 Erik Eppinger
Erik Eppinger Janosch Alexander David Gröning
Janosch Alexander David Gröning Andreas Stolz
Andreas Stolz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Catal., 05 August 2022
Sec. Biocatalysis
Volume 2 - 2022 | https://doi.org/10.3389/fctls.2022.952944
The conversion of rac-phenylglycinonitrile by different variants of the nitrilase from Pseudomonas fluorescens EBC191 (EC 3.5.5.1) was studied and the amounts and chiral composition of the formed phenylglycine and phenylglycine amide compared. Muteins that converted rac-phenylglycinonitrile to extraordinarily high amounts of phenylglycine or phenylglycine amide were tested for the chemoenzymatic enantioselective one-pot synthesis of (R)- and (S)-phenylglycine and (R)- and (S)-phenylglycine amide. The chemoenzymatic synthesis combined the initial step in the traditional chemical Strecker synthesis which results in the formation of rac-phenylglycinonitrile from benzaldehyde, cyanide, and ammonia with the enzymatic conversion of the formed nitrile by the nitrilase variants. The aminonitrile synthesis was optimized in order to obtain conditions which allowed under mildly alkaline conditions (pH 9.5) maximal yields of phenylglycinonitrile and the in-situ racemization of the compound. The racemic phenylglycinonitrile was directly converted under the alkaline conditions without any interposed purification step by cells of Escherichia coli overexpressing recombinant nitrilase variants. The application of a mutant of E. coli defect in a (S)-phenylglycine amide hydrolysing peptidase (E. coli JM109ΔpepA) expressing a highly reaction- and (R)-specific nitrilase variant allowed the synthesis of (R)-phenylglycine with ee-values ≥ 95% in yields up to 81% in relation to the initially added benzaldehyde. These yields indicated a dynamic kinetic resolution which involved the racemization of (S)- to (R)-phenylglycinonitrile under the used alkaline conditions with the concurrent hydrolysis of (R)-phenylglycinonitrile to (R)-phenylglycine. The addition of resting cells of E. coli JM109ΔpepA synthesizing an amide forming nitrilase variant to the final product of the Strecker synthesis and/or using E. coli strains with an intact aminopeptidase gene resulted in the preferred formation of (S)-phenylglycine amide, (R)-phenylglycine amide or (S)-phenylglycine.
Chiral non-proteinogenic amino acids are interesting products for the chemical industry because they find many applications as building blocks for the synthesis of various pharmaceuticals. There have been several approaches described in order to synthesize this class of compounds either by purely chemical means or by biosynthetic techniques (Bommarius et al., 2001; Nájera and Sansano, 2007; Servi et al., 2007; Walsh et al., 2013; Almhjell et al., 2018; Xue et al., 2018; Dennig et al., 2019). The quantitatively most important industrially produced chiral non-proteinogenic amino acids are presumably (R)-phenylglycine and (R)-4-hydroxyphenylglycine, which are intermediates in the commercial production of important semisynthetic β-lactam antibiotics (Grundmann and Fessner, 2008). Some of these antibiotics (ampicillin, cephalexin and amoxicillin) are included in the World Health Organisation’s “Model List of Essential Medicines”. The annual production of (R)-phenylglycine has been estimated to exceed 5,000 t per year (Vedha-Peters et al., 2006).
There are several enzymatic reactions known which allow the synthesis of the optically active forms of phenylglycine and/or phenylglycine amide. An enzymatic route to (S)-phenylglycine has been described which is based on the biosynthetic pathway of some natural peptide antibiotics, found in different streptomycetes. In these natural products (S)-phenylglycine is derived from the anabolic shikimate pathway with the intermediate formation of prephenate and 4-phenylpyruvate (Al Toma et al., 2015). The genes coding for the synthesis of (S)-phenylglycine have been cloned from the genomes of Streptomyces strains and recombinantly expressed in other actinomycetes (Moosmann et al., 2020). An alternative “synthetic” pathway for the enzymatic production of (S)-phenylglycine starts from mandelic acid which is enzymatically oxidized to phenylglyoxylate and converted in a subsequent enantioselective reductive amination reaction to (S)-phenylglycine. Recombinant E. coli strains which express this enzyme cascade produce (S)-phenylglycine with space-time yields up to almost 80 g 1−l d−1 (Resch et al., 2010; Tang et al., 2020).
The most important processes for the industrial production of (R)-phenylglycine start either with a diastereomeric salt crystallization of rac - phenylglycine or via the corresponding racemic hydantoin and use D-hydantoinases (± carbamoylases) for the synthesis of (R)-phenylglycine (Bommarius et al., 1992; Wegman et al., 2001b). In addition, synthetic pathways have been generated in E. coli which catalyse the conversion of (hydroxyl) phenylpyruvate via (hydroxyl)mandelate and (hydroxyl) phenylglyoxylate by a combination of a hydroxymandelate synthase, a hydroxymandelate oxidase (or a dehydrogenase) and a (di)hydroxyphenylglycine aminotransferase from different microbial sources (Müller et al., 2006; Liu et al., 2016; Tan et al., 2021). Zhou et al. (2017) demonstrated the synthesis of (R)-phenylglycine by recombinant E. coli strains from racemic mandelic acid, styrene, or L-phenylalanine in 3-8 steps via the intermediate formation of (S)-mandelic acid.
Several chemical methods have been described for the synthesis of (non-proteinogenic) amino acids. The Bucherer-Bergs reaction uses ketones (or aldehydes), cyanide, and ammonium carbonate as starting material for the synthesis of hydantoins, which can be used as precursors in the chemical or enzymatical synthesis of amino acids (Kalnik et al., 2021). In the Erlenmeyer-Plöchl synthesis an aromatic aldehyde and an aromatic carboxylic acid react in the presence of acetic acid anhydride to an azalactone which can be subsequently converted to an α-amino acid (Corti et al., 2021; Han et al., 2021). A wide range of non-proteinogenic α-amino acids are easily accessible chemically by the classical Strecker synthesis, which is based on the reaction of an aldehyde, ammonia and cyanide under alkaline conditions to the corresponding α-aminonitrile, which is subsequently hydrolysed under acid conditions to the amino acid (Kouznetsov and Galvis, 2018). The major disadvantage of the classical Strecker synthesis lies in the formation of the racemic amino acids and the necessity to use highly toxic cyanide. During the last years, several purely chemical routes have been described which try to overcome this limitation and induce enantioselectivity into the reaction often using thiourea-derived catalysts (Gröger, 2003; Ooi et al., 2006; Zuend et al., 2009; Wang et al., 2017; Kouznetsov and Galvis, 2018).
The coupling of the Strecker reaction with a subsequent enantioselective hydrolysis (or hydration) reaction allows in principle the stoichiometric conversion of an aldehyde (with ammonia and cyanide) to a chiral amino acid. This is due to the fact that under the alkaline conditions of the Strecker synthesis an equilibrium is maintained between the reaction substrates (aldehyde, ammonia, cyanide) and the product (α-aminonitrile). This enables the possibility of a dynamic kinetic resolution in the presence of an enantioselective catalyst (Chaplin et al., 2004).
The α-aminonitriles which are intermediately formed in the course of the Strecker reaction are substrates for nitrile converting enzymes. This has been repeatedly demonstrated with the substrate phenylglycinonitrile (PGN) for nitrilases and nitrile hydratases which hydrolyse and/or hydrate PGN to phenylglycine (PGA) and/or phenylglycine amide (PGAA). There are several reports which describe the enantioselective formation of phenylglycine amide or phenylglycine from phenylglycinonitrile by nitrile hydratase/amidase-systems or nitrilases (Wegman et al., 2000, 2001a; Ewert et al., 2008; Hensel et al., 2002; Wang and Lin, 2001; Qiu et al., 2014a, b; Eppinger and Stolz, 2019; Mareya et al., 2020).
It was recently shown that recombinant cells of E. coli which synthesize variants of the nitrilase from Pseudomonas fluorescens EBC191, convert rac-phenylglycinonitrile either to (R)-phenylglycine or to (R)- or (S)-phenylglycine amide. In addition, it was found that the whole cell catalysts were still active at pH 10.8 and that under these conditions phenylglycinonitrile racemizes with a significant rate (Eppinger and Stolz, 2019). Therefore, it was tested in the present publication if it was possible with this system to establish a productive coupling of the Strecker synthesis with a biological dynamic kinetic resolution.
E. coli JM109ΔpepA was used as host strain (Eppinger and Stolz, 2019).
The construction of plasmid pIK9 has been described before (Kiziak et al., 2005). Plasmid pIK9/pap encoded a chimeric nitrilase which basically represents a variant of the nitrilase from P. fluorescens EBC191 carrying 16 amino acid residues close to the catalytical centre analogously found in the nitrilase from Alcaligenes faecalis ATCC 8750. This chimeric nitrilase demonstrates with mandelonitrile a high reaction- and enantiospecificity for the formation of (R)-mandelic acid (Kiziak and Stolz, 2009).
Plasmid pEN15 carries the gene for the aminopeptidase PepA of E. coli cloned into pJOE2775 (Eppinger and Stolz, 2019).
The conditions for the cultivation of the recombinant strains and for the expression of the nitrilase (-variants) have been described before (Kiziak et al., 2005). The bacterial cultures were split in aliquots, flash-frozen in liquid nitrogen and kept at −70°C until usage. Prior to the biotransformation experiments, the aliquots were thawed on ice, the cells washed twice with a saline [0.9% (w/v) NaCl] and resuspended in saline to yield optical densities (OD600nm) of 25–100.
These techniques were performed as previously described (Sosedov and Stolz, 2015).
The reactions were performed in the smallest handable volumes in closed vessels in a fume hood using a portable cyanide alarm sensor (Dräger Pac 7000, Lübeck, Germany). In most experiments, the reaction mixtures (≤ 0.5 ml each) were composed of 100 mM benzaldehyde, 100 mM KCN, and a suitable source of ammonia in the tested buffer systems. The liquid volumes were incubated in tightly closed 2 ml glass vessels and mixed with small stirring bars at 750–1,500 rpm. After different time intervals aliquots (5 or 10 μl each) were taken and the reactions stopped by the addition of 45 µl or 90 µl of a mixture of methanol/water/1 M HCl (5:3:2 v/v/v).
