), pH 6.5. Before loading, the sample was diluted 3-fold to reduce the salt concentration and assure binding to the column. Elution was performed with a linear gradient of 0–1 M NaCl in 25 mM BES, pH 6.5, applied in 20 column volumes. The protein eluted as a single sharp peak at 250 mM NaCl, with a purity of at least 95%, as assessed by SDS-PAGE. Dynamic light scattering analysis was used to assure that the purified protein sample was sufficiently monodisperse and revealed an apparent molecular mass of the protein particles of 52 kDa, consistent with the presence of monomers.
Prior to crystallization screening, aliquots of the protein were incubated for 2 h with different combinations of ligands (Table 1), with the aim to trap the enzyme in different catalytic states. Crystallization screening was carried out at 20 °C using the sitting-drop vapour diffusion method, with the help of a Mosquito crystallization robot and applying different sparse-matrix crystal screens. Drops were dispensed in MRC-SD2 plates by mixing protein and reservoir solutions at two different ratios (1.25:0.75 and 0.75:1.25) to a final volume of 200 nL. Small, intergrown crystals appeared after 48 h at a few different crystallization conditions. The best crystals grew in solution containing 0.1 M acetic acid, pH 5.2, 200 mM NaCl, 7%–10% PEG3350, with all ligand combinations, but mostly appeared intergrown. Optimization was carried out by varying protein concentration, temperature, use of additives and changing the protein:reservoir ratio during drop mixing, as well as by exploring seeding, but the morphology of the crystals was not improved.
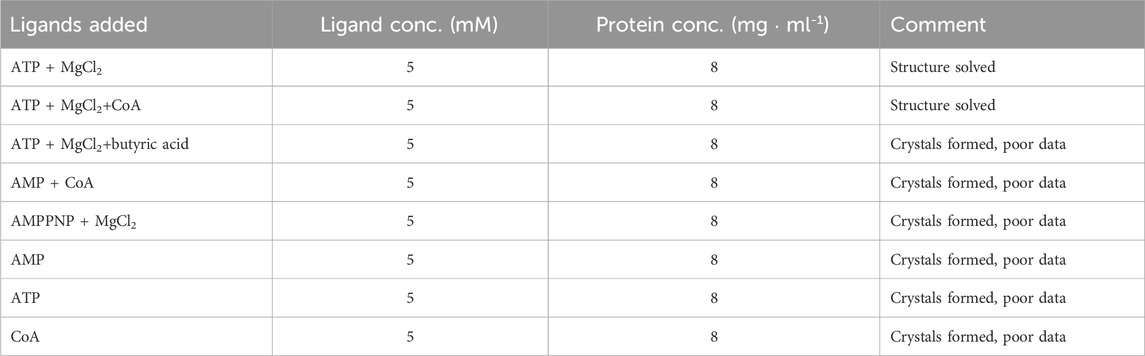
TABLE 1. Ligand combination used for co-crystallization experiments.
2.5 Crystal structure determination
Various crystals grown with different combinations of ligands were tested for X-ray diffraction at the Diamond Light Source (DLS) synchrotron in Oxfordshire, UK, and at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. Most crystals displayed very poor diffraction. A suitable X-ray diffraction data set to 2.8 Å resolution was collected at beamline i04 of the DLS for a MsACL crystal grown in the presence of 5 mM ATP and 5 mM MgCl2 using a crystallization solution containing 0.1 M acetic acid, pH 5.2, 200 mM NaCl and 8% PEG3350. At the same beamline, an additional data set to 3.1 Å resolution was collected with a similar MsACL crystal grown in the presence of 5 mM ATP, 5 mM MgCl2 and 5 mM CoA. Prior to data collection, the crystals were briefly soaked in a cryoprotectant solution containing the mother liquor and 30% v/v glycerol.
Diffraction data for the crystal obtained in the presence of ATP and MgCl2 were indexed and integrated using xia2-DIALS in space group P21, then scaled and merged with Aimless (Evans and Murshudov, 2013; Winter et al., 2018). The asymmetric unit was estimated to contain four protein molecules with a Matthews coefficient of 2.24 and a solvent content of 45%. Molecular replacement was carried out using Phaser, using a crystal structure of human ACSM2A as a search model (PDB ID: 3B7W, 33% sequence identity) (McCoy et al., 2007). We followed a 2-step molecular replacement strategy adapted for dealing with the intrinsic flexibility of the protein. First, a rotational and positional search was carried out only for the bulk N-terminal domains of the protein molecules in the asymmetric unit, followed by a rotational and positional search for the C-terminal domains. Subsequently, using the CCP4Cloud suite, phases were improved by density modification using Parrot and an initial protein model was automatically built with Buccaneer (Cowtan, 2006; Cowtan, 2010; Krissinel et al., 2018). Several iterations of restrained refinement with REFMAC5 (Murshudov et al., 2011) alternated by manual model building with Coot (Emsley and Cowtan, 2004) were necessary to improve the structure. We used an AlphaFold model of the protein, calculated using the ColabFold internet scripts (Mirdita et al., 2022) and Phenix. refine (Afonine et al., 2012), to improve the protein areas which were poorly resolved in the electron density maps. AMP molecules were placed in the active sites of the four molecules based on difference electron density maps. No ATP molecules or magnesium ions were found in the active sites. For one of the bound AMPs (in the active site of protein molecule B), the electron density map clearly indicated the presence of a small adduct attached to one of the phosphate oxygens, which we interpreted as an acetyl moiety. Thus, one acetyl-AMP was included during the final rounds of model building and refinement.
Diffraction data for the second crystal were autoprocessed with the autoPROC (Vonrhein et al., 2011) scripts installed at DLS. This crystal belonged to space group C2 with two protein molecules in the asymmetric unit and a solvent content of 51%. The structure was determined by molecular replacement using one of the MsACL molecules from the first crystal structure as a search model. The electron density maps clearly indicated the presence of bound acetyl-AMP and CoA in both protein molecules. CoA was added using Coot and the structure was completed using a few cycles of restrained refinement with REFMAC5 and model building with Coot. A summary of the crystallographic statistics is shown in Table 2. Figures were generated with PyMOL (Schrödinger, 2010).
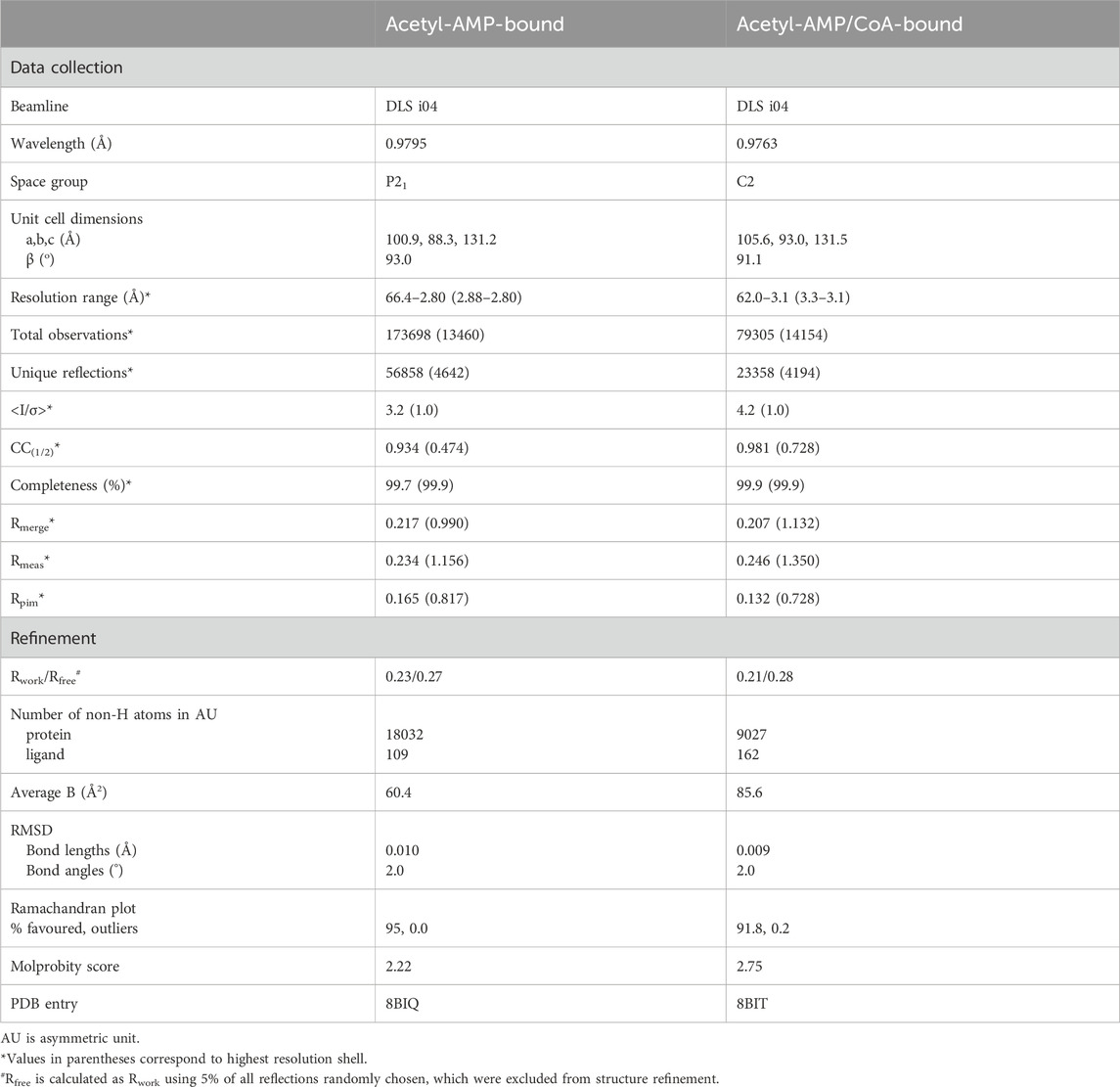
TABLE 2. Crystallographic data collection and refinement statistics for MsACL.
