 Amanda Silva de Miranda
Amanda Silva de Miranda Cintia D. F. Milagre
Cintia D. F. Milagre Frank Hollmann
Frank Hollmann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Catal., 10 May 2022
Sec. Biocatalysis
Volume 2 - 2022 | https://doi.org/10.3389/fctls.2022.900554
This article is part of the Research TopicReductions and Oxidations Catalyzed by Alcohol Dehydrogenases: Useful Biocatalytic Reactions in Organic SynthesisView all 3 articles
Alcohol dehydrogenases (ADHs) have become important catalysts for stereoselective oxidation and reduction reactions of alcohols, aldehydes and ketones. The aim of this contribution is to provide the reader with a timely update on the state-of-the-art of ADH-catalysis. Mechanistic basics are presented together with practical information about the use of ADHs. Current concepts of ADH engineering and ADH reactions are critically discussed. Finally, this contribution highlights some prominent examples and future-pointing concepts.
Redox reactions constitute a central theme of organic synthesis; particularly, the conversion of alcohols into aldehydes or ketones as well as the reverse reaction (i.e., reduction of carbonyl compounds into alcohols) play an important role also in industrial practice (Sheldon et al., 2002; Tojo and Fernández, 2006; Magano and Dunetz, 2012). Decades of intensive research have yielded a vast portfolio of efficient oxidation- and reduction procedures and –catalysts. Interestingly, chemical catalysts applicable for both, oxidation and reduction reactions are scarce. In contrast, alcohol dehydrogenases (ADHs, sometimes also termed carbonyl reductases, KREDs) are natural redox catalysts capable of catalysing the oxidation of alcohols and the reduction of carbonyl compounds. This versatility together with their often high selectivity makes ADHs powerful catalysts for the redox transformation of alcohols and carbonyl compounds. The past decades have seen enormous research efforts dedicated to the applicability of ADHs in organic synthesis. Earlier issues such as poor availability, narrow substrate scope or low economic attractiveness of ADH-catalyses reactions have been solved and ADHs are increasingly used on industrial scale (Hauer, 2020; Wu S. et al., 2021).
The aim of this contribution is to present the current state-of-the-art of ADHs in organic synthesis. It builds on and extends previous review articles covering various aspects of ADH catalysis (Li et al., 2021c; de Gonzalo and Paul, 2021; Hollmann et al., 2021; Musa et al., 2021; Musa, 2022).
ADHs catalyse the reversible oxidation of alcohols to the corresponding carbonyl products. Being a redox reaction by nature, the oxidation or reduction reaction has to be accompanied by a reduction or oxidation reaction of a stoichiometric cosubstrate. ADHs utilise nicotinamide cofactors for this (Figure 1). The nicotinamide cofactors exist in ribose-phosphorylated [NAD(P)] and ribose-non-phosphorylated (NAD) form; their basic physicochemical properties (e.g., redox potentials) are identical but, frequently, ADHs, depending on their role in the host organism, exhibit high selectivity for one of the two forms. As a rule of thumb, ADHs involved in anabolic pathways generally prefer NADP while metabolically relevant ADHs utilize NAD. As redox-cofactors, NAD(P) obviously exist in an oxidised [NAD(P)+] and a reduced [NAD(P)H] form. In essence, NAD(P)H and NAD(P)+ function as hydride ion donors or–acceptors, respectively.
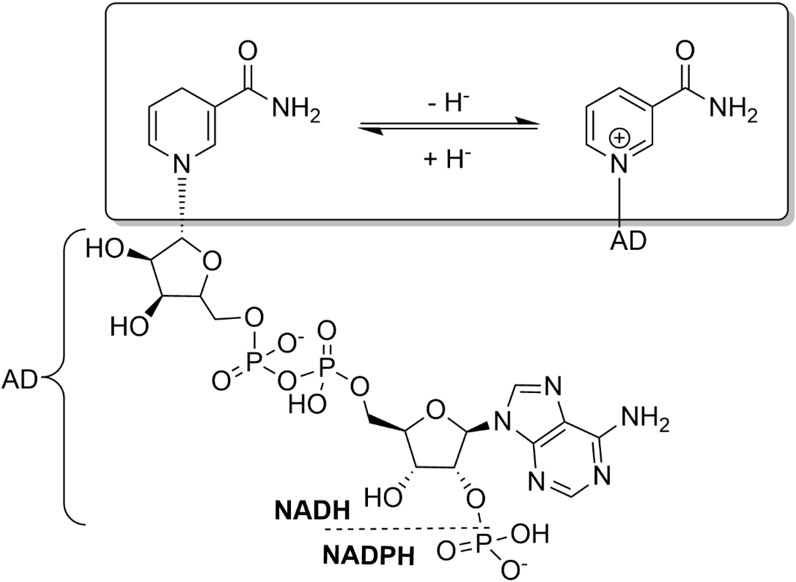
FIGURE 1. Structure and basic redox chemistry of the nicotinamide cofactors. The catalytically relevant nicotinamide moiety is highlighted. The ribose-phosphorylated (NADP) and ribose-non-phosphorylated (NAD) forms differ in their adenine dinucleotide (AD) moiety.
The catalytic mechanism of ADH-catalysed transformations comprises the binding of the starting material (alcohol or carbonyl compound) together with the nicotinamide cofactors to the enzyme active site followed by a hydride transfer between cofactor and substrate (Figure 2). Some ADHs contain a metal ion in their active site (also Fe ions), which, however, predominantly participates in coordination of the starting materials and does not fulfil redox activities. There are also metal-free ADHs, does not which principally follow the same hydride transfer mechanism (Zhou et al., 2020).
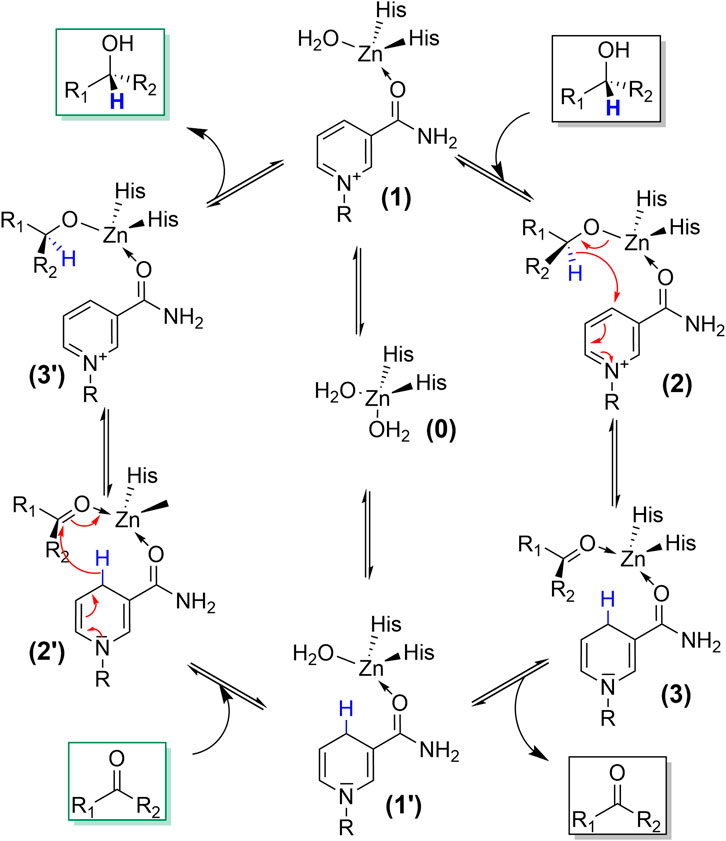
FIGURE 2. Simplified catalytic mechanism of ADH reactions. Upon binding of both substrates (e.g., NAD(P)+ and alcohol; 0->1->2) a hydride transfer occurs from the alcohol-carbon atom to the oxidised nicotinamide moiety yielding the Zn-coordinated carbonyl product and NAD(P)H (3). Both can dissociate from the active site yielding apo-ADH (0). Alternatively, only NAD(P)H stays bound and the reduced ADH can undergo a reductive conversion (1’->2’-> 3’-> 1).
One key-feature of ADH-catalysis is the reversibility of the reaction. Any given ADH catalyses both the NAD(P)H-driven reduction of carbonyl compounds to the corresponding alcohol and the NAD(P)+-driven oxidation of alcohols to the corresponding carbonyl products.
As both the carbonyl starting material and the nicotinamide moiety are chiral, four different stereochemical hydride transfer pathways are possible (Figure 3) (De Wildeman et al., 2007) Hydride transfers from the si-face of the prochiral ketone result in (R)-configured alcohols whereas hydride attacks from the re-face yield (S)-alcohols. In both cases the hydride transferred can stem from either the re- or si-face of the nicotinamide ring.
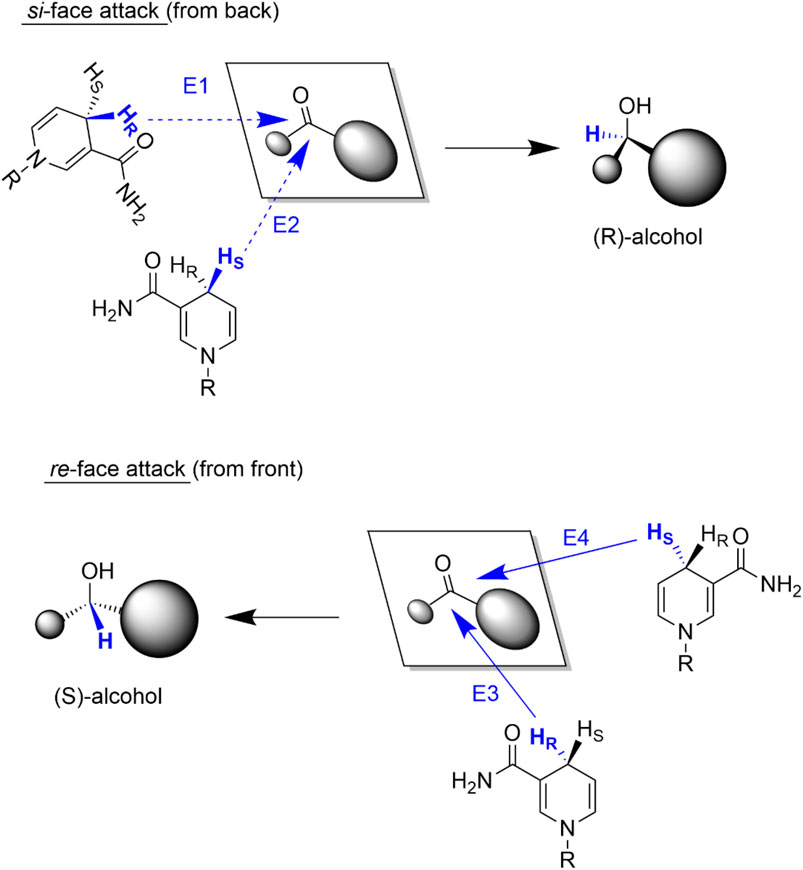
FIGURE 3. Possible stereochemical courses of the hydride transfer from NAD(P)H to the ketone. Attacks from the si-face of the ketone result in (R)-alcohols (E1 and E2) whereas hydride attacks from the re-face (E3 and E4) result in (S)-alcohols.
ADHs catalysing hydride addition from the re-face of the ketone (or abstraction of a hydride from (S)-alcohols) are termed Prelog-selective ADHs whereas those ADHs attacking from the si-face (or abstracting a hydride from the (R)-alcohol) are termed anti-Prelog ADHs. Hence, for any given enantiopreference a set of suitable ADHs is available (Table 1, vide infra) (Prelog, 1964)

TABLE 1. Selected examples for ADHs with (anti-) Prelog selectivity (de Gonzalo and Lavandera, 2021).
The selectivity, particularly the enantioselectivity of ADH-catalysis is predominantly determined by the geometrical composition of the active site and the structure of the starting material that control the binding of the substrate relative to the nicotinamide cofactor (Figure 3). Controlling this binding and the resulting selectivity is nowadays predominantly achieved through enzyme engineering (vide infra). There are, however, a range of other factors that may have a significant influence on the stereoselectivity of ADH-catalysed reactions.
As early as 1986, Keinan, Lamed and coworkers reported an influence of the reaction pH (and buffer) on the enantioselectivity of an ADH-catalysed reduction reaction (Keinan et al., 1986). Using the ADH from Thermus ethanolicus (TeADH) as model enzyme, Philips and coworkers identified the so-called racemic temperatures (TR) above and below which TeADH exhibits opposing stereoselectivity for a given substrate (Pham et al., 1989; Pham and Phillips, 1990). For the reduction of 2-butanone and 2-pentanone, for example, TR was determined to be 26oC and 77°C, respectively. Below these temperatures, (S)-alcohols were the predominant enantiomer whereas above TR the (R)-alcohol was formed. The enantioselectivity increased the further away the reaction temperature was from TR (above and below). A positive correlation between increasing temperature and (enantio)selectivity may appear counterintuitive as generally lower reaction temperatures are evaluated to increase selectivity. On condition the Curtin–Hammett principle applies to ADH-catalysed reactions, the enantioselectivity of a reaction (E) is the difference of the two Gibbs free energies of the transition states leading to the (R)- and (S)-product (ΔΔGǂ) and taking into account that ΔG is composed of an enthalpic (ΔH) and an entropic term (TΔS): ∆∆Gǂ = ∆∆Hǂ–T∆∆Sǂ (Eq. 1), a racemic temperature can be defined at which the reaction occurs non-enantioselectively (∆∆Gǂ = 0): TR = (∆∆Hǂ)/(∆∆Sǂ). Crossing TR, the sign of ∆∆Gǂ changes corresponding to a switch in enantioselectivity (Phillips, 1992).
Also pressure has been demonstrated to exhibit a measurable influence on the stereodiscrimination of enzymes (Patel and Phillips, 2014). Water soluble cosolvents can also exhibit some influence on the stereochemical outcome of ADH-catalysed redox reactions as exemplified by various authors (Fitzpatrick and Klibanov, 1991; Schumacher et al., 2006; Musa et al., 2007; Li et al., 2009; Nealon et al., 2015).
It should, however, be emphasised that the influence of these parameters generally is not pronounced enough to serve as convenient measure to control the stereoselectivity of ADH-reactions. Also, the molecular effect of, e.g., cosolvents today is by far not understood yet.
pH: ADHs generally exhibit two pH optima, one for the reductive direction and one for the oxidation reaction. As a rule of thumb, aldehyde/ketone reduction reactions are favoured in (slightly) acidic media whereas for the oxidation of alcohols alkaline media are favourable (Chang et al., 2009). Nevertheless, operating ADH reactions at neutral pH usually represents an acceptable compromise, especially if a substrate-coupled nicotinamide regeneration approach is used (vide infra). The pH of the reaction mixture also can have a significant influence on the stability of the nicotinamide cofactor: the reduced forms (NAD(P)H) are more stable in alkaline media whereas the oxidised forms (NAD(P)+) are more stable in acidic media (Chenault and Whitesides, 1987).
Temperature: The rate of ADH-catalysed reactions increases with the temperature of the reaction medium. A good approximation for the influence of the temperature on the rate of a chemical reaction is the Q10 = 2 value stating that the rate doubles if the reaction temperature is increased by 10°C. As a result, exponential increase of the rate of ADH-catalysed reactions with temperature is to be expected. However, also the thermal degradation of enzymes (such as ADHs) represents a chemical process accelerating with temperature. Hence, the optimal temperature for an ADH should be at the intersection of the (increasing) activity curve and the decreasing stability curve of an enzyme (Almeida and Marana, 2019).
Solvents: The effect of solvents on the activity/stability and selectivity of ADHs is difficult to predict. Different enzymes are differently affected by a given solvent whereas a given enzyme usually displays variable tolerance to different solvents. One study on the effect of water-immiscible organic solvents on three ADHs showed a higher or even increased stability in the presence of ethers whereas detrimental effects were observed for aromatic and aliphatic hydrocarbons and halogenated solvents (Villela Filho et al., 2003). Many ADHs exhibit decreased activity and stability in the presence of water-miscible organic solvents (Miroliaei and Nemat-Gorgani, 2002; Gröger et al., 2003; Schumacher et al., 2006; Li Y. et al., 2021). For instance, 1,4-dioxane and acetonitrile have been found to negatively affect stability and activity of LbADH but at the same time to also reduce substrate inhibition (Schumacher et al., 2006). Especially ADHs from thermophilic host organisms such as the ADH from Pyrococcus furiosus tend to exhibit higher solvent-stability compared to their mesophilic counterparts (Zhu et al., 2006b) but also ADHs from mesophilic origins can exhibit considerable robustness against water soluble cosolvents(Yan et al., 2021).
ADHs are ubiquitous in all three domains of life—Archea, Bacteria and Eukarya (Machielsen et al., 2006; Gaona-López et al., 2016; Thompson et al., 2018). In the early days of biocatalysis, naturally available enzymes have been used; the ADH from horse liver (HLADH) (Batelli and Stern, 1910; Lutwak-Mann, 1938) or the ADH from yeast (YADH) (de Smidt et al., 2008) being some prominent examples. With the advent of molecular biology and the possibility to recombinantly express enzymes of interest in well-characterised expression hosts, this situation has changed dramatically in the past 30 years (Bornscheuer et al., 2012). Today, recombinant expression of almost any gene of interest in robust and efficient expression hosts such as E. coli, S. cerevisiae or P. pastoris is possible and has become a standard technique. This has also made ADH production independent from seasonal changes as faced with plant sources, ethical issues as in case of animal-derived enzymes or fundamental issues as in case of (yet) unculturable microorganisms (Singh et al., 2016; Bodor et al., 2020). As a result, today, an enormous versatility of wild-type ADHs is available, which is also reflected by the variety of enzymes mentioned throughout this contribution. Various commercial suppliers also offer ADHs in their portfolios (Table 2).
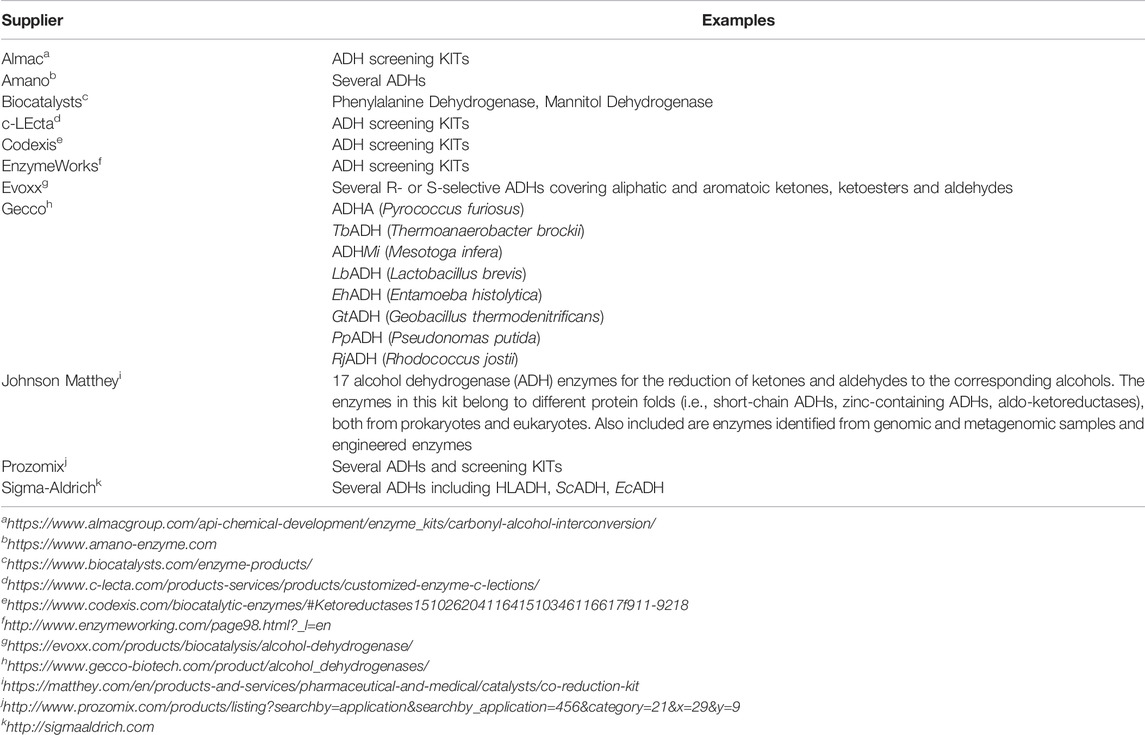
TABLE 2. Selection of commercial ADH suppliers.
The seemingly high costs of enzymes compared to “classical” chemical catalysts remains to be a persistent myth. Indeed, the prices found for small amounts of ADHs (and enzymes in general) often significantly exceed those of chemical components. It should, however, be kept in mind that enzyme production is subject to economy of scale (Tufvesson et al., 2010). Enzymes produced at small scale, where labour and equipment costs dominate the cost structure of enzyme production, tend to range at 10.000 € kg−1 and higher. However, if produced at scale, enzyme costs can go as low as 250 € kg−1. Furthermore, the cost contribution of a (bio)catalyst to the final product significantly depends on the catalyst’s performance in the chemical transformation of interest. For example, an ADH produced at large scale will be economically feasible to produce pharma products if approx. 20–30 kg product can be obtained per kg of ADH. An enzyme produced at smaller scale will have to generate one ton of product per kg of enzyme to meet the maximal cost contribution allowable for pharma products.
The formulation of the ADH catalyst is directly linked to its cost. Especially in the academic literature, often highly purified ADHs are reported. Enzyme purification, however, also in times of affinity tags still represents additional efforts and costs that can easily add up to more than five times of the enzyme fermentation costs (Tufvesson et al., 2010). Therefore, the majority of commercialised ADH preparations are crude cell extracts rather than purified ADHs. Provided that none of the “contaminating” metabolites and enzymes negatively influence the reaction of interest, crude extracts are usually more an advantage rather than a disadvantage as these preparations contain the nicotinamide cofactor thereby often making additional supplementation with NAD(P)+/NAD(P)H superfluous. Nevertheless, a commonly observed obstacle with crude enzyme preparations is the presence of endogeneous ADHs from the expression host, which may exhibit activity on the target starting material but not necessarily the desired selectivity. In such cases (partial) purification is almost inevitable.
Despite the vast natural diversity of ADHs accessible nowadays from commercial sources, strain collections or databases a given wild-type ADH may not meet the requirements for cost-efficient synthesis of the desired target product. Enzymes have evolved to meet the needs of their host organism for survival and not the needs of an organic chemist. Fortunately, protein engineering has made tremendous advances in the past 20 years (Fasan et al., 2019; Acevedo-Rocha et al., 2020). Adapted enzymes that fulfil the needs of organic chemists can be built by directed evolution or semi-rational design using diverse standard molecular biology techniques, computational modelling and bioinformatics. In directed evolution experiments, random mutations are introduced into the parent gene; the resulting enzyme variants are investigated and selected for improvements in the desired property, finally serving as parents for following evolutionary rounds. The random nature of this approach yields many variants with mutations in irrelevant positions for the desired property which is why huge mutant libraries have to be screened. This makes pure random directed evolutionary approaches very time- and resource-intensive. Smaller libraries of higher quality can be achieved by restricting the mutation sites based on structural or mechanistic information. This semi-rational approach needs prior knowledge (mostly crystal structures or homology models of the enzyme) to determine the locations for randomisation. In turn, the library sizes (as well as the screening efforts) are greatly reduced. Fully rational design of improved variants using in silico methods are yet in their infancy (Bornscheuer et al., 2012; Hammer et al., 2017; Li et al., 2018; Fasan et al., 2019; Yang et al., 2019; Acevedo-Rocha et al., 2020; Bell et al., 2021).
Next to engineering the biocatalyst itself, also engineering the reaction conditions, e.g., by adjusting the solvent composition or immobilising the biocatalysts can have a decisive effect on the practicability of an ADH-catalysed redox reaction.
Some selected examples of enzyme- and reaction engineering will be discussed in the following paragraphs.
Substrate scope enlargement. A broad substrate scope is a key requirement for an enzyme to be an effective biocatalyst to foster their application in organic synthesis. For instance, the secondary alcohol dehydrogenase from the thermophilic bacterium Thermoanaerobacter pseudoethanolicus ATCC 33223 (TeADH) is a very robust ADH exhibiting a rather narrow substrate scope (small substrates such as aliphatic ketones, Figure 4) (Musa et al., 2021). By analysing the active site architecture of wt-TeADH Phillips and co-workers identified Trp110 to interfere with the binding of sterically more demanding starting materials. Indeed, exchanging this amino acid residue for a smaller alanine residue resulted in a TeADH mutant accepting much larger substrates than the wild type while not being impaired in its thermal stability. A further mutant of this enzyme (W110A/I86A) accommodated even bulkier starting materials and also influenced the stereoselectivity of the enzyme (Figure 4) (Musa et al., 2018).
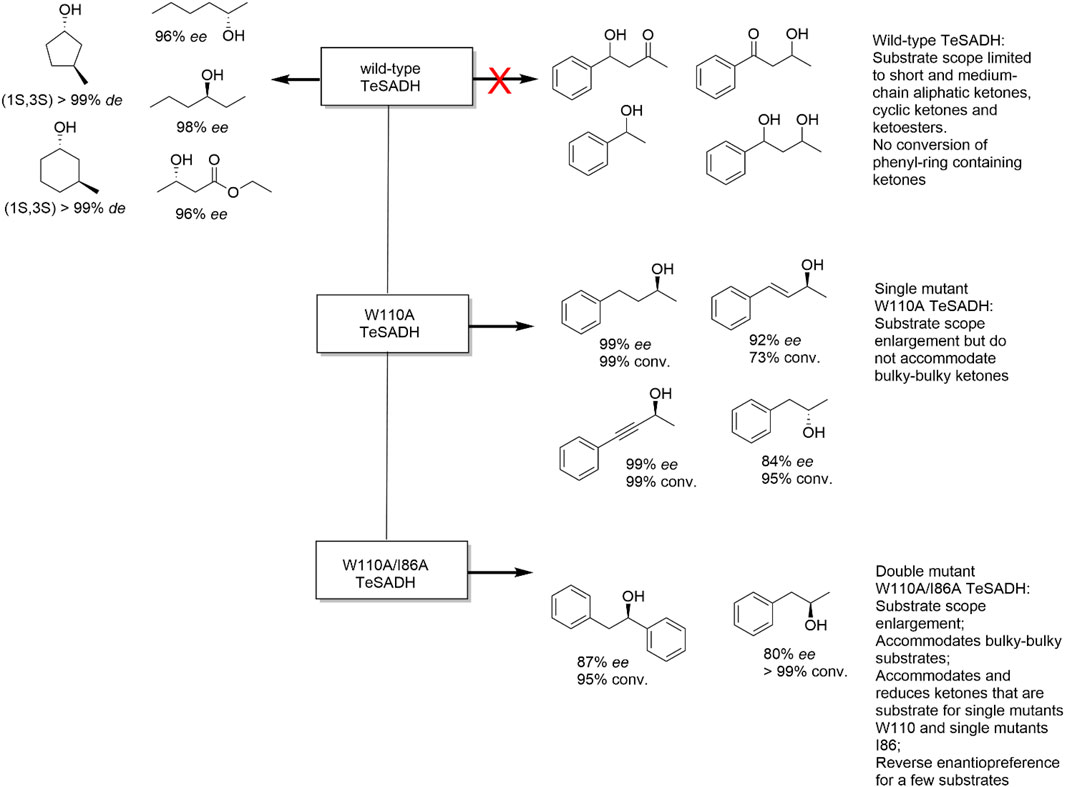
FIGURE 4. Substrate scope enlargement and reverse enantiopreference achieved with single and dual mutants from TeSADH (Thermoanaerobacter pseudoethanolicus secondary alcohol dehydrogenase).