Synthesis of rac-PGN: The reaction mixtures (1 ml) contained in tightly closed 2 ml glass vessels 150 mM KCN in 500 mM ammonium acetate/NH4OH-buffer (pH 9.5) at 40°C. The reactions were started by the addition of 50 mM benzaldehyde and the mixtures intensively stirred (1,500 rpm) for 120 min.
Synthesis of (R)-PGA and (S)-PGAA: The biotransformations were initiated by the addition of resting cells of different recombinant E. coli strains to the aqueous solutions of rac-PGN prepared in-situ as described above. For the synthesis of (R)-PGA resting cells of E. coli JM109ΔpepA (pIK9/pap) (final OD600nm = 1.3) and for the synthesis of (S)-PGAA cells of E. coli JM109ΔpepA (pIK9-W188K) (OD600nm = 0.5) were used. The same amounts of fresh cells as in the beginning were added after 180 and 360 min to the reaction vessels. The reactions were stopped after 24 h by the addition of 9 ml of a mixture of methanol/water/1 M HCl (5:3:2 v/v/v). The cells were removed by centrifugation [4,000 g, 15 min at 25°C] and the supernatants used for the further investigations.
Synthesis of (S)-PGA: The biotranformation was initiated after the completion of the chemical synthesis of rac-PGN by the addition of resting cells of E. coli JM109ΔpepA (pIK9-W188K-V193S) (final OD600nm = 1.5). The same amount of fresh cells was added after 180 and 360 min to the reaction vessel. After 24 h, the reaction mixtures were cooled down to 23°C, and supplemented with resting cells of E. coli JM109ΔpepA (pEN15) (60 μl, final OD600nm = 7.5). The mixtures were stirred (∼ 1,500 rpm) and the reactions terminated after 2 h by the addition of 9 ml of a solution consisting of methanol/water/1 M HCl (5:3:2 v/v/v). The further work up was performed as described above.
Synthesis of (R)-PGAA: The aqueous solution obtained by the Strecker synthesis was cooled down to 23°C and resting cells of E. coli JM109ΔpepA (pIK9-W188K) (OD600nm = 10) together with resting cells of E. coli JM109ΔpepA (pEN15) (35 μl, OD600nm = 10) were added. The reactions were terminated after 3 h and the product prepared as described above.
The chemical and biotransformation reactions were routinely analyzed by chiral and achiral HPLC. Two different HPLC methods were used.
For the achiral HPLC analysis a Pro C18 AQ column (Trentec Analysetechnik, Rutesheim, Germany) was used. The solvent system consisted of 5% (v/v) methanol, 1 g/l Na-hexane-1-sulfonate and 0.1% (v/v) formic acid in H2O. The standard flow rate was 1 ml/min. The average retention times (Rt) under these conditions were: benzaldehyde Rt = 11.5 min, mandelonitrile Rt = 8.8 min, phenylglycinonitrile Rt = 15.1 min, phenylglycine amide Rt = 6.5 min, and phenylglycine Rt = 3.0 min.
A Crownpak CR (+) column (Daicel, Illkirch Cedex, France) was applied for the separation of the enantiomers of phenylglycinonitrile, phenylglycine amide, and phenylglycine. The mobile phase (standard flow rate 1 ml/min) contained 16.3 g/l of 70% (v/v) perchloric acid in water. The column temperature was set to 35°C. The average retention times were for (R)- and (S)-phenylglycine amide Rt = 3.2 and 11.0 min, and for (R)- and (S)-phenylglycine Rt = 4.9 and 16.2 min, respectively. The commercially available racemic rac-phenylglycinonitrile showed under the same conditions two signals at Rt = 9.2 [for the (R)-enantiomer] and 11.9 min [for the (S)-enantiomer]. This chromatographic system did not allow the separation of the enantiomers of mandelonitrile, mandeloamide, and mandelic acid. The average retention times for rac-mandelic acid, rac-mandeloamide and rac-mandelonitrile were Rt = 8.5, 4.3, and 22.9 min, respectively.
The separated compounds were detected at a detector wavelength of 210 nm.
Salicylaldehyde, 5-nitro- and 3,5-dinitrosalicylaldehyde, 5-chloro- and 3,5-dichlorosalicylaldehyde were supplied by Merck, abcr and ICN Pharmaceuticals, respectively. 1-Butyl-3-methylimidazolium bis (trifluoromethylsulfonyl) imide (BMim NTf2) was obtained from Merck. Racemic mixtures of mandelonitrile were purchased from Sigma-Aldrich, mandelic acid and mandeloamide from Lancaster. The preparation of (S)-phenylglycinonitrile and the sources of the other commercially available chemicals have been described before (Eppinger and Stolz, 2019).
Previously, we studied the conversion of PGN by recombinant E. coli strains synthesizing the wild-type nitrilase from P. fluorescens EBC191 or the muteins Ala165Phe and Trp188Lys which formed from nitriles increased amounts of acids or amides, respectively. The whole cell catalysts synthesizing the Ala165Phe mutein converted rac-PGN to 97% of phenylglycine and 3% phenylglycine amide. The cells formed (R)-PGA with 93% ee (Eppinger and Stolz, 2019).
In the present study, it was tested if other variants could be generated which showed better catalytic efficiencies for the formation of (R)-PGA. Therefore, variants were tested which had been shown to produce from mandelonitrile (2-hydroxyphenylacetonitrile) increased amounts of mandelic acid and/or demonstrated with this substrate a higher degree of enantioselectivity. Cells of E. coli JM109 which expressed the nitrilase variant Trp59Arg produced from racemic mandelonitrile mainly (R)-mandelic acid with a significantly higher ee-value than the wild-type (Sosedov, 2013). Therefore, this variant and also the newly generated variants Trp59Ala, Trp59Phe and Trp59Lys were expressed in E. coli JM109ΔpepA and tested for the conversion of rac-PGN. The variant Trp59Arg demonstrated also with (R)-PGN an increased tendency for the formation of the (R)-enantiomer and a reduced amide formation. These effects were not observed with the other variants carrying mutations at this position (Figure 2).
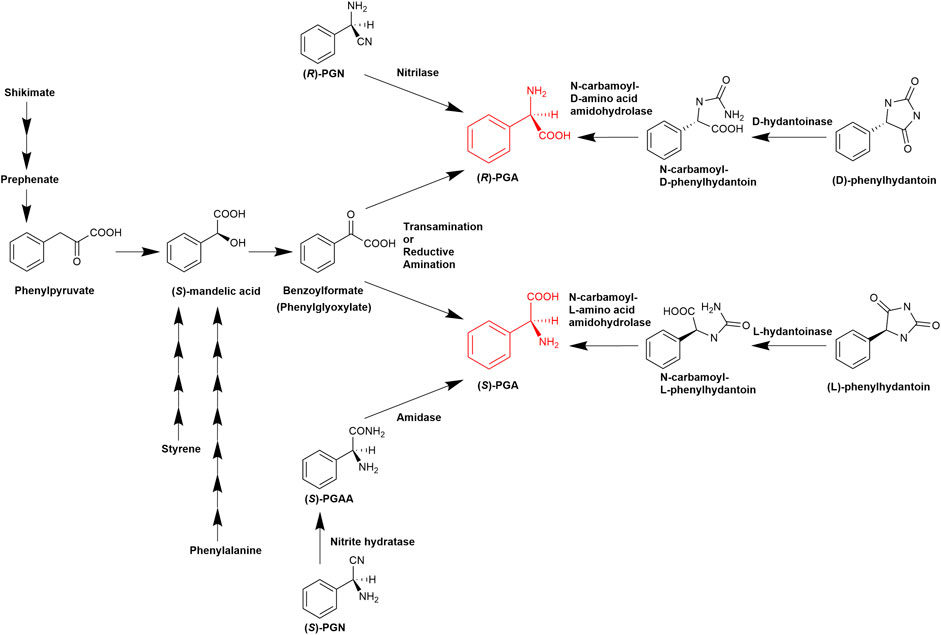
FIGURE 1. Established pathways for the enzymatic synthesis of (R)- or (S)-phenylglycine (Olivieri et al., 1979; Williams and Hendrix, 1992; Gokhale et al., 1996; Hensel et al., 2002; Mei et al., 2008; Al-Toma et al., 2015; Zhou et al., 2017. Liu et al., 2016; Eppinger and Stolz, 2019; Moosmann et al., 2020; Tang et al., 2020).
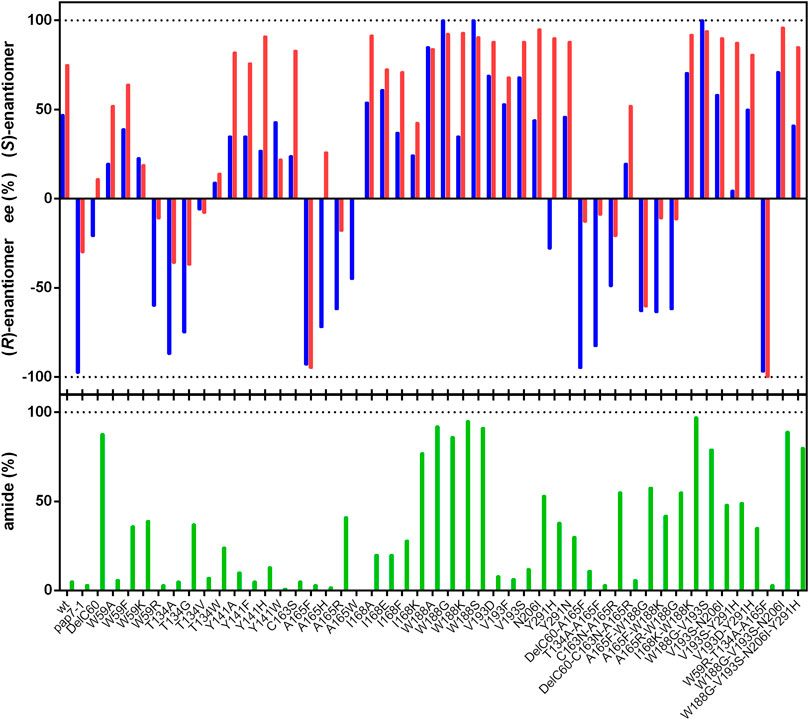
FIGURE 2. Conversion of rac-phenylglycinonitrile by variants of the nitrilase from P. fluorescens EBC191. The reaction mixtures contained in a final volume of 1 ml resting cells of E. coli JM109ΔpepA (pIK9-variant) (OD600nm = 0.1–2.5) in 100 mM potassium-phosphate buffer (pH 6.0). The biotransformation reactions were initiated by adding 100 µl of a rac-PGAA stock solution (100 mM in methanol), and incubated at 23°C and 750 rpm. After different time intervals, samples (90 µl) were taken and the reactions were terminated by the addition of 10 µl of 1 M HCl. The cells were removed by centrifugation (20,000 g, 2 min) and the supernatants were subsequently analyzed by chiral HPLC. The ee values and the degree of amide formation were determined at about 30% conversion. The ee-values for the formed PGA and PGAA are given in blue and red, respectively.
