
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Catal. , 08 March 2023
Sec. Biocatalysis
Volume 3 - 2023 | https://doi.org/10.3389/fctls.2023.1114536
 Sarah E. Cleary1*
Sarah E. Cleary1* Sofia Kazantzi2Joshua A. Trenchard2Martin Monedero2Jack W. Allman1
Sofia Kazantzi2Joshua A. Trenchard2Martin Monedero2Jack W. Allman1 Tara C. Lurshay1†Xu Zhao1
Tara C. Lurshay1†Xu Zhao1 Michael B. C. Kenny2
Michael B. C. Kenny2 Holly A. Reeve1*
Holly A. Reeve1*Introduction: This work was carried out to understand if a heterogeneous biocatalytic hydrogenation system could be implemented in a slurry mode continuous flow reactor, as a “slot-in” alternative to a metal/carbon catalyst with minimal process or chemical engineering development.
Method: Biocatalytic hydrogenation was compared to metal (Pd/C) catalysed hydrogenation for ketone to chiral alcohol conversion in both a continuous flow reactor (Coflore ACR, AM Technology) and analogous scaled down batch reactions.
Results and discussion: Initial results demonstrated that batch reactions can achieve high conversions in 30 min, with relatively low biocatalyst loadings, meeting critical criteria for operation as continuous slurry mode process. Further results demonstrated full conversion of quinuclidinone to (3R)-quinuclidinol in continuous flow under mild conditions (35°C, 2 bar H2). On intensification of the process to higher substrate loading (50 mM), conversion was similar to with Pd/C, however the biocatalytic system achieved far higher turnover frequency and total turnover number (65 min−1 and 20,000, respectively) than the metal system (0.16 min−1, 37). Comparison to an analogous batch reaction highlights that the biocatalytic system has promise for further optimisation and intensification in the scalable Coflore ACR. Overall, biocatalytic hydrogenation is shown to offer a decarbonised biocatalytic route and a “slot-in” replacement to metal catalysts for hydrogenation reactions in continuous flow reactors.
Catalytic hydrogenation reactions are prominent in the chemicals sector, with both ‘heterogeneous’ and ‘enantioselective’ hydrogenations accounting for a significant portion of patent filings between 2000–2020 (Stoffels et al., 2020). Heterogeneous metal-catalysed hydrogenation processes are well-established in chemical manufacturing, likely because they provide excellent atom economy and the ability to recover and reuse the catalyst.
Scaling up hydrogenation chemistry in continuous flow benefits from the higher interfacial area relative to batch processes (Mallia and Baxendale, 2016), allowing improved gas-liquid mass transfer rates. Relative to batch hydrogenation, an improvement can also be observed in catalyst continuity, in terms of predictability and minimisation of side reactions, owing to the continuous removal of reactants and products. Furthermore, improved safety is a key driver for adoption of continuous processes due to small operating volume for H2 handling, and similarly for better control over temperature and pressure requirements (Irfan et al., 2011).
However, there still remains room for improvement with implementation of hydrogenation processes, for example, decreasing the reliance on toxic and expensive precious metals, energy-demanding conditions, and flammable solvents, which currently combine to give a range of safety, environmental and economic limitations.
Heterogeneous biocatalytic hydrogenation is an alternative strategy that uses H2 gas as an atom economical reductant for redox biocatalyst pathways (Reeve et al., 2015), thus offering an alternative to precious metals for hydrogenation chemistry (see safety and sustainability comparisons in Supplementary Table S1, Supplementary Information). The biocatalyst system used in this report relies on co-immobilisation of a robust hydrogenase and NAD+ reductase on an electronically conducting carbon support. This support enables the transfer of electrons from H2 oxidation by the hydrogenase to then be used for NAD+ reduction to NADH by the NAD+ reductase (Figure 1A; see Supplementary Figure S1 in the Supplementary Information for details about the mechanism). This H2-driven NADH recycling strategy has previously been coupled to a variety of NADH-dependent oxidoreductase enzymes, such as alcohol dehydrogenase (carbonyl to alcohol), ene reductase (activated alkene to alkane), and amino acid dehydrogenase (ketone to amine) (Thompson et al., 2020b).
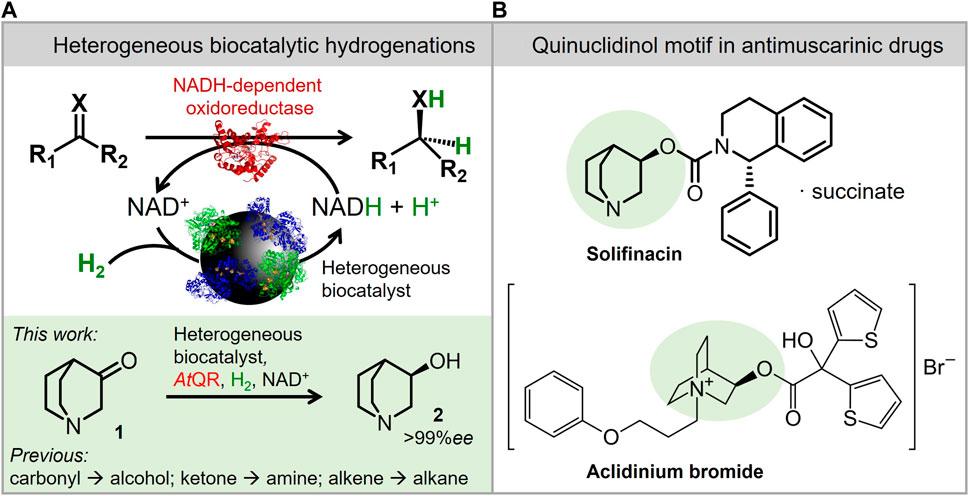
FIGURE 1. Heterogeneous biocatalytic hydrogenation as a strategy toward double-bond reductions. (A) Scheme to show the biocatalytic hydrogenation (see Supplementary Figure S1 in the ESI for mechanism details). This strategy was used here to reduce 3-quinuclidinone (1) to (3R)-quinuclidinol (2). (B) (3R)-Quinuclidinol (2, highlighted in green) is an important motif found in approved antimuscarinic drugs Solifinacin and aclidinium bromide.
Traditional redox biocatalysis and NADH recycling strategies rely on carbon-based reductants (glucose, formate, isopropanol) (Berenguer-Murcia and Fernandez-Lafuente, 2010), and a cofactor recycling enzyme that is typically handled in solution. This leads to generation of carbon-based waste and complicates widely applicable scale up strategies. The biocatalytic hydrogenation approach here decarbonises NADH-dependent biocatalysis minimising waste generation. Furthermore, the enzymes are immobilised on carbon giving them similar handling considerations to typical metal on carbon catalysts (i.e., Pd/C). To date, the biocatalyst system has been shown to operate in common hydrogenation equipment, with a focus on batch reactors and some early reports in continuous flow (Reeve et al., 2015; Zor et al., 2017; Thompson et al., 2020a; Thompson et al., 2020b; Poznansky et al., 2021).
In this report, we designed experiments to understand if this biocatalytic hydrogenation approach can ‘slot in’ to a proven, scalable continuous hydrogenation reactor and compare results to analogous metal-catalysed processes. We address parameters such as set-up protocol, temperature, pressure, catalyst loading, residence time, and catalyst selectivity for a case study reaction in continuous flow and provide further data in scaled-down batch reactions for comparison.