Improving/reversing enantioselectivity. Similar to the substrate scope, the enantioselectivity of an ADH is a property intimately linked to the geometry of the active site. The company Codexis has for long excelled in improving enzymes for various properties using the protein sequence activity relationship (ProSAR) algorithm (Fox et al., 2007). Using ProSAR, they, for example, improved the enantioselectivity of LkADH for tetrahydrothiophene-3-one from 63% to 99.3% (Liang et al., 2010b). By simultaneously challenging the mutant libraries with increasing thermal- and solvent-stress, the final mutant enzyme also showed significantly improved properties here. Starting from wt-Thermoethanolicus brokii ADH, Reetz and coworkers applied triple code saturation mutagenesis to engineer it into an S- or R-selective enzyme for tetrahydrotfuran-3-one and similar starting materials (Sun et al., 2016). Using this enzyme Reetz and his team also demonstrated that enantiodiscrimination of sites further away from the carbonyl group can be achieved (Agudo et al., 2013). Axially chiral 4-alkylidene cyclohexanone represent an almost impossible target for enantioselective reduction chemistry. Using combinatorial active-site saturation test (CAST)-based directed evolution, highly R- and S-selective mutants were obtained.
Another impressive example of engineering the selectivity of an ADH was reported recently by Ni and coworkers (Xu et al., 2018). (4-Chlorophenyl) (pyridin-2-yl)methanone (CPMK) is widely recognised as “difficult” substrate due to the similar steric demand of both substituents of the carbonyl group impeding differentiation of the prochiral substrate faces. The authors chose the ADH from Klyveromyces polysporus (KpADH) exhibiting modest enantioselectivity in the reduction of CPMK (84% ee). In a first step they identified possible amino acid residues in the active site (using a homology model of the enzyme) that may be involved in substrate binding. From those, potential hotspots were identified via a combined alanine- and tyrosine-scanning. The six hotspots were submitted to combinatorial mutagenesis using reduced codon alphabets. Overall, several KpADH mutants with significantly improved enantioselectivity (99.4% ee) and increased catalytic activity compared to the wt-enzyme were identified.
Catalytic activity improvement. Recently, Zheng and co-workers reported engineering the ADH from Lactobacillus kefir (LkADH) for improved activity for the synthesis of (S)-2-chloro-1-(2,4-dichlorophenyl) ethanol [(S)-TCPE] from the corresponding ketone starting material (Zheng et al., 2021). wt-LkADH showed high enantioselectivity but rather poor catalytic activity. A rational design approach was applied based on the structural characteristics of the LkADH substrate binding complex. Molecular docking calculations were used to generate the binding structure of ADH and 2,2′,4′-trichloroacetophenone (TCAP) and to identify the key residues responsible for LkADH activity. Additionally, molecular dynamic simulations of the LkADH-substrate binding complex demonstrated that the substrate binding pocket in wt-LkADH should be reconfigured to allow TCAP to be well accommodated in the correct orientation with the 2,4-dichlorophenyl group being located at the large binding pocket and the chloromethyl group at the small one. Based on these studies, three sites, specifically A94 (located at the large binding pocket), E145 (located at the small binding pocket) and S96 (located at the loop responsible for modulating the open and closed state of the binding pocket), were sequentially selected for site mutation and subsequently combined. The enantioselective triple mutant A94S/S96E/E145A displayed a 117- fold increase in relative activity compared to the wild-type enzyme.
Thermal and solvent stability improvement. Next to selectivity, robustness of a biocatalyst against hostile reaction conditions is one of the most desired properties in biocatalysis. Particularly, resistance against thermal inactivation and inactivation by organic solvents is of interest for the practical applicability of ADHs in organic synthesis.
Organic solvents can influence enzymes (ADHs) in many different ways (Li Y. et al., 2021). Water-immiscible solvents often inactivate enzymes at the solvent interface. The favourable interaction of hydrophobic, inner amino acids with the apolar organic solvent facilitates unfolding and thereby inactivation of the enzyme. Water-miscible solvents can interact in various ways with an enzyme and thereby influence its activity and stability: 1) the changed polarity of the solvent can alternate pKa values and thereby influence internal salt- or H-bridges relevant for catalysis/structural integrity of the enzyme; 2) disrupt hydrophobic interactions in the enzyme and 3) influence the enzyme flexibility by displacing water molecules.
Evolution of solvent-resistance is feasible as, e.g., demonstrated by coworkers of the company Codexis who engineered ADHs for broadened substrate scope and improved solvent stability (Liang et al., 2010a; Ma et al., 2010).
Interestingly, thermal stability and resistance against co-solvents often are correlated, possibly because of the similarities between the two protein unfolding mechanisms (Li S. F. et al., 2021). Nestl and Hauer pointed out the importance of flexible surface regions for stability suggesting to focus on these regions for improving thermal stability of an enzyme (Nestl and Hauer, 2014).
The B value approach developed by Reetz and coworkers addresses such regions by identifying flexible enzyme regions based on atomic displacement parameters of crystallographic data reflecting the smearing of electron densities due to thermal movement (Reetz et al., 2006).
Using an in silico method to identify promising residues for thermal stability improvement (FRESCO), Fraaije and coworkers recently reported an impressive stabilisation of ADH-A by 45°C without impairing the catalytic activity at ambient temperature (Aalbers et al., 2020). This is remarkable insofar as often increases in thermostability come with simultaneous decreases in the mutant’s enzyme activity at ambient temperature (Machielsen et al., 2006; Willies et al., 2010; Siddiqui, 2017).
In case of the tetrameric ADH from Leifsonia (LnADH) engineering of amino acids involved in the subunit contact areas proofed to be successful (Zhu et al., 2021).
Immobilisation has been (and remains) a very active subfield of ADH research. The main motivation to heterogenise enzymes including ADHs is to increase their robustness under hostile, non-natural but industrially relevant reaction conditions such as high reagent concentrations and elevated reaction temperatures (Hanefeld et al., 2009; Sheldon and Pereira, 2017). Further advantages of immobilised enzymes over their soluble counterparts are that they can be applied in continuous processes and/or in repetitive batch reactions, can easily be separated from the reaction mixtures and reused. Immobilised enzymes are also advantageous in multi-step cascade reactions if temporally or spatially separated reaction steps are necessary.
Principally, three immobilisation principles can be distinguished: 1) Entrapment, 2) Immobilisation to a carrier and 3) heterogenisation via crosslinking (Figure 5).
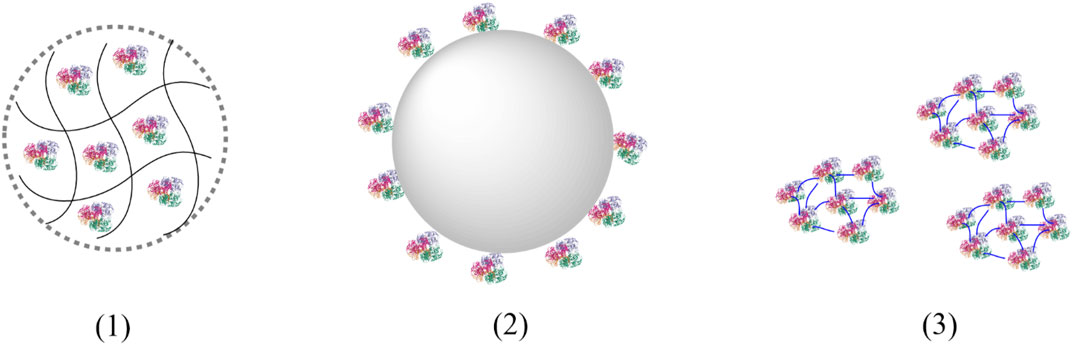
FIGURE 5. Immobilisation principles. (1) Entrapment of enzymes in a polymeric matrix; (2) Immobilisation of enzymes to a carrier; (3) Cross-linked enzymes aggregates (CLEAs).
Entrapment: Enzymes and also cells can be entrapped in both inorganic and organic polymeric matrices. This is most commonly achieved by preparing the carrier in the presence of the enzymes, so that they become embedded in the matrix lattice. Alternatively, enzymes can also be absorbed in pre-fabricated superabsorbent polymers. As a disadvantage of entrapment, leaching of enzymes is prone to occur, an issue that can be addressed by providing additional covalent binding or by increasing the protein size through cross-linking (Sheldon and Woodley, 2018). However, because of weaker binding forces involved in immobilisation, deactivation of the enzyme by conformational distortions is less likely in comparison with other approaches. Entrapment of ADHs with various gelating materials comprising silica gels (Musa et al., 2007; Nagy-Gyor et al., 2018; Liu et al., 2019; Nagy-Győr et al., 2019) polyvinyl alcohol (Krasnan et al., 2016; Petrovicova et al., 2018; Yildirim et al., 2019) poly ethyleneglycol (Schmieg et al., 2019) superabsorbers (Heidlindemann et al., 2014; Adebar and Groger, 2020), alginates (Milagre et al., 2005, 2006; He et al., 2017; Nasario et al., 2019), metal organic frameworks (MOFs) (Carucci et al., 2018; Ye et al., 2020) or liposomes (Yoshimoto et al., 2008) has been reported.
Immobilisation onto solid carriers. Solid carriers employed for immobilisation include inorganic materials, such as silica-based materials, kaolin or zeolites; organic materials, such as porous acrylic resins, polystyrene polymers and water-soluble natural polymers like cellulose and chitosan; and coordination polymers, particularly metal-organic frameworks (Sheldon and van Pelt, 2013). Inorganic carriers usually display high thermo-, chemo- and mechanical stability. They are also available as nanoparticles, including biocompatible gold nanoparticles and iron-based magnetic nanoparticles, which can offer advantages regarding enzyme loadings and mass transfer due to their high surface-to-volume ratios and controllable sizes (Sheldon and van Pelt, 2013). Some inorganic carriers, like mesoporous silica, present uniform pore diameter, high surface areas as well as high pore volume, so that relatively small enzymes can be immobilised both on the surface and inside the pores. For instance, a study on immobilisation of ADH showed that more enzymes were distributed inside the pores of a mesostructured cellular foam (MCFs) in comparison to ordered mesoporous silica particles such as SBA-15, suggesting MCF as a superior silica-based carrier for immobilisation of ADHs and similarly sized enzymes with higher loading (Zezzi do Valle Gomes and Palmqvist, 2017). Among organic polymers, synthetic acrylic resins are widely employed in enzyme immobilisation, including commercially available hydrophilic acrylic resins with mechanical and thermal stability and tolerance to organic solvents and to a wide pH range. Natural polymers have also been used and have the advantage of being biocompatible and biodegradable. They are mostly hydrophilic and thereby associated to a lower susceptibility to cause enzyme deactivation in comparison to hydrophobic carriers, which may induce detrimental conformational changes in proteins, leading to denaturation and activity loss.
Immobilisation onto solid carrier materials may occur via (reversible) physical adsorption based on electrostatic or hydrophobic interactions or (irreversibly) via covalent attachment. Physical adsorption can usually be achieved with the use of simple protocols not requiring chemical modification of the carrier or the enzyme. However, susceptibility to enzyme leaching, particularly in aqueous media, limits its applicability in most biocatalytic reactions under conditions employed in industrial processes. Nevertheless, some example of immobilisation of ADHs through adsorption for synthetic purposes have been reported, including immobilisation on raw inorganic materials, such as Al2O3 and TiO2 (Sigurdardóttir et al., 2019), carbomethyl dextran-coated magnetic nanoparticles (Vasic et al., 2020) and silica gel (Liu et al., 2019). Adsorption of ADHs through ionic binding onto polymers bearing cationic or anioninc groups has also been described (Dreifke et al., 2017). Importantly, electrostatic adsorption is a promising strategy for the immobilisation of negatively-charged phosphorylated cofactors, such as NADPH, within porous materials modified with positively charged groups (Benítez-Mateos et al., 2017; López-Gallego et al., 2017; Velasco-Lozano et al., 2017). Because of the reversible nature of such binding, the cofactor molecules are allowed to diffuse across the intra-porous space without leaving the support, thus being available for a co-immobilised enzyme.
Recombinant enzymes bearing a His-tag, i.e., a terminal sequence of six to nine histidine residues, can be non-covalently immobilised on polymers containing chelating groups loaded with metals such as Ni(II), Fe(III), Zn(II), Cu(II) and Co(II) (Sheldon and Pereira, 2017), polymers decorated with nitrile tri-acetic acid molecules and loaded with Ni(II) (Ni-NTA) (Homaei et al., 2013) and controlled porosity glass-based materials bearing chelated Fe(III), such as EziGTM (Homaei et al., 2013; Cassimjee et al., 2014; Thompson et al., 2019) being commonly used carriers. This strategy usually leads to relatively high immobilisation yields in short times and also offers the possibility of performing the immobilisation directly from a crude cell extract, since it also serves as a purification step. As a disadvantage, metal leaching may be an issue in large scale processes. Examples of immobilisation of ADHs by this approach comprehend co-immobilisation of ADH and GDH on sepharose charged with Ni(II) to give reusable bienzymatic heterogeneous biocatalysts with improved operational and storage stability for bioreductions in flow reactors in semi-continuous mode (Plž et al., 2020); immobilisation of Co(II)-containing magnetic beads for use in minituarised packed-bed flow reactors (Peschke et al., 2019) and co-immobilisation of ADHs together with non-ADH enzymes in Co(II)-charged agarose microbeads (Velasco-Lozano et al., 2020) and EziGTM (Böhmer et al., 2018) for cascade reactions.
Attaching enzymes to a carrier via covalent bonds provides a more stable binding in comparison to adsorption, preventing enzyme leaching and also leading to improved chemo- and thermostability due to multipoint attachment to the carrier. In addition, reactive groups on the surface of carriers used for this kind of immobilisation enable their modification by insertion of additional moieties or polymer-coating to confer them desirable properties such as hydrophilicity to improve enzyme stability (H. Orrego et al., 2020) On the other hand, conformational changes imposed to ADHs by covalent immobilisation and inappropriate enzyme orientation, together with mass transfer limitations, decreases activity retention and immobilisation efficiency. Interestingly, either improvement or decrease in stereoselectivity and even switch in enantiopreference have been reported to be caused by conformational changes after covalent immobilisation (Petkova et al., 2012). The covalent bonds between the enzyme and the carriers usually involve the reaction of nucleophilic amino acid residues on the surface of the protein, such as lysine or cysteine residues, with electrophilic moieties on the surface of the carrier, such as Michael acceptors and oxirane and formyl groups (Homaei et al., 2013). Attention must be given to the tolerance of the enzyme to the reaction conditions required for some immobilisation procedures, such as alkaline pH, and to possible cross-linking between particles resulting in a lower available surface area and thereby increasing mass transfer limitations.
Covalent immobilisation of ADHs have been achieved with epoxy resins (Figure 6A), (Sole et al., 2019) including the commercially available EupergitTM (Boller et al., 2002). As an example, Truppo and co-workers (Li et al., 2015) described the immobilisation of an ADH on a commercial amino-epoxy-functionalised poly methacrylate resin to give a heterogeneous biocatalyst with improved tolerance to organic solvents and good recyclability, which could be used to perform the synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)ethenone at 50g scale.
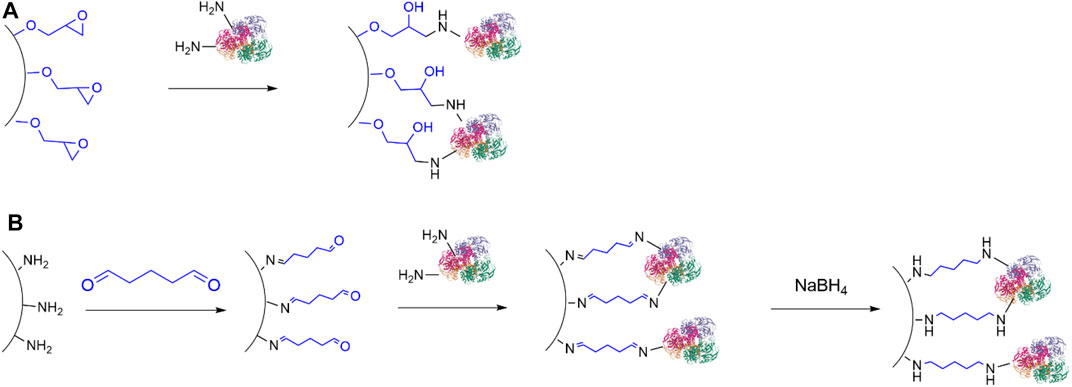
FIGURE 6. Immobilisation of enzymes on solid carriers via covalent bond. (A) Attachment of enzymes to epoxy resins; (B) Attachment of enzymes to carriers bearing formyl moieties.
Another commonly used method for covalent immobilisation of ADHs (and enzymes in general) relies on the treatment of amino-functionalised carriers with a di-aldehyde, typically glutaraldehyde, which is then followed by reaction with lysine residues, so that both carrier and enzymes become bonded to a spacer through imine groups (Figure 6B). Alternatively, the use of a carrier bearing formyl groups, such as glyoxyl-functionalised supports (Mateo et al., 2006; H. Orrego et al., 2020) obviates the addition of a di-aldehyde. Further reduction of the imines to secondary amine moieties with sodium borohydride or the milder 2-picoline borane (Orrego et al., 2018) provides a more stable attachment between the enzymes and the carrier, though this step is not always performed. Importantly, these immobilisation methods require alkaline conditions, so that denaturation of enzymes may occur. This strategy has been used to immobilise ADHs on amino-functionalised titania nanoparticles (Ghannadi et al., 2019), silicon carbide (Zeuner et al., 2018) and nanoporous silica (Engelmann et al., 2020).
Similarly, ADHs have been successfully immobilised in agarose bearing formyl groups (Mateo et al., 2006; Velasco-Lozano et al., 2017; H. Orrego et al., 2020). For instance, Serra and coworkers (Dall'Oglio et al., 2017) described the immobilisation of an ADH and GDH (for cofactor regeneration) in glyoxyl-agarose. The immobilisates were used as a blend in a packed-bed flow reactor, which could be operated for 15 days for continuous bioreduction of ketones with only a slight decrease in conversion, thus demostrating the robustness of the immobilised biocatalysts.
López-Gallego and coworkers reported a self-sufficient heterogeneous biocatalyst by coimmobilising both enzyme and cofactor on glyoxyl-agarose macroporous beads (Orrego et al., 2021). After covalent immobilisation of a thermophilic (S)-2-hydroxybutyryl-CoA dehydrogenase from Thermus thermophilus HB27 (Tt27-HBDH), the remaining formyl groups on the carrier allowed for a subsequent coating with polyethyleneimine (PEI) to give a cationic layer in which NADH molecules could be embedded and reversibly immobilised through ionic interactions. The resulting biocatalyst could be used for ten cycles to produce (S)-ethyl 3-hydroxybutyrate with a productivity in the range of 0.066–0.027 g L−1 h−1 and a TTN of 145 for NADH (in constrast to a TTN limited to 10 when soluble, non-coimmobilised NADH was used).
CLEAs: CLEAs are prepared through precipitation of the enzymes as physical aggregates in aqueous media, without disturbing their tertiary structure, with the aid of salts or non-ionic polymers or by the addition of an organic water-soluble solvent (Sheldon, 2019; Sheldon et al., 2021). The enzyme molecules in the aggregates are then covalently cross-linked by the addition of a di-aldehyde. Typically, ammonium sulphate is used as an aggregant and glutaraldehyde as a cross-linking agent. They can be added together in the enzyme solution, since physical aggregation takes place much faster than the cross-linking reaction. As an advantage, immobilisation through CLEAs do not require a carrier, thus resulting in lower cost and in high productivities and space-time yields in comparison to previously discussed strategies, since “activity dilution” due to the addition of a large amount of non-catalytic ballast is avoided. In addition, CLEAs are especially effective to improve stability of multimeric enzymes, have low susceptibility to enzyme leaching and usually present good diffusional properties due to their high porosity. Low mechanical robustness and the difficult to produce particles with uniform and suitable size, however, account for their disadvantages (Sheldon, 2019; Sheldon et al., 2021). An example of immobilisation of ADHs by producing CLEAs was provided by Shao and coworkers (Wu K. et al., 2021) who succeeded thereby in improving storage and thermo- and chemostability as well as tolerance to pH of a mutant short-chain dehydrogenase of Novosphingobium aromaticivorans. The immobilisate was used for the bioreduction of a chloroketone to give a chiral intermediate of the drug atazanavir with high stereoselectivity and a space-time yield of 226.6 g L−1 day−1 keeping 85.3% of conversion after being reused for six batches.
Two or more different enzymes can be immobilised in a single CLEA to give the so-called combi-CLEAs. This approach has been employed for the obtaining of bi-enzymatic immobilisates, including magnetic combi-CLEAs (Chen et al., 2018) bearing the main ADH and the cofactor regenerating enzyme (Ning et al., 2014; Zhang J. et al., 2020; Xu et al., 2020). As an example, Su et al. (2018) described the preparation of combi-CLEAs comprised of ketoreductase and GDH enzymes embedded with magnetic Fe3O4 nanoparticles and their use in the obtaining of 1.98 g of enantiopure ethyl (S)-4-chloro-3-hydroxybutyrate from its corresponding ketoester in 15 h with a TTN of NADH of 11.880. The bifunctional biocatalyst presented improved chemo- and mechanical stability, showing better recyclability than the non-magnetic combi-CLEAs.
Today, mostly aqueous reaction media are used for ADH-catalysis. Despite common perception, water is a sub-optimal solvent for many biocatalytic reactions. Many of the reagents of interest are rather hydrophobic and therefore rather poorly water-soluble. Today, a very common way around this challenge still is to utilise rather dilute reagent concentrations in the reaction mixtures (Holtmann and Hollmann, 2022). As a result product titres of less than 10 g L−1 and less are the rule rather than the exception in biocatalysis. This is unsustainable both from an economic (Huisman et al., 2010) and an environmental (Ni et al., 2014) point of view. Therefore, increasing the reagent concentrations represents a prime target en route to environmentally benign and economically attractive ADH-catalysed reactions.
Fortunately, in recent years, the pioneering works by Klibanov and coworkers are receiving a renewed interest from the research community (Zaks and Klibanov, 1984, 1985; Dordick et al., 1986). As early as the 1980s these authors demonstrated the applicability of various enzyme classes in non-aqueous media. Next to a larger choice of solvents and the increased solubility of the reagents of interest, non-aqueous media also offer advantages with respect to enzyme stability (especially at elevated temperatures). In case of ADH-catalysis the immobilisation of the nicotinamide cofactor within the ADH active site induced by the non-aqueous surrounding also eliminates a range of water-related inactivation mechanisms. Therefore, the number of ADH-reactions occurring in non-aqueous or micro-aqueous media is steadily increasing (de Gonzalo et al., 2007; Jakoblinnert et al., 2011; Musa and Phillips, 2011; Kara et al., 2014b; Heidlindemann et al., 2014; Spickermann et al., 2014; Popłoński et al., 2018).
In cases where non-aqueous reaction media are not straightforward to implement (e.g., because of issues with biocatalyst stability or –formulation) multiphase reaction systems offer an attractive alternative to improve the reagent payloads. Slurry-to-slurry reactions, for example, apply dispersions of solid, poorly water soluble reagents in aqueous (biocatalyst-containing) media. Provided the affinity of the enzyme for the starting material is sufficiently high (KM in the range of the equilibrium solubility or lower) the rate of the biocatalytic transformation is not impaired severely. An elegant example was reported by researchers from Codexis converting a very poorly starting ketone (solubility <0.5 g L−1) into the corresponding (not much more soluble) alcohol product; using the slurry-to-slurry concept substrate loadings of up to 100 g L−1 were converted efficiently into the desired product at 200 kg-scale (Liang et al., 2010a). An attractive feature of slurry-to-slurry reactions is of course that in principle a simple filtration step suffices for product isolation.
More commonly, two liquid phase systems are applied. Here, a hydrophobic, water-immiscible organic phase serves as substrate reservoir and product sink at the same time (Figure 7). 2LPS exhibit a range of advantages over simple monophasic reaction media): First, they enable overall higher reagent loadings and thereby increase the economic attractiveness and reduce water wastes. Furthermore, product separation from the catalysts is principally straightforward after phase separation (Wu et al., 2009; Kara et al., 2013c; Ou et al., 2019). Second, water-reactive substrates and products can (to some extent) be protected in the organic layer from, e.g., hydrolysis. Another attractive feature is that, in some cases the organic solvent can help to control the selectivity of multi-step reactions. For example, whole cell-catalysed oxidations of primary alcohols can be plagued by (undesired) overoxidation of the desired aldehyde due to the presence of endogeneous aldehyde dehydrogenases. This issue has been addressed using the 2 LPS approach, e.g., by Bühler et al. (2002), Bühler et al. (2003), Bühler and Schmid (2004) and Molinari et al. (1997), Gandolfi et al. (2001). In the presence of hydrophobic organic phases, the intermediate, hydrophobic aldehydes were efficiently extracted into the organic layer and were thereby not available for aldehyde-dehydrogenase-catalysed overoxidation to the acids.
