
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Catal., 23 September 2022
Sec. Biocatalysis
Volume 2 - 2022 | https://doi.org/10.3389/fctls.2022.867811
This article is part of the Research TopicRecent Advances in Biocatalysed Amine FormationView all 5 articles
 Jan-Moritz Sutter1
Jan-Moritz Sutter1 Daniel E. Mitchell2Marcel Schmidt1
Daniel E. Mitchell2Marcel Schmidt1 Michail N. Isupov2
Michail N. Isupov2 Jennifer A. Littlechild2*
Jennifer A. Littlechild2* Peter Schönheit1*
Peter Schönheit1*A branched chain aminotransferase from Thermoproteus tenax has been identified, cloned, over-expressed and biochemically characterised. A molecular modelling approach has been used to predict the 3D structure allowing its comparison with other related enzymes. This enzyme has high similarity to a previously characterised aminotransferase from Thermoproteus uzoniensis however its substrate specificity shows key differences towards the substrate α-ketoglutarate. Examination of the active sites of the two related enzymes reveals a single amino acid substitution of a glycine residue to a serine residue which could be responsible for this difference. When Gly104 in T. tenax was mutated to a serine residue and the resultant enzyme characterised, this single amino acid change resulted in a dramatic reduction in activity towards α-ketoglutarate with an 18-fold reduction in Vmax and a 20-fold Km increase, resulting in a 370-fold lower catalytic efficiency. Structural comparisons between the two related Thermoproteus enzymes and another branched chain aminotransferase from Geoglobus acetivorans has revealed that the serine residue affects the flexibility of a key loop involved in catalysis. This subtle difference has provided further insight into our understanding of the substrate specificity of these industrially important enzymes.
Aminotransferases (transaminases: EC 2.6.1) are pyridoxal 5-phosphate (PLP) dependent enzymes which reversibly catalyse the transfer of an amino group from an amino substrate to a ketone or aldehyde, resulting in the formation of a chiral amine (Hayashi 1995). The aminotransferases are key enzymes of amino acid metabolism, and can transfer a primary amino group from diverse amino acids and amines to a range of different keto acids, ketones and aldehydes (Cooper and Meister 1989). The reaction proceeds via two half reactions. Firstly, the primary amino group of the amino group donor is transferred to the PLP cofactor producing pyridoxamine 5-phosphate (PMP) and a ketone or aldehyde. In the second step the ketone (aldehyde) substrate accepts the amino group of PMP to yield an aminated product and regenerating the PLP (Hirotsu et al., 2005). This catalytic process can be inhibited by products and/or substrates (Hutson 2001; Yu et al., 2014). Most aminotransferases use α-ketoglutarate as their preferred acceptor substrate forming a glutamate product (Holeček, 2020). However, some have preference for a pyruvate acceptor to form an alanine product instead (Yonaha et al., 1987; Sayer et al., 2014). All aminotransferases function as homo-dimers or higher order oligomers built up from dimers, with the active site of each monomer in the catalytic dimer contributing essential amino acid residues to the active site (Eliot and Kirsch 2004).
The aminotransferases are extremely important in amino acid metabolism, and have been found to perform various functions beneficial to human health (Borgenvik et al., 2011; Sookoian and Pirola 2012). They have also attracted wide interest from the pharmaceutical industry due to their ability to produce asymmetrically pure chiral amines which are widely used as building blocks in drug synthesis (Ghislieri and Turner 2014; Wendisch 2020). For example, an engineered aminotransferase has been used in the production of the anti-diabetic drug sitagliptin (Savile et al., 2010). All aminotransferases have several advantages over conventional chemical synthesis, including excellent stereoselectivity, mild reaction conditions, elimination of toxic and expensive metal catalysts and reduction of the use of organic solvents (Deepankumar et al., 2014; Paul et al., 2014; Kelly et al., 2020). Since these enzymes make such useful biocatalysts, there has been much recent work undertaken into the structural understanding of their differing substrate specificity and selectivity based on their structural motifs and associated active sites (Sayer et al., 2013; Slabu et al., 2017).
Branched chain aminotransferases (BCATs) are a subfamily of aminotransferases with a preference for branched chain hydrophobic amino acid acceptors such as valine, leucine and isoleucine and they belong to the Pfam class IV of aminotransferase (Punta et al., 2012). Most aminotransferases have a class I PLP type fold which binds PLP so that the catalysis occurs on its si-face. The well studied Pfam class III aminotransferases are capable of catalysing an amino group transfer to substrates without an α-carboxyl group (ω-aminotransferases) and are (S)-selective (Sayer et al., 2013). Class IV aminotransferases belong to a different PLP fold class and catalyse the reaction on the re-face of the cofactor (Mehta et al., 1993). These properties allow a flexibility in the arrangement of the active site binding cavity, such that the α-carboxyl group of the amino acid can be bound differently to the PLP in the class IV enzymes when compared to other aminotransferases (Yonaha et al., 1987; Mehta et al., 1993). The aminotransferase class IV contains the BCATs, D-amino acid aminotransferases and (R)-selective amine aminotransferases. Fungal amine transaminase from Nectria haematococca has been characterized and shown to exhibit high activity towards both aromatic and aliphatic (R)-amines such as (R)- methylbenzylamine and 2(R)-aminohexane (Sayer et al., 2014; Jiang et al., 2015). However, the characterised BCATs from both eukaryotic and bacterial sources are most active against their natural branched chain amino acid substrates and corresponding keto acids (Conway and Hutson 2000; Venos et al., 2004). An analysis of the substrate specificity of all recently characterised BCATs has been carried out by the group of Vladimir Popov (Bezsudnova et al., 2017).
