 Valentina Marchini
Valentina Marchini Ana I. Benítez-Mateos
Ana I. Benítez-Mateos David Roura Padrosa
David Roura Padrosa Francesca Paradisi
Francesca Paradisi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Catal. , 26 November 2021
Sec. Biocatalysis
Volume 1 - 2021 | https://doi.org/10.3389/fctls.2021.790461
A novel fusion protein has been rationally designed, combining the hexameric glutamate dehydrogenase from Clostridium symbiosum with the dimeric formate dehydrogenase from Candida boidinii. The former enzyme consumes ammonia for the reductive amination of α-ketoglutarate using NADH, while the latter biocatalyst regenerates continuously the cofactor. This enzymes fusion opens new perspectives for the detection and the removal of ammonia. The bifunctional biocatalyst has been successfully created, expressed, and then characterized. The two fused protein domains retained identical properties and catalytic activity of the individual enzymes. Additionally, the immobilization on a methacrylate resin optimized the assembly providing a reusable and stable biocatalyst. This is an example of immobilization of a fusion protein, so that efficiency and sustainability of the process are enhanced. The immobilized biocatalyst could be recycled 10 times retaining still half of the initial activity. Such preparation outperforms the co-immobilized wild-type enzymes in the conversion of 300 mM of ammonia, which could be carried out also in continuous mode.
A fusion protein is a molecule consisting of two or more protein domains incorporated into one single complex. Naturally occurring fusion proteins are widespread, and their modular organization has been identified as an important evolutionary phenomenon (Béguin, 1999; Yu et al., 2015).
Artificial fusion proteins may be designed to achieve improved properties or new functionality (Yu et al., 2015). For this purpose, the genetic combination of two proteins to generate a bifunctional enzyme complex has evolved over the years (Lindbladh et al., 1992). The creation of one single protein presenting the biocatalytic activity of two distinct enzymes can dramatically simplify the phases of expression and purification with a significant impact on time, materials and costs spent for the manipulation. This feature translates into sustainable processes that are extremely attractive in line with the growing demand of environmental protection policies.
Furthermore, the very close proximity of the two protein domains yields a higher catalytic efficiency of the two sequential reactions. In fact, the molecule being produced from one enzyme could directly migrate to the active site of the second enzyme without first diffusing to the bulk environment, through substrate channeling (Wheeldon et al., 2016).
Fusion enzyme engineering is one of the promising approaches for the efficient in situ regeneration of cofactors for cost-effective biotransformations (Liu and Wang, 2007). As a matter of fact, the cofactor is too expensive to be applied in stoichiometric amounts, therefore a recycling system is always incorporated to enable a cofactor-dependent reaction in an economically feasible manner (Aalbers and Fraaije, 2019). Some examples of successful cases reporting the development of fusion systems possessing a protein domain to restore the cofactor have been highlighted in the literature (Prachayasittikul et al., 2006; Hölsch and Weuster-Botz, 2010; Sührer et al., 2014; Fang et al., 2015; Jiang and Fang, 2016; Aalbers and Fraaije, 2017; Zhang et al., 2017; Aalbers and Fraaije, 2019; Hartley et al., 2019). For example, the fusion between a NADP+- accepting mutant of formate dehydrogenase from Mycobacterium vaccae (MycFDH) and a ketone reductase (KR) showed beneficial effects in the asymmetric reduction of pentafluoroacetophenone in whole-cell biotransformations (Hölsch and Weuster-Botz, 2010). Later, the fusion of an alcohol dehydrogenase (ADH) with a cyclohexanone monooxygenase (CHMO) has been employed for the synthesis of Ɛ-caprolactone from cyclohexanol, exhibiting a better performance than the combined individual enzymes (Aalbers and Fraaije, 2017). However, only one of the six cloned variants of ADH/CHMO fusion protein maintained the original ADH activity, while the CHMO domain was active in all the cases. This work highlighted how genetic fusion does not always produce fully active enzymes, where structural changes and the orientation of the protein domains play a crucial role in the ability to form the assembled quaternary structure. Indeed, the oligomer interface of one domain may be hindered by the second protein, preventing the correct assembly needed for stability and activity (Aalbers and Fraaije, 2017).
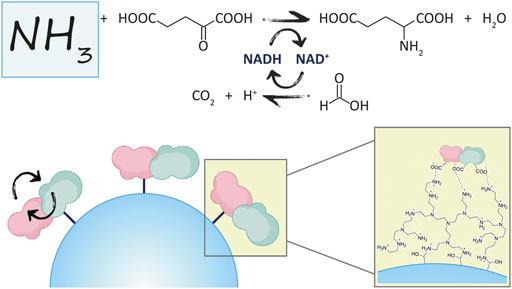
GRAPHICAL ABSTRACT
Fraaije and co-workers described the construction of three different ADHs fused with an NADPH oxidase (NOX). Each enzyme retained its catalytic properties and the coupling of the two proteins offered a rapid qualitative screening system for the detection of ADH/NOX oxidase activity to make large mutant libraries (Aalbers and Fraaije, 2019).
It is worthy to mention a further example, where enzymes were engineered to both retain and recycle their cofactor (NAD+ and ATP) with the final purpose of producing a drug precursor in a three-step continuous-flow reactor system. In this case, the fusion of three enzymes had little effect on the kinetic performance or thermal stability of each domain. Moreover, the enzyme fusions have been successfully immobilized with 86–98% yield and high recovered activity (Hartley et al., 2019).
Glutamate dehydrogenase (GluDH) from bovine liver is the enzyme of choice for the quantification of ammonia in solution. It catalyzes the reductive amination of α-ketoglutaric acid to glutamic acid using NADPH and ammonia, which is measured by monitoring the decrease in absorbance at 340 nm caused by the oxidation of the cofactor (Kun and Kearney, 1974). This enzyme has been successfully applied for the development of biosensors as well as the generation of the commercially available ammonia assay kits (Azmi et al., 2009). These systems are mainly used for the quantitative determination of ammonia in food, water and biological samples, such as blood and urine (Seshadri et al., 2006; Azmi et al., 2009). Since high concentrations of ammonia are toxic for humans (hyperammonemia) and for aquatic ecosystems, the quantification of ammonia becomes essential to diagnose an abnormality in nitrogen homeostasis in humans and is a key parameter for water quality, exploited for human consumption or use (like swimming pools), and for all the aquatic environments (Barsotti, 2001; Azmi et al., 2009; Mérian et al., 2014; Lin et al., 2019; Gogoi et al., 2021).
Ammonia commonly enters the environment through municipal, industrial, agricultural, and natural processes (Geadah, 1985). Ammonia concentrations in water vary seasonally and regionally. In natural waters, concentrations of total ammonia are generally less than 0.1 mg/L. Higher levels of ammonia normally indicate organic pollution (McNeely et al., 1979). In aquaculture systems, it is at most 5 mg/L (0.28 mM), while domestic sewage typically contains 20–40 mg/L of ammonia (1.1–2.2 mM). Dairy effluent, refinery effluent and mineral industry effluent may reach the 100–140 mg/L (5.5–7.8 mM) (Gogoi et al., 2021). However, many circumstances lead to a higher release of ammonia, like accidental spills, the use and disposal of cleansing agents that contain ammonia, urban runoff, accidental releases of ammonia-rich fertilizer, intensive farming, and the decomposition of livestock wasters (CEQG, 2011). For example, during the period from 1992 to 1995, groundwater samples at an old municipal landfill site in Grinsted (Denmark) contained ammonia levels higher than 1 g/L (>55 mM) (Kjeldsen et al., 1998). In 1991–1993, ammonia was found in the surface water collected at the Porto Alegre landfill (Brazil) at a maximum concentration of 1.63 g/L (90 mM), while the maximum amount of ammonia was 1.83 g/L (101 mM) in the leachate (Kuajara et al., 1997).
The methods to quantify ammonia are based normally on biosensors for concentrations up to 500 µmol/L, or assay kits (up to 800 µmol/L) (Azmi et al., 2009; Li et al., 2020), therefore the direct quantification of higher concentrations of ammonia is not possible.
On the other hand, there are biotransformations, for instance, where ammonia can be steadily produced on a millimolar scale as a by-product, so theoretically its continuous removal by a different enzyme can shift the reaction equilibrium towards the formation of the product of interest, increasing the overall biocatalytic efficiency of the desired reaction. Similarly, if the reagents and products of those biotransformations cannot be easily monitored and quantified, the accurate determination of ammonia via a coupled enzyme system employing GluDH can enable the assessment of the conversion rate of that reaction, by tracking α-ketoglutarate consumption and glutamate production.