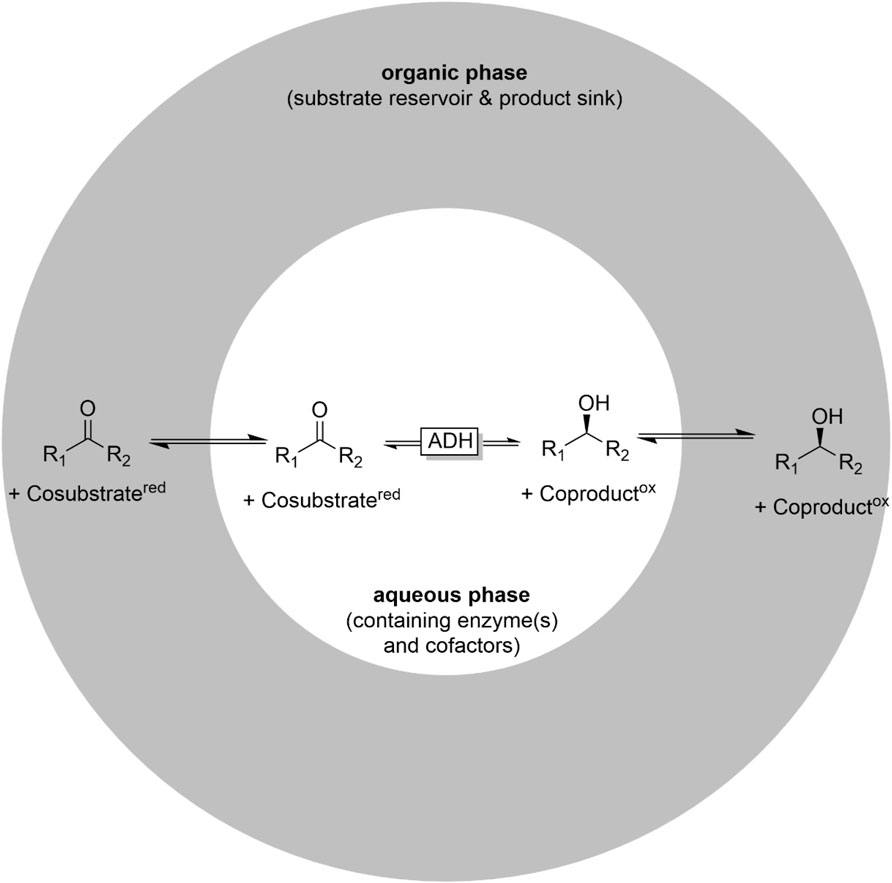
FIGURE 7. The two liquid phase system (2LPS) approach combining an organic phase (hydrophobic solvent containing the reagents in high concentrations) and an aqueous (biocatalyst-containing) reaction phase.
Ever since ADHs have moved into the focus of biocatalysis, cofactor regeneration strategies have been investigated (Chenault and Whitesides, 1987). The majority of ADHs considered today depend on either nicotinamide cofactor, either the non-phosphorylated (NAD+/NADH) or the phosphorylated (NADP+/NADPH) cofactor (Figure 1). Commercial nicotinamide cofactors are rather expensive, making their use in sub-stoichiometric amounts and in situ regeneration of the active redox state mandatory. Over the decades (Chenault and Whitesides, 1987), a broad variety of in situ regeneration systems has been developed, which has been reviewed extensively (Wichmann and Vasic-Racki, 2005; Kara et al., 2014a; Zhang and Hollmann, 2018). Therefore, in this contribution we focus on the most relevant ones with a particular focus on practicability and environmental impact.
The so-called substrate-coupled regeneration approach represents a very simple approach to regenerate NAD(P)H in ADH-catalysed reduction reactions (Wichmann and Vasic-Racki, 2005; Kara et al., 2013a). In this approach, an oxidisable cosubstrate (frequently isopropanol) is added to the reaction mixture enabling an overall biocatalytic variant of the Meerwein-Ponndorf-Verley reduction (Figure 8).
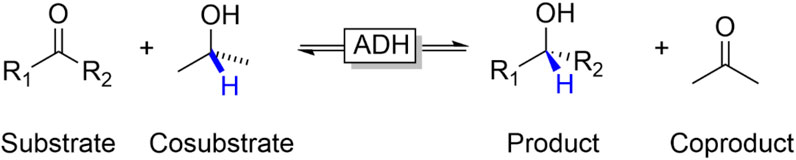
FIGURE 8. Substrate-coupled reduction of carbonyl groups (aldehydes, ketones) to the corresponding alcohols using a sacrificial alcohol cosubstrate (such as isopropanol) as stoichiometric reductant.
From a practical point-of-view this approach is very attractive as the production enzyme at the same time also serves as regeneration enzyme and that—in principle—the nicotinamide cofactor can stay bound to the enzyme active site. The latter is of interest for circumventing solvent-related degradation of the cofactor (Chenault and Whitesides, 1987) and enables using the enzyme under non aqueous conditions. Since the ADH reaction is reversible, the same approach can also be used to promote ADH-catalysed oxidation reactions (e.g., using isopropanol as cosubstrate).
On the downside, the reversibility of the reaction results in a rather unfavourable equilibrium of the overall reaction. Chemically, the substrate/cosubstrate couple is very similar to the product/coproduct couple (essentially consisting of an alcohol and a carbonyl compound) resulting in an equilibrium constant around one. To shift the equilibrium to the desired side, Le Chatelier measures such as the removal of one product from the reaction mixture can be taken. But most frequently, the cosubstrate is used in significant (more than 20-fold) excess to shift the equilibrium. “Smart cosubstrates” represent an interesting solution to this thermodynamic challenge. Kara et al., for example, developed a system comprising lactonisable diols as ‘smart cosubstrates’ (Figure 9) (Kara et al., 2013b; Zuhse et al., 2015; Huang et al., 2018).
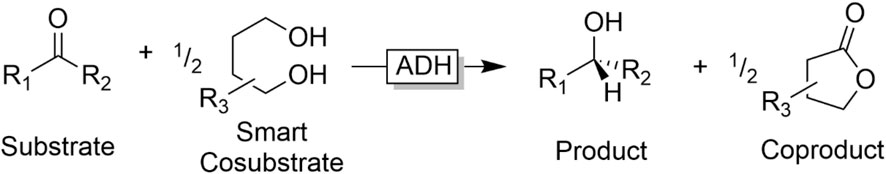
FIGURE 9. Smart Cosubstrates to turn ADH-catalysed reduction kinetically and thermodynamically irreversible.
“Smart cosubstrates” enable a dramatic reduction in the cosubstrate loading for several reasons: on the one hand, the lactone coproduct is very unreactive for thermodynamic and kinetic reasons. On the other hand, as the “smart cosubstrate” is oxidised twice by the ADH, only 0.5 equivalents are needed to obtain full conversion of the substrate.
The reversibility issue of ADH-catalysed reduction reactions is also frequently solved via the so-called enzyme-coupled NAD(P)H regeneration approach (Figure 10) (Wichmann and Vasic-Racki, 2005; Kara et al., 2013a) Here, a second, irreversible NAD(P)+-dependent oxidation reaction is used to regenerate the reduced nicotinamide cofactor. The most popular enzyme systems right now are glucose-dehydrogenase (Sorgedrager et al., 2008; Liang et al., 2010b; Huisman et al., 2010; Gong et al., 2017; Chen et al., 2021) and formate dehydrogenase (Shaked and Whitesides, 1980; Seelbach et al., 1996; Gröger et al., 2004; Jiang et al., 2020). Very recently, Vincent and coworkers reported significant advances in the use of hydrogenases (Reeve et al., 2012; Reeve et al., 2017; Zor et al., 2017; Zhao et al., 2021) enabling H2 as cosubstrates.
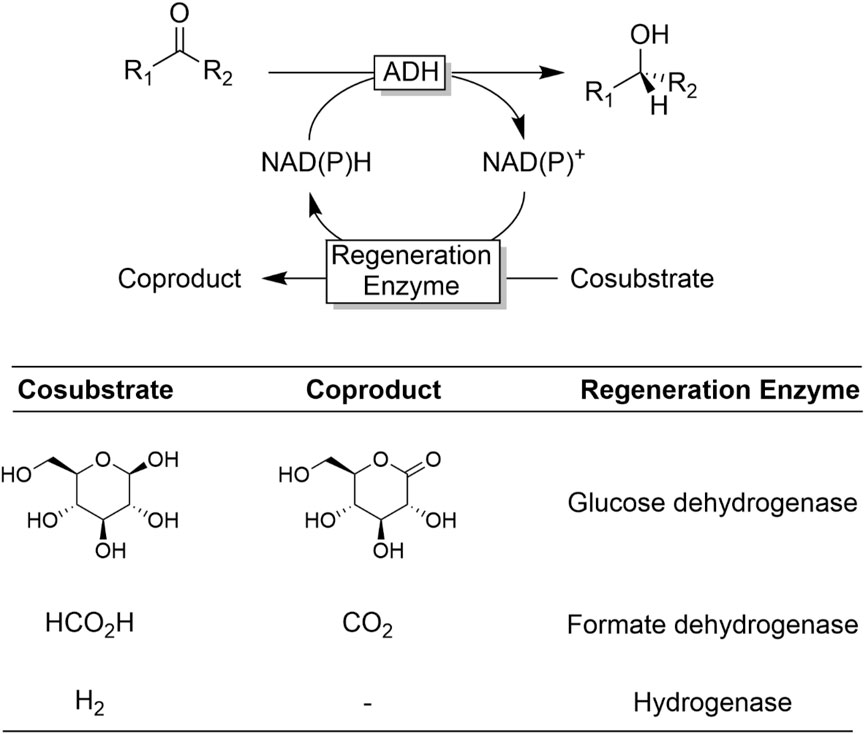
FIGURE 10. Selection of some common enzyme-coupled NAD(P)H regeneration approaches.
Regeneration of oxidised nicotinamide cofactors. Similarly to reductive ADH reactions, also for oxidative ADH processes, a range of enzyme- and substrate coupled approaches have been reported. The enzyme-coupled approach principally suffers from the same thermodynamic challenges as discussed above. Kroutil and coworkers therefore devised a “smart cosubstrate” approach based on α-halo substituted ketones as sacrificial electron acceptors (Lavandera et al., 2008b; Kurina-Sanz et al., 2009). The corresponding vic-halo alkanols are thermodynamically stabilised through intramolecular H-bonds thereby efficiently shifting the overall equilibrium.
In contrast to the regeneration of the reduced nicotinamide cofactors, the opposite reaction (regeneration of NAD(P)+ from NAD(P)H) is not plagued by regioselectivity issues and the aromatic NAD(P)+ can be formed. This is also why “chemical” regeneration systems represent a viable alternative to enzymatic counterparts. The preferred stoichiometric electron acceptor is molecular oxygen because of the high thermodynamic driving force of O2 reduction on the one hand and because of the favorable byproducts (H2O or H2O2, which is generally dismutated into O2 and H2O). Amongst the enzymatic NAD(P)+ regeneration systems, clearly the so-called NAD(P)H oxidases dominate (Riebel et al., 2002; Riebel et al., 2003; Jiang and Bommarius, 2004; Park et al., 2011; Gao et al., 2012; Dias Gomes et al., 2019; Anderson et al., 2021). But also some monooxygenases (at first sight unexpected) can be used for the regeneration of NAD(P)+ exploiting the (usually undesired) uncoupling reaction (Holtmann and Hollmann, 2016). For example, flavin-dependent monooxygenases (Ni et al., 2016) and ene reductases (Pesic et al., 2019) or heme-enzymes (Holec et al., 2016; Jia et al., 2017) can be used as NAD(P)+ regeneration catalysts.
Amongst the chemical NAD(P)+ regeneration catalysts redox-active dyes such as ABTS (Schröder et al., 2003; Aksu et al., 2009) or quinoid dyes such as Meldola’s blue (Kochius et al., 2012; Könst et al., 2013; Kochius et al., 2014) and others can be used. Most frequently used are the natural flavin cofactors (riboflavin, flavin adenine mononucleotide and flavin adenine dinucleotide) (Jones and Taylor, 1973; Jones and Taylor, 1976; Boratynski et al., 2010; Piantini et al., 2011; Boratynski et al., 2013), whose photoresponsiveness can be exploited to dramatically accelerate the oxidation of NAD(P)H to NAD(P)+ (Gargiulo et al., 2011; Rauch et al., 2017).
The stereoselective reduction of prochiral ketones is by far the most popular application of ADHs in organic synthesis. Especially in the synthesis of pharmaceutically active ingredients, ADHs are very popular due to their high selectivity (Raynbird et al., 2020). In the past years, thousands of publications have appeared reporting an ADH-catalysed ketoreduction reaction. Therefore, an exhaustive coverage of this vast literature landscape is not possible, and we restrict this passage to some representative examples and interesting concepts.
Ketones comprising one large and one small substituent at the carbonyl group are very common substrates for ADH-catalysed reductions. Especially methyl ketones are popular starting materials and a broad range of aliphatic (Sinha and Keinan, 1997), aromatic (Hamada et al., 2001; Stampfer et al., 2002; Hummel et al., 2003; Gonzalez-Martinez et al., 2019; Bandeira et al., 2020), and conjugated ketones (Schubert et al., 2001; Sgalla et al., 2007; Albarrán-Velo et al., 2020; González-Granda et al., 2021) have been reported. Figure 11 shows a representative selection of chiral alcohols obtained from the reduction of methyl ketones.
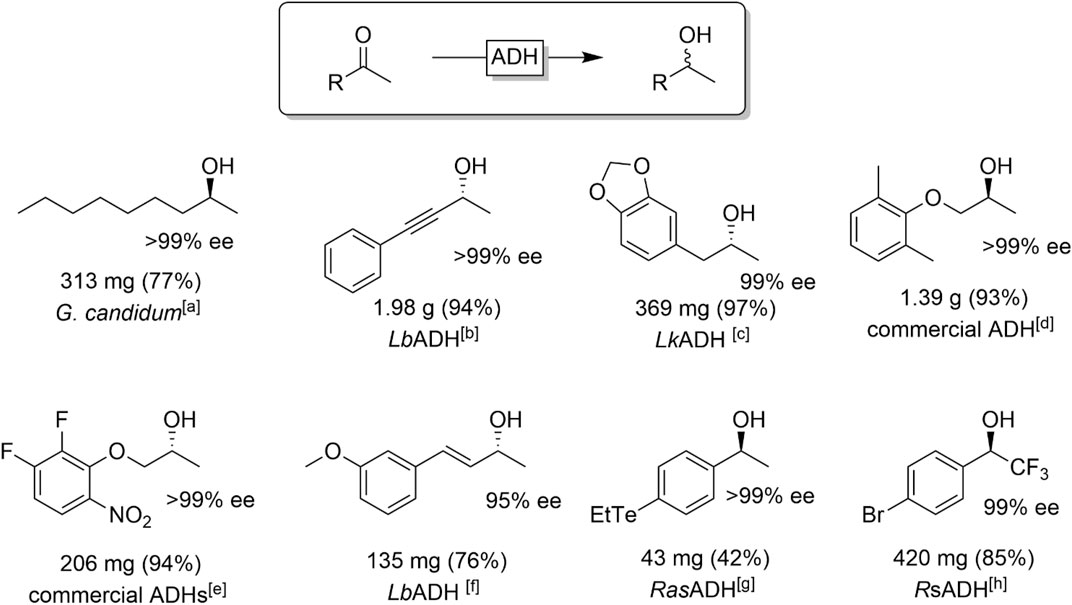
FIGURE 11. Selection of ADH-catalysed reductions of methyl ketones. [a] Geotrichum candidum acetone preparations (Nakamura and Matsuda, 1998), [b] LbADH (from Lactobacillus brevis) (Schubert et al., 2001), [c] LkADH (from Lactobacillus kefir) (Simon et al., 2014), [d] ADH from the commercial ADH screening KIT ’Chiralscreen OH′ (Nagai et al., 2018), [e]ADH-A (López-Iglesias et al., 2015), [f] LbADH (Albarrán-Velo et al., 2020) [g] RasADH (from Ralstonia sp.) (Bandeira et al., 2020), [h] RsADH (from Rhodococcus sp.) (Rosen et al., 2006).
Very recently, some of us proposed an extension of the ADH-catalysed stereoselective ketone reduction by a generation of the ketone starting material from non-functionalised starting materials (Xu et al., 2022). For this, we combined the peroxygenase-catalysed “through oxidation” of several alkylbenzenes to the corresponding acetophenones followed by the enantioselective reduction into the desired R- or S-alcohols. Instead of the peroxygenase also P450 monooxygenases (Both et al., 2016) or (photo)catalytic oxyfunctionalisation (Zhang et al., 2017; Gacs et al., 2019; Zhang et al., 2019; Albarrán-Velo et al., 2021) have been reported.
α-functionalised ketones represent another important class of ADH-starting materials (Figure 12). The resulting halohydrin products represent versatile building blocks via nucleophilic substitution reactions of the halides giving access to, e.g., epoxides (intramolecular substitution), amino alcohols or hydroxyl nitriles. Next to the often very high stereoselectivity of the reduction reaction another advantage of ADH-catalysis is that reductive dehalogenation reactions, frequently observed with transition metal catalysts, is generally not an issue (Erian et al., 2003; Mori et al., 2004).
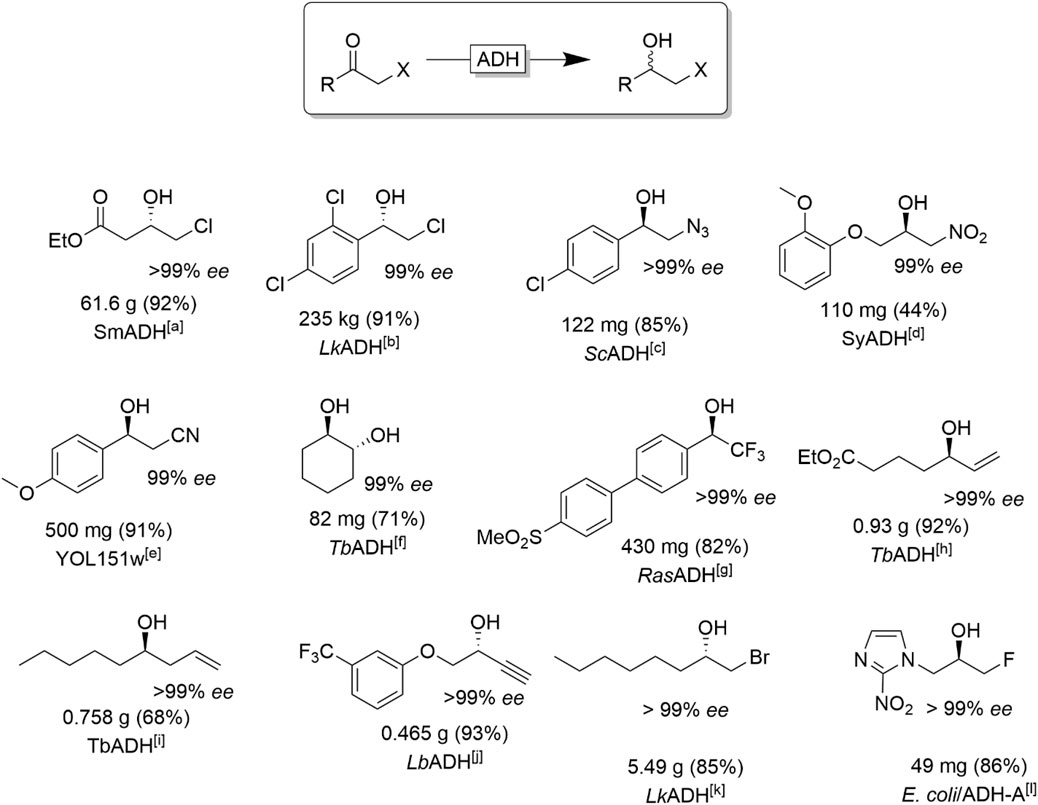
FIGURE 12. Selection of chiral α-functionalised alcohols obtained via stereoselective reduction of the corresponding ketone. [a] SmADH from Stenotrophomonas maltophilia (Yang et al., 2020), [b] engineered LkADH (from Lactobacillus kefir) (Zheng et al., 2021), [c] ScADH (from Saccharomyces cerevisiae) (Ankati et al., 2008), [d] (Wang et al., 2019), [e] YOL151w: Yeast ADH expressed in E. coli (Nowill et al., 2011), [f] (Zhang et al., 2018) [g] (Gonzalez-Martinez et al., 2019) [h] (Fischer and Pietruszka, 2012) [i] TbADH (from Thermoanaerobacter brokii) (Bisterfeld et al., 2017), [j] LbADH (from Lactobacillus brevis)(Holec et al., 2015). [k] engineered LkADH (from Lactobacillus kefir), (Berkessel et al., 2007), [l] overexpressed ADH-A (from Rhodococcus ruber) (Borzecka et al., 2013 ).
Several aryl, benzyl and alkyl chlorohydrins (Hanson et al., 2005; Poessl et al., 2005; Berkessel et al., 2007; Schrittwieser et al., 2009a; Schrittwieser et al., 2009b; Zheng et al., 2021), and, to a lesser extent, bromo-(Mangas-Sánchez et al., 2011) and fluorohydrins (Borzecka et al., 2013) have been synthesised on (semi-)preparative scale.
The large repertoire of halohydrins derived from ADH-catalysed reduction of the corresponding ketones (Figure 12) also includes propargylic chlorohydrins (Schubert et al., 2002) and compounds bearing functional groups such as esters (Yang et al., 2020), amino-protected groups (Patel, 2001; de Miranda et al., 2015) and heteroaryl moieties (Borzecka et al., 2013). Importantly, some relevant chlorohydrins have been produced by ADH in practical scale, such as the enantiopure ethyl (S)-chloroacetoacetate (Yang et al., 2020), a chiral synthetic intermediate of the “blockbuster” drug atorvastatin, and (S)-2-chloro-1-(2,4-dichlorophenyl) ethanol (Zheng et al., 2021), an intermediate of the antifungal agent luliconazole.
In addition of halo-substituted alcohols also ß-azido- (Edegger et al., 2006a; Cuetos et al., 2013), ß-nitro alcohols (Tentori et al., 2018; Wang et al., 2019) and ß-nitrile alcohols (Ankati et al., 2008) are accessible. Reduction of α-hydroxy carbonyls results in chiral vicinal diols (Kihumbu et al., 2002; Wachtmeister et al., 2014; Kulig et al., 2019; Muschallik et al., 2020). Chiral 2,2,2-trifluoro-1-arylethanols are important motifs in medicinal chemistry and drug development and therefore have also extensively been investigated as targets for ADH-catalysis (Rosen et al., 2006; Hussain et al., 2008; Adebar et al., 2019; Gonzalez-Martinez et al., 2019).
An interesting application of the ADH-catalysed reduction of α-haloketones was established by Kroutil and coworkers (Figure 13) (Schrittwieser et al., 2009a) In the first step ADH-catalysed reduction of α-chloro ketones gave access to enantiomerically pure β-chloro alcohols. The latter are substrates for halohydrine dehalogenases (Hhe’s) which reversibly dehalogenate them into epoxides. Utilising the reversibility of this step and shifting the equilibrium by an excess of alternative nucleophiles (such as azide or cyanide) a range of optically pure β-azido- and β-cyano-alcohols could be obtained in 50 mg scale.
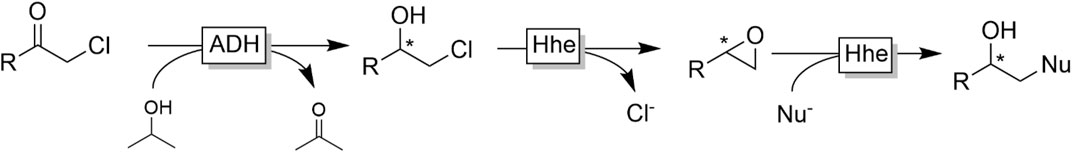
FIGURE 13. Bienzymatic cascade to transform α-chloroketones into β-substituted chiral alcohols combining stereoselective ADH-catalysed reduction of the chloroketone with a halohydrin dehalogenase (Hhe)-catalysed substitution of the chlorine with a nucleophile (occurring via an intermediate epoxide).
A very unconventional conversion of α-substituted carbonyl compounds was developed by Hyster and coworkers (Emmanuel et al., 2016; Biegasiewicz et al., 2018). Upon illumination with blue light, ADH-bound NAD(P)H can serve as single electron donor. In case of α-halo- or α-acetoxy-substituents, serving as leaving groups, light-induced dehalogention or deacetoxylation reactions have been observed (Figure 14).
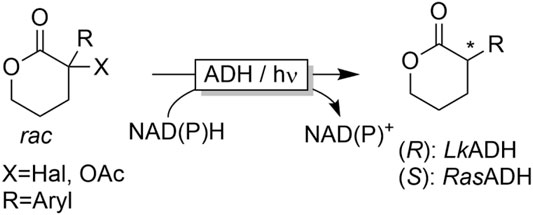
FIGURE 14. Photoenzymatic reductive dehalogenation of α-substituted carbonyl compounds.
Dicarbonyl starting materials have found significant attention in the past years as substrates for ADH-catalysed reduction reactions (Figure 15).
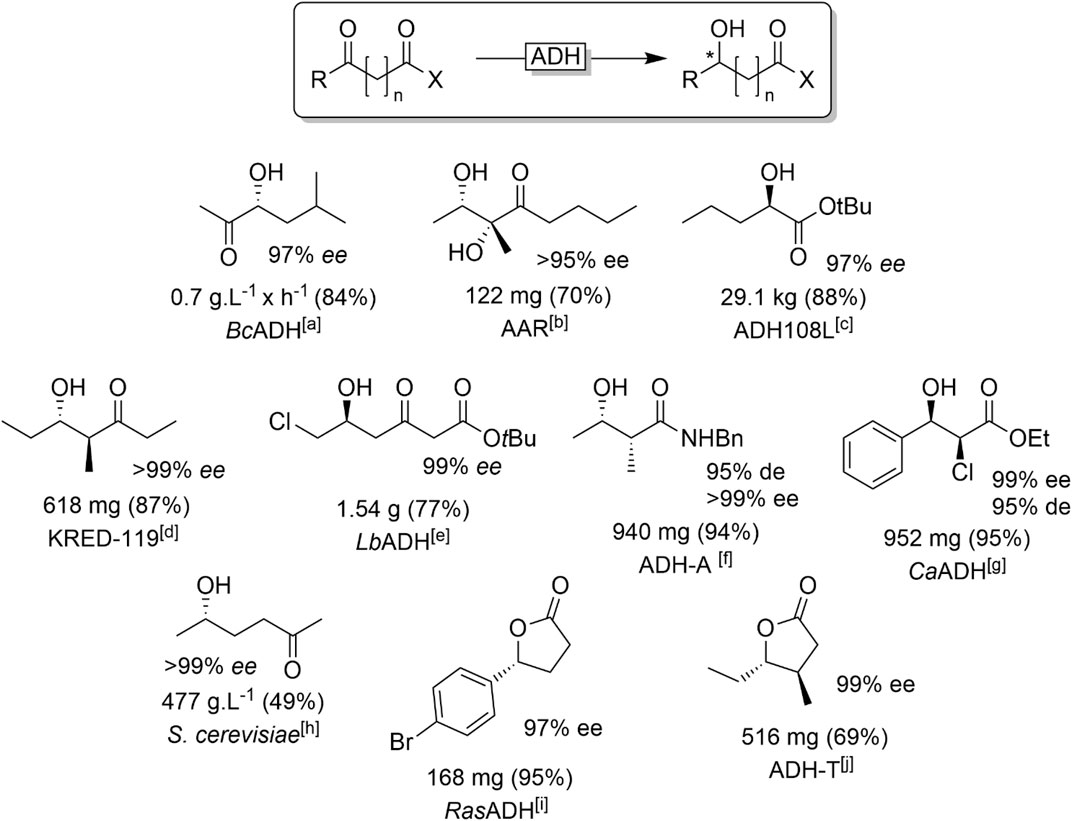
FIGURE 15. Representative examples of products obtained from ADH-catalysed reduction of dicarbonyl compounds. [a] BcADH (from Bacillus clausii) (Muschallik et al., 2020), [b] AAR (acetylacetoinreductase) (Di Carmine et al., 2018), [c] commercial ADH (Burns et al., 2017), [d] KRED-119: commercial ADH (Kalaitzakis et al., 2006), [e] LbADH (from Lactobacillus brevis) (Wolberg et al., 2000), [f] ADH-A (from Rhodococcus ruber) (Méndez-Sánchez et al., 2019), [g] CaADH (from Clostridium acetobutylicum) (Applegate et al., 2011), [h] Baker’s yeast (Katzberg et al., 2009), [i] RasADH (from Ralstonia) (Díaz-Rodríguez et al., 2014), [j] ADH-T (from Thermoanaerobacter sp.) (Kumru et al., 2018).