The first BCAT enzyme structurally solved was from Escherichia coli reported by Okada and co-workers (Okada et al., 2001) and to date there are currently around 60 structures deposited in the PDB database. Advances in genomic sequencing has resulted in an increasing number of archaeal BCATs being identified and biochemically and structurally characterised including those from T. uzoniensis, G. acetivorans and Archaeoglobus fulgidus (Boyko et al., 2016; Isupov et al., 2019). Enzymes from thermophilic organisms show thermostability, a property that is coupled to an increased resistance to organic solvents making them advantageous for industrial applications. The BCAT from T. uzoniensis, a hyperthermophilic crenarchaeon (Mardanov et al., 2011) was reported to have a very low activity towards α-ketoglutarate as the acceptor substrate which is in contrast to other BCATs (Bezsudnova et al., 2016).
In this study, we report the first characterisation of a BCAT from the hyperthermophilic archaeon T. tenax (TTX), which has potential for industrial biocatalysis due to its high thermal stability. In addition, a comparison is made between this BCAT with that from T. uzoniensis (TUZ), (Boyko et al., 2016) which despite having a very high sequence identity of 81% when compared to TTX retains its activity towards α-ketoglutarate. A further comparison has been made between TTX and the BCAT from G. acetivorans (GAC) for which a crystal structure is available with a bound α-ketoglutarate in the active site (43.46% sequence identity) (Isupov et al., 2019). The understanding of the BCAT active site with respect to its ability to use α-ketoglutarate as a substrate will aid the optimisation of transaminases for more efficient biocatalytic processes.
The putative BCAT from T. tenax, encoded by the gene TTX 0133, was expressed in E coli in a soluble form and purified to homogeneity. SDS-PAGE revealed a single band at 35 kDa which corresponds to the calculated molecular mass of the monomer of the His tagged protein (35 kDa) (Supplementary Figure S1A). The apparent molecular mass of the native enzyme by size exclusion chromatography was 155–165 kDa indicating the presence of an oligomeric state in solution that corresponds to a homohexamer, which is the oligomeric state of other related BCATs in solution and in the crystal structure of the TUZ. However, smaller amounts of homodimeric enzyme were also observed. The TTX enzyme catalyzed the transamination between L-alanine and a variety of acids, including branched chained oxoacids, alpha oxoacids and aromatic oxoacids (Table 1). With the branched chain oxoacids 3-methyl-2-oxobutanoic acid, 4-methyl-2-oxovalerate or 3-methyl-2-oxopentanoic acid the TTX showed a pronounced substrate inhibition (Figures 1A–C) with the reaction inhibited at concentrations higher than 2 mM 3-methyl-2-oxobutanoic acid, 1 mM 4-methyl-2-oxovaleric acid or 0.2 mM 3-methyl-2-oxopentanoic acid. About 20–50% of the activity was detected at 6 mM of these oxoacids.
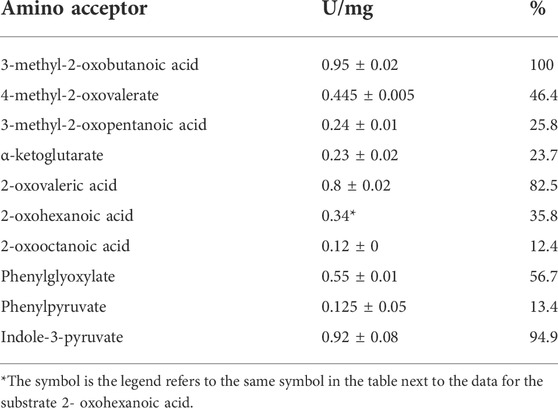
TABLE 1. Substrate specificity of recombinant BCAT TTX; activities were measured at 1 mM acid and given as a percentage of activity with 3-methyl-2-oxobutanoic acid which is taken as 100% the activities were mean values of two measurements, with exception of 2- oxohexanoic acid analysed as single measurement *.
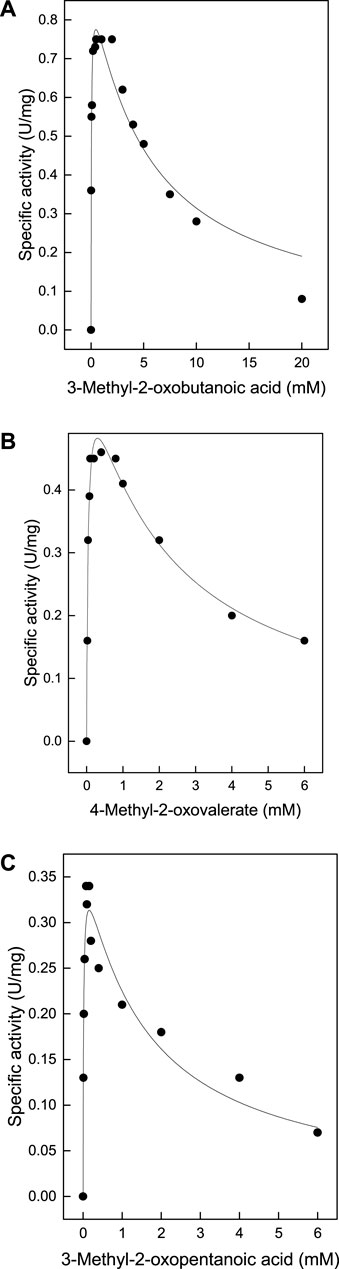
FIGURE 1. Substrate inhibition of TTX activity by methyl-oxoacids: TTX activity was measured in an continuous assay monitoring the oxidation of NADH at 340 nm. By coupling the methyl-oxoacid dependent formation of pyruvate from L-alanine using L-lactate dehydrogenase at 55°C. The data were fitted according to the equation [V=Vmax/(1+(Km/[S]0)+([S]0/Ki))] and the calculated standard errors were derived from the data fitting. (A) 3-methyl-2-oxobutanoic acid: Vmax 0.91 ± 0.05 U/mg, Km 0.041 ± 0 mM, Ki 5.2 ± 0.9 mM. (B) 4-methyl-2-oxovalerate:Vmax 0.62 ± 0.03 U/mg, Km 0.043 ± 0.006 mM, Ki 2.08 ± 0.3 mM (C) 3-methyl-2- oxopentanoic acid: Vmax 0379 ± 0.02 U/mg, Km 0.016 ± 0 mM, Ki 1.49 ± 0.34.
