 Bastien O. Burek
Bastien O. Burek Ayad W. H. Dawood2
Ayad W. H. Dawood2 Frank Hollmann
Frank Hollmann Andreas Liese
Andreas Liese Dirk Holtmann
Dirk Holtmann
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Catal. , 21 March 2022
Sec. Biocatalysis
Volume 2 - 2022 | https://doi.org/10.3389/fctls.2022.858706
This article is part of the Research Topic Horizons in Catalysis View all 9 articles
Enzyme catalysis, made tremendous progress over the last years in identification of new enzymes and new enzymatic reactivity’s as well as optimization of existing enzymes. However, the performance of the resulting processes is often still limited, e.g., in regard of productivity, realized product concentrations and the stability of the enzymes. Different topics (like limited specific activity, unfavourable kinetics or limited enzyme stability) can be addressed via enzyme engineering. On the other hand, there is also a long list of topics that are not addressable by enzyme engineering. Here typical examples are unfavourable reaction thermodynamics, selectivity in multistep reactions or low water solubility. These challenges can only be addressed through an adaption of the reaction system. The procedures of process intensification (PI) represent a good approach to reach most suitable systems. The general objective of PI is to achieve significant benefits in terms of capital and operating costs as well as product quality, waste, and process safety by applying innovative principles. The aim of the review is to show the current capabilities and future potentials of PI in enzyme catalysis focused on enzymes of the class of oxidoreductases. The focus of the paper is on alternative methods of energy input, innovative reactor concepts and reaction media with improved properties.
In enzyme catalysis, great success has been achieved in recent years in identification of new enzymes, optimization of existing enzymes using rational and evolutionary methods and in the establishment of new reactions, e.g., novel reactivity’s or enzymatic cascades. However, the performance of the resulting processes is often still limited, e.g., in regard of productivity, realized product concentrations and the stability of the enzymes. These parameters can usually be step by step improved through elaborate optimization. Very often these optimizations are not sufficient to achieve the necessary improvements. In order, to achieve the required economic and ecological key process indicators completely new approaches are often required. For comparable challenges in chemical process engineering, process intensification has proven itself as a method that brings about a paradigm shift and resulted very often in processes with an at least doubled process performance. Stankiewicz and Moulijn (2000) offers a comprehensive and widely applicable definition of PI: “Process intensification consists of the development of novel apparatuses and techniques that, compared to those commonly used today, are expected to bring dramatic improvements in manufacturing and processing, substantially decreasing equipment-size to production-capacity ratio, energy consumption, or waste production, and ultimately resulting in cheaper, sustainable technologies.” In additions, PI can be classified in terms of the underlying basic principles 1) time—improve kinetics, 2) space—maximize homogeneity 3) thermodynamics—maximize thermodynamic driving forces and transfer areas and 4) synergy—combine different unit operation in one reactor (Van Gerven and Stankiewicz, 2009). The general objective of PI is to achieve significant benefits in terms of capital and operating costs as well as product quality, waste and process safety by applying innovative principles. Therefore, all definitions can be simplified to “do more with less.” This can very often only be fulfilled by a paradigm shift that leads to the use of interdisciplinary techniques. In the last decades PI has been coined as a game changing concept within chemical engineering (Noorman et al., 2018). PI enables transition within chemical and biochemical engineering making it more fit for addressing global issues, such as climate change, limited petrochemical and natural resources, growing populations and higher environmental standards. The field of PI can be divided into two general areas 1) process-intensifying equipment, such as novel reactors, and intensive mixing, heat-transfer and mass-transfer devices and 2) process-intensifying methods, such as new or hybrid separations, integration of reaction and separation, multifunctional reactors, techniques using alternative energy sources (light, ultrasound, etc.) (Stankiewicz and Moulijn, 2000). It is often stated that application of PI principles to bioprocesses will be possible using the same, or at least similar, tools used in chemical engineering (Noorman et al., 2018). Certainly, there are many overlaps but also very strong differences. In chemical engineering, many PI approaches rely on heat transfer equipment, either as a unit operation with reduced size or as an integrated component of a multifunctional unit operation (e.g., microreactors). Heat transfer also plays a significant role in bioprocesses. However, since most of the bioprocesses are performed at moderate temperature (25–40°C) and with relatively diluted solutions (product concentration approx. 1%), different technical solutions compared to chemical engineering are necessary. The application of biocatalysts in aqueous milieus is a particular source of specific challenges. Most of the substrates are hardly soluble in aqueous systems, therefore, even at full conversion, very often only low product concentrations can be achieved. One of PI’s specific tasks in biocatalysis is to establish better solvents with an improved solubility of the substrates. Finally, it must be mentioned, that PI concerns by definition only engineering methods and novel equipment. Enzyme engineering, no matter how dramatic improvements it brings to existing technology, does not qualify as process intensification (Stankiewicz and Moulijn, 2000).
Optimization of enzymatic systems can be clearly differentiated into topics addressable either via enzyme engineering or PI and topics which can be optimized using both approaches together (Figure 1). For example, limited specific activity under the required reaction conditions, unfavourable kinetics at required high concentrations to yield a high space time yield, or even limited enzymes stability can clearly be addressed by enzyme engineering. On the other hand, there is also a long list of topics that are not addressable by enzyme engineering. These are all affected by the physicochemical properties of the reaction system itself. Typical examples are unfavourable reaction thermodynamics, selectivity in multistep reactions, low water solubility, high or low viscosity, high volatility or foaming. The latter ones, point to complex multiphase reaction systems, which specifically need detailed process engineering. However, this is not to be seen as a black or white world, there is also a multitude of grey tones available that need to be considered for optimal PI. In a true sense of interdisciplinary PI, often, a combination of enzyme and process engineering leads to the finally optimized processes. Foaming can be reduced by the integration of mechanical foam dissecting devices or the addition of chemicals like silicones. On the other hand, if one source of foaming are free proteins in the solution, also the surface properties of the protein can be fine-tuned by molecular engineering to reduce foaming. In the case of an inhibition, a complementary approach would be an improvement of the enzyme’s tolerance to the substrate by protein engineering and at the same time a selection of the best suited type of reactor, like fed batch in view of the substrate or a continuously operated stirred tank reactor under outflow conditions. Here, the substrate concentration is constantly minimized within the reactor, even if a high substrate concentration is fed continuously to the reactor. In the case of product inhibition also a complementary approach is available, while by means of enzyme engineering the inhibition by the product can be reduced, from the viewpoint of process engineering an in situ product removal is integrated, e.g., in situ crystallization, in situ distillation, in situ adsorption. In addition, there are also synergistic approaches. For example, in the case of thermodynamic limitation of a given reaction system, following Chatelier’s principle, an equilibrium can be shifted, if the substrate concentrations are significantly increased. Here an improvement of the substrate tolerance for a given enzyme can be yielded by enzyme engineering, which in consequence, enables a higher starting material concentration in the reactor leading to a shift of the thermodynamic equilibrium. In general, engineering of the biocatalyst is to be understood in terms of enhancing the transformation rate but does not have any effect on shifting the thermodynamic equilibrium.
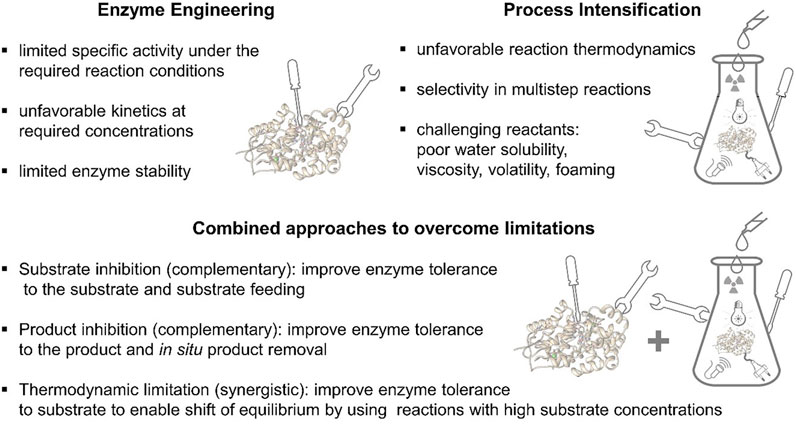
FIGURE 1. Differentiation and overlap between enzyme engineering and PI (detailed description in the text).
In consequence, there are three major steps resulting in process intensification by the combined approach of biotechnology, chemistry, and process engineering. In the first step A, the isolated catalyst, independent if it is a chemo- or a biocatalyst, has to be characterized and optimized in terms of its kinetic properties. At this step also the thermodynamic properties of the reaction system need to be known. In a second step B, this “naked” catalyst is incorporated in any microenvironment. This micro environment can be a surface, where the catalyst is immobilized on, or it could be the bulk reaction medium like an aqueous phase, an organic phase, a deep eutectic solvent, an ionic liquid or a gas phase. In this step, by means of enzyme as well as process engineering, the performance of the catalyst has to be optimized to the micro environment by optimizing the immobilization method or the enzyme structure to exhibit a high stability or activity in this micro environment. In the final step C this micro environment containing the catalyst is integrated into a reactor and with this into a process. This is a clear step of pure process engineering. Topics to be addressed here are type of reactor, multiphase flow behaviour, re-cyclization of the micro environment always considering limitations in multiphase mass transport. At this point also a possible in situ product removal comes into play considering a simultaneous integration into the reaction zone. In consequence, PI is always an interdisciplinary job where team members from the different disciplines need to cooperate from the beginning onwards. Finally, productivity of bioprocesses is often far from ideal to meet the large-scale need for food, drugs, biofuels and bio-based chemicals (Boodhoo et al., 2022). Due to the drastic improvements in process performances by applying PI, these limitations could be solved in the future.
The aim of the review is to show the current capabilities and future potentials of PI in enzyme catalysis. The focus of the paper (see also Table 1) is on alternative methods of energy input, innovative reactor concepts and reaction media with improved properties (compared to water-based systems). Furthermore the review focuses on the application of the oxidoreductases. This is based on the fact that in one study of 547 biocatalysis based patents filed between 2000 and 2015 more than two third were based on oxidoreductases (Buller et al., 2018). In addition to the PI approaches discussed herein, there are a wide number of additional approaches, see e.g., (Lye and Woodley, 1999; Bolivar et al., 2011; Wohlgemuth et al., 2015; Lorente-Arevalo et al., 2021; van der Wielen et al., 2021; Žnidaršič-Plazl, 2021; Boodhoo et al., 2022).
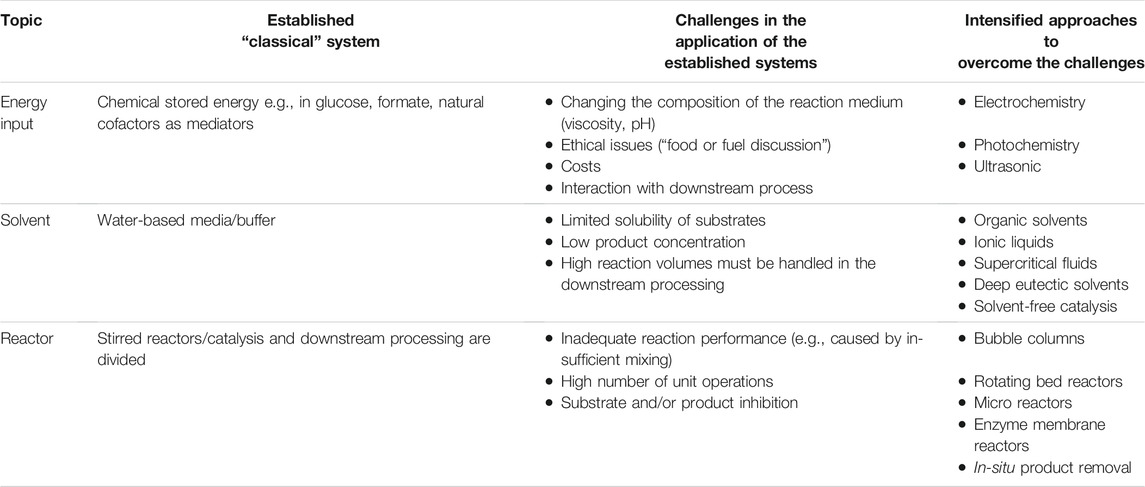
TABLE 1. Examples of process intensification approaches in enzyme catalysis.
The application of hydrolases (e.g., amidases, esterases, lipases, and proteases) has become well established in many bioproductions since decades (Wohlgemuth, 2010; Woodley, 2008). In the last years more and more oxidoreductases were characterized in detail and used in technical applications. Oxidoreductases consist of a large class of enzymes catalyzing the transfer of electrons from an electron donor (reductant) to an electron acceptor (oxidant) molecule, e.g., alcohol dehydrogenases, Baeyer–Villiger monooxygenases, cytochromes P450 monooxygenases, enoate reductases, peroxidases, and peroxygenases belong to this enzyme class. In order to transfer the redox equivalents the enzymes are using cofactors. Cofactors are low molecular compounds, which are responsible for the transfer of hydrogen, electrons or functional groups in enzyme-catalyzed reactions. Important cofactors are adenosine-5′-triphosphate (ATP), thiamin, flavin adenine dinucleotide (FAD), coenzyme A (CoA), pyrroloquinoline quinone (PQQ) and nicotinamide cofactors (nicotinamide adenine dinucleotide phosphate (NADPH) and nicotinamide adenine dinucleotide (NADH). By using oxidoreductase catalyzed reactions, the nicotinamides are the most commonly used cofactor (Wu et al., 2013). In technical applications an efficient and economical regeneration or substitution of the coenzymes is very often of particular significance. In vivo the oxidized and reduced forms of the cofactors are continuously regenerated within the cellular metabolism. In nature all procedures using oxidoreductases are combined with a second reaction—one for the biocatalytic reaction and one for cofactor regeneration. This principle is also applied in the established enzyme-coupled and substrate-coupled cofactor regeneration processes. Usually, the redox equivalents are provided by the conversion of substrates such as formate or glucose. Here PI focuses on the simplification of the established systems e.g., by using alternative energy sources. In general, different alternative sources and forms of energy can be applied in order to intensify a chemical or biochemical processes (Stankiewicz, 2006), e.g., energy of electric fields or energy of electromagnetic radiation (microwaves and light). Furthermore, a wide range of oxidoreductases utilize hydrogen peroxide as oxidant. The dependence of peroxidases and peroxygenases on simple peroxides only at first sight offers a trouble-free catalyst system to be used in organic synthesis (Holtmann et al., 2014a). Here, the hydrogen peroxide mediated inactivation of enzymes is one of the major issues on their way towards industrial applications (Holtmann et al., 2014a). There are several PI approaches to prevent or at least minimize inactivation by H2O2.
Photon driven enzymatic reactions became very prominent in the last decades as the use of light can be regarded as a sustainable energy source (Lee et al., 2013; Maciá-Agulló et al., 2015). Especially combining the often robust, good light absorbing and efficient chemical catalysts with the highly selective biocatalysts offers many advantages (Huang et al., 2020). However, it has to be carefully considered if the sole use of light necessarily leads to higher energy-efficient, safer, more compact, cleaner and therefore intensified processes. The use of light energy allows for mild and enzyme compatible reaction conditions and can be introduced contactless to the enzyme, but there are as well non-neglectable negative effects which can possibly occur, e.g., light may also be harmful to enzymes reducing the turnovers and thus the overall efficiency. Further, often very low apparent quantum yields are reached in the systems, therefore one has to assess if alternative power inputs are possible which might be more efficient. The scope of photoenzymatic reactions has expanded dramatically in recent years. For a better overview the different systems can be divided in regard of the energy transfer from light to the enzyme (Figure 2). It can either occur direct as in case of photoenzymes or indirect using a photocatalyst for the light to chemical energy conversion.
