 Tim Gerlach1,2
Tim Gerlach1,2 Jendrik Schain1Simone Söltl1
Jendrik Schain1Simone Söltl1 Morten M. C. H. van Schie1
Morten M. C. H. van Schie1 Fabienne Hilgers3Nora L. Bitzenhofer3
Fabienne Hilgers3Nora L. Bitzenhofer3 Thomas Drepper3
Thomas Drepper3 Dörte Rother1,2*
Dörte Rother1,2*- 1Institute of Bio- and Geosciences: Biotechnology (IBG-1), Forschungszentrum Jülich GmbH, Jülich, Germany
- 2Department of Biology, Aachen Biology and Biotechnology, RWTH Aachen University, Aachen, Germany
- 3Department of Biology, Institute of Molecular Enzyme Technology, Heinrich-Heine University Düsseldorf, Düsseldorf, Germany
Genetically encoded photosensitizers are able to produce reactive oxygen species upon illumination and are exploited in a wide range of applications, especially in the medical field. In this work, we envisioned to further apply these genetically encoded photosensitizers for the light-dependent control of single enzymes in multi-step biocatalysis. One of the challenges in the application of several enzymes in a cascade is the unwanted cross-reactivity of these biocatalysts on reaction intermediates when all enzymes are simultaneously present in the reaction. As one strategy to address this issue, we investigated whether the introduction of genetically encoded photosensitizers as fusion tags would allow the selective inactivation of enzymes after successful transformation by simply turning on light. We tested five different photosensitizers as molecular biological fusion tags to inactivate the pyruvate decarboxylase variant E469G/W543H from Acetobacter pasteurianus. Dimeric photosensitizer tags, like the flavin-binding fluorescent proteins from Bacillus subtilis and Pseudomonas putida showed the tendency to form insoluble protein aggregates in combination with the tetrameric carboligase. Enzyme activity was, to some extent, retained in these aggregates, but the handling of the insoluble aggregates proved to be unfeasible. Monomeric photosensitizer tags appeared to be much more suitable when fused to the tetrameric enzyme. In the dark, the singlet oxygen photosensitizing protein (SOPP3)-tagged carboligase retained 79% of its activity as compared to the unfused enzyme. Upon blue light exposure, the SOPP3 tag showed the best specific inactivation and enabled complete inactivation of the carboligase within 30 min. SOPP3 is thus seen as a promising photosensitizer tag to be applied in future multi-step enzyme cascades to overcome the challenge of cross-reactivity.
Introduction
Photosensitizers have the unique ability to locally produce short-lived reactive oxygen species (ROS) when irradiated with light of a specific wavelength (Jacobson et al., 2008). When a photosensitizer is bound to a particular ligand or antibody, the highly reactive ROS enable targeted inactivation of proximate biological structures as proteins, lipids, DNA and carbohydrates in a process reported as chromophore-assisted light inactivation (CALI) (Liao et al., 1994). Upon excitation of a photosensitizer in the presence of molecular oxygen, ROS generation can occur at the functional chromophore via two types. In the type I mechanism, an electron is transferred to oxygen leading to the formation of the superoxide radical anion, which can further react to hydrogen peroxide (H2O2) and the hydroxyl radical. The type II pathway involves an energy-transfer to molecular oxygen resulting in the production of singlet oxygen (1O2) (Abrahamse and Hamblin, 2016). Because of its energetically unfavorable state, 1O2 is highly reactive resulting in a lifetime of ∼4 µs and a diffusion range of ∼200 nm (Moan, 1990; Skovsen et al., 2005; Ogilby, 2010). In contrast, hydrogen peroxide, shows a lifetime of ∼1 ms and thus can diffuse over distances up to 5 µm (Reth, 2002). Originally, dyes as malachite green (Jay, 1988), fluorescein (Surrey et al., 1998) or curcumin (Bernd, 2014) were used to perform CALI. In a more recent approach, genetically encoded photosensitizers are used for the light-induced inactivation of proteins or cells. These types of photosensitizers can be produced within a living system and do not have to be added exogenously (Bulina et al., 2006a). So far, CALI has been used in various applications as in photodynamic therapy (PDT) for the destruction of cancer cells or diseased tissue (Robertson et al., 2009), for the modulation of specific protein activities (Surrey et al., 1998) or the light-driven inactivation of microbial cells (Endres et al., 2018; Hilgers et al., 2019; Hally et al., 2020). By fusing photosensitizers to target proteins, the efficient application of CALI was demonstrated in diverse approaches in vivo and in vitro (Takemoto, 2021). Another promising application for CALI would be the area of multi-step biocatalysis. With multiple enzymes used in a cascade, the synthesis of complex molecules from simple and optimally renewable starting compounds, is possible (Lopez-Gallego and Schmidt-Dannert, 2010; Erdmann et al., 2017). As enzymes often show very high chemo- and stereoselectivity with regard to product formation, they are very atom efficient catalysts, especially for the formation of chiral pharmaceutical compounds and fine chemicals (Ricca et al., 2011). However, with respect to the substrate range, many enzymes do not only accept one substrate but a number of natural and synthetic substrates. This behavior, also called promiscuity, gives enzymes immense strength for synthetic purposes (Bornscheuer and Kazlauskas, 2004). At the same time, this promiscuity can be a challenge in syntheses with multiple enzymatic steps, as an enzyme might not only bind its desired substrate, but could also accept one of the intermediates or the final product when present at the same time in the same reaction vessel. Undesired side-activity decreases the atom efficiency and increases the complexity of the reaction (Hult and Berglund, 2007). To overcome the issue of cross-reactivity, the enzyme activity has to be regulated in space, by separating the enzymatic steps in compartments, or in time, by adding the enzymes sequentially and removing the respective enzyme after successful transformation, which can be challenging and cost-intensive (Erdmann et al., 2017; Gruber et al., 2017; Claaßen et al., 2019). CALI could represent a suitable tool to selectively turn off enzyme activity in multistep biocatalysis by light when appropriate genetically encoded photosensitizers are fused to the target enzymes. Through genetic fusion, the photosensitizers are located closest to the target enzyme. This allows the CALI principle to be carried out in the most efficient way, since the high phototoxicity of the short-ranged ROS formed, especially singlet oxygen, can be optimally utilized (Lin et al., 2013). Detailed characterization of ROS species formed can be found in the literature (Pletnev et al., 2009; Takemoto et al., 2013; Wingen et al., 2014; Westberg et al., 2015, 2017; Endres et al., 2018; Hilgers et al., 2019; Onukwufor et al., 2020). Light is an attractive external stimulus, as it provides an orthogonal, non-invasive regulation approach with high spatial and temporal resolution (Willner and Rubin, 1996; Cruz et al., 2000; Mayer and Heckel, 2006; Lu et al., 2014). With CALI, the enzymes of a multi-step cascade could be inactivated after successful transformation, thus, cross-reactivity could be avoided (Claaßen et al., 2019). A disadvantage is that enzyme inactivation by CALI is not reversible, decreasing the economic efficiency of this concept. ROS exposure causes the proteins to suffer oxidative damage which results in amino acid modification, cleavage of the polypeptide chain and conversion of the proteins to derivatives (Stadtman, 2006; Sharma et al., 2012; Vatansever et al., 2013). Here, we tested CALI on the pyruvate decarboxylase variant E469G/W543H from Acetobacter pasteurianus (ApPDC-EGWH). This carboligase variant is a (S)-selective enzyme with the ability to produce phenylacetylcarbinol (PAC) from the inexpensive substrates benzaldehyde and pyruvate (Sehl et al., 2017). PAC, in turn, is a versatile compound which serves as a building block for pharmaceutically active molecules as nor(pseudo)ephedrines (Sehl et al., 2013, 2014). The variant ApPDC-EGWH has demonstrated high conversion of 3-hydroxybenzaldehyde in particular (Figure 1), which is a starting substrate for the formation of substituted tetrahydroisoquinolines (Erdmann et al., 2017; Sehl et al., 2017). To demonstrate the applicability of genetically encoded photosensitizers as light-responsive regulators for enzyme activities in biocatalysis, we created and purified five ApPDC-EGWH fusion enzymes carrying different photosensitizer domains. First, we compared the carboligation activity of the fusion enzymes in the dark to the untagged ApPDC-EGWH. Here, we aimed to exclude a negative influence of the tagged photosensitizers on the enzyme activity when not exposed to light. Subsequently, the inactivation potential of each fusion enzyme was determined, to evaluate which photosensitizer tag is most suitable for selective carboligase inactivation and therewith potentially suppressing cross-reactivity in a multi-step synthesis approach.
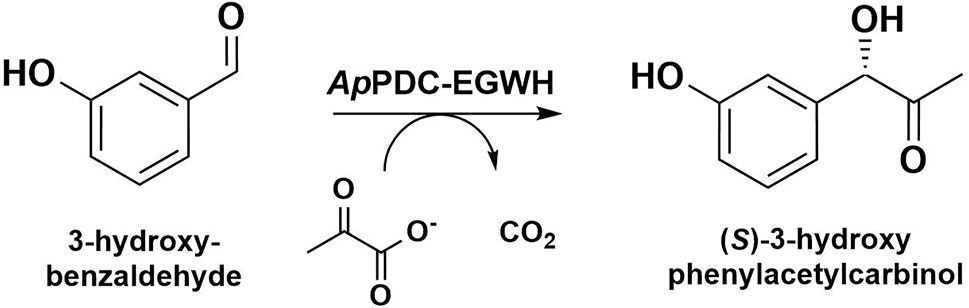
FIGURE 1. Carboligation of 3-hydroxybenzaldehyde and pyruvate to (S)-3-hydroxyphenylacetylcarbinol ((S)-3-OH-PAC) and CO2 catalyzed by the pyruvate decarboxylase variant E469G/W543H from Acetobacter pasteurianus (ApPDC-EGWH).