α-hydroxy acids have been synthesised from the corresponding keto acids (Zhu et al., 2006a; Pennacchio et al., 2008; Applegate et al., 2011; Burns et al., 2017). Starting from 1,2-diketo compounds, selective monoreduction to the corresponding α-hydroxyketone is challenging due to the frequently observed overreduction to the dialcohol (Hoyos et al., 2008; Monsalve et al., 2010; Pal et al., 2015) necessitating reaction engineering approaches to maximise the yield in the desired monoreduction product (Muschallik et al., 2020). Some new ADHs showing some potential for the selective monoreduction have been identified recently (Shanati et al., 2019). Similarly, β-hydroxy esters are accessible via various ADHs (Hummel et al., 2003; Muller, 2005; Zadlo et al., 2016; Wang et al., 2020) as well as ß-hydroxy ketones (Ludeke et al., 2009). The bioreduction of 1,4-diketones has been investigated for a long time already (Haberland et al., 2002a; Haberland et al., 2002b; Katzberg et al., 2009; Muller et al., 2010; Mourelle-Insua et al., 2018). Similar to 1,2-diketones, selective monoreduction so far has been achieved via kinetic control of the overall reaction.
α-substituted ß-diketones are prone to in situ racemisation of the enolisable C-H bond. This opens up the possibility of generating two chiral centers at the same time in a single ADH-catalysed reduction step provided the ADH exhibits stereoselectivity for both positions, i.e., preferentially converting one substrate isomer and performing the carbonyl reduction reaction stereoselectively (Ji et al., 2001; Kalaitzakis et al., 2006; Kalaitzakis and Smonou, 2010b; a; Giovannini et al., 2011; Kalaitzakis and Smonou, 2012; Méndez-Sánchez et al., 2019).
ɣ- and δ-ketoesters are attractive targets for ADH-catalysed reduction reactions as the corresponding alcohols easily (often spontaneously) undergo intramolecular esterification. The resulting lactones are common motifs in natural products (Korpak and Pietruszka, 2011; Fischer and Pietruszka, 2012; Classen et al., 2014; Díaz-Rodríguez et al., 2014; Kumru et al., 2018; Borowiecki et al., 2020). Starting from synthetically easily accessible substituted conjugated ketoesters bienzymatic cascades comprising ene reductases (ERs) and ADHs enable access to a broad range of chiral ɣ-butyrolactone products (Figure 16) (Korpak and Pietruszka, 2011; Classen et al., 2014; Brenna et al., 2015).
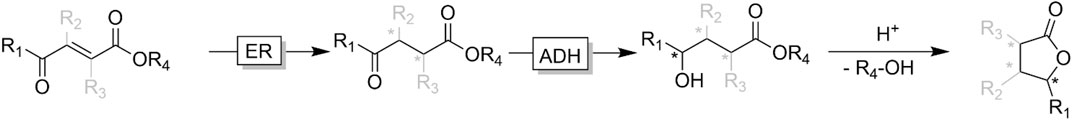
FIGURE 16. Bienzymatic cascade transforming conjugated ketoacid esters into lactones. In the first step, an ene reductase (ER) catalysed the reduction of the conjugated C=C-bond followed by ADH-catalysed keto reduction and acid-catalysed intramolecular lactonisation. For reasons of simplicity, enzyme cofactors and absolute stereochemistries have been omitted.
α,β-unsaturated carbonyl groups are popular starting materials for ADH-ER-cascades. Successful realisation of such cascades critically depends on the suitable selection of selective ADHs as principally both, the α,β-unsaturated starting material and the saturated carbonyl compound can be converted by ADHs. Hence, utilising unselective ADHs results in complex product mixtures (Paul et al., 2013) necessitating sequential arrangement of the individual reduction steps. Using the “right” combination of selective ADHs and ERs, however, enables efficient one-pot one-step cascades (Brenna et al., 2012a; b).
Finally, the reduction of so-called “bulky-bulky”-ketones, i.e., sterically very demanding starting materials, are worth discussing here. These substrates have been a challenge for stereoselective reductions mediated by ADHs for quite some time. Nevertheless, some useful enzymes have been reported comprising the ADHs from Sphingobium yanoikuyae (SyADH) (Lavandera et al., 2008a; Cuetos et al., 2012; Man et al., 2014), Ralstonia sp. (RasADH) (Lavandera et al., 2008a; Cuetos et al., 2012; Man et al., 2014), Sporobolomyces salmonicolor (SsADH) (Zhu and Hua, 2006; Li et al., 2009; Li et al., 2010; Chen et al., 2012) Kluyveromyces polysporus (KpADH) (Xu et al., 2018) Clostridium acetobutylicum (CaADH) (Applegate et al., 2011), hydroxysteroid dehydrogenases (Zhu et al., 2006a; Ferrandi et al., 2020) and enzymes from Kluyveromyces marxianus (Li et al., 2019) or Kluyveromyces polysporus (Xu et al., 2018; Zhou et al., 2020). A selection of bulky ketones reduced by ADHs is given in Figure 17.
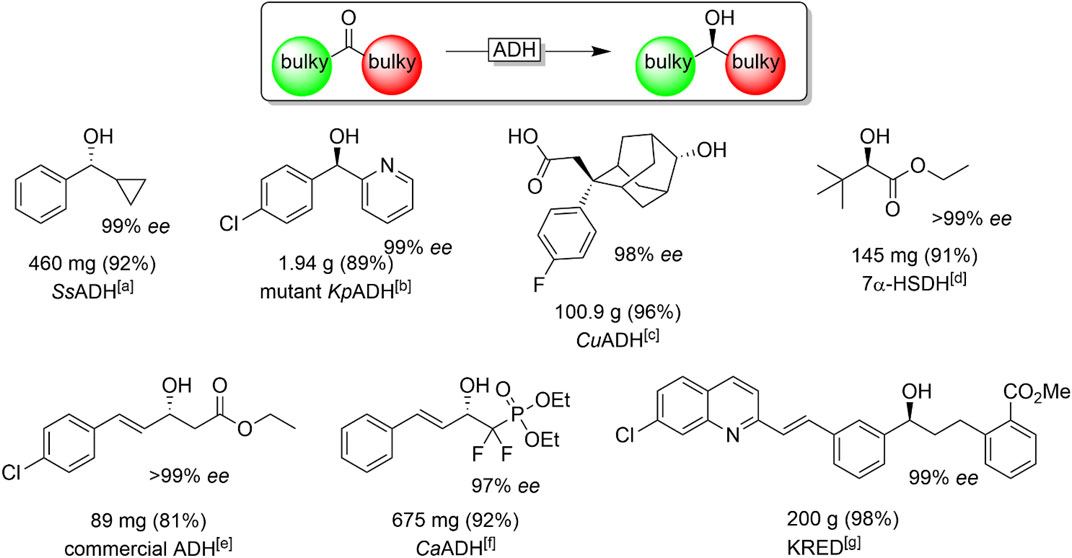
FIGURE 17. Chiral alcohols synthesised through ADH-catalysed asymmetric reduction of "bulky-bulky" ketones. [a] SsADH (from Sporobolomyces salmonicolor) (Zhu and Hua, 2006), [b] KpADH (mutant of the ADH from Kluyveromyces polysporus) (Xu et al., 2018), [c] CuADH (from Candida utilis) (Hanson et al., 2014), [d] 7-αHSDH (from Bacteroides fragilis) (Zhu et al., 2006a), [e] (Dai et al., 2013) [f] CaADH (from Clostridium acetobutylicum) (Panigrahi et al., 2015), [g] KRED: evolved ADH from the company CODEXIS (Liang et al., 2010a).
Reduction of aldehydes. In contrast to the reduction of ketones, aldehyde reductions are far less popular, which can largely be attributed to the fact that generally the reduction of an aldehyde group does not result in the formation of a chiral centre (chiral alcohol).
Nevertheless, some interesting applications of ADH-catalysed aldehyde reduction have been reported. Gröger and coworkers, for example, established an organo/biocatalytic cascade to access (R)-pantolactone from simple starting materials (Figure 18) (Heidlindemann et al., 2015) The ADH-step partially functioned as kinetic resolution thereby upgrading the comparably poor optical purity of the organocatalytic product.
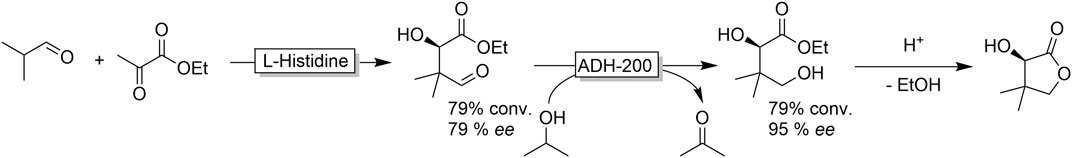
FIGURE 18. Cascade reaction combining organocatalytic (L-histidine-catalysed aldol reaction) C-C-bond formation and ADH-catalysed aldehyde reduction followed by acid-catalysed lactonisation to form (R)-pantolactone.
Hall and coworkers reported an ADH-catalysed, overall redox-neutral cascade reaction transforming dialdehydes into lactones (Tishchenko-type reaction, Figure 19) (Tassano et al., 2020).
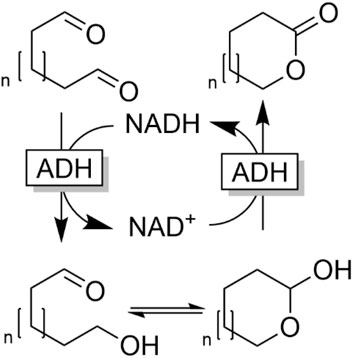
FIGURE 19. Tishchenko-type dismutation of dialdehydes by ADH-catalysed redox-neutral conversion of dialdehydes into lactones.
Also starting from dialdehydes Zhang et al. established an elegant synthesis of optically pure cyclic diols by combining the pyruvate decarboxylase (PDC)-catalysed aldol-type ring closure and ADH-catalysed reduction of the intermediate cyclic α-hydroxy ketone (Figure 20) (Zhang et al., 2018).
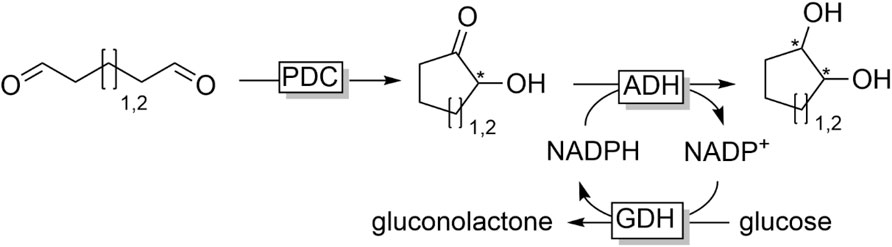
FIGURE 20. Combination of pyruvate decarboxylase-catalysed aldol-type ring closure of dialdehydes with ADH-catalysed reduction of the α-hydroxyketone intermediate.
ADHs often not only exhibit enantioselectivity with respect to the hydride transfer (yielding R- or S-alcohols) but can also discriminate between enantiomers at other positions. This has, for example, been exploited for the kinetic resolution of racemic aldehydes such as binaphthyls (Kawahara et al., 1988), biaryls (Yuan et al., 2010; Staniland et al., 2014) or ferrocenes (Yamazaki and Hosono, 1988). Aldehydes bearing a chiral centre in α-position represent attractive targets for stereoselective ADH-catalysed reduction especially under α-racemising conditions. Particularly the dynamic kinetic resolution of profene aldehydes to enantiomerically pure profene alcohols has been investigated by several groups (Giacomini et al., 2007; Friest et al., 2010; Rapp et al., 2021).
Reduction of acids. Carboxylic acids cannot be reduced by ADHs; the carboxylate group is thermodynamically and kinetically inert towards serving as hydride acceptor from NAD(P)H. Carboxylate reductases (CARs) circumvent this limitation by activating the carboxylate group as thioester (Winkler, 2018). Inspired by this strategy, we have evaluated the general possibility of reducing (chemically synthesised) thioesters with common ADHs (Figure 21) (Younes et al., 2017).
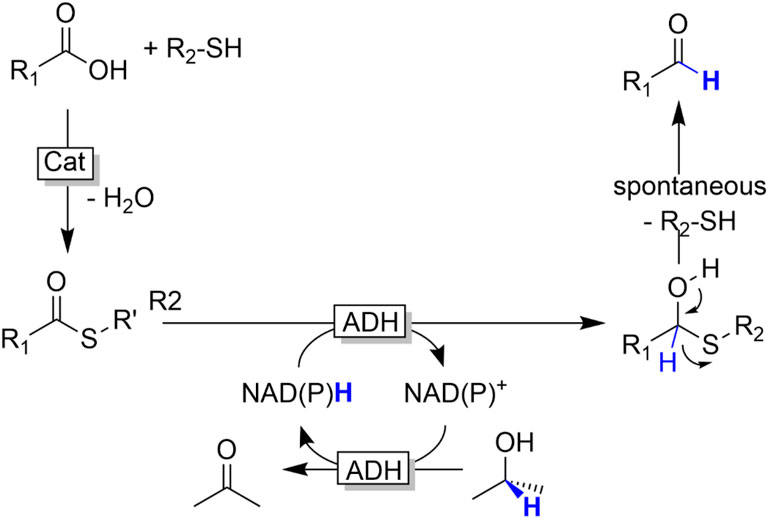
FIGURE 21. ADH-catalysed reduction of carboxylic acids activated as thioesters.
The oxidation of an alcohol to the corresponding carbonyl group is accompanied by the destruction of a (potential) chiral centre rather than the generation of one as in case of carbonyl reduction reactions. This seeming limitation has for a long time limited ADH-oxidation reactions to the kinetic resolution of racemic secondary alcohols. Some examples are shown in Figure 22.
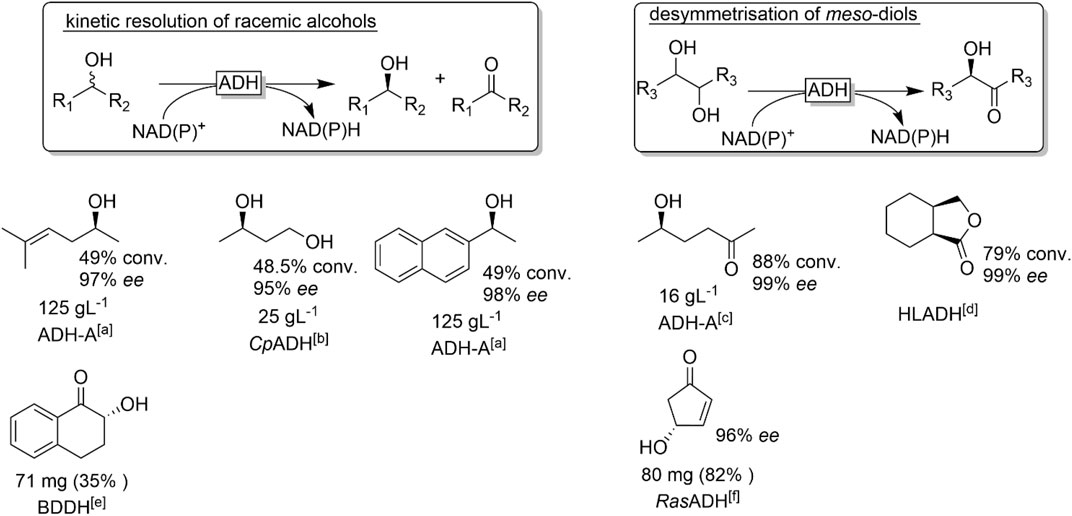
FIGURE 22. Representative examples of chiral alcohol oxidation products derived from the kinetic resolution of racemic alcohols or the desymmetrisation of meso-diols. [a] ADH-A (from Rhodococcus ruber) (Stampfer et al., 2002), [b] CpADH (from Candida parapsilosis) (Matsuyama et al., 2001) [c] (Edegger et al., 2006b) [d] via intramolecular lactonisation, HLADH: horse liver ADH (Irwin and Jones, 1977a; Irwin and Jones, 1977b) [e] 2,3-butanediol dehydrogenase from Bacillus subtilis (Zhang et al., 2013), [f] (Holec et al., 2015).
The intrinsic limitation of kinetic resolution reactions of having a theoretical yield of only 50% (provided the catalyst exhibits high enantioselectivity) can be overcome making use of the meso-trick. Here, a prochiral meso-alcohol is oxidised completely to (ideally) one product enantiomer. Obviously, the scope of this approach is rather limited to meso-starting materials. A more general approach to obtain enantiomerically pure alcohols has been proposed by Kroutil and coworkers by combining two enantiocomplementary ADHs (one for the kinetic resolution of the racemic alcohol and the other one for the stereoselective reduction of the ketone intermediate into the desired alcohol enantiomer). If both ADHs also exhibit exclusivity for either the phosphorylated and non-phosphorylated nicotinamide cofactor, the overall reaction can be conducted as a one-pot one-step reaction (Figure 23) (Voss et al., 2008a; Voss et al., 2008b; Voss et al., 2010).
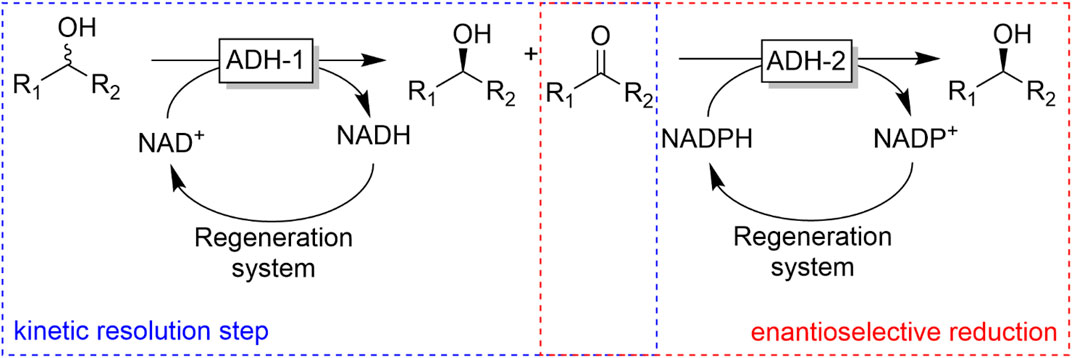
FIGURE 23. Bienzymatic deracemisation of racemic alcohols using two enantiocomplementary ADHs.
ADH-catalysed oxidations also play a key role in various multi-step cascades valorising simple starting materials into more complex products. For example, monooxygenase-catalysed hydroxylation reactions of non-functionalised C-H-bonds yield alcohols that can further be converted into the corresponding ketones (Figure 24A) (Müller et al., 2013; Staudt et al., 2013; Tavanti et al., 2017a; Tavanti et al., 2017b). Similarly, starting from alcohols, redox-neutral ADH-monooxygenase cascades producing lactones are possible (Figure 24B) (Schmidt et al., 2015a; Schmidt et al., 2015b; Scherkus et al., 2016; Scherkus et al., 2017; Wedde et al., 2017). Also combining both approaches has been demonstrated by Opperman and coworkers (Pennec et al., 2015).
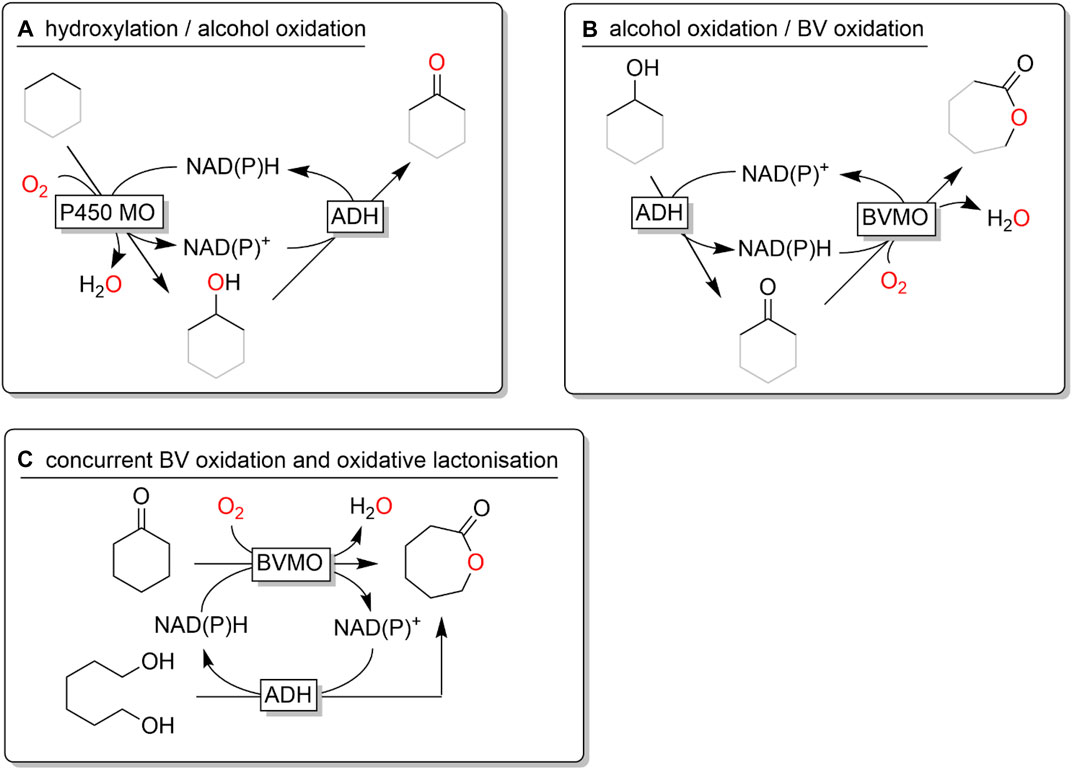
FIGURE 24. Examples of redox-neutral cascades involving ADH-catalysed alcohol oxidation reactions. (A) combining monooxygenase-catalysed hydroxylation with ADH-catalysed alcohol oxidation; (B) combining ADH-catalysed alcohol oxidation with monooxygenase-catalysed Baeyer-Villiger oxidation; (C) concurrent lactone formation comining monooxygenase-catalysed Baeyer-Viliger oxidation with ADH-catalysed oxidative lactonisation.
An interesting convergent cascade combining Baeyer-Villiger monooxygenase-catalysed synthesis of lactones with ADH-catalysed cofactor regeneration yielding the desired lactone as (co-)product has been proposed by Kara and coworkers (Figure 24C) (Bornadel et al., 2016; Huang et al., 2017).
Other cascades involving ADH-catalysed alcohol oxidations have also been reported in combination with ene-reductase-catalysed C=C-bond reductions (Gargiulo et al., 2012; Oberleitner et al., 2013), transaminase-catalysed (Tauber et al., 2013; Corrado et al., 2021) or amine reductase-catalysed (Tavanti et al., 2017a; Dennig et al., 2019) reductive aminations or obtaining the alcohol via hydratation of an existing C=C-bond (Koppireddi et al., 2016; Zhang W. et al., 2020; Cha et al., 2020).
As far as primary alcohols are concerned, ADHs generally catalyse the selective oxidation to the aldehyde stage. Further oxidation generally does not occur as the aldehyde proton is not abstractable as a hydride. If desired, this situation can be changed by transiently converting the aldehyde into its corresponding gem-diol (preferentially by adjusting the reaction pH to alkaline values). The latter does contain a hydridically abstractable H-atom and therefore can also be oxidised to the corresponding acid (Figure 25) (Könst et al., 2012).
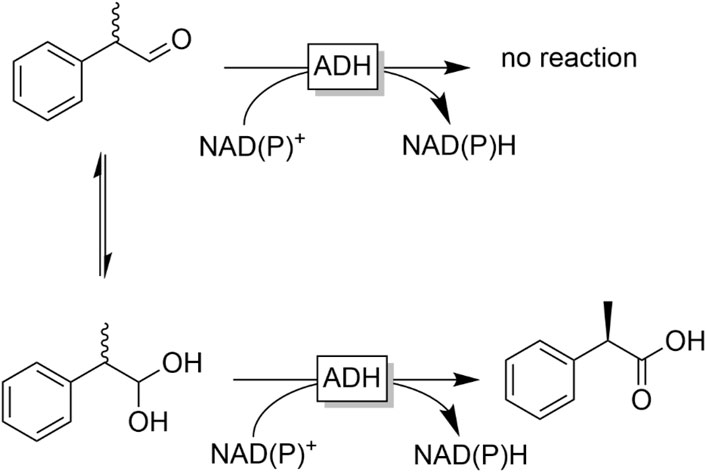
FIGURE 25. Oxidation of aldehyde-gem-diols to the corresponding acid. Under enolising conditions the reaction can also be performed as dynamic kinetic resolution of chiral α-substituted aldehydes. This strategy also allows for the full oxidation of alcohols to carboxylic acids as recently demonstrated by Paradisi and coworkers (Contente et al., 2020).
Biocatalytic reactions are frequently labelled as “green” (i.e., environmentally benign). To substantiate this claim, arguments such as enzymes being bio-based and bio-degradable (in contrast to transition metal catalysts), the mild reaction conditions (in contrast to frequently high reaction temperatures needed for chemical reactions) and the use of water as reaction medium (in contrast to organic solvents generally used in chemical reactions) are used.