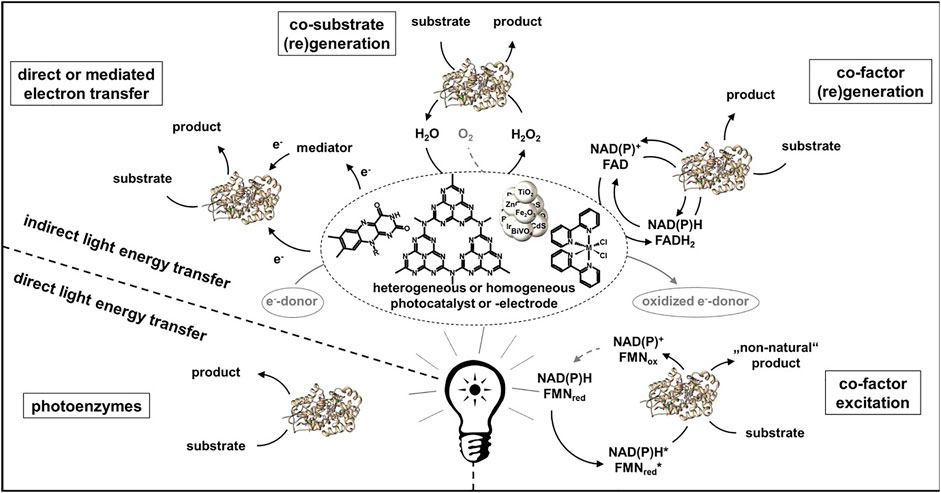
FIGURE 2. Overview of photoenzymatic systems ordered by type of energy transfer from light to the enzyme.
The four known naturally occurring photoenzymes are the photosystem (PS) (Gao et al., 2018), the cyclobutene pyrimidine dimer photolyase (Sancar, 2008), protochlorophyllide reductase (Kaschner et al., 2014) and the photodecarboxylase (Sorigué et al., 2017). Despite their natural function lyase for DNA repair and reductase in synthesis of chlorophyll, both are currently not used for photoenzymatic production. Regarding the PS, there are some attempts to utilize parts of it for artificial synthesis like photoelectrochemical water splitting (Mersch et al., 2015; Sokol et al., 2018). However, due to the complexity of the photosystem and it´s highly specific reaction (water oxidation and ATP synthesis) it is not to be expected to use this system recombinant for other applications. Of course, there are certain applications of microorganisms containing the PS like microalgae or cyanobacteria. Especially for the latter, there are many systems reported where the PS serves as a reduction equivalent delivery system for heterologous expressed enzymes like alcohol-dehydrogenases, ene-reductases or monooxygenases (Köninger et al., 2016; Böhmer et al., 2017; Hoschek et al., 2017; Sengupta et al., 2018). The photodecarboxylase recently earned a lot of attention for the conversion of fatty acids and smaller carboxylic acids to hydrocarbons and it can be expected that the scope of applications will increase in the nearest future (Huijbers et al., 2018; Xu et al., 2019; Zhang et al., 2019; Ma et al., 2020a; Amer et al., 2020; Zhang et al., 2020a; Ma et al., 2020b; Cha et al., 2020; Heyes et al., 2020; Lakavath et al., 2020).
An energy transfer via a photocatalyst can occur in different manners. In general, an electron is excited by light from the highest occupied (HOMO) to the lowest unoccupied molecular orbital (LUMO) in case of a sensitizer or from the valence (VB) to the conduction band (CB) in case of a semiconductor. The missing electron can be replaced via oxidation of an electron donor e.g., EDTA, formate or water. The electron donor is one of the highest waste factors in these systems, therefore atom economy has to be evaluated to match with PI criteria (Seel et al., 2018). Further, intermediates generated by organic donor oxidation like formaldehyde may affect the enzymes stability, therefore accumulation of such substances should be avoided (Burek et al., 2019a). Ideally water would serve as an electron donor, however this often leads to lower production rates and higher amounts of reactive oxygen species strongly reducing enzyme stability (Burek et al., 2019b). A transfer of the electron to an enzyme can either be directly, e.g., using a fluorescent dye like Eosin Y or rose bengal as photocatalyst in combination with P450 or Rieske Oxygenase (Park et al., 2015; Feyza Özgen et al., 2020). More common is an electron transfer via a mediator molecule like methyl viologen or flavinmononucleotid (FMN) often in combination with old yellow enzymes, monooxygenases or hydrogenases (Burai et al., 2012; Bachmeier et al., 2014; Mifsud et al., 2014; Honda et al., 2016; Lee et al., 2017).
Further, photocatalysis is often used for co-factor or co-substrate (re-) generation. For example, reductive regeneration of NAD(P)H has been carried out with a variety of photocatalysts which can drive enzymes like alcohol dehydrogenase (ADH), ene reductase, reductases, Baeyer-Villiger and other monooxygenases (Lee et al., 2009a; Lee et al., 2009b; Lee et al., 2012; Yadav et al., 2012; Yadav et al., 2014a; Yadav et al., 2014b; Choudhury et al., 2014; Huang et al., 2014). Since almost all photocatalysts perform one-electron reductions, which would lead to many unwanted side reactions of the intermediate NAD(P) radical, a redox mediator like pentamethylcyclopentadienyl rhodium (III) bipyridyl ([Cp*Rh (bpy)H2O]2+) is necessary. [Cp*Rh (bpy)H2O]2+ is often used due to its high regioselectivity in 1,4-NAD(P)H regeneration (Hollmann et al., 2001). Besides reductive also oxidative regeneration of NAD(P)+ by flavins or Ru (bipy) is known in combination with ADHs which consequential perform an oxidation of a alcohol (Ruppert and Steckhan, 1989; Gargiulo et al., 2011; Kochius et al., 2014a; Rauch et al., 2017). Even though a high number of different systems has been studied the reached turnover numbers for NAD(P)+ and product concentrations are way beyond the concurrent enzymatic regeneration systems. Major progression would be necessary to make photocatalytic NAD(P)+ regeneration to an applicable alternative.
As co-substrate hydrogen peroxide is often applied to peroxidases and peroxygenases. The photocatalytic generation of H2O2 by the reduction of molecular oxygen has been performed with a variety of sacrificial electron donors and photocatalysts (Churakova et al., 2013; Choi et al., 2017; Zhang et al., 2017; Burek et al., 2019b; van Schie et al., 2019; Yuan et al., 2020a; Hobisch et al., 2020). Regeneration of H2O2 via oxidation of water has as well successfully been performed, but the reached turnover numbers were significantly lower revealing the necessity of more efficient and selective two-electron water oxidation catalysts (Zhang et al., 2018). In general the reached turnovers fall back at least a factor of 2 compared to the alternative electrochemical or enzymatic in situ H2O2 generation systems. A novel concept which has recently been shown by Emmanuel et al. and Biegasiewicz et al. is the potential of exiting cofactors with photons to introduce “non-natural” catalytic activities in cofactor dependent enzymes. Excitation of reduced nicotinamide cofactors enabled enantioselective dehalogenation and deacetylation of α-substituted lactones (Emmanuel et al., 2016; Biegasiewicz et al., 2018). In combination with ene-reductase an excitation of the flavin-substrate charge-transfer complex within the active site led to asymmetric reductive cyclization via dehalogenation or ketone reduction (Biegasiewicz et al., 2019; Sandoval et al., 2019; Clayman and Hyster, 2020).
Details of the mentioned and further possible photoenzymatic reactions have recently been reviewed comprehensively (Lee et al., 2018; Schmermund et al., 2019; Meyer et al., 2021; Özgen et al., 2021). From a technical point of view, especially in case of directly utilizing sunlight, the control of the intensity and wavelengths is very often complex and requires assistive technologies. Another aspect is the broad wavelengths spectrum of the Sun, most of the light remains unused and only heats up the reaction vessels, especially when the light is concentrated. To avoid thermal inactivation of the enzymes cooling would be necessary which is a waste of energy and therefore stands in contrast to the PI criteria. Therefore, efficient artificial light sources like LEDs with narrow emission matching the necessary excitation energy, ideally powered by renewable energy, might offer a more effective control, and enable continuous operation even during the night. However, a lot of open questions remain regarding intensification of the described processes in general. Most of the photoenzymatic reactions are non-optimized or only partially in regard of the enzymatic turnover number and frequency. There are only few examples which are properly characterized regarding the incident photon flux, many even don´t characterize or name the used light source. This leads to difficulties in comparing those reactions as additionally different reactor types in each research group are used and light intensity and distribution inside the reactor remain unclear hampering further development. Furthermore, interactions between light source and enzyme like possible light mediated inactivation or between photocatalyst and enzyme e.g., ROS formation or oxidation of the enzyme which all might be the reason for low turnover numbers are rarely investigated. The probably biggest issue on the way to technical application remains in the poor light penetration depth into the reactor, especially on large-scale. Only a few approaches for up-scale e.g., using flow reactors or internal illumination concepts are known and still operate at early stages of development (Duong et al., 2021; Hobisch et al., 2021). As mentioned above and explained in more detail further below, especially the development of reactors shows the highest intensification potential for enzymatic reactions. So, to utilize the power of light most efficiently, technical concepts have to be developed or adapted and customized for the specific needs of each individual photoenzymatic reaction to reach intensified process conditions.
The central concept of electroenzymatic processes is to combine the advantages of enzymatic processes (especially the high specificity) with the advantages of electrochemistry (e.g., high atomic and energy efficiency) (Krieg et al., 2011). The research in the field can be divided into 3 main topics 1) basic investigations into the underlying mechanisms 2) biosensor applications and 3) electroenzymatic synthesis processes. The mechanistic studies are mostly aimed at gaining insight into the direct electron transfer (DET) between the active site of the enzyme and the electrode. In terms of PI, this would mean a considerable reduction in the number of process steps. However, the process performances (e.g., productivity, space-time-yield) of processes based on direct electron transfer are currently quite low, these processes are not considered in this review. In addition, in DET-based processes cytochrome c peroxidases, horseradish peroxidase, ferredoxin-NADP+reductase, FAD-dependent glucose oxidase and FAD-dependent glucose dehydrogenase are often applied (Adachi et al., 2020). However, these enzymes are not the focus of enzymatic synthesis processes. In the field of electroenzymatic sensors, the focus is on requirements such as sensitivity, selectivity, linearity and reproducibility. In the synthesis processes, these aspects have no or only a minor significance. As mentioned above, the combination of selective enzymatic productivity with electrochemical energy transfer is in the focus of electroenzymatic processes (Figure 3). The challenge of an efficient electron transfer is particularly challenging for enzymes of the oxidoreductase class. Here, cofactors can be regenerated electrochemically or co-substrates (mainly H2O2) can be generated. These process steps can then be combined with various enzymes (e.g., P450 and Baeyer-Villiger monooxygenasen, dehydrogenases, ene reductases). When different processes such as electrochemical and enzymatic processes are combined, the possibility of unwanted side reactions must always be taken into account. For example, by using a mediator based cofactor regeneration in combination with oxygen-dependent monooxygenases (e.g., P450s), there is competition between the desired mediator reduction and the unwanted oxygen reduction at the cathode (Tosstorff et al., 2014).
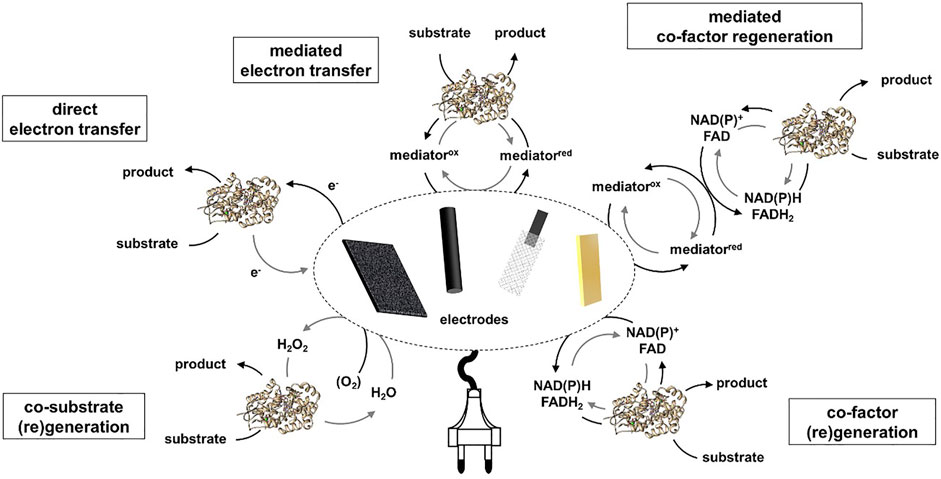
FIGURE 3. Overview of typical electroenzymatic processes.
The exploitation of new raw materials for chemical syntheses is also a subfield of PI. Here, electroenzymatic cascades for the conversion of CO2 are increasingly becoming a highly important field (Obert and Dave, 1999; Schlager et al., 2016; Yuan et al., 2018; Kuk et al., 2019; Szczesny et al., 2020; Zhang et al., 2021a; Zhang et al., 2021b). In general, CO2 reduction has the potential to address issues related to the needs of the chemical industry (abundant and cheap educts) and to environmental issues at the same time. A typical cascade to convert the CO2 via formate and formaldehyde to methanol needs 3 different enzymes, namely formate dehydrogenase (FDH), formaldehyde dehydrogenase, and alcohol dehydrogenase. Each of the energetically uphill reductions requires the supply of a stoichiometric amount of nicotinamide cofactor (NADH) to achieve catalytic turnover (Kuk et al., 2017). In 1999 Obert and Dave (1999) immobilised the enzymes in a sol-gel matrix and regenerated the cofactor electrochemically. Schlager et al. (2016) immobilised the 3 enzymes on an electrode and replaced the NADH by electrochemical cofactor substitution. Generation of methanol is observed for the six-electron reduction with Faradaic efficiencies of around 10%. In addition to the cascade reaction, the conversion of CO2 to formate was studied. Artificial cofactors or mediators such as methyl viologen were used to replace NADH in the FDH-driven reaction (Zhang et al., 2021b). A gas diffusion electrodes with a viologen-modified polymer and a tungsten-dependent FDH was applied to eliminate mass transport limitations (Szczesny et al., 2020). A molybdenum-dependent formate dehydrogenase was combined redox polymer (cobaltocene grafted to poly (allylamine). The resulting bio-cathode reduces CO2 to formate with a Faradaic efficiency up to 99 ± 5% (Yuan et al., 2018). A comparable system showed likewise Faradaic efficiencies and conversion rates up to 1.42 μmol h−1 and a turnover frequency of 976 h−1 (Kuk et al., 2019). In addition, CO2 can be electroenzymatically converted to ethylene and propene by using a vanadium nitrogenase in combination with a cobaltocenium mediator (Cai et al., 2018). A cascade containing immobilised NADPH-dependent imine reductase, oxygen-dependent diamine oxidase and NADP+-reducing hydrogenase was used in flow catalysis for the synthesis N-heterocycles from diamines (Al-Shameri et al., 2020a). The needed co-substrates H2 and O2 for the reaction are produced by electrolysis. The applicability of dissolved redox mediators for the cofactor regeneration has been shown several times. Nevertheless, the application of soluble mediators is not a very convenient strategy. The most important drawbacks of dissolved mediators in electro enzymatic biocatalytic processes are interferences during product purification, a limited reusability of the mediators and their cost-intensive elimination from wastewater. Therefore the use of immobilized mediators could have economic and ecological advantages in regard of PI (Kochius et al., 2012).