Materials and Methods
Vector Construction and Transformation
A pET28a vector (Merck, Darmstadt, Germany) was used for the construction of photosensitizer fusion genes. Details on the ApPDC-EGWH and the LbADH (alcohol dehydrogenase from Lactobacillus brevis) construct in a pET21a vector can be found in (Sehl et al., 2017) and in (Kulig et al., 2012), respectively and in the Supplementary Material. Information on the photosensitizers used as fusion tags (SOPP, SOPP3, EcFbFP, Pp2FbFP and SuperNova) are given elsewhere (Wingen et al., 2014; Endres et al., 2018; Hilgers et al., 2019) and in the Supplementary Material. Primers with specific overhangs for the cloning of the respective genes including sequences encoding N-terminal His6-tags were designed and ordered from Eurofins Genomics (Ebersberg, Germany). The expression plasmids were constructed with the Gibson Assembly® Cloning Kit (NEB, Frankfurt Germany) and verified by sequencing (LGC Genomics, Berlin, Germany). E. coli BL21(DE3) cells (Merck, Darmstadt, Germany) were transformed with the respective expression plasmids by adding 1 µl plasmid solution (100 ng μL−1) to 100 µl bacterial solution (OD600 = ∼12, in 80 mM CaCl2, 20% glycerol). For pET28a-SOPP3-ApPDC-EGWH only, E. coli Tuner™(DE3) cells (Merck, Darmstadt, Germany) were used. After 30 min incubation on ice, a heat shock was performed at 42°C for 40 s and the cells were stored on ice for 2 min. 900 μl S.O.C. medium (Thermo Fisher Scientific, Waltham, MA, United States) was added and the cells were incubated for 1 h at 37°C and 700 rpm in a thermomixer (comfort 5335r, Eppendorf, Hamburg, Germany). Finally, the cells were plated on lysogeny broth (LB) agar (with 50 μg ml−1 kanamycin, only for cells transformed with pET21a-ApPDC-EGWH 100 μg ml−1 ampicillin was used) and incubated overnight at 37°C.
Protein Production
Proteins were produced in shaking flasks with a filling volume of up to 15%. A single colony from the respective overnight plate was transferred to 50 ml LB medium (with 50 μg ml−1 kanamycin, for cells transformed with pET21a-ApPDC-EGWH 100 μg ml−1 ampicillin was used) and the preculture was cultivated overnight at 37°C and 150 rpm (Infors HT, Bottmingen, Switzerland). For the main cultures, LB medium was used except for the production of ApPDC-EGWH, LbADH and Pp2FbFP-ApPDC-EGWH which was done in auto induction (AI) medium. Details on the cultivation media are provided in the Supplementary Material. During production, cells containing light-sensitive proteins were handled under exclusion of light. LB cultivations lasted 24 h (induction with 0.5 mM IPTG, for E. coli Tuner™ (DE3) cells with 0.1 mM IPTG) and AI cultures were incubated for 48 h. Cultivations were started at 37°C (decreased to 20°C after 2 h) and 150 rpm (Infors HT, Bottmingen, Switzerland). After harvesting (Avanti J-20 XP, Rotor JLA-8.1000, Beckman Coulter, Brea, CA, United States; 30 min, 8,000 rpm, 4°C), cells containing light-sensitive proteins were stored under exclusion of light.
Determination of Enzyme Solubility and SDS-PAGE
Samples of 5 ml were taken at the end of each cultivation, centrifuged (20 min, 4°C, 4,000 rpm; Universal Kühlzentrifuge, Hettich, Tuttlingen, Germany) and the resulting pellets were frozen for 24 h. The pellets were thawed in 150 µl TRIS buffer (50 mM, pH 7.0) with 2.5 mM MgSO4, 0.1 mM thiamine diphosphate (ThDP), 1 mg ml−1 lysozyme (Merck, Darmstadt, Germany) and 5 units of benzonase (Merck, Darmstadt, Germany) for 20 min on ice and then centrifuged (25 min, 4°C, 14,000 rpm, Eppendorf 5417 R, Hamburg, Germany; soluble fraction). The insoluble proteins were solubilized by dissolving the pellet in 150 µl urea (7 M) with subsequent centrifugation (45 min, 20°C, 14,000 rpm, Eppendorf 5417 R, Hamburg, Germany; insoluble fraction). For SDS-PAGE analysis, 0.5 µl of both fractions containing soluble and insoluble proteins was mixed with 1.5 µl NuPAGE™ Reducing Reagent and 3.5 µl NuPAGE™ Sample Buffer (both Thermo Fisher Scientific, Waltham, MA, United States) and heated for 5 min at 90°C and 600 rpm (Thermomixer comfort 5335r, Eppendorf, Hamburg, Germany). The samples were analyzed by SDS-PAGE (NuPAGE™ 4–12% Bis-Tris gel; XCell SureLock Mini-Cell electrophoresis system, Thermo Fisher Scientific, Waltham, MA, United States; 200 V, 100 mA, 15 W, 50 min) and stained with Coomassie Brillant Blue G-250 (Thermo Fisher Scientific, Waltham, MA, United States) for 60 min. The gel was destained for another 60 min using water.
Protein Purification and Storage
For the purification and storage of light-active fusion enzymes, light exposure was limited to an absolute minimum. Therefore, columns used for purification as well as the lyophilizer and respective solutions were wrapped with aluminium foil and windows were covered. The untagged ApPDC-EGWH was purified and stored under normal light conditions (electric and sunlight). Frozen cells containing respective enzymes were thawed (30% (w/v)) in TRIS buffer (50 mM, pH 7.0, 2.5 mM MgSO4, 0.1 mM ThDP) with 1 mg ml−1 lysozyme (Merck, Darmstadt, Germany) and 10 units ml−1 benzonase (Merck, Darmstadt, Germany) for 30 min on ice. The cells were disrupted by ultrasonication (Digital Sonifier 450, Emerson Electric Co., Ferguson, MO, United States) on ice for a total sonication time of 5 min (intervals of 2 s sonication pulse and 8 s pause) at 60% intensity. After centrifugation (Avanti J-20 XP, Rotor JA-10, Beckman Coulter, Brea, CA, United States; 40 min, 15,000 rpm, 4°C), the supernatant was applied to a Ni-NTA Superflow resin (Qiagen, Hilden, Germany), pre-equilibrated with TRIS buffer (50 mM, pH 7.5, 2.5 mM MgSO4, 0.1 mM ThDP) using an ÄKTA pure chromatography system (GE Healthcare, Bosten, MA, United States). After a washing step with an appropriate buffer (50 mM TRIS buffer, pH 7.5, 2.5 mM MgSO4, 0.1 mM ThDP, 25 mM imidazole), the His6-tagged proteins were eluted (50 mM TRIS buffer, pH 7.5, 2.5 mM MgSO4, 0.1 mM ThDP, 300 mM imidazole). Relevant protein samples were pooled and desalted on a HiTrap™ Sephadex G-25 resign (GE Healthcare, Bosten, MA, United States), pre-equilibrated with TRIS buffer (10 mM, pH 7.5, 2.5 mM MgSO4, 0.1 mM ThDP). The protein concentration of the pooled relevant samples was adjusted to 1 mg ml−1 with water to improve the lyophilization process. Protein concentrations were determined in 1:100 (v/v) dilution according to Bradford (Bradford, 1976), the respective buffer was used as blank. After freezing the protein solution overnight, it was submitted to lyophilization (Alpha 1-4 LD Plus, Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany) for 3 d. After lyophilization, the light-sensitive fusion constructs were stored under exclusion of light. In case of the insoluble EcFbFP-ApPDC-EGWH and Pp2FbFP-ApPDC-EGWH, the pellets obtained after cell disruption and centrifugation were subjected to two washing steps with TRIS buffer (50 mM, pH 7.5, 2.5 mM MgSO4, 0.1 mM ThDP) and lyophilization was applied as described above.
Activity Tests with Photosensitizer Fusion Enzymes
Initial rate determination (without light exposure) of the photosensitizer-tagged and the untagged carboligase was performed using the reaction from 3-hydroxybenzaldehyde to (S)-3-hydroxyphenylacetylcarbinol ((S)-3-OH-PAC) (Figure 1). The transformations were performed on 1 ml scale in 50 mM HEPES buffer (pH 7.5) containing 2.5 mM MgSO4, 0.1 mM ThDP, 40 mM 3-hydroxybenzaldehyde and 200 mM pyruvate (sodium salt). The enzymes SOPP-ApPDC-EGWH (5.0 mg ml−1), SOPP3-ApPDC-EGWH (0.3 mg ml−1), EcFbFP-ApPDC-EGWH (24.8 mg ml−1), Pp2FbFP-ApPDC-EGWH (2.47 mg ml−1) and SuperNova-ApPDC-EGWH (2.0 mg ml−1) were applied in a purified, lyophilized formulation. Protein concentrations where determined using the Bradford assay (Bradford, 1976). Samples were incubated for a duration of 30 min at 22°C and 850 rpm in a thermomixer (comfort 5335r, Eppendorf, Hamburg, Germany). Initial rate determinations were performed under dark conditions in 1.5 ml amber-colored reaction tubes. The activity test of SOPP3-ApPDC-EGWH (0.5 mg ml−1; purified and lyophilized) at different light conditions (details are given in Table 2) was done for the same reaction conditions except the exposure time was set to 60 min. Incubation in illuminated setups was done using a stirred 1.5 ml quartz glass cuvette (Hellma GmbH & Co. KG, Müllheim, Deutschland; 600 rpm). The light intensities of the blue and orange LED and of the normal light conditions were determined at room temperature using the PM100D energy meter with an S302C sensor (Thorlabs, Newton, NJ, United States). The sensor was positioned at the exact position of the reaction setup (instead of the cuvette) and aligned with the respective light source. Intensities of the blue and orange LED and of the normal light conditions were calculated using the pre-installed sensor software by setting 450 nm or 610 nm as the calibration wavelength, respectively. A detailed description of the light intensity measurement is given in the Supplementary Material (Supplementary Figure S3). Unless stated otherwise, experiments were carried out as three technical replicates. The enzymatic reaction in samples taken during the carboligase reactions was quenched 1:20 (v/v) in 100% acetonitrile. Product formation was analyzed via HPLC which is described in the following. The activity is given in U µmolenzyme−1, which is defined as the amount of enzyme in µmol, which catalyzes the formation of 1 μmol (S)-3-OH-PAC per minute. The activity test with soluble crude cell extract and the insoluble protein fraction of the EcFbFP-ApPDC-EGWH production (Figure 5A) was done for the reaction from benzaldehyde to (S)-phenylacetylcarbinol ((S)-PAC). For the activity test with soluble crude cell extract and the insoluble protein fraction of the Pp2FbFP-ApPDC-EGWH production (Figure 5B), the above mentioned reaction from 3-hydroxybenzaldehyde to (S)-3-OH-PAC was applied. In both cases, the amount of lyophilized soluble and insoluble protein was equivalent to 500 mg wet cell weight.