Formate dehydrogenase (FDH) catalyzes the oxidation of formic acid to CO2 using NAD+ as cofactor and is often used in the recycling of NADH due to its highly favorable thermodynamic equilibrium, and the inertness of the substrate and reaction product (Bolivar et al., 2007). Indeed, this enzyme has been already applied in the generation of fusion proteins such as in combination to a phenylalanine dehydrogenase (PheDH) and to a leucine dehydrogenase (LeuDH) for yielding an efficient cofactor regeneration system (Jiang and Fang, 2016; Zhang et al., 2017). GluDH however, has never been implemented in such enzyme fusions.
The reaction of interest in this work is shown in Figure 1, where GluDH is coupled with the FDH for the removal of ammonia, and in-situ regeneration of the cofactor.

FIGURE 1. Coupled reaction where GluDH and FDH are involved.
Here, the NAD(H)-dependent glutamate dehydrogenase from bacteria Clostridium symbiosum (GluDH) and the formate dehydrogenase from yeast Candida boidinii (FDH) have been chosen as candidates for the rational design of a novel fusion protein.
The new complex was fully characterized and later immobilized to enhance the efficiency of the bifunctional system, which was additionally proven by performing biotransformations.
Chemicals, reagents, and medium component, unless stated otherwise, were obtained as analytical grade from Sigma-Aldrich and Fisher Scientific. All the materials and kits employed for the cloning were acquired from New England Biolabs (NEB). Primers were synthetized by Microsynth AG; NADH and NAD+ were purchased from Apollo Scientific Ltd., while immobilization supports were kindly provided by Resindion S.r.l and Purolite Ltd.
In silico analysis have been performed to predict the assembly of the fusion protein, when a short peptide linker of GSGGGGSAS is integrated between the two protein domains. The two 3D-structures for GluDH (PDBid: 2YHF) and the formate dehydrogenase (PDBid: 5DNA) were constructed using UCSF Chimera to mimic the hexameric form, known to be the predominant with size exclusion chromatography (Pettersen et al., 2004). After mimicking the assembly shown in Supplementary Figure S6, the loop to connect both proteins was modelled using the DaReus server (Karami et al., 2019). The final PDB file has been obtained after local minimization with openMM software, which is a toolkit for molecular simulation using high performance GPU code (Eastman et al., 2017).
The pET28b(+) vector harboring the formate dehydrogenase gene was used as template for PCR amplification (Fwd: 5′-CTCAGGGCATTGCTTGGTAAGAGCTCAAACTCGAGCACCACCAC-3’; Rev: 5′-GCTAGCGC TGCCACCACCGCCGGATCCTTTTTTATCGTGTTTAC-3′). The glutamate dehydrogenase gene was amplified from pRSETb-GluDH to obtain the insert fragments (Fwd: 5′-GGCGGTGGTGGCAGCGCTAGCATGAGCAAGTATGTTGACAG- 3′; Rev: 5′-TGGTGGTGGTGCTCGAGTTTGAGCTCTTACCAAGCAATGCCCTGAG-3′). The primers were designed to incorporate a short peptide linker in the fusion protein (italics) with flanking regions containing restriction sites (underlined). BamHI and NheI have been chosen for this purpose, since they are not contained in any other site of the entire construction and the translated amino acids follow the requirement described in the previous statement. Indeed, BamHI restriction site encodes glycine (GGA) and serine (TCC), while NheI translates to alanine (GCT) and serine (AGC). The length of the linker resulted to be a compromise where the distancing between the two protein domains was effectively supplied, and the primers involved in the gene cloning were not too long for being inefficient. The linker consisted of the following protein sequence: Gly-Ser-Gly-Gly-Gly-Gly-Ser-Ala-Ser (9 amino acid residues). The genetic construction was designed to include two additional restriction sites at the very beginning of FDH gene (EcoRI) and at the very end of GluDH DNA sequence (SacI). This creates a modular and versatile system which allows the exchange of the genes to develop other fusion proteins using FDH as a cofactor recycling system, as well as the incorporation of a third fusion domain which would be involved in the catalytic reaction system.
The genes were amplified by PCR using Q5 High-Fidelity DNA polymerase and the obtained fragments (insert and vector) were used for cloning the fusion construct with the Gibson Assembly Cloning kit. To transform host cells with the assembled product, 2 µl of the Gibson Assembly reaction mixture were added to the chemically competent E. coli DH5α cells (included in the kit), following the protocol given by the supplier. After overnight growth on a LB agar plate supplemented with 50 µg/ml of kanamycin, colonies were picked and grown in liquid LB with kanamycin (50 µg/ml). Then the plasmid was isolated (Thermo Scientific GeneJET Plasmid Miniprep kit) and sent for sequencing to confirm the correct genetic construction.
Through the Gibson assembly method, the linker and the glutamate dehydrogenase sequence have been joined with the sequence of the formate dehydrogenase to successfully assemble a double-stranded fully sealed DNA sequence of the recombinant fusion protein.
The transformed E. coli BL21 (DE3) cells containing the plasmid were cultured at 37°C in 5 ml of LB media supplemented with kanamycin (50 µg/ml) for an overnight. Subsequently, the culture was inoculated into 300 ml of fresh LB broth including kanamycin (50 µg/ml) and incubated at 37°C under shaking. At an optical density (OD600) of 0.5–0.6, the flasks were subjected to a cold-shock stage for 30 min. Afterwards, the overexpression of recombinant fusion protein was induced by the addition of 0.1 mM of IPTG and the culture was grown at 20°C for an overnight. Cells were harvested by centrifugation (4,500 rpm, 20 min, 4°C).
Cells were first resuspended in loading buffer (50 mM potassium phosphate buffer pH 7.5, 300 mM NaCl, 30 mM imidazole) and the resulting suspension subjected to sonication to disrupt cells (5 s pulse on and 10 s off for 8 min). Cell debris was removed by centrifugation (14,500 rpm for 45 min at 4°C) to yield a cell-free extract which was filtered (0.45 µm pore size). The affinity chromatography was performed using an AKTA Start system, equilibrated with the loading buffer. The crude extract was loaded (0.5 ml/min flow rate) onto a 5 ml His-trap Ni-affinity column (GE Healthcare) collecting the flow-through and washed with the loading buffer until the non-specific proteins were completely eluted. The collected solution was then applied to the column a second time to ensure that all the proteins were effectively bound, and the column was washed again. After that, an isocratic wash step with only 10% of elution buffer (50 mM potassium phosphate buffer pH 7.5, 300 mM NaCl, 300 mM imidazole) was performed to remove any non-specific protein still bound onto the column. Finally, recombinant proteins were eluted with 100% of elution buffer and dialyzed at 4°C under mild stirring, against storage buffer (50 mM potassium phosphate buffer pH 7.5). The dialysis was performed for an overnight replacing the buffer at least 2 times.
The purity of the obtained proteins was tested by 12% (w/v) SDS-PAGE and the concentration was quantified by Bradford assay.
One unit of activity was determined as the amount of enzyme needed to produce or consume 1 µmol of NADH per minute at 25°C. The activity assay was performed in a 96-well plate and detected by UV absorbance at 340 nm (ɛ340 = 6.22 mM−1 cm−1). Each protein domain was evaluated singularly (GluDH: 10 mM α-ketoglutaric acid disodium salt and 200 mM ammonium chloride for reductive amination, 40 mM L-glutamic acid monosodium salt for oxidative deamination, in potassium phosphate buffer 50 mM pH 7.5; FDH: 100 mM sodium formate in potassium phosphate 50 mM pH 7.5). The assay was started by the addition of the cofactor in the reaction environment (0.2 mM of NADH or 1.0 mM of NAD+ as final concentration). The specific activity was calculated in U/mg considering the overall concentration of the fusion protein.