2.6 Docking simulations
Target ligands for docking were designed using the JSME structure editor (Bienfait and Ertl, 2013) and energy minimized using AVOGADRO (Hanwell et al., 2012). The protein molecule in the 2.8 Å crystal structure of MsACL with a bound acetyl-AMP was used as a template for the receptor. The acetyl-AMP was removed from the crystal structure and mutation W259G was introduced using Coot. The MsACL-W259G protein model and simulation cell for docking were further prepared using YASARA (Krieger and Vriend, 2014). Both the receptor and the ligand were subjected to energy minimization in YASARA prior to the simulations. Docking simulations were carried out with AutoDock Vina using the macro “dock_run.mcr” provided with the YASARA software (Trott and Olson, 2010; Eberhardt et al., 2021). The number of docking runs was set to 250 and the protein model was kept rigid during the docking, except for the side chains of solvent-accessible residues at the acyl-binding pocket (i.e., residues V/T238, W254, A255, K256, W/G259, A327, F/Y350; residues are numbered considering the N-terminal His6-tag). Docked ligand poses were automatically clustered using a cut-off RSMD of 2 Å. Manual rescoring was carried out to select the best poses, ensuring that the binding mode of the AMP moieties in the docked acyl-AMP ligands was similar to that observed in the crystal structure.
2.7 UHPLC-MS analysis
Analysis of lactams by UHPLC-MS (APCI) was performed using a Luna Omega Polar-C18 (Phenomenex) column (100 × 2.1 mm; 1.6 μm). Analytical yields were calculated using calibration curves obtained in duplicate with γ-butyrolactam, δ-valerolactam and ε-caprolactam done in presence of various amounts of lactams incubated and treated in same conditions as the biocatalytic reactions for lactam synthesis.
UHPLC-MS (ESI) analysis of N-methylamides was performed using a Kinetex EVO-C18 (Phenomenex) column (100 × 2.1 mm; 1.7 μm. Analytical yields were calculated using calibration curves measured in duplicate with synthesized N-methylamides incubated and treated in the same conditions as the biocatalytic reaction for amide bond synthesis.
2.8 Biocatalytic reactions
For reaction with MsACL and mutants, both MnCl2 and MgCl2 can be used interchangeably. Reaction mixtures with ω-amino acid substrates were carried out in mixtures (100 μL) containing 5 mM ω-amino acid, 5 mM ATP and 5 mM MnCl2 in 50 mM 3-(N-morpholino)propane sulfonic acid (MOPS) buffer (pH 8.5) with 2 mg/mL of purified enzyme. The reactions were stirred at 400 rpm in an Eppendorf ThermoMixer at 60 °C for 24 h. Negative controls without enzyme or substrate were performed in parallel. Aliquots of 30 μL were withdrawn, quenched with 2 μL of 6M HCl solution and diluted with 110 μL CH3OH. After filtration on a 0.22 μm filter, the sample was analysed by UHPLC-MS using conditions described above for lactam detection.
Reactions with carboxylic acid substrates (50 μL) contained 5 mM carboxylic acid, 5 mM ATP, 5 mM MgCl2 and 50 mM methylamine in 50 mM phosphate buffer (pH 8). Reactions were started by adding 0.2 mg/mL of purified enzyme. The mixtures were stirred at 400 rpm in a ThermoMixer at 60 °C for 24 h. Negative controls without enzyme or substrate were performed in parallel. Aliquots of 30 μL were withdrawn, quenched with 2 μL 6M HCl and diluted with 100 μL H2O. After filtration on a 0.22 μm filter, samples were analysed by UHPLC-MS.
2.9 Activity assays and determination of kinetic parameters
All reactions were conducted in duplicate. Specific activities of MsACL and variants for the carboxylic acid substrates were determined by a NADH-pyrophosphate coupled assay (Sigma-Aldrich). Pyrophosphate (PPi) formation is coupled to the consumption of NADH by four enzymatic steps allowing spectrophotometric monitoring at 340 nm (Gulick et al., 2003). The reactions were performed at 60 °C in Tris-HCl buffer, pH 7.5, in a final volume of 60 μL containing 5 mM substrate, 2 mM ATP, 5 mM MnCl2 and 50 mM methylamine. Tris-HCl buffer at pH 7.5 gave a lower absorbance background than other buffers or pH values (data not shown). Reactions were initiated by addition of the appropriate amount of enzyme. Aliquots (15 µL) were withdrawn at different times and transferred to a 10 mm cuvette containing 85 μL of the NADH-pyrophosphate assay mixture previously heated at 37 °C. Monitoring of NADH disappearance at 340 nm was used to determine the specific activity of the enzyme, according to the supplier’s instructions.
Kinetic parameters of MsACL and variants for the ω-amino carboxylic acid substrates and ATP were determined using the same conditions, with varying concentrations of ω-amino carboxylic acid and without methylamine. Reaction mixtures (40 μL) were preincubated at 60 °C for 3 min prior to addition of enzymes, after which incubation was continued for 30 min at 60 °C. After cooling on ice, the reaction mixture was transferred to a 10 mm cuvette containing 110 μL of the NADH-pyrophosphate assay mixture. Initial rates of NADH disappearance were measured at 37 °C and 340 nm. Michaelis-Menten kinetic parameters were calculated from initial rates obtained with varying concentrations of substrate and saturating or optimal concentrations of the other substrates.
3 Results
3.1 Crystal structures of MsACL in the thioesterification state with bound acetyl-AMP and CoA
In order to trap MsACL in a functionally relevant state, protein crystallization screening was performed in the presence of different combinations of the ligands ATP, AMP, CoA and Mg2+. Crystals grew in acetate buffer, pH 5.2, in the presence of ATP and MgCl2 or in the presence of ATP, MgCl2 and CoA, allowing two structures to be determined at 2.8 Å and 3.1 Å resolution, respectively (Table 2; Figure 2). The crystal structure of MsACL obtained by co-crystallization with ATP and MgCl2 belongs to space group P21 and contains four protein molecules in the asymmetric unit (solvent content of 45%). MsACL adopts the characteristic two-domain fold of ATP-dependent ACLs (Kochan et al., 2009; Shah et al., 2009), consisting of a large N-terminal domain (residues 16–449; sequence numbering includes the N-terminal (His)6 tag) and a small C-terminal domain (residues 455-570), connected by a small hinge region (residues 450-455). A structural comparison of MsACL with other ACLs shows that the ligase is in the thioester-forming conformational state (Kochan et al., 2009). Surprisingly, inspection of the electron density map indicated that three protein molecules in the asymmetric unit contain a bound AMP molecule, instead of ATP, while one protein molecule has an acetyl-AMP bound in the active site (Figure 2B). Thus, it appears that the enzyme converted the acetate and ATP in the crystallization solution to acetyl-AMP prior to forming crystals. The apparent presence of AMP in three protein molecules may be an artifact of the low resolution of the X-ray diffraction data, which results in the small acetyl group being poorly resolved in the electron density map. The crystal structure of MsACL obtained by co-crystallization with ATP, MgCl2 and CoA also revealed enzymatic conversion of acetate and ATP to acetyl-AMP. This 3.1 Å crystal structure belongs to space group C2 and contains two protein molecules in the asymmetric unit (solvent content of 45%). The overall conformations of the protein molecules are identical to those in the 2.8 Å structure, but in addition to acetyl-AMP each protein molecule contains a bound molecule of CoA (Figure 2C), confirming that MsACL was trapped in a functionally relevant state.
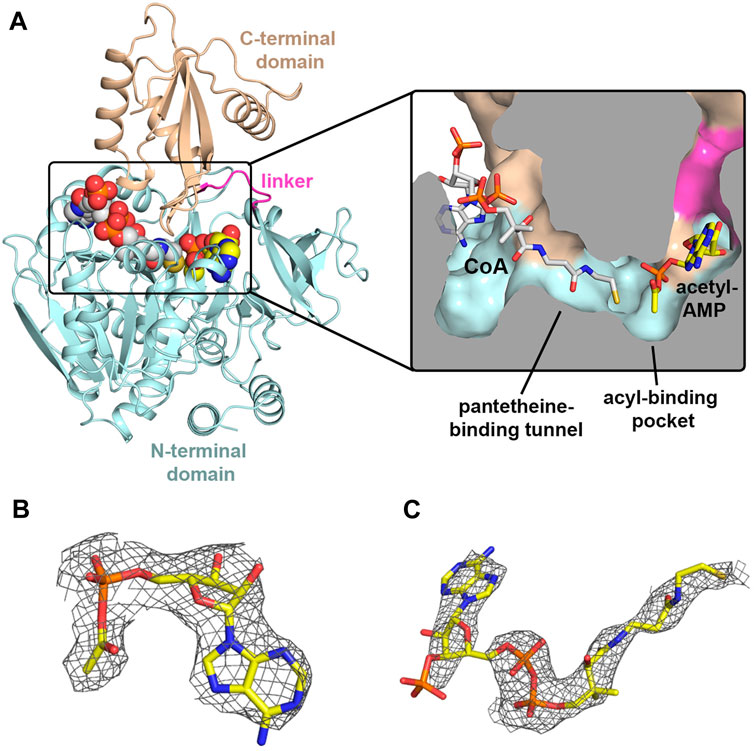
FIGURE 2. Crystal structures of MsACl. (A) Cartoon representation of the 3.1 Å MsACL structure with bound CoA and acetyl-AMP molecules shown as spheres. Oxygens are colored in red, nitrogens in blue, and the carbons of CoA and acetyl-AMP in grey and yellow, respectively. The inset shows a cut-through surface representation of the CoA and acetyl-AMP binding pockets with the two ligands represented by sticks. (B) Acetyl-AMP and its associated electron density in the 2.8 Å MsACL structure. (C) CoA and its associated electron density in the 3.1 Å MsACL structure. Ligands are shown as sticks. The electron densities were calculated as Fo-Fc difference omit Fourier maps, and contoured at 2σ.