Such qualitative arguments, however, are too short-sighted. First of all, one should be aware that any chemical transformation, irrespective if it is enzyme-catalysed or based on “traditional” chemical technologies, represents an environmental burden. Reactions consume energy to heat/cool and stir/pump the reaction mixtures, which still causes greenhouse gas emissions albeit indirectly and not always obvious for the user. The production of enzymes consumes resources and also biocatalytic reactions generate wastes (Tieves et al., 2019). Finally, water can be considered as green solvent only until after its usage as then it is generally contaminated with reactants and cannot be simply disposed into the environment and has to be considered as waste (Holtmann and Hollmann, 2022). Overall, a chemical transformation does not per se turn environmentally if performed using an enzyme catalyst.
Enzymatic reactions can exhibit significant environmental benefits (i.e., they can be greener) over traditional chemical syntheses. But it is not enough just to repeat the boilerplate arguments mentioned above. A quantitative, comparative assessment of the environmental impact of alternative routes is necessary to claim greenness. Ideally, such comparisons include the entire production chain from raw materials to the final product, as full life cycle assessments typically aim at (Eissen et al., 2009; Tufvesson et al., 2012). The extensive data basis required for such LCAs usually significantly goes beyond the possibilities of lab researchers. Therefore, simplified mass-based metrics such as Sheldon’s E-Factor (Sheldon et al., 2022) (E for Environmental) and its derivates (Sheldon, 2017; 2018) represent a doable compromise to compare different synthetic routes based on their waste generation.
We encourage the readers of this contribution to utilise the E-Factor to assess the wastes generated in their own reactions and to utilise the result as a starting point for further improvements. In other words, an honest E-Factor analysis will reveal the real issues of a given reaction instead of perceived ones and can serve as a guiding principle to plan new experiments. A “bad” E-Factor can be a good starting point for improvements.
Alcohol dehydrogenases can nowadays be considered as established catalysis in organic synthesis. ADHs enable the selective oxidation of alcohols (and aldehydes) as well as reduction of aldehydes and ketones on preparative scale under mild reaction conditions.
Limitations stressed in the past such as limited substrate scope, limited stability or their dependence on costly nicotinamide cofactors have been solved in the past decades. Protein engineering has become a standard technique to tailor the substrate scope, selectivity and stability of a given ADH. Also ADH stability under non-natural conditions (such as high reagent concentrations or elevated temperatures) does not represent a preparative hurdle any more as, e.g., engineered ADH mutants are available and/or reaction engineering measures (such as immobilisation of the enzymes) can be utilised.
So far, ADHs have been valued mostly for their enantioselectivity giving access to optically pure chiral fine chemicals and pharmaceutical intermediates. We do expect that the scope of ADH-catalysis will expand also to the synthesis of commodity and bulk chemicals. The tighter cost requirements for such products will necessitate to focus more on performance indicators such as space-time yields, final product titres and turnover numbers of the enzymes and cofactors. Given the rapid development of ADH catalysis in the past, we are convinced that soon ADHs will have become indispensable catalysts for all chemical synthesis routes.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Financial support of the Sao Paulo Research Foundation—FAPESP (grants #2014/50249-8), National Institute of Science and Technology—INCT BioNat, (CNPQ grant #465637/2014-0, FAPESP grant #2014/50926-0, Brazil).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Brazilian authors also thank CAPES for maintaining the CAPES Periodical Portal.
Aalbers, F. S., Fürst, M. J., Rovida, S., Trajkovic, M., Gómez Castellanos, J. R., Bartsch, S., et al. (2020). Approaching Boiling Point Stability of an Alcohol Dehydrogenase through Computationally-Guided Enzyme Engineering. eLife 9. doi:10.7554/eLife.54639
Acevedo-Rocha, C. G., Hollmann, F., Sanchis, J., and Sun, Z. (2020). A Pioneering Career in Catalysis: Manfred T. Reetz. ACS Catal. 10, 15123–15139. doi:10.1021/acscatal.0c04108
Adebar, N., Choi, J. E., Schober, L., Miyake, R., Iura, T., Kawabata, H., et al. (2019). Overcoming Work‐Up Limitations of Biphasic Biocatalytic Reaction Mixtures through Liquid‐Liquid Segmented Flow Processes. ChemCatChem 11 (23), 5788–5793. doi:10.1002/cctc.201901107
Adebar, N., and Gröger, H. (2020). Heterogeneous Catalysts "on the Move": Flow Chemistry with Fluid Immobilised (Bio)Catalysts. Eur. J. Org. Chem. 2020, 6062–6067. doi:10.1002/ejoc.202000705
Agudo, R., Roiban, G.-D., and Reetz, M. T. (2013). Induced Axial Chirality in Biocatalytic Asymmetric Ketone Reduction. J. Am. Chem. Soc. 135 (5), 1665–1668. doi:10.1021/ja3092517
Aksu, S., Arends, I. W. C. E., and Hollmann, F. (2009). A New Regeneration System for Oxidized Nicotinamide Cofactors. Adv. Synth. Catal. 351 (9), 1211–1216. doi:10.1002/adsc.200900033
Albarrán‐Velo, J., Gotor‐Fernández, V., and Lavandera, I. (2021). Markovnikov Wacker‐Tsuji Oxidation of Allyl(hetero)arenes and Application in a One‐Pot Photo‐Metal‐Biocatalytic Approach to Enantioenriched Amines and Alcohols. Adv. Synth. Catal. 363 (16), 4096–4108. doi:10.1002/adsc.202100351
Albarrán-Velo, J., Gotor-Fernández, V., and Lavandera, I. (2020). One-pot Two-step Chemoenzymatic Deracemization of Allylic Alcohols Using Laccases and Alcohol Dehydrogenases. Mol. Catal. 493, 111087. doi:10.1016/j.mcat.2020.111087
Almeida, V. M., and Marana, S. R. (2019). Optimum Temperature May Be a Misleading Parameter in Enzyme Characterization and Application. PLoS One 14 (2), e0212977. doi:10.1371/journal.pone.0212977
Anderson, S. R., Bommarius, B. R., Woodley, J. M., and Bommarius, A. S. (2021). Sparged but Not Stirred: Rapid, ADH-NADH Oxidase Catalyzed Deracemization of Alcohols in a Bubble Column. Chem. Eng. J. 417, 127909. doi:10.1016/j.cej.2020.127909
Ankati, H., Yang, Y., Zhu, D., Biehl, E. R., and Hua, L. (2008). Synthesis of Optically Pure 2-Azido-1-Arylethanols with Isolated Enzymes and Conversion to Triazole-Containing β-Blocker Analogues Employing Click Chemistry. J. Org. Chem. 73 (16), 6433–6436. doi:10.1021/jo8009616
Applegate, G. A., Cheloha, R. W., Nelson, D. L., and Berkowitz, D. B. (2011). A New Dehydrogenase from Clostridium Acetobutylicum for Asymmetric Synthesis: Dynamic Reductive Kinetic Resolution Entry into the Taxotère Side Chain. Chem. Commun. 47 (8), 2420–2422. doi:10.1039/C0CC04585C
Bandeira, P. T., Gotor-Fernández, V., and Piovan, L. (2020). Stereoselective Bioreduction of Telluro-Acetophenones to Optically Active Hydroxy Tellurides. Eur. J. Org. Chem. 2020 (9), 1129–1135. doi:10.1002/ejoc.201901841
Bell, E. L., Finnigan, W., France, S. P., Green, A. P., Hayes, M. A., Hepworth, L. J., et al. (2021). Biocatalysis. Nat. Rev. Methods Prim. 1 (1), 46. doi:10.1038/s43586-021-00044-z
Benítez-Mateos, A. I., San Sebastian, E., Ríos-Lombardía, N., Morís, F., González-Sabín, J., and López-Gallego, F. (2017). Asymmetric Reduction of Prochiral Ketones by Using Self-Sufficient Heterogeneous Biocatalysts Based on NADPH-dependent Ketoreductases. Chem. Eur. J. 23 (66), 16843–16852. doi:10.1002/chem.201703475
Berkessel, A., Rollmann, C., Chamouleau, F., Labs, S., May, O., and Gröger, H. (2007). Practical Two-step Synthesis of an Enantiopure Aliphatic Terminal (S)-Epoxide Based on Reduction of Haloalkanones with "Designer Cells". Adv. Synth. Catal. 349 (17-18), 2697–2704. doi:10.1002/adsc.200700244
Biegasiewicz, K. F., Cooper, S. J., Emmanuel, M. A., Miller, D. C., and Hyster, T. K. (2018). Catalytic Promiscuity Enabled by Photoredox Catalysis in Nicotinamide-dependent Oxidoreductases. Nat. Chem. 10 (7), 770–775. doi:10.1038/s41557-018-0059-y
Bisterfeld, C., Holec, C., Böse, D., Marx, P., and Pietruszka, J. (2017). Chemoenzymatic Total Synthesis of the Proposed Structures of Putaminoxins B and D. J. Nat. Prod. 80 (5), 1563–1574. doi:10.1021/acs.jnatprod.7b00101
Bodor, A., Bounedjoum, N., Vincze, G. E., Erdeiné Kis, Á., Laczi, K., Bende, G., et al. (2020). Challenges of Unculturable Bacteria: Environmental Perspectives. Rev. Environ. Sci. Biotechnol. 19 (1), 1–22. doi:10.1007/s11157-020-09522-4
Böhmer, W., Knaus, T., and Mutti, F. G. (2018). Hydrogen-Borrowing Alcohol Bioamination with Coimmobilized Dehydrogenases. ChemCatChem 10 (4), 731–735. doi:10.1002/cctc.201701366
Boller, T., Meier, C., and Menzler, S. (2002). EUPERGIT Oxirane Acrylic Beads: How to Make Enzymes Fit for Biocatalysis. Org. Process Res. Dev. 6 (4), 509–519. doi:10.1021/op015506w
Boratyński, F., Kiełbowicz, G., and Wawrzeńczyk, C. (2010). Lactones 34 [1]. Application of Alcohol Dehydrogenase from Horse Liver (HLADH) in Enantioselective Synthesis of δ- and Ɛ-Lactones. J. Mol. Catal. B Enzym. 65 (1-4), 30–36. doi:10.1016/j.molcatb.2010.01.025
Boratyński, F., Smuga, M., and Wawrzeńczyk, C. (2013). Lactones 42. Stereoselective Enzymatic/microbial Synthesis of Optically Active Isomers of Whisky Lactone. Food Chem. 141 (1), 419–427. doi:10.1016/j.foodchem.2013.02.106
Bornadel, A., Hatti-Kaul, R., Hollmann, F., and Kara, S. (2016). Enhancing the Productivity of the Bi-enzymatic Convergent Cascade for Ɛ-Caprolactone Synthesis through Design of Experiments and a Biphasic System. Tetrahedron 72, 7222–7228. doi:10.1016/j.tet.2015.11.054
Bornscheuer, U. T., Huisman, G. W., Kazlauskas, R. J., Lutz, S., Moore, J. C., and Robins, K. (2012). Engineering the Third Wave of Biocatalysis. Nature 485 (7397), 185–194. doi:10.1038/nature11117
Borowiecki, P., Telatycka, N., Tataruch, M., Żądło‐Dobrowolska, A., Reiter, T., Schühle, K., et al. (2020). Biocatalytic Asymmetric Reduction of γ‐Keto Esters to Access Optically Active γ‐Aryl‐γ‐butyrolactones. Adv. Synth. Catal. 362 (10), 2012–2029. doi:10.1002/adsc.201901483
Borzęcka, W., Lavandera, I., and Gotor, V. (2013). Biocatalyzed Synthesis of Both Enantiopure Fluoromisonidazole Antipodes. Tetrahedron Lett. 54 (37), 5022–5025. doi:10.1016/j.tetlet.2013.07.013
Both, P., Busch, H., Kelly, P. P., Mutti, F. G., Turner, N. J., and Flitsch, S. L. (2016). Whole-Cell Biocatalysts for Stereoselective C−H Amination Reactions. Angew. Chem. Int. Ed. 55 (4), 1511–1513. doi:10.1002/anie.201510028
Brenna, E., Gatti, F. G., Monti, D., Parmeggiani, F., and Sacchetti, A. (2012a). Cascade Coupling of Ene Reductases with Alcohol Dehydrogenases: Enantioselective Reduction of Prochiral Unsaturated Aldehydes. ChemCatChem 4 (5), 653–659. doi:10.1002/cctc.201100418
Brenna, E., Gatti, F. G., Monti, D., Parmeggiani, F., and Sacchetti, A. (2012b). Productivity Enhancement of CC Bioreductions by Coupling the In Situ Substrate Feeding Product Removal Technology with Isolated Enzymes. Chem. Commun. 48 (1), 79–81. doi:10.1039/C1CC16014A
Brenna, E., Gatti, F. G., Monti, D., Parmeggiani, F., Sacchetti, A., and Valoti, J. (2015). Substrate-engineering Approach to the Stereoselective Chemo-Multienzymatic Cascade Synthesis of Nicotiana Tabacum Lactone. J. Mol. Catal. B Enzym. 114, 77–85. doi:10.1016/j.molcatb.2014.12.011
Bühler, B., Bollhalder, I., Hauer, B., Witholt, B., and Schmid, A. (2003). Use of the Two-Liquid Phase Concept to Exploit Kinetically Controlled Multistep Biocatalysis. Biotechnol. Bioeng. 81 (6), 683–694. doi:10.1002/bit.10512
Bühler, B., and Schmid, A. (2004). Process Implementation Aspects for Biocatalytic Hydrocarbon Oxyfunctionalization. J. Biotechnol. 113 (1-3), 183–210. doi:10.1016/j.jbiotec.2004.03.027
Bühler, B., Witholt, B., Hauer, B., and Schmid, A. (2002). Characterization and Application of Xylene Monooxygenase for Multistep Biocatalysis. Appl. Environ. Microbiol. 68 (2), 560–568. doi:10.1128/AEM.68.2.560-568.2002
Burns, M., Martinez, C. A., Vanderplas, B., Wisdom, R., Yu, S., and Singer, R. A. (2017). A Chemoenzymatic Route to Chiral Intermediates Used in the Multikilogram Synthesis of a Gamma Secretase Inhibitor. Org. Process Res. Dev. 21 (6), 871–877. doi:10.1021/acs.oprd.7b00096
Carucci, C., Bruen, L., Gascón, V., Paradisi, F., and Magner, E. (2018). Significant Enhancement of Structural Stability of the Hyperhalophilic ADH from Haloferax Volcanii via Entrapment on Metal Organic Framework Support. Langmuir 34 (28), 8274–8280. doi:10.1021/acs.langmuir.8b01037
Cha, H. J., Hwang, S. Y., Lee, D. S., Kumar, A. R., Kwon, Y. U., Voss, M., et al. (2020). Whole‐Cell Photoenzymatic Cascades to Synthesize Long‐Chain Aliphatic Amines and Esters from Renewable Fatty Acids. Angew. Chem. Int. Ed. 59 (18), 7024–7028. doi:10.1002/anie.201915108
Chang, A., Scheer, M., Grote, A., Schomburg, I., and Schomburg, D. (2009). BRENDA, AMENDA and FRENDA the Enzyme Information System: New Content and Tools in 2009. Nucleic Acids Res. 37, D588–D592. doi:10.1093/nar/gkn820
Chen, C., Xuan, Y., Chen, Q., Ni, G.-W., Pan, J., and Xu, J.-H. (2021). Asymmetric Reduction of 2-Chloro-3-Oxo-Ester into Enantiomerically High Pure Diltiazem Precursor by a Candida Ketoreductase. Mol. Catal. 510, 111670. doi:10.1016/j.mcat.2021.111670
Chen, X., Gao, X., Wu, Q., and Zhu, D. (2012). Synthesis of Optically Active Dihydrocarveol via a Stepwise or One-Pot Enzymatic Reduction of (R)- and (S)-carvone. Tetrahedron Asymmetry 23 (10), 734–738. doi:10.1016/j.tetasy.2012.05.019
Chen, Y., Jiang, Q., Sun, L., Li, Q., Zhou, L., Chen, Q., et al. (2018). Magnetic Combined Cross-Linked Enzyme Aggregates of Ketoreductase and Alcohol Dehydrogenase: An Efficient and Stable Biocatalyst for Asymmetric Synthesis of (R)-3-Quinuclidinol with Regeneration of Coenzymes In Situ. Catalysts 8 (8), 334. doi:10.3390/catal8080334
Chenault, H. K., and Whitesides, G. M. (1987). Regeneration of Nicotinamide Cofactors for Use in Organic Synthesis. Appl. Biochem. Biotechnol. 14 (2), 147–197. doi:10.1007/bf02798431
Classen, T., Korpak, M., Schölzel, M., and Pietruszka, J. (2014). Stereoselective Enzyme Cascades: An Efficient Synthesis of Chiral γ-Butyrolactones. ACS Catal. 4 (5), 1321–1331. doi:10.1021/cs5000262
Contente, M. L., Fiore, N., Cannazza, P., Roura Padrosa, D., Molinari, F., Gourlay, L., et al. (2020). Uncommon Overoxidative Catalytic Activity in a New Halo‐tolerant Alcohol Dehydrogenase. ChemCatChem 12 (22), 5679–5685. doi:10.1002/cctc.202001112
Corrado, M. L., Knaus, T., and Mutti, F. G. (2021). High Regio‐ and Stereoselective Multi‐enzymatic Synthesis of All Phenylpropanolamine Stereoisomers from β‐Methylstyrene. Chembiochem 22 (13), 2345–2350. doi:10.1002/cbic.202100123
Cuetos, A., Bisogno, F. R., Lavandera, I., and Gotor, V. (2013). Coupling Biocatalysis and Click Chemistry: One-Pot Two-step Convergent Synthesis of Enantioenriched 1,2,3-Triazole-Derived Diols. Chem. Commun. 49 (26), 2625–2627. doi:10.1039/C3CC38674K
Cuetos, A., Rioz-Martínez, A., Bisogno, F. R., Grischek, B., Lavandera, I., de Gonzalo, G., et al. (2012). Access to Enantiopure α-Alkyl-β-hydroxy Esters through Dynamic Kinetic Resolutions Employing Purified/Overexpressed Alcohol Dehydrogenases. Adv. Synth. Catal. 354 (9), 1743–1749. doi:10.1002/adsc.201200139
Dai, Z., Guillemette, K., and Green, T. K. (2013). Stereoselective Synthesis of Aryl γ,δ-unsaturated β-hydroxyesters by Ketoreductases. J. Mol. Catal. B Enzym. 97, 264–269. doi:10.1016/j.molcatb.2013.09.003
Dall'Oglio, F., Contente, M. L., Conti, P., Molinari, F., Monfredi, D., Pinto, A., et al. (2017). Flow-based Stereoselective Reduction of Ketones Using an Immobilized Ketoreductase/glucose Dehydrogenase Mixed Bed System. Catal. Commun. 93, 29–32. doi:10.1016/j.catcom.2017.01.025
de Gonzalo, G., and Lavandera, I. (2021). Biocatalysis for Practitioners: Techniques, Reactions and Applications. Weinheim: Wiley VCH.
de Gonzalo, G., Lavandera, I., Faber, K., and Kroutil, W. (2007). Enzymatic Reduction of Ketones in "Micro-aqueous" Media Catalyzed by ADH-A from Rhodococcus Ruber. Org. Lett. 9 (11), 2163–2166. doi:10.1021/ol070679c
de Gonzalo, G., and Paul, C. E. (2021). Recent Trends in Synthetic Enzymatic Cascades Promoted by Alcohol Dehydrogenases. Curr. Opin. Green Sustain. Chem. 32, 100548. doi:10.1016/j.cogsc.2021.100548
de Smidt, O., du Preez, J. C., and Albertyn, J. (2008). The Alcohol Dehydrogenases ofSaccharomyces Cerevisiae: a Comprehensive Review. Fems Yeast Res. 8 (7), 967–978. doi:10.1111/j.1567-1364.2008.00387.x
De Wildeman, S. M. A., Sonke, T., Schoemaker, H. E., and May, O. (2007). Biocatalytic Reductions: From Lab Curiosity to "First Choice". Acc. Chem. Res. 40 (12), 1260–1266. doi:10.1021/ar7001073
Dennig, A., Blaschke, F., Gandomkar, S., Tassano, E., and Nidetzky, B. (2019). Preparative Asymmetric Synthesis of Canonical and Non‐canonical α‐amino Acids through Formal Enantioselective Biocatalytic Amination of Carboxylic Acids. Adv. Synth. Catal. 361 (6), 1348–1358. doi:10.1002/adsc.201801377
de Miranda, A. S., Simon, R. C., Grischek, B., de Paula, G. C., Horta, B. A. C., de Miranda, L. S. M., et al. (2015). Chiral Chlorohydrins from the Biocatalyzed Reduction of Chloroketones: Chiral Building Blocks for Antiretroviral Drugs. ChemCatChem 7(6), 984–992. doi:10.1002/cctc.201403023
Dias Gomes, M., Bommarius, B. R., Anderson, S. R., Feske, B. D., Woodley, J. M., and Bommarius, A. S. (2019). Bubble Column Enables Higher Reaction Rate for Deracemization of ( R,S )‐1‐Phenylethanol with Coupled Alcohol Dehydrogenase/NADH Oxidase System. Adv. Synth. Catal. 361 (11), 2574–2581. doi:10.1002/adsc.201900213
Díaz-Rodríguez, A., Borzęcka, W., Lavandera, I., and Gotor, V. (2014). Stereodivergent Preparation of Valuable γ- or δ-Hydroxy Esters and Lactones through One-Pot Cascade or Tandem Chemoenzymatic Protocols. ACS Catal. 4 (2), 386–393. doi:10.1021/cs4010024
Di Carmine, G., Bortolini, O., Massi, A., Müller, M., Bernacchia, G., Fantin, G., et al. (2018). Enzymatic Cross‐Benzoin‐Type Condensation of Aliphatic Aldehydes: Enantioselective Synthesis of 1‐Alkyl‐1‐hydroxypropan‐2‐ones and 1‐Alkyl‐1‐hydroxybutan‐2‐ones. Adv. Synth. Catal. 360 (21), 4132–4141. doi:10.1002/adsc.201800357
Dordick, J. S., Marletta, M. A., and Klibanov, A. M. (1986). Peroxidases Depolymerize Lignin in Organic Media but Not in Water. Proc. Natl. Acad. Sci. U.S.A. 83 (17), 6255–6257. doi:10.1073/pnas.83.17.6255
Dreifke, M., Brieler, F. J., and Fröba, M. (2017). Immobilization of Alcohol Dehydrogenase from E. coli onto Mesoporous Silica for Application as a Cofactor Recycling System. ChemCatChem 9 (7), 1197–1210. doi:10.1002/cctc.201601288
Edegger, K., Gruber, C. C., Poessl, T. M., Wallner, S. R., Lavandera, I., Faber, K., et al. (2006a). Biocatalytic Deuterium- and Hydrogen-Transfer Using Over-expressed ADH-'A': Enhanced Stereoselectivity and2H-Labeled Chiral Alcohols. Chem. Commun. (22), 2402–2404. doi:10.1039/b602487d
Edegger, K., Mang, H., Faber, K., Gross, J., and Kroutil, W. (2006b). Biocatalytic Oxidation of Sec-Alcohols via Hydrogen Transfer. J. Mol. Catal. A Chem. 251 (1-2), 66–70. doi:10.1016/j.molcata.2006.02.007
Eissen, M., Geisler, G., Bühler, B., Fischer, C., Hungerbühler, K., Schmid, A., et al. (2009). “Mass Balances and Life Cycle Assessment,” in Green Chemistry Metrics: Measuring and Monitoring Sustainable Processes. Editors Alexei. Lapkin, and D. J. C. Constable (Chichester, UK: John Wiley & Sons).