As described in the introduction, saving energy is a major focus of PI. Here, electroenzymatic 200% cells (also paired electrolysis) with value-added reactions at both electrodes could contribute in a particularly important ways. The combination of an enzymatic electrosynthesis with an enzymatic fuel cell results in a highly valuable usage of energy to produce chemicals (Chen et al., 2020). A hydrogenase was used in the anode chamber. A cascade of a nitrogenase and a diaphorase catalysed the conversion of N2 to NH3 and NADH recycling. The produced NH3 and NADH were reacted in the cathode with leucine dehydrogenase to synthesize L-norleucine. In summary, the conversion of nitrogen gas to chiral amino acids, powered by H2 oxidation was realized in this electroenzymatic cascade in a 200% cell (Chen et al., 2020). A hydrogenase bio-anode was also coupled with a bio-cathode with the alkane monooxygenase from Pseudomonas putida (alkB) (Yuan et al., 2020b). A membrane-less paired electrolysis was used to oxidise glucose to gluconic acid. At the anode a immobilized glucose oxidase was used in combination with the mediator tetrathiafulvalene, while at the cathode a cascade of glucose oxidase and horseradish peroxidase was immobilised (Varničić et al., 2015). The space time yield of gluconic acid achieved at a glucose conversion of 47% was 18.2 g h−1 cm−2. The recent developments of electroenzymatic cascades are summarized in (Hickey et al., 2018).
The multiple types of electroenzymatic reactions also necessitate the use of various types of reactors, e.g., parallelized screening reactors (Ley et al., 2013a; Ley et al., 2013b), 2-phase reactors (Dong et al., 2020), fixed-bed reactors (Lütz et al., 2007; Kochius et al., 2014b; Kochius et al., 2014c), micro-fluidic systems (Fisher et al., 2013) or micro-reactors (Rodríguez-Hinestroza et al., 2017). As in all further electrochemical processes, the scale-up of the electro-enzymatic reactions is a challenge. Here, solid and fluidised bed electrodes as well as processes with gas diffusion electrodes (Holtmann et al., 2014b; Horst et al., 2016; Bormann et al., 2019) seem to be particularly feasible to fulfil the requirements of PI. Computational methods such as design of experiments (Tosstorff et al., 2017) or modelling in combination with simulations (Bormann et al., 2021) appear to be particularly useful for an accelerated implementation of electroenzymatic processes in industry. Furthermore, sustainability of the electroenzymatic process should be evaluated (Varničić, 2020).
Besides electro- or photochemical power inputs there are several other possibilities to transfer energy to an enzyme. For example, chemically stored energy can be delivered to enzymes in the form of small molecules like hydrogen peroxide (Burek et al., 2019c) or molecular hydrogen (Lubitz et al., 2014; Al-Shameri et al., 2020b), which both can as well be produced electro- or photochemically as mentioned above. Molecular hydrogen can as well be used to produce H2O2 in the so-called direct synthesis with molecular oxygen on the surface of a noble metal catalyst which can be directly coupled to a peroxygenase catalysed reaction (Karmee et al., 2009; Freakley et al., 2019). Further, H2 has been used to reduce anthraquinone separately which then can be used in an oxygen rich atmosphere to produce H2O2 enabling a lipase mediated epoxidation of monoterpenes (Ranganathan et al., 2015). Furthermore, technical devices have been used for an energy input to power enzyme catalysis. For example, ultrasound has been used piezocatalytically with bismuth oxychloride (Yoon et al., 2020) as well as atmospheric plasma techniques (Yayci et al., 2020a; Yayci et al., 2020b) both to generate H2O2 in situ for peroxygenase catalyzed C-H-functionalization. Comparably, gamma radiation has been used to produce H2O2 coupled with peroxygenases enabling nuclear waste driven enzymatic catalysis (Zhang et al., 2020b). Also, microwave radiation is known to enhance the performance of some (thermophilic or thermostable) enzymes, however, due to the accompanying (local) heating of the reaction solution it has a negative impact on the stability of many enzymes and sometimes showed no effect (Lin et al., 2005; Rejasse et al., 2007; Young et al., 2008). As microwave radiation might lead to conformal changes in the enzymes structure it is nearly impossible to predict the effect on an enzyme (Porcelli et al., 1997). Furthermore, microwave radiation is only an assistive power input and doesn´t drive enzyme catalysis directly.
In order to facilitate process intensification, the discipline of bioengineering aims for this adaption of an enzymatic process to the demanded operation conditions, commonly by varying the reactor-setup (Woodley, 2017). Certainly, other individual process relevant aspects like down-stream-processing, up-scaling, biocatalyst engineering and formulation (e.g., enzyme immobilisation) are certainly valid and important approaches to achieve higher yields of the target product (Strube et al., 2018). But indeed, it is the reactor itself which has the major effect on the overall process efficiency. For instance, a carefully chosen reactor can promote a specific kinetic profile of the used catalyst or can influence the thermodynamic position of a utilized reaction. So, the reactor represents the central element of a certain process and should be the first target of process optimization and intensification, not least because some of the aspects given above can be integrated in a certain reactor concept. Therefore, process intensification is more than only parameter optimization, it means rather the introduction of structural elements and novel reactor concepts for an economical (and sometimes ecological) improvement.
Indeed, no other PI approach is similarly represented in literature like the in situ product removal (ISPR) as well as co-product removal (Lye and Woodley, 1999; Satyawali et al., 2017). As tool of process intensification ISPR (or in situ extraction) has been investigated already in the 1970, predominantly in context with chemical processes. In the early 1980’s this concept was started to be transferred to the field of biotransformation, in the beginning mainly for the production of (low molecular) pharmaceuticals and food additives (Freeman et al., 1993). Downstream processing is a relevant cost factor for industrial processes in order to obtain high yields and purity of the aimed product (Lye and Woodley, 1999; Woodley et al., 2008; Hamideh et al., 2015). But the main reasons for the application of ISPR techniques are certainly product inhibition of the respective enzyme, detrimental effect of toxic products, avoiding of side products by a long-term presence of reactants or shifting the thermodynamic equilibrium of the target reaction (Woodley et al., 2008). There are several approaches to realize ISPR. For instance, there is the integration of definite and modular separation units into the actual reactor setup [like membranes, (Börner et al., 2015; Uthoff and Gröger, 2018)] or the inclusion of a subsequent complexing as well as crystallization reaction to withdraw the respective compound from the reaction system (Buque-Taboada et al., 2004; Buque-Taboada et al., 2006; Hülsewede et al., 2018). But in fact, ISPR by multiphasic reactor concepts is the most utilized approach and intensively represented in literature. Therefore, this is declared as main focus of this chapter to present significant achievements in ISPR, in particular those including multiphasic reactor design. For further insights in the field of ISPR the reader is referred to recent reviews (Woodley, 2017; Fellechner et al., 2019; Lindeque and Woodley, 2019; Woodley, 2020).
Reactive distillation is a popular and commonly used technique for reaction intensification in the chemical industry (Harmsen, 2007; Kiss et al., 2019). The integration of a thermal separation of reactants (on the basis of different boiling points) into the current process bears large potential for equilibrium-limited reactions. The only requirement for a suitable reaction system is a differentiation of the reactants by the boiling temperature (Kiss et al., 2019). Unfortunately, the biocatalytic applicability of reactive distillation columns is still limited, mainly due to the lower thermal stability of biocatalysts (Heils et al., 2012). Heils et al. (2012) could show the applicability of CalB in reactive distillation columns. This approach was further optimized in a subsequent work, on the one side towards a spray-coating immobilization technique to allow reproducible coating also of larger surfaces and on the other side towards the full integration of an enzymatic process into a reactive distillation approach, namely the kinetic resolution of racemic 2-pentanol in a transesterification reaction of e.g., ethyl butyrate (Heils et al., 2015). The recent publication from these authors presented computer-guided reaction design using an in silico preselection tool under consideration of physicochemical properties of the respective reactants (Kühn et al., 2017). The best option in terms of lack of azeotropes contained racemic 2-pentanol in combination with ethyl butyrate and propyl butyrate as substrates. This contribution represented the first notable application of an enzyme in a reactive distillation setup, not least due to the high stability of the silica-gel coating and less enzyme leaching but also because of the reactor setup in the form of a compartmentalized and temperature-controlled distillation column under reduced pressure (operation conditions: 75°C and 80 mbar). The authors presented for the first time a fractional separation of 1-propanol as co-product and (S)-2-pentanol as target product from the chosen enantioselective transesterification reaction directly from the distillation stream. However, the presented reactive distillation approach in combination with the coating-driven immobilization of the biocatalyst has been proven as very promising albeit this is limited yet to the application of CalB.
In order to intensify mass transfer in enzymatic reactions aerated columns are frequently used. Buergler et al. (2020) shows an intensification of the enzymatic hydroxylation of nonactivated hydrocarbons such as dodecanonic acid. The aerated aeration column was used as reactor bearing an aqueous phase and the immobilized cytochrome P450 BM3, resulting in a three-phase gas-liquid-solid system. Under optimized conditions with 80 mM C12:0 substrate concentration 90% conversion and 82% isolated yield after ∼28 h of incubation were realized. A bubble column was also used in combination with an unspecific peroxygenases (Perz et al., 2020). The gaseous substrate butane was converted to 2-butanol in a bubble column reactor. The process was scaled up to 2 L and coupled with continuous extraction, resulting in the production of 115 mmol 2-butanol and 70 mmol butanone (as over oxidation product).
Albeit the majority of biocatalytic transformations are performed in batch, continuously operated reactors are already widely used (Lindeque and Woodley, 2019; De Santis et al., 2020; Cardoso Marques et al., 2021). The underlying principle, namely generating a constant stream of reactants is considered as attractive, e.g., in the context of avoidance of enzyme inhibition or straight forward downstream processing.
The major key advantage of continuous flow reactors is definitely the high adjustability to the needs and demands of the respective transformation (Leemans Martin et al., 2020). This is mostly related to conversion and thermodynamic considerations and refers operationally very often to variation of residence time and/or enzyme concentration. Recently, Lindeque and Woodley (2019) published a valuable contribution on this topic in which batch and continuous operation is compared and evaluated in terms of API production, considering characteristics and application of the mentioned reactors.
Since the last decade continuous microflow systems started to attract some attention in biocatalysis as miniaturized reactors (Fernandes, 2010; Marques and Fernandes, 2011), albeit microfluidics was already intensively used before in chemical catalysis but also in biocatalyst screening, prototype process development or lab-on-a-chip approaches (Marques and Fernandes, 2011; Wohlgemuth et al., 2015; Tamborini et al., 2018; Žnidaršič-Plazl, 2019). The most important characteristics of those systems are for instance the significant smaller dimensions, a significant enhanced surface-to-volume-ratio within the reactor, shorter reaction times and very often higher biocatalyst stability. Those characteristics (which partially overlapping with those of a plug-flow reactor or a continuously operated batch reactor) provide in consequence substantial benefits over batch reactors (Lindeque and Woodley, 2019). Reaction conditions are drastically better controllable mainly regarding heat and mass transfer, fluid flow, reaction time or reactant concentration at any position in the reactor which is especially important for multiphase systems (Strniša et al., 2019; Žnidaršič-Plazl, 2019). Furthermore, the modular character of flow reactors allows a sequential setup of cascade reactions, e.g., chemoenzymatically and/or in combination with in line downstream processing (Žnidaršič-Plazl, 2017; Rudroff et al., 2018; Žnidaršič-Plazl, 2019). In fact, mass transfer limited transformations do benefit massively from the operation mode stated above since very often higher productivities and a reduced waste or energy consumption is easily reachable using microflow systems (Marques and Fernandes, 2011; Tamborini et al., 2018). Karande et al. (2010) and Karande et al. (2011) investigated as a mass transfer limited model reaction the reduction of 1-heptaldehyde to 1-heptanol in a multiphasic environment (formed by aqueousand organic liquids) catalysed by a thermostable alcohol dehydrogenase in segmented flow micro reactor. The effect of capillary diameter, flow velocity as well as the ratio of different phases and set those in relation to the applied enzyme or substrate concentration was investigated, the optimisation resulted in a productivity of 48 mMproduct h−1.
One of the main issues when using gaseous substrates is definitely the low concentration of dissolved substrate (e.g., O2) in the (enzyme containing) aqueous phase (Bolivar and Nidetzky, 2013). Therefore, the intensification of the transport of O2 from the supplying gas phase into the liquid phase is one ongoing research topic in multiphase biotransformations for that microflow systems were stated out as possible solution. Bolivar et al. (2019) presented a pressurized microflow reactor in order to increase the concentration of dissolved oxygen in the liquid phase. It was shown that the available concentration of oxygen was increased 170-fold to 43 mM at 34 bar. By using glucose oxidase and d-amino acid oxidase as catalyst and glucose and d-methionine as substrates, product concentrations up to 80 mM (α‐keto‐acid as oxidative deamination product of d-methionine) were realized. The given examples showed that miniaturization in the form of microflow reactors offers tremendous possibilities for an intensification of multiphasic liquid-liquid and gas-liquid reactions. As explained in the last section the way how contact between different phases is generated has an immense effect on mass transfer but also on enzyme activity and stability. Therefore, reaction conditions can be applied which are still conducive for the aimed enzyme reaction, e.g., in terms of residence times (Woodley, 2017).
In continuous catalysis immobilized (or heterogeneous) catalysts are mostly the preferred formulation. This is especially valid for biocatalysts and the advantages are obvious: often higher enzyme stability and easier downstream processing (Liese and Hilterhaus, 2013). Despite the efforts and achievements in the field of enzyme immobilization this point will probably remain as one additional issue in reaction (or enzyme) optimization, depending on the utilized enzyme and the demanded reaction conditions (e.g., high temperatures, high substrate concentrations, presence of organic solvents, etc.). The right reactor setup can make enzyme immobilisation (and thus labour-intensive investigations beforehand) obsolete. This is for instance true for enzyme membrane reactors (Rios et al., 2004). Enzyme membrane reactors and their application were recently reviewed, also in terms of process intensification (Satyawali et al., 2017). According to Satyawali et al. (2017) they show often higher efficiency against the respective batch reactor, the possibility of automatization and control of the molecular size of the product during ISPR. Also, limitations were mentioned, like enzyme loss through membranes, low concentration of product in permeate (effort in DSP) or membrane fouling (Rios et al., 2004; Prieto et al., 2007; Prieto et al., 2008). However, Adebar and Gröger et al. presented an attractive alternative to membrane reactors when enzyme immobilization is not suitable, namely the usage of homogeneous catalysts in the form of a fluidized moving bed reactor (Adebar and Gröger, 2020). The enzymes were entrapped in a superadsorber-gel matrix (consisting of polymerized polyacrylic acid) which can be considered as a compartmentalized and feedable mobile phase (micro-environment). Thus, this approach combines the advantages of catalyst stabilization by immobilisation and the advantageous properties of homogeneous catalyst utilization in a fluid (aqueous) phase.
The majority of all biocatalytic reactions proceed in liquid solution. Some exceptions such as biocatalytic gas-solid phase reactions have been reported (Trivedi et al., 2006a; Trivedi et al., 2006b) but are currently not developed very far. By far, water (or buffered aqueous solutions) represents the solvent of choice. On the one hand, it is generally accepted that water represents the natural (and therefore most suitable) solvent for enzymes (Holtmann and Hollmann, 2022). On the other hand; the believe that water is a green solvent prevails. Water, is a suitable solvent for polar and ionic compounds whereas its dissolution power with hydrophobic compounds, which also represent a significant proportion of the compounds of interest, is very limited. As a consequence, substrate concentrations used in biocatalytic experiments tend to be low. A comparison of typical starting material concentrations used in chemo- and biocatalysis is shown in Figure 4 [the data obtained from publications in ChemCatChem (vol. 12 (2020), issues 1–12)]. Of the biocatalysis manuscripts inspected, almost 50% used substrate concentration of 10 mM or lower. Roughly 12% of the manuscripts used 100 mM or higher. This is strikingly different compared to chemocatalysis reports where only a minority of 13 uses starting concentrations below 100 mM. Almost 20% of all contributions utilised more than 1 M concentrations or even “neat” (i.e., devoid of additional solvents) reaction conditions.