3.2 Homology with acyl/aroyl-ACLs
To establish the overall structural relationships between MsACL and related CoA ligases with known structures, a BLAST search was performed against the protein sequences in the Protein Data Bank. A total of 59 unique proteins were identified, 36 being annotated as acyl/aryl-CoA ligases (sequence identities with MsACL ranging between 22%–38%), while the others are luciferases or adenylation domains of nonribosomal peptide synthetases. A selection of five acyl/aroyl-CoA ligases homologous to MsACL is listed in Tables 3, 4. The selected ACLs, with a sequence identity to MsACL of 30%–38%, have been crystallized in the thioester-forming state, showing similar overall Cα-backbone conformations. These enzymes are well characterized in terms of substrate preference. The closest MsACL homolog (38% sequence identity) is the acyl-CoA ligase from Methanosarcina acetivorans (MaACL, PDB entry 3ETC.) (Shah et al., 2009), which belongs to the medium chain ACL class and utilizes 2-methylbutyrate as the preferred substrate (Meng et al., 2010). The human acyl-CoA synthetase ACSM2A (here abbreviated as HsACL, PDB entry 2WD9) also belongs to the medium chain ACLs, but has a wider substrate scope as it is able to convert longer fatty acids, up to decanoic acid (C10), and aromatic carboxylated substrates like ibuprofen. The other homologs include the acetyl-CoA ligase from Salmonella enterica (SeACL, PDB entry 1PG4) (Gulick et al., 2003), a short chain ACL which converts small carboxylic acids up to propionate, and the aryl-CoA ligases from Rhodopseudomonas palustris (RpACL, PDB entry 4RM3) and Stigmatella aurantiaca (SaACL, PDB entry 4WV3), which constitute a special class of ACLs dedicated to the conversion of aromatic carboxylic acids. The AMP and CoA binding sites of MsACL, and in particular its acyl-binding pocket, were compared to these five homologous ACLs, the results of which are described in the next three sections.
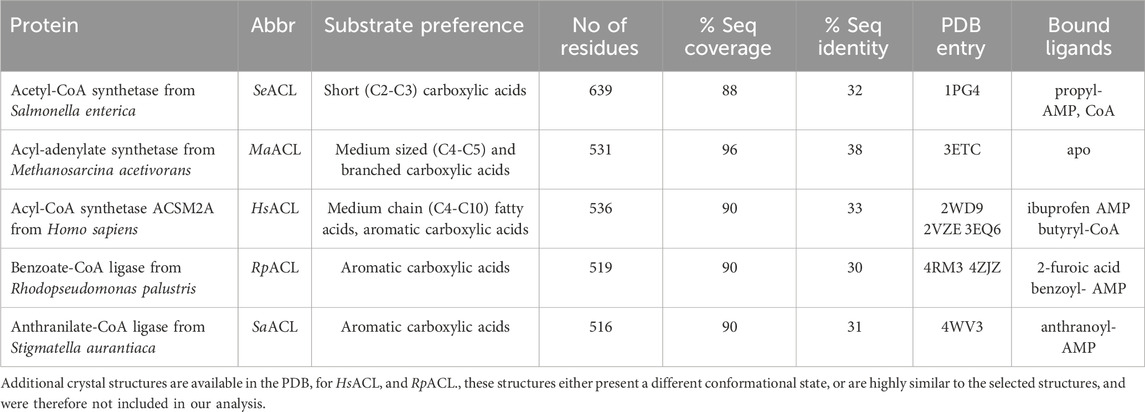
TABLE 3. Selected ACLs in thioesterification state showing homology to MsACL.

TABLE 4. Similarities in C⍺-backbone conformations of selected ACLs compared to MsACLb.
3.3 AMP binding site
The AMP binding site of MsACL is positioned at the domain interface near the hinge region with residues from mainly the N-terminal domain contributing to binding of the nucleotide (Figures 2, 3). Residues D349 and F350 form hydrogen bonds with the adenine base, D435 and Q353 with the hydroxyls of the ribose and T243, R461 and K456 with the α-phosphate group. Most of these AMP-binding residues are highly conserved in the related ACLs. K456 forms an additional hydrogen bond with the carbonyl oxygen of the carboxyl-phosphate linkage between AMP and the acetyl group. It is assumed that this conserved lysine residue in the C-terminal domain is vital for enhancing the reactivity of the carboxyl-phosphate linkage for a nucleophilic attack by the thiol group of CoA (Branchini et al., 2000; Kochan et al., 2009; Shah et al., 2009; Thornburg et al., 2015).
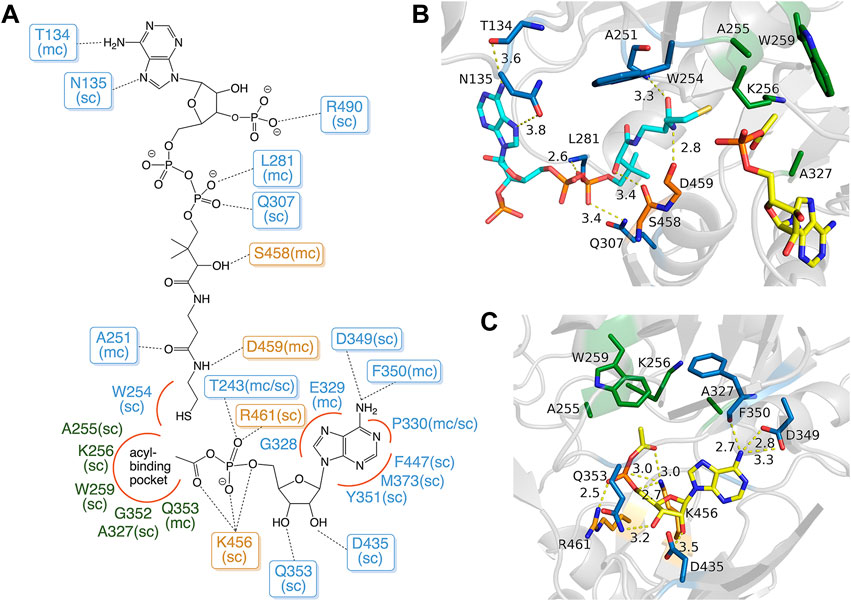
FIGURE 3. Binding modes of acetyl-AMP and CoA in MsACL. (A) Schematic diagram showing the binding interactions with residues of MsACL, as based on analysis of the crystal structures. Residues from the N-terminal domain are indicated with blue and green labels, residues from the C-terminal domain with orange labels. Dashed lines indicate hydrogen bonds. Red curves indicate favorable van der Waals contacts. Abbreviations used: sc, side chain; mc, main chain. (B) Close-up view of CoA and acetyl-AMP binding sites in the 3.1 Å MsACL structure. (C) Close-up view of the acetyl-AMP binding site in the 2.8 Å MsACL structure. Distances in angstroms are indicated with dashed lines.
3.4 CoA binding site
The CoA binding site of MsACL is located next to the AMP binding site at the domain interface and consists of an open pocket for binding the adenosine-diphosphate moiety and a tunnel that leads to the active site for binding the pantetheine moiety of CoA. Among the five MsACL homologs, only the SeACL structure contains a bound CoA, while one of the structures determined for HsACL has a bound butyryl-CoA product (PDB entry 3EQ6). While the binding modes of the CoA pantetheine moieties are highly similar in these structures, the interactions with the adenosine-diphosphate moiety differ significantly. The only conserved interaction with this part of CoA is a salt bridge formed between an arginine residue (R490 in MsACL, Figure 3) and the 3′-phosphate group attached to the ribose. It should be noted, however, that the low resolution and partial disorder of the electron density observed for CoA in the 3.1 Å MsACL crystal structure prohibited an unambiguous identification of all binding interactions. The pantetheine moiety of CoA appears to be bound in an extended conformation with the thiol group closely approaching the acetyl-AMP carboxyl-phosphate linkage. The side chain of W254 at the end of the pantetheine binding tunnel is in van der Waals contact with the thiol group. All ACLs contain an aromatic amino acid residue at this position, forming a constriction that guides the CoA thiol group to optimally approach the acyl-adenylate intermediate for a nucleophilic attack (Kochan et al., 2009).
3.5 Acyl binding pocket
The MsACL acyl binding pocket is buried within the N-terminal domain and is constituted by the side chains of amino acid residues A255, K256, W259 and A327, and the main chain atoms of G352 and Q353 (Figures 2, 3). It has a shallow shape and is largely hydrophobic. W259 forms the pocket “floor” while the other residues form the pocket “walls”. A sequence and structure comparison of the acyl-binding pocket in MsACL with the five related ACLs is presented in Table 5; Figures 4, 5. While all acyl-binding pockets are hydrophobic, they show differences in shape and depth, in line with the differences in substrate preferences among the ACLs. The highest similarity is observed with the acyl-binding pocket of MaACL (Figure 5A), which is nearly identical to that of MsACL. Five of the six acyl pocket forming residues in MsACL are identical to those in MaACL, including the floor-forming tryptophan, with the only difference being the replacement of A255 in MsACL by a glycine residue in MaACL. It’s unclear whether a thiocarbamate linkage is present between residues K256 and C299 of MsACL, similar as observed between residues K256 and C289 in the crystal structure of MaACL (Shah et al., 2009). While the side chains of these residues exhibit close proximity, the assessment of a potential linkage is hindered by limitations in the quality and resolution of the MsACL electron density maps. Overall, the high similarity in shape and chemistry of their acyl binding pockets points to a close relationship in biological function and substrate preference of MsACL and MaACL.