The selected case study process was for the reduction of 3-quinuclidinone (1) to (3R)-quinuclidinol (2), a motif found in approved antimuscarinic drugs (Naito et al., 2005; Montuschi and Ciabattoni, 2015) such as Solifinacin [approved for overactive bladder (Maniscalco et al., 2006)] and aclidinium bromide [approved for COPD (Woods et al., 2013)], both illustrated in Figure 1B. 3-Quinuclidinol (2) has previously been prepared via asymmetric Ru-catalysed hydrogenation in batch (Arai et al., 2010), including at multi-kilogram scale (Tsutsumi et al., 2009) with pressures of 15–50 bar H2 used and between 88% and 97% ee achieved. Elevated pressures can be avoided when preparing 2, and this has been reported with excellent (>99.9%) enantiomeric excess using whole-cell biocatalysis (Zhang et al., 2013), including a multi-kilogram synthesis (Chen et al., 2019) both of which used ambient pressure at 30°C. The whole-cell systems used glucose-driven NADH recycling, which created stoichiometric gluconolactone waste and necessitated maintaining pH by adding NaOH. We have previously prepared 2 at laboratory scale using our heterogeneous biocatalyst to supply NADH to an isolated 3-quinuclidinone reductase “AtQR” (Thompson et al., 2020a; Rowbotham et al., 2020), yielding product at >99% ee. The pH-neutral H2-driven cofactor recycling meant that there was no need for continuous pH monitoring.
AM Technology has developed three dynamically-mixed continuous flow reactors covering R&D-, pilot- and production-scales. The Coflore ACR is suitable for laboratory-based process research and development with a variable total system volume from 30 to 90 mL. The Coflore ACR reactor has previously been used to handle solids (Chuzeville et al., 2022) as well as for O2-dependent biocatalytic oxidation of glucose (Toftgaard Pedersen et al., 2017) wherein the ACR offered an improved gas mass transfer rate compared to batch. The solids handling nature of the pilot scale Coflore ATR has also been characterised where the influence of palladium-coated carbon particles on agitator motion was investigated where the solid catalyst had no negative impact on the agitation method (Rice et al., 2022).
These properties made the Coflore suite a promising scale-up strategy, and here we report the translation of our heterogeneous biocatalytic hydrogenation for generation of (3R)-quinuclidinol, from small scale batch reactions to the Coflore ACR flow reactor.
Buffer salts (Sigma Aldrich), NAD+ (Prozomix), carbon black particles (Black Pearls 2000 “BP 2000”, Cabot Corporation), activated charcoal (Darco®, −100 mesh, Sigma Aldrich), Pd/C powder (5R452, 5% palladium on Activated Carbon Paste, Type 452, Johnson Matthey), Pd/C pellets (1% palladium, Type 783 pellet, 1–3 mm, Johnson Matthey), quinuclidinone-HCl (1, Sigma Aldrich), quinuclidinol (2 as both R and racemic mixture, Sigma Aldrich) were all purchased and used as received. All aqueous solutions were prepared with deionised water. 1H NMR spectra were obtained at room temperature (298 K) on a Bruker Advance III HD nanobay (400 MHz) using 90 vol% reaction mixture and 10 vol% D2O. The Bruker proc_1d processing algorithm was used, and a manual phase correction was followed by a multipoint baseline correction. Chiral-phase GC-FID was used as described in S2.3 (Supplementary Information). UV-visible spectra were recorded by a Jenway 7205 spectrophotometer using a quartz cuvette (path length 1 cm). Thin-layer chromatography (TLC) analysis was carried out using silica gel on aluminium plates. Visualisation of the TLC plates was achieved using I2.
The hydrogenase (E. coli hydrogenase 1, “Hyd1”, molecular weight 100 kDa) was produced and isolated following published protocols (Ramirez et al., 2022). Subsequently, the enzyme was buffer-exchanged into Tris-HCl buffer (20 mM Tris-HCl pH 7.2, 350 mM NaCl, 0.02% Triton X, 1 mM DTT), concentrated to 14.7 mg/mL and stored at −80°C. The NAD+ reducing soluble hydrogenase from Ralstonia eutropha (“SH”) was isolated and purified following published protocols (Lauterbach and Lenz, 2013), then stored at −80°C with a concentration of 21.6 mg/mL in Tris-HCl (100 mM, pH 8.0). This enzyme retains its native H2 oxidation activity but significantly benefits from improved activity and stability via the co-immobilised Hyd1 (Poznansky et al., 2021). The NADH-dependent 3-quinuclidinone reductase from Agrobacterium tumefaciens (AtQR) was heterologously produced in E. coli as previously described (Rowbotham et al., 2020) but was used in this report as the soluble lysate without further purification. The resulting solution of AtQR soluble lysate had a total protein concentration of 143 mg/mL.
Carbon black (BP 2000) was suspended in Tris-HCl buffer (50 mM, pH 8.0, 22°C) to make a 20 mg/mL slurry in a 15 mL centrifuge tube. Larger carbon agglomerates were dispersed using a pipette, then sonicated 3 × 15 min which gave an ink-like slurry. This slurry was allowed to cool in an ice block. Solutions of hydrogenase and NAD+ reductase were mixed together, then the required volume of BP2000 slurry was added to achieve the indicated mass ratio of 1:7:7 hydrogenase/NAD+ reductase/carbon black. The enzyme and carbon slurry was incubated at 4°C for 1 h. The percent of protein immobilisation onto carbon black was determined by taking 100 µL aliquots of the catalyst slurry, centrifuging for 2 min (10,000 × g) and analysing the supernatant by UV-visible spectrometry (see details in S2.1 in the Supplementary Information). All procedures carried out on the bench in aerobic conditions.
Upon full enzyme adsorption, the catalyst was portioned into aliquot samples as needed, and stored at −20°C.
Solutions of hydrogenase and NAD+ reductase were mixed together, then the required mass of activated charcoal powder was added to achieve a mass ratio of 1:7:14, hydrogenase/NAD+ reductase/charcoal. This enzyme and carbon slurry was incubated at 4°C for 1 h. The percent of enzyme immobilisation onto activated charcoal was determined by taking 100 µL aliquots of the catalyst slurry, centrifuging for 2 min (10,000 × g) and analysing the supernatant by UV-visible spectrometry (see details in S2.1 in the Supplementary Information). All procedures carried out on the bench in aerobic conditions.
Upon full enzyme adsorption, the catalyst was portioned into aliquot samples as needed, and stored at −20°C.
Separate slurries (20 mg/mL) of carbon black and activated charcoal were sonicated and dispersed in buffer following the description above. In separate centrifuge tubes, 100 µg NAD+ reductase was mixed with 100 µg of the designated carbon material slurry (either carbon black or activated charcoal) in a total volume of 20 µL. The NAD+ reductase was immobilised by mixing at 600 r.p.m. in a Thermoblock shaker at 4°C for 1 h, then slurries were centrifuged for 3 min (10,000 × g). The supernatant was removed and analysed by UV-vis to measure the remaining “unabsorbed protein concentration” using the absorbance at 280 nm (see results corresponding with “0” wash cycles in Results; see details in S2.1 in the Supplementary Information for information on calculating the protein adsorption).
From here, the catalysts were subjected to “washing cycles”, by suspending in 10 µL of solution (either 25 mM Tris-HCl, or 10 mM quinuclidinone in 25 mM Tris-HCl) and mixing at 600 r.p.m. on the Thermoblock shaker at 30°C for 1 h. The slurries were then centrifuged for 3 min (10,000 × g). The supernatant was removed and analysed by UV-vis to measure the remaining ‘unabsorbed protein concentration’ using the absorbance at 280 nm. Washing cycles were repeated thrice and analysed for unabsorbed protein in the supernatant, shown in Figure 2.