TTX also exhibited significant activity towards α-ketoglutarate (Table 1), which in contrast to branched chain oxoacids did not show substrate inhibition up to 60 mM (Figure 2A). By utilizing α-ketoglutarate, TTX differed from TUZ which did not show measurable activity towards α-ketoglutarate (Bezsudnova et al., 2016) or very low activities (Figure 3).
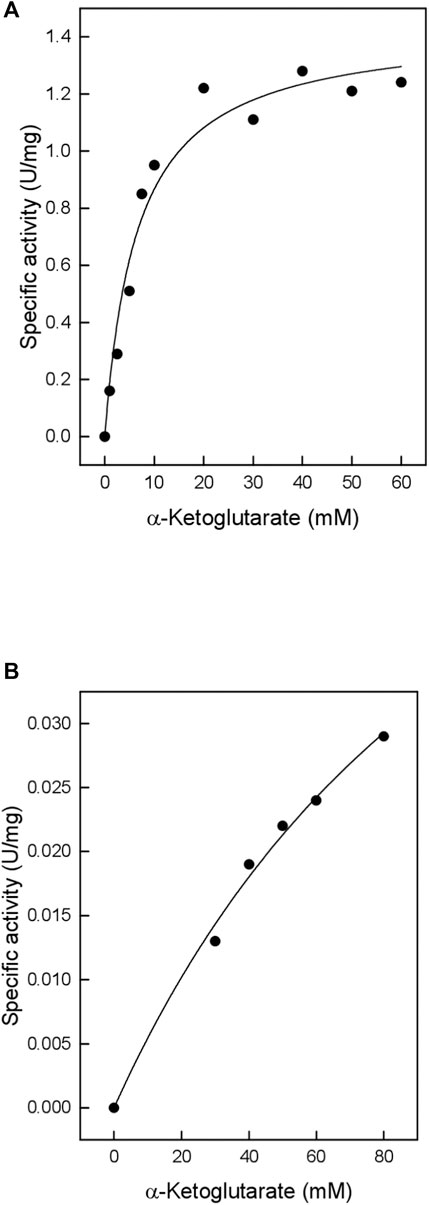
FIGURE 2. Catalytic activity of the TTX wild type and the G104S mutant with α-ketoglutarate (A) Wild type (B) G104S mutant. The kinetic constants were fitted to the Michaelis-Menten equation, and the calculated standard errors were derived from the data fitting. (A): Vmax 1.44 ± 0.07 U/mg; Km 6.59 ± 1.2 mM. (B): Vmax 0.078 ± 0.016 U/mg; Km 132.4 ± 39.4 mM.
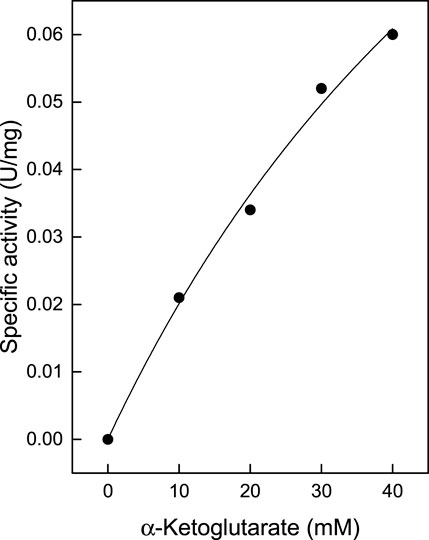
FIGURE 3. Catalytic activity of TUZ wildtype with α-ketoglutarate. The kinetic constants (Vmax: 0.18 ± 0.05 U/mg; Km: 83.5 ± 31.3 mM) were fitted to the Michaelis Menten kinetics and the calculated standard errors were derived from the data fitting.
The enzyme was active over a wide pH range with an optimum activity at pH 8.0; 70% of the activity was measured at pH 7.5 and 80% at pH 9.5 (Supplementary Figure S2). The thermostability was investigated by the pre-incubation of the enzyme at different temperatures for up to 120 min with subsequent analysis of the residual BCAT activity using the standard assay. More than 90% residual activity was detected after preincubation at 65°C or 75°C for 120 min; preincubation at 85°C for 120 min resulted in 65% residual activity (Figure 4). This high temperature stability makes TTX of interest for industrial applications.
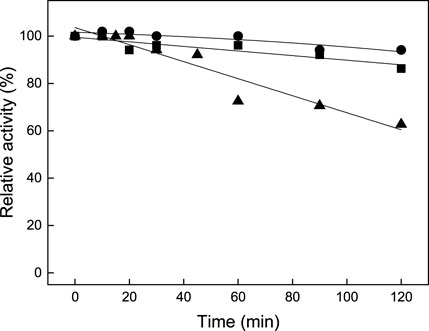
FIGURE 4. Graph showing the temperature stability of TTX BCAT at 65°C (●), 75°C (■), and 85°C (▲). 100% activity corresponds to 0.52 U/mg. The activities were mean values of two measurements.
Previously the detailed TUZ biochemical characterization and its crystal structure were reported (Boyko et al., 2016). The TUZ enzyme has a sequence identity of 81% when compared to TTX. A comparison of both enzymes revealed strong similarities but also several differences. Both enzymes showed a broad substrate specificity where they were active towards branched-chain amino acids and or their corresponding oxoacids with a substrate inhibition by these oxoacids. Despite the high similarity between the two BCATs they showed an interesting difference in their substrate specificity. The TTX used α-ketoglutarate as the amino acceptor which is also reported for most characterized BCATs, whereas TUZ displayed negligible activity towards α-ketoglutarate. Due to the high sequence and structural similarity between TTX and other BCATs a 3D structural model was constructed using AlphaFold 2 (Jumper et al., 2021) to enable a detailed comparison of the substrate binding sites of the TUZ and TTX enzymes.