TABLE 5. Residues at the MsACL acyl-binding pocket and corresponding residues in related ACLs.

FIGURE 4. Sequence alignment of related structural segments forming the acyl-binding pockets in MsACL and its homologs. Residues forming the pocket walls are colored in orange, the pocket floor forming tryptophan in MsACL, SeACL, HsACL and MaACL is colored in red.
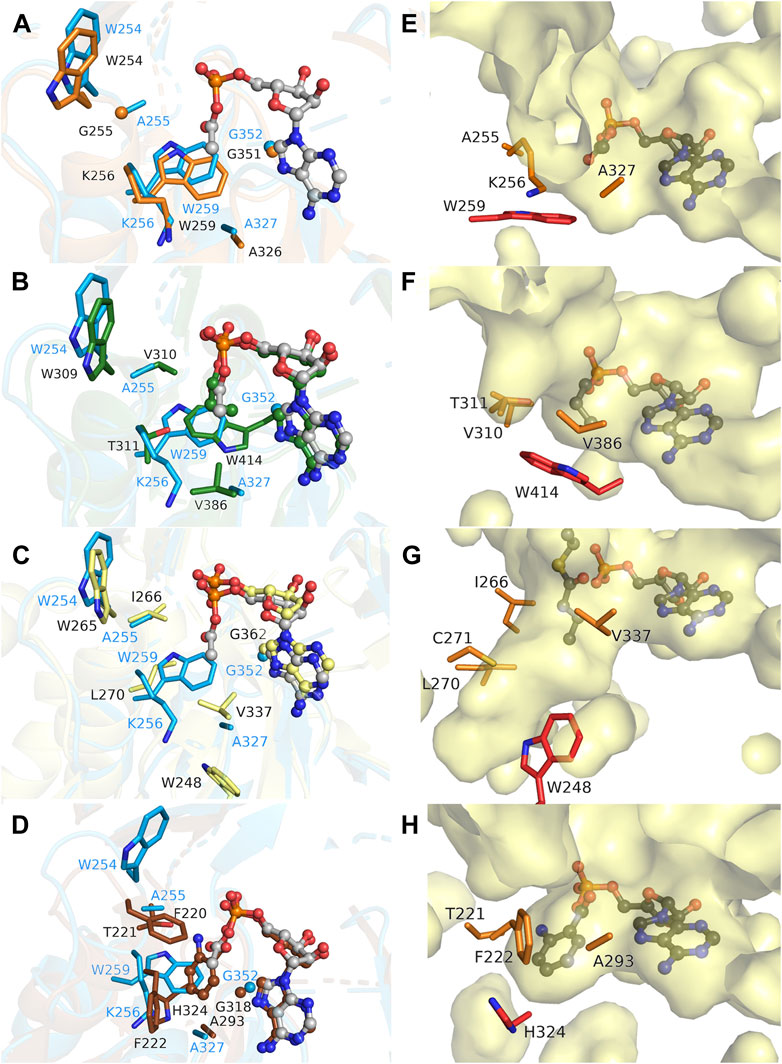
FIGURE 5. Comparison of the acyl binding pockets of MsACL and related ACLs. (A) Structural overlay of the acyl-binding sites of MsACL (blue, with bound acetyl-AMP in ball-and-sticks) and MaACL (PDB 3ETC., orange, apo). Side chains forming the pocket are depicted as sticks. (B). As (A), comparing MsACL to SeACL (1PG4, green, with bound propyl-AMP). (C) As (A), comparing MsACL to HsACL (3EQ6, yellow, with bound AMP). (D) As (A), comparing MsACL to SaACL (4WV3, brown, with bound anthranoyl-AMP). (E–H) Surface representations of the acyl binding pockets in MsACL, SeACL (1PG4), HsACL (3EQ6) and SaACL (4WV3), respectively, including the acyl-AMP as indicated except for HsACL that shows AMP and butytylCoA bound.
Significant differences are observed with the acyl-binding pockets of the other ACLs. In SeACL, a different tryptophan (Trp414) forms the floor of the acyl binding pocket, as the equivalent of G352 of MsACL, which truncates the acyl-binding pocket at a smaller depth. (Figures 5B, F). Additionally, A255 and A327 of MsACL are replaced by two valine residues in SeACL, thus creating a very small acyl binding pocket explaining why SeACL prefers acetate and propionate as substrates (Gulick, 2009; Shah et al., 2009). In HsACL, a leucine residue is located at the equivalent position of W259 in MsACL, opening up the acyl binding pocket and increasing the pocket depth (Figures 5C, G), consistent with its ability to bind and convert longer substrates. Similarly, in RpACL and SaACL, W259 is replaced by a glycine residue, enabling the formation of a deeper pocket for binding aromatic carboxyl acid substrates (Figures 5D, H). A histidine residue (H333 in RpACL, H324 in SaACL) acts as pocket floor in these two ACLs.
3.6 Docking and mutant design
As evident from the crystal structure analysis, a deepening of the acyl binding pocket is necessary to expand the substrate scope of MsACL towards ⍵-amino acids. To create such a deeper pocket, we considered changing W259 to a glycine residue. A similar substitution of the pocket floor forming tryptophan in short-chain ACLs drastically expanded their substrate preference from acetate and propionate to octanoate (Ingram-Smith et al., 2006; Sofeo et al., 2019). To predict the effect of a W259G mutation in MsACL, as well as to suggest additional mutations, we docked a variety of substrates in a W259G mutant model and examined their binding modes. Initial docking performed with 4-aminobutyric acid (1), 5-aminopentanoic acid (2) and 6-aminohexanoic acid (3) (Figure 7A) resulted in a large variety of docking poses, many of which presented unproductive binding modes.
We then switched to docking the adenylated intermediates of the ⍵-amino acids, which has the advantage that the reliability of the docked poses could be assessed by comparing them to the crystallographic binding mode of acetyl-AMP, ensuring that the AMP moiety and the acyl-phosphate linkage are similarly bound as in the crystal structures. Docking of 4-aminobutyryl-AMP in the MsACL W259G model did not result in poses that could explain how the ω-amino group would be stabilized in the enlarged acyl-binding pocket. In fact, more favorable docking poses of this intermediate were obtained in the wild-type MsACL structure, some of which included a cation-pi interaction between the ω-amino group and W259, thus questioning whether a W259G mutation would be beneficial for converting 4-aminobutyric acid. In contrast, reasonable docking poses of 5-aminopentanoyl-AMP and 6-aminohexanoyl-AMP were only obtained using the MsACL W259G model, due to the enlargement of the acyl-binding pocket (Figure 6). The best docking pose of 5-aminopentanoyl-AMP positioned its amino group close to the backbones of Q353 and T356, allowing the formation of hydrogen bonds with these residues (Figure 6A). Docking of 6-aminohexanoyl-AMP resulted in a low energy binding pose with the amino group pointing deep into the space created by the W259G mutation (Figure 6B), allowing the formation of hydrogen bonds with the side chain of T234 and the backbone amine of A358. The pose positions the C6 carbon of the amino-acyl moiety close to the Cα-carbon of G259, enabling a favorable van der Waals interaction, further indicating the potential of the W259G mutation to improve binding and conversion of longer ⍵-amino acids.
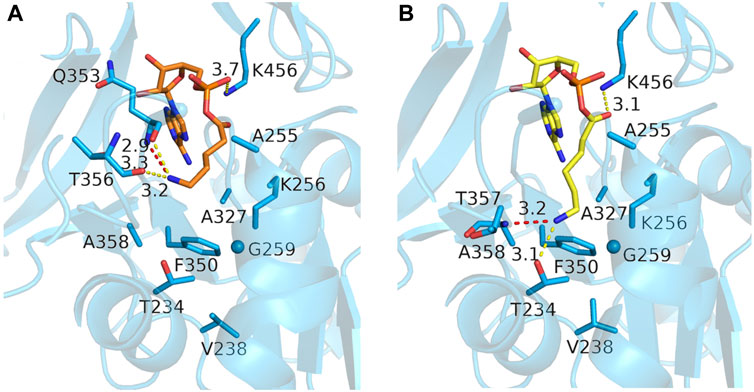
FIGURE 6. Docking poses of two ω-amino acid-AMP adducts in MsACL W259G. (A) Model of MsACL W259G with docked 5-aminopentanoic-AMP (orange). (B) Docked 6-aminohexanoic-AMP (yellow) in W259G model.
Furthermore, the W259G mutation, by providing access to the side chains of T234, V238, F350, and A358, identifies these residues as additional candidates for site-directed mutagenesis. Such mutations can be strategically designed to introduce additional polar groups, aiding in the stabilization of the ω-amino group within the target substrates. We reasoned that introducing mutations V238T and F350Y would be advantageous since they incorporate polar groups while exerting minimal impact on the steric properties of the side chains. T234 was not considered for mutation considering its potential importance for stabilizing the amino group in 6-amino-hexanoyl-AMP. Likewise, residue A327 was also excluded for mutation, as it is highly conserved in medium-chain ACLs and plays a crucial role in shaping the upper part of the acyl-binding pocket. Substituting it with a larger residue would lead to a reduction in the acyl-binding pocket’s size, which was deemed undesirable (Reger et al., 2007; Sofeo et al., 2019).