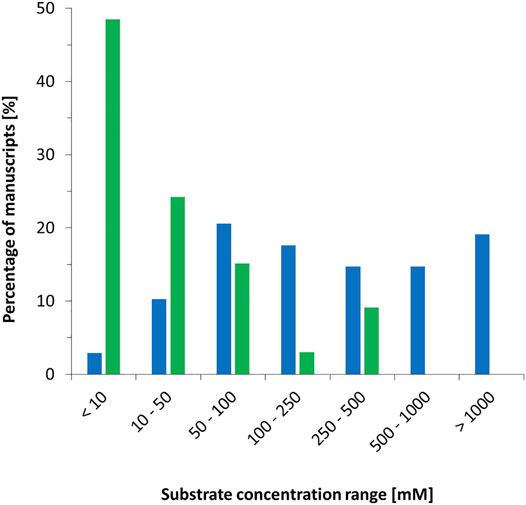
FIGURE 4. Substrate concentrations reported in publications in ChemCatChem [vol. 12 (2020), issues 1–12]. Green: for biocatalytic contributions (33 in total), blue: for chemocatalytic contributions in liquid phase (68: in total).
While chemists tend to choose the solvent most suitable for the reaction of interest, biotechnologists still largely tend to use water. The resulting rather dilute reaction mixtures are not attractive neither from an environmental (Ni et al., 2014) nor an economic (Tufvesson et al., 2010; Tufvesson et al., 2011) point-of-view. This strong preference for aqueous reaction media is astonishing as since decades, suitable strategies to increase the substrate loading exist. Some of them will be discussed in more detail in the following in the context of PI.
“The best solvent is no solvent” (Sheldon, 2005). Such biocatalysis systems would perfectly fulfil the above mentioned requirements, in particular a substantial decrease in equipment-size, energy consumption, and waste production).
Especially hydrolase-catalysed esterification reactions using liquid alcohols and carboxylic acids are well-established on industrial scale. For example, the lipase-catalysed synthesis of cosmetic esters using a commercially available immobilised lipase operates smoothly on a >1,000 ton a−1 (own estimations) scale without the need of additional solvents and minimal consumption of the enzyme (Ansorge-Schumacher and Thum, 2013). Because of their substrate scope, lipases are generally believed to be predestined for no-aqueous reaction media. However, already in 1984 Zaks and Klibanow reported organic media as solvents for different enzyme classes (Zaks and Klibanov, 1984; Zaks and Klibanov, 1985). Next to higher substrate loadings and significantly increased thermal stability of the biocatalysts also some interesting changes in the reactivity (Dordick et al., 1986) were observed (Gupta et al., 2013). Non-aqueous (or micro-aqueous) reactions are still largely restricted to lipase reactions (Scheme 1A) but the interest in using mirco-aqueous, non-aqueous or even neat reaction conditions increases steadily. In addition to increasing the reagent concentration considerably, some additional advantages are worth being highlighted. Hydroxynitrile lyases (HNLs) are preferentially used under non-aqueous conditions to suppress the non-catalysed addition of HCN (or its anion) to the carbonyl group yielding racemic product [Scheme 1B, (Paravidino et al., 2010; Scholz et al., 2012)]. In case of ADH-catalysed redox reactions, non-aqueous media circumvent the diffusion of the nicotinamide cofactor and thereby further protect it from aqueous degradation [Scheme 1C, (Kara et al., 2014; Jakoblinnert et al., 2011; Hibino and Ohtake, 2013; Erdmann et al., 2014; Hijazi et al., 2019)]. More recently, also reductive aminations [Scheme 1D, (Böhmer, 2020)] and oxyfunctionalisations in non-aqueous media have been reported [Scheme 1E, (Rauch et al., 2019)]. Also more elaborated cascades of enzymes and enzymes with chemocatalysts have been reported [Scheme 1F, (Jakoblinnert and Rother, 2014; Gómez Baraibar, 2016; Wachtmeister et al., 2016)] have been reported.
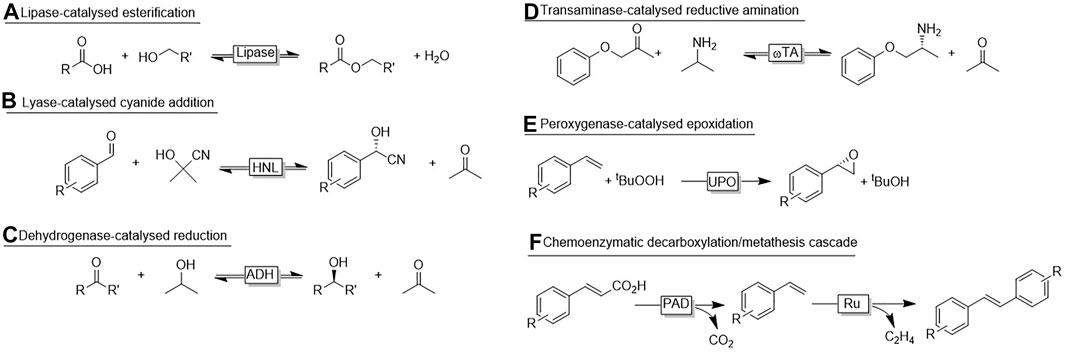
SCHEME 1. Examples for biocatalytic transformations in non-aqueous or micro-aqueous media.
(Bio)catalytic reactions not necessarily have to occur in homogeneous phase. Various biocatalytic multiphase reactions have been reported in the past years. The two liquid phase system (2LPS) approach is probably the best known one to increase the overall loading of hydrophobic reagents for biocatalytic reactions. The reagents partition between the hydrophobic organic phase and the aqueous (biocatalyst-containing) aqueous layer (Scheme 2).
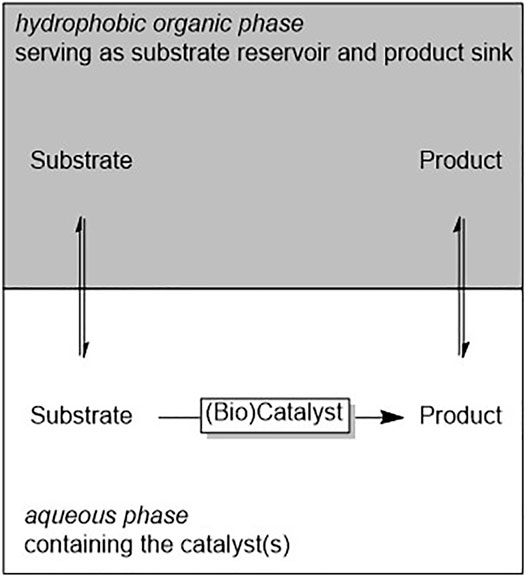
SCHEME 2. General representation of the two liquid phase system (2LPS) approach wherein a hydrophobic organic phase serves as substrate reservoir and product sink. The reagents partition between both phases.
2LPS can be used to control the concentration of toxic reagents in the aqueous phase and thereby minimise inhibitory effects on whole cells (Park et al., 2007). Hydrophobic reagents will preferentially partition into the organic phase and thereby will be removed from the reactive organic layer. This can be exploited to control the selectivity of the oxidation of primary alcohols (Bühler, 2003a; Bühler, 2003b) or the reduction of carboxylic acids to the aldehyde stage (Ni et al., 2012) and thereby simply prevent over-oxidation or over-reduction. In situ extraction of reactive products can also help to minimise undesired hydrolysis of epoxides (Park et al., 2007) or lactones (Kara et al., 2013).
An interesting application of the 2LPS approach was reported by Kragl and coworkers (Scheme 3) (Eckstein et al., 2004; Pfruender et al., 2004). ADH-catalysed reduction reactions, especially if simple alcohols such as isopropanol are used as stoichiometric reductants (in a Meerwein-Ponndrof-Verley-type reduction reaction) are often plague by product inhibition. As an organic layer the authors used the ionic liquid [BMIM][(CF3SO2)2N] which exhibits a significantly higher affinity to acetone compared to isopropanol thereby reducing the in situ concentration of acetone in the aqueous, enzyme-containing phase and therefore reducing product inhibition and accelerating the overall rate of the ADH-catalysed reduction reaction.
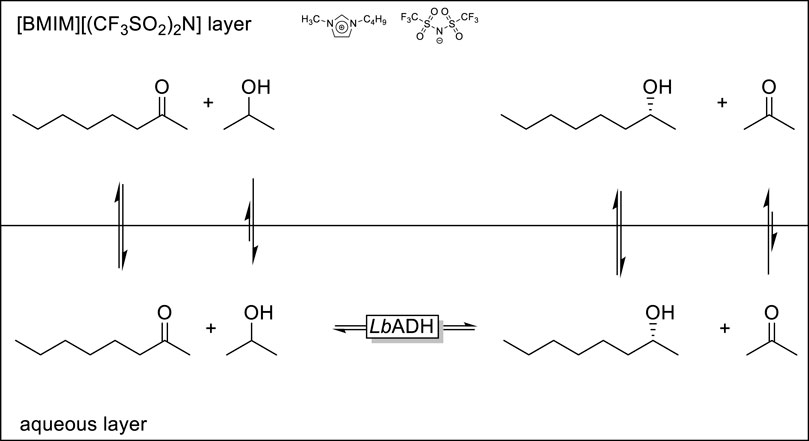
SCHEME 3. Selective in situ removal of a reaction by-product using the 2LPS approach to minimise product inhibition of a LbADH (ADH from Lactobacillus brevis) catalysed reduction reaction.
Liquid-solid reactions are less common but may offer some advantages over fully homogeneous reaction systems. If, for example the product is solid and precipitates from the reaction mixture, simple filtration may represent an attractive DSP method (Buque-Taboada et al., 2005; Schmidt et al., 2017). A very nice example how in situ product crystallisation can help to circumvent undesired side reactions is the synthesis of poorly water soluble Amoxicillin by Penicillin acylase-catalysed amidation of carboxylic acid esters (Bruggink et al., 1998). The enzyme also catalyses the undesired hydrolysis of the product yielding thermodynamically stable salts. As the product, however, is poorly soluble, it also is not available for the hydrolysis reaction.
There is an ongoing interest in reducing the environmental impact of solvents (Jessop, 2011) and replacing ecologically questionable solvents of the past with new (neoteric) alternatives. Ionic liquids (ILs) represented the first wave of this development [reviewed in (Potdar et al., 2015; Xu et al., 2016)]. Especially their negligible vapour pressure resulting in almost no evaporation losses and atmospheric pollution motivated a first hype around the turn of the millennium. Later, the enthusiasm for ILs cooled down significantly with more and more publications focussing on toxicological issues such as aquatic toxicity (Thuy Pham et al., 2010). Another concern about ILs is their often rather complex synthesis. The multitude of synthesis steps involved suggests that the synthesis of ILs may be rather resource-demanding thereby casting doubt on their environmental benignity (Jessop, 2011). More recently, (natural) deep eutectic solvents [(NA)DES] are moving into the centre of attention (Pätzold et al., 2019a). Today, the (NA)DES as solvents for biocatalysis are still in the explorative phase and future will tell if the promise of lower environmental impact and being bio-based can be held and if current issues such as viscosity can be solved. The studies on the DES mostly comprise proof-of-principle studies, comparisons of process performance with alternative solvents are often missing. Milker et al. (2019) investigated the aldolase activity of a lipase with 4-nitrobenzaldehyde (4-NBA) and acetone in different deep eutectic solvents. The authors concluded that the best process performance was realized by using acetone as co-solvent (and substrate) without the addition of a DES. Nevertheless, some technical-relevant applications of DES as solvents are worth mentioning here. The esterification of fatty acids with carbohydrates for example, is a notorious challenge for biocatalysis due to the solvent incompatibility of the hydrophobic fatty acid (esters) and the highly polar carbohydrates (Siebenhaller et al., 2017; Siebenhaller et al., 2018; Delavault et al., 2021). Such dual use as substrate and solvent at the same time (2-in1 concept) of DES has also been reported with menthol-based DES and the synthesis of menthol esters (Hümmer et al., 2018; Pätzold et al., 2019b). Here both DES compounds acted as substrates and reaction solvent in the lipase-catalyzed esterification. In addition, it was shown that a subsequent recycling and re-use of the excess DES compound (menthol) is possible (Pätzold et al., 2019c).
In general, PI enables the transition of the chemical as well as biochemical engineering field and make it more fit for addressing global issues, such as climate change, limited petrochemical and natural resources, growing populations and higher environmental standards. Through PI, biotech companies strive to enhance enzymatic and other production by reducing energy consumption, increasing reaction rates, reducing wasted energy and costs associated with waste products, improving purification steps, reducing equipment size, increasing safety and operational simplicity, etc. In doing so, companies can increase the sustainability of their activities. It is important for us to emphasize that in order to establish the various PI technologies, it is always important to show the specific improvements on the basis of key performance indicators (e.g., space-time-yield, energy consumption). Here it is also important that the complete process chains must always be considered and not just isolated reaction steps. Additional aspects in terms of PI were not or very briefly discussed in this review, e.g., automatization and in situ process control. The gapless monitoring of a certain process will be a very essential topic in industrial catalysis, especially if an automatized of self-regulated adjustment of reaction parameters can take place in real time. This concept of a smart reactor came up recently in some publications (Hu et al., 2020; Spille et al., 2020). From our point of view it is indeed very likely that this aspect of PI becomes more and more trendsetting in prospective biocatalysis. In the last years more specified terms of PI (e.g., as reaction intensification, transport intensification, and in particular local and global process intensification) have been adopted in chemical engineering. It can be expected that these more specific definitions will also become common in biocatalysis. Local intensification is defined here as the classical approach of PI based on the use of techniques and methods for the drastic improvement of the efficiency of a single unit or device (see most of the examples below), (Portha et al., 2014). When PI focuses on single units the interactions among all units within the process are ignored and the impact of local intensification of a single unit can be very limited, resulting in weak improvement of the whole process. A holistic process-based intensification (or a global intensification) will result in the full exploitation of PI’s potential.
AL, DH and FH conceived the review article all authors contributed to the manuscript composition.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adachi, T., Kitazumi, Y., Shirai, O., and Kano, K. (2020). Recent Progress in Applications of Enzymatic Bioelectrocatalysis. Catalysts 10 (12), 1413. doi:10.3390/catal10121413
Adebar, N., and Gröger, H. (2020). Heterogeneous Catalysts "on the Move": Flow Chemistry with Fluid Immobilised (Bio)Catalysts. Eur. J. Org. Chem. 2020 (38), 6062–6067. doi:10.1002/ejoc.202000705
Al-Shameri, A., Petrich, M. C., Junge Puring, K., Apfel, U. P., Nestl, B. M., and Lauterbach, L. (2020). Powering Artificial Enzymatic Cascades with Electrical Energy. Angew. Chem. Int. Ed. Engl. 59 (27), 10929–10933. doi:10.1002/anie.202001302
Al-Shameri, A., Willot, S. J.-P., Paul, C. E., Hollmann, F., and Lauterbach, L. (2020). H2as a Fuel for Flavin- and H2O2-dependent Biocatalytic Reactions. Chem. Commun. 56 (67), 9667–9670. doi:10.1039/d0cc03229h
Amer, M., Wojcik, E. Z., Sun, C., Hoeven, R., Hughes, J. M. X., Faulkner, M., et al. (2020). Low Carbon Strategies for Sustainable Bio-Alkane Gas Production and Renewable Energy. Energy Environ. Sci. 13 (6), 1818–1831. doi:10.1039/d0ee00095g
Ansorge-Schumacher, M. B., and Thum, O. (2013). Immobilised Lipases in the Cosmetics Industry. Chem. Soc. Rev. 42 (15), 6475–6490. doi:10.1039/c3cs35484a
Bachmeier, A., Murphy, B. J., and Armstrong, F. A. (2014). A Multi-Heme Flavoenzyme as a Solar Conversion Catalyst. J. Am. Chem. Soc. 136 (37), 12876–12879. doi:10.1021/ja507733j
Biegasiewicz, K. F., Cooper, S. J., Emmanuel, M. A., Miller, D. C., and Hyster, T. K. (2018). Catalytic Promiscuity Enabled by Photoredox Catalysis in Nicotinamide-dependent Oxidoreductases. Nat. Chem 10 (7), 770–775. doi:10.1038/s41557-018-0059-y
Biegasiewicz, K. F., Cooper, S. J., Gao, X., Oblinsky, D. G., Kim, J. H., Garfinkle, S. E., et al. (2019). Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science 364 (6446), 1166–1169. doi:10.1126/science.aaw1143
Böhmer, S., Köninger, K., Gomez-Baraibar, A., Bojarra, S., Mügge, C., Schmidt, S., et al. (2017). Enzymatic Oxyfunctionalization Driven by Photosynthetic Water-Splitting in the Cyanobacterium Synechocystis Sp. PCC 6803. Catalysts 7 (8).