The threonine residue in position 134 in the nitrilase from P. fluorescens EBC191 is homologous to Trp138 in the amidase from Pseudomonas aeruginosa which also belongs to the nitrilase superfamily. The exchange Trp138Gly resulted in the amidase from P. aeruginosa in the ability to convert larger substrates (Karmali et al., 2001). Therefore, the nitrilase variants Thr134Gly, Thr134Ala, Thr134Val, and Thr134Trp were constructed. The exchange of Thr134 against the smaller Gly or Ala residues resulted in the preferred formation of (R)-PGA. Unfortunately, the Thr134Gly variant also formed substantial amounts of PGAA, this was not found with the Thr134Ala variant (Figure 2).
Previously, it was found that the mutein Ala165Phe formed a large surplus of (R)-PGA from racemic PGN (Eppinger and Stolz, 2019). Therefore, the hydrolysis of rac-PGN by the variants Ala165His, Ala165Arg, and Ala165Trp was studied and it was found that they were less selective than Ala165Phe (Figure 2).
The described single point mutations did not result in nitrilase variants with higher enantioselectivities or acid forming abilities than the previously studied Ala165Phe variant. Therefore, it was tested if the introduction of additional mutations resulted in improved variants. Thus, a previously constructed chimeric enzyme variant [NitA(pap)] was tested which converted rac-mandelonitrile with a very high degree of enantioselectivity to (R)-mandelic acid (Kiziak and Stolz, 2009). In this chimeric enzyme 16 amino acid residues close to the catalytical active Cys164 were replaced by the homologous sequence from the nitrilase of Alcaligenes faecalis ATCC8750 (Kiziak and Stolz, 2009). This resulted in the replacement of the sequence motif SVYGEGDGSDLAVHDTTLGRLGALCCAEHIQPLSKYAMYAQHEQV (Cys164 shown in bold) by TVFGEGYARDLIVSDTELGRVGALCCWEHLSPLSKYALYSQHEAI (modified aa residues underlined). NitA(pap) demonstrated from all tested variants under the applied standard conditions (10 mM rac-PGN, about 30% conversion) the highest degree of enantioselectivity for the formation of (R)-PGA (Figure 2).
In the following the kinetic constants of the recombinant E. coli strains were compared that either expressed the wild-type nitrilase or the variants Ala165Phe or NitA(pap). The biocatalysts were incubated with different concentrations of rac-PGN (0.5–10 mM) and the conversion of (R)- and (S)-PGN analyzed by chiral HPLC. The cells which expressed the wild-type enzyme converted (S)-PGN with a maximal reaction rate about 2.2-fold higher than the corresponding rate for (R)-PGN (Figure 3). The Km-values for (S)- and (R)-PGN were calculated as 0.37 ± 0.03 mM and 0.20 ± 0.05 mM, respectively. The experiments suggested that the increased (R)-selectivity of the Ala165Phe und NitA(pap) variants is mainly caused by a pronounced decrease (compared to the wild-type) in the reaction velocity and affinity for the (S)-enantiomer of PGN. Furthermore, the variants Ala165Phe or NitA(pap) showed a significantly lower affinity for (S)-PGN than for (R)-PGN (Figure 3). This indicated that with the variants the highest degree of (R)-selectivity can be obtained by using comparatively low concentrations of rac-PGN.
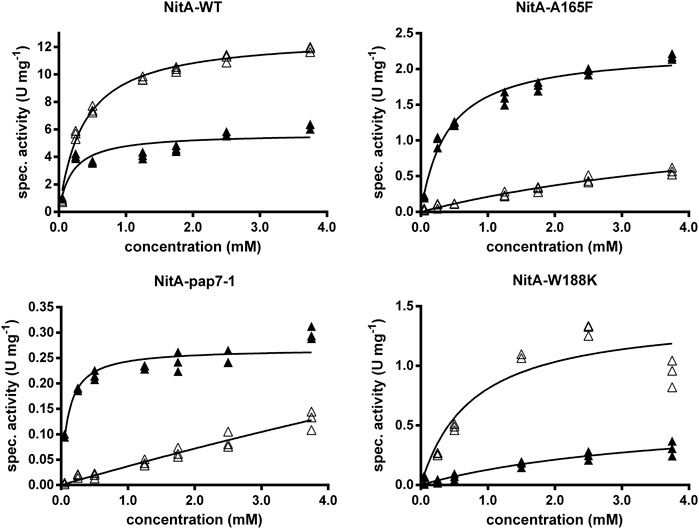
FIGURE 3. Conversion of different concentrations of rac-PGN by resting cells of E. coli JM109 expressing different nitrilase variants. The reaction mixtures contained in 1 ml 100 mM Na/K-phosphate buffer (pH 6.0) and resting cells of the recombinant E. coli cells (OD600nm = 0.01–0.1). The reactions were started by the addition of different concentrations of rac-PGN (0.5–10 mM; from 10–100 mM methanolic stock solutions). The reaction mixtures were shaken with 750 rpm at 23°C and the reactions started by the addition of the substrate. Six aliquots (90 µl each) were taken and the reactions terminated by the addition of 10 µl of 1 M HCl and subsequent centrifugation (2 min, 14,000 rpm). The concentrations of (R)-PGN and (S)-PGN in the supernatants were determined by chiral HPLC and the enzyme activities for the conversion of (R)-PGN (▲) and (S)-PGN (∆) calculated. The results from three independent reactions are shown.
It was previously reported that under our standard conditions the wild-type enzyme formed from rac-PGN about 8% PGAA with a slight preference for the (S)-enantiomer (42% ee). The variant Trp188Lys converted rac-PGN to more than 95% of PGAA (and only 5% PGA) with a pronounced preference for (S)-PGAA (93% ee) (Eppinger and Stolz, 2019).
In the present study the conversion of different concentrations of rac-PGN by the Trp188Lys variant was studied (Figure 3) and it was found that (S)-PGN was the preferred substrate compared to (R)-PGN in respect of the calculated vmax-values (1.4 ± 0.2 vs. 0.6 ± 0.2 U/mg protein) and also Km-values (0.8 ± 0.3 vs. 3.9 ± 1.7 mM).
The analysis of the large set of variants generated in the present study showed that almost all variants had the same enantiopreference for the formation of PGA and PGAA and those variants which preferentially formed (R)-PGA also formed preferentially (R)-PGAA. In addition, all variants which formed from rac-PGN more PGAA than PGA synthesized preferentially the (S)-enantiomers (Figure 2). Therefore, it was tested if the combination of a mutation which resulted in the preferred synthesis of the (R)-enantiomer (e.g., Ala165Phe) with mutations that result in a “high amide phenotype” (e.g., Trp188X) could result in variants with the ability to form in excess (R)-PGAA. Therefore, the double mutants Ala165Phe/Trp188Gly, Ala165Phe/Trp188Lys and Ala165Arg/Trp188Gly were constructed. This demonstrated only a partial additivity of the two phenotypic traits. Thus, for the double mutant Ala165Phe/Trp188Gly the preferred formation of PGAA (57% of the totally formed products) with an ee for (R)-PGAA of 63% was found. The other variants demonstrated lower degrees of amide formation and also of (R)-selectivity.
In the classical Strecker synthesis equimolar amounts of an aldehyde and KCN are incubated under alkaline conditions in water with ammonia ions in excess (Becker, 1986). In the present study, it was attempted to work with synthetically relevant substrate concentrations but to minimize the dangerous effects of cyanide. Therefore, 100 mM KCN and 1 M NH4Cl were incubated in rather small volumes (≤ 0.5 ml) with an equimolar amount of benzaldehyde (corresponding to the added KCN). Benzaldehyde does not completely dissolve in water under these conditions [6.95 g/l (∼66 mM) of benzaldehyde dissolve in water at 25°C; Yalkowsky et al., 2010].
In order to test if an efficient Strecker synthesis is possible under the “biocompatible” reaction conditions previously defined (pH ≤ 10.8; Eppinger and Stolz 2019), benzaldehyde (corresponding to a concentration of 100 mM), KCN (100 mM), and NH4Cl (1 M) were added to aqueous Na-carbonate buffers with different pH-values (pH 8.5–10.5). The reaction mixtures were vigorously stirred and aliquots (10 μl each) taken at different time points, diluted with 90 μl of a mixture of methanol/water/HCl (in order to completely dissolve the residual benzaldehyde) and analyzed by HPLC. This demonstrated that under all tested conditions in addition to PGN also mandelonitrile was formed (Figure 4 and Figure 5). At all tested pH-values, initially a large surplus of mandelonitrile was found and only a significantly delayed formation of PGN. It took ≥90 min until the initially formed mandelonitrile was completely converted. This suggested that the (initial) formation of mandelonitrile is kinetically controlled while the formation of the PGN is thermodynamically controlled. A similar observation has been previously made for the synthesis of aminoacetonitrile in an aqueous system containing formaldehyde, hydrogen cyanide, and ammonia (Moutou et al., 1995).
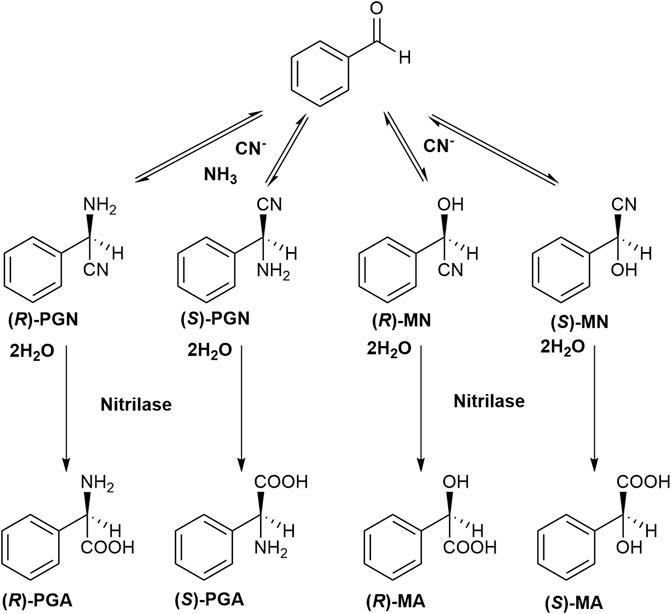
FIGURE 4. Formation of phenylglycinonitrile and mandelonitrile from benzaldehyde and cyanide (± ammonia) under the conditions of the Strecker synthesis and subsequent hydrolysis of the nitriles by a nitrilase. PGN phenylglycinonitrile, PGA phenylglycine, MN mandelonitrile, MA mandelic acid.