In conclusion, based on the MsACL crystal structures and the docking results, W259G, V238T and F350Y were pointed out as the most promising mutations for enhancing the activity of MsACL towards ω-amino acid substrates, in particular towards 6-aminohexanoic acid, the precursor of ε-caprolactam.
3.7 Activity of mutants with ω-amino acids
To establish the effect of the proposed mutations, three single MsACL mutants, W259G, V238T, F350Y and two double mutants, W259G/V238T and W259G/F350Y, were constructed by site-directed mutagenesis and expressed in E. coli. Activities of the purified enzymes were examined with 4-aminobutyric acid (1), 5-aminopentanoic acid (2) and 6-aminohexanoic acid (3) (Figure 7A; Figure 8).
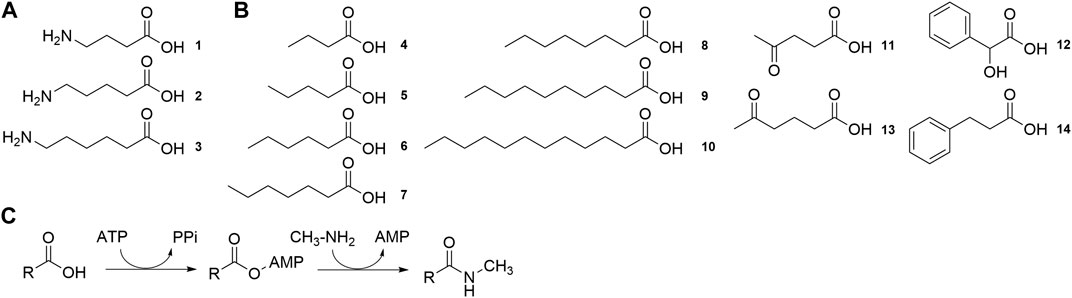
FIGURE 7. Substrates tested for conversion by MsACL (A) Structure of ω-amino acid substrates for lactam synthesis. (B). Structure of carboxylic acid substrates for the synthesis of N-methylamide derivatives. (C). Reaction for the synthesis of N-methylamide derivatives.
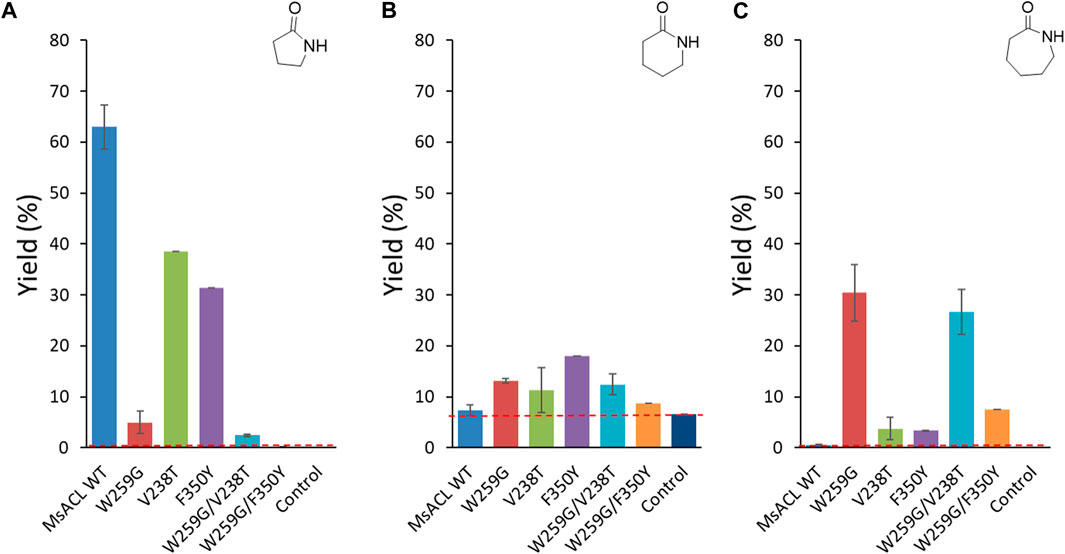
FIGURE 8. Analytical yields of lactam formation from ω-amino acids catalyzed by WT and mutant enzymes. (A) γ-butyrolactam. (B) δ-valerolactam. (C) ε-caprolactam. The dotted red line indicates the yield of spontaneous cyclisation. Reaction conditions: 5 mM ω-amino acid (1-3), 5 mM ATP, 5 mM MnCl2 in 50 mM MOPS buffer (pH 8.5), 2 mg/mL of purified enzyme, 400 rpm at 60 °C for 24 h. Control reactions correspond to samples without enzyme. Analytical yields were deduced from calibration curves by UHPLC-MS (APCI).
In case of formation of γ-butyrolactam from 4-aminobutyric acid, the best analytical yield was obtained with the wild-type enzyme (∼63% yield) (Figure 8A). The tryptophan to glycine substitution at the pocket floor appeared to be detrimental for this activity as none of the W259G mutants allowed significant formation of γ-butyrolactam. Reduced activity with short carboxylic acids due to the W259G mutation was previously observed with small-chain ACLs, indicating that drastic enlargement of the binding pocket reduced acceptance of 4-aminobutyric acid (1) (Hawkins et al., 2014). Single mutants with replacement of V238 and F350 by similar polar, 238T and 350Y respectively, residues partially retained the wild-type activity with 4-aminobutyric acid (39% and 31% lactam yield, respectively).
With longer amino acids 2 and 3, no enzymatic conversion was observed with the wild-type enzyme. However, with 5-aminopentanoic acid (2) significant spontaneous formation of δ-valerolactam occurred in the control reaction without enzyme (6.5% yield) (Figure 8B). For this substrate, yields were hardly increased with enzyme, for all variants. Some conversion was observed with the F350Y single mutant (18% yield), which was designed to introduce an interaction with the ω-amino group of the substrate. The single V238T mutation, which was designed for the same purpose, had only a slight positive effect on δ-valerolactam formation (11% yield), as did the single mutation W259G (13% yield). Combination of the two positive mutations in the W259G/V238T double mutant did not lead to any improvement (12% yield), whereas in the W259G/F350Y double mutant, the positive effects cancelled each other out leading to 8.7% yield.
For the formation of ε-caprolactam from 6-aminohexanoic acid (3) (Figure 8C), the best performing variant was the single mutant W259G, which gave 30% yield. This mutation was intended to improve binding with long substrates by suppressing the steric clash with W259. For the W259G/V238T and W259G/F350Y double mutants, we observed the same trend as for the formation of δ-valerolactam: the W259G/V238T variant converted 6-aminohexanoic acid with a similar yield of 27%, whereas with W259G/F350Y the positive effect introduced by the W259G mutation is almost abolished by the F350Y mutation, resulting in a low yield of 7.5%. The single mutants V238T and F350Y converted substrate 3 with low efficiency (±3.5% yield), albeit still higher than wild-type.
Overall, regarding formation of 6 and 7-member lactams we observed beneficial effects for each of the single mutations, W259G, V238T and F350Y, but in different proportions compared to wild-type MsACL. Unfortunately, for all three substrates we did not observe an additive effect of combining mutations.
These results prompted us to investigate a possible synergistic effect when combining three mutations. As the F350Y substitution had a negative effect when combined with W259G, we also mutated position F350 with non-aromatic polar and apolar groups. Three triple mutants W259G/V238T/F350T, W259G/V238T/F350A and W259G/V238T/F350V were constructed by site-directed mutagenesis and expressed in E. coli. In parallel, we also constructed the corresponding single mutants F350T, F350A and F350V.
Of the single mutations, F350V showed the highest relative yield for the formation of γ-butyrolactam (Figure 9A) and δ-valerolactam (Figure 9B) compared to W259G, while none of them allowed significant ε-caprolactam formation (Figure 9C). With the W259G/V238T/F350X triple mutants, low formation of γ-butyrolactam was observed, as expected because of the enlargement of the pocket with W259G mutation. For the formation δ-valerolactam, the combination of the W259G/V238T mutations with the F350X mutation gave an increased yield of up to 200% in case of the W259G/V238T/F350V variant, compared to the W259G mutant. For the formation of ε-caprolactam, the triple mutations led to a lower conversion, particularly when F350T and F350V were added to W259G/V238T since this suppressed the benefits of the other mutations.
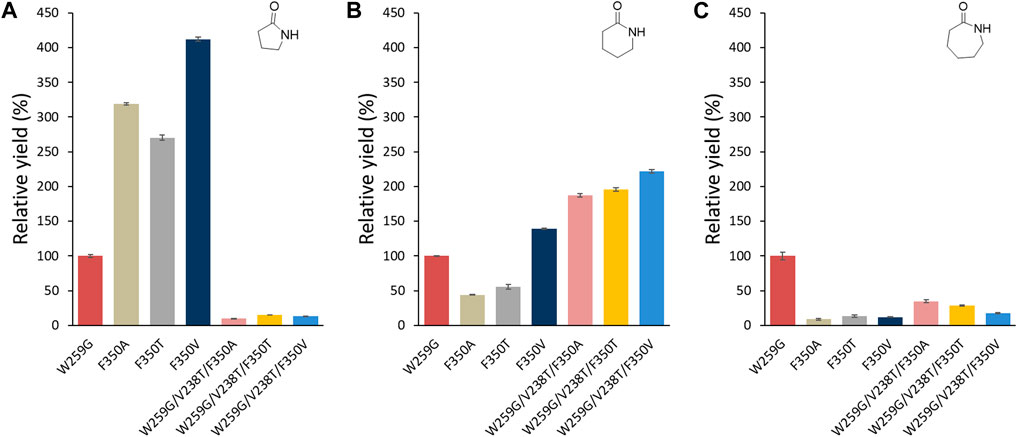
FIGURE 9. Relative analytical yields of lactam formation from ω-amino acids catalyzed by MsACL enzyme variants compared to W259G mutant. Results were normalized to 100% for W259G analytical yield. Reaction conditions: 5 mM amino acid (1-3), 5 mM ATP, 5 mM MnCl2 in 50 mM MOPS buffer (pH 8.5), 2 mg/mL of purified enzyme, 400 rpm at 60 °C for 24 h. Control reactions correspond to samples without enzyme. Analytical yields were deduced from calibration curves by UHPLC-MS (APCI). 100% yield with W259G mutant corresponds to 5%, 13.2% and 30% in (A–C), respectively.