The kinetic constants were found in triplicate by measuring the dependence of initial rate on substrate concentration at 25°C in potassium phosphate buffer 50 mM (pH 8.0 for GluDH and pH 7.5 for FDH). For oxidative deamination reaction of GluDH, glutamic acid concentration was varied from 0.1 to 100 mM in the presence of 1 mM NAD+. For the reverse reaction, α-ketoglutaric acid concentration was ranging between 0.05 and 20 mM at a fixed concentration of 200 mM ammonium chloride; ammonium chloride was varied from 1 to 200 mM at 10 mM α-ketoglutaric acid. In both cases, the concentration of NADH was 0.35 mM. The kinetic constants for formic acid in the reaction of FDH were found by using a range between 1 and 100 mM formate concentration at 1 mM NAD+. The cofactor affinity was also evaluated by changing the concentration of NADH from 0.01 to 0.4 mM (10 mM α-ketoglutaric acid, 200 mM ammonium chloride) and 0.1–1.0 mM for NAD+ (40 mM glutamic acid for GluDH and 100 mM formic acid for FDH). Reactions were made by adding an appropriate amount of enzyme to the substrate solution and incubating the mixture for at least 10 min to allow the enzyme to adjust to the environment. The measurement started after mixing the cofactor and following the change in absorbance at 340 nm.
The stability and activity assays at different temperatures and pH were executed in triplicate by incubating the biocatalyst solution at different temperatures (pH 7.5) or at chosen pH values (temperature 25°C) and withdrawing samples at regular times for proceeding with specific activity assays of the fusion protein in parallel with the original GluDH and FDH. The activity of the enzyme was also checked using the previous protocol at several pH and temperature conditions. The reagents were prepared with the chosen pH or incubated at the chosen temperature before proceeding with the activity tests. The results were fitted in a graph showing the trend of the stability/activity of both glutamate and formate DH at chosen temperature or pH values.
The purified fusion protein was applied to a gel filtration column (Superdex 200 10/300 GL, GE Healthcare), which was equilibrated with buffer (50 mM Tris/HCl, 150 mM NaCl, pH 7.5) and mounted onto an ÄKTA Pure instrument (GE Healthcare). The flow rate was set at 0.5 ml/min. A calibration curve was made by plotting the elution times of the following protein standards: carbonic anhydrase (29 KDa), albumin (66 KDa), alcohol dehydrogenase from yeast (150 KDa), ß-amylase from sweet potato (200 KDa), apoferritin from horse spleen (443 KDa), thyroglobulin bovine (669 KDa). Blue dextran (2,000 KDa) was employed to determine the void volume and to check column packing. The elution of the standard proteins and the sample was followed by absorbance readings at 280 nm. The molecular weight of the fusion protein was calculated through the elution time fitted in the equation obtained from the calibration curve.
The CsGluDH-pRSETb and the CbFDH-pET28b were separately used to transform E. coli BL21 STAR, which were then streaked onto a LB agar plate containing the antibiotic (100 µg/ml ampicillin for GluDH and 50 µg/ml kanamycin for FDH). A colony from each plate was inserted in 300 ml autoinduction media containing the proper antibiotic and incubated at 37°C for 8 h. Afterwards, the Erlenmeyer flasks were incubated on ice for 30 min (cold shock) and left at 30°C (GluDH) or 25°C (FDH) for the overnight growth. Cells were harvested by centrifugation and pellet were resuspended with loading buffer to proceed with the purification (same buffers as fusion protein). The cells suspensions were sonicated and centrifuged to obtain cell extracts that were filtered and loaded onto a 1 ml His-trap FF crude Ni-affinity column on the ÄKTA start for the purification. The purity of the protein solutions was then checked by SDS-PAGE and quantified by Bradford assay. The activity was measured as described for the fusion protein.
The chosen support was left in incubation with the enzyme solubilized in the appropriate buffer. Typically, 1 g of support was added to 1 ml of a solution containing the desired amount of protein and incubated at room temperature under mild agitation. The protein load is defined as the amount of protein that has been immobilized per Gram of carrier (mg/g).
Small amounts of supernatant were taken over time to detect enzymatic activity, that was compared to the activity of the initial enzyme solution that was offered to the carrier (control sample). To better confirm the full immobilization, the protein concentration was then checked by Bradford assay and SDS-PAGE. When the maximum achievable yield was obtained, the support was washed.
The activity assay was performed by adding 5–10 mg of immobilized enzyme to a 2 ml reaction mixture containing substrates and cofactor and monitoring it using the same procedure as for the activity assay of the free enzyme. The aim is to determine the expressed activity in U/g (units of enzyme catalyzing the formation of 1 µmol of product per minute per Gram of support), the specific activity in U/mg (expressed activity divided by the amount of protein loaded to the carrier) and the recovered activity, defined as a percentage calculated from the ratio between the specific activity of the immobilized enzyme and the specific activity of the free enzyme.
All the procedures are done at room temperature, if not otherwise stated, under mild agitation. The washing steps are performed with deionized water. All the resins are stored at 4°C after preparation or immobilization.
For the activation of epoxy resin to aldehyde groups, 1 g of support was mixed with 10 ml of 100 mM H2SO4 and incubated overnight. The support was washed, incubated with 10 ml of 30 mM NaIO4 for 2 h and washed again. For the immobilization, the protein solution was prepared in 100 mM NaHCO3 buffer pH 10. The support was then washed, and the Schiff bases were reduced by incubating the support with 10 mg of NaBH4 in 10 ml of 100 mM NaHCO3 pH 10 for 30 min at 4°C (Guisán, 1988).
For the covalent immobilization on epoxy resin by first interaction with a metal, 1 g of resin was incubated with 2 ml of modification buffer (100 mM sodium borate, 2 M iminodiacetic acid, 50 mM phosphate buffer pH 8) for 2 h. After washing, the support was incubated with 5 ml of metal buffer (1 M NaCl, 5 mg/ml CoCl2 in 50 mM phosphate buffer pH 6) for 2 h. The support was then washed and incubated with the protein solution in phosphate buffer 50 mM pH 8.5. The support was then washed with 3 ml of desorption buffer (50 mM EDTA, 500 mM NaCl in 50 mM phosphate buffer pH 7) to remove the metal and rinsed with deionized water. The support was left in 4 ml blocking buffer (3 M glycine in 20 mM phosphate buffer pH 8.5) for an overnight and then washed (Mateo et al., 2007).
For the covalent immobilization on epoxy groups by interaction with ethylenediamine (EDA), 1 g of the epoxy resin was incubated with 6 ml of 2% (v/v) EDA in 100 mM NaHCO3 buffer pH 8.5 for 2 h. The support was then washed (water, 1 M NaCl and again with water). The protein solution was prepared in 5 mM phosphate buffer pH 7. After being washed, the immobilize enzyme was incubated with 1 ml of 5 mM NaHCO3 buffer pH 9 for at least 2 h. Afterwards, the support was washed and left in 4 ml blocking buffer (prepared as previously described) for an overnight (Trobo-Maseda et al., 2020).
For the coating of epoxy and glyoxyl resin with polyethyleneimine (PEI), a solution of 10 mg/ml of PEI 60 kDa was prepared in 100 mM NaHCO3 buffer pH 10 and 10 ml were incubated with 1 g of support for an overnight. After that, the epoxy resin was washed and stored, while the glyoxyl resin was both washed and incubated with 1 mg of NaBH4 in 10 ml of 100 mM NaHCO3 buffer at pH 10 for 30 min at 4°C. The protein solution was prepared in 5 mM phosphate buffer pH 7. After immobilization, the resin was washed and stored (Mateo et al., 2000; Velasco-Lozano et al., 2017).
For the qualitative assay with picrylsulfonic acid, an adapted version of a previously reported protocol was used (Guisán, 1988). 200 µl of 100 mM NaHCO3 buffer pH 10 were added to 20 mg of resin. Then, 20 µl of picrylsulfonic acid were inserted and left in incubation for 10 min. The support was filtered, washed 3 times with 500 µl of saturated NaCl and 3 times with 500 µl of 100 mM NaHCO3 buffer pH 10.
The EP400/SS resin was firstly activated with glyoxyl groups and then coated with PEI 60 KDa. Several protein loadings were tested: 1, 5 and 10 mg per Gram of resin.
Different strategies of immobilization were chosen, using the resins ReliSorb EP400/SS (Resindion S.R.L.) and Lifetech ECR8204F, ECR8304F, ECR8285 (Purolite Ltd.). The resins EP400/SS and ECR8204F were activated to obtain aldehyde groups. A part of them was employed for the covalent immobilization on glyoxyl groups, while the other part was functionalized with PEI, 60 KDa for the respective immobilization. The same epoxy resins were also modified with iminodiacetic acid and cobalt, with EDA or with PEI for the relative immobilizations. The biocatalysts were also immobilized directly to Lifetech ECR8304F and ECR8285 (protein eluted in 5 mM phosphate buffer pH 7). All the carriers presenting amino groups were qualitatively analyzed using picrylsulfonic acid as reagent.