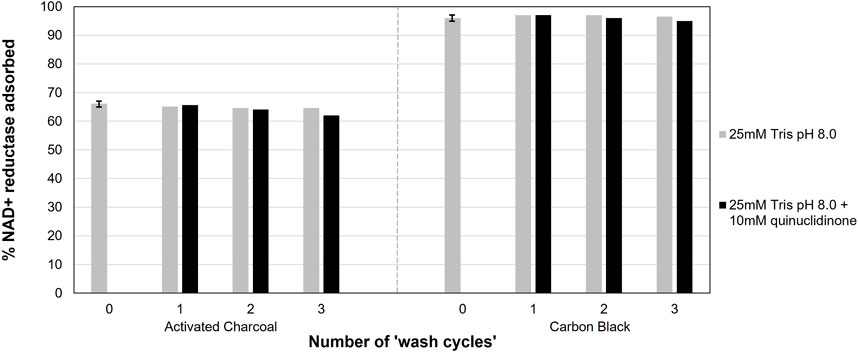
FIGURE 2. Percentage of NAD+ reductase (100 µg) retained on 100 µg of carbon in the presence of Tris-HCl (grey), or Tris-HCl with 10 mM quinuclidinone (black) after wash cycles. The bars that correspond with “0 wash cycles” were performed in duplicate, with the range represented as error bars (±1% NAD+ reductase adsorbed in both cases). See 2.3 for details about the immobilising and washing procedure.
A round bottom flask equipped with a stir bar was sealed with a rubber septum, two needles were inserted, and the flask was placed under an active flow of N2. The flask was placed in a room temperature water bath, then the flask was charged with a solution (2–5 mL) that contained five or 50 mM quinuclidinone-HCl (1) and 1 mM NAD+ in Tris-HCl (25 mM, pH 8.0, 25°C). This was sparged with N2 for 30 min by inserting the gas inlet needle into the solution. The gas line was switched from N2 to H2, and the reaction solution was sparged under H2 for 10 min. The H2 inlet needle was then moved out of the solution and into the flask headspace in order to maintain an H2 atmosphere (1 bar). The water bath was warmed to either 25 or 35°C (as noted in Table 2). A 0.2 mL portion of this solution was used to aid the transfer of the heterogeneous biocatalyst via syringe and needle into the reaction flask to the catalyst loading required. A further 0.2 mL portion of the resulting slurry was then used to aid the transfer of AtQR via syringe and needle into the reaction flask to give desired concentration. This slurry was then stirred under a steady flow of H2 (1 bar) at the designated temperature. At the indicated time, a 1 mL portion of the slurry was then centrifuged for 2 min (10,000 × g) and 450 µL of the resulting supernatant was combined with 50 µL of D2O in an NMR spectroscopy tube and taken for analysis by 1H NMR spectroscopy (see S2.2 in the Supplementary Information for details). Activity, total turnover number (TTN) and turnover frequency (TOF) are calculated using the total amount of adsorbed protein, assuming all immobilised enzyme is active.
An analogous set-up protocol was use for metal-catalysed reactions. A round bottom flask equipped with a stir bar was sealed with a rubber septum, two needles were inserted, and the flask was placed under an active flow of N2 via the needles. The flask was placed in a room temperature water bath, then the flask was charged with a 6 mL solution that contained 5 mM quinuclidinone-HCl (1) in deionised water, then this was stirred and sparged with N2 for 30 min by inserting the gas inlet needle into the solution. The gas line was switched from N2 to H2, and the stirred reaction solution was sparged under H2 for 10 min. The H2 inlet needle was then moved out of the solution and into the flask headspace in order to maintain an H2 atmosphere (1 bar). The water bath was then warmed to 35°C (as noted in Table 2). The septum was quickly removed and 5 wt% Pd/C catalyst (1.2 mg) was added to the stirred reaction solution, then the septum and gas inlet and outlet needs were replaced. This slurry was then stirred under a steady flow of H2 (1 bar) for 30 min. A 1 mL portion of the slurry was then centrifuged for 2 min (10,000 × g) and 450 µL of the resulting supernatant was combined with 50 µL of D2O in an NMR spectroscopy tube and taken for analysis by 1H NMR spectroscopy (see S2.2 in the Supplementary Information for details).
All continuous hydrogenation reactions were run in the reactor set up depicted in Supplementary Figure S2 (Supplementary Information). Details of the set up are described below.
Prior to start up, the reactor system was flooded with deionised water. An inertion procedure was conducted whereby 3 bar N2 was applied to the system and vented repeatedly (three iterations). Once inert, the H2 lines were purged by metering a flowrate of 50 SmL/min (standard temperature pressure, STP mL/min) through the mass flow controller to vent.
The liquid feed containing reagents was prepared as described in the results section and stirred in a 1 L jacketed glass vessel (ID 150 mm) using a 50 mm diameter four-bladed 45° PTFE coated pitch blade impeller driven by a IKA Eurostar 40 overhead stirrer. A continuous sweep of N2 was metered into the feed vessel at 50 mL/min. The designated portion of heterogeneous catalyst slurry was added to this vessel, followed by AtQR for the biocatalytic system (as a 143 mg/mL solution). The mixture was stirred at 200 r.p.m. for 5 min to ensure homogeneity.
The slurry feed was pumped using a Masterflex peristaltic pump with Norprene LS14 tubing, at the desired flowrate (Supplementary Table S2, Supplementary Information) into the first reactor cell of the Coflore Agitated Cell Reactors (ACR or ACX) through a length of 1/8ʺ PTFE tubing to achieve the desire process conditions. Both the feed vessel and reactor cell block temperature were set to 35°C using a Julabo Presto A40 heat transfer unit, and thermocouples were located within the feed vessel, and at cells two, and ten to monitor the process temperatures.
Hydrogen gas was fed to the system from a 10 L H2 cylinder into an Alicat scientific mass flow controller (MC-1SLPM). The 15 SmL/min flowrate setpoint of hydrogen was fed into the first cell of the Coflore ACR through 1/8ʺ PTFE tubing.
Both H2 and the suspension were dosed into the first cell 1. Both an ACR block and ACX block were trialed with high shear agitators (10% volume) and 50% volume agitators respectively. An ACR block is designed such that a single 4 mm diameter channel connects each of the ten reactor cells. A Coflore ACX block is designed such that two 4 mm diameter channels connect each of the ten reactor cells whilst maintaining a constant reactor cell configuration. Due to the additional channel connections present in the Coflore ACX, the block was expected to minimise gas phase holdup whilst maintaining a constant liquid volume through the change of agitator volumes.
The outlet of the Coflore reactor was connected to a sampling arrangement where a 3 mL polypropene syringe was isolated from the process through a 1/8ʺ manual ball valve. Samples (0.5–1 mL portions) of slurry were collected for analysis, then centrifuged for 2 min (10,000 × g). A 450 µL sample of the resulting supernatant was combined with 50 µL of D2O in an NMR spectroscopy tube and taken for analysis by 1H NMR spectroscopy (see S2.2 in the Supplementary Information for details). A 50 µL sample from Table 3, entry three was also adjusted to pH 12 using 3 M aqueous NaOH, extracted into chloroform, then analysed using chiral-phase GC-FID (see S2.3 in the Supplementary Information for details). Activity, total turnover number (TTN) and turnover frequency (TOF) are calculated using the total amount of adsorbed protein, assuming all immobilised enzyme is active.
All material produced was directed from the sampling arrangement to a glass and stainless-steel collection vessel. Gas was disengaged from the suspension within the vessel, the excess gas flow vented through an Equilibar ZF1 back pressure regulator to an extraction system whilst the suspension was intermittently drained.
Prior to start up, the system was flooded with deionised water. An inertion procedure was conducted whereby 3 bar N2 was applied to the system and vented repeatedly (three iterations). Once inert, the H2 lines were purged by metering a flowrate of 50 SmL/min through the mass flow controller to vent.