Inactivation of Photosensitizer Fusion Enzymes
Inactivation experiments (with blue/orange light exposure) were carried out in a stirred 1.5 ml quartz glass cuvette (Hellma GmbH & Co. KG, Müllheim, Deutschland; 600 rpm). Inactivation assays were performed for the above mentioned reaction from 3-hydroxybenzaldehyde to (S)-3-OH-PAC, except that 50 mM HEPES buffer (pH 6.5) was used. The enzymes SOPP-ApPDC-EGWH (3.0 mg ml−1), SOPP3-ApPDC-EGWH (1.0 mg ml−1), EcFbFP-ApPDC-EGWH (3.6 mg ml−1), Pp2FbFP-ApPDC-EGWH (4.0 mg ml−1) and SuperNova-ApPDC-EGWH (1.0 mg ml−1) where applied in a purified, lyophilized formulation. Protein concentrations where determined using the Bradford assay (Bradford, 1976). A single blue LED (λmax = 450 nm, royal blue, XP-E2 SMD-LED, Star-PCB, Cree, Durham, NC, United States; ∼60 mW cm−2) was applied for the illumination of fusion enzymes. Orange light (λmax = 610 nm, amber, NCSA219B-V1 SMD-Star-PCB, Nichia, Anan, Japan; ∼40 mW cm−2) was used only for the SuperNova-tagged fusion enzyme. Further specifications on the LEDs are given in the Supplementary Material (Supplementary Table S7). The illumination setup was cooled with ice to compensate for a large temperature increase, nevertheless, during 30 min of LED illumination the temperature gradually increased from 20°C to 24°C. For the sake of comparison, the corresponding dark control was exposed to the same temperature changes. The enzymatic reaction in samples taken during the carboligase reactions was quenched 1:20 (v/v) in 100% acetonitrile. Product formation was analyzed via HPLC as described in the following. Control experiments with the ApPDC-EGWH in blue and orange light are shown in the Supplementary Material (Supplementary Figure S2A,B).
Inactivation Effect of Photosensitizer Fusion Enzymes on an Additional Alcohol Dehydrogenase in the Same Reaction Solution
Initial rate assays with the LbADH were performed using the transformation from benzaldehyde to benzyl alcohol (Figure 9.). The enzymatic reactions were performed on 0.5 ml scale in a 50 mM potassium-phosphate buffer (pH 7) containing 1 mM MgCl2, 10 mM NADPH, 10 mM benzaldehyde and 0.01 mg ml−1 LbADH. Lyophilized LbADH after purification was obtained from elsewhere (Kulig et al., 2012). Protein concentrations where determined using the Bradford assay (Bradford, 1976). Samples were incubated for 2 min at 22°C and 850 rpm in a thermomixer (comfort 5335r, Eppendorf, Hamburg, Germany). Experiments were carried out as three technical replicates. For light experiments with carboligase fusion enzymes, 0.1 mg ml−1 LbADH and 1 mg ml−1 of the respective purified and lyophilized fusion enzymes were illuminated for 10 min in 50 mM potassium-phosphate buffer (pH 7, with 1 mM MgCl2) prior to the enzymatic reaction, using blue or orange LEDs as described above. Then, the initial rate of the respective illuminated LbADH was determined. The enzymatic reaction in samples taken during LbADH reactions was quenched 1:9 (v/v) in a solution of 50% acetonitrile and 50% water (v/v). Product formation was analyzed via HPLC which is described in the following. Control experiments with the LbADH in blue and orange light are shown in the Supplementary Material (Supplementary Figure S2C,D).
HPLC Analytics
Samples were analyzed using a 1260 Infinity Quaternary LC system (Agilent Technologies, Inc., Santa Clara, CA, United States) equipped with an 1260 diode array detector and a Chiralpak IE column (85325, Daicel, Osaka, Japan). For carboligase samples, analysis was carried out isocratically with a flow rate of 0.7 ml min−1 at 20°C for 10 min using a solvent mixture of acetonitrile and water (25:75; v/v). 5 μL Samples were injected and the absorption of (S)-3-OH-PAC was detected at 220.4 nm with a retention time of 6.2 min, while (R)-3-OH-PAC was detected with a retention time of 6.85 min. The absorption of 3-hydroxybenzaldehyde was detected at 220.4 nm with a retention time of 8.8 min. For LbADH samples, analysis was carried out isocratically with a flow rate of 1 ml min−1 at 20°C for 10 min using a solvent mixture of acetonitrile and water (50:50; v/v). 10 μL Samples were injected and the absorption of benzyl alcohol was detected at 215 nm with a retention time of 4.1 min. Calibrations with (S)-3-OH-PAC, 3-hydroxybenzaldehyde and benzyl alcohol are shown in the Supplementary Material (Supplementary Figure S1).
Results and Discussion
Construction of Photosensitizer Enzyme Fusions
In a first step, the ApPDC-EGWH was fused to five genetically encoded photosensitizers resulting in corresponding photosensitizer-enzyme fusion constructs (Figure 2.). Mainly, flavin mononucleotide-binding fluorescent proteins were tested as fusion tags, which originate from blue light receptors of light-oxygen voltage (LOV) domains from plants or bacteria (Table 1) (Drepper et al., 2007; Shu et al., 2011). These are the singlet oxygen photosensitizing proteins SOPP and SOPP3 (Westberg et al., 2017), the flavin-binding fluorescent protein (FbFP) from Bacillus subtilis codon-optimized for Escherichia coli (EcFbFP) and the FbFP from Pseudomonas putida (Pp2FbFP; Wingen et al., 2014). In addition, one member of the green fluorescent protein (GFP)-like photosensitizer family (SuperNova) was used (Takemoto et al., 2013). SOPP3 is one of the strongest genetically encoded singlet oxygen producers currently known (Westberg et al., 2017), but it has been shown that SOPP3 is conditionally able to produce very high amounts of H2O2 as well (Hilgers et al., 2019). While EcFbFP produces mainly H2O2 and only small quantities of 1O2, Pp2FbFP is producing higher amounts of 1O2 than H2O2 (Endres et al., 2018). For SuperNova it is suggested that predominantly H2O2 via the type I pathway is produced (Pletnev et al., 2009; Hilgers et al., 2019). Other sources report, that both types of ROS, but in comparison to KillerRed (the dimeric form of SuperNova) more 1O2 and less H2O2 are produced by SuperNova (Bulina et al., 2006a, 2006b; Takemoto et al., 2013). Thus, the selected photosensitizers show a variety in ROS production with different amounts of produced 1O2 (ranked according to literature: SOPP3 > SOPP > Pp2FbFP > EcFbFP > SuperNova) and H2O2 (ranked according to literature: SOPP3 > SOPP > EcFbFP > Pp2FbFP > SuperNova). Considering the position of the C- and N-termini in the crystal structure of the ApPDC (PDB ID: 2VBI), the photosensitizer tags were fused to the N-terminus of the enzyme. In ApPDCs, four monomers form an active tetrameric enzyme (Rother neé Gocke et al., 2011). As the C-termini of the monomers in the quaternary structure of the ApPDC are in close proximity to each other, it was predicted, that the introduction of tags might impair proper folding of the tetrameric enzyme. Five fusion constructs coding for SOPP-ApPDC-EGWH, SOPP3-ApPDC-EGWH, EcFbFP-ApPDC-EGWH, Pp2FbFP-ApPDC-EGWH and SuperNova-ApPDC-EGWH were cloned and subsequently produced in E. coli.
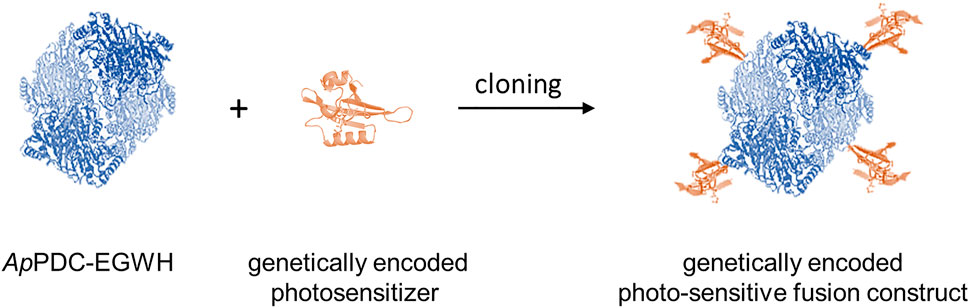
FIGURE 2. Schematic representation of the construction of genetically encoded fusions between photosensitizers (symbolized by the structure of SOPP) and the enzyme ApPDC-EGWH. Orange: monomeric photosensitizer subunits; blue: tetrameric ApPDC-EGWH.