3.8 Kinetic parameters
Kinetic parameters were determined only for the most significant results, MsACL with 4-amino butyric acid (1) and mutants showing improved activities compared to the wild-type MsACL, W259G and W259G/V238T with 6-aminohexanoic acid (3). The results showed improved activities compared to the wild-type MsACL (Table 6). The catalytic efficiencies for ATP were also increased by mutation W259G. Notably, we observed a 100-fold higher kcat/KM for variants W259G and W259G/V238T compared to the wild-type enzyme. With amino acid substrate 3, the kinetic parameters are similar for both mutants W259G and W259G/V238T, with very low affinities as indicated by the high KM values.
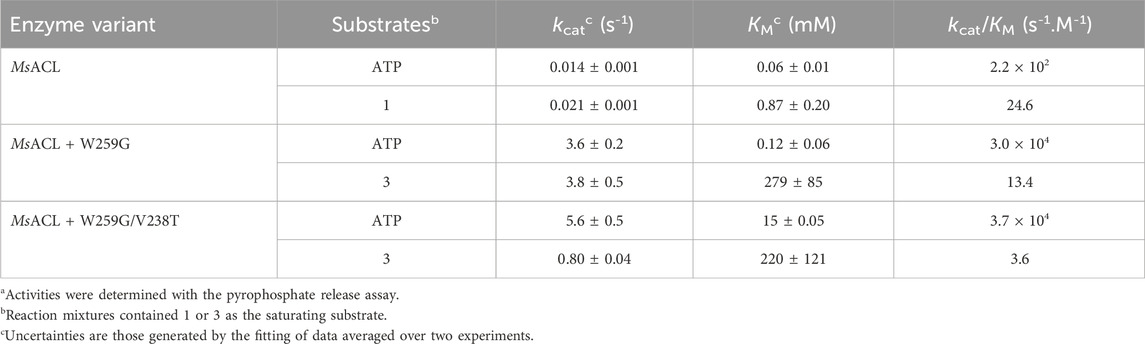
TABLE 6. Kinetic data for MsACL with 4-aminobutyric acid (1) and variants W259G and W259G/V238T with 6-aminohexanoic acid (3)a.
3.9 Mutant substrate scope
To explore their substrate range, we measured the activity of the six MsACL variants, including wild-type, with a range of carboxylic acids with varying chain length up to C12 (4-10) and with several keto and aryl derivatives (11-14) (Figure 7B). The ligase activities were assayed in model reactions containing methylamine and formation of N-methylamide derivatives was monitored by UHPLC-MS (Figure 7C). Due to differences in the mass spectrometry response of the amide products, activities can only be compared between different enzyme variants for the same substrate, whereas comparison between different substrates with the same enzyme are qualitative.
As expected, wild-type MsACL was the most active enzyme for conversion of the shorter carboxylic acid 4. Wild-type MsACL was also able to convert carboxylic acid 5, and the W259G substitution did not significantly alter this activity, both in the single mutant and the double W259G/V238T and W259G/F350Y variants. In contrast, with substrates 6–9 a significantly higher activity was observed for the W259G variant and the double mutants compared to wild-type. Interestingly, for these substrates a beneficial trend of the W259G mutation on amide formation seems to appear with increasing chain length of the carboxylic acid (Table 7; Figure 9). With the longer acyl chain substrates 7, 8 and 9, no amide formation or only traces of product were observed with the wild-type enzyme, and the expected amide products were only observed with variants containing the W259G mutation. No amide formation was detected with the longer C-12 substrate 10, for all MsACL variants. Furthermore, the single V238T and F350Y mutations did not significantly alter the activity of MsACL towards the carboxylic acids, except with substrate 6 where they resulted in a somewhat enhanced activity. Combined with the W259 mutation, the V238T and F350Y mutations have no substantial positive effect on the conversion of most linear carboxylic acids to N-methylamides, as yields were very similar for W259G/V238T, W259G/F350Y and W259G variants (Figure 10).
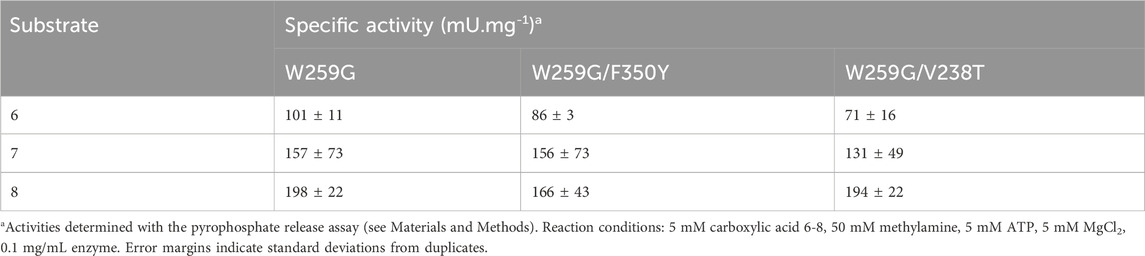
TABLE 7. Activities of MsACL variants with C-6 to C-8 carboxylic acids.
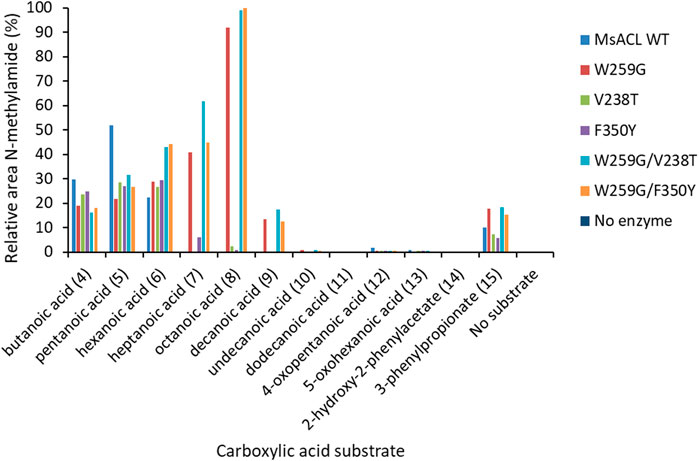
FIGURE 10. Methylamide synthesis observed with wild-type and variants of MsACL. Formation of N-methylamide compounds was determined by UHPLC-MS and relative peaks areas are shown, normalized to 100% for the best reaction. Reaction conditions: 5 mM substrate, 5 mM ATP, 5 mM MgCl2, 50 mM MeNH2 in 50 mM phosphate buffer (pH 8) and 0.2 mg/mL of purified enzyme. Incubation at 60°C for 24 h.
The UHPLC-MS analysis of reaction mixtures with the substituted carboxylic acids 11–14 showed that the patterns of amide formation for wild-type MsACL and the variants were very similar, indicating that none of the mutations was beneficial. We also notice that no amide formation was detected when keto acids 11 and 13 were tested as substrate, whereas with their non-functionalized counterparts 5 and 6 formation of amides was observed with all enzymes. This indicates that none of the variants was capable of accommodating keto substrates, including the W259G variants with substitution of V238 and F350 by similar hindered polar residues, V238T and F350Y, respectively.
3.10 Activity towards C6-C8 carboxylic acids
Specific activities were determined for C6 to C8 carboxylic acids with MsACL variants, using the pyrophosphate release assay (Table 7). The activities were assayed by monitoring pyrophosphate release with the NADH/PPi assays. Overall, the results were consistent with the conversions observed by analysis of the N-methylamides by UHPLC-MS. For a given substrate, the specific activities are very similar for all three mutants, indicating that the second mutation, F350Y or V238T, has almost no effect. From these data, we conclude that the mutants are, as targeted, more active towards C8 than C6 carboxylic acids.
4 Discussion
Access to lactams by enzymatic synthesis is highly desirable to improve the environmental sustainability of their chemical production, but examples of biochemical synthesis are rare. It typically relies on acyclic precursors such as ω-amino acids (Vivienne Barker et al., 1999; Ladkau et al., 2011; Stavila and Loos, 2013; Zhang et al., 2017; Huang et al., 2018; Mourelle-Insua et al., 2018; Gordillo Sierra and Alper, 2020; Sarak et al., 2021; Qin et al., 2022; Jiang et al., 2023). Unfortunately, the lack of highly efficient and stable enzymes capable of cyclizing these substrates is a major obstacle (Stavila and Loos, 2013; Hevilla et al., 2021). To date, only few enzymes have been reported that support the conversion of ω-amino acids into lactams, most notably a native ACL from Streptomyces aizunensis named ORF26 (Zhang et al., 2017) and, more recently, a group of bacterial carboxylic acid reductases belonging to the same ANL enzyme superfamily as MsACL (Qin et al., 2022). While the application of these enzymes in lactam synthesis gave promising results, product yields are generally low and the enzymes do not show high thermostability.