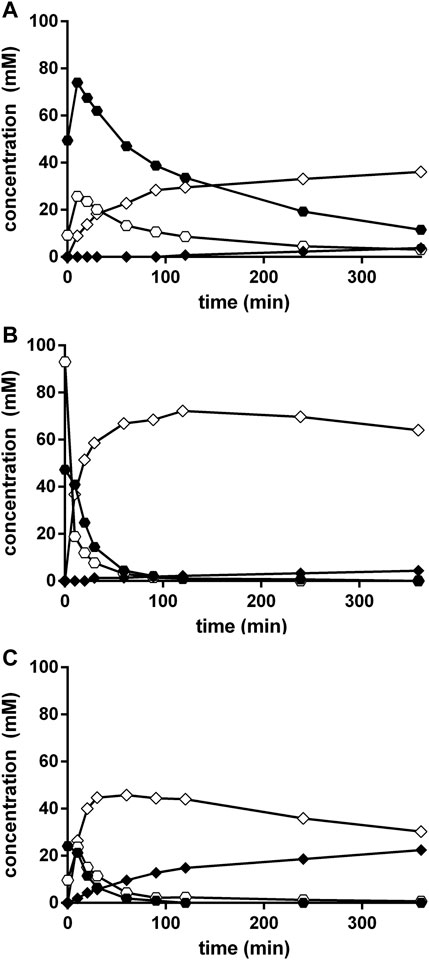
FIGURE 5. (Chemical) conversion of benzaldehyde, ammonia, and cyanide in Na-carbonate-buffer at different pH-values. The reaction mixtures (300 μl) contained at 23°C in 2 ml glass vessels 100 mM benzaldehyde, 100 mM KCN, and 1 M NH4Cl in 100 mM Na2CO3/NaHCO3-buffer at pH 8.5 (A), pH 9.7 (B), or pH 10.5 (C). The liquid volumes were mixed with small stirring bars at about 750 rpm. After different time intervals aliquots (10 μl each) were taken and the reactions stopped by the addition of 90 μl of a mixture of methanol/water/1 M HCl (5:3:2 v/v/v). The concentrations of benzaldehyde (○), mandelonitrile (•), phenylglycinonitrile (♢), and phenylglycinamide (♦) were determined by HPLC.
The comparison of the reactions conducted at different pH-values demonstrated that the concentrations of the initially formed mandelonitrile decreased with increasing pH-values (Figure 5). Furthermore, at pH 10.5 significant amounts of PGAA were formed (Figure 5C). The highest concentration of PGN was detected at pH 9.7 (Figure 5B).
In the following, the influence of the temperature and pH on the formation of PGN and PGAA were analyzed in greater detail and it was found that at all tested pH-values a temperature rise resulted in increased rates of PGN formation. Furthermore, at all tested temperatures, the highest rates of PGN formation were found at pH-values of 9.5–10 (Figure 6A). This was due to the formation of significant amounts of PGAA at pH-values > pH 9.5 (Figure 6B).
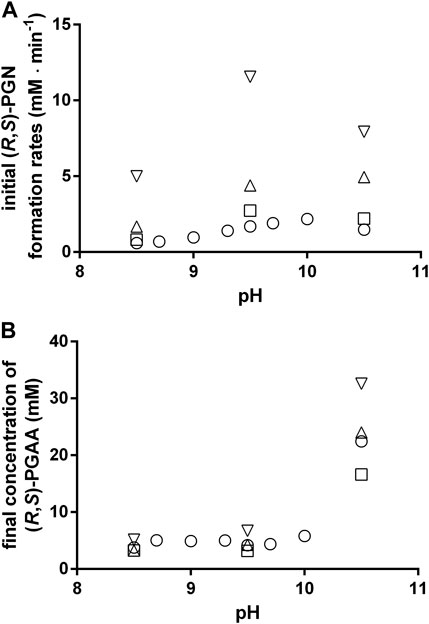
FIGURE 6. Chemical formation of phenylglycinonitrile (A) and phenylglycine amide (B) at different pH-values and different temperatures. The reactions were performed as described in the legend of Figure 2. The reaction mixtures contained at the indicated pH-values 100 mM benzaldehyde, 100 mM KCN, and 1 M NH4Cl in 100 mM Na2CO3/NaHCO3-buffers. The experiments were performed at the following temperatures: 23°C (○), 30°C (□), 40°C (△) and 50°C (▽). (A) The formation rates of PGN were calculated from the increase in the PGN concentration (measured by HPLC) at the beginning of the experiments. (B) The concentrations of the formed PGAA after the complete conversion of the added benzaldehyde were determined by HPLC.
The observed formation of mandelonitrile as by-product complicated the intended biotransformation reaction as mandelonitrile is also a substrate for the nitrilase from P. fluorescens EBC191 (Kiziak et al., 2005). This would result in the aspired system in the formation of a mixture of PGA and mandelic acid. Therefore, different buffer systems were tested in order to try to suppress the formation of mandelonitrile and/or PGAA. Initially, the NH4Cl in the buffer system described above was replaced by equimolar amounts of NH4OH. This resulted at pH 9.5 approximately in the doubling of the formation of PGAA. If the Na-carbonate/NH4Cl-system was replaced by (NH4)2CO3 (500 mM, pH 9.5), significant amounts of 5-phenylhydantoin were formed. (The formation of hydantoins from aldehydes and KCN in the presence of (NH4)2CO3 has been previously described; Meusel and Gütschow, 2004; Monteiro et al., 2016).
Subsequently, a reaction mixture was tested which contained 100 mM benzaldehyde and 100 mM KCN in a buffer consisting of 0.5 M NH4-acetate plus 0.45 M NH4OH which showed a pH-value of about pH 9.5. This system showed a more pronounced buffering capacity than the previously tested ones. Although the formation of PGN was at 30°C slightly slower compared to the Na-carbonate buffer with the same pH, less PGAA (and no detectable 5-phenylhydantoin) was formed (Table 1).
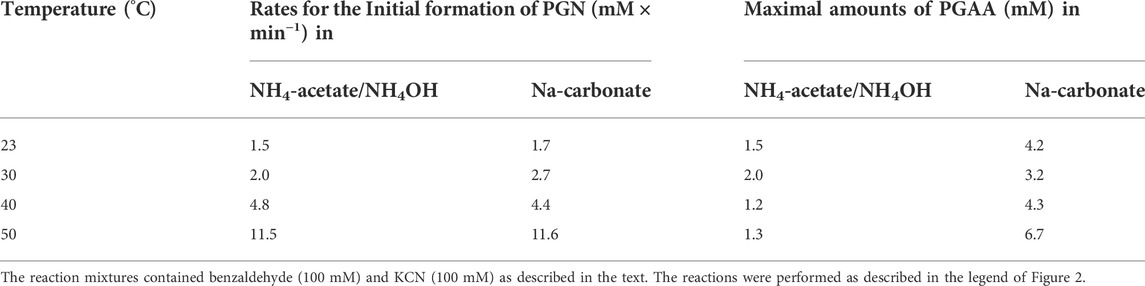
TABLE 1. Rates for the formation of PGN and amounts of PGAA formed in different buffer systems (at pH 9.5) and at different temperatures.
The effect of an increased ratio of KCN to benzaldehyde was tested in order to increase the yield of PGN. Thus, KCN and benzaldehyde were added to the 0.5 M NH4-acetate plus 0.45 M NH4OH-buffer (pH 9.5) in ratios of 1:1, 1.5:1, and 3:1. Furthermore, at each time-point always a complete reaction mixture was worked up in order to minimize the analytical problems related to the low solubility of benzaldehyde (see above). Although the increase in the relative proportion of KCN resulted initially in an increased formation of mandelonitrile, in the reaction mixtures which contained KCN:benzaldehyde in a ratio of 3:1 after 90 min almost stoichiometrical amounts of PGN were formed from benzaldehyde (Figure 7).
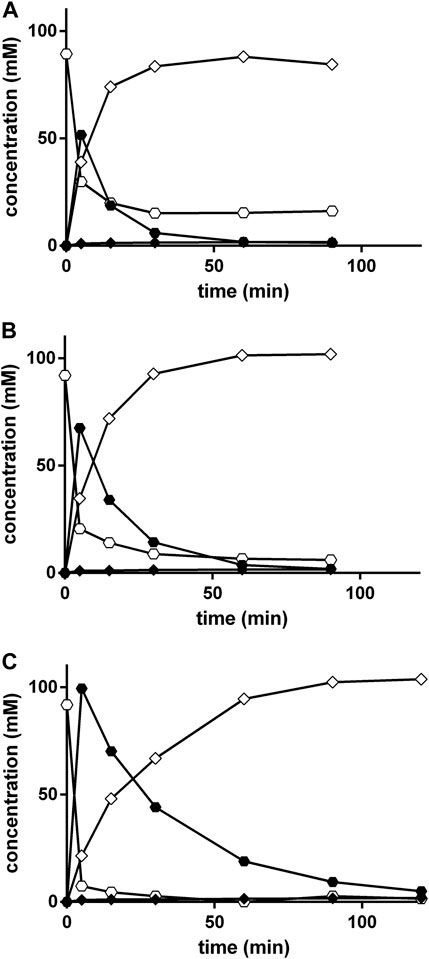
FIGURE 7. Strecker synthesis in NH4-acetate/NH4OH-buffer at pH 9.5 using different KCN to benzaldehyde ratios. The reaction mixtures (300 μl) contained at 40°C in 3 ml glass vessels 100 mM benzaldehyde and varying KCN concentrations [(A), 100 mM, (B) 150 mM and (C), 300 mM] in 0.5 M NH4-acetate/NH4OH-buffer (about 0.95 M of ammonia-ions) at pH 9.5. The liquid volumes were mixed with small stirring bars at about 750 rpm. A separate experiment was performed for each time point. At the indicated times, the reactions were stopped in the individual vessels by the addition of 2.7 ml of a mixture of methanol/water/1 M HCl (5:3:2 v/v/v) and the mixtures were stirred at room temperature with 750 rpm for further 30 min. The concentrations of benzaldehyde (○), mandelonitrile (•), phenylglycinonitrile (♢), and phenylglycine amide (♦) were determined by HPLC.