The immobilization yield and the recovered activity were calculated, by following the same conditions as for the activity assays (oxidative deamination direction for GluDH).
For the co-immobilization, ReliSorb EP400/SS was firstly treated to get glyoxyl groups and after that, it was coated with PEI 60 KDa. The two enzymes were incubated with the carrier both simultaneously and stepwise adding firstly GluDH and then FDH, and vice versa, to compare any difference in recovered activity. Different amounts of enzymes as well as different ratios were loaded.
The immobilized protein was tested in terms of operational stability by performing consecutive biotransformations and checking the decrease of relative rate of conversion for each cycle of reaction.
The biotransformations were prepared in 1 ml total volume containing 50 mM α-ketoglutaric acid disodium salt, 200 mM ammonium chloride, 200 mM sodium formate and 1 mM NADH in potassium phosphate buffer 50 mM pH 7.5, adding 10–40 mg of immobilized biocatalyst. The reaction was left in incubation at 37°C under agitation for 30 min, then the supernatant was taken for sample preparation and subsequent GC-FID analysis.
Enough free or immobilized biocatalyst was added to a 2 ml tube containing 1 ml of reaction mixture (50/300/400 mM α-ketoglutaric acid disodium salt, 200/400/300 mM ammonium chloride, 200/400 mM sodium formate and 1 mM NADH in 50 mM potassium phosphate buffer pH 7.5). A control without enzyme was run in parallel. The mixture was then incubated at 37°C under agitation (150 RPM) and aliquots were taken over time.
Samples were prepared and derivatized following the available protocol (Walsh et al., 2014) employing methyl chloroformate as reagent, in combination with methanol and pyridine. The reaction components (derivatized glutamic acid and derivatized α-ketoglutaric acid) were extracted in chloroform and submitted to the GC-FID analysis (temperature of inlet: 250°C, FID detector: 275°C). The column oven temperature was initially held at 75°C for 1 min, then increased to 200°C with a ramp of 25°C per minute and a final hold of 4 min, using nitrogen as gas carrier with a flow of 6.5 ml/min and CP-Chirasil-Dex CB 25m x 250 µm x 0.25 µm as GC column.
The production of glutamic acid and the consumption of α-ketoglutaric acid were monitored and compared between free and immobilized biocatalytic reaction to assess the efficiency of the system.
The genetic construction of the recombinant fusion protein has been developed using the original plasmid encoding the FDH from Candida boidinii (pET28b-FDH) as a template, which contains an N-terminal His-tag (Schütte et al., 1976; Padrosa et al., 2021). The gene of the GluDH from Clostridium symbiosum was inserted downstream of FDH DNA sequence (with removal of the FDH stop codon), to enable the correct folding of the GluDH C-terminal domain which loops into the internal cavity of the hexameric complex when the quaternary structure is formed (Rice et al., 1985; Syed et al., 1991; Baker et al., 1992). The construct (N-His6-tag)-FDH-linker-GluDH is shown in Figure 2.
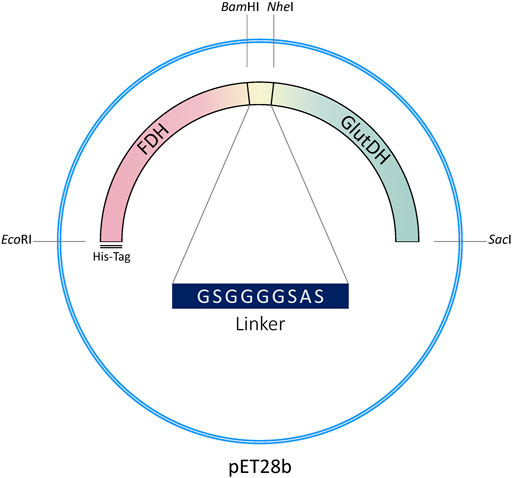
FIGURE 2. Genetic construction of the recombinant fusion protein.
A 9 aa flexible linker was inserted between the two protein domains to facilitate folding (Chen et al., 2013). Linkers are in fact ubiquitously observed in naturally occurring multidomain proteins with the function of maintaining the necessary distance to reduce steric hindrance and/or permit favorable domain-domain interaction between the two protein moieties (Yu et al., 2015).
The linker has been designed to include two flanking restriction sites to facilitate direct modification of the genetic sequence (length of the linker, flexibility, amino acids involved, etc.) and versatility of the construct (see Materials and Methods).
Computational studies have been performed to probe the rationale behind the gene design, before proceeding with the experimental development of the fusion protein. The PDB file resulting from the in-silico analyses supported the insertion of such linker to facilitate the correct assembly of each subunit. As shown in Figure 3, the six subunits are favored to assemble with the hexameric GluDH located in the center (green color) of the quaternary structure. The fused FDH subunits are exposed to the bulk to form three assembled dimers (pink color) enabling proper folding without any major distortion.
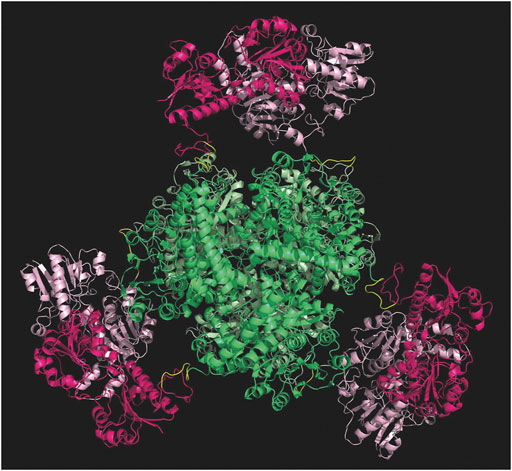
FIGURE 3. Modelling of the hexamer fusion protein construct created with molecular modelling and minimized with openMM (Eastman et al., 2017). GluDH assembly in green and FDH dimers are in pink. Graphic visualization and editing in Pymol (Schrödinger and DeLano, 2020).
The FDH-GluDH fused gene was successfully created via Gibson assembly and confirmed by sequence analysis.
Several conditions to maximize overexpression have been tested (data not shown). LB media induced with 0.1 mM of IPTG at 20°C for an overnight yielded the best results. The correct, soluble expression of the large fusion protein (93 KDa per monomer with a total of 558 KDa if hexamer), was facilitated by a lower concentration of inducer together with a lower incubation and expression temperature. At 20°C, the undesired formation of inclusion bodies was not observed. Following IMAC purification and dialysis, a yield of 40–60 mg of protein per liter of culture were obtained (see ESI paragraph 1 for further details).
The fusion protein yield was comparable to the results obtained with the individual enzymes. Indeed, similar expression levels between fused and free enzymes were also reported in the literature for other fusion proteins (Aalbers and Fraaije, 2017; Aalbers and Fraaije, 2019).
The specific activity of the fusion protein was determined for both catalytic domains (GluDH and FDH) and compared with the activity of the single enzymes. The monomeric molecular mass of the free enzymes (GluDH 53 and FDH 43 KDa) is about half than the fusion protein (93 KDa), therefore the results are reported in Table 1 normalized for concentration of each individual enzyme (see paragraph 2 in the Supplementary Material for calculations).

TABLE 1. Specific activity of the two protein domains compared to the normalized specific activity of the wild-type FDH and GluDH (shown only as reference values without the standard deviations).
The two domains exhibited very similar activity to the wild-type free proteins with an average of 86.4% of GluDH and an 80% of FDH activity. This outcome is consistent with a previous report where the FDH moiety retained less parental activity than partner enzyme (Zhang et al., 2017). Likewise, around an 80% of retained activity was attained by the FDH of that fusion protein, which was presenting the flexible linker (GSSSS)2, very similar to the linker of the current study.
The predicted assembly suggested by the molecular modelling has been therefore validated experimentally. The subunits of the two domains were clearly assembled without any major distortion, meaning that the GluDH possessed a hexameric quaternary structure as the wild-type, while the FDH presented a dimeric configuration. The size exclusion chromatography further reinforced the accuracy of the molecular model, showing a predominant hexameric and octameric form with traces of assembled 2 and 12 monomers (see paragraph 3 in the Supplementary Material for more details). The kinetic constants for all substrates were determined and the results are shown in Table 2. The assays of GluDH were performed at pH 8.0 in order to provide a better evaluation of the results of this work with respect to previously reported values (Sharkey and Engel, 2009), as far as the WT GluDH. The calculated results were found to be comparable. On the contrary, the specific activity was measured at pH 7.5 as compromise between stability/activity of both domains of the fusion protein. The different pH numbers were the reason why the values presented in Table 1 for GluDH were dissimilar from the values of the same enzyme shown in Table 2.