The liquid feed was prepared as described and stirred in a 1 L jacketed glass vessel (ID 150 mm) using a 50 mm diameter four-bladed 45° PTFE coated pitch blade impeller driven by a IKA Eurostar 40 overhead stirrer. A continuous sweep of N2 was metered into the feed vessel at 50 mL/min. The designated portion of reaction solution was added, then the mixture was stirred at 200 r.p.m. for 5 min to ensure homogeneity.
The liquid feed was pumped using an Elados EMP II E60 solenoid driven diaphragm pump, at the desired flowrate (Table 5) into the first reactor cell of the Coflore ACR through a length of 1/8ʺ PTFE tubing to achieve the desire process conditions. Both the feed vessel and reactor cell block temperature were set to 35°C using a Julabo Presto A40 heat transfer unit, and thermocouples were located within the feed vessel, and at cells two, and ten to monitor the process temperatures.
Hydrogen gas was fed to the system from a 10 L H2 cylinder into an Alicat scientific mass flow controller (MC-1SLPM). The 15 SmL/min flowrate setpoint of hydrogen was fed into the first cell of the Coflore ACR through 1/8ʺ PTFE tubing.
Both H2 and the solution were dosed into the first cell 1. The ACX block was fitted with agitating catalyst baskets. The outlet of the Coflore reactor was connected to a sampling arrangement where a 3 mL polypropene syringe was isolated from the process through a 1/8ʺ manual ball valve. Samples (0.5–1 mL portions) of solution were collected for analysis. A 450 µL sample was combined with 50 µL of D2O in an NMR spectroscopy tube and taken for analysis by 1H NMR spectroscopy (see S2.2 in the Supplementary Information for details). A 50 µL sample from Table 5, entry 2 was also adjusted to pH 12 using 3 M aqueous NaOH, extracted into chloroform, then analysed using chiral-phase GC-FID (see S2.3 in the Supplementary Information for details).
All material produced was directed from the sampling arrangement to a glass and stainless-steel collection vessel. Gas was disengaged from the suspension within the vessel, the excess gas flow vented through an Equilibar ZF1 back pressure regulator to an extraction system whilst the suspension was intermittently drained.
When planning to translate our hydrogenation biocatalyst into a slurry reactor, we first sought suitable conditions that were likely to lead to high conversion and enable lowered catalyst loading. The ACR/ACX flow reactor has previously handled nanoparticles which had an average particle size of 70 nm (Chuzeville et al., 2022).
A variety of carbon materials have been proven as suitable supports for the biocatalytic hydrogenation system (Reeve et al., 2015; Thompson et al., 2020b), which enables us to take advantage of the different properties (e.g., dispersion, suspension, enzyme adsorption). For example, enzymes immobilised on carbon black nanopowder offer excellent dispersion when mixed in batch. In a previous report, enzymes adsorbed onto −100 mesh activated charcoal particles are suitable in packed bed flow reactors without any carbon leaching (Poznansky et al., 2021), whereas nanomaterials can leak through the packed bed frits. An advantage of dynamically-mixed slurry processes is the possibility of using high surface area nanoparticles as a (this report) compared to packed bed reactors that typically require confined mm sized materials. Advantageously the reactor used in this work allows operation with catalyst in slurry or confined mode.
Initially, we examined the ability of the two carbon types for immobilisation of hydrogenase and NAD+ reductase via simple absorption (Table 1, with details described in Section 2.1 in the Supplementary Information). In the case of activated charcoal, when the mass ratio of enzyme to carbon was 2:1, only 52% of the enzyme adsorbed, whereas when the mass ratio of enzyme to carbon was decreased to 0.5:1, 65% was adsorbed (entries 1–2). Carbon black had a superior ability to adsorb the enzymes, even at high (3:1) mass ratio of enzyme to carbon black, resulting in 72% enzyme adsorption, and a moderate 1:1 mass ratio of enzyme to carbon resulted in 96%–98% adsorption (tested at two different scales, entries 3–5). This high enzyme loading on the nano sized carbon material suggests that translation into continuous reactor with high activity should be prioritised as a slurry process.
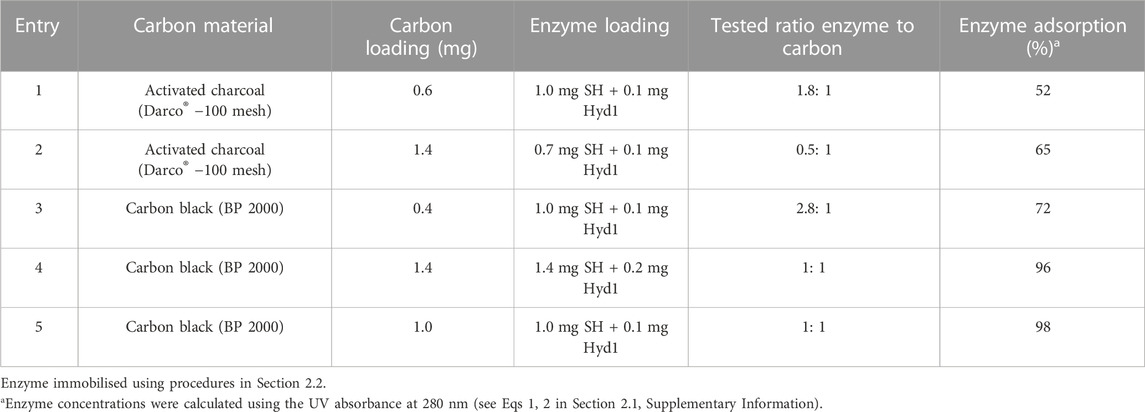
TABLE 1. Extent of enzyme immobilisation on carbon materials.
We next sought to determine if the NAD+ reductase leaches from the carbon after immobilisation (see procedure details in Section 2.3). The NAD+ reductase was first immobilised onto the two carbon materials. The slurry was then centrifuged and the supernatant was analysed for protein in solution, which showed NAD+ reductase immobilisation that in line with those in Table 1 (shown as 0 wash cycles, Figure 2). Subsequently, the immobilised enzyme on carbon aliquots were subjected to washing cycles with Tris-HCl buffer with or without presence of quinuclidinone solution (to mimic reaction conditions, procedure described in detail in Section 2.3). The supernatants were analysed for NAD+ reductase leaching between washes. This information was used to calculate the percent of enzyme applied that was retained on the carbon materials.
The results in Figure 2 show minimal leaching, significantly, with negligible (<0.5%) NAD+ reductase lost in the absence quinuclidinone. In the presence of 10 mM quinuclidinone and 25 mM Tris-HCl, only 5% (activated charcoal) or 2% (carbon black) of the NAD+ reductase had leached. The quinuclidinone may interfere with some of the interactions that allow the enzyme to stay adsorbed to the carbon materials, however the quantity lost over the three wash cycles supported moving forwards with a carbon black slurry process.
The heterogeneous biocatalysts were next used for the hydrogenation of 3-quinuclidinone (1) in scale-down batch (Figure 3). Here, we sought to achieve key reaction parameters required for translation into continuous flow slurry process, including short reaction times and low (0.2–0.6 mg/mL) catalyst loading.
FIGURE 3. Heterogeneous biocatalytic hydrogenation of 1 in scale-down batch reactions. Photography credit: Charlie Flounders Photography©.
The activated charcoal and carbon black biocatalyst systems were compared in parallel for the ability to drive the biocatalytic hydrogenation of 1 to 2 following the procedure detailed in Section 2.4. Solutions containing 3-quinuclidinone-HCl (5 mM) and 1 mM NAD+ (1 mM) in Tris-HCl (25 mM, pH 8.0, 25 °C) were mixed with heterogeneous biocatalyst (0.2 mg/mL) and AtQR (0.05 mg/mL) under a steady flow of H2 (1 bar) at 25°C for 30 min.