Emmanuel, M. A., Greenberg, N. R., Oblinsky, D. G., and Hyster, T. K. (2016). Accessing Non-natural Reactivity by Irradiating Nicotinamide-dependent Enzymes with Light. Nature 540 (7633), 414–417. doi:10.1038/nature20569
Engelmann, C., Ekambaram, N., Johannsen, J., Fellechner, O., Waluga, T., Fieg, G., et al. (2020). Enzyme Immobilization on Synthesized Nanoporous Silica Particles and Their Application in a Bi‐enzymatic Reaction. ChemCatChem 12 (8), 2245–2252. doi:10.1002/cctc.201902293
Engelmark Cassimjee, K., Kadow, M., Wikmark, Y., Svedendahl Humble, M., Rothstein, M. L., Rothstein, D. M., et al. (2014). A General Protein Purification and Immobilization Method on Controlled Porosity Glass: Biocatalytic Applications. Chem. Commun. 50 (65), 9134–9137. doi:10.1039/c4cc02605e
Erian, A., Sherif, S., and Gaber, H. (2003). The Chemistry of α-Haloketones and Their Utility in Heterocyclic Synthesis. Molecules 8 (11), 793–865. doi:10.3390/81100793
Fasan, R., Jennifer Kan, S. B., and Zhao, H. (2019). A Continuing Career in Biocatalysis: Frances H. Arnold. ACS Catal. 9 (11), 9775–9788. doi:10.1021/acscatal.9b02737
Ferrandi, E. E., Bertuletti, S., Monti, D., and Riva, S. (2020). Hydroxysteroid Dehydrogenases: An Ongoing Story. Eur. J. Org. Chem. 2020, 4463–4473. doi:10.1002/ejoc.202000192
Fischer, T., and Pietruszka, J. (2012). Alcohol Dehydrogenase‐Catalyzed Synthesis of Enantiomerically Pure δ ‐Lactones as Versatile Intermediates for Natural Product Synthesis. Adv. Synth. Catal. 354 (13), 2521–2530. doi:10.1002/adsc.201200258
Fitzpatrick, P. A., and Klibanov, A. M. (1991). How Can the Solvent Affect Enzyme Enantioselectivity? J. Am. Chem. Soc. 113 (8), 3166–3171. doi:10.1021/ja00008a054
Fox, R. J., Davis, S. C., Mundorff, E. C., Newman, L. M., Gavrilovic, V., Ma, S. K., et al. (2007). Improving Catalytic Function by ProSAR-Driven Enzyme Evolution. Nat. Biotechnol. 25 (3), 338–344. doi:10.1038/nbt1286
Friest, J. A., Maezato, Y., Broussy, S., Blum, P., and Berkowitz, D. B. (2010). Use of a Robust Dehydrogenase from an Archael Hyperthermophile in Asymmetric Catalysis−Dynamic Reductive Kinetic Resolution Entry into (S)-Profens. J. Am. Chem. Soc. 132 (17), 5930–5931. doi:10.1021/ja910778p
Gacs, J., Zhang, W., Knaus, T., Mutti, F. G., Arends, I. W. C. E., and Hollmann, F. (2019). A Photo-Enzymatic Cascade to Transform Racemic Alcohols into Enantiomerically Pure Amines. Catalysts 9, 305. doi:10.3390/catal9040305
Gandolfi, R., Ferrara, N., and Molinari, F. (2001). An Easy and Efficient Method for the Production of Carboxylic Acids and Aldehydes by Microbial Oxidation of Primary Alcohols. Tetrahedron Lett. 42 (3), 513–514. doi:10.1016/S0040-4039(00)02008-6
Gao, H., Tiwari, M. K., Kang, Y. C., and Lee, J.-K. (2012). Characterization of H2O-Forming NADH Oxidase from Streptococcus Pyogenes and its Application in L-Rare Sugar Production. Bioorg. Med. Chem. Lett. 22 (5), 1931–1935. doi:10.1016/j.bmcl.2012.01.049
Gaona-López, C., Julián-Sánchez, A., and Riveros-Rosas, H. (2016). Diversity and Evolutionary Analysis of Iron-Containing (Type-III) Alcohol Dehydrogenases in Eukaryotes. PLoS One 11 (11), e0166851. doi:10.1371/journal.pone.0166851
Gargiulo, S., Arends, I. W. C. E., and Hollmann, F. (2011). A Photoenzymatic System for Alcohol Oxidation. ChemCatChem 3 (2), 338–342. doi:10.1002/cctc.201000317
Gargiulo, S., Opperman, D. J., Hanefeld, U., Arends, I. W. C. E., and Hollmann, F. (2012). A Biocatalytic Redox Isomerisation. Chem. Commun. 48, 6630–6632. doi:10.1039/C2CC31947K
Ghannadi, S., Abdizadeh, H., Miroliaei, M., and Saboury, A. A. (2019). Immobilization of Alcohol Dehydrogenase on Titania Nanoparticles to Enhance Enzyme Stability and Remove Substrate Inhibition in the Reaction of Formaldehyde to Methanol. Ind. Eng. Chem. Res. 58 (23), 9844–9854. doi:10.1021/acs.iecr.9b01370
Giacomini, D., Galletti, P., Quintavalla, A., Gucciardo, G., and Paradisi, F. (2007). Highly Efficient Asymmetric Reduction of Arylpropionic Aldehydes by Horse Liver Alcohol Dehydrogenase through Dynamic Kinetic Resolution. Chem. Commun. (39), 4038–4040. doi:10.1039/B712290J
Giovannini, P. P., Fantin, G., Massi, A., Venturi, V., and Pedrini, P. (2011). Enzymatic Diastereo- and Enantioselective Synthesis of α-alkyl-α,β-dihydroxyketones. Org. Biomol. Chem. 9 (23), 8038–8045. doi:10.1039/C1OB05928A
Gong, X.-M., Zheng, G.-W., Liu, Y.-Y., and Xu, J.-H. (2017). Identification of a Robust Carbonyl Reductase for Diastereoselectively Building Syn-3,5-Dihydroxy Hexanoate: a Bulky Side Chain of Atorvastatin. Org. Process Res. Dev. 21 (9), 1349–1354. doi:10.1021/acs.oprd.7b00194
González‐Granda, S., Lavandera, I., and Gotor‐Fernández, V. (2021). Alcohol Dehydrogenases and N‐Heterocyclic Carbene Gold(I) Catalysts: Design of a Chemoenzymatic Cascade towards Optically Active β,β‐Disubstituted Allylic Alcohols. Angew. Chem. Int. Ed. 60 (25), 13945–13951. doi:10.1002/anie.202015215
González‐Martínez, D., Gotor, V., and Gotor‐Fernández, V. (2019). Chemoenzymatic Synthesis of an Odanacatib Precursor through a Suzuki‐Miyaura Cross‐Coupling and Bioreduction Sequence. ChemCatChem 11 (23), 5800–5807. doi:10.1002/cctc.201901351
Gröger, H., Hummel, W., Buchholz, S., Drauz, K., Nguyen, T. V., Rollmann, C., et al. (2003). Practical Asymmetric Enzymatic Reduction through Discovery of a Dehydrogenase-Compatible Biphasic Reaction Media. Org. Lett. 5 (2), 173–176. doi:10.1021/ol0272139
Gröger, H., Hummel, W., Rollmann, C., Chamouleau, F., Hüsken, H., Werner, H., et al. (2004). Preparative Asymmetric Reduction of Ketones in a Biphasic Medium with an (S)-alcohol Dehydrogenase under In Situ-cofactor-recycling with a Formate Dehydrogenase. Tetrahedron 60 (3), 633–640. doi:10.1016/j.tet.2003.11.066
Haberland, J., Hummel, W., Daussmann, T., and Liese, A. (2002a). New Continuous Production Process for Enantiopure (2R,5R)-Hexanediol. Org. Process Res. Dev. 6 (4), 458–462. doi:10.1021/op020023t
Haberland, A. Kriegesmann, E. Wolfr, J., Kriegesmann, A., Wolfram, E., Hummel, W., and Liese, A. (2002b). Diastereoselective Synthesis of Optically Active (2 R ,5 R )-hexanediol. Appl. Microbiol. Biotechnol. 58 (5), 595–599. doi:10.1007/s00253-002-0936-5
Hamada, H., Miura, T., Kumobayashi, H., Matsuda, T., Harada, T., and Nakamura, K. (2001). Asymmetric Synthesis of (R)-2-chloro-1-(m-chlorophenyl)ethanol Using Acetone Powder of Geotrichum Candidum. Biotechnol. Lett. 23 (19), 1603–1606. doi:10.1023/A:1011922823367
Hammer, S. C., Knight, A. M., and Arnold, F. H. (2017). Design and Evolution of Enzymes for Non-natural Chemistry. Curr. Opin. Green Sustain. Chem. 7, 23–30. doi:10.1016/j.cogsc.2017.06.002
Hanefeld, U., Gardossi, L., and Magner, E. (2009). Understanding Enzyme Immobilisation. Chem. Soc. Rev. 38 (2), 453–468. doi:10.1039/b711564b
Hanson, R. L., Goldberg, S., Goswami, A., Tully, T. P., and Patel, R. N. (2005). Purification and Cloning of a Ketoreductase Used for the Preparation of Chiral Alcohols. Adv. Synthesis Catal. 347 (7-8), 1073–1080. doi:10.1002/adsc.200505045
Hanson, R. L., Goldberg, S. L., Guo, Z., Tully, T. P., Goswami, A., Ye, X.-Y., et al. (2014). Enzymatic Reduction of Adamantanones to Chiral Adamantanol Intermediates for the Synthesis of 11-β-Hydroxysteroid Dehydrogenase Inhibitors. Org. Process Res. Dev. 18 (8), 960–968. doi:10.1021/op5002098
Hauer, B. (2020). Embracing Nature's Catalysts: A Viewpoint on the Future of Biocatalysis. ACS Catal. 10, 8418–8427. doi:10.1021/acscatal.0c01708
He, Y., Ding, Y., Ma, C., Di, J., Jiang, C., and Li, A. (2017). One-pot Conversion of Biomass-Derived Xylose to Furfuralcohol by a Chemo-Enzymatic Sequential Acid-Catalyzed Dehydration and Bioreduction. Green Chem. 19 (16), 3844–3850. doi:10.1039/c7gc01256j
Heidlindemann, M., Hammel, M., Scheffler, U., Mahrwald, R., Hummel, W., Berkessel, A., et al. (2015). Chemoenzymatic Synthesis of Vitamin B5-Intermediate (R)-Pantolactone via Combined Asymmetric Organo- and Biocatalysis. J. Org. Chem. 80 (7), 3387–3396. doi:10.1021/jo502667x
Heidlindemann, M., Rulli, G., Berkessel, A., Hummel, W., and Gröger, H. (2014). Combination of Asymmetric Organo- and Biocatalytic Reactions in Organic Media Using Immobilized Catalysts in Different Compartments. ACS Catal. 4 (4), 1099–1103. doi:10.1021/cs4010387
Holec, C., Neufeld, K., and Pietruszka, J. (2016). P450 BM3 Monooxygenase as an Efficient NAD(P)H-Oxidase for Regeneration of Nicotinamide Cofactors in ADH-Catalysed Preparative Scale Biotransformations. Adv. Synth. Catal. 358 (11), 1810–1819. doi:10.1002/adsc.201600241
Holec, C., Sandkuhl, D., Rother, D., Kroutil, W., and Pietruszka, J. (2015). Chemoenzymatic Synthesis towards the Active Agent Travoprost. ChemCatChem 7 (19), 3125–3130. doi:10.1002/cctc.201500587
Hollmann, F., Opperman, D. J., and Paul, C. E. (2021). Biocatalytic Reduction Reactions from a Chemist's Perspective. Angew. Chem. Int. Ed. 60 (11), 5644–5665. doi:10.1002/anie.202001876
Holtmann, D., and Hollmann, F. (2022). Is Water the Best Solvent for Biocatalysis? Mol. Catal. 517, 112035. doi:10.1016/j.mcat.2021.112035
Holtmann, D., and Hollmann, F. (2016). The Oxygen Dilemma: A Severe Challenge for the Application of Monooxygenases? ChemBioChem 17, 1391–1398. doi:10.1002/cbic.201600176
Homaei, A. A., Sariri, R., Vianello, F., and Stevanato, R. (2013). Enzyme Immobilization: an Update. J. Chem. Biol. 6 (4), 185–205. doi:10.1007/s12154-013-0102-9
H. Orrego, A., Millán-Linares, M. D. C., Pedroche, J., Guisán, J. M., Rocha-Martin, J., et al. (2020). High Stabilization of Enzymes Immobilized on Rigid Hydrophobic Glyoxyl-Supports: Generation of Hydrophilic Environments on Support Surfaces. Catalysts 10, 676. doi:10.3390/catal10060676
H. Orrego, A., Romero-Fernández, M., Millán-Linares, M., Yust, M., Guisán, J., and Rocha-Martin, J. (2018). Stabilization of Enzymes by Multipoint Covalent Attachment on Aldehyde-Supports: 2-Picoline Borane as an Alternative Reducing Agent. Catalysts 8 (8), 333. doi:10.3390/catal8080333
Hoyos, P., Sansottera, G., Fernández, M., Molinari, F., Sinisterra, J. V., and Alcántara, A. R. (2008). Enantioselective Monoreduction of Different 1,2-Diaryl-1,2-Diketones Catalysed by Lyophilised Whole Cells from Pichia Glucozyma. Tetrahedron 64 (34), 7929–7936. doi:10.1016/j.tet.2008.06.019
Huang, L., Romero, E., Ressmann, A. K., Rudroff, F., Hollmann, F., Fraaije, M. W., et al. (2017). Nicotinamide Adenine Dinucleotide-dependent Redox-Neutral Convergent Cascade for Lactonizations with Type II Flavin-Containing Monooxygenase. Adv. Synth. Catal. 359 (12), 2142–2148. doi:10.1002/adsc.201700401
Huang, L., Sayoga, G. V., Hollmann, F., and Kara, S. (2018). Horse Liver Alcohol Dehydrogenase-Catalyzed Oxidative Lactamization of Amino Alcohols. ACS Catal. 8, 8680–8684. doi:10.1021/acscatal.8b02355
Huisman, G. W., Liang, J., and Krebber, A. (2010). Practical Chiral Alcohol Manufacture Using Ketoreductases. Curr. Opin. Chem. Biol. 14 (2), 122–129. doi:10.1016/j.cbpa.2009.12.003
Hummel, W., Abokitse, K., Drauz, K., Rollmann, C., and Gröger, H. (2003). Towards a Large-Scale Asymmetric Reduction Process with Isolated Enzymes: Expression of an (S)-Alcohol Dehydrogenase in E.Coli and Studies on the Synthetic Potential of This Biocatalyst. Adv. Synthesis Catal. 345 (1‐2), 153–159. doi:10.1002/adsc.200390001
Hussain, W., Pollard, D. J., Truppo, M., and Lye, G. J. (2008). Enzymatic Ketone Reductions with Co-factor Recycling: Improved Reactions with Ionic Liquid Co-solvents. J. Mol. Catal. B Enzym. 55 (1), 19–29. doi:10.1016/j.molcatb.2008.01.006
Irwin, A. J., and Jones, J. B. (1977a). Asymmetric Syntheses via Enantiotopically Selective Horse Liver Alcohol Dehydrogenase Catalyzed Oxidations of Diols Containing a Prochiral Center. J. Am. Chem. Soc. 99 (2), 556–561. doi:10.1021/ja00444a040
Irwin, A. J., and Jones, J. B. (1977b). Regiospecific and Enantioselective Horse Liver Alcohol Dehydrogenase Catalyzed Oxidations of Some Hydroxycyclopentanes. J. Am. Chem. Soc. 99 (5), 1625–1630. doi:10.1021/ja00447a057
Jakoblinnert, A., Mladenov, R., Paul, A., Sibilla, F., Schwaneberg, U., Ansorge-Schumacher, M. B., et al. (2011). Asymmetric Reduction of Ketones with Recombinant E. coli Whole Cells in Neat Substrates. Chem. Commun. 47 (44), 12230–12232. doi:10.1039/C1CC14097C
Ji, A., Wolberg, M., Wandrey, C., Müller, M., and Hummel, W. (2001). Dynamic Kinetic Resolution of Tert-Butyl 4-Methyl-3,5-Dioxohexanoate through Enzymatic Reduction. Chem. Commun. (1), 57–58. doi:10.1039/B007448I
Jia, H.-Y., Zong, M.-H., Yu, H.-L., and Li, N. (2017). Dehydrogenase-Catalyzed Oxidation of Furanics: Exploitation of Hemoglobin Catalytic Promiscuity. ChemSusChem 10 (18), 3524–3528. doi:10.1002/cssc.201701288
Jiang, H.-W., Chen, Q., Pan, J., Zheng, G.-W., and Xu, J.-H. (2020). Rational Engineering of Formate Dehydrogenase Substrate/Cofactor Affinity for Better Performance in NADPH Regeneration. Appl. Biochem. Biotechnol. 192 (2), 530–543. doi:10.1007/s12010-020-03317-7
Jiang, R., and Bommarius, A. S. (2004). Hydrogen Peroxide-Producing NADH Oxidase (Nox-1) from Lactococcus Lactis. Tetrahedron Asymmetry 15 (18), 2939–2944. doi:10.1016/j.tetasy.2004.07.057
Jones, J. B., and Taylor, K. E. (1976). Nicotinamide Coenzyme Regeneration. Flavin Mononucleotide (Riboflavin Phosphate) as an Efficient, Economical, and Enzyme-Compatible Recycling Agent. Can. J. Chem. 54 (19), 2969–2973. doi:10.1139/v76-420
Jones, J. B., and Taylor, K. E. (1973). Use of Pyridinium and Flavin Derivatives for Recycling of Catalystic Amounts of NAD+ during Preparative-Scale Horse Liver Alchohol Dehydrogenase-Catalysed Oxidations of Alcohols. J. Chem. Soc. Chem. Commun., 205–206. doi:10.1039/c39730000205
Kalaitzakis, D., Rozzell, J. D., Smonou, I., and Kambourakis, S. (2006). Synthesis of Valuable Chiral Intermediates by Isolated Ketoreductases: Application in the Synthesis of α-Alkyl-β-hydroxy Ketones and 1,3-Diols. Adv. Synth. Catal. 348 (14), 1958–1969. doi:10.1002/adsc.200606185
Kalaitzakis, D., and Smonou, I. (2010b). A Two-step, One-Pot Enzymatic Synthesis of 2-Substituted 1,3-Diols. J. Org. Chem. 75 (24), 8658–8661. doi:10.1021/jo101519t
Kalaitzakis, D., and Smonou, I. (2012). Chemoenzymatic Synthesis of Stegobinone and Stegobiol, Components of the Natural Sex Pheromone of the Drugstore Beetle (Stegobium Paniceum L.). Eur. J. Org. Chem. 2012 (1), 43–46. doi:10.1002/ejoc.201101319
Kalaitzakis, D., and Smonou, I. (2010a). Highly Diastereoselective Synthesis of 2-Substituted-1,3-Diols Catalyzed by Ketoreductases. Tetrahedron 66 (48), 9431–9439. doi:10.1016/j.tet.2010.09.096
Kara, S., Schrittwieser, J. H., Hollmann, F., and Ansorge-Schumacher, M. B. (2014a). Recent Trends and Novel Concepts in Cofactor-dependent Biotransformations. Appl. Microbiol. Biotechnol. 98 (4), 1517–1529. doi:10.1007/s00253-013-5441-5
Kara, S., Schrittwieser, J. H., and Hollmann, F. (2013a). “Strategies for Cofactor Regeneration in Biocatalyzed Reductions,” in Synthetic Methods for Biologically Active Molecules: Exploring the Potential of Bioreductions. Editor E. Brenna (Wiley VCH). doi:10.1002/9783527665785.ch08
Kara, S., Spickermann, D., Schrittwieser, J. H., Leggewie, C., Van Berkel, W. J. H., Arends, I. W. C. E., et al. (2013b). More Efficient Redox Biocatalysis by Utilising 1,4-butanediol as a 'smart Cosubstrate'. Green Chem. 15, 330–335. doi:10.1039/c2gc36797a
Kara, S., Spickermann, D., Schrittwieser, J. H., Weckbecker, A., Leggewie, C., Arends, I. W. C. E., et al. (2013c). Access to Lactone Building Blocks via Horse Liver Alcohol Dehydrogenase-Catalyzed Oxidative Lactonization. ACS Catal. 3, 2436–2439. doi:10.1021/cs400535c
Kara, S., Spickermann, D., Weckbecker, A., Leggewie, C., Arends, I. W. C. E., and Hollmann, F. (2014b). Bioreductions Catalyzed by an Alcohol Dehydrogenase in Non-aqueous Media. ChemCatChem 6 (4), 973–976. doi:10.1002/cctc.201300841
Katzberg, M., Wechler, K., Müller, M., Dünkelmann, P., Stohrer, J., Hummel, W., et al. (2009). Biocatalytical Production of (5S)-Hydroxy-2-Hexanone. Org. Biomol. Chem. 7, 304–314. doi:10.1039/B816364B
Kawahara, K., Matsumoto, M., Hashimoto, H., and Miyano, S. (1988). Kinetic Resolution of 2-Formyl-1,1′-Binaphthyls by Baker's-Yeast Reduction of the Formyl Function. Chem. Lett. 17 (7), 1163–1164. doi:10.1246/cl.1988.1163
Keinan, E., Hafeli, E. K., Seth, K. K., and Lamed, R. (1986). Thermostable Enzymes in Organic Synthesis. 2. Asymmetric Reduction of Ketones with Alcohol Dehydrogenase from Thermoanaerobium Brockii. J. Am. Chem. Soc. 108 (1), 162–169. doi:10.1021/ja00261a026
Kihumbu, D., Stillger, T., Hummel, W., and Liese, A. (2002). Enzymatic Synthesis of All Stereoisomers of 1-Phenylpropane-1,2-Diol. Tetrahedron Asymmetry 13 (10), 1069–1072. doi:10.1016/s0957-4166(02)00247-1
Kochius, S., Magnusson, A. O., Hollmann, F., Schrader, J., and Holtmann, D. (2012). Immobilized Redox Mediators for Electrochemical NAD(P)+ Regeneration. Appl. Microbiol. Biotechnol. 93 (6), 2251–2264. doi:10.1007/s00253-012-3900-z
Kochius, S., Ni, Y., Kara, S., Gargiulo, S., Schrader, J., Holtmann, D., et al. (2014). Light-Accelerated Biocatalytic Oxidation Reactions. ChemPlusChem 79 (11), 1554–1557. doi:10.1002/cplu.201402152
Könst, P., Kara, S., Kochius, S., Holtmann, D., Arends, I. W. C. E., Ludwig, R., et al. (2013). Expanding the Scope of Laccase-Mediator Systems. ChemCatChem 5 (10), 3027–3032. doi:10.1002/cctc.201300205
Könst, P., Merkens, H., Kara, S., Kochius, S., Vogel, A., Zuhse, R., et al. (2012). Oxidation von Aldehyden mit Alkoholdehydrogenasen. Angew. Chem. 124, 10052–10055. doi:10.1002/ange.201203219
Koppireddi, S., Seo, J.-H., Jeon, E.-Y., Chowdhury, P. S., Jang, H.-Y., Park, J.-B., et al. (2016). Combined Biocatalytic and Chemical Transformations of Oleic Acid to ω-Hydroxynonanoic Acid and α,ω-Nonanedioic Acid. Adv. Synth. Catal. 358 (19), 3084–3092. doi:10.1002/adsc.201600216
Korpak, M., and Pietruszka, J. (2011). Chemoenzymatic One-Pot Synthesis of γ-Butyrolactones. Adv. Synth. Catal. 353, 1420–1424. doi:10.1002/adsc.201100110
Krasňan, V., Stloukal, R., Rosenberg, M., and Rebroš, M. (2016). Immobilization of Cells and Enzymes to LentiKats®. Appl. Microbiol. Biotechnol. 100(6), 2535–2553. doi:10.1007/s00253-016-7283-4
Kroutil, W., Voss, C., and Gruber, C. (2010). Deracemisation of Secondary Alcohols via Biocatalytic Stereoinversion. Synlett 2010 (7), 991–998. doi:10.1055/s-0029-1219567
Kulig, J., Sehl, T., Mackfeld, U., Wiechert, W., Pohl, M., and Rother, D. (2019). An Enzymatic 2‐Step Cofactor and Co‐Product Recycling Cascade towards a Chiral 1,2‐Diol. Part I: Cascade Design. Adv. Synth. Catal. 361 (11), 2607–2615. doi:10.1002/adsc.201900187
Kumru, C., Classen, T., and Pietruszka, J. (2018). Enantioselective, Catalytic One‐Pot Synthesis of γ ‐Butyrolactone‐Based Fragrances. ChemCatChem 10 (21), 4917–4926. doi:10.1002/cctc.201801040
Kurina-Sanz, M., Bisogno, F. R., Lavandera, I., Orden, A. A., and Gotor, V. (2009). Promiscuous Substrate Binding Explains the Enzymatic Stereo- and Regiocontrolled Synthesis of Enantiopure Hydroxy Ketones and Diols. Adv. Synth. Catal. 351 (11-12), 1842–1848. doi:10.1002/adsc.200900218
Lavandera, I., Kern, A., Ferreira-Silva, B., Glieder, A., de Wildeman, S., and Kroutil, W. (2008a). Stereoselective Bioreduction of Bulky-Bulky Ketones by a Novel ADH from Ralstonia Sp. J. Org. Chem. 73 (15), 6003–6005. doi:10.1021/jo800849d
Lavandera, I., Kern, A., Resch, V., Ferreira-Silva, B., Glieder, A., Fabian, W. M. F., et al. (2008b). One-Way Biohydrogen Transfer for Oxidation of Sec-Alcohols. Org. Lett. 10 (11), 2155–2158. doi:10.1021/ol800549f
Li, G., Wang, J.-B., and Reetz, M. T. (2018). Biocatalysts for the Pharmaceutical Industry Created by Structure-Guided Directed Evolution of Stereoselective Enzymes. Bioorg. Med. Chem. 26 (7), 1241–1251. doi:10.1016/j.bmc.2017.05.021
Li, H., Moncecchi, J., and Truppo, M. D. (2015). Development of an Immobilized Ketoreductase for Enzymatic (R)-1-(3,5-Bis(trifluoromethyl)phenyl)ethanol Production. Org. Process Res. Dev. 19 (7), 695–700. doi:10.1021/op5003215
Li, H., Yang, Y., Zhu, D., Hua, L., and Kantardjieff, K. (2010). Highly Enantioselective Mutant Carbonyl Reductases Created via Structure-Based Site-Saturation Mutagenesis. J. Org. Chem. 75 (22), 7559–7564. doi:10.1021/jo101541n
Li, H., Zhu, D., Hua, L., and Biehl, E. R. (2009). Enantioselective Reduction of Diaryl Ketones Catalyzed by a Carbonyl Reductase fromSporobolomyces Salmonicolorand its Mutant Enzymes. Adv. Synth. Catal. 351 (4), 583–588. doi:10.1002/adsc.200900045
Li, S. F., Xie, J. Y., Qiu, S., Xu, S. Y., Cheng, F., Wang, Y. J., et al. (2021a). Semirational Engineering of an Aldo-Keto Reductase Km AKR for Overcoming Trade‐offs between Catalytic Activity and Thermostability. Biotech Bioeng. 118 (11), 4441–4452. doi:10.1002/bit.27913
Li, Y., Zhang, R., and Xu, Y. (2021b). Structure-based Mechanisms: On the Way to Apply Alcohol Dehydrogenases/reductases to Organic-Aqueous Systems. Int. J. Biol. Macromol. 168, 412–427. doi:10.1016/j.ijbiomac.2020.12.068