The aspired dynamic kinetic resolution required the racemization of the PGN formed in the course of the “biocompatible Strecker reaction” and the simultaneous enzymatic turn-over of one of the enantiomers of PGN. Furthermore, the racemization has to be significantly faster than the conversion of the nitrile in order to obtain high enantiomeric excesses (Faber, 2018).
Previously, it was shown that (S)-PGN racemized in Na-carbonate buffer at pH-values ≥ 10 with significant rates (Eppinger and Stolz, 2019). In order to obtain a better coordination of the Strecker synthesis, chemical racemization and the enzymatic reaction, the racemization of (S)-PGN in the NH4-acetate/NH4OH-buffer was analyzed and compared with the racemization reaction in the Na-carbonate buffer. Therefore, a solution of (S)-PGN was prepared as described previously (Eppinger and Stolz, 2019) and incubated in the NH4-acetate/NH4OH-buffer at pH 9.5, 10.0, or 10.5. These experiments demonstrated that also in the NH4-acetate/NH4OH-buffer increased pH-values resulted in significantly increased racemization rates (Figure 8). A comparison of the racemization rates suggested that the racemization occurred at pH 9.5 with similar rates in both buffer systems.
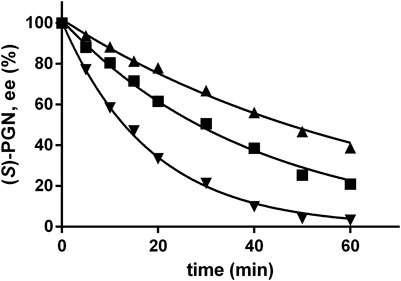
FIGURE 8. Spontaneous racemization of phenylglycinonitrile in NH4-acetate/NH4OH-buffer at different pH-values. The reaction mixtures (100 μl) contained at 40°C in 2 ml glass vessels about 1 mM of (S)-PGN in 500 mM NH4-acetate/NH4OH-buffer with the indicated pH-values. The liquid volumes were mixed with small stirring bars at about 750 rpm. After different time intervals aliquots (2 μl each) were taken and the reactions stopped by the addition of 18 µl of a mixture of methanol/water/1 M HCl (5:3:2 v/v/v). The concentrations of (R)- and (S)-PGN were determined by chiral HPLC and the ee-value of the remaining (S)-PGN at pH 9,5 (▲), pH 10 (■), and 10.5 (▼) calculated.
The racemization of (S)-PGN was accompanied by a significant decrease in the total concentration of (R)- plus (S)-PGN. This was presumably related to the intermediate formation of the corresponding imine in the course of the racemization reaction, as imines easily hydrolyse in aqueous solutions to the corresponding oxo-compounds (Figure 9). In order to minimize this unwanted side reaction, the decomposition of (S)-PGN was analyzed in both buffer systems at pH 9.5, 10, or 10.5 and 23, 30, 40, or 50°C. These experiments demonstrated that the decomposition rates were strongly influenced by the temperature (and that the tested pH-values had only a weak effect). Thus, at 40°C a half-live of rac-PGN of about 70 min and at 23°C of approximately 470 min was found.
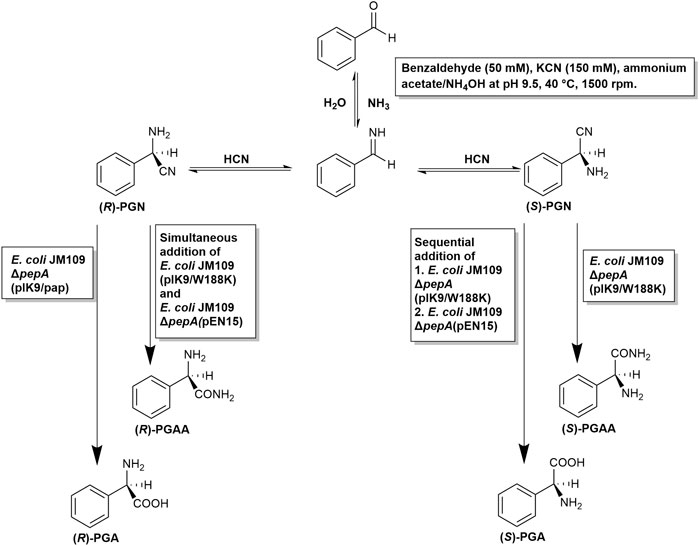
FIGURE 9. Developed chemoenzymatic synthesis of enantiomerically enriched (R)- and (S)-phenylglycine and (R)-and (S)-phenylglycine amide from benzaldehyde, cyanide, and ammonia.
For α-amino acid esters increased racemization rates in the presence of aromatic aldehydes via the formation of the corresponding Schiff bases have been described (Schichl et al., 2008). Mechanistically, this type of reaction is also possible for aminonitriles. Therefore, the racemization of (S)-PGN (1 mM) was also tested at pH 9.5 (500 mM NH4-acetate/NH4OH-buffer) in the presence of salicylaldehyde, 5-chloro-, 5-nitro-, 3,5-dichloro- or 3,5-dinitrosalicylaldehyde (10 mol% each) at 23 and 40°C, but no significant effect was found.
For the biotransformation reactions E. coli JM109ΔpepA (pIK9/pap) was used. The host strain E. coli JM109ΔpepA carries a mutation in the gene coding for the aminopeptidase PepA. The usage of this host strain facilitated the analysis of the reactions as PepA is able to hydrolyse (S)-PGAA which is a potential side product of the nitrilase reaction (Eppinger and Stolz, 2019). Plasmid pIK9/pap codes for a variant of the nitrilase from P. fluorescens EBC191 which converted other substituted phenylacetonitriles with a high reaction- and pronounced (R)-specificity to the corresponding acids (Kiziak and Stolz, 2009).
Initially, it was tested if nitrilase-active resting cells of E. coli JM109ΔpepA (pIK9/pap) were able to convert rac-PGN under the optimized conditions of the Strecker synthesis. Thus it was found that at about 30% substrate conversion (R)-PGA was formed with an ee-value of ≥ 95% (Figure 10).
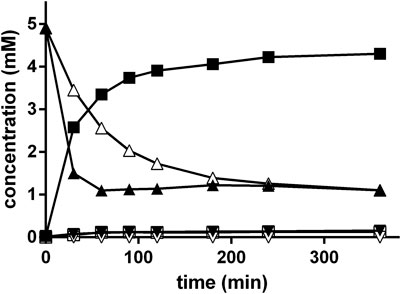
FIGURE 10. Conversion of rac-PGN by resting cells of E. coli JM109ΔpepA (pIK9/pap) under the conditions of the Strecker synthesis. The reaction mixture contained at 40°C in a final volume of 1 ml in a 2 ml glass vessel resting cells of E. coli JM109ΔpepA (pIK9/pap) (OD600nm = 0.5) in 500 mM NH4-acetate/NH4OH buffer (pH 9.5). The reaction was started by the addition of 0.1 ml of 100 mM rac-PGN (in methanol). The reaction was mixed with a small stirring bar at about 1,500 rpm. At the indicated time intervals samples (10 µl each) were taken and diluted with 90 µl of methanol/water/1 M HCl (5:3:2 v/v/v). The cell debris was removed by centrifugation (20,000 g, 2 min). The concentrations of (R)-phenylglycinonitrile (▲), (S)-phenylglycinonitrile (Δ), (R)-phenylglycine (■), (S)-phenylglycine (□), (R)-phenylglycine amide (▼) and (S)-phenylglycine amide (▽) were determined by chiral HPLC.
The experiments described above in order to establish a “biocompatible Strecker synthesis” always resulted in the initial formation of significant amounts of mandelonitrile. It was therefore decided to temporally resolve the chemical formation of the aminonitrile from the following biotransformation. Thus, the chemosynthesis was performed in a NH4-acetate/NH4OH-buffer (pH 9.5) with a mixture of 100 mM benzaldehyde, 300 mM KCN and (in total) 1 M ammonium. The reaction mixture was stirred (1,500 rpm) at 40°C and the reaction monitored by HPLC. After 2 h, the initially formed mandelonitrile was completely converted and the benzaldehyde almost quantitatively converted to rac-PGN. Then, resting cells of E. coli JM109ΔpepA (pIK9/pap) were added and the reaction mixtures further stirred. Aliquots were taken at different time intervals and the supernatants analyzed by chiral and achiral HPLC. The reaction analysis demonstrated that indeed almost exclusively (R)-PGA was formed with an ee-value of 94% and a yield of 67% (in correlation to the initial concentration of benzaldehyde) (Figure 11).
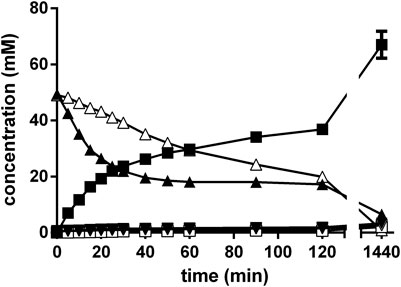
FIGURE 11. Chemoenzymatic synthesis of (R)-PGA using a combination of a chemical Strecker synthesis and hydrolysis of the formed PGN with E. coli JM109ΔpepA (pIK9/pap). The reaction mixture (1 ml) contained in 2 ml glass vessels 100 mM benzaldehyde and 300 mM KCN in 500 mM NH4-acetate/NH4OH (pH 9.5). It was mixed at 40°C with a small stirring bars at about 1,500 rpm. The Strecker synthesis was started by the addition of benzaldehyde. After the completion of the chemical step (120 min), the biotransformation reaction was initiated by the addition of the cell suspension (55 μl, resulting in an OD600nm = 5 in the experiment). Aliquots (10 μl each) were removed after different times and the reactions stopped by adding 90 μl of methanol/water/1 M HCl (5:3:2 v/v/v). The cells were removed by centrifugation (20,000 g, 2 min at room temperature) and the concentrations of (R)-phenylglycinonitrile (▲), (S)-phenylglycinonitrile (Δ), (R)-phenylglycine (■), (S)-phenylglycine (□), (R)-phenylglycine amide (▼) and (S)-phenylglycine amide (▽) determined by chiral HPLC.