The reactions were analysed for conversion to 2 using 1H NMR spectroscopy (see results in Table 2). Entries 1 and 2 show that the catalyst formulated using carbon black had marginally higher (84%) conversion per mg biocatalyst compared with that which was formulated using activated charcoal (79%), although due to the catalyst formulation, a higher enzyme turnover frequency (TOF) was observed using activated charcoal. We also observed improved mixing using the carbon black particles, which did not settle as rapidly as the activated charcoal. The particle size is likely a contributing factor to this, where the carbon black “BP 2000” particle size is on the nanometer scale (Zhao et al., 2021) and the −100 mesh activated charcoal is on the micron scale. Conversion to 2 in 30 min was further improved to 90% at elevated reaction temperature (35°C, entry 3, Table 2). Although performed at modest reactant loadings, previous reports on this biocatalyst system in batch have used 24 h reaction times and this is an initial step to achieving high conversion in a timeframe suitable for translation into flow, whilst maintaining relatively low carbon loadings that are suitable for slurry processes.
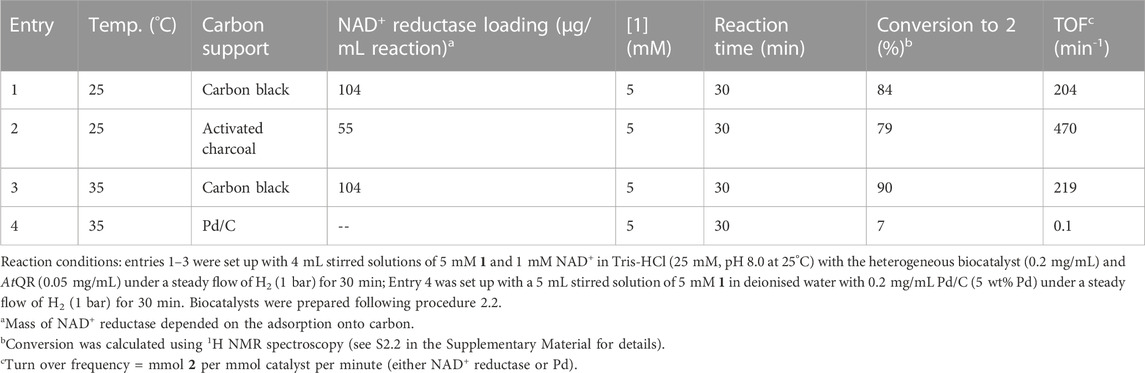
TABLE 2. Effect of varying different parameters on the rate of conversion of 1 in batch.
A comparison Pd/C-catalysed hydrogenation (entry 4) was run using analogous reaction conditions (to entry 3, 0.2 mg/mL catalyst loading, 35°C, 1 bar H2), though deionised water was used in place of buffer in order to avoid additives that could poison the Pd. After 30 min, a 7% conversion was determined using 1H NMR spectroscopy, and this equates to a Pd TOF of 0.1 min-1. This low conversion and catalyst activity could be explained by the likelihood that the Pd requires higher H2 availability usually achieved via elevated temperature and pressure, improved mixing, or a different solvent. For example, Pd/C has previously been used in tandem with a chiral-rhodium catalyst to hydrogenate 1 (84% yield, 58% ee) using intensified conditions: 75°C and 10–14 bar H2 in methanol (Brieden, 1996). It’s worth noting that such conditions requiring elevated pressures complicate translation into a continuous slurry process.
High conversions to 2 using the biocatalytic hydrogenation strategy in an aqueous, ambient batch reactions provided the groundwork to move the system into a continuous slurry flow process. Here the focus is on understanding if the biocatalyst system can simply “slot-in” without significant chemical or process engineering.
In comparing the protocols for biocatalytic hydrogenation of 1 compared to typically metal-catalysed hydrogenations, a key difference to be addressed is in the solvent, moving from organic solvents for metals to aqueous media for biocatalytic with implications for H2 solubility. For example, the solubility of hydrogen at 35°C is ca. 8 times lower (when comparing by molality) in water than for methanol under comparable pressures (Descamps et al., 2005). We expected that using the Coflore ACR would help overcome this rate limitation due to the three-fold increase regarding mass transfer coefficients (Toftgaard Pedersen et al., 2017), opening up opportunities to use aqueous solvent. The use of water in place of methanol or other toxic, flammable organic solvents is desirable because it lowers overall health and safety risks of a hydrogenation process (Prat et al., 2014; Byrne et al., 2016).
To test the Coflore ACR with an aqueous hydrogenation reaction, we took the biocatalyst formulated on the carbon black nanopowder and the 35°C reaction temperature forward to translate to continuous flow. With a typical particle size range of 20–50 nm as measured through transmission electron microscopy (Weingarth et al., 2014), and a specific surface area of 1272 m2/g as measured through nitrogen gas sorption and BET analysis (or 1949 m2/g using the Langmuir model [Zhao et al., 2021)], risk reduction in sedimentation, as well as a higher activity per catalyst mass were expected when compared to larger, lower surface area carbon catalyst materials (see comparable data for a variety of Pd/C catalysts in Zhao et al., 2021 supporting information). Reliable slurry feed at flow rates as low as 0.4 mL/min was possible using a Masterflex peristaltic pump when using Norprene LS14 flexible tubing.
The continuous biocatalytic hydrogenation was set up as shown in Figure 4 (see details in Section 2.5), where a slurry feed that contained 1 (5 mM), NAD+ (1 mM), heterogeneous biocatalyst (0.15 mg/mL), and AtQR (0.05–0.40 mg/mL) in Tris-HCl buffer (75 mM, pH 8.0 at 25°C) was stirred in a jacketed vessel. This slurry was pumped into the ACR where it as mixed with H2 at 35°C at various residence times (tRes) achieving 2 bar overall system pressure. Reaction slurries were then collected in a vessel, with samples taken for NMR spectroscopy analysis after every reactor volume. Additional details for process is shown in Supplementary Table S2 (Supplementary Information).
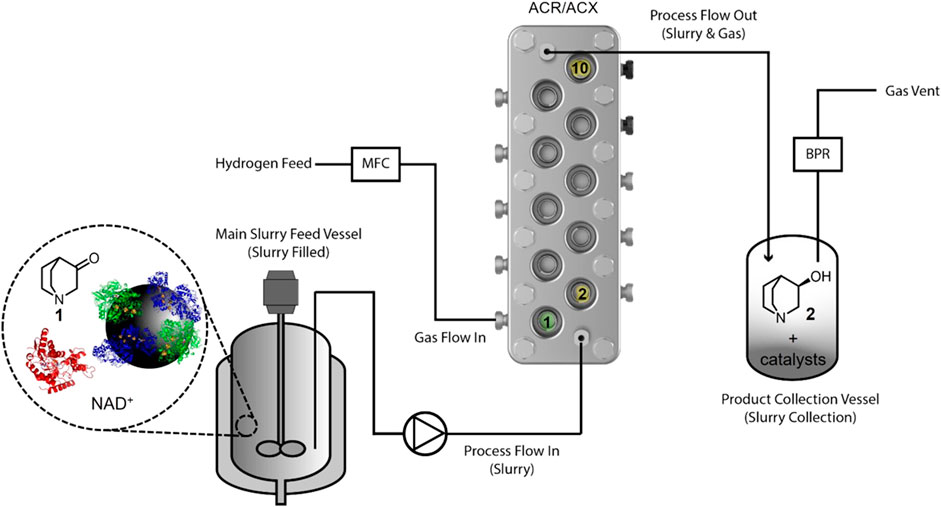
FIGURE 4. Continuous biocatalytic hydrogenation of 3-quinuclidinone (1) using an agitated reactor (ACR or ACX). The slurry feed was stirred in a jacketed vessel and pumped into the agitated reactor. Both the H2 and the suspension were dosed into the first cell “1”. The reactor temperature was monitored at cells 2 and 10.