Based on the crystal structure of various different BCATs it was predicted that the specific orientation and chemical properties of the residues in positions 102–106 (T. uzoniensis numbering) were responsible for the difference observed regarding the substrate specificity towards α-ketoglutarate (Okada et al., 2001; Bezsudnova et al., 2016). The residues Ile103 and Leu105 are part of the “A-pocket” which makes up half of the active site which is lined with the conserved residues Arg91, Tyr29, Phe34, Trp120, and Tyr124 as well as the non-conserved residues Leu101 and Leu103 which are in line with the predicted BCAT motif 107 ZGZ Z: valine, isoleucine, leucine or methionine (Höhne et al., 2010). The two so called BCAT motifs can also be clearly observed, motif 1: 31-YxxxxF[ED]Gx[KR], and motif 2: 95-YxR … 107-[LMVI]G[VL] (the numbering corresponds to BCAT from E. coli where they were first found). The striking difference between TUZ and other BCATs is the substitution of the glycine in this motif for a serine (Bezsudnova et al., 2016). A primary structure alignment with other structurally solved archaeal BCATs that show high activity towards α-ketoglutarate (Figure 5), has revealed that the glycine residue at the position equivalent to residue 104 of TUZ is well conserved. Moreover, this substitution is the only difference between TUZ and TTX within the active site region as demonstrated by the superimposition of the crystal structure of the active site of TUZ and the AlphaFold 2 modelled active site of TTX (Figures 6A,B).
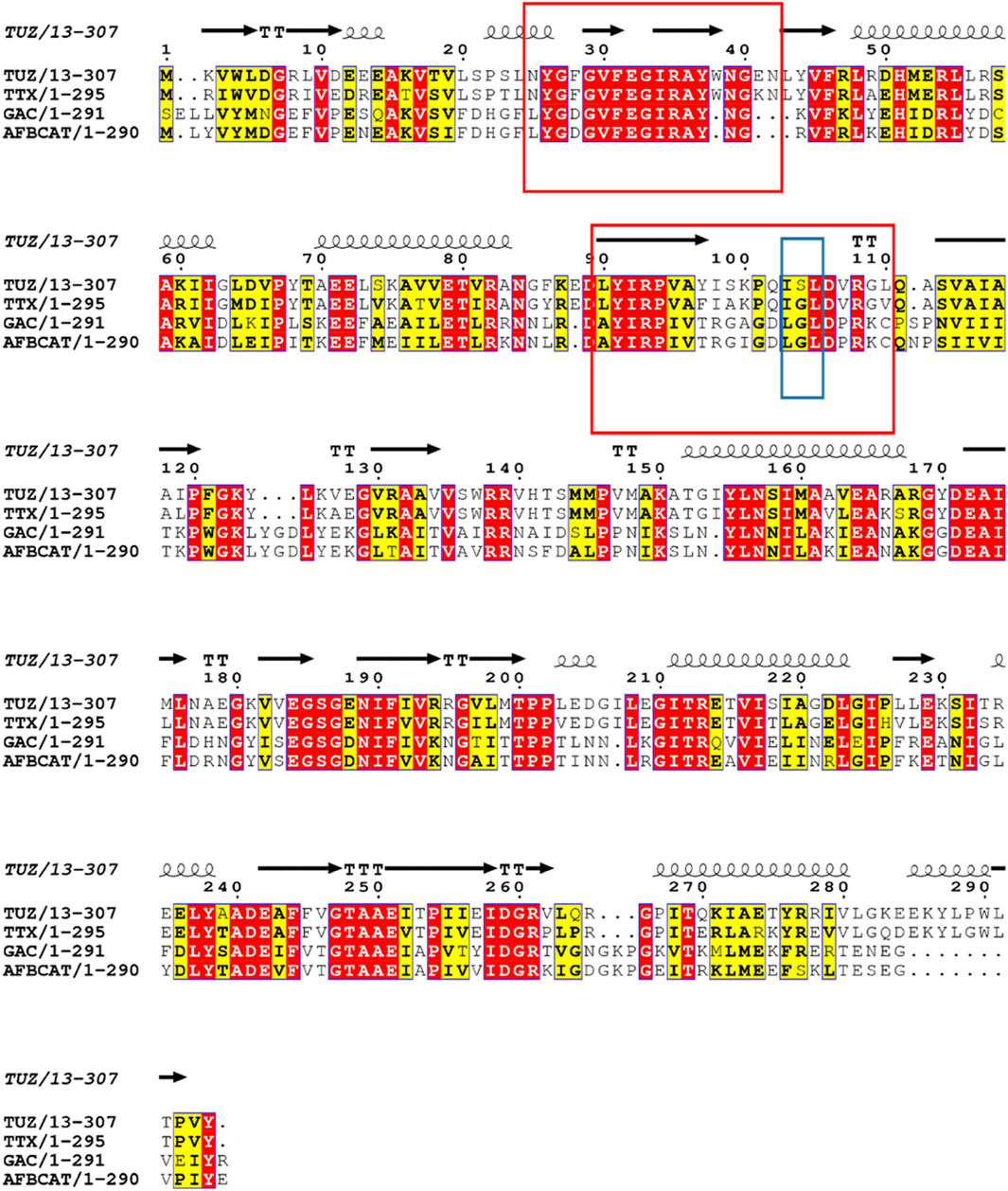
FIGURE 5. Sequence alignment of TTX, TUZ and other archaeal BCATs with high activity towards α-ketoglutarate, from G. acetivorans (GAC), A. fulgidus (AFBCAT). Residue 104 is highlighted in a blue box for clarity and shows the switch from glycine to serine in TUZ. The two conserved motifs characteristic for the BCATs are shown in red boxes. Conserved residues are shown in red. The secondary structural elements found in the crystal structure of TUZ are shown above the alignment as coils and arrows. The alignment was prepared using ESPript3x (Robert and Gouet 2014).