It was already stated above that benzaldehyde did not completely dissolve in water under the conditions applied. Therefore, in the following it was analyzed if the relative yields of (R)-PGA obtained could be increased by using a decreased initial concentration of benzaldehyde (50 mM) or adding different water-miscible or water-immiscible organic solvents or a water immiscible ionic liquid. Furthermore, the biocatalysts were repeatedly added to the reaction mixtures. In all these experiments (R)-PGA was formed with a high degree of enantioselectivity. Furthermore, it became evident that the decrease in the starting concentration of benzaldehyde and the repeated addition of the biocatalyst indeed resulted in an increased relative yield of (R)-PGA, but that the addition of the organic solvents did not show any significant effects (Table 2). The average yields of (R)-PGA (in relationship to the initially added benzaldehyde) in the crude reaction mixtures were about 80%. This clearly demonstrated that indeed a dynamic kinetic resolution occurred. Furthermore, only ≤ 10% of the benzaldehyde used were converted to mandelic acid and mandelic acid amide (MAA) (Table 2).
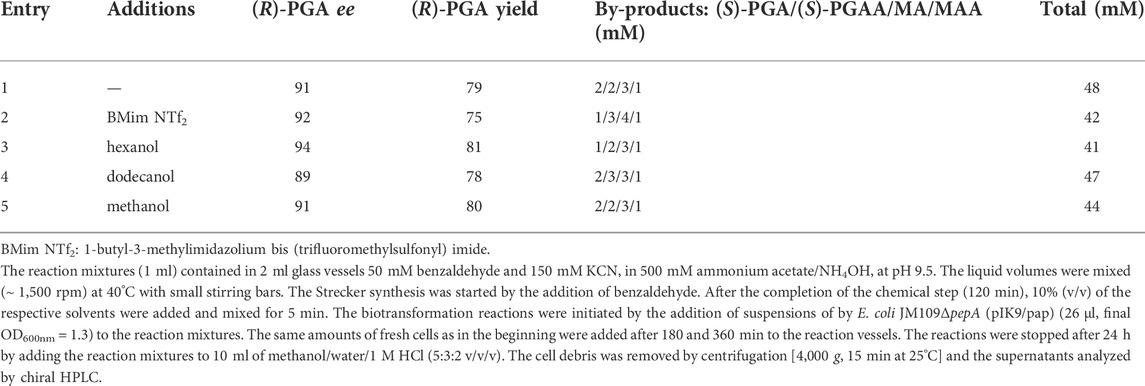
TABLE 2. Chemoenzymatic enantioselective synthesis of (R)-phenylglycine from benzaldehyde, ammonia and KCN by a combination of a Strecker synthesis and a biotransformation using E. coli JM109ΔpepA (pIK9/pap).
Variants of the nitrilase from P. fluorescens EBC191 which harbor mutations in Trp188 show an increased tendency to form amides from α-substituted nitriles (Sosedov and Stolz, 2015; Brunner et al., 2018). It was previously shown that E. coli JM109 (pIK9/Trp188Lys) converted racemic PGN to more than 95% PGAA with a large surplus of (S)-PGAA (93% ee) (Eppinger and Stolz, 2019). For the synthesis of (S)-PGAA, first PGN was prepared as described above by performing the Strecker synthesis using benzaldehyde, cyanide, and ammonia in a ratio of 1:3:20. Subsequently, resting cells of E. coli JM109ΔpepA (pIK9/Trp188Lys) and some further evolved amide producing nitrilase variants were added and the reactions analyzed by HPLC (Table 3). The recombinant cells of E. coli JM109ΔpepA (pIK9/Trp188Lys) converted 68% of the initially added benzaldehyde mainly to (S)-PGAA with an ee-value of about 80%. In addition, also small amounts of (S)-PGA, (R)-PGAA and mandelic acid amide were formed (Table 3, entry 2). The utilization of the other amide forming nitrilase variants did not result in significant improvements (Table 3, entries 3 + 4).
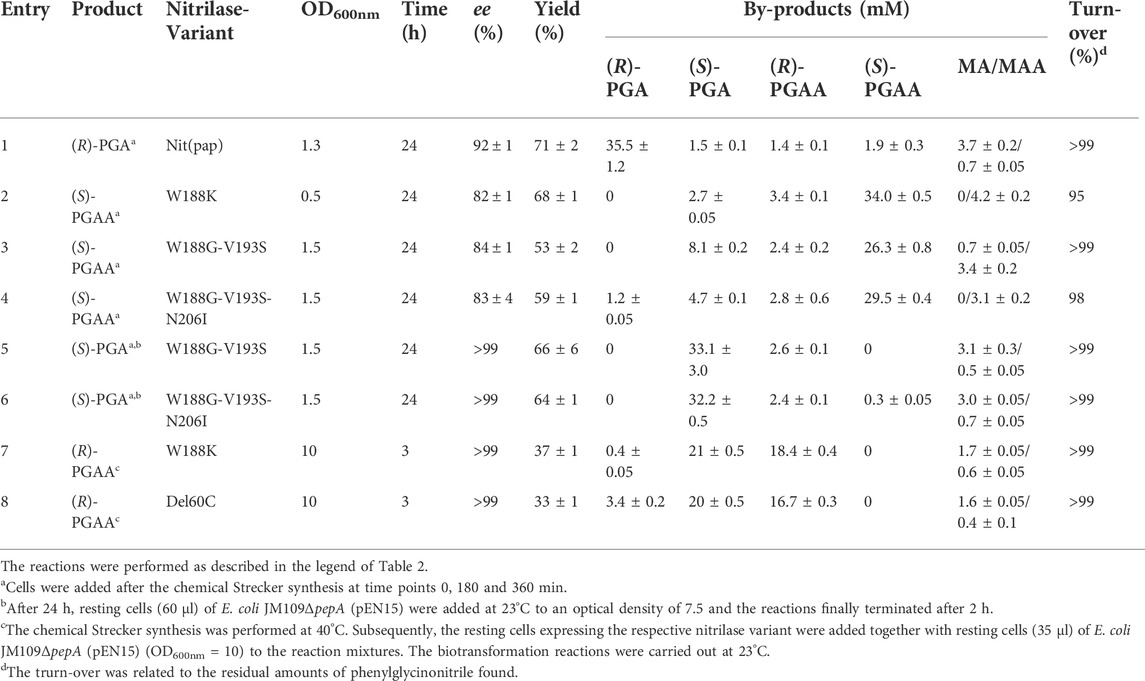
TABLE 3. Enantioselective synthesis of (R)- and (S)-phenylglycine and (R)- and (S)-phenylglycinamide from benzaldehyde, KCN and ammonia by using different variants of the nitrilase from P. fluorescens EBC191.
The change of the host strain also allowed the synthesis of the other enantiomers of PGA and PGAA. Thus, the synthesis of (S)-PGA could be accomplished by the combination of an amide forming nitrilase variant with an aminopeptidase positive E. coli host strain, e.g., the wild-type strain of E. coli JM109 or the complemented strain E. coli JM109ΔpepA (pEN15) (Eppinger and Stolz, 2019). In this construct, the nitrilase variant forms preferentially (S)-PGAA and the (S)-PGAA is converted to (S)-PGA by the highly enantioselective aminopeptidase PepA (Eppinger and Stolz 2019). The combination of the (S)-specific amide-forming nitrilase variants with the aminopeptidase allowed the formation of (S)-PGA with a very high enantiomeric excess (Table 3, entries 5 + 6).
The simultaneous expression of a preferentially amide forming nitrilase variant (with a low degree of enantioselectivity) together with an aminopeptidase activity allowed also the preferred formation of (R)-PGAA, as the highly enantioselective aminopeptidase will convert the (S)-PGAA formed by the nitrilase variant almost completely to (S)-PGA and thus leaves the (R)-PGAA. This concept was experimentally verified by converting the product of the Strecker synthesis to (R)-PGAA with a high enantiomeric excess (Table 3, entries 7 + 8).
The comparative analysis of the large set of muteins performed in the course of the present study (Figure 2) showed that mutations at several positions could influence the composition of the products formed from rac-PGN. Thus, an increased enantioselectivity for the formation of (R)-PGA was found for the variants Trp59Arg, Thr134Ala, and Ala165Phe. The involvement of these amino acid residues in the enantioselectivity of the nitrilase from P. fluorescens EBC191 agrees well with a recent study about the conversion of 2-methoxymandelonitrile by a nitrilase from Burkholderia cenocepacia BCJ2315 (Scott et al., 2022). In this study, docking models were presented which suggested that Trp59, Thr134, and Trp165 (analogous numbering of the homologous amino acid residues in both nitrilases) are involved in the enantiodiscrimination of the enzyme bound substrate enantiomers. The comparison of the data set also demonstrated that those variants which formed an increased proportion of PGAA from PGN in general also converted mandelonitrile to larger amounts of mandeloamide (Kiziak et al., 2007; Kiziak and Stolz, 2009; Sosedov and Stolz, 2015).
There are also several possible applications for the amide forming enzyme variants. Thus, for the synthesis of semisynthetic β-lactams it is necessary to activate the phenylglycine(s) prior to the coupling with the amino-groups of the β-lactam nuclei. This can be achieved by the formation of the respective acid chlorides, esters, or amides. Therefore, also (R)-PGAA is a very interesting target molecule as side-chain donor for the synthesis of semisynthetic β-lactam antibiotics (Wegman et al., 2001a). The synthesis of (R)-PGAA from rac-PGN by different Rhodococcus strains harbouring nitrile hydratase/amidase systems has been described and it was repeatedly found that the rac-PGN was hydrated by rather unspecific nitrile hydratases to rac-PGAA, which was subsequently converted by the action of highly specific amidases to (R)-PGAA and (S)-PGA (Wegman et al., 2000, 2001a; Hensel et al., 2002; Ewert et al., 2008). In the present manuscript it was shown that basically the same products can be obtained by the coupling of an amide forming nitrilase variant with an aminopeptidase activity intrinsically present in E. coli strains. This might be very beneficial under the conditions of the Strecker synthesis as many nitrile hydratases are rather sensitive against elevated cyanide concentrations (Gerasimova et al., 2004; van Pelt et al., 2009). Furthermore, the preferred formation of amides with an (S)-configuration by the “Trp188X-variants” also allows (e.g., in combination with an amidase) the access to (S)-acids.