The results in Table 3 show conversions to 2 during the steady state using different continuous flow parameters. For the reaction summarised in entry 1, a Coflore ACR block was initially used in conjunction with 10 high shear agitators resulting in a 90 mL reactor assembly. The suspension feed contained 5 mM 1, 0.15 mg/mL heterogeneous biocatalyst and 0.05 mg/mL AtQR. The slurry flow rate was set such that the liquid phase tRes in the agitated reactor was 15 min. Under these conditions, a 35% conversion to 2 was reached during steady state, with catalyst TOF (286 min−1) similar to that achieved in batch suggesting that the biocatalyst system had good compatibility with the slurry continuous mode.
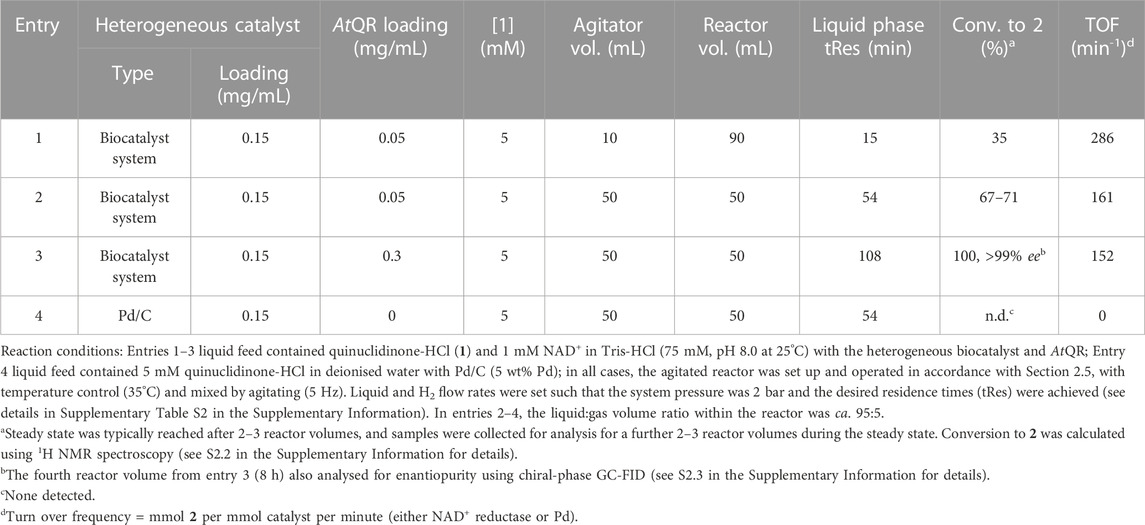
TABLE 3. Hydrogenation of 1 in continuous flow slurry reactors.
For the reactions summarised in entries 2–4 (Table 3), the tRes was increased to 54 min in order to achieve higher conversions. The high shear agitators were substituted for 50% volume agitators, which decreased the reactor volume from 90 mL to 50 mL. The Coflore ACR block was substituted for an ACX block to minimise the gas phase holdup, whilst maintaining a constant liquid phase volume, and minimisation of working volumes. At steady state, a 67%–71% conversion was achieved (entry 2). We estimate the NAD+ reductase TOF of 161 min-1 based on concentration of 2 generated, although a build up of NADH was observed, suggesting that the reaction was limited here by AtQR loading. Regardless, this catalyst activity is still on the same order of magnitude as those achieved in batch (see values in Table 2).
For comparison, a slurry feed that contained 1 (5 mM) and 5% Pd/C (0.15 mg/mL) in deionised water was pumped through the ACX reactor such that tRes was 54 min at 35°C and under 2 bar H2. Under these conditions, there was no detection of 2 (entry 4, Table 3). Similar to the low conversion when Pd/C was used these mild temperature and pressure conditions in batch, it is likely that harsher conditions or higher catalyst loading would be required for Pd-catalysed hydrogenation of 1. The Pd mass loading was 7.5 μg/mL (ca. 0.8 mmol/mL), whereas the immobilised enzyme loading was six orders of magnitude smaller per presumed ‘active site’ (ca. 5 × 10−7 mmol/mL).
Results in entry three show that on doubling tRes to 108 min and increasing AtQR loading to 0.3 mg/mL, 100% conversion to 2 was achieved at steady state. The sample collected at 8 h (4 reactor volumes) was also analysed by chiral-phase GC-FID, and only the R-enantiomer of 2 was detected (>99% ee).
Although these results are promising, the substrate loading is low. Therefore the reaction was next intensified to 50 mM 1 with increased heterogenous biocatalyst (0.45 mg/mL) and AtQR (0.40 mg/mL) loading (entry 1, Table 4). Continuous process was carried out with 108 min slurry tRes, 35°C and 2 bar H2. During steady state, a 18%–20% conversion to 2 was achieved (entry 5), showing a step improvement to provide 10 mM 2 in continuous flow.
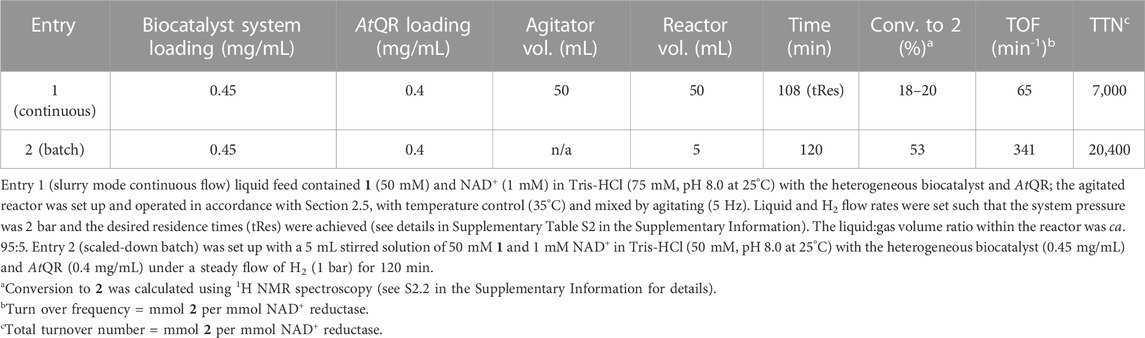
TABLE 4. Continuous slurry process comparison to batch at higher [1] (50 mM).
A comparable batch reaction was also carried out (entry 2), leading to higher conversion (53%, 26.5 mM 2) and catalyst activity (341 min−1). Given the low TOF for entry 1 (65 min−1), the slurry mode continuous process is likely to benefit from improved H2 availability which could be achieved via improved mixing, use of solvent or increased pressure.
For operation of continuous hydrogenation in Coflore reactors, metal/C catalysts can be handled in packed basket agitators. This prevents a need to pump slurries under higher pressure conditions, and advantageously retains the catalyst in the reactor system.
To demonstrate the Pd/C hydrogenation of 1 under increased pressure more typically used for metal-catalysed hydrogenations, the reactor set up was adjusted. The conditions (10 bar H2) required that an Elados EMP II E60 solenoid driven diaphragm pump be used in place of the peristaltic pump that was used with the more mild slurry reactions. This in turn necessitated using Pd/C in packed catalyst basket agitators rather than pumping through the ACX as a slurry, as the Pd/C particles increase the risk of blockages in the diaphragm pump check valves or settling in the diaphragm chamber.