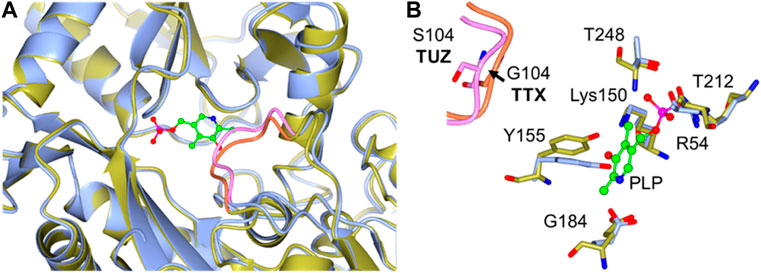
FIGURE 6. (A) A Cartoon representation of the superimposition of the TTX and TUZ BCATs active sites, TUZ (blue) and TTX (gold). The loop containing Gly104 (TTX) to Ser104 (TUZ) substitution is highlighted in pink (TUZ) and orange (TTX) respectively. (B) Close-up of the active site superposition. The Gly to Ser change in the loop shown in pink, for TUZ (PDB ID: 5CE8) and in the loop displayed in orange for TTX. Active site residues are shown as stick models with carbon atoms coloured in blue for TUZ and gold for TTX. For clarity an arrow is added highlighting movement of the loop between glycine and serine. The cofactor PLP is shown as a green ball and stick model. The images were prepared using CCP4MG (McNicholas et al., 2011).
Comparing the active sites using the crystal structures of the G. acetivorans (GAC) aminotransferase, which was reported with a molecule of α-ketoglutarate bound in the active site, with the TUZ aminotransferase active site, a loop movement is observed away from the active site when the serine residue is present (Figure 7A). We cannot eliminate the possibility that this movement could be due to a conformational change resulting from the bound ligand. However, this movement is also seen in the structural comparison of TUZ and the AlphaFold 2 modelled structure of the TTX enzyme.
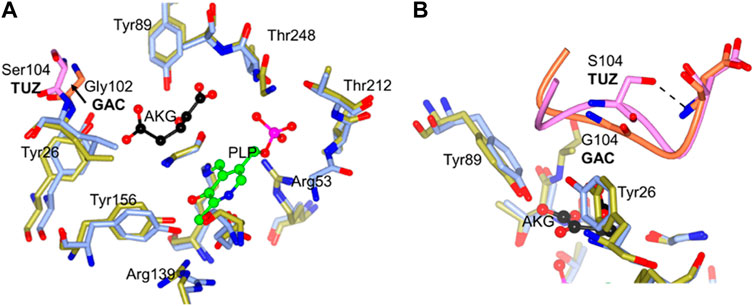
FIGURE 7. (A) The active site superposition of TUZ (PDB ID: 5CE8) and GAC (PBD ID: 5E25). Active site residues of TUZ are shown as sticks with carbons in blue, those of GAC in gold. Ser104 and the loop from TUZ shown in pink and Gly102 and associated loop from GAC shown in orange. For clarity an arrow is added highlighting movement of the loop between glycine and serine. (B) Close up of key 102–106 loop superposition between TUZ and TUZ (PDB ID: 5CE8) and GAC (PBD ID: 5E25). Active site residues of TUZ are shown as sticks with carbons in blue, those of GAC in gold. Ser104 and the loop from TUZ shown in pink and Gly102 and associated loop from GAC shown in orange. A dashed shows the hydrogen bond between Ser104 and main chain nitrogen of Asp106. The cofactor PLP is shown as a ball and stick model (carbons in green) and an acceptor substrate α-ketoglutarate is shown as ball and stick with carbons in black. The image was prepared using CCP4MG (McNicholas, et al., 2011).
A more recent structure of TUZ in complex with L-Norvaline (Boyko et al., 2020) clearly shows a hydrophobic pocket formed by side chains of I103, L105 and Y124 responsible for binding of the side chain of the branched chain amino acid substrate. The substrate is thereby positioned favourably for catalysis. This hydrophobic pocket in TUZ has no binding site for a distal carboxyl group of Glu and α-ketoglutarate which makes these two substrates a poor nitrogen donor or acceptor, respectively. The side chain of Ser104 forms a hydrogen bond to the main chain nitrogen of Asp106 (Figure 7B), fixing the main chain conformation of Ser104. The S104G replacement allows more conformational freedom in the main chain conformation of this residue. This likely results in positioning of its main chain nitrogen so it can bind the α-ketoglutarate distal carboxyl, fixing Glu/α-ketoglutarate in the extended catalytic conformation favourable for the amino group transfer.
In order to conclusively demonstrate that the residue Ser 104 has an influence on the specificity for α-ketoglutarate, the residue Gly104 from TTX has been mutated to Ser104 to mimic the active site of TUZ, since the TTX enzyme demonstrates the highest sequence similarity. The resulting mutant TTX G104S was expressed in a soluble form and purified to homogeneity. SDS-PAGE revealed a single monomer band at 35 kDa (Supplementary Figure S1B). The gel filtration profile (Supplementary Figure S3) revealed the same oligomeric state as the wild type TTX corresponding to a hexamer and a small amount of dimer. The mutant enzyme catalyzed the transamination between L-alanine and the branched-chain oxoacids 3-methyl-2-oxobutanoic acid (Vmax 1.7 U/mg, Km 0.019), and 4-methyl- 2-oxovalerate (Vmax 2.1 U/mg, Km 0.069 mM) with higher Vmax values but similar Km values as compared to the wild-type enzyme. However, the activity towards α-ketoglutarate was significantly changed compared to the wild—type enzyme; the Vmax of the TTX G104S mutant towards α-ketoglutarate was 18-fold reduced and the Km increased 20-fold resulting in a 370-fold lower catalytic efficiency Vmax/Km (Figure 2).
The results obtained confirm the predicted role of Ser104 in substrate specificity by the lack of α- ketoglutarate activity observed for TUZ and the presence of activity for TTX. Using the purified TUZ donated from the laboratory of Vladimir Popov, we repeated the activity measurement towards the α-ketoglutarate substrate and verified the lack of activity as depicted in Figure 3 (Bezsudnova et al., 2016).