Böhmer, W. (2020). Continuous Flow Bioamination of Ketones in Organic Solvents at Controlled Water Activity Using Immobilized ω-Transaminases. Adv. Synth. Catal. 362 (9), 1858–1867.
Bolivar, J. M., Mannsberger, A., Thomsen, M. S., Tekautz, G., and Nidetzky, B. (2019). Process Intensification for O 2 ‐dependent Enzymatic Transformations in Continuous Single‐phase Pressurized Flow. Biotechnol. Bioeng. 116 (3), 503–514. doi:10.1002/bit.26886
Bolivar, J. M., and Nidetzky, B. (2013). Multiphase Biotransformations in Microstructured Reactors: Opportunities for Biocatalytic Process Intensification and Smart Flow Processing. Green. Process. Synth. 2 (6), 541–559. doi:10.1515/gps-2013-0091
Bolivar, J. M., Wiesbauer, J., and Nidetzky, B. (2011). Biotransformations in Microstructured Reactors: More Than Flowing with the Stream? Trends Biotechnol. 29 (7), 333–342. doi:10.1016/j.tibtech.2011.03.005
Boodhoo, K. V. K., Flickinger, M. C., Woodley, J. M., and Emanuelsson, E. A. C. (2022). Bioprocess Intensification: A Route to Efficient and Sustainable Biocatalytic Transformations for the Future. Chem. Eng. Process. - Process Intensification 172, 108793. doi:10.1016/j.cep.2022.108793
Bormann, S., Hertweck, D., Schneider, S., Bloh, J. Z., Ulber, R., Spiess, A. C., et al. (2021). Modeling and Simulation‐based Design of Electroenzymatic Batch Processes Catalyzed by Unspecific Peroxygenase from A. Aegerita. Biotechnol. Bioeng. 118 (1), 7–16. doi:10.1002/bit.27545
Bormann, S., Schie, M. M. C. H., De Almeida, T. P., Zhang, W., Stöckl, M., Ulber, R., et al. (2019). H 2 O 2 Production at Low Overpotentials for Electroenzymatic Halogenation Reactions. ChemSusChem 12 (21), 4759–4763. doi:10.1002/cssc.201902326
Börner, T., Rehn, G., Grey, C., and Adlercreutz, P. (2015). A Process Concept for High-Purity Production of Amines by Transaminase-Catalyzed Asymmetric Synthesis: Combining Enzyme Cascade and Membrane-Assisted ISPR. Org. Process Res. Development 19 (7), 793–799.doi:10.1021/acs.oprd.5b00055
Bruggink, A., Roos, E. C., and de Vroom, E. (1998). Penicillin Acylase in the Industrial Production of β-Lactam Antibiotics. Org. Process. Res. Dev. 2 (2), 128–133. doi:10.1021/op9700643
Buergler, M. B., Dennig, A., and Nidetzky, B. (2020). Process Intensification for Cytochrome P450 BM3‐catalyzed Oxy‐functionalization of Dodecanoic Acid. Biotechnol. Bioeng. 117 (8), 2377–2388. doi:10.1002/bit.27372
Bühler, B. (2003). Chemical Biotechnology for the Specific Oxyfunctionalization of Hydrocarbons on a Technical Scale. Biotechnol. Bioeng. 82 (7), 833–842.
Bühler, B. (2003). Use of the Two-Liquid Phase Concept to Exploit Kinetically Controlled Multistep Biocatalysis. Biotechnol. Bioeng. 81 (6), 683–694.
Buller, R., Hecht, K., Mirata, M., and Meyer, H-P. (2018). “An Appreciation of Biocatalysis in the Swiss Manufacturing Environment,” in Biocatalysis: An Industrial Perspective (Cambridge and London, UK: The Royal Society of Chemistry), 1–43. doi:10.1039/9781782629993-00001
Buque-Taboada, E. M., Straathof, A. J. J., Heijnen, J. J., and van der Wielen, L. A. M. (2006). In Situ product Recovery (ISPR) by Crystallization: Basic Principles, Design, and Potential Applications in Whole-Cell Biocatalysis. Appl. Microbiol. Biotechnol. 71 (1), 1–12. doi:10.1007/s00253-006-0378-6
Buque-Taboada, E. M., Straathof, A. J. J., Heijnen, J. J., and van der Wielen, L. A. M. (2004). In Situ product Removal Using a Crystallization Loop in Asymmetric Reduction of 4-oxoisophorone bySaccharomyces Cerevisiae. Biotechnol. Bioeng. 86 (7), 795–800. doi:10.1002/bit.20093
Buque-Taboada, E. M., Straathof, J., Joseph, J., Luuk, A., and Wielen, V. (2005). Microbial Reduction and In Situ Product Crystallization Coupled with Biocatalyst Cultivation during the Synthesis of 6R-Dihydrooxoisophorone. Adv. Synth. Catal. 347 (7-8), 1147–1154. doi:10.1002/adsc.200505024
Burai, T. N., Panay, A. J., Zhu, H., Lian, T., and Lutz, S. (2012). Light-Driven, Quantum Dot-Mediated Regeneration of FMN to Drive Reduction of Ketoisophorone by Old Yellow Enzyme. ACS Catal. 2 (4), 667–670. doi:10.1021/cs300085h
Burek, B. O., Bormann, S., Hollmann, F., Bloh, J. Z., and Holtmann, D. (2019). Hydrogen Peroxide Driven Biocatalysis. Green. Chem. 21 (12), 3232–3249. doi:10.1039/c9gc00633h
Burek, B. O., de Boer, S. R., Tieves, F., Zhang, W., van Schie, M., Bormann, S., et al. (2019). Photoenzymatic Hydroxylation of Ethylbenzene Catalyzed by Unspecific Peroxygenase: Origin of Enzyme Inactivation and the Impact of Light Intensity and Temperature. ChemCatChem 11 (13), 3093–3100. doi:10.1002/cctc.201900610
Burek, B. O., Timm, J., Bahnemann, D. W., and Bloh, J. Z. (2019). Kinetic Effects and Oxidation Pathways of Sacrificial Electron Donors on the Example of the Photocatalytic Reduction of Molecular Oxygen to Hydrogen Peroxide over Illuminated Titanium Dioxide. Catal. Today 335, 354–364. doi:10.1016/j.cattod.2018.12.044
Cai, R., Milton, R. D., Abdellaoui, S., Park, T., Patel, J., Alkotaini, B., et al. (2018). Electroenzymatic C-C Bond Formation from CO2. J. Am. Chem. Soc. 140 (15), 5041–5044. doi:10.1021/jacs.8b02319
Cardoso Marques, M. P., Lorente-Arevalo, A., and Bolivar, J. M. (2021). Biocatalysis in Continuous-Flow Microfluidic Reactors. Adv. Biochem. Eng. Biotechnol. doi:10.1007/10_2020_160
Cha, H. J., Hwang, S. Y., Lee, D. S., Kumar, A. R., Kwon, Y. U., Voß, M., et al. (2020). Whole‐Cell Photoenzymatic Cascades to Synthesize Long‐Chain Aliphatic Amines and Esters from Renewable Fatty Acids. Angew. Chem. Int. Ed. 59 (18), 7024–7028. doi:10.1002/anie.201915108
Chen, H., Prater, M. B., Cai, R., Dong, F., Chen, H., and Minteer, S. D. (2020). Bioelectrocatalytic Conversion from N2 to Chiral Amino Acids in a H2/α-Keto Acid Enzymatic Fuel Cell. J. Am. Chem. Soc. 142 (8), 4028–4036. doi:10.1021/jacs.9b13968
Choi, D. S., Ni, Y., Fernández-Fueyo, E., Lee, M., Hollmann, F., and Park, C. B. (2017). Photoelectroenzymatic Oxyfunctionalization on Flavin-Hybridized Carbon Nanotube Electrode Platform. ACS Catal. 7 (3), 1563–1567. doi:10.1021/acscatal.6b03453
Choudhury, S., Baeg, J.-O., Park, N.-J., and Yadav, R. K. (2014). A Solar Light-Driven, Eco-Friendly Protocol for Highly Enantioselective Synthesis of Chiral Alcohols via Photocatalytic/biocatalytic Cascades. Green. Chem. 16 (9), 4389–4400. doi:10.1039/c4gc00885e
Churakova, E., Arends, I. W. C. E., and Hollmann, F. (2013). Increasing the Productivity of Peroxidase-Catalyzed Oxyfunctionalization: A Case Study on the Potential of Two-liquid-phase Systems. ChemCatChem 5 (2), 565–568. doi:10.1002/cctc.201200490
Clayman, P. D., and Hyster, T. K. (2020). Photoenzymatic Generation of Unstabilized Alkyl Radicals: An Asymmetric Reductive Cyclization. J. Am. Chem. Soc. 142 (37), 15673–15677. doi:10.1021/jacs.0c07918
De Santis, P., Meyer, L.-E., and Kara, S. (2020). The Rise of Continuous Flow Biocatalysis - Fundamentals, Very Recent Developments and Future Perspectives. React. Chem. Eng. 5 (12), 2155–2184. doi:10.1039/d0re00335b
Delavault, A., Ochs, K., Gorte, O., Syldatk, C., Durand, E., and Ochsenreither, K. (2021). Microwave-Assisted One-Pot Lipid Extraction and Glycolipid Production from Oleaginous Yeast Saitozyma Podzolica in Sugar Alcohol-Based Media. Molecules 26 (2), 470. doi:10.3390/molecules26020470
Dong, F., Chen, H., Malapit, C. A., Prater, M. B., Li, M., Yuan, M., et al. (2020). Biphasic Bioelectrocatalytic Synthesis of Chiral β-Hydroxy Nitriles. J. Am. Chem. Soc. 142 (18), 8374–8382. doi:10.1021/jacs.0c01890
Dordick, J. S., Marletta, M. A., and Klibanov, A. M. (1986). Peroxidases Depolymerize Lignin in Organic media but Not in Water. Proc. Natl. Acad. Sci. 83 (17), 6255–6257. doi:10.1073/pnas.83.17.6255
Duong, H. T., Wu, Y., Sutor, A., Burek, B. O., Hollmann, F., and Bloh, J. Z. (2021). Intensification of Photobiocatalytic Decarboxylation of Fatty Acids for the Production of Biodiesel. ChemSusChem 14 (4), 1053–1056. doi:10.1002/cssc.202002957
Eckstein, M., Villela Filho, M., Liese, A., and Kragl, U. (2004). Use of an ionic liquid in a two-phase system to improve an alcohol dehydrogenase catalysed reductionElectronic supplementary information (ESI) available: experimental section. See http://www.rsc.org/suppdata/cc/b4/b401065e/. Chem. Commun. (9), 1084–1085. doi:10.1039/b401065e
Emmanuel, M. A., Greenberg, N. R., Oblinsky, D. G., and Hyster, T. K. (2016). Accessing Non-natural Reactivity by Irradiating Nicotinamide-dependent Enzymes with Light. Nature 540 (7633), 414–417. doi:10.1038/nature20569
Erdmann, V., Mackfeld, U., Rother, D., and Jakoblinnert, A. (2014). Enantioselective, Continuous (R)- and (S)-2-butanol Synthesis: Achieving High Space-Time Yields with Recombinant E. coli Cells in a Micro-aqueous, Solvent-free Reaction System. J. Biotechnol. 191, 106–112. doi:10.1016/j.jbiotec.2014.06.032
Fellechner, O., Blatkiewicz, M., and Smirnova, I. (2019). Reactive Separations for In Situ Product Removal of Enzymatic Reactions: A Review. Chem. Ingenieur Technik 91 (11), 1522–1543. doi:10.1002/cite.201900027
Fernandes, P. (2010). Miniaturization in Biocatalysis. Ijms 11 (3), 858–879. doi:10.3390/ijms11030858
Feyza Özgen, F., Runda, M. E., Burek, B. O., Wied, P., Bloh, J. Z., Kourist, R., et al. (2020). Artificial Light-Harvesting Complexes Enable Rieske Oxygenase Catalyzed Hydroxylations in Non-photosynthetic Cells. Angew. Chem. Int. Edition 59 (10), 3982–3987. doi:10.1002/anie.201914519
Fisher, K., Mohr, S., Mansell, D., Goddard, N. J., Fielden, P. R., and Scrutton, N. S. (2013). Electro-enzymatic Viologen-Mediated Substrate Reduction Using Pentaerythritol Tetranitrate Reductase and a Parallel, Segmented Fluid Flow System. Catal. Sci. Technol. 3 (6), 1505–1511. doi:10.1039/c3cy20720j
Freakley, S. J., Kochius, S., van Marwijk, J., Fenner, C., Lewis, R. J., Baldenius, K., et al. (2019). A Chemo-Enzymatic Oxidation cascade to Activate C-H Bonds with In Situ Generated H2O2. Nat. Commun. 10 (1), 4178. doi:10.1038/s41467-019-12120-w
Freeman, A., Woodley, J. M., and Lilly, M. D. (1993). In Situ Product Removal as a Tool for Bioprocessing. Nat. Biotechnol. 11 (9), 1007–1012. doi:10.1038/nbt0993-1007
Gao, J., Wang, H., Yuan, Q., and Feng, Y. (2018). Structure and Function of the Photosystem Supercomplexes. Front. Plant Sci. 9 (357), 357. doi:10.3389/fpls.2018.00357
Gargiulo, S., Arends, I. W. C. E., and Hollmann, F. (2011). A Photoenzymatic System for Alcohol Oxidation. ChemCatChem 3 (2), 338–342. doi:10.1002/cctc.201000317
Gómez Baraibar, Á. (2016). A One-Pot Cascade Reaction Combining an Encapsulated Decarboxylase with a Metathesis Catalyst for the Synthesis of Bio-Based Antioxidants. Angew. Chem. Int. Edition 55 (47), 14823–14827.