Our study showcases the power of structure-based engineering to adapt a thermostable native ACL for the chemo-enzymatic conversion of ω-amino acids into lactams using ATP and divalent cation as cofactors. The crystal structures of MsACL obtained in this study establish the roles of various protein residues in binding the acetyl-AMP intermediate and the CoA cosubstrate, which are largely similar to those in other ACLs despite low overall sequence identities. In particular, the crystal structures allowed for a detailed analysis of the acyl binding pocket of MsACL and to relate its steric and chemical features to the substrate preferences of the enzyme. Residue W259 at the pocket floor was found to play a crucial role in limiting the range of acyl substrates which can be converted by MsACL. Wild-type MsACL is able to adenylate 4-aminobutyric acid, which then spontaneously converts into γ-butyrolactam, but it cannot adenylate the longer substrates 5-aminopentanoic acid and 6-aminohexanoic acid. This substrate preference is in accordance with previous results, showing that wild-type MsACL is able to use 4-hydroxybutanoic acid as substrate but cannot convert longer hydroxylated fatty acids (Hawkins et al., 2013; Hawkins et al., 2014; Lelièvre et al., 2020). The W259G mutation enlarges the acyl-binding pocket, enabling the binding and adenylation of 5-aminopentanoic acid and 6-aminohexanoic acid, with subsequent conversion into the corresponding lactam, with the beneficial effect for lactam conversion being more pronounced for the longer ω-amino acid. The detrimental effect of the W259G mutation on activity toward 4-aminobutyric acid suggests that, in the wild-type enzyme, W259 plays a crucial role in binding this substrate, perhaps via formation of a cation-π interaction with the ω-amino group. On the contrary, this mutation is mandatory for activity toward 6-aminohexanoic acid, as the other single variants, V238T and F350-substituted mutants (F350Y/A/T/V), gave only very modest conversion. Disappointingly, no beneficial synergetic effect was observed with double (W259G/F350Y and W259G/V238T) and triple mutants (W259G/V238T/F350X). As expected, the W259G mutation also expands the substrate scope of MsACL towards longer fatty acids, up to dodecanoic acid (C10), with a maximal beneficial effect observed for conversion of octanoic acid (C8). On the other hand, conversion of pentanoic acid (C5) is negatively affected by the W259G mutation, supporting the general notion that the substrate preferences of ACLs are closely linked to the size and shape of their acyl-binding pockets (Gulick et al., 2003; Ingram-Smith et al., 2006; Reger et al., 2007; Shah et al., 2009; Meng et al., 2010; Thornburg et al., 2015).
It should be emphasized that although the MsACL mutants show enhanced performance with 5-aminopentanoic acid and 6-aminohexanoic acid, the rates of lactam production remain low. The kinetic parameters for the adenylation reaction indicate low activity and affinity but the latter is not necessarily detrimental for use in applied biocatalysis. A few examples demonstrate that enzymes with poor affinity can very well be useful, as high substrate and product concentrations can be reached without an inhibition effect (Mayol et al., 2016). Nevertheless, substantial further improvement of the catalytic rate of MsACL, through design or screening of additional mutants, will be required for this enzyme to become a viable candidate for biotechnological production of lactams. Importantly, however, our study reveals the potential of enzyme engineering efforts within the CoA ligase family, enabling the enzymatic production of diverse amides and lactams. Exploring ACL mutants through structural studies, docking, and computational enzyme redesign holds promise for a better understanding of the substrate specificity mechanism of these enzymes and for enhancing the efficiency of naturally thermostable ACLs in lactam synthesis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.wwpdb.org/, 8BIQ and 8BIT.
Author contributions
NC: Writing–original draft, Writing–review and editing. CL: Writing–review and editing, Writing–original draft. OT: Writing–review and editing, Writing–original draft. AF-J: Writing–review and editing. CV-V: Writing–review and editing, Writing–original draft. DJ: Writing–original draft, Writing–review and editing. A-MT: Writing–original draft, Writing–review and editing. AZ: Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Part of this project has received funding from the European Union’s Horizon 2020 Programme (Marie Curie Actions-ITN ES-Cat) under GA No. 722610, which supported NC Additional support was obtained from the Commissariat à l’énergie atomique et aux énergies alternatives (CEA), the Centre National de la Recherche Scientifique (CNRS) and the University Evry Paris Saclay.
Acknowledgments
The authors thank J.-L. Petit and A. Debard for their help in mutant production, P. Sirvain for protein purification, O. Maciejak (Université Evry Paris Saclay) for NMR assistance, the Region Ile de France for financial support of the 600 MHz spectrometer. The Diamond Light Source (DLS) and European Synchrotron Radiation Facility (ESRF) are acknowledged for provision of synchrotron radiation facilities and the authors thank the beamline scientists for assistance in using DLS beamline i04 and ESRF beamline MASSIF-1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2024.1360129/full#supplementary-material
References
Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., et al. (2012). Towards automated crystallographic structure refinement with phenix.refine. Phenix.Refine. Acta Crystallogr. Sect. D. Biol. Crystallogr. 68 (4), 352–367. doi:10.1107/S0907444912001308
Bienfait, B., and Ertl, P.JSME (2013). JSME: a free molecule editor in JavaScript. J. Cheminform. 5 (1), 24. doi:10.1186/1758-2946-5-24
Branchini, B. R., Murtiashaw, M. H., Magyar, R. A., and Anderson, S. M. (2000). The role of lysine 529, a conserved residue of the acyl-adenylate-forming enzyme superfamily, in firefly luciferase. Biochemistry 39 (18), 5433–5440. doi:10.1021/bi9928804
Caruano, J., Muccioli, G. G., and Robiette, R. (2016). Biologically active γ-lactams: synthesis and natural sources. Org. Biomol. Chem. 14 (43), 10134–10156. doi:10.1039/C6OB01349J
Chen, Y., Li, T. L., Lin, X., Li, X., Li, X. D., and Guo, Z. (2017). Crystal structure of the thioesterification conformation of Bacillus subtilis O-Succinylbenzoyl-CoA synthetase reveals a distinct substrate-binding mode. J. Biol. Chem. 292 (29), 12296–12310. doi:10.1074/jbc.M117.790410
Chen, Y., Sun, Y., Song, H., and Guo, Z. (2015). Structural basis for the ATP-dependent configuration of adenylation active site in Bacillus subtilis o-succinylbenzoyl-CoA synthetase. J. Biol. Chem. 290 (39), 23971–23983. doi:10.1074/jbc.M115.676304
Clark, L., Leatherby, D., Krilich, E., Ropelewski, A. J., and Perozich, J. (2018). Silico analysis of class I adenylate-forming enzymes reveals family and group-specific conservations. PLoS One 13 (9), 203218. doi:10.1371/journal.pone.0203218
Cowtan, K. (2006). The buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. D. Biol. Crystallogr. 62 (9), 1002–1011. doi:10.1107/S0907444906022116
Cowtan, K. (2010). Recent developments in classical density modification. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66 (4), 470–478. doi:10.1107/S090744490903947X
D’Ambrosio, H. K., and Derbyshire, E. R. (2020). Investigating the role of class I adenylate-forming enzymes in natural product biosynthesis. ACS Chem. Biol. 15 (1), 17–27. doi:10.1021/acschembio.9b00865
Eberhardt, J., Santos-Martins, D., Tillack, A. F., and Forli, S. (2021). AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61 (8), 3891–3898. doi:10.1021/acs.jcim.1c00203
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D. Biol. Crystallogr. 60 (12), 2126–2132. doi:10.1107/S0907444904019158
Evans, P. R., and Murshudov, G. N. (2013). How good are my data and what is the resolution? Acta Crystallogr. Sect. D. Biol. Crystallogr. 69 (7), 1204–1214. doi:10.1107/S0907444913000061
Gordillo Sierra, A. R., and Alper, H. S. (2020). Progress in the metabolic engineering of bio-based lactams and their ω-amino acids precursors. Biotechnol. Adv. 43, 107587. doi:10.1016/j.biotechadv.2020.107587
Gulick, A. M. (2009). Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4 (10), 811–827. doi:10.1021/cb900156h
Gulick, A. M., Starai, V. J., Horswill, A. R., Homick, K. M., and Escalante-Semerena, J. C. (2003). The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5‘-propylphosphate and coenzyme A. Biochemistry 42 (10), 2866–2873. doi:10.1021/bi0271603
Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeersch, T., Zurek, E., and Hutchison, G. R. (2012). Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4 (1), 17. doi:10.1186/1758-2946-4-17
Hawkins, A. B., Adams, M. W. W., and Kelly, R. M. (2014). Conversion of 4-hydroxybutyrate to acetyl coenzyme A and its anapleurosis in the Metallosphaera sedula 3-hydroxypropionate/4-hydroxybutyrate carbon fixation pathway. Appl. Environ. Microbiol. 80 (8), 2536–2545. doi:10.1128/AEM.04146-13