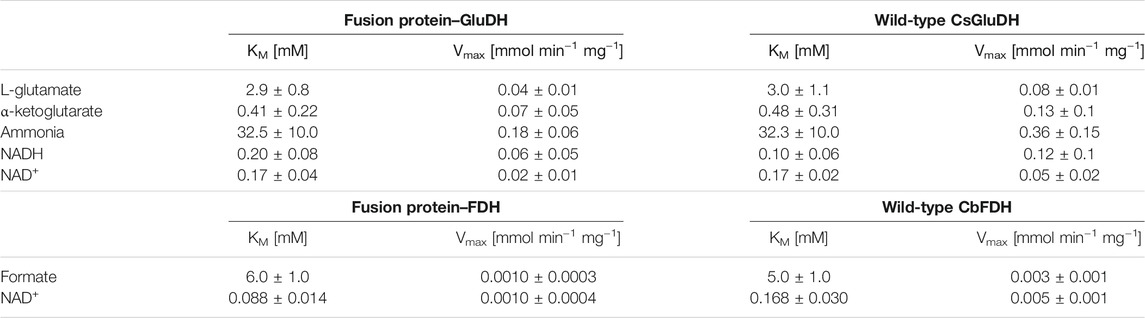
TABLE 2. Kinetic constants for the fusion protein, the glutamate- and the formate dehydrogenase.
The resulting KM values were comparable between the fusion protein and the wild-type enzymes; the affinity to the substrates has not been affected where the fusion of the two proteins could have destabilized the quaternary structure. The FDH domain showed a lower KM value towards NAD+ compared with that of the free FDH, while the GluDH had a decreased affinity for the NADH after the fusion. The same alteration of the cofactor kinetic parameters was found in the LeuDH-FDH fusion protein as well as in the KR fused with the mutant formate dehydrogenase MycFDH (Sührer et al., 2014; Zhang et al., 2017). It has been suggested that the different affinity values towards the cofactor were caused by moiety-moiety interactions or the proximity of two cofactor binding domains in the fusion enzymes (Zhang et al., 2017).
The maximum velocities (Vmaxs) of the fusion protein were consistently half of those of GluDH, and 3 to 5 times lower than FDH, but this simply reflects the fact that the weight of the fusion protein is approximately twice that of the wild-type enzymes, therefore also the rate of conversion is conserved. The bifunctional protein retained the overall catalytic properties of the individual enzymes, as seen for some fused enzymes. In other cases, however, such as for some ADH-CHMO fusion proteins, the catalytic efficiency was dramatically diminished, possibly due to the orientation of the two domains or the fusion itself that hindered the association between the subunits to form the oligomers. It is in fact possible that one protein domain covers the oligomerization interface of its fusion partner, preventing the multimeric assembly, essential for the activity (Aalbers and Fraaije, 2017). In the case of the GluDH-FDH fusion protein, the domains were able to interact properly without any structural impact.
The fusion protein presented the same profiles as the wild-type enzymes for both pH and temperature activity/stability (details in paragraph two of ESI). Furthermore, the FDH moiety acquired a cooperative behavior that is typical of the GluDH but does not occur in the WT FDH. The fusion, therefore, led to a tight connection between the two enzymes where one domain is dependent to the other for structural modifications induced by temperature shift, for example.
To further probe the applicability of this new assembly, the fusion protein was immobilized on a solid support. Preliminary immobilization tests on the individual WT enzymes, based on a trial-and-error approach, showed that all the immobilization strategies based on covalent bonds were exhibiting almost no recovered activity (Supplementary Figures S11, 12). Attempts of immobilization on the resins ECR8204F, ECR8304F, ECR8285 from Purolite did not result in any improvement, giving very low activity with either covalent, hydrophobic, or ionic interactions. Instead, the best option for both WT GluDH and WT FDH was the immobilization on ReliSorb EP400/SS (Resindion) through ionic bonds with polyethyleneimine (PEI) polymer, that covered all the surface of the support. In this case, the WT GluDH showed 85% recovered activity, while the WT FDH exhibited a recovered activity of 100% (at 1 mg/g of loaded protein) with a complete immobilization yield. Therefore, the same support (EP400/SS) and chemical strategy (ionic interactions with PEI) have been applied for the co-immobilization of the WT enzymes, both as simultaneous addition of the two protein solutions and as sequential approaches by first immobilizing one of the two enzymes and then the second one, and vice versa. No decrease from the previously found 85 and 100% recovered activity was detected when the immobilization was performed sequentially, firstly with the binding of the GluDH followed by the addition of the FDH.
Recently, a python-based GUI-application called CapiPy has been developed to assist in protein immobilization, which supplies information to rationalize the choice of immobilization strategy (Roura Padrosa et al., 2021).
CapiPy offered an initial evaluation of the amino acid residues available on the surface of the enzyme through a detailed analysis of the hexameric structure, whose model was given by minimizing a manually assembled hexameric structure followed by minimization with openMM (Eastman et al., 2017). The final quaternary structure is shown in Figure 4, where it is evident that negative charged groups (red) are spread throughout the surface of the hexamer, offering many points of interactions with positively charged groups. Therefore, the functionalization of a support with amino groups would offer easy interaction with the aspartate and glutamate clusters of the fusion protein.
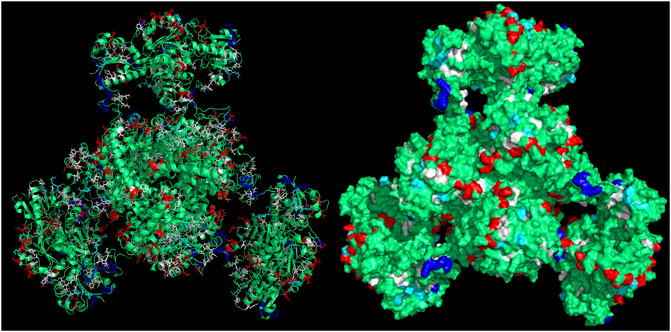
FIGURE 4. 3D structure of the assembled fusion protein after minimization with outputs retrieved from CapiPy tool after the analysis of the structure (Roura Padrosa et al., 2021). Visualized and edited in Pymol (Schrödinger and DeLano, 2020). Left: cartoon and licorice sticks for colored amino acids Right: surface visualization.
The polymethacrylate epoxy resin ReliSorb EP400/SS has been selected as a support, and it provides a highly porous structure. The material of the resin offers a robust support to the enzyme, while the porosity gives many points of anchoring. Polyethyleneimine (PEI) has been used to coat the carrier, to enhance the enzyme binding through the high number of amino groups introduced and create a more hydrophilic environment.
The enzyme was efficiently bound through ionic interactions between the amino groups of PEI and the exposed carboxyl groups of the enzyme (aspartate and the glutamate residues). This favors the retention of enzyme flexibility which is essential for its activity since it undergoes a conformational change from an “active” to an “inactive” form, where the six subunits have the capacity for cooperative interaction (Syed and Engel, 1990; Wang and Engel, 1995).
A complete immobilization yield was achieved, with no remaining protein in the supernatant. As indicated in Figure 5, a recovered activity of 50 and 100% for GluDH and FDH, respectively, was obtained with a loading of 1 mg of protein per Gram of support. Differently from the sequential co-immobilization of the WT enzymes, the two domains of the fusion protein have been simultaneously immobilized since their fusion did not allow for a sequential approach. Increasing the loading to 5 mg/g of support caused a slight decrease to 35% for GluDH and 85% for FDH in recovered activity. This was further reduced at 10 mg/g of loading (30% for GluDH and 60% for FDH) indicating a diffusion limitation issue.
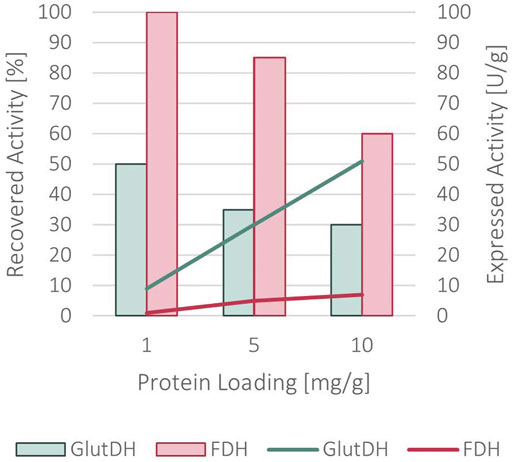
FIGURE 5. Recovered activity (in bars) and expressed activity (in lines) after the immobilization of the fused protein onto ReliSorb EP400/SS, loading an increasing amount of protein.