Interestingly, only a few BCATs which share high sequence identity (over 55%) to the Thermoproteus enzyme have a glycine at position equivalent to 104. Other BCATs have a Thr, Ser, Ala or Asn residue instead at this position. Additionally, BCATs with reported activity towards α-ketoglutarate (from E. coli, Thermus thermophilus, Pseudomonas aeruginosa) all have a glycine residue in this position. Assuming the activity towards α-ketoglutarate depends on the presence of the glycine residue at this position, it would appear that metabolism of branched chain amino acids in Thermoproteaceae and some other archaeal families does not involve α-ketoglutarate/glutamate.
In summary, the identification of the serine 104 residue has important implications when determining which substrates a particular BCAT transaminase is likely to display activity towards. We report here the first biochemical characterization of a BCAT from T. tenax which shows very high sequence and structural similarity to a previously characterized BCAT from T. uzoniensis but has a different substrate specificity towards the substrate α-ketoglutarate. This difference has been demonstrated to be consistent with the substitution of a glycine residue to a serine residue on a loop close to the active site of the enzyme. The importance of this residue has been confirmed by site-directed mutagenesis where the G104S mutant of the BCAT from T. tenax leads to a large decrease in activity towards α-ketoglutarate. This provides valuable information for the rational design of the substrate binding pocket of BCAT aminotransferases and their use as biocatalysts for production of specific chiral amines of critical importance for the pharmaceutical industry.
The TTX 0133 gene (uniprot accession number G4RMH9) was amplified using DNA from the type strain T. tenax Kra1 (DSM 2078) by PCR using Pwo polymerase (peqlab) with the specific primers (TTX0133s: GATTGATGTTCGGCATATGAGGATCTG, TTX0133as: GCTTTCTCGAGTATAGACTAATACACCG) and cloned into pET28a (Novagene). The resulting plasmid was transformed into E. coli Rosetta (DE3)-pLysS (Stratagene). For expression of recombinant TTX 0133 wild type and TTX G104S mutant cells were grown at 37°C in 1.2 L of LB medium supplemented with kanamycin (34 μg ml−1) and chloramphenicol (30 μg ml−1) The 1.2 L culture was inoculated with an overnight culture to an optical density at 600 nm of 0.3 and incubated up to an optical density of 1.2 at 600 nm. Expression was induced by the addition of 1 mM isopropyl -D-1-thiogalactopyranoside. After 5 h of further growth, cells were harvested by centrifugation (max 9800 × g at 18°C) and stored at −20°C. The E. coli pellet was suspended in 50 mM Tris-HCl, pH 8.2, containing 300 mM NaCl and 10 mM imidazole (buffer A). Cell disruption was performed by sonication on ice using a Branson Sonifier 250 at a duty cycle control of 30% and an output control of 3 followed by centrifugation (100,000 × g at 4°C). E. coli proteins in the supernatant were precipitated by 20 min incubation at 70°C and centrifuged (100000 × g at 4°C). The supernatant was applied to a 1 ml nickel-nitrilotriacetic acid (Ni-NTA) column (Qiagen) that was equilibrated in buffer A using the AKTA FPLC system. After a washing step, protein was eluted by stepwise increasing the imidazole concentration up to 500 mM. Fractions containing the highest enzyme activity were applied to a Superdex 200 HiLoad gel filtration column (1.6 × 60 cm) that had been equilibrated with 50 mM Tris-HCl, pH 7.5, containing 500 mM NaCl and 0.05 mM PLP. Protein elution was performed with an isocratic flow at 1 ml min−1 for determination of the native molecular mass of TTX, the gel filtration column was calibrated by using HWM and LWM kits (GE Healthcare) as specified by the manufacturer. The protein purity was analyzed by SDS/PAGE (12%) and protein concentration was determined by the Bradford method. Purified BCAT from T. uzoniensis TUZ was donated from the laboratory of Vladimir Popov (Russian Academy of Science, Moscow).
The substrate specificity of the TTX BCAT was measured using a continuous assay monitoring the oxidation of NADH at 340 nm by coupling the amino acceptor 3-methyl-2-oxobutanoic acid dependent formation of pyruvate from L-alanine using L-lactate dehydrogenase at 55°C. One unit of enzyme activity corresponded to the formation of 1 µmol pyruvate per min. The standard assay mixture contained 0.1 M Tris-HCl, pH 8.0 (at 55°C), 0.1 mM PLP, 5 mM 3-methyl-2-oxobutanoic acid, 10 mM L-alanine, 0.3 mM NADH, 2 U Llactate dehydrogenase (Roche) and TTX BCAT enzyme. Substrate specificity of recombinant BCAT TTX for various amino acceptors were assayed each at 1 mM concentration, 3-methyl-2-oxobutanoic acid, 4-methyl-2-oxovalerate, 3-methyl-2-oxopentanoic acid, α-ketoglutarate, 2-oxovaleric acid, 2-oxohexanoic acid, 2-oxooctanoic acid, phenylglyoxylate, phenylpyruvate and indole-3-pyruvate.
The kinetic constants Vmax and Km values for all the methyl oxoacids 3-methyl-2-oxobutanoic acid, 4-methyl-2-oxovalerate, 3-methyl-2-oxopentanoic acid, which showed substrate inhibition within the range of 6–20 mM for the TTX BCAT wild type and the TTX G105S mutant were calculated by fitting the data according to the equation V=Vmax/(1+(Km/[S]0)+([S]0/Ki)) (Schellenberger, 1989) using the Origin 2015 software to generate the calculated standard errors. The kinetic constants for α-ketoglutarate for the TTX BCAT wild type and the TTX G105S mutant, which does not show substrate inhibition, were calculated by fitting the data with the Origin 2015 software using the hyperbolic function according to the standard Michaelis-Menten equation.