Catalyst basket agitators (ACR-CB-Ha) were packed with Pd/C pellets (1 wt% Pd) and fitted into the ACX (see Supplementary Figure S4 in the Supplementary Information for diagram). A solution of 1 (5 mM) in deionised water was then pumped through the agitated (5 Hz) ACX such that a 56 min tRes was achieved at 35°C under 10 bar H2 (results summarised in entry 1, Table 5). When the catalyst basket agitators were completely filled with Pd/C pellets (5.34 g total catalyst, 68 mL reactor volume), full conversion to 2 was determined during the steady state. Despite the excellent conversion, we note that this equates to <3 total Pd turnover numbers owing to the high catalyst loading.
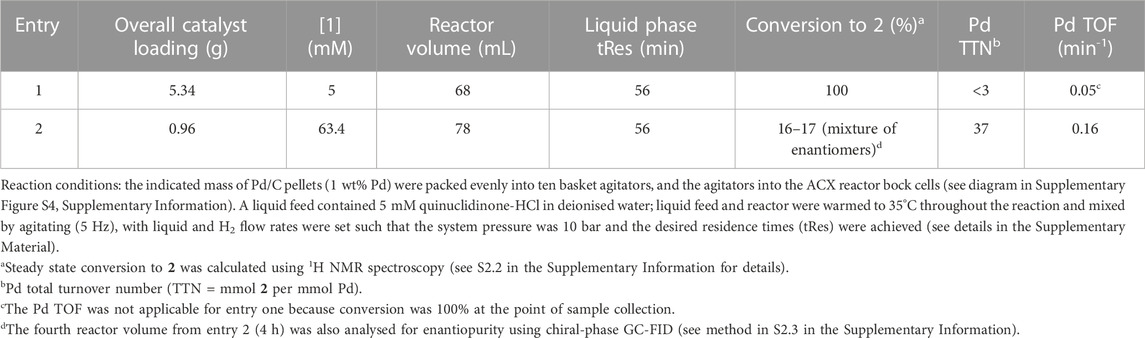
TABLE 5. Continuous hydrogenation of 1 by Pd/C pellets in packed bed agitators.
In a second experiment the overall Pd/C loading to 0.96 g (78 mL reactor volume) and increased the [1] to 63.7 mM, in order to overall lower the mass ratio of catalyst: substrate. As catalytic activity is commonly surface area-limited (mass transfer is limited to the number of catalyst active sites), equalising catalyst loading based on this metric allows a more direct comparison of activity, cost, and performance. In this case, the conversion to 2 averaged to 17% during steady state (entry 2). This equates to 10.8 mM 2 generated and 37 total Pd turnovers (Pd TOF = 0.16 min−1). This Pd activity is similar to the Pd activity achieved during the batch reaction that was performed at 35°C and 1 bar H2 (see entry 4, Table 2). A sample from the fourth reactor volume (4 h) was also analysed using chiral-phase GC-FID, which revealed that a mixture of both enantiomers were generated by the metal catalyst.
We have demonstrated that a heterogenous biocatalytic hydrogenation process can be developed for high conversion in short reaction times, providing a basis for translation into continuous flow with slurry feeds. This system was shown to “slot-in” to the Coflore continuous flow reactor, and achieve high conversions of ketone reduction to (3R)-quinuclidinol, with comparable activity to scale down batch reactions without any need for process or chemical engineering development or optimisation. The processes benefit from operating at mild temperature (35°C) and pressure (2 bar) in aqueous conditions and were able to achieve 100% conversion, albeit at low substrate concentration, retaining >99% ee expected with a biocatalytic system. Under analogous conditions, Pd/C did not lead to any ketone reduction in slurry mode continuous flow, likely due to use of ambient conditions for ease of comparison. This ability for the biocatalytic hydrogenation system to operate under mild conditions is likely to have implication for lower energy demands and improved safety metrics at scale.
On intensification of substrate loading, the result show that the biocatalyst performed well in scale-down reactions, and more optimisation steps can be explored with the continuous set up to enhance the biocatalyst activity (solvent, pressure, mixing). We were encouraged to observe similar conversions for the bio- and metal-catalyst systems at higher substrate loading, despite a significant different catalyst loadings that lead to TTNs of >20,000 for biocatalyst and ca. 37 for the palladium. This further suggests that improvements could be achieved using the biocatalyst system in the packed basket agitators, where there would be more opportunity to maximise the enzyme capabilities at high TTN and take advantage of the proven multi-day stability of the biocatalyst system (Poznansky et al., 2021).
Overall, biocatalytic hydrogenation provides a route to decarbonisng biocatalysis by removing requirement for carbon-based reductants, and can provide “slot-in” replacement to metal catalysts for hydrogenation reactions in continuous flow reactors.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
SC, TL, and HR designed the scale-down batch experiments. SC, TL, and XZ carried out batch experiments. JT designed the continuous flow equipment setup. JT and MM designed the continuous flow experiments. SC, SK, JT, JA, and MM carried out continuous flow experiments. All authors analysed data, discussed the results and assisted in the preparation of the manuscript. SC and HR wrote the paper.
Research at HydRegen was supported in part by Innovate UK grant 85087.
We are grateful to Miguel A. Ramirez (HydRegen Ltd.) for supplying the enzymes. We thank Prof. Kylie Vincent (U. Oxford) for visitor agreement to use the Bruker Advance III HD nanobay and ThermoScientific Trace 1310 GC and to Sofia Helin (U. Oxford) for help with establishing GC protocols. We thank Prof. Frans L. Muller (University of Leeds) for providing the Pd/C pellets for immobilised catalyst experiments.
HydRegen is a limited company commercialising biocatalytic hydrogenation tools. AM Technology is a limited company providing flow reactor technology including the Coflore ACR. This work was carried out using internal company resources, with HydRegen providing catalyst and biocatalysis expertise and AM Technology providing access and expertise for continuous flow reactors. This research was not part of a commercial relationship and was carried out for R&D purposes only.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2023.1114536/full#supplementary-material
Arai, N., Akashi, M., Sugizaki, S., Ooka, H., Inoue, T., and Ohkuma, T. (2010). Asymmetric hydrogenation of bicyclic ketones catalyzed by BINAP/IPHAN-Ru(II) complex. Org. Lett. 12, 3380–3383. doi:10.1021/ol101200z
Berenguer-Murcia, A., and Fernandez-Lafuente, R. (2010). New trends in the recycling of NAD(P)H for the design of sustainable asymmetric reductions catalyzed by dehydrogenases. Curr. Org. Chem. 14, 1000–1021. doi:10.2174/138527210791130514
Brieden, W. (1996). Method for the preparation of optically active 3-hydroxyquinuclidine. Available at https://patents.google.com/patent/EP0785198A1/en (Accessed November 18, 2022).