Gupta, M. N., Mukherjee, J., and Malhotra, D. (2013). Use of High Activity Enzyme Preparations in Neat Organic Solvents for Organic Synthesis. Universal Org. Chem. 1 (1). doi:10.7243/2053-7670-1-1
Hamideh, V., Eskandari, M., Sobhani, H. V., Berenjian, A., Song, Y., Jafarizadeh-Malmiri, H., et al. (2015). Process Intensification for Production and Recovery of Biological Products. Am. J. Biochem. Biotechnol. 11 (1). doi:10.3844/ajbbsp.2015.37.43
Harmsen, G. J. (2007). Reactive Distillation: The Front-Runner of Industrial Process Intensification. Chem. Eng. Process. Process Intensification 46 (9), 774–780. doi:10.1016/j.cep.2007.06.005
Heils, R., Jensen, J.-H., Wichert, S., Behrens, N., Fabuel-Ortega, M., Liese, A., et al. (2015). Enzymatic Reactive Distillation: Kinetic Resolution of Rac-2-Pentanol with Biocatalytic Coatings on Structured Packings. Ind. Eng. Chem. Res. 54 (38), 9458–9467. doi:10.1021/acs.iecr.5b02802
Heils, R., Sont, A., Bubenheim, P., Liese, A., and Smirnova, I. (2012). Integration of Enzymatic Catalysts in a Reactive Distillation Column with Structured Packings. Ind. Eng. Chem. Res. 51 (35), 11482–11489. doi:10.1021/ie300837v
Heyes, D. J., Lakavath, B., Hardman, S. J. O., Sakuma, M., Hedison, T. M., and Scrutton, N. S. (2020). Photochemical Mechanism of Light-Driven Fatty Acid Photodecarboxylase. ACS Catal. 10 (12), 6691–6696. doi:10.1021/acscatal.0c01684
Hibino, A., and Ohtake, H. (2013). Use of Hydrophobic Bacterium Rhodococcus Rhodochrous NBRC15564 Expressed Thermophilic Alcohol Dehydrogenases as Whole-Cell Catalyst in Solvent-free Organic media. Process Biochem. 48 (5), 838–843. doi:10.1016/j.procbio.2013.03.022
Hickey, D. P., Gaffney, E. M., and Minteer, S. D. (2018). Electrometabolic Pathways: Recent Developments in Bioelectrocatalytic Cascades. Top. Curr. Chem. (Z) 376 (6), 43. doi:10.1007/s41061-018-0221-4
Hijazi, M., Spiekermann, P., Krumm, C., and Tiller, J. C. (2019). Poly(2‐oxazoline)s Terminated with 2,2′‐imino Diacetic Acid Form Noncovalent Polymer-Enzyme Conjugates that Are Highly Active in Organic Solvents. Biotechnol. Bioeng. 116 (2), 272–282. doi:10.1002/bit.26877
Hobisch, M., Schie, M. M. C. H., Kim, J., Røjkjær Andersen, K., Alcalde, M., Kourist, R., et al. (2020). Solvent‐Free Photobiocatalytic Hydroxylation of Cyclohexane. ChemCatChem 12 (16), 4009–4013. doi:10.1002/cctc.202000512
Hobisch, M., Spasic, J., Malihan‐Yap, L., Barone, G. D., Castiglione, K., Tamagnini, P., et al. (2021). Internal Illumination to Overcome the Cell Density Limitation in the Scale‐up of Whole‐Cell Photobiocatalysis. ChemSusChem 14 (15), 3219–3225. doi:10.1002/cssc.202100832
Hollmann, F., Schmid, A., and Steckhan, E. (2001). The First Synthetic Application of a Monooxygenase Employing Indirect Electrochemical NADH Regeneration. Angew. Chem. Int. Ed. 40 (1), 169–171. doi:10.1002/1521-3773(20010105)40:1<169::aid-anie169>3.0.co;2-t
Holtmann, D., Fraaije, M. W., Arends, I. W. C. E., Opperman, D. J., and Hollmann, F. (2014). The Taming of Oxygen: Biocatalytic Oxyfunctionalisations. Chem. Commun. 50 (87), 13180–13200. doi:10.1039/c3cc49747j
Holtmann, D., and Hollmann, F. (2022). Is Water the Best Solvent for Biocatalysis? Mol. Catal. 517, 112035. doi:10.1016/j.mcat.2021.112035
Holtmann, D., Krieg, T., Getrey, L., and Schrader, J. (2014). Electroenzymatic Process to Overcome Enzyme Instabilities. Catal. Commun. 51, 82–85. doi:10.1016/j.catcom.2014.03.033
Honda, Y., Hagiwara, H., Ida, S., and Ishihara, T. (2016). Application to Photocatalytic H2Production of a Whole-Cell Reaction by RecombinantEscherichia coliCells Expressing [FeFe]-Hydrogenase and Maturases Genes. Angew. Chem. Int. Ed. 55 (28), 8045–8048. doi:10.1002/anie.201600177
Horst, A. E. W., Bormann, S., Meyer, J., Steinhagen, M., Ludwig, R., Drews, A., et al. (2016). Electro-enzymatic Hydroxylation of Ethylbenzene by the Evolved Unspecific Peroxygenase of Agrocybe Aegerita. J. Mol. Catal. B: Enzymatic 133, S137–S142. doi:10.1016/j.molcatb.2016.12.008
Hoschek, A., Bühler, B., and Schmid, A. (2017). Overcoming the Gas-Liquid Mass Transfer of Oxygen by Coupling Photosynthetic Water Oxidation with Biocatalytic Oxyfunctionalization. Angew. Chem. Int. Ed. 56 (47), 15146–15149. doi:10.1002/anie.201706886
Hu, X., Karnetzke, J., Fassbender, M., Drücker, S., Bettermann, S., Schroeter, B., et al. (2020). Smart Reactors - Combining Stimuli-Responsive Hydrogels and 3D Printing. Chem. Eng. J. 387, 123413. doi:10.1016/j.cej.2019.123413
Huang, J., Antonietti, M., and Liu, J. (2014). Bio-inspired Carbon Nitride Mesoporous Spheres for Artificial Photosynthesis: Photocatalytic Cofactor Regeneration for Sustainable Enzymatic Synthesis. J. Mater. Chem. A. 2 (21), 7686–7693. doi:10.1039/c4ta00793j
Huang, X., Cao, M., and Zhao, H. (2020). Integrating Biocatalysis with Chemocatalysis for Selective Transformations. Curr. Opin. Chem. Biol. 55, 161–170. doi:10.1016/j.cbpa.2020.02.004
Huijbers, M. M. E., Zhang, W., Tonin, F., and Hollmann, F. (2018). Light-Driven Enzymatic Decarboxylation of Fatty Acids. Angew. Chem. Int. Ed. 57 (41), 13648–13651. doi:10.1002/anie.201807119
Hülsewede, D., Tänzler, M., Mildner, A., and Süss, P. (2018). Development of an In Situ-Product Crystallization (ISPC)-Concept to Shift the Reaction Equilibria of Selected Amine Transaminase-Catalyzed Reactions. Eur. J. Org. Chem. 2018 (18), 2130–2133. doi:10.1002/ejoc.201800323
Hümmer, M., Kara, S., Liese, A., Huth, I., Schrader, J., Holtmanm, D., et al. (2018). Synthesis of (-)-menthol Fatty Acid Esters in and from (-)-menthol and Fatty Acids – Novel Concept for Lipase Catalyzed Esterification Based on Eutectic Solvents. Mol. Catal. 458, 67–72. doi:10.1016/j.mcat.2018.08.003
Jakoblinnert, A., Mladenov, R., Paul, A., Sibilla, F., Schwaneberg, U., Ansorge-Schumacher, M. B., et al. (2011). Asymmetric Reduction of Ketones with Recombinant E. coli Whole Cells in Neat Substrates. Chem. Commun. 47 (44), 12230–12232. doi:10.1039/c1cc14097c
Jakoblinnert, A., and Rother, D. (2014). A Two-step Biocatalytic cascade in Micro-aqueous Medium: Using Whole Cells to Obtain High Concentrations of a Vicinal Diol. Green. Chem. 16 (7), 3472–3482. doi:10.1039/c4gc00010b
Jessop, P. G. (2011). Searching for green Solvents. Green. Chem. 13 (6), 1391–1398. doi:10.1039/c0gc00797h
Kara, S., Spickermann, D., Schrittwieser, J. H., Weckbecker, A., Leggewie, C., Arends, I. W. C. E., et al. (2013). Access to Lactone Building Blocks via Horse Liver Alcohol Dehydrogenase-Catalyzed Oxidative Lactonization. ACS Catal. 3 (11), 2436–2439. doi:10.1021/cs400535c
Kara, S., Spickermann, D., Weckbecker, A., Leggewie, C., Arends, I. W. C. E., and Hollmann, F. (2014). Bioreductions Catalyzed by an Alcohol Dehydrogenase in Non-aqueous Media. ChemCatChem 6 (4), 973–976. doi:10.1002/cctc.201300841
Karande, R., Schmid, A., and Buehler, K. (2010). Enzyme Catalysis in an Aqueous/Organic Segment Flow Microreactor: Ways to Stabilize Enzyme Activity. Langmuir 26 (11), 9152–9159. doi:10.1021/la9048727
Karande, R., Schmid, A., and Buehler, K. (2011). Miniaturizing Biocatalysis: Enzyme-Catalyzed Reactions in an Aqueous/Organic Segmented Flow Capillary Microreactor. Adv. Synth. Catal. 353 (13), 2511–2521. doi:10.1002/adsc.201100394
Karmee, S. K., Roosen, C., Kohlmann, C., Lütz, S., Greiner, L., and Leitner, W. (2009). Chemo-enzymatic cascade Oxidation in Supercritical Carbon Dioxide/water Biphasic media. Green. Chem. 11 (7), 1052–1055. doi:10.1039/b820606f
Kaschner, M., Loeschcke, A., Krause, J., Minh, B. Q., Heck, A., Endres, S., et al. (2014). Discovery of the First Light-dependent Protochlorophyllide Oxidoreductase in Anoxygenic Phototrophic Bacteria. Mol. Microbiol. 93 (5), 1066–1078. doi:10.1111/mmi.12719
Kiss, A. A., Jobson, M., and Gao, X. (2019). Reactive Distillation: Stepping up to the Next Level of Process Intensification. Ind. Eng. Chem. Res. 58 (15), 5909–5918. doi:10.1021/acs.iecr.8b05450
Kochius, S., Magnusson, A. O., Hollmann, F., Schrader, J., and Holtmann, D. (2012). Immobilized Redox Mediators for Electrochemical NAD(P)+ Regeneration. Appl. Microbiol. Biotechnol. 93 (6), 2251–2264. doi:10.1007/s00253-012-3900-z
Kochius, S., Ni, Y., Kara, S., Gargiulo, S., Schrader, J., Holtmann, D., et al. (2014). Light-Accelerated Biocatalytic Oxidation Reactions. ChemPlusChem 79 (11), 1554–1557. doi:10.1002/cplu.201402152
Kochius, S., Paetzold, M., Scholz, A., Merkens, H., Vogel, A., Ansorge-Schumacher, M., et al. (2014). Enantioselective Enzymatic Synthesis of the α-hydroxy Ketone (R)-acetoin from Meso-2,3-Butanediol. J. Mol. Catal. B: Enzymatic 103, 61–66. doi:10.1016/j.molcatb.2013.08.016
Kochius, S., Park, J. B., Ley, C., Könst, P., Hollmann, F., Schrader, J., et al. (2014). Electrochemical Regeneration of Oxidised Nicotinamide Cofactors in a Scalable Reactor. J. Mol. Catal. B: Enzymatic 103, 94–99. doi:10.1016/j.molcatb.2013.07.006
Köninger, K., Gómez Baraibar, Á., Mügge, C., Paul, C. E., Hollmann, F., Hollmann, F., et al. (2016). Recombinant Cyanobacteria for the Asymmetric Reduction of C=C Bonds Fueled by the Biocatalytic Oxidation of Water. Angew. Chem. Int. Ed. Engl. 55 (18), 5582–5585. doi:10.1002/anie.201601200
Krieg, T., Hüttmann, S., Mangold, K.-M., Schrader, J., and Holtmann, D. (2011). Gas Diffusion Electrode as Novel Reaction System for an Electro-Enzymatic Process with Chloroperoxidase. Green. Chem. 13 (10), 2686–2689. doi:10.1039/c1gc15391a
Kühn, S., Sluyter, G., Heils, R., Stobener, A., Kleber, J., Liese, A., et al. (2017). In Situ Separation of the Chiral Target Compound (S)-2-Pentanol in Biocatalytic Reactive Distillation. Ind. Eng. Chem. Res. 56 (22), 6451–6461. doi:10.1021/acs.iecr.7b01163
Kuk, S. K., Gopinath, K., Singh, R. K., Kim, T.-D., Lee, Y., Choi, W. S., et al. (2019). NADH-free Electroenzymatic Reduction of CO2 by Conductive Hydrogel-Conjugated Formate Dehydrogenase. ACS Catal. 9 (6), 5584–5589. doi:10.1021/acscatal.9b00127
Kuk, S. K., Singh, R. K., Nam, D. H., Singh, R., Lee, J.-K., and Park, C. B. (2017). Photoelectrochemical Reduction of Carbon Dioxide to Methanol through a Highly Efficient Enzyme Cascade. Angew. Chem. Int. Ed. 56 (14), 3827–3832. doi:10.1002/anie.201611379
Lakavath, B., Hedison, T. M., Heyes, D. J., Shanmugam, M., Sakuma, M., Hoeven, R., et al. (2020). Radical-based Photoinactivation of Fatty Acid Photodecarboxylases. Anal. Biochem. 600, 113749. doi:10.1016/j.ab.2020.113749
Lee, H. Y., Ryu, J., Kim, J. H., Lee, S. H., and Park, C. B. (2012). Biocatalyzed Artificial Photosynthesis by Hydrogen-Terminated Silicon Nanowires. ChemSusChem 5 (11), 2129–2132. doi:10.1002/cssc.201200251
Lee, S. H., Choi, D. S., Kuk, S. K., and Park, C. B. (2018). Photobiocatalysis: Activating Redox Enzymes by Direct or Indirect Transfer of Photoinduced Electrons. Angew. Chem. Int. Ed. 57 (27), 7958–7985. doi:10.1002/anie.201710070
Lee, S. H., Choi, D. S., Pesic, M., Lee, Y. W., Paul, C. E., Hollmann, F., et al. (2017). Cofactor-Free, Direct Photoactivation of Enoate Reductases for the Asymmetric Reduction of C=C Bonds. Angew. Chem. Int. Ed. 56 (30), 8681–8685. doi:10.1002/anie.201702461
Lee, S. H., Kim, J. H., and Park, C. B. (2013). Coupling Photocatalysis and Redox Biocatalysis toward Biocatalyzed Artificial Photosynthesis. Chem. Eur. J. 19 (14), 4392–4406. doi:10.1002/chem.201204385
Lee, S. H., Nam, D. H., Kim, J. H., Baeg, J.-O., and Park, C. B. (2009). Eosin Y-Sensitized Artificial Photosynthesis by Highly Efficient Visible-Light-Driven Regeneration of Nicotinamide Cofactor. Chembiochem 10 (10), 1621–1624. doi:10.1002/cbic.200900156
Lee, S. H., Nam, D. H., and Park, C. B. (2009). Screening Xanthene Dyes for Visible Light-Driven Nicotinamide Adenine Dinucleotide Regeneration and Photoenzymatic Synthesis. Adv. Synth. Catal. 351 (16), 2589–2594. doi:10.1002/adsc.200900547
Leemans Martin, L., Peschke, T., Venturoni, F., and Mostarda, S. (2020). Pharmaceutical Industry Perspectives on Flow Chemocatalysis and Biocatalysis. Curr. Opin. Green Sustainable Chem. 25, 100350. doi:10.1016/j.cogsc.2020.04.011
Ley, C., Schewe, H., Ströhle, F. W., Ruff, A. J., Schwaneberg, U., Schrader, J., et al. (2013). Coupling of Electrochemical and Optical Measurements in a Microtiter Plate for the Fast Development of Electro Enzymatic Processes with P450s. J. Mol. Catal. B: Enzymatic 92, 71–78. doi:10.1016/j.molcatb.2013.03.019
Ley, C., Zengin Çekiç, S., Kochius, S., Mangold, K.-M., Schwaneberg, U., Schrader, J., et al. (2013). An Electrochemical Microtiter Plate for Parallel Spectroelectrochemical Measurements. Electrochimica Acta 89, 98–105. doi:10.1016/j.electacta.2012.10.151
Liese, A., and Hilterhaus, L. (2013). Evaluation of Immobilized Enzymes for Industrial Applications. Chem. Soc. Rev. 42 (15), 6236–6249. doi:10.1039/c3cs35511j
Lin, S.-S., Wu, C.-H., Sun, M.-C., Sun, C.-M., and Ho, Y.-P. (2005). Microwave-assisted Enzyme-Catalyzed Reactions in Various Solvent Systems. J. Am. Soc. Mass. Spectrom. 16 (4), 581–588. doi:10.1016/j.jasms.2005.01.012
Lindeque, R., and Woodley, J. (2019). Reactor Selection for Effective Continuous Biocatalytic Production of Pharmaceuticals. Catalysts 9 (3), 262. doi:10.3390/catal9030262
Lorente-Arevalo, A., Ladero, M., and Bolivar, J. M. (2021). Intensification of Oxygen-dependent Biotransformations Catalyzed by Immobilized Enzymes. Curr. Opin. Green Sustainable Chem. 32, 100544. doi:10.1016/j.cogsc.2021.100544
Lubitz, W., Ogata, H., Rüdiger, O., and Reijerse, E. (2014). Hydrogenases. Chem. Rev. 114 (8), 4081–4148. doi:10.1021/cr4005814
Lütz, S., Vuorilehto, K., and Liese, A. (2007). Process Development for the Electroenzymatic Synthesis of (R)-methylphenylsulfoxide by Use of a 3-dimensional Electrode. Biotechnol. Bioeng. 98 (3), 525–534.