Li, Z., Wang, Z., Wang, Y., Wu, X., Lu, H., Huang, Z., et al. (2019). Substituent Position‐Controlled Stereoselectivity in Enzymatic Reduction of Diaryl‐ and Aryl(heteroaryl)methanones. Adv. Synth. Catal. 361 (8), 1859–1865. doi:10.1002/adsc.201801543
Li, Z., Yang, H., Liu, J., Huang, Z., and Chen, F. (2021c). Application of Ketoreductase in Asymmetric Synthesis of Pharmaceuticals and Bioactive Molecules: An Update (2018-2020). Chem. Rec. 21 (7), 1611–1630. doi:10.1002/tcr.202100062
Liang, J., Lalonde, J., Borup, B., Mitchell, V., Mundorff, E., Trinh, N., et al. (2010a). Development of a Biocatalytic Process as an Alternative to the (−)-DIP-Cl-Mediated Asymmetric Reduction of a Key Intermediate of Montelukast. Org. Process Res. Dev. 14 (1), 193–198. doi:10.1021/op900272d
Liang, J., Mundorff, E., Voladri, R., Jenne, S., Gilson, L., Conway, A., et al. (2010b). Highly Enantioselective Reduction of a Small Heterocyclic Ketone: Biocatalytic Reduction of Tetrahydrothiophene-3-One to the Corresponding (R)-Alcohol. Org. Process Res. Dev. 14 (1), 188–192. doi:10.1021/op9002714
Liu, X.-h., Du, X., Feng, J.-r., Wu, M.-B., Lin, J.-p., Guan, J., et al. (2019). Co-immobilization of Short-Chain Dehydrogenase/Reductase and Glucose Dehydrogenase for the Efficient Production of (±)-Ethyl Mandelate. Catal. Lett. 149 (6), 1710–1720. doi:10.1007/s10562-019-02727-5
López-Gallego, F., Jackson, E., and Betancor, L. (2017). Heterogeneous Systems Biocatalysis: The Path to the Fabrication of Self-Sufficient Artificial Metabolic Cells. Chem. Eur. J. 23 (71), 17841–17849. doi:10.1002/chem.201703593
López-Iglesias, M., Busto, E., Gotor, V., and Gotor-Fernández, V. (2015). Chemoenzymatic Asymmetric Synthesis of 1,4-Benzoxazine Derivatives: Application in the Synthesis of a Levofloxacin Precursor. J. Org. Chem. 80 (8), 3815–3824. doi:10.1021/acs.joc.5b00056
Lüdeke, S., Richter, M., and Müller, M. (2009). Stereoselective Synthesis of Three Isomers of Tert-Butyl 5-Hydroxy-4-Methyl-3-Oxohexanoate through Alcohol Dehydrogenase-Catalyzed Dynamic Kinetic Resolution. Adv. Synth. Catal. 351 (1-2), 253–259. doi:10.1002/adsc.200800619
Lutwak-Mann, C. (1938). Alcohol Dehydrogenase of Animal Tissues. Biochem. J. 32, 1364–1374. doi:10.1042/bj0321364
Ma, S. K., Gruber, J., Davis, C., Newman, L., Gray, D., Wang, A., et al. (2010). A Green-By-Design Biocatalytic Process for Atorvastatin Intermediate. Green Chem. 12 (1), 81–86. doi:10.1039/b919115c
Machielsen, R., Uria, A. R., Kengen, S. W. M., and van der Oost, J. (2006). Production and Characterization of a Thermostable Alcohol Dehydrogenase that Belongs to the Aldo-Keto Reductase Superfamily. Appl. Environ. Microbiol. 72 (1), 233–238. doi:10.1128/AEM.72.1.233-238.2006
Magano, J., and Dunetz, J. R. (2012). Large-Scale Carbonyl Reductions in the Pharmaceutical Industry. Org. Process Res. Dev. 16 (6), 1156–1184. doi:10.1021/op2003826
Man, H., Kędziora, K., Kulig, J., Frank, A., Lavandera, I., Gotor-Fernández, V., et al. (2014). Structures of Alcohol Dehydrogenases from Ralstonia and Sphingobium Spp. Reveal the Molecular Basis for Their Recognition of 'Bulky-Bulky' Ketones. Top. Catal. 57 (5), 356–365. doi:10.1007/s11244-013-0191-2
Mangas-Sánchez, J., Busto, E., Gotor-Fernández, V., Malpartida, F., and Gotor, V. (2011). Asymmetric Chemoenzymatic Synthesis of Miconazole and Econazole Enantiomers. The Importance of Chirality in Their Biological Evaluation. J. Org. Chem. 76 (7), 2115–2122. doi:10.1021/jo102459w
Mateo, C., Palomo, J. M., Fuentes, M., Betancor, L., Grazu, V., López-Gallego, F., et al. (2006). Glyoxyl Agarose: A Fully Inert and Hydrophilic Support for Immobilization and High Stabilization of Proteins. Enzyme Microb. Technol. 39 (2), 274–280. doi:10.1016/j.enzmictec.2005.10.014
Matsuyama, A., Yamamoto, H., Kawada, N., and Kobayashi, Y. (2001). Industrial Production of (R)-1,3-butanediol by New Biocatalysts. J. Mol. Catal. B Enzym. 11 (4-6), 513–521. doi:10.1016/S1381-1177(00)00032-1
Méndez‐Sánchez, D., Mourelle‐Insua, Á., Gotor‐Fernández, V., and Lavandera, I. (2019). Synthesis of α‐Alkyl‐β‐Hydroxy Amides through Biocatalytic Dynamic Kinetic Resolution Employing Alcohol Dehydrogenases. Adv. Synth. Catal. 361 (11), 2706–2712. doi:10.1002/adsc.201900317
Milagre, H. M. S., Milagre, C. D. F., Moran, P. J. S., Santana, M. H. A., and Rodrigues, J. A. R. (2006). Asymmetric Bioreduction of Ethyl 3-Halo-2-Oxo-4-Phenylbutanoate by Saccharomyces cerevisiae Immobilized in Ca-Alginate Beads with Double Gel Layer. Org. Process Res. Dev. 10 (3), 611–617. doi:10.1021/op0502497
Milagre, H. M. S., Milagre, C. D. F., Moran, P. J. S., Santana, M. H. A., and Rodrigues, J. A. R. (2005). Reduction of Ethyl Benzoylformate Mediated by Saccharomyces cerevisiae Entrapped in Alginate Fibers with Double Gel Layers in a Continuously Operated Reactor. Enzyme Microb. Technol. 37 (1), 121–125. doi:10.1016/j.enzmictec.2005.02.011
Miroliaei, M., and Nemat-Gorgani, M. (2002). Effect of Organic Solvents on Stability and Activity of Two Related Alcohol Dehydrogenases: a Comparative Study. Int. J. Biochem. Cell Biol. 34 (2), 169–175. doi:10.1016/S1357-2725(01)00109-1
Molinari, F., Aragozzini, F., Cabral, J. M. S., and Prazeres, D. M. F. (1997). Continuous Production of Isovaleraldehyde through Extractive Bioconversion in a Hollow-Fiber Membrane Bioreactor. Enzyme Microb. Technol. 20 (8), 604–611. doi:10.1016/s0141-0229(96)00205-0
Monsalve, L. N., Cerrutti, P., Galvagno, M. A., and Baldessari, A. (2010). Rhodotorula Minuta-mediated Bioreduction of 1,2-diketones. Biocatal. Biotransformation 28 (2), 137–143. doi:10.3109/10242420903515445
Mori, T., Tomikawa, D., Katano, Y., Kubo, J., and Morikawa, Y. (2004). Reduction of Chloroacetone over Silica-Supported Noble Metal Catalysts. Appl. Catal. A General 271 (1), 77–84. doi:10.1016/j.apcata.2004.02.048
Mourelle-Insua, Á., De Gonzalo, G., Lavandera, I., and Gotor-Fernández, V. (2018). Stereoselective Enzymatic Reduction of 1,4-Diaryl-1,4-Diones to the Corresponding Diols Employing Alcohol Dehydrogenases. Catalysts 8 (4), 150. doi:10.3390/catal8040150
Müller, C. A., Akkapurathu, B., Winkler, T., Staudt, S., Hummel, W., Gröger, H., et al. (2013). In VitroDouble Oxidation Ofn-Heptane with Direct Cofactor Regeneration. Adv. Synth. Catal. 355 (9), 1787–1798. doi:10.1002/adsc.201300143
Müller, M. (2005). Chemoenzymatic Synthesis of Building Blocks for Statin Side Chains. Angew. Chem. Int. Ed. 44 (3), 362–365. doi:10.1002/anie.200460852
Müller, M., Katzberg, M., Bertau, M., and Hummel, W. (2010). Highly Efficient and Stereoselective Biosynthesis of (2S,5S)-Hexanediol with a Dehydrogenase from Saccharomyces cerevisiae. Org. Biomol. Chem. 8 (7), 1540–1550. doi:10.1039/b920869k
Musa, M. M. (2022). Alcohol Dehydrogenases with Anti ‐Prelog Stereopreference in Synthesis of Enantiopure Alcohols. ChemistryOpen 11 (4), e202100251. doi:10.1002/open.202100251
Musa, M. M., Bsharat, O., Karume, I., Vieille, C., Takahashi, M., and Hamdan, S. M. (2018). Expanding the Substrate Specificity of Thermoanaerobacter Pseudoethanolicus Secondary Alcohol Dehydrogenase by a Dual Site Mutation. Eur. J. Org. Chem. 2018 (6), 798–805. doi:10.1002/ejoc.201701351
Musa, M. M., and Phillips, R. S. (2011). Recent Advances in Alcohol Dehydrogenase-Catalyzed Asymmetric Production of Hydrophobic Alcohols. Catal. Sci. Technol. 1 (8), 1311–1323. doi:10.1039/c1cy00160d
Musa, M. M., Vieille, C., and Phillips, R. S. (2021). Secondary Alcohol Dehydrogenases from Thermoanaerobacter Pseudoethanolicus and Thermoanaerobacter Brockii as Robust Catalysts. ChemBioChem 22 (11), 1884–1893. doi:10.1002/cbic.202100043
Musa, M. M., Ziegelmann-Fjeld, K. I., Vieille, C., Zeikus, J. G., and Phillips, R. S. (2007). Xerogel-Encapsulated W110A Secondary Alcohol Dehydrogenase fromThermoanaerobacter Ethanolicus Performs Asymmetric Reduction of Hydrophobic Ketones in Organic Solvents. Angew. Chem. Int. Ed. 46 (17), 3091–3094. doi:10.1002/anie.200604615
Muschallik, L., Molinnus, D., Jablonski, M., Kipp, C. R., Bongaerts, J., Pohl, M., et al. (2020). Synthesis of α-hydroxy Ketones and Vicinal (R,R)-diols by Bacillus Clausii DSM 8716T Butanediol Dehydrogenase. RSC Adv. 10 (21), 12206–12216. doi:10.1039/D0RA02066D
Nagai, T., Sakurai, S., Natori, N., Hataoka, M., Kinoshita, T., Inoue, H., et al. (2018). Synthesis of Enantiomerically Enriched Drug Precursors and an Insect Pheromone via Reduction of Ketones Using Commercially Available Carbonyl Reductase Screening Kit "Chiralscreen OH". Bioorg. Med. Chem. 26 (7), 1304–1313. doi:10.1016/j.bmc.2017.03.067
Nagy‐Győr, L., Abaházi, E., Bódai, V., Sátorhelyi, P., Erdélyi, B., Balogh‐Weiser, D., et al. (2018). Co‐immobilized Whole Cells with ω‐Transaminase and Ketoreductase Activities for Continuous‐Flow Cascade Reactions. ChemBioChem 19 (17), 1845–1848. doi:10.1002/cbic.201800286
Nagy-Győr, L., Lăcătuş, M., Balogh-Weiser, D., Csuka, P., Bódai, V., Erdélyi, B., et al. (2019). How to Turn Yeast Cells into a Sustainable and Switchable Biocatalyst? On-Demand Catalysis of Ketone Bioreduction or Acyloin Condensation. ACS Sustain. Chem. Eng. 7 (24), 19375–19383. doi:10.1021/acssuschemeng.9b03367
Nakamura, K., and Matsuda, T. (1998). Asymmetric Reduction of Ketones by the Acetone Powder of Geotrichum Candidum. J. Org. Chem. 63 (24), 8957–8964. doi:10.1021/jo9812779
Nasário, F., Moran, P., and Rodrigues, J. A. (2019). Deracemization of Sec-Alcohols through Sequential Application of C. Albicans and Ketoreductases. J. Braz. Chem. Soc. 30 (4), 772–779. doi:10.21577/0103-5053.20180205
Nealon, C. M., Musa, M. M., Patel, J. M., and Phillips, R. S. (2015). Controlling Substrate Specificity and Stereospecificity of Alcohol Dehydrogenases. ACS Catal. 5 (4), 2100–2114. doi:10.1021/cs501457v
Nestl, B. M., and Hauer, B. (2014). Engineering of Flexible Loops in Enzymes. ACS Catal. 4 (9), 3201–3211. doi:10.1021/cs500325p
Ni, Y., Fernández-Fueyo, E., Baraibar, A. G., Ullrich, R., Hofrichter, M., Yanase, H., et al. (2016). Peroxygenase-catalyzed Oxyfunctionalization Reactions Promoted by the Complete Oxidation of Methanol. Angew. Chem. Int. Ed. 55, 798–801. doi:10.1002/anie.201507881
Ni, Y., Holtmann, D., and Hollmann, F. (2014). How Green Is Biocatalysis? to Calculate Is to Know. ChemCatChem 6 (4), 930–943. doi:10.1002/cctc.201300976
Ning, C., Su, E., Tian, Y., and Wei, D. (2014). Combined Cross-Linked Enzyme Aggregates (Combi-CLEAs) for Efficient Integration of a Ketoreductase and a Cofactor Regeneration System. J. Biotechnol. 184, 7–10. doi:10.1016/j.jbiotec.2014.05.004
Nowill, R. W., Patel, T. J., Beasley, D. L., Alvarez, J. A., Jackson, E., Hizer, T. J., et al. (2011). Biocatalytic Strategy toward Asymmetric β-hydroxy Nitriles and γ-amino Alcohols. Tetrahedron Lett. 52 (19), 2440–2442. doi:10.1016/j.tetlet.2011.03.009
Oberleitner, N., Peters, C., Muschiol, J., Kadow, M., Sass, S., Bayer, T., et al. (2013). An Enzymatic Toolbox for Cascade Reactions: A Showcase for an In Vivo Redox Sequence in Asymmetric Synthesis. ChemCatChem 5 (12), 3524–3528. doi:10.1002/cctc.201300604
Orrego, A. H., Andrés-Sanz, D., Velasco-Lozano, S., Sanchez-Costa, M., Berenguer, J., Guisan, J. M., et al. (2021). Self-sufficient Asymmetric Reduction of β-ketoesters Catalysed by a Novel and Robust Thermophilic Alcohol Dehydrogenase Co-immobilised with NADH. Catal. Sci. Technol. 11 (9), 3217–3230. doi:10.1039/D1CY00268F
Ou, X.-Y., Wu, X.-L., Peng, F., Xu, P., Zhang, S.-Y., Zong, M.-H., et al. (2019). Highly Efficient Asymmetric Reduction of 2-octanone in Biphasic System by Immobilized Acetobacter Sp. CCTCC M209061 Cells. J. Biotechnol. 299, 37–43. doi:10.1016/j.jbiotec.2019.04.024
Pal, M., Srivastava, G., Sharma, A. N., Kaur, S., and Jolly, R. S. (2015). Biocatalyzed Asymmetric Reduction of Benzils to Either Benzoins or Hydrobenzoins: pH Dependent Switch. Catal. Sci. Technol. 5 (8), 4017–4028. doi:10.1039/C5CY00158G
Panigrahi, K., Applegate, G. A., Malik, G., and Berkowitz, D. B. (2015). Combining a Clostridial Enzyme Exhibiting Unusual Active Site Plasticity with a Remarkably Facile Sigmatropic Rearrangement: Rapid, Stereocontrolled Entry into Densely Functionalized Fluorinated Phosphonates for Chemical Biology. J. Am. Chem. Soc. 137 (10), 3600–3609. doi:10.1021/jacs.5b00022
Park, J. T., Hirano, J.-I., Thangavel, V., Riebel, B. R., and Bommarius, A. S. (2011). NAD(P)H Oxidase V from Lactobacillus Plantarum (NoxV) Displays Enhanced Operational Stability Even in Absence of Reducing Agents. J. Mol. Catal. B Enzym. 71 (3-4), 159–165. doi:10.1016/j.molcatb.2011.04.013
Patel, J. M., and Phillips, R. S. (2014). Effects of Hydrostatic Pressure on Stereospecificity of Secondary Alcohol Dehydrogenase from Thermoanaerobacter Ethanolicus Support the Role of Solvation in Enantiospecificity. ACS Catal. 4 (2), 692–694. doi:10.1021/cs4010997
Patel, R. N. (2001). Enzymatic Synthesis of Chiral Intermediates for Drug Development. Adv. Synth. Catal. 343 (6-7), 527–546. doi:10.1002/1615-4169(200108)343:6/7<527::aid-adsc527>3.0.co;2-i
Paul, C. E., Gargiulo, S., Opperman, D. J., Lavandera, I., Gotor-Fernández, V., Gotor, V., et al. (2013). Mimicking Nature: Synthetic Nicotinamide Cofactors for C═C Bioreduction Using Enoate Reductases. Org. Lett. 15 (1), 180–183. doi:10.1021/ol303240a
Pennacchio, A., Pucci, B., Secundo, F., La Cara, F., Rossi, M., and Raia, C. A. (2008). Purification and Characterization of a Novel Recombinant Highly Enantioselective Short-Chain NAD(H)-Dependent Alcohol Dehydrogenase from Thermus Thermophilus. Appl. Environ. Microbiol. 74 (13), 3949–3958. doi:10.1128/AEM.00217-08
Pennec, A., Hollmann, F., Smit, M. S., and Opperman, D. J. (2015). One-pot Conversion of Cycloalkanes to Lactones. ChemCatChem 7 (2), 236–239. doi:10.1002/cctc.201402835
Peschke, T., Bitterwolf, P., Rabe, K. S., and Niemeyer, C. M. (2019). Self‐Immobilizing Oxidoreductases for Flow Biocatalysis in Miniaturized Packed‐Bed Reactors. Chem. Eng. Technol. 42 (10), 2009–2017. doi:10.1002/ceat.201900073
Pesic, M., Willot, S. J.-P., Fernández-Fueyo, E., Tieves, F., Alcalde, M., and Hollmann, F. (2019). Multienzymatic In Situ Hydrogen Peroxide Generation Cascade for Peroxygenase-Catalysed Oxyfunctionalisation Reactions. Z. für Naturforsch. C 74, 101–104. doi:10.1515/znc-2018-0137
Petkova, G. A., Záruba, K., and Král, V. (2012). Synthesis of Silica Particles and Their Application as Supports for Alcohol Dehydrogenases and Cofactor Immobilizations: Conformational Changes that Lead to Switch in Enzyme Stereoselectivity. Biochimica Biophysica Acta (BBA) - Proteins Proteomics 1824 (6), 792–801. doi:10.1016/j.bbapap.2012.03.010
Petrovičová, T., Markošová, K., Hegyi, Z., Smonou, I., Rosenberg, M., and Rebroš, M. (2018). Co-Immobilization of Ketoreductase and Glucose Dehydrogenase. Catalysts 8 (4), 168. doi:10.3390/catal8040168
Pham, V. T., and Phillips, R. S. (1990). Effects of Substrate Structure and Temperature on the Stereospecificity of Secondary Alcohol Dehydrogenase from Thermoanaerobacter Ethanolicus. J. Am. Chem. Soc. 112 (9), 3629–3632. doi:10.1021/ja00165a057
Pham, V. T., Phillips, R. S., and Ljungdahl, L. G. (1989). Temperature-dependent Enantiospecificity of Secondary Alcohol Dehydrogenase from Thermoanaerobacter Ethanolicus. J. Am. Chem. Soc. 111 (5), 1935–1936. doi:10.1021/ja00187a089
Phillips, R. S. (1992). Temperature Effects on Stereochemistry of Enzymatic Reactions. Enzyme Microb. Technol. 14 (5), 417–419. doi:10.1016/0141-0229(92)90013-e
Piantini, U., Schrader, J., Wawrzun, A., and Wüst, M. (2011). A Biocatalytic Route towards Rose Oxide Using Chloroperoxidase. Food Chem. 129 (3), 1025–1029. doi:10.1016/j.foodchem.2011.05.068
Plž, M., Petrovičová, T., and Rebroš, M. (2020). Semi-Continuous Flow Biocatalysis with Affinity Co-immobilized Ketoreductase and Glucose Dehydrogenase. Molecules 25 (18), 4278. doi:10.3390/molecules25184278
Poessl, T. M., Kosjek, B., Ellmer, U., Gruber, C. C., Edegger, K., Faber, K., et al. (2005). Non-Racemic Halohydrinsvia Biocatalytic Hydrogen-Transfer Reduction of Halo-Ketones and One-Pot Cascade Reaction to Enantiopure Epoxides. Adv. Synthesis Catal. 347 (14), 1827–1834. doi:10.1002/adsc.200505094
Popłoński, J., Reiter, T., and Kroutil, W. (2018). Biocatalytic Racemization Employing TeSADH: Substrate Scope and Organic Solvent Compatibility for Dynamic Kinetic Resolution. ChemCatChem 10 (4), 763–768. doi:10.1002/cctc.201701395
Prelog, V. (1964). Specification of the Stereospecificity of Some Oxido-Reductases by Diamond Lattice Sections. Pure Appl. Chem. 9, 119–130. doi:10.1351/pac196409010119
Rapp, C., Pival-Marko, S., Tassano, E., Nidetzky, B., and Kratzer, R. (2021). Reductive Enzymatic Dynamic Kinetic Resolution Affording 115 G/L (S)-2-phenylpropanol. BMC Biotechnol. 21 (1), 58. doi:10.1186/s12896-021-00715-5
Rauch, M., Schmidt, S., Arends, I. W. C. E., Oppelt, K., Kara, S., and Hollmann, F. (2017). Photobiocatalytic Alcohol Oxidation Using LED Light Sources. Green Chem. 19, 376–379. doi:10.1039/C6GC02008A
Raynbird, M. Y., Sampson, J. B., Smith, D. A., Forsyth, S. M., Moseley, J. D., and Wells, A. S. (2020). Ketone Reductase Biocatalysis in the Synthesis of Chiral Intermediates toward Generic Active Pharmaceutical Ingredients. Org. Process Res. Dev. 24 (6), 1131–1140. doi:10.1021/acs.oprd.0c00120
Reetz, M. T., Carballeira, J. D., and Vogel, A. (2006). Iterative Saturation Mutagenesis on the Basis of B Factors as a Strategy for Increasing Protein Thermostability. Angew. Chem. Int. Ed. 45 (46), 7745–7751. doi:10.1002/anie.200602795
Reeve, H. A., Ash, P. A., Park, H., Huang, A., Posidias, M., Tomlinson, C., et al. (2017). Enzymes as Modular Catalysts for Redox Half-Reactions in H2-Powered Chemical Synthesis: from Biology to Technology. Biochem. J. 474, 215–230. doi:10.1042/bcj20160513
Reeve, H. A., Lauterbach, L., Ash, P. A., Lenz, O., and Vincent, K. A. (2012). A Modular System for Regeneration of NADcofactors Using Graphite Particles Modified with Hydrogenase and Diaphorase Moieties. Chem. Commun. 48 (10), 1589–1591. doi:10.1039/C1CC14826E
Riebel, B. R., Gibbs, P. R., Wellborn, W. B., and Bommarius, A. S. (2003). Cofactor Regeneration of Both NAD+ from NADH and NADP+ from NADPH:NADH Oxidase from Lactobacillus Sanfranciscensis. Adv. Synthesis Catal. 345 (6-7), 707–712. doi:10.1002/adsc.200303039
Riebel, B. R., Gibbs, P. R., Wellborn, W. B., and Bommarius, A. S. (2002). Cofactor Regeneration of NAD + from NADH: Novel Water‐Forming NADH Oxidases. Adv. Synth. Catal. 344 (10), 1156–1168. doi:10.1002/1615-4169(200212)344:10<1156::AID-ADSC1156>3.0.CO;2-#
Rosen, T. C., Feldmann, R., Dünkelmann, P., and Daußmann, T. (2006). Bioreductive Synthesis of Perfluorinated Chiral Alcohols. Tetrahedron Lett. 47 (28), 4803–4806. doi:10.1016/j.tetlet.2006.05.061
Scherkus, C., Schmidt, S., Bornscheuer, U. T., Gröger, H., Kara, S., and Liese, A. (2016). A Fed-Batch Synthetic Strategy for a Three-step Enzymatic Synthesis of Poly-ϵ-Caprolactone. ChemCatChem 8 (22), 3446–3452. doi:10.1002/cctc.201600806
Scherkus, C., Schmidt, S., Bornscheuer, U. T., Gröger, H., Kara, S., and Liese, A. (2017). Kinetic Insights into ϵ-caprolactone Synthesis: Improvement of an Enzymatic Cascade Reaction. Biotechnol. Bioeng. 114 (6), 1215–1221. doi:10.1002/bit.26258
Schmidt, S., Büchsenschütz, H. C., Scherkus, C., Liese, A., Gröger, H., and Bornscheuer, U. T. (2015a). Biocatalytic Access to Chiral Polyesters by an Artificial Enzyme Cascade Synthesis. ChemCatChem 7 (23), 3951–3955. doi:10.1002/cctc.201500823
Schmidt, S., Scherkus, C., Muschiol, J., Menyes, U., Winkler, T., Hummel, W., et al. (2015b). An Enzyme Cascade Synthesis of ε-Caprolactone and its Oligomers. Angew. Chem. Int. Ed. 54 (9), 2784–2787. doi:10.1002/anie.201410633
Schmieg, B., Döbber, J., Kirschhöfer, F., Pohl, M., and Franzreb, M. (2019). Advantages of Hydrogel-Based 3D-Printed Enzyme Reactors and Their Limitations for Biocatalysis. Front. Bioeng. Biotechnol. 6. doi:10.3389/fbioe.2018.00211
Schrittwieser, J. H., Lavandera, I., Seisser, B., Mautner, B., and Kroutil, W. (2009a). Biocatalytic Cascade for the Synthesis of Enantiopure β-Azidoalcohols and β-Hydroxynitriles. Eur. J. Org. Chem. 2009, 2293–2298. doi:10.1002/ejoc.200900091
Schrittwieser, J. H., Lavandera, I., Seisser, B., Mautner, B., Lutje Spelberg, J. H., and Kroutil, W. (2009b). Shifting the Equilibrium of a Biocatalytic Cascade Synthesis to Enantiopure Epoxides Using Anion Exchangers. Tetrahedron Asymmetry 20 (4), 483–488. doi:10.1016/j.tetasy.2009.02.035
Schröder, I., Steckhan, E., and Liese, A. (2003). In Situ NAD+ Regeneration Using 2,2'-Azinobis(3-Ethylbenzothiazoline-6-Sulfonate) as an Electron Transfer Mediator. J. Electroanal. Chem. 541, 109–115. doi:10.1016/S0022-0728(02)01420-1
Schubert, T., Hummel, W., Kula, M.-R., and Müller, M. (2001). Enantioselective Synthesis of Both Enantiomers of Various Propargylic Alcohols by Use of Two Oxidoreductases. Eur. J. Org. Chem. 2001 (22), 4181–4187. doi:10.1002/1099-0690(200111)2001:22<4181::aid-ejoc4181>3.0.co;2-t
Schubert, T., Hummel, W., and Müller, M. (2002). Highly Enantioselective Preparation of Multifunctionalized Propargylic Building Blocks. Angew. Chem. Int. Ed. 41 (4), 634–637. doi:10.1002/1521-3773(20020215)41:4<634::aid-anie634>3.0.co;2-u