The thermostability of the TTX wild-type was measured between 65 and 85°C. The protein (10.5–21 µg) was incubated for up to 120 min at the respective temperature. Samples were chilled on ice and remaining BCAT activity was determined using the standard assay conditions. The pH optimum was determined with 29–172 µg protein, standard conditions and the following buffers each at a concentration of 0.1 M, MES (pH 5.5, 6.0, 6.5), Bis-Tris (pH 6.5, 7.0), Tris-HCl (pH 7.4, 7.8), glycylglycine (pH 8.0, 8.5, 9.0, 9.5). BCAT activity and kinetic constants of TTX for 3-methyl-2-oxobutanoic acid (up to 20 mM), 4-methyl-2- oxovalerate (up to 6 mM), 3-methyl-2-oxopentanoic acid (up to 6 mM and α-ketoglutarate (up to 60 mM) were determined with 10 mM L-alanine and 10.7–21.4 µg protein using standard conditions with 0.1 M glycylglycine pH 8.0 (at 55°C) as buffer. The substrate specificity was tested with 1 mM amino acceptors, 10.7–47.2 µg protein using standard conditions with 0.1 M glycylglycine pH 8.0 (at 55°C) as buffer.
The Q5 site-directed mutagenesis kit (NEB) was used with primers (TTX0133muts: GCCGCAGATATCGCTCGACGTCAG; TTX0133mutAs: TTCGCTATAAACGCCACAG) and protocols predicted from NEBaseChanger. The Gly104 from TTX was changed to serine using pET28a-TTX as a template. The modified BCAT TTX G104S was expressed, purified and measured as described for the wild type.
A structure for TTX BCAT was produced using AlphaFold2 (Jumper et al., 2021). Comparisons between different structures were undertaken using CCP4MG (McNicholas et al., 2011) and UCSF Chimera (Pettersen et al., 2004).
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
PS and JL initiated the project; MS and J-MS cloned and expressed the genes, and biochemically characterized the wildtype and mutant enzyme. DM, MI, and JL carried out the structural analyses, PS, JL, J-MS, DM, and MI wrote the manuscript.
This work was funded in the UK by BBSRC grant BB/BB/L002035/1 and in Germany by BMBF grant 031A222 as part of the EU ERA-IB THERMOGENE grant agreement ID: 291814.
PS, J-MS, and MS acknowledge the University of Kiel. DM acknowledges EPSRC grant EP/T002875/1 and the University of Exeter. We acknowledge financial support by Land Schleswig-Holstein within the funding programme Open Access Publikationfonds.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2022.867811/full#supplementary-material
Bezsudnova, E. Y., Stekhanova, T. N., Suplatov, D. A., Mardanov, A. V., Ravin, N. V., and Popov, V. O. (2016). Experimental and computational studies on the unusual substrate specificity of branched-chain amino acid aminotransferase from Thermoproteus uzoniensis. Arch. Biochem. Biophys. 1, 27–36. doi:10.1016/j.abb.2016.08.009
Bezsudnova, E. Y., Boyko, K. M., and Popov, V. O. (2017). Properties of bacterial and archaeal branched-chain amino acid aminotransferases. Biochem. Mosc. 13, 1572–1591. doi:10.1134/S0006297917130028
Borgenvik, M., Apró, W., and Blomstrand, E. (2011). Intake of branched-chain amino acids influences the levels of MAFbx mRNA and MuRF-1 total protein in resting and exercising human muscle. Am. J. Physiology-Endocrinology Metabolism 302, 510–521. doi:10.1152/ajpendo.00353.2011
Boyko, K. M., Stekhanova, T. N., Nikolaeva, A. Y., Mardanov, A. V., Rakitin, A. L., Ravin, N. V., et al. (2016). First structure of archaeal branched-chain amino acid aminotransferase from Thermoproteus uzoniensis specific for L-amino acids and R-amines. Extremophiles 20, 215–225. doi:10.1007/s00792-016-0816-z
Boyko, K. M., Nikolaeva, A. Y., Timofeev, V. I., Popov, V. O., and Bezsudnova, E. Y. (2020). Three-dimensional structure of branched-chain amino acid transaminase from Thermoproteus uzoniensis in complex with N-Norvaline. Crystallogr. Rep. 65, 740–743. doi:10.1134/S1063774520040045
Conway, M. E., and Hutson, S. M. (2000). “Mammalian branched-chain aminotransferases,” in Methods in Enzymology (London: Academic Press), 355–365. doi:10.1016/s0076-6879(00)24245-6
Cooper, A. J. L., and Meister, A. (1989). The discovery and scope of enzymatic transamination. Biochimie 71, 387–404. doi:10.1016/0300-9084(89)90169-7
Deepankumar, K., Shon, M., Nadarajan, S. P., Shin, G., Mathew, S., Ayyadurai, N., et al. (2014). Enhancing thermostability and organic solvent tolerance of ω-transaminase through global incorporation of fluorotyrosine. Adv. Syn. Catalysis 356, 993–998. doi:10.1002/adsc.201300706
Eliot, A. C., and Kirsch, J. F. (2004). Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415. doi:10.1146/annurev.biochem.73.011303.074021
Ghislieri, D., and Turner, N. J. (2014). Biocatalytic approaches to the synthesis of enantiomerically pure chiral amines. Top. Catal. 57, 284–300. doi:10.1007/s11244-013-0184-1
Hayashi, H. (1995). Pyridoxal enzymes: Mechanistic diversity and uniformity. J. Biochem. 118, 463–473. doi:10.1093/oxfordjournals.jbchem.a124931
Hirotsu, K., Goto, M., Okamoto, A., Miyahara, I., and Miyahara, I. (2005). Dual substrate recognition of aminotransferases. Chem. Rec. 5, 160–172. doi:10.1002/tcr.20042
Höhne, M., Schätzle, S., Jochens, H., Robins, K., and Bornscheuer, U. T. (2010). Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 6, 807–813. doi:10.1038/nchembio.447
Holeček, M. (2020). Branched-chain amino acids and branched-chain keto acids in hyperammonemic states: Metabolism and as supplements. Metabolites 10, 324. doi:10.3390/metabo10080324