The present study demonstrated that it is in principle possible to directly combine the Strecker synthesis with the biocatalytical hydrolysis or hydration of the aminonitrile formed as product of the Strecker reaction. Nevertheless, several problems have been identified which could hinder a dynamic kinetic resolution of the aminonitrile and thus the quantitative synthesis of enantiopure amino acids. Thus, in order to proceed with a sufficient velocity the racemization reaction required in all tested buffer systems pH-values ≥ 9.5. Although in the present study it was shown that resting cells of E. coli JM109 could be used at a pH of 9.5, this pH-value is clearly critical for E. coli strains. Thus, some derivatives of E. coli lost their viability at pH 9.0 (Zilberstein et al., 1980, 1984). On the other hand, for the wild-type strain E. coli FRAG1 it was shown that it was able to keep its internal pH sufficiently low at an external pH of 9.0 (Slonczewski et al., 1981) and E. coli W3110 and E. coli ETEC even grow at pH 9.5 (Vanhauteghem et al., 2013; Kumar and Doerrler, 2015). This indicated that biotransformations with E. coli at a pH of 9.5 should be feasible even for longer reaction times but might require a rather strict pH-control.
The major problem of the studied concept is caused by the Strecker reaction itself which initially results in the rapid formation of the corresponding hydroxynitrile which is only slowly replaced by the thermodynamically more stable aminonitrile. This temporal succession of the formed products was largely independent from the applied buffer system and has also been observed during the Strecker synthesis of other aminonitriles (Moutou et al., 1995) and thus might be inherent to the Strecker synthesis. This problem might be tackled by a timely separation of the Strecker synthesis and the biotransformation of the aminonitrile (as done in the present study). Unfortunately, this prevents a direct productive coupling of the Strecker synthesis with the hydrolysis of the aminonitriles as in the course of the ongoing chemical reaction the hydroxynitrile and the aminonitrile are always simultaneously present. Furthermore, it is also generally assumed that the racemization of the α-aminonitriles under alkaline conditions occurs via the corresponding α-hydroxynitriles. The simultaneous presence of the α-amino- and α-hydroxynitriles during the Strecker synthesis and the racemization of the α-aminontriles is highly relevant as the nitrilase from P. fluorescens (and presumably also other arylacetonitrilases) convert both α-aminontriles and α-hydroxynitriles. A possible solution for this problem might be the screening or selection for nitrilases or nitrilase variants which only convert α-aminontriles but not α-hydroxynitriles.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
EE and AS conceived and designed the research. EE And JADG conducted experiments. EE, JADG, and AS analyzed data and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al Toma, R. S., Brieke, C., Cryle, M. J., and Süssmuth, R. D. (2015). Structural aspects of phenylglycines, their biosynthesis and occurrence in peptide natural products. Nat. Prod. Rep. 32, 1207–1235. doi:10.1039/c5np00025d
Almhjell, P. J., Boville, C. E., and Arnold, F. H. (2018). Engineering enzymes for noncanonical amino acid synthesis. Chem. Soc. Rev. 47, 8980–8997. doi:10.1039/c8cs00665b
Bommarius, A. S., Drauz, K., Groeger, U., and Wandrey, C. (1992). “Membrane bioreactors for the production of enantiomerically pure α-amino acids,” in Chirality in industry. Editors A. N. Collins, G. N. Sheldrake, and J. Crosby (Chichester, England: John Wiley & Sons).
Bommarius, A. S., Schwarm, M., and Drauz, K. (2001). Comparison of different chemoenzymatic process routes to enantiomerically pure amino acids. Chimia 55, 50–59.
Brunner, S., Eppinger, E., Fischer, S., Gröning, J., and Stolz, A. (2018). Conversion of aliphatic nitriles by the arylacetonitrilase from Pseudomonas fluorescens EBC191. World J. Microbiol. Biotechnol. 34, 91. doi:10.1007/s11274-018-2477-9
Chaplin, J., Levin, M. D., Morgan, B., Farid, N., Li, J., Zhu, Z., et al. (2004). Chemoenzymatic approaches to the dynamic kinetic asymmetric synthesis of aromatic amino acids. Tetrahedron Asymmetry 15, 2793–2796. doi:10.1016/j.tetasy.2004.07.060
Corti, V., Riccioli, R., Martinelli, A., Sandri, S., Fochi, M., Bernardi, L., et al. (2021). Stereodivergent entry to β-branched β-trifluoromethyl α-amino acid derivatives by sequential catalytic asymmetric reactions. Chem. Sci. 12, 10233–10241. doi:10.1039/d1sc01442k
Dennig, A., Blaschke, F., Gandomkar, S., Tassano, E., and Nidetzky, B. (2019). Preparative asymmetric synthesis of canonical and non-canonical α-amino acids through formalenantioselective biocatalytic amination of carboxylic acids. Adv. Synth. Catal. 361, 1348–1358. doi:10.1002/adsc.201801377
Eppinger, E., and Stolz, A. (2019). Conversion of phenylglycinonitrile by recombinant Escherichia coli cells synthesizing variants of the arylacetonitrilase from Pseudomonas fluorescens EBC191. Appl. Microbiol. Biotechnol. 103, 6737–6746. doi:10.1007/s00253-019-09957-y
Ewert, C., Lutz-Wahl, S., and Fischer, L. (2008). Enantioselective conversion of α-arylnitriles by Klebsiella oxytoca. Tetrahedron Asymmetry 19, 2573–2578. doi:10.1016/j.tetasy.2008.11.014
Gerasimova, T., Novikov, A., Osswald, S., and Yanenko, A. (2004). Screening, characterization and application of cyanide-resistant nitrile hydratases. Eng. Life Sci. 4, 543–546. doi:10.1002/elsc.200402160
Gokhale, D. V., Bastawde, K. B., Patil, S. G., Kalkote, U. R., Joshi, R. R., Joshi, R. A., et al. (1996). Chemoenzymatic synthesis of (D)-phenylglycine using hydantoinase of Pseudomonas desmolyticum resting cells. Enzyme Microb. Technol. 18, 353–357. doi:10.1016/0141-0229(95)00127-1
Gröger, H. (2003). Catalytic enantioselective Strecker reactions and analogous syntheses. Chem. Rev. 103, 2795–2828. doi:10.1021/cr020038p
Grundmann, P., and Fessner, W. D. (2008). One-pot, regioselective synthesis of substituted arylglycines for kinetic resolution by penicillin G acylase. Adv. Synth. Catal. 350, 1729–1735. doi:10.1002/adsc.200800203
Han, J., Lyutenko, N. V., Sorochinsky, A. E., Okawara, A., Konno, H., White, S., et al. (2021). Tailor-made amino acids in pharmaceutical industry: Synthetic approaches to aza-tryptophan derivatives. Chem. A Eur. J. 27, 17510–17528. doi:10.1002/chem.202102485
Hensel, M., Lutz-Wahl, S., and Fischer, L. (2002). Stereoselective hydration of (R, S)-phenylglycine nitrile by new whole cell biocatalysts. Tetrahedron Asymmetry 13, 2629–2633. doi:10.1016/s0957-4166(02)00751-6
Kalnik, M., Gabko, P., Bella, M., and Koóš, M. (2021). The Bucherer-Bergs multicomponent synthesis of hydantoins-excellence in simplicity. Molecules 26, 4024. doi:10.3390/molecules26134024
Karmali, A., Pacheco, R., Tata, R., and Brown, P. (2001). Substitutions of Thr-103-Ile and Trp-138-Gly in amidase from Pseudomonas aeruginosa are responsible for altered kinetic properties and enzyme instability. Mol. Biotechnol. 17, 201–212. doi:10.1385/mb:17:3:201
Kiziak, C., Conradt, D., Stolz, A., Mattes, R., and Klein, J. (2005). Nitrilase from Pseudomonas fluorescens EBC191: Cloning and heterologous expression of the gene and biochemical characterization of the recombinant enzyme. Microbiology 151, 3639–3648. doi:10.1099/mic.0.28246-0
Kiziak, C., Klein, J., and Stolz, A. (2007). Influence of different carboxyterminal mutations on the substrate-reaction-and enantiospecifity of the arylacetonitrilase from Pseudomonas fluorescens EBC191. Protein Eng. Des. Sel. 20, 385–396. doi:10.1093/protein/gzm032
Kiziak, C., and Stolz, A. (2009). Identification of amino acid residues which are responsible for the enatioselectivity and amide formation capacity of the arylacetonitrilase from Pseudomonas fluorescens EBC191. Appl. Environ. Microbiol. 75, 5592–5599. doi:10.1128/aem.00301-09
Kouznetsov, V. V., and Galvis, C. E. P. (2018). Strecker reaction and α-amino nitriles: Recent advances in their chemistry, synthesis, and biological properties. Tetrahedron 74, 773–810. doi:10.1016/j.tet.2018.01.005
Kumar, S., and Doerrler, W. T. (2015). Escherichia coli YqjA, a member of the conserved DedA/Tvp38 membrane protein family, is a putative osmosensing transporter required for growth at alkaline pH. J. Bacteriol. 197, 2292–2300. doi:10.1128/jb.00175-15
Liu, S. P., Liu, R. X., Mao, J., Zhang, L., Ding, Z. Y., Gu, Z. H., et al. (2016). Structural-based screening of L-phenylglycine aminotransferase using L-phenylalanine as the optimal amino donor: Recycling of L-phenylalanine to produce L-phenylglycine. Biotechnol. Bioprocess Eng. 21, 153–159. doi:10.1007/s12257-015-0714-8
Mareya, T. M., Coady, T. M., O´Reilly, C., Kinsella, M., Coffey, L., Lennon, C. M., et al. (2020). Process optimisation studies and aminonitrile substrate evaluation of Rhodococcus erythropolis SET1, a nitrile hydrolysing bacterium. ChemistryOpen 9, 512–520. doi:10.1002/open.202000088
Mei, Y. Z., He, B. F., and Ouyang, P. K. (2008). Enzymatic production of L-amino acids from the corresponding DL-5-substituted hydantoins by Bacillus fordii MH602. World J. Microbiol. Biotechnol. 24, 375–381. doi:10.1007/s11274-007-9485-5
Meusel, M., and Gütschow, M. (2004). Recent developments in hydantoin chemistry. ChemInform 36, 391–443. doi:10.1002/chin.200509253
Monteiro, J. L., Pieber, B., Corrêa, A. G., and Kappe, C. O. (2016). Continuous synthesis of hydantoins: Intensifying the Bucherer-Berg reaction. Synlett 27, 83–87.