TABLE 1. LOV- and GFP-based photosensitizers with their origin, maximal excitation wavelength (λmax), singlet oxygen quantum yields (ΦΔ) and H2O2 production levels. afrom Westberg et al., 2015, bfrom Endres et al., 2018, cfrom Westberg et al., 2017, dfrom Hilgers et al., 2019, efrom Wingen et al., 2014, ffrom Takemoto et al., 2013, gfrom Onukwufor et al., 2020; SOG = singlet oxygen generator, YtvA/SB2 = bacterial blue light receptors.
Production of Photosensitizer Enzyme Fusions
Production of soluble photosensitizer fusion enzymes proved to be difficult and was not possible for all constructs (Figure 3). In E. coli BL21(DE3), only low production levels of SOPP-ApPDC-EGWH were observed after SDS-PAGE analysis, as compared to the untagged ApPDC-EGWH, while for the SOPP3 fusion variant, at first, soluble protein could not be detected at all. Changing the expression host to E. coli Tuner™(DE3) cells resulted in a reasonable production of SOPP3-ApPDC-EGWH. The fusion enzyme SuperNova-ApPDC-EGWH revealed to give the best production of all soluble fusion enzymes. In contrast, for the EcFbFP and Pp2FbFP fusion enzymes mainly production of insoluble protein could be detected. Several approaches to increase the solubility were investigated, amongst others (data not shown): (i) Changing the expression host to E. coli Tuner™(DE3), (ii) testing different expression media (LB, AI, terrific broth) with (iii) additives as cofactors, betaine or sorbitol, (iv) varying cultivation parameters as the total cultivation time (24, 48, 72 h), the expression temperature (15°C, 20°C) and the IPTG concentration used for induction (0.1, 0.25, 0.5 mM). Even (v) the co-expression of chaperones, did not shift the expression of both fusion enzymes towards a higher degree of soluble protein production. As none of the optimization strategies enhanced the soluble production of the fusion enzymes, we believe that not a misfolding prevented the production of soluble fusion proteins, but we rather produced insoluble protein aggregates (Figure 4). Because soluble protein production was not observed for the EcFbFP-ApPDC-EGWH and Pp2FbFP-ApPDC-EGWH constructs (Figure 3.), the soluble part of the crude cell extracts and the respective insoluble protein fraction, were subjected to qualitative activity assays under dark conditions to search for possible retained activity (Figure 5). The activity test with soluble crude cell extract and the insoluble protein fraction of the EcFbFP-ApPDC-EGWH production (Figure 5A) was done for a test-reaction setup from benzaldehyde and pyruvate to (S)-phenylacetylcarbinol ((S)-PAC) and CO2. However, for the activity test with soluble crude cell extract and the insoluble protein fraction of the Pp2FbFP-ApPDC-EGWH production (Figure 5B), the reaction from 3-hydroxybenzaldehyde and pyruvate to (S)-3-hydroxyphenylacetylcarbinol ((S)-3-OH-PAC) and CO2 was applied. These activity tests show preliminary results from very early project phases where no respective calibration was available, thus, the relative peak areas are presented. In the case of the soluble crude cell extracts, a small amount of activity was detected for the soluble EcFbFP fusion construct (Figure 5A), while no activity was observed for the soluble Pp2FbFP construct (Figure 5B). However, in case of the insoluble protein fractions, both constructs showed significant enzyme activity over the course of 6 h. It has been reported, that insoluble proteins could maintain activity to a certain extent when aggregated, if the active protein structure is preserved (Krauss et al., 2017). Intentional protein aggregation/immobilization can occur by introducing a suitable tag via molecular biological fusion, which can result in the formation of catalytically-active inclusion bodies (CatIBs). Although their activity is significantly reduced compared to soluble protein preparations, advantages are gained with respect to long term stability and production protocols (Diener et al., 2016). Most likely, a similar principle is relevant for the constructs presented here. Native EcFbFP and Pp2FbFP are dimers, whereas the other photosensitizers used in this study are monomers. When a dimeric photosensitizer fuses to a protein, aggregation is possible. If the enzyme to which the naturally dimeric photosensitizer is fused is a monomer, the photosensitizer could cause a fusion between two photosensitizer-enzyme constructs, as this would achieve the preferred quaternary structure of the photosensitizer dimer. Most likely this has no impact on the solubility. But for enzymes with four or more subunits, the fusion to a dimeric photosensitizer might lead to the formation of an insoluble three-dimensional protein aggregate as proposed in Figure 4. This principle is e.g. known from enzyme immobilization, when enzymes are cross-linked to each other forming a mesh (Sheldon and van Pelt, 2013). Multimerization of proteins via the fused photosensitizer tag was observed before for KillerRed (Bulina et al., 2006a; 2006b). Thus, protein engineering was applied to the photosensitizer and the result was a monomeric variant of KillerRed, called SuperNova (Takemoto et al., 2013). This is the reason why SuperNova was also applied in this study. Nevertheless, EcFbFP-ApPDC-EGWH and Pp2FbFP-ApPDC-EGWH are active and the formation of insoluble protein aggregates could be promising if immobilization is intended. As the design of monomeric variants of EcFbFP and Pp2FbFP exceeds the scope of this work, the insoluble protein aggregates of EcFbFP-ApPDC-EGWH and Pp2FbFP-ApPDC-EGWH are evaluated in this study.
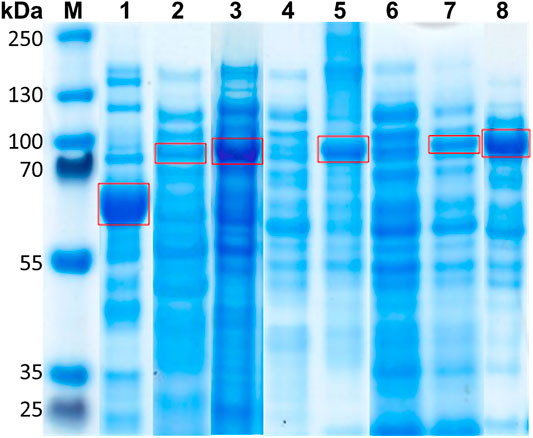
FIGURE 3. SDS-PAGE analysis of the soluble and insoluble protein content during enzyme production in E. coli after cell disruption. (1) Soluble fraction of ApPDC-EGWH production (61 kDa). (2) Soluble fraction of SOPP-ApPDC-EGWH production (75 kDa). (3) Soluble fraction of SOPP3-ApPDC-EGWH production (74 kDa). (4) Soluble and (5) insoluble fraction of EcFbFP-ApPDC-EGWH production (78 kDa). (6) Soluble and (7) insoluble fraction of Pp2FbFP-ApPDC-EGWH production (79 kDa). (8) Soluble fraction of SuperNova-ApPDC-EGWH production (89 kDa). (M) Marker PageRulerTM Plus Prestained Protein Ladder (Thermo Fisher Scientific, Waltham, MA, United States). Target protein bands are indicated by a red box. 5.5 µL of the appropriately prepared enzyme solution was applied to the gel.
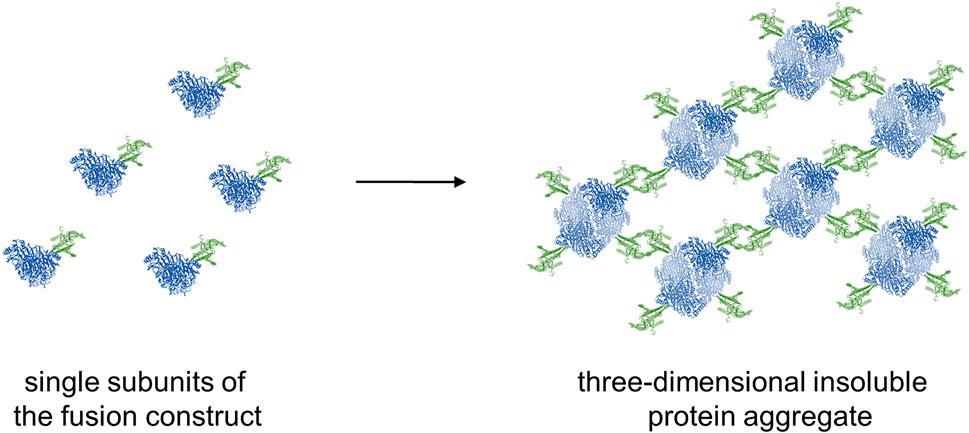
FIGURE 4. Schematic representation of the formation of insoluble three-dimensional protein aggregates by the combination of a dimeric photosensitizer with a tetrameric enzyme. Green: dimeric photosensitizers (EcFbFP or Pp2FbFP); blue: tetrameric ApPDC-EGWH.