Schumacher, J., Eckstein, M., and Kragl, U. (2006). Influence of Water-Miscible Organic Solvents on Kinetics and Enantioselectivity of the (R,-specific Alcohol Dehydrogenase fromLactobacillus Brevis. Biotechnol. J. 1 (5), 574–581. doi:10.1002/biot.200600039
Seelbach, K., Riebel, B., Hummel, W., Kula, M.-R., Tishkov, V. I., Egorov, A. M., et al. (1996). A Novel, Efficient Regenerating Method of NADPH Using a New Formate Dehydrogenase. Tetrahedron Lett. 37 (9), 1377–1380. doi:10.1016/0040-4039(96)00010-X
Sgalla, S., Fabrizi, G., Cirilli, R., Macone, A., Bonamore, A., Boffi, A., et al. (2007). Chiral (R)- and (S)-allylic Alcohols via a One-Pot Chemoenzymatic Synthesis. Tetrahedron Asymmetry 18 (23), 2791–2796. doi:10.1016/j.tetasy.2007.10.043
Shaked, Z. e., and Whitesides, G. M. (1980). Enzyme-catalyzed Organic Synthesis: NADH Regeneration by Using Formate Dehydrogenase. J. Am. Chem. Soc. 102 (23), 7104–7105. doi:10.1021/ja00543a038
Shanati, T., Lockie, C., Beloti, L., Grogan, G., and Ansorge-Schumacher, M. B. (2019). Two Enantiocomplementary Ephedrine Dehydrogenases from Arthrobacter Sp. TS-15 with Broad Substrate Specificity. ACS Catal. 9 (7), 6202–6211. doi:10.1021/acscatal.9b00621
Sheldon, R. A., Arends, I. W. C. E., ten Brink, G.-J., and Dijksman, A. (2002). Green, Catalytic Oxidations of Alcohols. Acc. Chem. Res. 35 (9), 774–781. doi:10.1021/ar010075n
Sheldon, R. A., Basso, A., and Brady, D. (2021). New Frontiers in Enzyme Immobilisation: Robust Biocatalysts for a Circular Bio-Based Economy. Chem. Soc. Rev. 50 (10), 5850–5862. doi:10.1039/D1CS00015B
Sheldon, R. A., Bode, M. L., and Akakios, S. G. (2022). Metrics of Green Chemistry: Waste Minimization. Curr. Opin. Green Sustain. Chem. 33, 100569. doi:10.1016/j.cogsc.2021.100569
Sheldon, R. A. (2018). Metrics of Green Chemistry and Sustainability: Past, Present, and Future. ACS Sustain. Chem. Eng. 6 (1), 32–48. doi:10.1021/acssuschemeng.7b03505
Sheldon, R. A., and Pereira, P. C. (2017). Biocatalysis Engineering: the Big Picture. Chem. Soc. Rev. 46 (10), 2678–2691. doi:10.1039/c6cs00854b
Sheldon, R. A. (2017). The E Factor 25 Years on: the Rise of Green Chemistry and Sustainability. Green Chem. 19 (1), 18–43. doi:10.1039/C6GC02157C
Sheldon, R. A., and van Pelt, S. (2013). Enzyme Immobilisation in Biocatalysis: Why, what and How. Chem. Soc. Rev. 42 (15), 6223–6235. doi:10.1039/c3cs60075k
Sheldon, R. A., and Woodley, J. M. (2018). Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 118 (2), 801–838. doi:10.1021/acs.chemrev.7b00203
Sheldon, R. (2019). CLEAs, Combi-CLEAs and 'Smart' Magnetic CLEAs: Biocatalysis in a Bio-Based Economy. Catalysts 9 (3), 261. doi:10.3390/catal9030261
Siddiqui, K. S. (2017). Defying the Activity-Stability Trade-Off in Enzymes: Taking Advantage of Entropy to Enhance Activity and Thermostability. Crit. Rev. Biotechnol. 37 (3), 309–322. doi:10.3109/07388551.2016.1144045
Sigurdardóttir, S. B., Lehmann, J., Grivel, J. C., Zhang, W., Kaiser, A., and Pinelo, M. (2019). Alcohol Dehydrogenase on Inorganic Powders: Zeta Potential and Particle Agglomeration as Main Factors Determining Activity during Immobilization. Colloids Surfaces B Biointerfaces 175, 136–142. doi:10.1016/j.colsurfb.2018.11.080
Simon, R. C., Busto, E., Richter, N., Belaj, F., and Kroutil, W. (2014). Chemoenzymatic Synthesis of Enantiomerically Puresyn-Configured 1-Aryl-3-Methylisochroman Derivatives. Eur. J. Org. Chem. 2014 (1), 111–121. doi:10.1002/ejoc.201301429
Singh, R., Kumar, M., Mittal, A., and Mehta, P. K. (2016). Microbial Enzymes: Industrial Progress in 21st Century. 3 Biotech. 6 (2), 174. doi:10.1007/s13205-016-0485-8
Sinha, S. C., and Keinan, E. (1997). Total Synthesis of (+)-Aspicilin. The Naked Carbon Skeleton Strategy vs the Bioorganic Approach. J. Org. Chem. 62 (2), 377–386. doi:10.1021/jo9614811
Solé, J., Brummund, J., Caminal, G., Schürman, M., Álvaro, G., and Guillén, M. (2019). Ketoisophorone Synthesis with an Immobilized Alcohol Dehydrogenase. ChemCatchem 11 (19), 4862–4870. doi:10.1002/cctc.201901090
Sorgedrager, M. J., van Rantwijk, F., Huisman, G. W., and Sheldon, R. A. (2008). Asymmetric Carbonyl Reductions with Microbial Ketoreductases. Adv. Synth. Catal. 350 (14-15), 2322–2328. doi:10.1002/adsc.200800200
Spickermann, D., Kara, S., Barackov, I., Hollmann, F., Schwaneberg, U., Duenkelmann, P., et al. (2014). Alcohol Dehydrogenase Stabilization by Additives under Industrially Relevant Reaction Conditions. J. Mol. Catal. B Enzym. 103, 24–28. doi:10.1016/j.molcatb.2013.11.015
Stampfer, W., Kosjek, B., Moitzi, C., Kroutil, W., and Faber, K. (2002). Biocatalytic Asymmetric Hydrogen Transfer. Angew. Chem. Int. Ed. 41 (6), 1014–1017. doi:10.1002/1521-3773(20020315)41:6<1014::aid-anie1014>3.0.co;2-6
Staniland, S., Yuan, B., Giménez-Agulló, N., Marcelli, T., Willies, S. C., Grainger, D. M., et al. (2014). Enzymatic Desymmetrising Redox Reactions for the Asymmetric Synthesis of Biaryl Atropisomers. Chem. Eur. J. 20 (41), 13084–13088. doi:10.1002/chem.201404509
Staudt, S., Burda, E., Giese, C., Müller, C. A., Marienhagen, J., Schwaneberg, U., et al. (2013). Direktoxidation von Cycloalkanen zu Cycloalkanonen mit Sauerstoff in Wasser. Angew. Chem. 125 (8), 2415–2419. doi:10.1002/ange.201204464
Su, E., Meng, Y., Ning, C., Ma, X., and Deng, S. (2018). Magnetic Combined Cross-Linked Enzyme Aggregates (Combi-CLEAs) for Cofactor Regeneration in the Synthesis of Chiral Alcohol. J. Biotechnol. 271, 1–7. doi:10.1016/j.jbiotec.2018.02.007
Sun, Z., Lonsdale, R., Ilie, A., Li, G., Zhou, J., and Reetz, M. T. (2016). Catalytic Asymmetric Reduction of Difficult-To-Reduce Ketones: Triple-Code Saturation Mutagenesis of an Alcohol Dehydrogenase. ACS Catal. 6 (3), 1598–1605. doi:10.1021/acscatal.5b02752
Tassano, E., Merusic, K., Buljubasic, I., Laggner, O., Reiter, T., Vogel, A., et al. (2020). Regioselective Biocatalytic Self-Sufficient Tishchenko-type Reaction via Formal Intramolecular Hydride Transfer. Chem. Commun. 56, 6340–6343. doi:10.1039/D0CC02509G
Tauber, K., Fuchs, M., Sattler, J. H., Pitzer, J., Pressnitz, D., Koszelewski, D., et al. (2013). Artificial Multi-Enzyme Networks for the Asymmetric Amination Ofsec-Alcohols. Chem. Eur. J. 19 (12), 4030–4035. doi:10.1002/chem.201202666
Tavanti, M., Mangas-Sanchez, J., Montgomery, S. L., Thompson, M. P., and Turner, N. J. (2017a). A Biocatalytic Cascade for the Amination of Unfunctionalised Cycloalkanes. Org. Biomol. Chem. 15, 9790–9793. doi:10.1039/C7OB02569F
Tavanti, M., Parmeggiani, F., Castellanos, J. R. G., Mattevi, A., and Turner, N. J. (2017b). One-Pot Biocatalytic Double Oxidation of α-Isophorone for the Synthesis of Ketoisophorone. ChemCatChem 9 (17), 3338–3348. doi:10.1002/cctc.201700620
Tentori, F., Brenna, E., Colombo, D., Crotti, M., Gatti, F., Ghezzi, M., et al. (2018). Biocatalytic Approach to Chiral β-Nitroalcohols by Enantioselective Alcohol Dehydrogenase-Mediated Reduction of α-Nitroketones. Catalysts 8 (8), 308. doi:10.3390/catal8080308
Thompson, C. E., Freitas, L. B., and Salzano, F. M. (2018). Molecular Evolution and Functional Divergence of Alcohol Dehydrogenases in Animals, Fungi and Plants. Genet. Mol. Biol. 41 (1Suppl. l), 341–354. doi:10.1590/1678-4685-GMB-2017-0047
Thompson, M. P., Derrington, S. R., Heath, R. S., Porter, J. L., Mangas-Sanchez, J., Devine, P. N., et al. (2019). A Generic Platform for the Immobilisation of Engineered Biocatalysts. Tetrahedron 75 (3), 327–334. doi:10.1016/j.tet.2018.12.004
Tieves, F., Tonin, F., Fernández-Fueyo, E., Robbins, J. M., Bommarius, B., Bommarius, A. S., et al. (2019). Energising the E-Factor: The E+-factor. Tetrahedron 75, 1311–1314. doi:10.1016/j.tet.2019.01.065
Tojo, G., and Fernández, M. (2006). Oxidation of Alcohols to Aldehydes and Ketones. Springer. doi:10.1002/chemv.201700026
Tufvesson, L. M., Tufvesson, P., Woodley, J. M., and Börjesson, P. (2012). Life Cycle Assessment in Green Chemistry: Overview of Key Parameters and Methodological Concerns. Int. J. Life Cycle Assess. 18, 431–444. doi:10.1007/s11367-012-0500-1
Tufvesson, P., Lima-Ramos, J., Nordblad, M., and Woodley, J. M. (2010). Guidelines and Cost Analysis for Catalyst Production in Biocatalytic Processes. Org. Process Res. Dev. 15 (1), 266–274. doi:10.1021/op1002165
Vasić, K., Knez, Ž., and Leitgeb, M. (2020). Immobilization of Alcohol Dehydrogenase from Saccharomyces cerevisiae onto Carboxymethyl Dextran-Coated Magnetic Nanoparticles: a Novel Route for Biocatalyst Improvement via Epoxy Activation. Sci. Rep. 10 (1), 19478. doi:10.1038/s41598-020-76463-x
Velasco‐Lozano, S., Benítez‐Mateos, A. I., and López‐Gallego, F. (2017). Co‐immobilized Phosphorylated Cofactors and Enzymes as Self‐Sufficient Heterogeneous Biocatalysts for Chemical Processes. Angew. Chem. Int. Ed. 56 (3), 771–775. doi:10.1002/anie.201609758
Velasco‐Lozano, S., Santiago‐Arcos, J., Mayoral, J. A., and López‐Gallego, F. (2020). Co‐immobilization and Colocalization of Multi‐Enzyme Systems for the Cell‐Free Biosynthesis of Aminoalcohols. ChemCatChem 12 (11), 3030–3041. doi:10.1002/cctc.201902404
Villela Filho, M., Stillger, T., Müller, M., Liese, A., and Wandrey, C. (2003). Is Log P a Convenient Criterion to Guide the Choice of Solvents for Biphasic Enzymatic Reactions? Angew. Chem. Int. Ed. 42 (26), 2993–2996. doi:10.1002/anie.200351089
Voss, C. V., Gruber, C. C., Faber, K., Knaus, T., Macheroux, P., and Kroutil, W. (2008a). Orchestration of Concurrent Oxidation and Reduction Cycles for Stereoinversion and Deracemisation of Sec-Alcohols. J. Am. Chem. Soc. 130 (42), 13969–13972. doi:10.1021/ja804816a
Voss, C. V., Gruber, C. C., and Kroutil, W. (2008b). Deracemization of Secondary Alcohols through a Concurrent Tandem Biocatalytic Oxidation and Reduction. Angew. Chem. Int. Ed. 47 (4), 741–745. doi:10.1002/anie.200703296
Wachtmeister, J., Jakoblinnert, A., Kulig, J., Offermann, H., and Rother, D. (2014). Whole-Cell Teabag Catalysis for the Modularisation of Synthetic Enzyme Cascades in Micro-aqueous Systems. ChemCatChem 6 (4), 1051–1058. doi:10.1002/cctc.201300880
Wang, H.-Y., Tang, J.-W., Peng, P., Yan, H.-J., Zhang, F.-L., and Chen, S.-X. (2020). Development of a Novel Chemoenzymatic Process for (S)-1-(Pyridin-4-yl)-1,3-propanediol. Org. Process Res. Dev. 24 (12), 2890–2897. doi:10.1021/acs.oprd.0c00403
Wang, Z., Wu, X., Li, Z., Huang, Z., and Chen, F. (2019). Ketoreductase Catalyzed Stereoselective Bioreduction of α-nitro Ketones. Org. Biomol. Chem. 17 (14), 3575–3580. doi:10.1039/C9OB00051H
Wedde, S., Rommelmann, P., Scherkus, C., Schmidt, S., Bornscheuer, U. T., Liese, A., et al. (2017). An Alternative Approach towards Poly-ε-Caprolactone through a Chemoenzymatic Synthesis: Combined Hydrogenation, Bio-Oxidations and Polymerization without the Isolation of Intermediates. Green Chem. 19, 1286–1290. doi:10.1039/C6GC02529C
Wichmann, R., and Vasic-Racki, D. (2005). “Cofactor Regeneration at the Lab Scale,” in Technology Transfer in Biotechnology: From Lab to Industry to Production (Berlin: Springer-Verlag Berlin), 225–260. doi:10.1007/b98911
Willies, S., Isupov, M., and Littlechild, J. (2010). Thermophilic Enzymes and Their Applications in Biocatalysis: a Robust Aldo‐keto Reductase. Environ. Technol. 31 (10), 1159–1167. doi:10.1080/09593330.2010.490857
Winkler, M. (2018). Carboxylic Acid Reductase Enzymes (CARs). Curr. Opin. Chem. Biol. 43, 23–29. doi:10.1016/j.cbpa.2017.10.006
Wolberg, M., Hummel, W., Wandrey, C., and Müller, M. (2000). Highly Regio- and Enantioselective Reduction of 3,5-dioxocarboxylates. Angew. Chem. Int. Ed. 39 (23), 4306–4308. doi:10.1002/1521-3773(20001201)39:23<4306::aid-anie4306>3.0.co;2-g
Wu, K., Hu, X., Yang, Z., Huang, J., Wang, X., and Shao, L. (2021a). Efficient Synthesis of an Antiviral Drug Intermediate Using Overexpressed Short‐chain Dehydrogenase and Cross‐linked Enzyme Aggregates Stabilization. J. Chem. Technol. Biotechnol. 96 (3), 714–722. doi:10.1002/jctb.6584
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K., and Bornscheuer, U. T. (2021b). Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 60 (1), 88–119. doi:10.1002/anie.202006648
Wu, X., Wang, Y., Ju, J., Chen, C., Liu, N., and Chen, Y. (2009). Enantioselective Synthesis of Ethyl (S)-2-hydroxy-4-phenylbutyrate by Recombinant Diketoreductase. Tetrahedron Asymmetry 20 (21), 2504–2509. doi:10.1016/j.tetasy.2009.10.036
Xu, G.-C., Wang, Y., Tang, M.-H., Zhou, J.-Y., Zhao, J., Han, R.-Z., et al. (2018). Hydroclassified Combinatorial Saturation Mutagenesis: Reshaping Substrate Binding Pockets of KpADH for Enantioselective Reduction of Bulky-Bulky Ketones. ACS Catal. 8 (9), 8336–8345. doi:10.1021/acscatal.8b02286
Xu, M.-Q., Li, F.-L., Yu, W.-Q., Li, R.-F., and Zhang, Y.-W. (2020). Combined Cross-Linked Enzyme Aggregates of Glycerol Dehydrogenase and NADH Oxidase for High Efficiency In Situ NAD+ Regeneration. Int. J. Biol. Macromol. 144, 1013–1021. doi:10.1016/j.ijbiomac.2019.09.178
Xu, X., Brasselet, H., Jongkind, E. P. J., Alcalde, M., Paul, C. E., and Hollmann, F. (2022). A Peroxygenase‐Alcohol Dehydrogenase Cascade Reaction to Transform Ethylbenzene Derivatives into Enantioenriched Phenylethanols. ChemBioChem 23 (6), e202200017. doi:10.1002/cbic.202200017
Yamazaki, Y., and Hosono, K. (1988). Stereoselectivity of Horse Liver Alcohol Dehydrogenase Catalyzed Oxidoreduction of an Organometallic Meso Diol and the Corresponding Dialdehyde. Tetrahedron Lett. 29 (45), 5769–5770. doi:10.1016/S0040-4039(00)82187-5
Yan, J., Wang, X., Li, F., Yang, L., Shi, G., Sun, W., et al. (2021). Biocatalytic Preparation of a Key Intermediate of Antifungal Drugs Using an Alcohol Dehydrogenase with High Organic Tolerance. Tetrahedron Lett. 84, 153455. doi:10.1016/j.tetlet.2021.153455
Yang, K. K., Wu, Z., and Arnold, F. H. (2019). Machine-learning-guided Directed Evolution for Protein Engineering. Nat. Methods 16 (8), 687–694. doi:10.1038/s41592-019-0496-6
Yang, Z., Ye, W., Xie, Y., Liu, Q., Chen, R., Wang, H., et al. (2020). Efficient Asymmetric Synthesis of Ethyl (S)-4-Chloro-3-hydroxybutyrate Using Alcohol Dehydrogenase SmADH31 with High Tolerance of Substrate and Product in a Monophasic Aqueous System. Org. Process Res. Dev. 24 (6), 1068–1076. doi:10.1021/acs.oprd.0c00088
Ye, N., Kou, X., Shen, J., Huang, S., Chen, G., and Ouyang, G. (2020). Metal‐Organic Frameworks: A New Platform for Enzyme Immobilization. ChemBioChem 21 (18), 2585–2590. doi:10.1002/cbic.202000095
Yildirim, D., Alagöz, D., Toprak, A., Tükel, S., and Fernandez-Lafuente, R. (2019). Tuning Dimeric Formate Dehydrogenases Reduction/oxidation Activities by Immobilization. Process Biochem. 85, 97–105. doi:10.1016/j.procbio.2019.07.001
Yoshimoto, M., Sato, M., Yoshimoto, N., and Nakao, K. (2008). Liposomal Encapsulation of Yeast Alcohol Dehydrogenase with Cofactor for Stabilization of the Enzyme Structure and Activity. Biotechnol. Prog. 24 (3), 576–582. doi:10.1021/bp070392e
Younes, S. H. H., Ni, Y., Schmidt, S., Kroutil, W., and Hollmann, F. (2017). Alcohol Dehydrogenases Catalyze the Reduction of Thioesters. ChemCatChem 9, 1389–1392. doi:10.1002/cctc.201700165
Yuan, B., Page, A., Worrall, C. P., Escalettes, F., Willies, S. C., McDouall, J. J. W., et al. (2010). Biocatalytic Desymmetrization of an Atropisomer with Both an Enantioselective Oxidase and Ketoreductases. Angew. Chem. Int. Ed. 49 (39), 7010–7013. doi:10.1002/anie.201002580
Zadlo, A., Schrittwieser, J. H., Koszelewski, D., Kroutil, W., and Ostaszewski, R. (2016). Enantioselective Reduction of Ethyl 3-Oxo-5-Phenylpentanoate with Whole-Cell Biocatalysts. Eur. J. Org. Chem. 2016 (5), 1007–1011. doi:10.1002/ejoc.201501460
Zaks, A., and Klibanov, A. M. (1984). Enzymatic Catalysis in Organic Media at 100°C. Science 224 (4654), 1249–1251. doi:10.1126/science.6729453
Zaks, A., and Klibanov, A. M. (1985). Enzyme-catalyzed Processes in Organic Solvents. Proc. Natl. Acad. Sci. U.S.A. 82 (10), 3192–3196. doi:10.1073/pnas.82.10.3192
Zeuner, B., Ma, N., Berendt, K., Meyer, A. S., Andric, P., Jørgensen, J. H., et al. (2018). Immobilization of Alcohol Dehydrogenase on Ceramic Silicon Carbide Membranes for Enzymatic CH3 OH Production. J. Chem. Technol. Biotechnol. 93 (10), 2952–2961. doi:10.1002/jctb.5653
Zezzi do Valle Gomes, M., and Palmqvist, A. E. C. (2017). Influence of Operating Conditions and Immobilization on Activity of Alcohol Dehydrogenase for the Conversion of Formaldehyde to Methanol. New J. Chem. 41 (19), 11391–11397. doi:10.1039/C7NJ02028G
Zhang, J., Pu, T., Xu, Y., Huang, J., Chen, D., and Shao, L. (2020a). Synthesis of Dehydroepiandrosterone by Co‐immobilization of Keto Reductase and Glucose Dehydrogenase. J. Chem. Technol. Biotechnol. 95 (9), 2530–2536. doi:10.1002/jctb.6438
Zhang, J., Xu, T., and Li, Z. (2013). Enantioselective Biooxidation of Racemictrans-Cyclic Vicinal Diols: One-Pot Synthesis of Both Enantiopure (S,S)-Cyclic Vicinal Diols and (R)-α-Hydroxy Ketones. Adv. Synth. Catal. 355 (16), 3147–3153. doi:10.1002/adsc.201300301
Zhang, W., Bariotaki, A., Smonou, I., and Hollmann, F. (2017). Visible-light-driven Photooxidation of Alcohols Using Surface-Doped Graphitic Carbon Nitride. Green Chem. 19, 2096–2100. doi:10.1039/C7GC00539C
Zhang, W., Fueyo, E. F., Hollmann, F., Martin, L. L., Pesic, M., Wardenga, R., et al. (2019). Combining Photo-Organo Redox- and Enzyme Catalysis Facilitates Asymmetric C-H Bond Functionalization. Eur. J. Org. Chem. 2019 (1), 80–84. doi:10.1002/ejoc.201801692
Zhang, W., and Hollmann, F. (2018). Nonconventional Regeneration of Redox Enzymes - a Practical Approach for Organic Synthesis? Chem. Commun. 54, 7281–7289. doi:10.1039/C8CC02219D
Zhang, W., Lee, J.-H., Younes, S. H. H., Tonin, F., Hagedoorn, P.-L., Pichler, H., et al. (2020b). Photobiocatalytic Synthesis of Chiral Secondary Fatty Alcohols from Renewable Unsaturated Fatty Acids. Nat. Commun. 11 (1), 2258. doi:10.1038/s41467-020-16099-7
Zhang, Y., Yao, P., Cui, Y., Wu, Q., and Zhu, D. (2018). One‐Pot Enzymatic Synthesis of Cyclic Vicinal Diols from Aliphatic Dialdehydes via Intramolecular C−C Bond Formation and Carbonyl Reduction Using Pyruvate Decarboxylases and Alcohol Dehydrogenases. Adv. Synth. Catal. 360 (21), 4191–4196. doi:10.1002/adsc.201800455
Zhao, X., Cleary, S. E., Zor, C., Grobert, N., Reeve, H. A., and Vincent, K. A. (2021). Chemo-bio Catalysis Using Carbon Supports: Application in H2-Driven Cofactor Recycling. Chem. Sci. 12 (23), 8105–8114. doi:10.1039/D1SC00295C
Zheng, C., Wang, Z., Wang, Q., Wang, S., Lao, S., He, J., et al. (2021). Efficient Preparation of the Chiral Intermediate of Luliconazole with Lactobacillus Kefir Alcohol Dehydrogenase through Rational Rearrangement of the Substrate Binding Pocket. Mol. Catal. 509, 111639. doi:10.1016/j.mcat.2021.111639
Zhou, J., Xu, G., and Ni, Y. (2020). Stereochemistry in Asymmetric Reduction of Bulky-Bulky Ketones by Alcohol Dehydrogenases. ACS Catal. 10 (19), 10954–10966. doi:10.1021/acscatal.0c02646
Zhu, D., and Hua, L. (2006). Enantioselective Enzymatic Reductions of Sterically Bulky Aryl Alkyl Ketones Catalyzed by a NADPH-dependent Carbonyl Reductase. J. Org. Chem. 71 (25), 9484–9486. doi:10.1021/jo061571y
Zhu, D., Malik, H. T., and Hua, L. (2006b). Asymmetric Ketone Reduction by a Hyperthermophilic Alcohol Dehydrogenase. The Substrate Specificity, Enantioselectivity and Tolerance of Organic Solvents. Tetrahedron Asymmetry 17 (21), 3010–3014. doi:10.1016/j.tetasy.2006.10.042
Zhu, D., Stearns, J. E., Ramirez, M., and Hua, L. (2006). Enzymatic Enantioselective Reduction of α-ketoesters by a Thermostable 7α-Hydroxysteroid Dehydrogenase from Bacteroides Fragilis. Tetrahedron 62 (18), 4535–4539. doi:10.1016/j.tet.2006.02.041
Zhu, L., Song, Y., Chang, C., Ma, H., Yang, L., Deng, Z., et al. (2021). Engineering Leifsonia Alcohol Dehydrogenase for Thermostability and Catalytic Efficiency by Enhancing Subunit Interactions. ChemBioChem 22 (22), 3178–3183. doi:10.1002/cbic.202100431
Zor, C., Reeve, H. A., Quinson, J., Thompson, L. A., Lonsdale, T. H., Dillon, F., et al. (2017). H2-Driven Biocatalytic Hydrogenation in Continuous Flow Using Enzyme-Modified Carbon Nanotube Columns. Chem. Commun. 53 (71), 9839–9841. doi:10.1039/C7CC04465H
Keywords: alcohol dehydrogenases (ADH), oxidation reactions, reduction reactions, enantioselective synthesis, alcohols, ketones, aldehydes, biocatalysis
Citation: de Miranda AS, Milagre CDF and Hollmann F (2022) Alcohol Dehydrogenases as Catalysts in Organic Synthesis. Front. Catal. 2:900554. doi: 10.3389/fctls.2022.900554
Received: 20 March 2022; Accepted: 08 April 2022;
Published: 10 May 2022.
Edited by:
Jesús Fernández Lucas, European University of Madrid, SpainReviewed by:
Vicente Gotor-Fernández, University of Oviedo, SpainCopyright © 2022 de Miranda, Milagre and Hollmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda Silva de Miranda, YXNtaXJhbmRhQHVmbWcuYnI=; Cintia D. F. Milagre, Y2ludGlhLm1pbGFncmVAdW5lc3AuYnI=; Frank Hollmann, Zi5ob2xsbWFubkB0dWRlbGZ0Lm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.