Lye, G. J., and Woodley, J. M. (1999). Application of In Situ Product-Removal Techniques to Biocatalytic Processes. Trends Biotechnol. 17 (10), 395–402. doi:10.1016/s0167-7799(99)01351-7
Ma, Y., Zhang, X., Li, Y., Li, P., Hollmann, F., and Wang, Y. (2020). Production of Fatty Alcohols from Non-edible Oils by Enzymatic cascade Reactions. Sustainable Energ. Fuels 4 (8), 4232–4237. doi:10.1039/d0se00848f
Ma, Y., Zhang, X., Zhang, W., Li, P., Li, Y., Hollmann, F., et al. (2020). Photoenzymatic Production of Next Generation Biofuels from Natural Triglycerides Combining a Hydrolase and a Photodecarboxylase. ChemPhotoChem 4 (1), 39–44. doi:10.1002/cptc.201900205
Maciá-Agulló, J. A., Corma, A., and Garcia, H. (2015). Photobiocatalysis: The Power of Combining Photocatalysis and Enzymes. Chem. – A Eur. J. 21 (31), 10940–10959.
Marques, M. P. C., and Fernandes, P. (2011). Microfluidic Devices: Useful Tools for Bioprocess Intensification. Molecules 16 (10), 8368–8401. doi:10.3390/molecules16108368
Mersch, D., Lee, C.-Y., Zhang, J. Z., Brinkert, K., Fontecilla-Camps, J. C., Rutherford, A. W., et al. (2015). Wiring of Photosystem II to Hydrogenase for Photoelectrochemical Water Splitting. J. Am. Chem. Soc. 137 (26), 8541–8549. doi:10.1021/jacs.5b03737
Meyer, L.-E., Eser, B. E., and Kara, S. (2021). Coupling Light with Biocatalysis for Sustainable Synthesis-Very Recent Developments and Future Perspectives. Curr. Opin. Green Sustainable Chem. 31, 100496. doi:10.1016/j.cogsc.2021.100496
Mifsud, M., Gargiulo, S., Iborra, S., Arends, I. W. C. E., Hollmann, F., and Corma, A. (2014). Photobiocatalytic Chemistry of Oxidoreductases Using Water as the Electron Donor. Nat. Commun. 5 (1), 3145. doi:10.1038/ncomms4145
Milker, S., Pätzold, M., Bloh, J. Z., and Holtmann, D. (2019). Comparison of Deep Eutectic Solvents and Solvent-free Reaction Conditions for Aldol Production. Mol. Catal. 466, 70–74. doi:10.1016/j.mcat.2019.01.012
Ni, Y., Hagedoorn, P.-L., Xu, J.-H., Arends, I. W. C. E., and Hollmann, F. (2012). A Biocatalytic Hydrogenation of Carboxylic Acids. Chem. Commun. 48 (99), 12056–12058. doi:10.1039/c2cc36479d
Ni, Y., Holtmann, D., and Hollmann, F. (2014). How Green Is Biocatalysis? to Calculate Is to Know. ChemCatChem 6 (4), 930–943. doi:10.1002/cctc.201300976
Noorman, H. J., van Winden, W., Heijnen, J. J., and van der Lans, R. G. J. M. (2018). “CHAPTER 1. Intensified Fermentation Processes and Equipment,” in Intensification of Biobased Processes (Cambridge and London, UK: The Royal Society of Chemistry), 1–41. doi:10.1039/9781788010320-00001
Obert, R., and Dave, B. C. (1999). Enzymatic Conversion of Carbon Dioxide to Methanol: Enhanced Methanol Production in Silica Sol−Gel Matrices. J. Am. Chem. Soc. 121 (51), 12192–12193. doi:10.1021/ja991899r
Özgen, F. F., Runda, M. E., and Schmidt, S. (2021). Photo-biocatalytic Cascades: Combining Chemical and Enzymatic Transformations Fueled by Light. ChemBioChem 22 (5), 790–806.
Paravidino, M., Sorgedrager, M. J., Orru, R. V. A., and Hanefeld, U. (2010). Activity and Enantioselectivity of the Hydroxynitrile Lyase MeHNL in Dry Organic Solvents. Chem. Eur. J. 16 (25), 7596–7604. doi:10.1002/chem.201000487
Park, J.-B., Bühler, B., Panke, S., Witholt, B., and Schmid, A. (2007). Carbon Metabolism and Product Inhibition Determine the Epoxidation Efficiency of Solvent-tolerantPseudomonas Sp. Strain VLB120ΔC. Biotechnol. Bioeng. 98 (6), 1219–1229. doi:10.1002/bit.21496
Park, J. H., Lee, S. H., Cha, G. S., Choi, D. S., Nam, D. H., Lee, J. H., et al. (2015). Cofactor-free Light-Driven Whole-Cell Cytochrome P450 Catalysis. Angew. Chem. Int. Ed. 54 (3), 969–973. doi:10.1002/anie.201410059
Pätzold, M., Burek, B. O., Liese, A., Bloh, J. Z., and Holtmann, D. (2019). Product Recovery of an Enzymatically Synthesized (−)-menthol Ester in a Deep Eutectic Solvent. Bioproc. Biosyst. Eng. 42 (8), 1385–1389. doi:10.1007/s00449-019-02125-6
Pätzold, M., Liese, A., Holtmanm, D., and Weimer, A. (2019). Optimization of Solvent-free Enzymatic Esterification in Eutectic Substrate Reaction Mixture. Biotechnol. Rep. 22, e00333. doi:10.1016/j.btre.2019.e00333
Pätzold, M., Siebenhaller, S., Kara, S., Liese, A., Syldatk, C., and Holtmann, D. (2019). Deep Eutectic Solvents as Efficient Solvents in Biocatalysis. Trends Biotechnol. 37 (9), 943–959. doi:10.1016/j.tibtech.2019.03.007
Perz, F., Bormann, S., Ulber, R., Alcalde, M., Bubenheim, P., Hollmann, F., et al. (2020). Enzymatic Oxidation of Butane to 2‐Butanol in a Bubble Column. ChemCatChem 12 (14), 3666–3669. doi:10.1002/cctc.202000431
Pfruender, H., Amidjojo, M., Kragl, U., and Weuster-Botz, D. (2004). Efficient Whole-Cell Biotransformation in a Biphasic Ionic Liquid/Water System. Angew. Chem. Int. Ed. 43 (34), 4529–4531. doi:10.1002/anie.200460241
Porcelli, M., Cacciapuoti, G., Fusco, S., Massa, R., d'Ambrosio, G., Bertoldo, C., et al. (1997). Non-thermal Effects of Microwaves on Proteins: Thermophilic Enzymes as Model System. FEBS Lett. 402 (2), 102–106. doi:10.1016/s0014-5793(96)01505-0
Portha, J.-F., Falk, L., and Commenge, J.-M. (2014). Local and Global Process Intensification. Chem. Eng. Process. Process Intensification 84, 1–13. doi:10.1016/j.cep.2014.05.002
Potdar, M., Kelso, G., Schwarz, L., Zhang, C., and Hearn, M. (2015). Recent Developments in Chemical Synthesis with Biocatalysts in Ionic Liquids. Molecules 20 (9), 16788–16816. doi:10.3390/molecules200916788
Prieto, C. A., Guadix, A., González-Tello, P., and Guadix, E. M. (2007). A Cyclic Batch Membrane Reactor for the Hydrolysis of Whey Protein. J. Food Eng. 78 (1), 257–265. doi:10.1016/j.jfoodeng.2005.09.024
Prieto, C. A., Guadix, E. M., and Guadix, A. (2008). Influence of Temperature on Protein Hydrolysis in a Cyclic Batch Enzyme Membrane Reactor. Biochem. Eng. J. 42 (3), 217–223. doi:10.1016/j.bej.2008.06.018
Ranganathan, S., Gärtner, T., Wiemann, L. O., and Sieber, V. (2015). A One Pot Reaction cascade of In Situ Hydrogen Peroxide Production and Lipase Mediated In Situ Production of Peracids for the Epoxidation of Monoterpenes. J. Mol. Catal. B: Enzymatic 114, 72–76. doi:10.1016/j.molcatb.2014.12.008
Rauch, M. C. R., Tieves, F., Paul, C. E., Arends, I. W. C. E., Alcalde, M., and Hollmann, F. (2019). Peroxygenase‐Catalysed Epoxidation of Styrene Derivatives in Neat Reaction Media. ChemCatChem 11 (18), 4519–4523. doi:10.1002/cctc.201901142
Rauch, M., Schmidt, S., Arends, I. W. C. E., Oppelt, K., Kara, S., and Hollmann, F. (2017). Photobiocatalytic Alcohol Oxidation Using LED Light Sources. Green. Chem. 19 (2), 376–379. doi:10.1039/c6gc02008a
Rejasse, B., Lamare, S., Legoy, M. D., and Besson, T. (2007). Influence of Microwave Irradiation on Enzymatic Properties: Applications in Enzyme Chemistry. J. Enzyme Inhib. Med. Chem. 22 (5), 518–526. doi:10.1080/14756360701424959
Rios, G. M., Belleville, M. P., Paolucci, D., and Sanchez, J. (2004). Progress in Enzymatic Membrane Reactors – a Review. J. Membr. Sci. 242 (1), 189–196. doi:10.1016/j.memsci.2003.06.004
Rodríguez-Hinestroza, R. A., Rossmery, A., and Carmen, L. (2017). HLADH-catalyzed Synthesis of β-amino Acids, Assisted by Continuous Electrochemical Regeneration of NAD+ in a Filter Press Microreactor. Chem. Eng. Sci. 158, 196–207.
Rudroff, F., Mihovilovic, M. D., Gröger, H., Snajdrova, R., Iding, H., and Bornscheuer, U. T. (2018). Opportunities and Challenges for Combining Chemo- and Biocatalysis. Nat. Catal. 1 (1), 12–22. doi:10.1038/s41929-017-0010-4
Ruppert, R., and Steckhan, E. (1989). Efficient Photoelectrochemical In-Situ Regeneration of NAD(P)+ Coupled to Enzymatic Oxidation of Alcohols. J. Chem. Soc. Perkin Trans. 2 2 (7), 811–814. doi:10.1039/p29890000811
Sancar, A. (2008). Structure and Function of Photolyase and In Vivo Enzymology: 50th Anniversary. J. Biol. Chem. 283 (47), 32153–32157. doi:10.1074/jbc.r800052200
Sandoval, B. A., Kurtoic, S. I., Chung, M. M., Biegasiewicz, K. F., and Hyster, T. K. (2019). Photoenzymatic Catalysis Enables Radical‐Mediated Ketone Reduction in Ene‐Reductases. Angew. Chem. Int. Ed. 58 (26), 8714–8718. doi:10.1002/anie.201902005
Satyawali, Y., Vanbroekhoven, K., and Dejonghe, W. (2017). Process Intensification: The Future for Enzymatic Processes? Biochem. Eng. J. 121, 196–223. doi:10.1016/j.bej.2017.01.016
Schlager, S., Dumitru, L. M., Haberbauer, M., Fuchsbauer, A., Neugebauer, H., Hiemetsberger, D., et al. (2016). Electrochemical Reduction of Carbon Dioxide to Methanol by Direct Injection of Electrons into Immobilized Enzymes on a Modified Electrode. ChemSusChem 9 (6), 631–635. doi:10.1002/cssc.201501496
Schmermund, L., Jurkaš, V., Özgen, F. F., Barone, G. D., Büchsenschütz, H. C., Winkler, C. K., et al. (2019). Photo-Biocatalysis: Biotransformations in the Presence of Light. ACS Catal. 9 (5), 4115–4144. doi:10.1021/acscatal.9b00656
Schmidt, S., Pedroso de Almeida, T., Rother, D., and Hollmann, F. (2017). Towards Environmentally Acceptable Synthesis of Chiral α-hydroxy Ketones via Oxidase-Lyase Cascades. Green. Chem. 19 (5), 1226–1229. doi:10.1039/c7gc00020k
Scholz, K. E., Okrob, D., Kopka, B., Grünberger, A., Pohl, M., Jaeger, K.-E., et al. (2012). Synthesis of Chiral Cyanohydrins by Recombinant Escherichia coli Cells in a Micro-aqueous Reaction System. Appl. Environ. Microbiol. 78 (14), 5025–5027. doi:10.1128/aem.00582-12
Seel, C. J., Králík, A., Hacker, M., Frank, A., König, B., and Gulder, T. (2018). Atom-Economic Electron Donors for Photobiocatalytic Halogenations. ChemCatChem 10 (18), 3960–3963. doi:10.1002/cctc.201800886
Sengupta, A., Pakrasi, H. B., and Wangikar, P. P. (2018). Recent Advances in Synthetic Biology of Cyanobacteria. Appl. Microbiol. Biotechnol. 102 (13), 5457–5471. doi:10.1007/s00253-018-9046-x
Sheldon, R. A. (2005). Green Solvents for Sustainable Organic Synthesis: State of the Art. Green. Chem. 7 (5), 267–278. doi:10.1039/b418069k
Siebenhaller, S., Kirchhoff, J., Kirschhöfer, F., Brenner-Weiß, G., Muhle-Goll, C., Luy, B., et al. (2018). Integrated Process for the Enzymatic Production of Fatty Acid Sugar Esters Completely Based on Lignocellulosic Substrates. Front. Chem. 6 (421), 421. doi:10.3389/fchem.2018.00421
Siebenhaller, S., Hajek, T., Muhle-Goll, C., Himmelsbach, M., Luy, B., Kirschhöfer, F., et al. (2017). Beechwood Carbohydrates for Enzymatic Synthesis of Sustainable Glycolipids. Bioresour. Bioproc. 4 (1), 25. doi:10.1186/s40643-017-0155-7
Sokol, K. P., Robinson, W. E., Warnan, J., Kornienko, N., Nowaczyk, M. M., Ruff, A., et al. (2018). Bias-free Photoelectrochemical Water Splitting with Photosystem II on a Dye-Sensitized Photoanode Wired to Hydrogenase. Nat. Energ. 3 (11), 944–951. doi:10.1038/s41560-018-0232-y
Sorigué, D., Légeret, B., Cuiné, S., Blangy, S., Moulin, S., Billon, E., et al. (2017). An Algal Photoenzyme Converts Fatty Acids to Hydrocarbons. Science 357 (6354), 903–907. doi:10.1126/science.aan6349
Spille, C., Lyberis, A., Maiwald, M. I., Herzog, D., Hoffmann, M., Emmelmann, C., et al. (2020). SMART‐Reactors: Tailoring Gas Holdup Distribution by Additively Manufactured Lattice Structures. Chem. Eng. Technol. 43 (10), 2053–2061. doi:10.1002/ceat.202000211
Stankiewicz, A. (2006). Energy Matters. Chem. Eng. Res. Des. 84 (7), 511–521. doi:10.1205/cherd.05214
Stankiewicz, A. I., and Moulijn, J. A. (2000). Process Intensification:Transforming Chemical Engineering. Chem. Eng. Prog. 96 (1), 22–33.