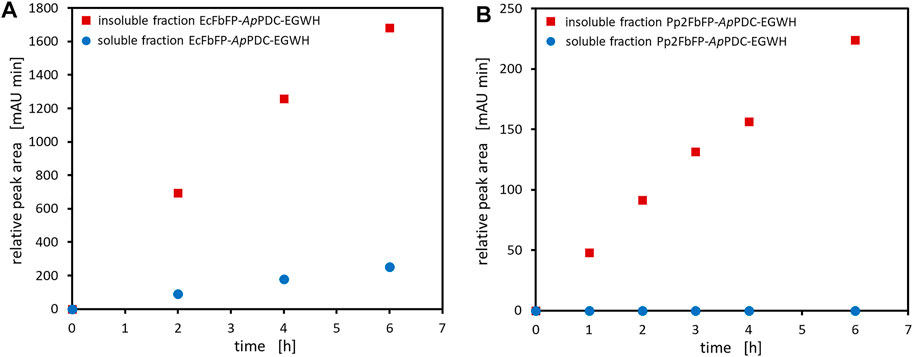
FIGURE 5. Activity test of the soluble and insoluble protein fraction after cell disruption of samples taken during the production of EcFbFP-ApPDC-EGWH (A) and Pp2FbFP-ApPDC-EGWH (B). General reaction conditions: Incubation on 1 ml scale at 22°C and 850 rpm for 6 h in a thermoshaker using 50 mM HEPES buffer (pH 7.5), 2.5 mM MgSO4, 0.1 mM ThDP and 200 mM pyruvate (sodium salt) with (A) 35 mM benzaldehyde and (B) 40 mM 3-hydroxybenzaldehyde. The amount of lyophilized soluble and insoluble protein was equivalent to 500 mg wet cell weight.
Activity of Photosensitizer-Tagged Enzymes and the Effect of Different Light Conditions on it
Besides the proof of activity for the dimeric photosensitizer forming aggregates (Figure 5), the initial rates of all photosensitizer fusion enzymes were measured to determine the effect of the different photosensitizer tags on the enzyme activity. The aim for the fusion constructs was to achieve as high activity as possible compared to untagged ApPDC-EGWH under dark conditions to have powerful fusion constructs for the application in syntheses. An optimal fusion construct would show similar (or even higher) activity than the untagged enzyme without light exposure but very quick inactivation upon light exposure. Initial rate experiments were performed under dark conditions to avoid any potential ROS formation by remaining light sources in the lab. To correct for the different weight of the fusion proteins, the enzyme activity in U µmolenzyme−1 was calculated to enable a comparison on a molar basis (Figure 6). Thus, the displayed activities correspond to an amount of 1 µmol of each fusion enzyme. The initial rate was determined for the mentioned carboligation from 3-hydroxybenzaldehyde to (S)-3-OH-PAC. Since the correct amount of 3-hydroxybenzaldehyde is difficult to detect, as the substance may adhere to the reaction vessel or pipette tips, we decided to determine enzyme activities based on product formation, as this is a more accurate measure. Compared to the activity of the untagged ApPDC-EGWH, the SOPP variant only appeared to retain roughly a third of its activity. This loss in activity was not unexpected for the fusion enzyme, as immobilization or tagging of enzymes has been reported to have a negative effect in some cases due to reduced enzyme flexibility and active site accessibility (Spahn and Minteer, 2008). Interestingly, the initial rate activity of the fusion construct SOPP3-ApPDC-EGWH was much closer to that of the unfused carboligase. With 79% remaining activity, this was a very promising fusion candidate for further inactivation experiments. As SOPP and SOPP3 are structurally very close (Westberg et al., 2015, 2017), a similar effect of the tags on the carboligase was expected and generally observed. The activities of the EcFbFP and Pp2FbFP variants are low as expected after the experiments shown in Figure 5. Additionally, they are less reliable, as the insoluble protein aggregates affected the formation of inhomogeneous reaction solutions, which complicates the sampling during the experiments. As a high residual activity is the aim, fusions with multimeric photosensitizers are thus not recommended when the target enzyme has four or more subunits as in case of the ApPDC-EGWH. Unexpectedly, the fusion of SuperNova had a beneficial effect on the carboligase as an activity increase of 25% was observed. This is interesting as SuperNova (29 kDa) is roughly twice the size compared to the four LOV-based photosensitizers (13—19 kDa). Fortunately, all the fusion proteins tested in this study were active to a certain extent while the carboligase fusions to SOPP3 and SuperNova showed the highest activities. In addition, it should be emphasized that in all activity measurements only the (S)-stereoisomer of 3-OH-PAC was detectable. This demonstrates a highly selective production of the (S)-enantiomer by all photosensitizer fusion enzymes (enantiomeric excess of >99%), which is in accordance with literature of the unfused carboligase (Sehl et al., 2017). Conclusively, the photosensitizer tags do not negatively affect the enzymes stereoselectivity. As the photosensitizer fusion proteins are engineered to be sensitive to light, the effect of naturally occurring light (e.g. electric light in the laboratory and sunlight) on the protein activity was determined. Therefore, five general light conditions in different setups were tested (Table 2; Figure 7). After 60 min reaction time, product formations observed under dark and shady conditions revealed similar results within the experimental error. However, for both the experiments under low light and full light conditions, the product formation was drastically reduced. As expected and demanded the blue light fraction of the sunlight appeared to have a significant detrimental effect on the photosensitizer fused proteins, even with the electric lights switched off. Upon illumination with a blue LED (λmax = 450 nm, ∼60 mW cm−2), product formation diminished already after 30 min under non-optimized conditions. For the first time, this demonstrates the potential of genetically encoded photosensitizers for light-mediated inactivation of the fused enzyme in biocatalysis approaches. Furthermore, as the sunlight appeared to already have a strong effect on the photosensitizer, leading to enzyme inactivation, all of the protein purifications and experimental preparations were performed under shady conditions. This includes coverage of the windows and electric lights switched off during experiments. In a next step, all photosensitizer fusion constructs were illuminated, to comparatively evaluate the inactivation potential of each construct.
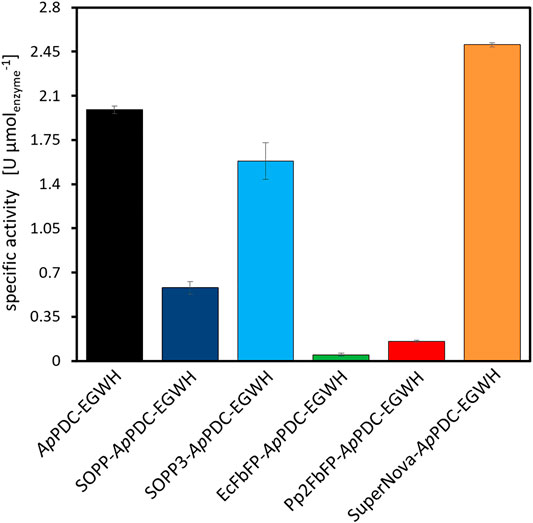
FIGURE 6. Initial rate of the photosensitizer fusion enzymes in dark conditions compared to the untagged ApPDC-EGWH. For fusions with EcFbFP and Pp2FbFP the activity of the insoluble protein aggregates was determined, apart from this soluble (fusion) enzymes were used. General reaction conditions: 40 mM 3-hydroxybenzaldehyde, 200 mM pyruvate (sodium salt) and 50 mM HEPES buffer (pH 7.5) with 2.5 mM MgSO4 and 0.1 mM ThDP. Incubation on 1 ml scale at 22°C and 850 rpm in a thermomixer. The displayed enzyme activities in U µmolenzyme−1 correspond to an amount of 1 µmol of each purified and lyophilized fusion enzyme. The error bars describe standard deviation of the technical triplicates.

TABLE 2. Light conditions tested in different setups with respective light intensities.
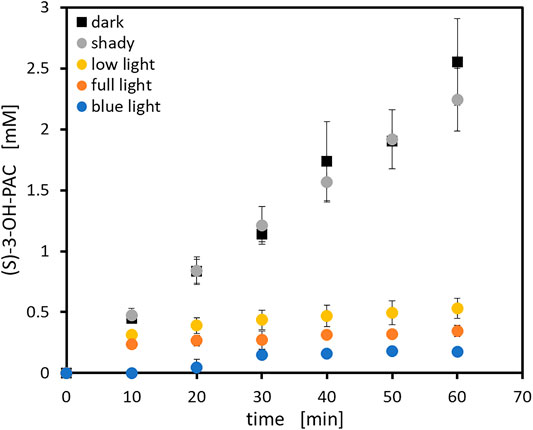
FIGURE 7. Activity test of SOPP3-ApPDC-EGWH (0.5 mg ml−1; purified and lyophilized) at different light conditions (details are given in Table 2) and 60 min of exposure time. General reaction conditions: 40 mM 3-hydroxybenzaldehyde, 200 mM pyruvate (sodium salt) and 50 mM HEPES buffer (pH 7.5) with 2.5 mM MgSO4 and 0.1 mM ThDP. Incubation on 1 ml scale at 22°C and 850 rpm in a thermomixer (dark conditions) or in a stirred quartz-glass cuvette (illuminated setups). The error bars describe standard deviation of technical triplicates.