Hawkins, A. S., Han, Y., Bennett, R. K., Adams, M. W. W., and Kelly, R. M. (2013). Role of 4-hydroxybutyrate-CoA synthetase in the CO2 fixation cycle in thermoacidophilic archaea. J. Biol. Chem. 288 (6), 4012–4022. doi:10.1074/jbc.M112.413195
Hevilla, V., Sonseca, A., Echeverría, C., Muñoz-Bonilla, A., and Fernández-García, M. (2021). Enzymatic synthesis of polyesters and their bioapplications: recent advances and perspectives. Macromol. Biosci. 2021 (10), 2100156. doi:10.1002/mabi.202100156
Huang, L., Sayoga, G. V., Hollmann, F., and Kara, S. (2018). Horse liver alcohol dehydrogenase-catalyzed oxidative lactamization of amino alcohols. ACS Catal. 8 (9), 8680–8684. doi:10.1021/acscatal.8b02355
Ingram-Smith, C., Woods, B. I., and Smith, K. S. (2006). Characterization of the acyl substrate binding pocket of acetyl-CoA synthetase. Biochemistry 45 (38), 11482–11490. doi:10.1021/bi061023e
Jiang, N., Du, X., and Zheng, L. (2023). Highly efficient synthesis of chiral lactams by using a ω-transaminase from Bacillus megaterium and its mutant enzymes. Mol. Catal. 547, 113364. doi:10.1016/j.mcat.2023.113364
Kochan, G., Pilka, E. S., von Delft, F., Oppermann, U., and Yue, W. W. (2009). Structural snapshots for the conformation-dependent Catalysis by human medium-chain acyl-coenzyme A synthetase ACSM2A. J. Mol. Biol. 388 (5), 997–1008. doi:10.1016/j.jmb.2009.03.064
Krieger, E., and Vriend, G. (2014). YASARA view—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 30 (20), 2981–2982. doi:10.1093/bioinformatics/btu426
Krissinel, E., and Henrick, K. (2004). Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D. Biol. Crystallogr. 60 (12), 2256–2268. doi:10.1107/S0907444904026460
Krissinel, E., Uski, V., Lebedev, A., Winn, M., and Ballard, C. (2018). Distributed computing for macromolecular crystallography. Acta Crystallogr. Sect. D. Struct. Biol. 74 (2), 143–151. doi:10.1107/S2059798317014565
Ladkau, N., Hermann, I., Bühler, B., and Schmid, A. (2011). Enzyme-catalyzed laurolactam synthesis via intramolecular amide bond formation in aqueous solution. Adv. Synth. Catal. 353 (13), 2501–2510. doi:10.1002/adsc.201100396
Lelièvre, C. M., Balandras, M., Petit, J., Vergne-Vaxelaire, C., and Zaparucha, A. (2020). ATP regeneration system in chemoenzymatic amide bond formation with thermophilic CoA ligase. ChemCatChem 12 (4), 1184–1189. doi:10.1002/cctc.201901870
Li, Z., and Nair, S. K. (2015). Structural basis for specificity and flexibility in a plant 4-coumarate:CoA ligase. Structure 23 (11), 2032–2042. doi:10.1016/j.str.2015.08.012
Mayol, O., David, S., Darii, E., Debard, A., Mariage, A., Pellouin, V., et al. (2016). Asymmetric reductive amination by a wild-type amine dehydrogenase from the thermophilic bacteria petrotoga mobilis. Catal. Sci. Technol. 6 (20), 7421–7428. doi:10.1039/C6CY01625A
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40 (4), 658–674. doi:10.1107/S0021889807021206
Meng, Y., Ingram-Smith, C., Cooper, L. L., and Smith, K. S. (2010). Characterization of an archaeal medium-chain acyl coenzyme A synthetase from Methanosarcina acetivorans. J. Bacteriol. 192 (22), 5982–5990. doi:10.1128/JB.00600-10
Mirdita, M., Schütze, K., Moriwaki, Y., Heo, L., Ovchinnikov, S., and Steinegger, M. (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19 (6), 679–682. doi:10.1038/s41592-022-01488-1
Mourelle Insua, Á., Zampieri, L. A., Lavandera, I., and Gotor-Fernández, V. (2018). Conversion of Γ- and Δ-keto esters into optically active lactams. Transaminases in cascade processes. Adv. Synth. Catal. 360 (4), 686–695. doi:10.1002/adsc.201701304
Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D. Biol. Crystallogr. 67 (4), 355–367. doi:10.1107/S0907444911001314
Perchat, N., Saaidi, P.-L., Darii, E., Pellé, C., Petit, J.-L., Besnard-Bonnet, M., et al. (2018). Elucidation of the trigonelline degradation pathway reveals previously undescribed enzymes and metabolites. Proc. Natl. Acad. Sci. U. S. A. 115, E4358–E4367. doi:10.1073/pnas.1722368115
Qin, Z., Zhang, X., Sang, X., Zhang, W., Qu, G., and Sun, Z. (2022). Carboxylic acid reductases enable intramolecular lactamization reactions. Green Synth. Catal. 3 (3), 294–297. doi:10.1016/j.gresc.2022.05.009
Reger, A. S., Carney, J. M., and Gulick, A. M. (2007). Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 46 (22), 6536–6546. doi:10.1021/bi6026506
Sarak, S., Sung, S., Jeon, H., Patil, M. D., Khobragade, T. P., Pagar, A. D., et al. (2021). An integrated cofactor/Co-product recycling cascade for the biosynthesis of nylon monomers from cycloalkylamines. Angew. Chem. Int. Ed. 60 (7), 3481–3486. doi:10.1002/anie.202012658
Schmelz, S., and Naismith, J. H. (2009). Adenylate-forming enzymes. Curr. Opin. Struct. Biol. 19 (6), 666–671. doi:10.1016/j.sbi.2009.09.004
Schrödinger, L. L. C. 2010, The PyMOL molecular graphics system. New York, NY, USA: Schrodinger Inc.
Shah, M. B., Ingram-Smith, C., Cooper, L. L., Qu, J., Meng, Y., Smith, K. S., et al. (2009). The 2.1 Å crystal structure of an acyl-CoA synthetase from Methanosarcina acetivorans reveals an alternate acyl-binding pocket for small branched acyl substrates. Proteins Struct. Funct. Bioinforma. 77 (3), 685–698. doi:10.1002/prot.22482
Sibikin, I., and Karger-Kocsis, J. (2018). Toward industrial use of anionically activated lactam polymers: past, present and future. Adv. Ind. Eng. Polym. Res. 1 (1), 48–60. doi:10.1016/j.aiepr.2018.06.003
Sofeo, N., Hart, J. H., Butler, B., Oliver, D. J., Yandeau-Nelson, M. D., and Nikolau, B. J. (2019). Altering the substrate specificity of acetyl-CoA synthetase by rational mutagenesis of the carboxylate binding pocket. ACS Synth. Biol. 8 (6), 1325–1336. doi:10.1021/acssynbio.9b00008
Stavila, E., and Loos, K. (2013). Synthesis of lactams using enzyme-catalyzed aminolysis. Tetrahedron Lett. 54 (5), 370–372. doi:10.1016/j.tetlet.2012.10.133
Stockmann, P. N., Van Opdenbosch, D., Poethig, A., Pastoetter, D. L., Hoehenberger, M., Lessig, S., et al. (2020). Biobased chiral semi-crystalline or amorphous high-performance polyamides and their scalable stereoselective synthesis. Nat. Commun. 11 (1), 509. doi:10.1038/s41467-020-14361-6
Thornburg, C. K., Wortas-Strom, S., Nosrati, M., Geiger, J. H., and Walker, K. D. (2015). Kinetically and crystallographically guided mutations of a benzoate CoA ligase (BadA) elucidate mechanism and expand substrate permissivity. Biochemistry 54 (40), 6230–6242. doi:10.1021/acs.biochem.5b00899
Trott, O., and Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31 (2), 455–461. doi:10.1002/jcc.21334
Vergne-Vaxelaire, C., Bordier, F., Fossey, A., Besnard-Bonnet, M., Debard, A., Mariage, A., et al. (2013). Nitrilase activity screening on structurally diverse substrates: providing biocatalytic tools for organic synthesis. Adv. Synth. Catal. 355, 1763–1779. doi:10.1002/adsc.201201098
Vivienne Barker, C., Page, M. I., Korn, S. R., and Monteith, M. (1999). Esterase catalysed enantioselective ring closure. Chem. Commun. 8, 721–722. doi:10.1039/a901204d
Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., et al. (2011). Data processing and analysis with the AutoPROC toolbox. Acta Crystallogr. D. Biol. Crystallogr. 67 (4), 293–302. doi:10.1107/S0907444911007773
Winnacker, M., and Rieger, B. (2016). Biobased polyamides: recent advances in basic and applied research. Macromol. Rapid Commun. 37 (17), 1391–1413. doi:10.1002/marc.201600181
Winter, G., Waterman, D. G., Parkhurst, J. M., Brewster, A. S., Gildea, R. J., Gerstel, M., et al. (2018). DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D. Struct. Biol. 74 (2), 85–97. doi:10.1107/S2059798317017235
Wu, R., Reger, A. S., Lu, X., Gulick, A. M., and Dunaway-Mariano, D. (2009). The mechanism of domain alternation in the acyl-adenylate forming ligase superfamily member 4-chlorobenzoate: coenzyme A ligase. Biochemistry 48 (19), 4115–4125. doi:10.1021/bi9002327
Keywords: CoA ligase, enzyme structure, protein engineering, lactams, chemoenzymatic synthesis, biocatalysis
Citation: Capra N, Lelièvre C, Touré O, Fossey-Jouenne A, Vergne-Vaxelaire C, Janssen DB, Thunnissen A-MWH and Zaparucha A (2024) Adapting an acyl CoA ligase from Metallosphaera sedula for lactam formation by structure-guided protein engineering. Front. Catal. 4:1360129. doi: 10.3389/fctls.2024.1360129
Received: 22 December 2023; Accepted: 05 February 2024;
Published: 04 March 2024.
Edited by:
Tarek Rouissi, Université du Québec, CanadaReviewed by:
Xiang Sheng, Chinese Academy of Sciences (CAS), ChinaJohn-Paul Bacik, Hauptman-Woodward Medical Research Institute, United States
Bekir Engin Eser, Aarhus University, Denmark
Copyright © 2024 Capra, Lelièvre, Touré, Fossey-Jouenne, Vergne-Vaxelaire, Janssen, Thunnissen and Zaparucha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andy-Mark W. H. Thunnissen, YS5tLncuaC50aHVubmlzc2VuQHJ1Zy5ubA==; Anne Zaparucha, YW5uZS56YXBhcnVjaGFAZ2Vub3Njb3BlLmNucy5mcg==
†These authors have contributed equally to this work