Strniša, F., Urbic, T., Žnidaršič-Plazl, P., and Plaz, I. (2019). “Process Intensification and Miniaturization of Chemical and Biochemical Processes,” in Computer Aided Chemical Engineering. Editor A. A. Kiss (Elsevier), 1801–1806. doi:10.1016/B978-0-12-818634-3.50301-5
Strube, J., Ditz, R., Kornecki, M., Huter, M., Schmidt, A., Thiess, H., et al. (2018). Process Intensification in Biologics Manufacturing. Chem. Eng. Process. - Process Intensification 133, 278–293. doi:10.1016/j.cep.2018.09.022
Szczesny, J., Ruff, A., Oliveira, A. R., Pita, M., Pereira, I. A. C., De Lacey, A. L., et al. (2020). Electroenzymatic CO2 Fixation Using Redox Polymer/Enzyme-Modified Gas Diffusion Electrodes. ACS Energ. Lett. 5 (1), 321–327. doi:10.1021/acsenergylett.9b02436
Tamborini, L., Fernandes, P., Paradisi, F., and Molinari, F. (2018). Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 36 (1), 73–88. doi:10.1016/j.tibtech.2017.09.005
Thuy Pham, T. P., Cho, C.-W., and Yun, Y.-S. (2010). Environmental Fate and Toxicity of Ionic Liquids: A Review. Water Res. 44 (2), 352–372. doi:10.1016/j.watres.2009.09.030
Tosstorff, A., Dennig, A., Ruff, A. J., Schwaneberg, U., Sieber, V., Mangold, K.-M., et al. (2014). Mediated Electron Transfer with Monooxygenases-Insight in Interactions between Reduced Mediators and the Co-substrate Oxygen. J. Mol. Catal. B: Enzymatic 108, 51–58. doi:10.1016/j.molcatb.2014.06.011
Tosstorff, A., Kroner, C., Opperman, D. J., Hollmann, F., and Holtmann, D. (2017). Towards Electroenzymatic Processes Involving Old Yellow Enzymes and Mediated Cofactor Regeneration. Eng. Life Sci. 17 (1), 71–76. doi:10.1002/elsc.201600158
Trivedi, A. H., Spiess, A. C., Daussmann, T., and Büchs, J. (2006). Effect of Additives on Gas-phase Catalysis with Immobilised Thermoanaerobacter Species Alcohol Dehydrogenase (ADH T). Appl. Microbiol. Biotechnol. 71 (4), 407–414. doi:10.1007/s00253-005-0169-5
Trivedi, A. H., Spiess, A. C., Daussmann, T., and Buchs, J. (2006). Study on Mesophilic and Thermophilic Alcohol Dehydrogenases in Gas-phase Reaction. Biotechnol. Prog. 22 (2), 454–458. doi:10.1021/bp050316g
Tufvesson, P., Fu, W., Jensen, J. S., and Woodley, J. M. (2010). Process Considerations for the Scale-Up and Implementation of Biocatalysis. Food Bioproducts Process. 88 (1), 3–11. doi:10.1016/j.fbp.2010.01.003
Tufvesson, P., Lima-Ramos, J., Nordblad, M., and Woodley, J. M. (2011). Guidelines and Cost Analysis for Catalyst Production in Biocatalytic Processes. Org. Process. Res. Dev. 15 (1), 266–274. doi:10.1021/op1002165
Uthoff, F., and Gröger, H. (2018). Asymmetric Synthesis of 1-Phenylethylamine from Styrene via Combined Wacker Oxidation and Enzymatic Reductive Amination. J. Org. Chem. 83 (16), 9517–9521. doi:10.1021/acs.joc.8b01247
van der Wielen, L. A. M., Mussatto, S. I., and van Breugel, J. (2021). Bioprocess Intensification: Cases that (Don't) Work. New Biotechnol. 61, 108–115. doi:10.1016/j.nbt.2020.11.007
Van Gerven, T., and Stankiewicz, A. (2009). Structure, Energy, Synergy, Time-The Fundamentals of Process Intensification. Ind. Eng. Chem. Res. 48 (5), 2465–2474. doi:10.1021/ie801501y
van Schie, M. M. C. H., Zhang, W., Tieves, F., Choi, D. S., Park, C. B., Burek, B. O., et al. (2019). Cascading G-C3n4 and Peroxygenases for Selective Oxyfunctionalization Reactions. ACS Catal. 9 (8), 7409–7417. doi:10.1021/acscatal.9b01341
Varničić, M. (2020). Selectivity and Sustainability of Electroenzymatic Process for Glucose Conversion to Gluconic Acid. Catalysts 10 (3).
Varničić, M., Vidaković-Koch, T., and Sundmacher, K. (2015). Gluconic Acid Synthesis in an Electroenzymatic Reactor. Electrochimica Acta 174, 480–487.
Wachtmeister, J., Jakoblinnert, A., and Rother, D. (2016). Stereoselective Two-step Biocatalysis in Organic Solvent: Toward All Stereoisomers of a 1,2-Diol at High Product Concentrations. Org. Process. Res. Dev. 20 (10), 1744–1753. doi:10.1021/acs.oprd.6b00232
Wohlgemuth, R. (2010). Biocatalysis-key to Sustainable Industrial Chemistry. Curr. Opin. Biotechnol. 21 (6), 713–724. doi:10.1016/j.copbio.2010.09.016
Wohlgemuth, R., Plazl, I., Žnidaršič-Plazl, P., Gernaey, K. V., and Woodley, J. M. (2015). Microscale Technology and Biocatalytic Processes: Opportunities and Challenges for Synthesis. Trends Biotechnol. 33 (5), 302–314. doi:10.1016/j.tibtech.2015.02.010
Woodley, J. M. (2017). Bioprocess Intensification for the Effective Production of Chemical Products. Comput. Chem. Eng. 105, 297–307. doi:10.1016/j.compchemeng.2017.01.015
Woodley, J. M., Bisschops, M., Straathof, A. J. J., and Ottens, M. (2008). Future Directions Forin-Situ Product Removal (ISPR). J. Chem. Technol. Biotechnol. 83 (2), 121–123. doi:10.1002/jctb.1790
Woodley, J. M. (2020). New Frontiers in Biocatalysis for Sustainable Synthesis. Curr. Opin. Green Sustainable Chem. 21, 22–26. doi:10.1016/j.cogsc.2019.08.006
Woodley, J. M. (2008). New Opportunities for Biocatalysis: Making Pharmaceutical Processes Greener. Trends Biotechnol. 26 (6), 321–327. doi:10.1016/j.tibtech.2008.03.004
Wu, H., Tian, C., Song, X., Liu, C., Yang, D., and Jiang, Z. (2013). Methods for the Regeneration of Nicotinamide Coenzymes. Green. Chem. 15 (7), 1773–1789. doi:10.1039/c3gc37129h
Xu, J., Hu, Y., Fan, J., Arkin, M., Li, D., Peng, Y., et al. (2019). Light‐Driven Kinetic Resolution of α‐Functionalized Carboxylic Acids Enabled by an Engineered Fatty Acid Photodecarboxylase. Angew. Chem. Int. Ed. 58 (25), 8474–8478. doi:10.1002/anie.201903165
Xu, P., Zheng, G.-W., Du, P.-X., Zong, M.-H., and Lou, W.-Y. (2016). Whole-Cell Biocatalytic Processes with Ionic Liquids. ACS Sustainable Chem. Eng. 4 (2), 371–386. doi:10.1021/acssuschemeng.5b00965
Yadav, R. K., Baeg, J.-O., Kumar, A., Kong, K.-j., Oh, G. H., and Park, N.-J. (2014). Graphene-BODIPY as a Photocatalyst in the Photocatalytic-Biocatalytic Coupled System for Solar Fuel Production from CO2. J. Mater. Chem. A. 2 (14), 5068–5076. doi:10.1039/c3ta14442a
Yadav, R. K., Baeg, J.-O., Oh, G. H., Park, N.-J., Kong, K.-j., Kim, J., et al. (2012). A Photocatalyst-Enzyme Coupled Artificial Photosynthesis System for Solar Energy in Production of Formic Acid from CO2. J. Am. Chem. Soc. 134 (28), 11455–11461. doi:10.1021/ja3009902
Yadav, R. K., Oh, G. H., Park, N.-J., Kumar, A., Kong, K.-j., and Baeg, J.-O. (2014). Highly Selective Solar-Driven Methanol from CO2 by a Photocatalyst/Biocatalyst Integrated System. J. Am. Chem. Soc. 136 (48), 16728–16731. doi:10.1021/ja509650r
Yayci, A., Baraibar, Á. G., Krewing, M., Fueyo, E. F., Hollmann, F., Alcalde, M., et al. (2020). Plasma‐Driven In Situ Production of Hydrogen Peroxide for Biocatalysis. ChemSusChem 13 (8), 2072–2079. doi:10.1002/cssc.201903438
Yayci, A., Dirks, T., Kogelheide, F., Alcalde, M., Hollmann, F., Awakowicz, P., et al. (2020). Microscale Atmospheric Pressure Plasma Jet as a Source for Plasma‐Driven Biocatalysis. ChemCatChem 12 (23), 5893–5897. doi:10.1002/cctc.202001225
Yoon, J., Kim, J., Tieves, F., Zhang, W., Alcalde, M., Hollmann, F., et al. (2020). Piezobiocatalysis: Ultrasound-Driven Enzymatic Oxyfunctionalization of C-H Bonds. ACS Catal. 10 (9), 5236–5242. doi:10.1021/acscatal.0c00188
Young, D. D., Nichols, J., Kelly, R. M., and Deiters, A. (2008). Microwave Activation of Enzymatic Catalysis. J. Am. Chem. Soc. 130 (31), 10048–10049. doi:10.1021/ja802404g
Yuan, B., Mahor, D., Fei, Q., Wever, R., Alcalde, M., Zhang, W., et al. (2020). Water-Soluble Anthraquinone Photocatalysts Enable Methanol-Driven Enzymatic Halogenation and Hydroxylation Reactions. ACS Catal. 10 (15), 8277–8284. doi:10.1021/acscatal.0c01958
Yuan, M., Abdellaoui, S., Chen, H., Kummer, M. J., Malapit, C. A., You, C., et al. (2020). Selective Electroenzymatic Oxyfunctionalization by Alkane Monooxygenase in a Biofuel Cell. Angew. Chem. Int. Ed. 59 (23), 8969–8973. doi:10.1002/anie.202003032
Yuan, M., Sahin, S., Cai, R., Abdellaoui, S., Hickey, D. P., Minteer, S. D., et al. (2018). Creating a Low‐Potential Redox Polymer for Efficient Electroenzymatic CO 2 Reduction. Angew. Chem. Int. Ed. 57 (22), 6582–6586. doi:10.1002/anie.201803397
Zaks, A., and Klibanov, A. M. (1984). Enzymatic Catalysis in Organic Media at 100°C. Science 224 (4654), 1249–1251. doi:10.1126/science.6729453
Zaks, A., and Klibanov, A. M. (1985). Enzyme-catalyzed Processes in Organic Solvents. Proc. Natl. Acad. Sci. 82 (10), 3192–3196. doi:10.1073/pnas.82.10.3192
Zhang, W., Burek, B. O., Fernández-Fueyo, E., Alcalde, M., Bloh, J. Z., and Hollmann, F. (2017). Selective Activation of C−H Bonds in a Cascade Process Combining Photochemistry and Biocatalysis. Angew. Chem. Int. Ed. 56 (48), 15451–15455. doi:10.1002/anie.201708668
Zhang, W., Fernández-Fueyo, E., Ni, Y., van Schie, M., Gacs, J., Renirie, R., et al. (2018). Selective Aerobic Oxidation Reactions Using a Combination of Photocatalytic Water Oxidation and Enzymatic Oxyfunctionalizations. Nat. Catal. 1 (1), 55–62. doi:10.1038/s41929-017-0001-5
Zhang, W., Lee, J.-H., Younes, S. H. H., Tonin, F., Hagedoorn, P.-L., Pichler, H., et al. (2020). Photobiocatalytic Synthesis of Chiral Secondary Fatty Alcohols from Renewable Unsaturated Fatty Acids. Nat. Commun. 11 (1), 2258. doi:10.1038/s41467-020-16099-7
Zhang, W., Liu, H., van Schie, M. M. C. H., Hagedoorn, P.-L., Alcalde, M., Denkova, A. G., et al. (2020). Nuclear Waste and Biocatalysis: A Sustainable Liaison? ACS Catal. 10 (23), 14195–14200. doi:10.1021/acscatal.0c03059
Zhang, W., Ma, M., Huijbers, M. M. E., Filonenko, G. A., Pidko, E. A., van Schie, M., et al. (2019). Hydrocarbon Synthesis via Photoenzymatic Decarboxylation of Carboxylic Acids. J. Am. Chem. Soc. 141 (7), 3116–3120. doi:10.1021/jacs.8b12282
Zhang, Z., Li, J., Ji, M., Liu, Y., Wang, N., Zhang, X., et al. (2021). Encapsulation of Multiple Enzymes in a Metal-Organic Framework with Enhanced Electro-Enzymatic Reduction of CO2 to Methanol. Green. Chem. 23 (6), 2362–2371. doi:10.1039/d1gc00241d
Zhang, Z., Vasiliu, T., Li, F., Laaksonen, A., Mocci, F., and Ji, X. (2021). Electrochemically Driven Efficient Enzymatic Conversion of CO2 to Formic Acid with Artificial Cofactors. J. CO2 Utilization 52, 101679. doi:10.1016/j.jcou.2021.101679
Žnidaršič-Plazl, P. (2019). The Promises and the Challenges of Biotransformations in Microflow. Biotechnol. J. 14 (8), e1800580. doi:10.1002/biot.201800580
Žnidaršič-Plazl, P. (2021). Biocatalytic Process Intensification via Efficient Biocatalyst Immobilization, Miniaturization, and Process Integration. Curr. Opin. Green Sustainable Chem. 32, 100546.
Keywords: biocatalysis, process intensification, energy input in biocatalysis, reactor design, solvent, electrobiocatalysis
Citation: Burek BO, Dawood AWH, Hollmann F, Liese A and Holtmann D (2022) Process Intensification as Game Changer in Enzyme Catalysis. Front. Catal. 2:858706. doi: 10.3389/fctls.2022.858706
Received: 20 January 2022; Accepted: 01 March 2022;
Published: 21 March 2022.
Edited by:
Fernando López Gallego, CIC biomaGUNE, SpainReviewed by:
Juan M. Bolivar, Complutense University of Madrid, SpainCopyright © 2022 Burek, Dawood, Hollmann, Liese and Holtmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank Hollmann, Zi5ob2xsbWFubkB0dWRlbGZ0Lm5s; Dirk Holtmann, ZGlyay5ob2x0bWFubkBsc2UudGhtLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.