Inactivation of the Photosensitizer Fusion Enzymes
For the inactivation of the photosensitizer fusion enzymes a blue LED (λmax = 450 nm, ∼60 mW cm−2) was used for the LOV-based photosensitizer tags (Figures 8A–E) and an orange LED (λmax = 610 nm, ∼40 mW cm−2) for SuperNova-ApPDC-EGWH (Figure 8F). Since we observed virtually complete inactivation of SOPP3-ApPDC-EGWH after 30 min of blue light irradiation in the previously described experiment with different light conditions, we chose a reaction time of 30 min for the inactivation of all fusion enzymes. However, the amount of product present in the reaction system after 30 min of inactivation time was formed mainly at the beginning of each reaction where the inactivation is not yet far advanced. In order to identify the degree of inactivation at the end of each reaction, the increase in product between the end point of each reaction after 30 min and the previous measurement point after 20 min reaction time was determined. The degree of inactivation was then calculated by comparing this value from the experiment with blue light exposure with the value from the respective experiment in dark conditions. The results show each photosensitizer to inactivate the attached ApPDC-EGWH to a certain extent (Figures 8B–F). The degree of inactivation is equally high for SOPP-ApPDC-EGWH (94%) and for SOPP3-ApPDC-EGWH (92%) after 30 min of light exposure, which confirmed the inactivation seen for SOPP3-ApPDC-EGWH before (Figure 7). SOPP and SOPP3 are both reported to be strong ROS producers (Westberg et al., 2015; Westberg et al., 2017; Endres et al., 2018; Hilgers et al., 2019). For EcFbFP-ApPDC-EGWH, an inactivation tendency was observed, though the handling issues of the insoluble protein aggregates make it hard to draw a precise conclusion. The fusion enzymes Pp2FbFP-ApPDC-EGWH (38%) and SuperNova-ApPDC-EGWH (42%) both show less effective inactivation kinetics. A lower inactivation performance of Pp2FbFP-ApPDC-EGWH was expected, as the insoluble protein aggregates minimize the light transmission into the reaction medium and Pp2FbFP was reported to produce lower amounts of ROS, especially of H2O2 (Endres et al., 2018). Moreover, as the enzyme showed low activity, a high amount of protein lyophilisate was required, this limited light transmission, repeatedly. Regarding SuperNova, previous work showed this tag to exhibit a low type I photosensitizing activity which led to a less efficient inactivation of bacterial cells as compared to other photosensitizers (Hilgers et al., 2019). In additional tests, we confirmed, that the seen inactivation is purely mediated by the photosensitizer tags, as neither the untagged carboligase nor any of the reaction components (especially (S)-3-OH-PAC and 3-hydroxybenzaldehyde) appeared to be sensitive to blue or orange illumination (Supplementary Figure S2A,B). In summary, SOPP-ApPDC-EGWH and SOPP3-ApPDC-EGWH displayed the best inactivation performance close to full inactivation while the aggregate-forming fusion proteins and the SuperNova construct performed less than half as efficient. Genetic fusion of the photosensitizer to the target enzyme resulted in successful inactivation of the target enzyme by effectively utilizing the locally produced ROS and especially the high phototoxic effect of 1O2 (Sharma et al., 2012). Thus, by applying a single LED setup for illumination, the time necessary to achieve complete inactivation of the carboligase was 30 min. To enable complete inactivation as quickly as possible, preferably within a few minutes, further studies should be devoted to optimizing the inactivation setup. Conceivable setups are those, in which the light intensity can be regulated or LEDs are positioned around the sample instead of exposing only from one side.
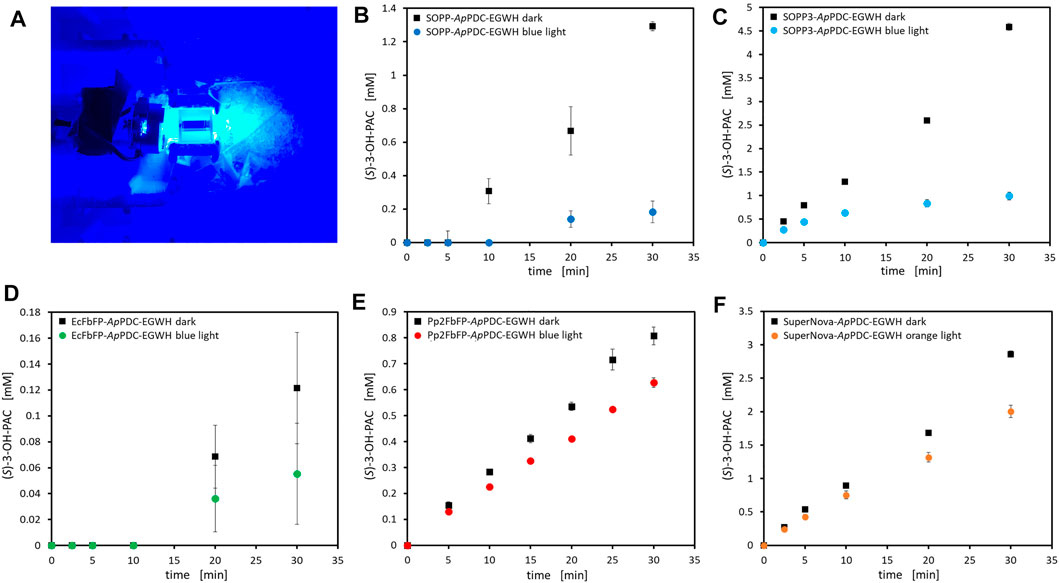
FIGURE 8. Use of a single blue LED (λmax = 450 nm, ∼60 mW cm−2; (A)) or a single orange LED (λmax = 610 nm, ∼40 mW cm−2; only for SuperNova-ApPDC-EGWH) for the 30 min lasting light inactivation of the purified and lyophilized fusion enzymes SOPP-ApPDC-EGWH (3.0 mg ml−1; (B)), SOPP3-ApPDC-EGWH (1.0 mg ml−1; (C)), EcFbFP-ApPDC-EGWH (3.6 mg ml−1; (D)), Pp2FbFP-ApPDC-EGWH (4.0 mg ml−1; (E)) and SuperNova-ApPDC-EGWH (1.0 mg ml−1; (F)) and the corresponding reactions in the dark. General reaction conditions: 40 mM 3-hydroxybenzaldehyde, 200 mM pyruvate (sodium salt) and 50 mM HEPES buffer (pH 6.5) with 2.5 mM MgSO4, 0.1 mM ThDP. Incubation on 1 ml scale at 22°C and 850 rpm in a thermomixer (dark conditions) or in a stirred quartz-glass cuvette (illuminated setups). The error bars describe standard deviation of technical triplicates.
The Effect of Illuminated Photosensitizer Fusion Enzymes on Other Enzymes in a Reaction System
The unspecific inactivation potential of the carboligase fusion enzymes was determined by illuminating photosensitizer fusion enzymes for 10 min in presence of the alcohol dehydrogenase from Lactobacillus brevis (LbADH) in the same reaction vessel, before the relative initial rate of the LbADH was measured. Specifically, in this experiment we were interested in whether free ROS can affect other enzymes in the same reaction system. Thus, the ADH represents another compound (or enzyme) in a reaction solution which could be passively affected by free ROS produced by the carboligase fusion enzymes upon illumination. During the previous inactivation experiment almost full inactivation was achieved after 30 min of blue light exposure (92% for the SOPP3-tagged carboligase), but a high degree of inactivation (74%) was already present after 10 min. Therefore, for the evaluation of the unspecific inactivation an approach lasting 10 min was chosen. In our opinion 10 min is a suitable inactivation time that can be achieved in an optimized inactivation setup. The LbADH activity was determined for the conversion of benzaldehyde to benzyl alcohol (involving Mg2+ and NADPH; Figure 9). The activity of the LbADH in dark conditions without addition of a carboligase fusion enzyme served as control. Similar to the ApPDC-EGWH, neither the LbADH alone nor respective reaction components (especially benzaldehyde and benzyl alcohol) were sensitive to blue or orange illumination (Supplementary Figure S2C,D). In the presence of Pp2FbFP-ApPDC-EGWH and SuperNova-ApPDC-EGWH, the LbADH activity is decreased by ∼10% (Figure 10). It is in accordance to the results shown in Figures 8E–F, that Pp2FbFP and SuperNova in fusion with the ApPDC-EGWH have only low CALI potential after 10 min of light exposure and beyond. The LbADH inactivation mediated by SOPP-ApPDC-EGWH, SOPP3-ApPDC-EGWH and EcFbFP-ApPDC-EGWH (20–30%) is slightly higher, which might be caused by higher production levels of H2O2 which was reported for SOPP, SOPP3 and EcFbFP before (Endres et al., 2018; Hilgers et al., 2019). H2O2 can travel longer distances than 1O2 as it has a half-live of ∼1 ms (Reth, 2002). Thus, the undesired inactivation is depending on the H2O2 amount produced and probably also on the sensitivity of the present enzymes in the reaction system. Nevertheless, the passive inactivation of the ADH by the ROS produced by the fusion enzymes under exposure to light is considerably lower than is the case with direct inactivation of the fusion enzymes. Compared to the previous inactivation experiment (Figure 8C), where SOPP3-ApPDC-EGWH was already inactivated by 74% after 10 min of blue light illumination, the passive inactivation of the LbADH after 10 min of joint exposure with SOPP3-ApPDC-EGWH is significantly lower at about 30%. This is most likely due to the fact that photosensitizers always produce different types of ROS and it has been shown in the literature that the phototoxicity of photosensitizers is greatly enhanced by the production of 1O2 in particular (Hilgers et al., 2019). In order to further evaluate the selectivity of the inactivation mediated by the fusion enzymes, it should be highlighted, that the photosensitizer fusions were used in 10-fold excess compared to the LbADH to see the effect in this experiment. This high surplus would normally not take place in a multi-step synthesis approach. As mentioned earlier, an exposure time of 10 min should be undercut in future optimizations. This would further reduce H2O2 formation and therewith undesired enzyme inactivation.
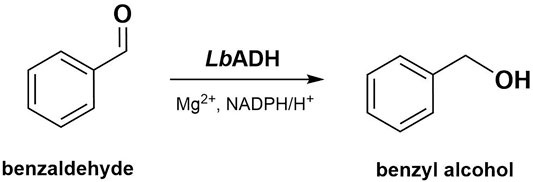
FIGURE 9. Schematic conversion of benzaldehyde to benzyl alcohol catalyzed by the LbADH involving NADPH and Mg2+.