Hutson, S. (2001). “Structure and function of branched chain aminotransferases,” in Progress in nucleic acid research and molecular biology (London: Academic Press), 175–206. doi:10.1016/S0079-6603(01)70017-7
Isupov, M. N., Boyko, K. M., Sutter, J-M., James, P., Sayer, C., Schmidt, M., et al. (2019). Thermostable branched-chain amino acid transaminases from the archaea Geoglobus acetivorans and Archaeoglobus fulgidus: Biochemical and structural characterization. Front. Bioeng. Biotechnol. 7, 7. doi:10.3389/fbioe.2019.00007
Jiang, J., Chen, X., Zhang, D., Wu, Q., and Zhu, D. (2015). Characterization of (R)-selective amine transaminases identified by in silico motif sequence blast. Appl. Microbiol. Biotechnol. 99, 2613–2621. doi:10.1007/s00253-014-6056-1
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kelly, S. A., Mix, S., Moody, T. S., and Gilmore, B. F. (2020). Transaminases for industrial biocatalysis: Novel enzyme discovery. Appl. Microbiol. Biotechnol. 104, 4781–4794. doi:10.1007/s00253-020-10585-0
Mardanov, A. V., Gumerov, V. M., Beletsky, A. V., Prokofeva, M. I., Beletsky, E. A., Prokofeva, N. V., et al. (2011). Complete genome sequence of the thermoacidophilic crenarchaeon Thermoproteus uzoniensis 768-20. J. Bacteriol. 193, 156–3157. doi:10.1128/jb.00409-11
McNicholas, S., Potterton, E., Wilson, K. S., and Noble, M. E. M. (2011). Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D. Biol. Crystallogr. D67, 386–394. doi:10.1107/S0907444911007281
Mehta, P. K., Hale, T. F., and Christen, P. (1993). Aminotransferases: Demonstration of homology and division into evolutionary subgroups. Eur. J. Biochem. 214, 549–561. doi:10.1111/j.1432-1033.1993.tb17953.x
Okada, K., Hirotsu, K., Hayashi, H., and Kagamiyama, H. (2001). Structures of Escherichia coli branched-chain amino acid aminotransferase and its complexes with 4-methylvalerate and 2-methylleucine: Induced fit and substrate recognition of the enzyme. Biochemistry 40, 7453–7463. doi:10.1021/bi010384l
Paul, C. E., Rodríguez-Mata, M., Busto, E., Lavandera, I., Gotor-Fernández, V., Gotor, V., et al. (2014). Transaminases applied to the synthesis of high added-value Enantiopure amines. Organic Process Res. Develop. 18, 788–792. doi:10.1021/op4003104
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. doi:10.1002/pro.3943
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2012). The Pfam protein families database. Nucleic Acids Res. 40, 290–301. doi:10.1093/nar/gkr1065
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, 320–324. doi:10.1093/nar/gku316
Savile, C. K., Janey, J. M., Mundorff, E. C., Moore, J. C., Tam, S., Jarvis, W. R., et al. (2010). Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329, 305–309. doi:10.1126/science.1188934
Sayer, C., Isupov, M. N., Westlake, A., and Littlechild, J. A. (2013). Structural studies of Pseudomonas and Chromobacterium ω-aminotransferases provide insights into their differing substrate specificity. Acta Crystallogr. D. Biol. Crystallogr. D69, 564–576. doi:10.1107/S0907444912051670
Sayer, C., Martinez-Torres, R. J., Richter, N., Isupov, M. N., Hailes, H. C., Littlechild, J. A., et al. (2014). The substrate specificity, enantioselectivity and structure of the (R)-selective amine : Pyruvate transaminase from Nectria haematococca. FEBS J. 281, 2240–2253. Epub 20140407. doi:10.1111/febs.12778
Slabu, I., Galman, J. L., Lloyd, R. C., and Turner, N. J. (2017). Discovery, engineering, and synthetic application of transaminase biocatalysts. ACS Catal. 7, 8263–8284. doi:10.1021/acscatal.7b02686
Sookoian, S., and Pirola, C. J. (2012). Alanine and aspartate aminotransferase and glutamine-cycling pathway: Their roles in pathogenesis of metabolic syndrome. World J. Gastroenterol. 18, 3775–3781. doi:10.3748/wjg.v18.i29.3775
Venos, E. S., Knodel, M. H., Radford, C. L., and Berger, B. J. (2004). Branched-chain amino acid aminotransferase and methionine formation in Mycobacterium tuberculosis. BMC Microbiol. 4, 39. doi:10.1186/1471-2180-4-39
Wendisch, V. F. (2020). Metabolic engineering advances and prospects for amino acid production. Metab. Eng. 58, 17–34. doi:10.1016/j.ymben.2019.03.008
Yonaha, K., Toyama, S., and Soda, K. (1987). omega-Amino acid-pyruvate aminotransferase. Methods Enzymol. 143, 500–504. doi:10.1016/0076-6879(87)43090-5
Keywords: (R)- selective aminotransferase, branched chain, substrate specificity, enzyme kinetics, protein structure, industrial biocatalysis
Citation: Sutter J-M, Mitchell DE, Schmidt M, Isupov MN, Littlechild JA and Schönheit P (2022) Substrate specificity of branched chain amino acid aminotransferases: The substitution of glycine to serine in the active site determines the substrate specificity for α-ketoglutarate. Front. Catal. 2:867811. doi: 10.3389/fctls.2022.867811
Received: 01 February 2022; Accepted: 12 August 2022;
Published: 23 September 2022.
Edited by:
Per Berglund, KTH Royal Institute of Technology, SwedenReviewed by:
Ioannis V. Pavlidis, University of Crete, GreeceCopyright © 2022 Sutter, Mitchell, Schmidt, Isupov, Littlechild and Schönheit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer A. Littlechild, Si5BLkxpdHRsZWNoaWxkQGV4ZXRlci5hYy51aw==; Peter Schönheit, cGV0ZXIuc2Nob2VuaGVpdEBpZmFtLnVuaS1raWVsLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.