Byrne, F. P., Jin, S., Paggiola, G., Petchey, T. H. M., Clark, J. H., Farmer, T. J., et al. (2016). Tools and techniques for solvent selection: Green solvent selection guides. Sustain. Chem. Process. 4, 7. doi:10.1186/s40508-016-0051-z
Chen, Q., Xie, B., Zhou, L., Sun, L., Li, S., Chen, Y., et al. (2019). A tailor-made self-sufficient whole-cell biocatalyst enables scalable enantioselective synthesis of (R)-3-Quinuclidinol in a high space-time yield. Org. Process Res. Dev. 23, 1813–1821. doi:10.1021/acs.oprd.9b00004
Chuzeville, L., Boury, F., Duday, D., Anand, R., Moretto, E., and Thomann, J.-S. (2022). Eco-friendly processes for the synthesis of amorphous calcium carbonate nanoparticles in ethanol and their stabilisation in aqueous media †. Green Chem. 24, 1270–1284. doi:10.1039/d1gc03396d
Descamps, C., Coquelet, C., Bouallou, C., and Richon, D. (2005). Solubility of hydrogen in methanol at temperatures from 248.41 to 308.20 K. Thermochim. Acta 430, 1–7. doi:10.1016/j.tca.2004.12.001
Irfan, M., Glasnov, T. N., and Kappe, C. O. (2011). Heterogeneous catalytic hydrogenation reactions in continuous-flow reactors. ChemSusChem 4, 300–316. doi:10.1002/cssc.201000354
Lauterbach, L., and Lenz, O. (2013). Catalytic production of hydrogen peroxide and water by oxygen-tolerant [NiFe]-Hydrogenase during H2 cycling in the presence of O2. J. Am. Chem. Soc. 135, 17897–17905. doi:10.1021/ja408420d
Mallia, C. J., and Baxendale, I. R. (2016). The use of gases in flow synthesis. Org. Process Res. Dev. 20, 327–360. doi:10.1021/acs.oprd.5b00222
Maniscalco, M., Singh-Franco, D., Wolowich, W. R., and Torres-Colón, R. (2006). Solifenacin succinate for the treatment of symptoms of overactive bladder. Clin. Ther. 28, 1247–1272. doi:10.1016/j.clinthera.2006.09.017
Montuschi, P., and Ciabattoni, G. (2015). Bronchodilating drugs for chronic obstructive pulmonary disease: Current status and future trends. J. Med. Chem. 58, 4131–4164. doi:10.1021/jm5013227
Naito, R., Yonetoku, Y., Okamoto, Y., Toyoshima, A., Ikeda, K., and Takeuchi, M. (2005). Synthesis and antimuscarinic properties of quinuclidin-3-yl 1,2,3,4-Tetrahydroisoquinoline-2-carboxylate derivatives as novel muscarinic receptor antagonists. J. Med. Chem. 48, 6597–6606. doi:10.1021/jm050099q
Poznansky, B., Cleary, S. E., Thompson, L. A., Reeve, H. A., and Vincent, K. A. (2021). Boosting the productivity of H2-driven biocatalysis in a commercial hydrogenation flow reactor using H2 from water electrolysis. Front. Chem. Eng. 3, 35. doi:10.3389/fceng.2021.718257
Prat, D., Hayler, J., and Wells, A. (2014). A survey of solvent selection guides. Green Chem. 16, 4546–4551. doi:10.1039/c4gc01149j
Ramirez, M. A., Joseph Srinivasan, S., Cleary, S. E., Todd, P. M. T., Reeve, H. A., and Vincent, K. A. (2022). H2-Driven reduction of flavin by hydrogenase enables cleaner operation of nitroreductases for nitro-group to amine reductions. Front. Catal. 2, 906694. doi:10.3389/fctls.2022.906694
Reeve, H. A., Lauterbach, L., Lenz, O., and Vincent, K. A. (2015). Enzyme-Modified particles for selective biocatalytic hydrogenation by hydrogen-driven NADH recycling. ChemCatChem 7, 3480–3487. doi:10.1002/cctc.201500766
Rice, H. P., He, Y., Muller, F. L., Bayly, A. E., Ashe, R., Karras, A., et al. (2022). Physical and numerical characterisation of an agitated tubular reactor (ATR) for intensification of chemical processes. Chem. Eng. Process. Intensif. 179, 109067. doi:10.1016/j.cep.2022.109067
Rowbotham, J. S., Ramirez, M. A., Lenz, O., Reeve, H. A., and Vincent, K. A. (2020). Bringing biocatalytic deuteration into the toolbox of asymmetric isotopic labelling techniques. Nat. Commun. 11, 1454. doi:10.1038/s41467-020-15310-z
Stoffels, M. A., Klauck, F. J. R., Hamadi, T., Glorius, F., and Leker, J. (2020). Technology trends of catalysts in hydrogenation reactions: A patent landscape analysis. Adv. Synth. Catal. 362, 1258–1274. doi:10.1002/adsc.201901292
Thompson, L. A., Rowbotham, J. S., Nicholson, J. H., Ramirez, M. A., Zor, C., Reeve, H. A., et al. (2020a). Rapid, heterogeneous biocatalytic hydrogenation and deuteration in a continuous flow reactor. ChemCatChem 12, 3913–3918. doi:10.1002/cctc.202000161
Thompson, L. A., Rowbotham, J. S., Reeve, H. A., Zor, C., Grobert, N., and Vincent, K. A. (2020b). Biocatalytic hydrogenations on carbon supports. Methods Enzym. 630, 303–325. doi:10.1016/bs.mie.2019.10.017
Toftgaard Pedersen, A., Sutherland, E., Rehn, G., Ashe, R., and Woodley, J. M. (2017). Characterization of a continuous agitated cell reactor for oxygen dependent biocatalysis. Biotechnol. Bioeng. 114, 1222–1230. doi:10.1002/bit.26267
Tsutsumi, K., Katayama, T., Utsumi, N., Murata, K., Arai, N., Kurono, N., et al. (2009). Practical asymmetric hydrogenation of 3-quinuclidinone catalyzed by the XylSkewphos/PICA-ruthenium(II) complex. Org. Process Res. Dev. 13, 625–628. doi:10.1021/op9000302
Weingarth, D., Drumm, R., Foelske-Schmitz, A., Kötz, R., and Presser, V. (2014). An electrochemical in situ study of freezing and thawing of ionic liquids in carbon nanopores. Phys. Chem. Chem. Phys. 16, 21219–21224. doi:10.1039/c4cp02727b
Woods, J. A., Nealy, K. L., and Barrons, R. W. (2013). Aclidinium bromide: An alternative long-acting inhaled anticholinergic in the management of chronic obstructive pulmonary disease. Ann. Pharmacother. 47, 1017–1028. doi:10.1345/aph.1s002
Zhang, W.-X., Xu, G.-C., Huang, L., Pan, J., Yu, H.-L., and Xu, J.-H. (2013). Highly efficient synthesis of (R)-3-Quinuclidinol in a space-time-yield of 916 g L-1 d-1 using a new bacterial reductase ArQR. Org. Lett. 15, 4917–4919. doi:10.1021/ol402269k
Zhao, X., Cleary, S. E., Zor, C., Grobert, N., Reeve, H. A., and Vincent, K. A. (2021). Chemo-bio catalysis using carbon supports: Application in H2-driven cofactor recycling. Chem. Sci. 12, 8105–8114. doi:10.1039/d1sc00295c
Keywords: biocatalytic hydrogenation, cofactor (NADH) regeneration, asymmetric reduction, green chemistry, continuous process
Citation: Cleary SE, Kazantzi S, Trenchard JA, Monedero M, Allman JW, Lurshay TC, Zhao X, Kenny MBC and Reeve HA (2023) Preparation of (3R)-quinuclidinol using heterogeneous biocatalytic hydrogenation in a dynamically-mixed continuous flow reactor. Front. Catal. 3:1114536. doi: 10.3389/fctls.2023.1114536
Received: 02 December 2022; Accepted: 22 February 2023;
Published: 08 March 2023.
Edited by:
Luciana Rocha Barros Gonçalves, Federal University of Ceara, BrazilReviewed by:
Jan von Langermann, University of Rostock, GermanyCopyright © 2023 Cleary, Kazantzi, Trenchard, Monedero, Allman, Lurshay, Zhao, Kenny and Reeve. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah E. Cleary, c2FyYWhAaHlkcmVnZW5veGZvcmQuY29t; Holly A. Reeve, aG9sbHlAaHlkcmVnZW5veGZvcmQuY29t
†Present address: Tara C. Lurshay, Department of Chemistry, University of Oxford, Oxford, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.