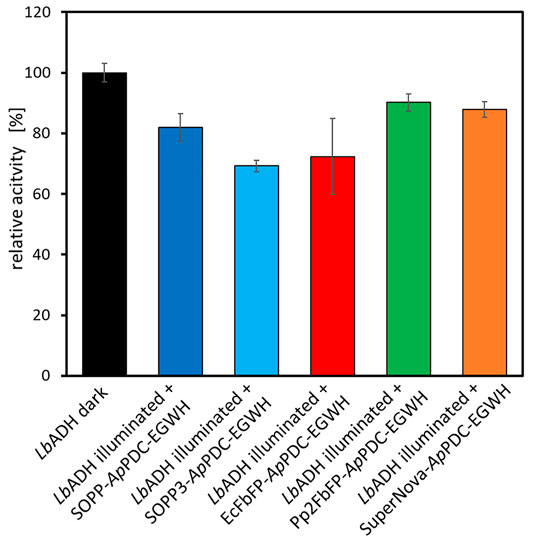
FIGURE 10. Relative activity of the LbADH (0.01 mg ml−1; purified and lyophilized) after illumination at a concentration of 0.1 mg ml−1 in combination with SOPP-ApPDC-EGWH, SOPP3-ApPDC-EGWH, EcFbFP-ApPDC-EGWH, Pp2FbFP-ApPDC-EGWH or SuperNova-ApPDC-EGWH (1 mg ml−1 each; purified and lyophilized). Illumination conditions: 50 mM potassium phosphate buffer (pH 7) with 1 mM MgCl2 for 10 min with a single blue (λmax = 450 nm, ∼60 mW cm−2) or orange LED (λmax = 610 nm, ∼40 mW cm−2; only for SuperNova-ApPDC-EGWH) in a stirred quartz-glass cuvette. General reaction conditions: 10 mM benzaldehyde, 10 mM NADPH and 50 mM potassium phosphate buffer (pH 7) with 1 mM MgCl2. Incubation on 1 ml scale at 22°C and 850 rpm for 2 min in a thermomixer (dark conditions). The activity of the LbADH in the dark and without addition of any photosensitizer fusion enzymes served as control. The error bars describe standard deviation of technical triplicates.
Conclusion
In this work we aim for the construction of active photosensitizer fusion enzymes which show a high degree of inactivation after illumination. We envision these fusion proteins to be a suitable tool to prevent cross-reactivity in multi-step enzyme cascades, which will be shown in another publication. Hence, we fused the photosensitizers SOPP, SOPP3, EcFbFP, Pp2FbFP and SuperNova to the carboligase variant ApPDC-EGWH. SOPP3 proved to be the most promising of the tags tested, for the reasons of soluble expression of the fusion protein, high enzyme activity of the fusion construct in the dark and efficient inactivation upon specific light exposure. The initial rate of the SOPP3 fusion enzyme is similar to the untagged ApPDC-EGWH in the dark. Of all tags tested, only the SuperNova fusion enzyme showed an increase in activity in the dark as compared to the unfused carboligase, however, the SuperNova tag demonstrated only limited CALI activity. SOPP3-ApPDC-EGWH, along with the SOPP-fused variant revealed the best inactivation performance and they could be inactivated by over 90% within 30 min of blue light illumination. Furthermore it was observed, that dimeric photosensitizers as EcFbFP and Pp2FbFP are not suited as tags for enzymes consisting of four or more subunits as the formation of insoluble protein aggregates minimizes the enzyme activity and clearly complicates the handling of the fusion enzymes. Efficient use and photo-induced inactivation of SOPP3-ApPDC-EGWH in an enzyme cascade is currently conducted and optimized in our lab. Additionally, on the long run this method can enable self-controlled regulation of activity by switching on/off a blue light source, e.g., if an in-line monitoring of substrate decrease and product increase is present. Further, selective enzyme regulation might be available in micro-scale chips (Gruenberger et al., 2013; Burmeister et al., 2019) and whole-cell biocatalysis (Jakoblinnert and Rother, 2014; Wachtmeister and Rother, 2016), which is currently challenging to achieve.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
TG, JS, and SS designed and performed the experiments. TG wrote the paper, TD, MvS, FH, NB, and DR helped to draft the manuscript, gave advice for certain techniques and supervised the research. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This project was funded by the European Research Council in frame of ERC starting grant 757320 “Light Controlled Synthetic Enzyme Cascades”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Doris Hahn for assisting with the construction of the photosensitizer fusion genes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2022.835919/full#supplementary-material
References
Abrahamse, H., and Hamblin, M. R. (2016). New Photosensitizers for Photodynamic Therapy. Biochem. J. 473, 347–364. doi:10.1042/BJ20150942
Bernd, A. (2014). Visible Light And/or UVA Offer a strong Amplification of the Anti-tumor Effect of Curcumin. Phytochem. Rev. 13, 183–189. doi:10.1007/s11101-013-9296-2
Bornscheuer, U. T., and Kazlauskas, R. J. (2004). Catalytic Promiscuity in Biocatalysis: Using Old Enzymes to Form New Bonds and Follow New Pathways. Angew. Chem. Int. Ed. 43, 6032–6040. doi:10.1002/anie.200460416
Bradford, M. M. (1976). A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 72, 248–254. doi:10.1016/0003-2697(76)90527-3
Bulina, M. E., Chudakov, D. M., Britanova, O. V., Yanushevich, Y. G., Staroverov, D. B., Chepurnykh, T. V., et al. (2006a). A Genetically Encoded Photosensitizer. Nat. Biotechnol. 24, 95–99. doi:10.1038/nbt1175
Bulina, M. E., Lukyanov, K. A., Britanova, O. V., Onichtchouk, D., Lukyanov, S., and Chudakov, D. M. (2006b). Chromophore-assisted Light Inactivation (CALI) Using the Phototoxic Fluorescent Protein KillerRed. Nat. Protoc. 1, 947–953. doi:10.1038/nprot.2006.89
Burmeister, A., Hilgers, F., Langner, A., Westerwalbesloh, C., Kerkhoff, Y., Tenhaef, N., et al. (2019). A Microfluidic Co-cultivation Platform to Investigate Microbial Interactions at Defined Microenvironments. Lab. Chip 19, 98–110. doi:10.1039/C8LC00977E
Claaßen, C., Gerlach, T., and Rother, D. (2019). Stimulus‐Responsive Regulation of Enzyme Activity for One‐Step and Multi‐Step Syntheses. Adv. Synth. Catal. 361, 2387–2401. doi:10.1002/adsc.201900169
Cruz, F. G., Koh, J. T., and Link, K. H. (2000). Light-Activated Gene Expression. J. Am. Chem. Soc. 122, 8777–8778. doi:10.1021/ja001804h
Diener, M., Kopka, B., Pohl, M., Jaeger, K.-E., and Krauss, U. (2016). Fusion of a Coiled-Coil Domain Facilitates the High-Level Production of Catalytically Active Enzyme Inclusion Bodies. ChemCatChem 8, 142–152. doi:10.1002/cctc.201501001
Drepper, T., Eggert, T., Circolone, F., Heck, A., Krauß, U., Guterl, J.-K., et al. (2007). Reporter Proteins for In Vivo Fluorescence without Oxygen. Nat. Biotechnol. 25, 443–445. doi:10.1038/nbt1293
Endres, S., Wingen, M., Torra, J., Ruiz-González, R., Polen, T., Bosio, G., et al. (2018). An Optogenetic Toolbox of LOV-Based Photosensitizers for Light-Driven Killing of Bacteria. Sci. Rep. 8, 15021. doi:10.1038/s41598-018-33291-4
Erdmann, V., Lichman, B. R., Zhao, J., Simon, R. C., Kroutil, W., Ward, J. M., et al. (2017). Enzymatic and Chemoenzymatic Three‐Step Cascades for the Synthesis of Stereochemically Complementary Trisubstituted Tetrahydroisoquinolines. Angew. Chem. Int. Ed. 56, 12503–12507. doi:10.1002/anie.201705855
Gruber, P., Marques, M. P. C., O'Sullivan, B., Baganz, F., Wohlgemuth, R., and Szita, N. (2017). Conscious Coupling: The Challenges and Opportunities of Cascading Enzymatic Microreactors. Biotechnol. J. 12, 1700030. doi:10.1002/biot.201700030
Gruenberger, A., Probst, C., Heyer, A., Wiechert, W., Frunzke, J., and Kohlheyer, D. (2013). Microfluidic Picoliter Bioreactor for Microbial Single-Cell Analysis: Fabrication, System Setup, and Operation. J. Vis. Exp. 82, 50560. doi:10.3791/50560
Hally, C., Delcanale, P., Nonell, S., Viappiani, C., and Abbruzzetti, S. (2020). Photosensitizing Proteins for Antibacterial Photodynamic Inactivation. Transl. Biophotonics 2, 1–11. doi:10.1002/tbio.201900031
Hilgers, F., Bitzenhofer, N. L., Ackermann, Y., Burmeister, A., Grünberger, A., Jaeger, K.-E., et al. (2019). Genetically Encoded Photosensitizers as Light-Triggered Antimicrobial Agents. Ijms 20, 4608. doi:10.3390/ijms20184608
Hult, K., and Berglund, P. (2007). Enzyme Promiscuity: Mechanism and Applications. Trends Biotechnol. 25, 231–238. doi:10.1016/j.tibtech.2007.03.002
Jacobson, K., Rajfur, Z., Vitriol, E., and Hahn, K. (2008). Chromophore-assisted Laser Inactivation in Cell Biology. Trends Cel. Biol. 18, 443–450. doi:10.1016/j.tcb.2008.07.001
Jakoblinnert, A., and Rother, D. (2014). A Two-step Biocatalytic cascade in Micro-aqueous Medium: Using Whole Cells to Obtain High Concentrations of a Vicinal Diol. Green. Chem. 16, 3472–3482. doi:10.1039/C4GC00010B