Moosmann, D., Mokeev, V., Kulik, A., Osipenkov, N., Kocadinc, S., Ort-Winklbauer, R., et al. (2020). Genetic engineering approaches for the fermentative production of phenylglycines. Appl. Microbiol. Biotechnol. 104, 3433–3444. doi:10.1007/s00253-020-10447-9
Moutou, G., Taillades, J., Bénefice-Malouet, S., Commeyras, A., Messina, G., Mansani, R., et al. (1995). Equilibrium of α-aminoacetonitrile formation from formaldehyde, hydrogen cyanide and ammonia in aqueous solution: Industrial and prebiotic significance. J. Phys. Org. Chem. 8, 721–730. doi:10.1002/poc.610081105
Müller, U., van Assema, F., Gunsior, M., Orf, S., Kremer, S., Schipper, D., et al. (2006). Metabolic engineering of the E. coli L-phenylalanine pathway for the production of D-phenylglycine (D-Phg). Metab. Eng. 8, 196–208. doi:10.1016/j.ymben.2005.12.001
Nájera, C., and Sansano, J. M. (2007). Catalytic asymmetric synthesis of α-amino acids. Chem. Rev. 107, 4584–4671. doi:10.1021/cr050580o
Olivieri, R., Fascetti, E., Angelini, L., and Degen, L. (1979). Enzymatic conversion of N-carbamoyl-D-amino acids to D-amino acids. Enzyme Microb. Technol. 1, 201–204. doi:10.1016/0141-0229(79)90030-9
Ooi, T., Uematsu, Y., and Maruoka, K. (2006). Asymmetric Strecker reaction of aldimines using aqueous potassium cyanide by phase-transfer catalysis of chiral quaternary ammonium salts with a tetranaphthyl backbone. J. Am. Chem. Soc. 128, 2548–2549. doi:10.1021/ja058066n
Qiu, J., Su, E., Wang, W., and Wei, D. (2014b). High yield synthesis of D-phenylglycine and its derivatives by nitrilase mediated dynamic kinetic resolution in aqueous 1-octanol biphasic system. Tetrahedron Lett. 55, 1448–1451. doi:10.1016/j.tetlet.2014.01.044
Qiu, J., Su, E. Z., Wang, H. L., Cai, W. W., Wang, W., Wei, D. Z., et al. (2014a). Cloning, overexpression, and characterization of a high enantioselective nitrilase from Sphingomonas wittichii RW1 for asymmetric synthesis of (R)-phenylglycine. Appl. Biochem. Biotechnol. 173, 365–377. doi:10.1007/s12010-014-0845-y
Resch, V., Fabian, W. M. F., and Kroutil, W. (2010). Deracemisation of mandelic acid to optically pure non-natural L-phenylglycine via a redox-neutral biocatalytic cascade. Adv. Synth. Catal. 352, 993–997. doi:10.1002/adsc.200900891
Schichl, D. A., Enthaler, S., Holla, W., Riermeier, T., Kragl, U., Beller, M., et al. (2008). Dynamic kinetic resolution of α-amino acid esters in the presence of aldehydes. Eur. J. Org. Chem. 2008, 3506–3512. doi:10.1002/ejoc.200800253
Scott, M. E., Wang, X., Humphreys, L. D., Geier, M. J., Kannan, B., Chan, J., et al. (2022). Enzyme optimization and process development for a scalable synthesis of (R)-2-methoxymandelic acid. Org. Process Res. Dev. 26, 849–858. doi:10.1021/acs.oprd.1c00250
Servi, S., Tessaro, D., and Pedrocchi-Fantoni, G. (2007). Chemo-enzymatic deracemization methods for the preparation of enantiopure non-natural α-amino acids. Coord. Chem. Rev. 252, 715–726. doi:10.1016/j.ccr.2007.09.012
Slonczewski, J. L., Rosen, B. P., Alger, J. R., and Macnab, R. M. (1981). pH homeostasis in Escherichia coli: Measurement by 31P nuclear magnetic resonance of methylphosphonate and phosphate. Proc. Natl. Acad. Sci. U. S. A. 78, 6271–6275. doi:10.1073/pnas.78.10.6271
Sosedov, O. (2013). Steigerung der Nitril-Hydratase-Aktivität der Nitrilase aus Pseudomonas fluorescens EBC191. Stuttgart: University of Stuttgart. [Dissertation].
Sosedov, O., and Stolz, A. (2015). Improvement of the amides forming capacity of the arylacetonitrilase from Pseudomonas fluorescens EBC191 by site-directed mutagenesis. Appl. Microbiol. Biotechnol. 99, 2623–2635. doi:10.1007/s00253-014-6061-4
Tan, X., Zhang, S., Song, W., Liu, J., Gao, C., Chen, X., et al. (2021). A multi-enzyme cascade for efficient production of D-p-hydroxyphenylglycine from L-tyrosine. Bioresour. Bioprocess. 8, 41. doi:10.1186/s40643-021-00394-2
Tang, C. D., Shi, H.-L., Jia, Y.-Y., Li, X., Wang, L.-F., and Xu, J.-H. (2020). High level and enantioselective production of L-phenylglycine from racemic mandelic acid by engineered Escherichia coli using response surface methodology. Enzyme Microb. Technol. 136, 109513. doi:10.1016/j.enzmictec.2020.109513
van Pelt, S., van Rantwijk, F., and Sheldon, R. A. (2009). Synthesis of aliphatic (S)-α-hydroxycarboxylic amides using a one-pot bienzymatic cascade of immobilized oxynitrilase and nitrile hydratase. Adv. Synth. Catal. 351, 397–404. doi:10.1002/adsc.200800625
Vanhauteghem, D., Janssens, G. P. J., Lauwaerts, A., Sys, S., Boyen, F., Cox, E., et al. (2013). Exposure to the proton scavenger glycine under alkaline conditions induces Escherichia coli viability loss. PLoS One 8, e60328. doi:10.1371/journal.pone.0060328
Vedha-Peters, K., Gunawardana, M., Rozzell, J. D., and Novick, S. J. (2006). Creation of a broad-range and highly stereoselective D-amino acid dehydrogenase for the one-step synthesis of D-amino acids. J. Am. Chem. Soc. 128, 10923–10929. doi:10.1021/ja0603960
Walsh, C. T., O'Brien, R. V., and Khosla, C. (2013). Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. 52, 7098–7124. doi:10.1002/anie.201208344
Wang, H., Wang, K., Ren, Y., Li, N., Tang, B., Zhao, G., et al. (2017). Asymmetric Strecker reactions catalyzed by thiourea phosphonium and ammonium salts. Adv. Synth. Catal. 359, 1819–1824. doi:10.1002/adsc.201700029
Wang, M.-X., and Lin, S.-J. (2001). Highly efficient and enantioselective synthesis of L-arylglycines and D-arylglycine amides from biotransformations of nitriles. Tetrahedron Lett. 42, 6925–6927. doi:10.1016/s0040-4039(01)01439-3
Wegman, M. A., Heinemann, U., Stolz, A., van Rantwijk, F., and Sheldon, R. A. (2000). Stereoretentive nitrile hydratase catalysed hydration of D-phenylglycine nitrile. Org. Process Res. Dev. 4, 318–322. doi:10.1021/op000055l
Wegman, M. A., Heinemann, U., van Rantwijk, F., Stolz, A., and Sheldon, R. A. (2001a). Hydrolysis of D, L-phenylglycine nitrile by new bacterial cultures. J. Mol. Catal. B Enzym. 11, 249–253. doi:10.1016/s1381-1177(00)00089-8
Wegman, M. A., Janssen, M. H. A., van Rantwijk, F., and Sheldon, R. A. (2001b). Towards biocatalytic synthesis of β-lactam antibiotics. Adv. Synth. Catal. 343, 559–576. doi:10.1002/1615-4169(200108)343:6/7<559::aid-adsc559>3.0.co;2-z
Williams, R. M., and Hendrix, J. A. (1992). Asymmetric synthesis of arylglycines. Chem. Rev. 92, 889–917. doi:10.1021/cr00013a007
Xue, Y.-P., Cao, C., and Zheng, Y.-G. (2018). Enzymatic asymmetric synthesis of chiral amino acids. Chem. Soc. Rev. 47, 1516–1561. doi:10.1039/c7cs00253j
Yalkowsky, S. H., He, Y., and Jain, P. (2010). Handbook of aqueous solubility data. 2 nd. Boca Raton, Fl: CRC Press, 367.
Zhou, J., Wu, S., and Li, Z. (2017). One-pot enantioselective synthesis of D-phenylglycines from racemic mandelic acids, styrenes, or biobased L-phenylalanine via cascade biocatalysis. Adv. Synth. Catal. 359, 4305–4316. doi:10.1002/adsc.201700956
Zilberstein, D., Agman, V., Schuldiner, S., and Padan, E. (1984). Escherichia coli intracellular pH, membrane potential, and cell growth. J. Bacteriol. 158, 246–252. doi:10.1128/jb.158.1.246-252.1984
Zilberstein, D., Padan, E., and Schuldiner, S. (1980). A single locus in Escherichia coli governs growth in alkaline pH and on carbon sources whose transport is sodium dependent. FEBS Lett. 116, 177–180. doi:10.1016/0014-5793(80)80637-5
Keywords: biotranformations, recombinant biocatalysts, chemoenzymatic synthesis, dynamic kinetic resolution, Strecker synthesis, nitrilase
Citation: Eppinger E, Gröning JAD and Stolz A (2022) Chemoenzymatic enantioselective synthesis of phenylglycine and phenylglycine amide by direct coupling of the Strecker synthesis with a nitrilase reaction. Front. Catal. 2:952944. doi: 10.3389/fctls.2022.952944
Received: 25 May 2022; Accepted: 30 June 2022;
Published: 05 August 2022.
Edited by:
Daniela Remonatto, São Paulo State University, BrazilReviewed by:
Paweł Borowiecki, Warsaw University of Technology, PolandCopyright © 2022 Eppinger, Gröning and Stolz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Stolz, YW5kcmVhcy5zdG9sekBpbWIudW5pLXN0dXR0Z2FydC5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.