The enzyme was trialed first for its potential application in the synthesis of glutamic acid at different concentrations of starting material (α-ketoglutarate) and sub-stoichiometric amounts of NADH (1:10, 1:50, 1:300). In this case, the ammonia and formate substrates are kept in excess with respect to the α-ketoglutaric acid (see Materials and Methods). The biotransformation was performed with the immobilized fusion protein, which was compared to the results attained from the biocatalytic reaction done with the free bifunctional protein. Initially, a 5 mg/g immobilized enzyme with 10 mM of α-ketoglutarate and 1 mM NADH yielded 72% molar conversion in 30 min, reaching completion in less than 1 hour. The same results have been seen for an equal amount of soluble protein in the same conditions of biotransformation.
At a 50 mM scale of α-ketoglutaric acid (1 mM NADH), the free protein and the immobilized form appeared to biocatalyse similarly the reaction, showing analogous conversion rates (Figure 6) with equal amounts of biocatalyst. As a matter of fact, both forms showed a complete conversion in less than 4 h, demonstrating the high efficiency of the system. No significant limitation due to the less efficient FDH was notable. Under these conditions, the operational stability of the immobilized protein was tested through repeated usage, showing that 50% of the activity was still retained after 10 cycles (Supplementary Figure S9).
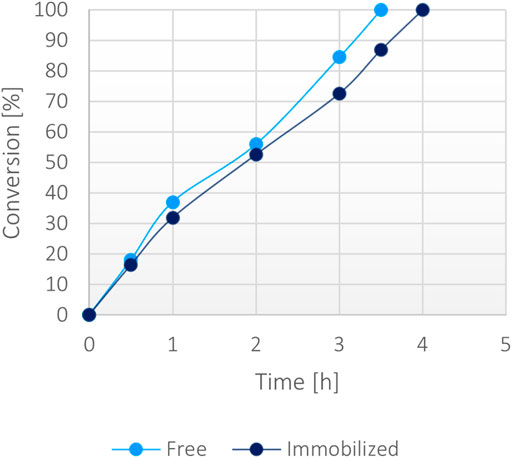
FIGURE 6. Percentage of conversion of 20 mg of immobilized fusion protein (5 mg/g) versus 0.1 mg/ml of the soluble form with 50 mM α-ketoglutarate, 200 mM ammonium, 200 mM formate, 1 mM NADH at 37°C.
Then, a large-scale biotransformation of 300 mM of α-ketoglutaric acid (Figure 7) with still just 1 mM of NADH was set up to test the cost-effectiveness and to push the self-sustainability of the system. Full conversion was obtained in less than 40 h using the immobilized enzyme, while the soluble biocatalyst achieved 70% conversion within the same timeframe. Some protein precipitation was noted in the reaction environment with the free form, causing a decrease of activity over time. The loss of stability was probably caused by the presence of high substrates concentration in the solution (300 mM α-ketoglutarate disodium salt, 400 mM ammonium chloride, 400 mM sodium formate and 1 mM NADH). However, the immobilized enzyme worked efficiently reaching a >99% conversion, suggesting that the immobilization effectively enhanced the stability of the bifunctional system over time even at higher substrates concentration.
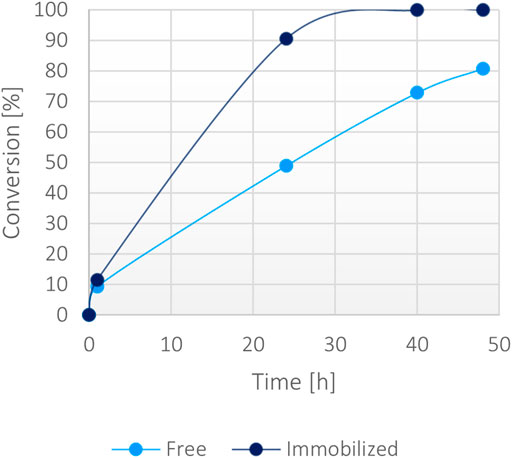
FIGURE 7. Percentage of conversion of 15 mg of immobilized fusion protein (10 mg/g) versus 0.15 mg/ml of the soluble form with 300 mM α-ketoglutarate, 400 mM ammonium, 400 mM formate, 1 mM NADH at 37°C.
The GluDH-FDH fused system could be envisaged as a tool to remove ammonia, for example in contaminated waters following spillages. To better evaluate the efficiency of the system in such potential application, different experiments where the ammonia must be fully depleted have been designed. The removal of ammonia was determined by measuring the consumption of the α-ketoglutarate (substrate provided in excess) combined with the rate of production of the glutamate, since the ammonia employed for the amination is stoichiometric with the two consumed/formed components.
Firstly, to compare the performance of GluDH alone and the GluDH in the fused assembly, with or without the assistance of the FDH, a low amount of ammonia (15 mM) was supplemented with an excess of α-ketoglutarate (30 mM) and the catalytic efficiency tested in the absence or presence of the formate substrate, with either stoichiometric or catalytic amounts of NADH. The reactions were monitored at 1, 2, and 24 h but, in all cases, after 1 h no further change in the ammonia elimination was observed. The fusion protein emerged to be more efficient (Table 3), achieving full depletion of ammonia only when the FDH is activated (entry 2). This result is particularly encouraging as at 15 mM, the concentration of ammonia is significantly below the calculated KM value (Table 2). In contrast, the GluDH in both fused and wild-type form achieved only 80% conversion, in the absence of FDH activation, even when the NADH was provided in a stoichiometric amount (entry 1).
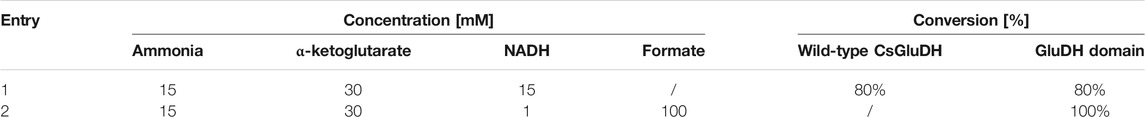
TABLE 3. Percentage of conversion of 15 mM ammonia without (first line) and with (second line) NADH recycling.
While the fusion protein faired very well with low concentration of ammonia, the robustness of the system was tested under significantly harsher conditions. A second set of reactions was set up with 300 mM ammonia together with a moderate excess of α-ketoglutarate (400 mM) and formate (400 mM), and 1 mM NADH. Surprisingly, only the immobilized fusion protein showed a >99% conversion in 48 h (Figure 8). The soluble fusion protein appeared to have a slower pace but achieved almost 80% in 72 h. To complete the comparative evaluation, the individual WT enzymes were also included in this experiment. Both a combination of free forms and a co-immobilized version (all calculated to be in the same ratio and concentration of the fusion protein) were trialed. Surprisingly, the combination of the wild-type soluble proteins reached only 20% of conversion, suggesting a stability issue of at least one of the two enzymes, reinforcing the hypothesis that the fused assembly infers overall a higher structural stability to the catalysts. This is much more apparent in high ionic strength conditions. The co-immobilized enzymes however, also showed improved performance, mimicking the behavior of the free fusion protein, confirming the general higher stability of immobilized systems.
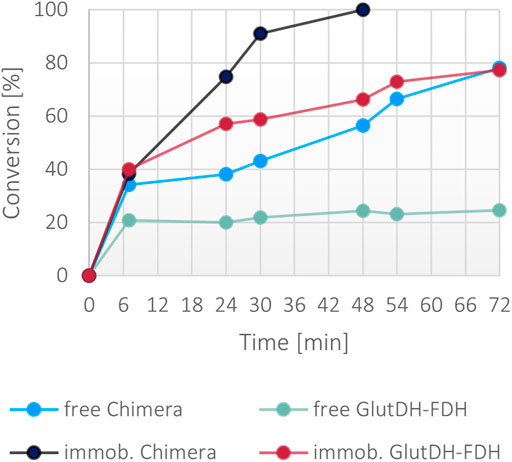
FIGURE 8. Percentage of conversion of the fusion protein and the individual enzymes in both soluble and immobilized form with 300 mM ammonium, 400 mM α-ketoglutarate, 400 mM formate, 1 mM NADH. The final concentration of the fusion protein in both forms was 0.1 mg/ml, the final concentration of the free and co-immobilized individual enzymes was 0.057 mg/ml for GluDH and 0.046 mg/ml for FDH.