Jay, D. G. (1988). Selective Destruction of Protein Function by Chromophore-Assisted Laser Inactivation. Proc. Natl. Acad. Sci. 85, 5454–5458. doi:10.1073/pnas.85.15.5454
Krauss, U., Jäger, V. D., Diener, M., Pohl, M., and Jaeger, K.-E. (2017). Catalytically-active Inclusion Bodies-carrier-free Protein Immobilizates for Application in Biotechnology and Biomedicine. J. Biotechnol. 258, 136–147. doi:10.1016/j.jbiotec.2017.04.033
Kulig, J., Simon, R. C., Rose, C. A., Husain, S. M., Häckh, M., Lüdeke, S., et al. (2012). Stereoselective Synthesis of Bulky 1,2-Diols with Alcohol Dehydrogenases. Catal. Sci. Technol. 2, 1580–1589. doi:10.1039/c2cy20120h
Liao, J. C., Roider, J., and Jay, D. G. (1994). Chromophore-Assisted Laser Inactivation of Proteins Is Mediated by the Photogeneration of Free Radicals. Proc. Natl. Acad. Sci. 91, 2659–2663. doi:10.1073/pnas.91.7.2659
Lin, J. Y., Sann, S. B., Zhou, K., Nabavi, S., Proulx, C. D., Malinow, R., et al. (2013). Optogenetic Inhibition of Synaptic Release with Chromophore-Assisted Light Inactivation (CALI). Neuron 79, 241–253. doi:10.1016/j.neuron.2013.05.022
Lopez-Gallego, F., and Schmidt-Dannert, C. (2010). Multi-enzymatic Synthesis. Curr. Opin. Chem. Biol. 14, 174–183. doi:10.1016/j.cbpa.2009.11.023
Lu, Y., Sun, W., and Gu, Z. (2014). Stimuli-Responsive Nanomaterials for Therapeutic Protein Delivery. J. Controlled Release 194, 1–19. doi:10.1016/j.jconrel.2014.08.015
Mayer, G., and Heckel, A. (2006). Biologically Active Molecules with a "Light Switch". Angew. Chem. Int. Ed. 45, 4900–4921. doi:10.1002/anie.200600387
Moan, J. (1990). On the Diffusion Length of Singlet Oxygen in Cells and Tissues. J. Photochem. Photobiol. B: Biol. 6, 343–344. doi:10.1016/1011-1344(90)85104-5
Ogilby, P. R. (2010). Singlet Oxygen: There Is Indeed Something New under the Sun. Chem. Soc. Rev. 39, 3181–3209. doi:10.1039/b926014p
Onukwufor, J. O., Trewin, A. J., Baran, T. M., Almast, A., Foster, T. H., and Wojtovich, A. P. (2020). Quantification of Reactive Oxygen Species Production by the Red Fluorescent Proteins KillerRed, SuperNova and mCherry. Free Radic. Biol. Med. 147, 1–7. doi:10.1016/j.freeradbiomed.2019.12.008
Pletnev, S., Gurskaya, N. G., Pletneva, N. V., Lukyanov, K. A., Chudakov, D. M., Martynov, V. I., et al. (2009). Structural Basis for Phototoxicity of the Genetically Encoded Photosensitizer KillerRed. J. Biol. Chem. 284, 32028–32039. doi:10.1074/jbc.M109.054973
Reth, M. (2002). Hydrogen Peroxide as Second Messenger in Lymphocyte Activation. Nat. Immunol. 3, 1129–1134. doi:10.1038/ni1202-1129
Ricca, E., Brucher, B., and Schrittwieser, J. H. (2011). Multi-enzymatic cascade Reactions: Overview and Perspectives. Adv. Synth. Catal. 353, 2239–2262. doi:10.1002/adsc.201100256
Robertson, C. A., Evans, D. H., and Abrahamse, H. (2009). Photodynamic Therapy (PDT): A Short Review on Cellular Mechanisms and Cancer Research Applications for PDT. J. Photochem. Photobiol. B: Biol. 96, 1–8. doi:10.1016/j.jphotobiol.2009.04.001
Rother neé Gocke, D., Kolter, G., Gerhards, T., Berthold, C. L., Gauchenova, E., Knoll, M., et al. (2011). S-selective Mixed Carboligation by Structure-Based Design of the Pyruvate Decarboxylase from Acetobacter Pasteurianus. ChemCatChem 3, 1587–1596. doi:10.1002/cctc.201100054
Sehl, T., Hailes, H. C., Ward, J. M., Wardenga, R., von Lieres, E., Offermann, H., et al. (2013). Two Steps in One Pot: Enzyme cascade for the Synthesis of Nor(pseudo)ephedrine from Inexpensive Starting Materials. Angew. Chem. Int. Ed. 52, 6772–6775. doi:10.1002/anie.201300718
Sehl, T., Hailes, H. C., Ward, J. M., Menyes, U., Pohl, M., and Rother, D. (2014). Efficient 2-step Biocatalytic Strategies for the Synthesis of All Nor(pseudo)ephedrine Isomers. Green. Chem. 16, 3341–3348. doi:10.1039/C4GC00100A
Sehl, T., Bock, S., Marx, L., Maugeri, Z., Walter, L., Westphal, R., et al. (2017). Asymmetric Synthesis of (S)-phenylacetylcarbinol - Closing a gap in C-C Bond Formation. Green. Chem. 19, 380–384. doi:10.1039/C6GC01803C
Sharma, P., Jha, A. B., Dubey, R. S., and Pessarakli, M. (2012). Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Bot. 2012, 1–26. doi:10.1155/2012/217037
Sheldon, R. A., and van Pelt, S. (2013). Enzyme Immobilisation in Biocatalysis: Why, what and How. Chem. Soc. Rev. 42, 6223–6235. doi:10.1039/c3cs60075k
Shu, X., Lev-Ram, V., Deerinck, T. J., Qi, Y., Ramko, E. B., Davidson, M. W., et al. (2011). A Genetically Encoded Tag for Correlated Light and Electron Microscopy of Intact Cells, Tissues, and Organisms. Plos Biol. 9, e1001041–10. doi:10.1371/journal.pbio.1001041
Skovsen, E., Snyder, J. W., Lambert, J. D. C., and Ogilby, P. R. (2005). Lifetime and Diffusion of Singlet Oxygen in a Cell. J. Phys. Chem. B 109, 8570–8573. doi:10.1021/jp051163i
Spahn, C., and Minteer, S. (2008). Enzyme Immobilization in Biotechnology. Eng 2, 195–200. doi:10.2174/187221208786306333
Stadtman, E. R. (2006). Protein Oxidation and Aging. Free Radic. Res. 40, 1250–1258. doi:10.1080/10715760600918142
Surrey, T., Elowitz, M. B., Wolf, P.-E., Yang, F., Nédélec, F., Shokat, K., et al. (1998). Chromophore-assisted Light Inactivation and Self-Organization of Microtubules and Motors. Proc. Natl. Acad. Sci. 95, 4293–4298. doi:10.1073/pnas.95.8.4293
Takemoto, K., Matsuda, T., Sakai, N., Fu, D., Noda, M., Uchiyama, S., et al. (2013). SuperNova, a Monomeric Photosensitizing Fluorescent Protein for Chromophore-Assisted Light Inactivation. Sci. Rep. 3, 1–7. doi:10.1038/srep02629
Takemoto, K. (2021). Optical Manipulation of Molecular Function by Chromophore-Assisted Light Inactivation. Proc. Jpn. Acad. Ser. B: Phys. Biol. Sci. 97, 197–209. doi:10.2183/pjab.97.011
Vatansever, F., de Melo, W. C. M. A., Avci, P., Vecchio, D., Sadasivam, M., Gupta, A., et al. (2013). Antimicrobial Strategies Centered Around Reactive Oxygen Species - Bactericidal Antibiotics, Photodynamic Therapy, and beyond. FEMS Microbiol. Rev. 37, 955–989. doi:10.1111/1574-6976.12026
Wachtmeister, J., and Rother, D. (2016). Recent Advances in Whole Cell Biocatalysis Techniques Bridging from Investigative to Industrial Scale. Curr. Opin. Biotechnol. 42, 169–177. doi:10.1016/j.copbio.2016.05.005
Westberg, M., Holmegaard, L., Pimenta, F. M., Etzerodt, M., and Ogilby, P. R. (2015). Rational Design of an Efficient, Genetically Encodable, Protein-Encased Singlet Oxygen Photosensitizer. J. Am. Chem. Soc. 137, 1632–1642. doi:10.1021/ja511940j
Westberg, M., Bregnhøj, M., Etzerodt, M., and Ogilby, P. R. (2017). No Photon Wasted: An Efficient and Selective Singlet Oxygen Photosensitizing Protein. J. Phys. Chem. B 121, 9366–9371. doi:10.1021/acs.jpcb.7b07831
Willner, I., and Rubin, S. (1996). Control of the Structure and Functions of Biomaterials by Light. Angew. Chem. Int. Ed. Engl. 35, 367–385. doi:10.1002/anie.199603671
Keywords: cross-reactivity, enzyme activity regulation, photosensitizers, photo-regulation, multi-step biocatalysis, synthetic enzyme cascades
Citation: Gerlach T, Schain J, Söltl S, van Schie MMCH, Hilgers F, Bitzenhofer NL, Drepper T and Rother D (2022) Photo-Regulation of Enzyme Activity: The Inactivation of a Carboligase with Genetically Encoded Photosensitizer Fusion Tags. Front. Catal. 2:835919. doi: 10.3389/fctls.2022.835919
Received: 15 December 2021; Accepted: 04 February 2022;
Published: 16 March 2022.
Edited by:
Sandy Schmidt, University of Groningen, NetherlandsReviewed by:
Wuyuan Zhang, Tianjin Institute of Industrial Biotechnology (CAS), ChinaChristoph Winkler, University of Graz, Austria
Florian Rudroff, Vienna University of Technology, Austria
Copyright © 2022 Gerlach, Schain, Söltl, van Schie, Hilgers, Bitzenhofer, Drepper and Rother. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dörte Rother, ZG8ucm90aGVyQGZ6LWp1ZWxpY2guZGU=