Despite the general improved stability attained with the immobilization method, the conversion rates achieved in 48 h with the immobilized fusion protein and the co-immobilized WT enzymes are clearly divergent (>99 versus 65%, respectively) with 1.5-fold higher rate for the bifunctional protein. The performance in working conditions provides a suitable method of comparison of the catalytic efficiency of the two systems (with and without the fusion) where the new enzyme is more effective timewise.
Looking at the literature, also the PheDH-FDH fusion protein showed a 1.5-fold higher molecular conversion rate than the individual enzymes, due to the improved cofactor regeneration process, where the product of the first reaction can be quickly transferred to the second enzyme, which leads to a lower time of diffusion (Jiang and Fang, 2016).
Consequently, the bifunctional enzyme gained remarkable enhancements, especially in the performance, reducing the reaction duration and intensifying the overall conversion degree. The fusion protein results a superior biocatalyst than the soluble or co-immobilized wild-type enzymes under several points, from the time- and cost-effectiveness production (including transformation, expression, purification, immobilization steps) to the enhanced biocatalytic reaction.
The GluDH from Clostridium symbiosum and the FDH from Candida boidinii have been successfully fused to yield one active, stable, and self-sufficient protein. The new bifunctional enzyme has been created overcoming all the possible drawbacks that may appear during the production of a fusion protein. Moreover, the close proximity of the two biocatalysts, given by the direct linkage, offered a great enzymatic tool for the application in small and high scale conversion of ammonia and α-ketoglutarate. The fusion protein was also immobilized, providing a highly efficient biocatalyst, that can be reused for several cycles of reaction. In the future, the design and development of a protein possessing the activity of two different enzymes may surpass the already advantageous co-immobilization for a more industrially appealing solution, that fulfill the demand for sustainable processes. The novel fusion protein has proved to remove ammonia efficiently in experimental conditions, as proof of concept. The treatment of environmental samples and the coupling to other reaction systems where ammonia is the by-product are still potential applications by now. Nevertheless, this work provides a useful starting point for further studies and future applications.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
FP conceptualized the idea and supervised the project, VM performed the experiments and the characterization studies, and wrote the initial draft; DRP performed the MD analysis, AIB-M designed and supervised the genetic experiments and the immobilization studies. All the authors discussed the results and reviewed the manuscript.
SNSF grant number 200021_192274/1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank SNSF (200021_192274) for funding this research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2021.790461/full#supplementary-material
Aalbers, F. S., and Fraaije, M. W. (2017). Coupled Reactions by Coupled Enzymes: Alcohol to Lactone cascade with Alcohol Dehydrogenase-Cyclohexanone Monooxygenase Fusions. Appl. Microbiol. Biotechnol. 101, 7557–7565. doi:10.1007/s00253-017-8501-4
Aalbers, F. S., and Fraaije, M. W. (2019). Design of Artificial Alcohol Oxidases: Alcohol Dehydrogenase-NADPH Oxidase Fusions for Continuous Oxidations. ChemBioChem 20, 51–56. doi:10.1002/cbic.201800421
Azmi, N. E., Ahmad, M., Abdullah, J., Sidek, H., Heng, L. Y., and Karuppiah, N. (2009). Biosensor Based on Glutamate Dehydrogenase Immobilized in Chitosan for the Determination of Ammonium in Water Samples. Anal. Biochem. 388, 28–32. doi:10.1016/j.ab.2009.02.005
Baker, P. J., Britton, K. L., Engel, P. C., Farrants, G. W., Lilley, K. S., and Rice, D. W., (1992). Subunit Assembly and Active Site Location in the Structure of Glutamate Dehydrogenase. Proteins Struct. Funct. Bioinforma. 12, 75–86. doi:10.1002/prot.340120109
Barsotti, R. J. (2001). Measurement of Ammonia in Blood. J. Pediatr. 138, S11–S20. doi:10.1067/mpd.2001.111832
Béguin, P. (1999). Hybrid Enzymes. Curr. Opin. Biotechnol. 10, 336–340. doi:10.1016/S0958-1669(99)80061-5
Bolivar, J. M., Wilson, L., Ferrarotti, S. A., Fernandez-Lafuente, R., Guisan, J. M., and Mateo, C. (2007). Evaluation of Different Immobilization Strategies to Prepare an Industrial Biocatalyst of Formate Dehydrogenase from Candida Boidinii. Enzyme Microb. Technol. 40, 540–546. doi:10.1016/j.enzmictec.2006.05.009
CEQG (2011). Canadian Water Quality Guidelines for the Protection of Aquatic Life. Canada: Canadian Council of Ministers of the Environment (CCME). https://ccme.ca/en/current-activities/canadian-environmental-quality-guidelines.
Chen, X., Zaro, J. L., and Shen, W. C. (2013). Fusion Protein Linkers: Property, Design and Functionality. Adv. Drug Deliv. Rev. 65, 1357–1369. doi:10.1016/j.addr.2012.09.039
Eastman, P., Swails, J., Chodera, J. D., McGibbon, R. T., Zhao, Y., Beauchamp, K. A., et al. (2017). OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. Plos Comput. Biol. 13, e1005659. doi:10.1371/journal.pcbi.1005659
Fang, B., Jiang, W., Zhou, Q., and Wang, S. (2015). Codon-Optimized NADH Oxidase Gene Expression and Gene Fusion with Glycerol Dehydrogenase for Bienzyme System with Cofactor Regeneration. PLoS One 10, 1–15. doi:10.1371/journal.pone.0128412
Geadah, M. L. (1985). National Inventory of Natural and Anthropogenic Sources and Emissions of Ammonia. Environ. Canada, Environ. Prot. Serv.
Gogoi, M., Bhattacharya, P., Kumar Sen, S., Mukherjee, I., Bhushan, S., and Chaudhuri, S. R. (2021). Aquaculture Effluent Treatment with Ammonia Remover Bacillus Albus (ASSF01). J. Environ. Chem. Eng. 9, 105697. doi:10.1016/j.jece.2021.105697
Guisán, J. M. (1988). Aldehyde-agarose Gels as Activated Supports for Immobilization-Stabilization of Enzymes. Enzyme Microb. Technol. 10, 375–382. doi:10.1016/0141-0229(88)90018-X
Hartley, C. J., Williams, C. C., Scoble, J. A., Churches, Q. I., North, A., French, N. G., et al. (2019). Engineered Enzymes that Retain and Regenerate Their Cofactors Enable Continuous-Flow Biocatalysis. Nat. Catal. 211 (2), 1006–1015. doi:10.1038/s41929-019-0353-0
Hölsch, K., and Weuster-Botz, D. (2010). Enantioselective Reduction of Prochiral Ketones by Engineered Bifunctional Fusion Proteins. Biotechnol. Appl. Biochem. 56, 131–140. doi:10.1042/ba20100143
Jiang, W., and Fang, B. S. (2016). Construction and Evaluation of a Novel Bifunctional Phenylalanine–Formate Dehydrogenase Fusion Protein for Bienzyme System with Cofactor Regeneration. J. Ind. Microbiol. Biotechnol. 43, 577–584. doi:10.1007/s10295-016-1738-6
Karami, Y., Rey, J., Postic, G., Murail, S., Tufféry, P., and De Vries, S. J. (2019). DaReUS-Loop: a Web Server to Model Multiple Loops in Homology Models. Nucleic Acids Res. 47, W423–W428. doi:10.1093/nar/gkz403
Kjeldsen, P., Bjerg, P. L., Rügge, K., Christensen, T. H., and Pedersen, J. K. (1998). Characterization of an Old Municipal Landfill (Grindsted, Denmark) as a Groundwater Pollution Source: Landfill Hydrology and Leachate Migration. Waste Manag. Res. 16, 14–22. doi:10.1177/0734242X9801600103
Kuajara, O., Sanchez, J. C. D., Ballestrin, R. A., and Teixeira, E. C. (1997). Environmental Monitoring of the North Porto Alegre Landfill, Brazil. Water Environ. Res. 69, 1170–1177. doi:10.2175/106143097x125920
Kun, E., and Kearney, E. B. (1974). Ammonia. Methods Enzym. Anal. 4, 1802–1806. Available at: https://linkinghub.elsevier.com/retrieve/pii/B9780120913046500318. doi:10.1016/b978-0-12-091304-6.50031-8
Li, D., Xu, X., Li, Z., Wang, T., and Wang, C. (2020). Detection Methods of Ammonia Nitrogen in Water: A Review. Trac - Trends Anal. Chem. 127, 115890. doi:10.1016/j.trac.2020.115890
Lin, K., Zhu, Y., Zhang, Y., and Lin, H. (2019). Determination of Ammonia Nitrogen in Natural Waters: Recent Advances and Applications. Trends Environ. Anal. Chem. 24, e00073. doi:10.1016/j.teac.2019.e00073
Lindbladh, C., Persson, M., Bülow, L., and Mosbach, K. (1992). Characterization of a Recombinant Bifunctional Enzyme, Galactose Dehydrogenase/bacterial Luciferase, Displaying an Improved Bioluminescence in a Three‐enzyme System. Eur. J. Biochem. 204, 241–247. doi:10.1111/j.1432-1033.1992.tb16630.x
Liu, W., and Wang, P. (2007). Cofactor Regeneration for Sustainable Enzymatic Biosynthesis. Biotechnol. Adv. 25, 369–384. doi:10.1016/j.biotechadv.2007.03.002
Mateo, C., Abian, O., Fernandez-Lafuente, R., and Guisan, J. M. (2000). Reversible Enzyme Immobilization via a Very strong and Nondistorting Ionic Adsorption on Support-Polyethylenimine Composites. Biotechnol. Bioeng. 68, 98–105. doi:10.1002/(sici)1097-0290(20000405)68:1<98:aid-bit12>3.0.co;2-t
Mateo, C., Palomo, J. M., Fernandez-Lorente, G., Guisan, J. M., and Fernandez-Lafuente, R. (2007). Improvement of Enzyme Activity, Stability and Selectivity via Immobilization Techniques. Enzyme Microb. Technol. 40, 1451–1463. doi:10.1016/j.enzmictec.2007.01.018
McNeely, R. N., Neimanis, V. P., and Dwyer, L. (1979). Water Quality Sourcebook: A Guide to Water Quality Parameters. University of Illinois in Urbana-Champaign: Inland Waters Directorate, Water Quality Branch.
Mérian, T., Redon, N., Zujovic, Z., Stanisavljev, D., Wojkiewicz, J. L., and Gizdavic-Nikolaidis, M. (2014). Ultra Sensitive Ammonia Sensors Based on Microwave Synthesized Nanofibrillar Polyanilines. Sensors Actuators, B Chem. 203, 626–634. doi:10.1016/j.snb.2014.07.004
Padrosa, D. R., Nissar, Z., and Paradisi, F. (2021). Efficient Amino Donor Recycling in Amination Reactions: Development of a New Alanine Dehydrogenase in Continuous Flow and Dialysis Membrane Reactors. Catalysts 11, 520. doi:10.3390/catal11040520
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Prachayasittikul, V., Ljung, S., Isarankura-Na-Ayudhya, C., and Bülow, L. (2006). NAD(H) Recycling Activity of an Engineered Bifunctional Enzyme Galactose Dehydrogenase/lactate Dehydrogenase. Int. J. Biol. Sci. 2, 10–16. doi:10.7150/ijbs.2.10
Rice, D. W., Hornby, D. P., and Engel, P. C. (1985). Crystallization of an NAD+-dependent Glutamate Dehydrogenase from Clostridium Symbiosum. J. Mol. Biol. 181, 147–149. doi:10.1016/0022-2836(85)90334-1
Roura Padrosa, D., Marchini, V., and Paradisi, F. (2021). CapiPy: Python-Based GUI-Application to Assist in Protein Immobilization. Bioinformatics 37, 2761–2762. doi:10.1093/bioinformatics/btab030
Schrödinger, L., and DeLano, W. (2020). PyMOL. http://www.pymol.org/pymol
Schütte, H., Flossdorf, J., Sahm, H., and Kula, M. R. (1976). Purification and Properties of Formaldehyde Dehydrogenase and Formate Dehydrogenase from Candida Boidinii. Eur. J. Biochem. 62, 151–160. doi:10.1111/j.1432-1033.1976.tb10108.x
Seshadri, R. M., Klein, J. D., Smith, T., Sands, J. M., Handlogten, M. E., Verlander, J. W., et al. (2006). Changes in Subcellular Distribution of the Ammonia Transporter, Rhcg, in Response to Chronic Metabolic Acidosis. Am. J. Physiol. - Ren. Physiol. 290, 397–408. doi:10.1152/ajprenal.00459.2005
Sharkey, M. A., and Engel, P. C. (2009). Modular Coenzyme Specificity: A Domain-Swopped Chimera of Glutamate Dehydrogenase. Proteins Struct. Funct. Bioinforma. 77, 268–278. doi:10.1002/prot.22433
Sührer, I., Haslbeck, M., and Castiglione, K. (2014). Asymmetric Synthesis of a Fluoxetine Precursor with an Artificial Fusion Protein of a Ketoreductase and a Formate Dehydrogenase. Process. Biochem. 49, 1527–1532. doi:10.1016/j.procbio.2014.06.001
Syed, S. E. H., and Engel, P. C. (1990). A pH-dependent Activation-Inactivation Equilibrium in Glutamate Dehydrogenase of Clostridium Symbiosum. Biochem. J. 271, 351–355. doi:10.1042/bj2710351
Syed, S. E. H., Engel, P. C., and Parker, D. M. (1991). Functional Studies of a Glutamate Dehydrogenase with Known Three-Dimensional Structure: Steady-State Kinetics of the Forward and Reverse Reactions Catalysed by the NAD+-dependent Glutamate Dehydrogenase of Clostridium Symbiosum. BBA - Gen. Subj. 1115, 123–130. doi:10.1016/0304-4165(91)90020-H
Trobo-Maseda, L., Orrego, A. H., Romero-Fernández, M., Guisan, J. M., and Rocha-Martín, J. (2020). Immobilization of Enzymes on Hetero-Functional Supports: Physical Adsorption Plus Additional Covalent Immobilization. Methods Mol. Biol. 2100, 159–174. doi:10.1007/978-1-0716-0215-7_10
Velasco-Lozano, S., Benítez-Mateos, A. I., and López-Gallego, F. (2017). Co-Immobilized Phosphorylated Cofactors and Enzymes as Self-Sufficient Heterogeneous Biocatalysts for Chemical Processes. Angew. Chem. - Int. Ed. 56, 771–775. doi:10.1002/anie.201609758
Walsh, R. G., He, S., and Yarnes, C. T. (2014). Compound-specific δ13C and δ15N Analysis of Amino Acids: A Rapid, Chloroformate-Based Method for Ecological Studies. Rapid Commun. Mass. Spectrom. 28, 96–108. doi:10.1002/rcm.6761
Wang, X. G., and Engel, P. C. (1995). Positive Cooperativity with Hill Coefficients of up to 6 in the Glutamate Concentration Dependence of Steady-State Reaction Rates Measured with Clostridial Glutamate Dehydrogenase and the Mutant A163G at High pH. Biochemistry 34, 11417–11422. doi:10.1021/bi00036a014
Wheeldon, I., Minteer, S. D., Banta, S., Barton, S. C., Atanassov, P., and Sigman, M. (2016). Substrate Channelling as an Approach to cascade Reactions. Nat. Chem. 8, 299–309. doi:10.1038/nchem.2459
Yu, K., Liu, C., Kim, B. G., and Lee, D. Y. (2015). Synthetic Fusion Protein Design and Applications. Biotechnol. Adv. 33, 155–164. doi:10.1016/j.biotechadv.2014.11.005
Keywords: fusion protein, protein engineering, ammonia removal, glutamate dehydrogenase, formate dehydrogenase, enzyme immobilization
Citation: Marchini V, Benítez-Mateos AI, Roura Padrosa D and Paradisi F (2021) Fusion of Glutamate Dehydrogenase and Formate Dehydrogenase Yields a Bifunctional Efficient Biocatalyst for the Continuous Removal of Ammonia. Front. Catal. 1:790461. doi: 10.3389/fctls.2021.790461
Received: 06 October 2021; Accepted: 08 November 2021;
Published: 26 November 2021.
Edited by:
Bettina Bommarius, Georgia Institute of Technology, United StatesReviewed by:
Carine Vergne-Vaxelaire, Université Paris-Saclay, FranceCopyright © 2021 Marchini, Benítez-Mateos, Roura Padrosa and Paradisi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Paradisi, ZnJhbmNlc2NhLnBhcmFkaXNpQHVuaWJlLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.