 Laurine Ducrot1
Laurine Ducrot1 Megan Bennett2
Megan Bennett2 Adam A. Caparco3
Adam A. Caparco3 Julie A. Champion3Andreas S. Bommarius3
Julie A. Champion3Andreas S. Bommarius3 Anne Zaparucha1
Anne Zaparucha1 Gideon Grogan2
Gideon Grogan2 Carine Vergne-Vaxelaire1*
Carine Vergne-Vaxelaire1*- 1Génomique Métabolique, Genoscope, Institut François Jacob, CEA, CNRS, Univ Evry, Université Paris-Saclay, Evry, France
- 2York Structural Biology Laboratory, Department of Chemistry, University of York, York, United Kingdom
- 3School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA, United States
Small optically active molecules, and more particularly short-chain chiral amines, are key compounds in the chemical industry and precursors of various pharmaceuticals. Their chemo-biocatalytic production on a commercial scale is already established, mainly through lipase-catalyzed resolutions leading to ChiPros™ products among others. Nevertheless, their biocatalytic synthesis remains challenging for very short-chain C4 to C5 amines due to low enantiomeric excess. To complement the possibilities recently offered by transaminases, this work describes alternative biocatalytic access using amine dehydrogenases (AmDHs). Without any protein engineering, some of the already described wild-type AmDHs (CfusAmDH, MsmeAmDH, MicroAmDH, and MATOUAmDH2) were shown to be efficient for the synthesis of hydroxylated or unfunctionalized small 2-aminoalkanes. Conversions up to 97.1% were reached at 50 mM, and moderate to high enantioselectivities were obtained, especially for (S)-1-methoxypropan-2-amine (98.1%), (S)-3-aminobutan-1-ol (99.5%), (3S)-3-aminobutan-2-ol (99.4%), and the small (S)-butan-2-amine (93.6%) with MsmeAmDH. Semi-preparative scale-up experiments were successfully performed at 150 mM substrate concentrations for the synthesis of (S)-butan-2-amine and (S)-1-methoxypropan-2-amine, the latter known as “(S)-MOIPA”. Modeling studies provided some preliminary results explaining the basis for the challenging discrimination between similarly sized substituents in the active sites of these enzymes.
Introduction
Short-chain chiral amines with carbon chain lengths ranging from C1 to C5 have a wide range of applications in the chemical industry as precursors of pharmaceuticals and agrochemicals (Wu et al., 2021). For example, the butan-2-amine substructure is present in the herbicide Bromacil commercialized as the racemate and in the drug candidate XL888. When substituted with a hydroxyl group, the resulting highly functionalized chiral amino alcohols serve as chiral auxiliaries or ligands in various asymmetric syntheses (Ager et al., 1996). Key examples of these important structural moieties are L-valinol and (R)-3-aminobutan-1-ol ((R)-ABOL), intermediates in the synthesis of HIV integrase inhibitors Elvitegravir (Japan Tobacco, Gilead Sciences) and Dolutegravir (GSK, Pfizer, Shionogi Limited) respectively, (S)-MOIPA, a key chiral element within the herbicide Outlook® (BASF), or 2-aminopropan-1-ol, the main building block of the antiarrhythmic agent Mexiletine (Figure 1).
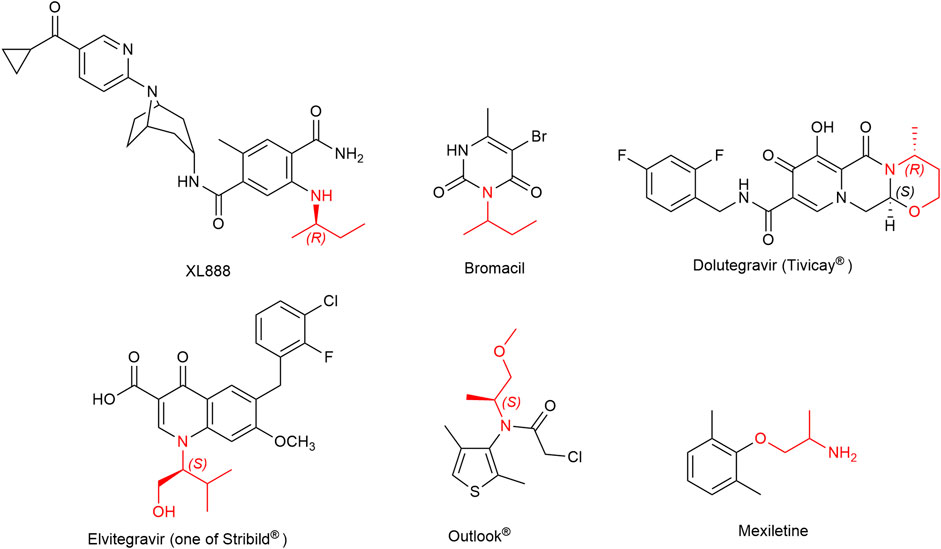
FIGURE 1. Structures of some active components of pharmaceuticals and agrochemicals containing short chiral amines. The chiral amino moieties are highlighted in red.
All of these key small chemical entities are industrially produced by well-established traditional synthetic routes, but green alternatives such as biocatalytic access are topics of great interest in the current context of waste reduction and sustainable chemistry. The ChiPros™ process of BASF based on lipase-catalyzed acetylation is highly efficient for the kinetic resolution of many alkyl amines, such as (S)-1-phenylbutylamine or (S)-2-aminononane, with E values of >1,000. Nevertheless, in the case of small alkyl amines, E values are much lower (Ebutan-2-amine = 8, E(R)-ABOL << 10, E2-aminopentane = 50, and E3-methylbutan-2-amine = 80) due to the similar size of the R-groups, and their resolution requires more expensive acyl donors or solvents. Therefore, biocatalyzed enantioselective syntheses of these molecules are particularly challenging. Some success has already been reported with transaminases (TAs). BASF has completed the process development of (R)-ABOL 2.0 with ee values of >99.9% (Ditrich, 2020). Kroutil and colleagues have produced both enantiomers of Mexiletine in up to >99% ee by deracemization using ω-TA transaminase chemistry (Koszelewski et al., 2009), and (R)- or (S)-butan-2-amine has been efficiently synthesized by continuous-flow chemistry using covalently immobilized transaminases *RTA-43 and HEwT_F84W, respectively (Heckmann et al., 2021). ω-Transaminases have also been proved to be useful tools for the asymmetric amination of α-hydroxy ketones, either alone or in cascade reactions for the synthesis of chiral vicinal amino alcohols (Gomm and O'Reilly, 2017; Slabu et al., 2017; Patil et al., 2018a). The requirement of transaminases for the addition of an excess of the amino donor that is sacrificed during the process alongside in situ by-product removal or in situ product removal for example using supported liquid membrane (Rehn et al., 2016) pushed the search for alternative enzymes. Enzymes performing proper reductive amination have emerged as promising biocatalysts for the synthesis of optically active amines. Thanks to the recent discovery of native Amine Dehydrogenases (AmDHs) and Reductive Aminases (RedAms), such enzymes are no longer restricted to engineered Amino Acid Dehydrogenases (engAADHs) (Ye et al., 2015; Patil et al., 2018b; Cheng et al., 2020; Ducrot et al., 2020; Cosgrove et al., 2021; Mutti and Knaus, 2021). Although they are described to be active toward a wide range of carbonyl-containing compounds, only a few examples are reported for the synthesis of small chiral alkyl amines and small amino alcohols. These include the engineered L-AmDH-TV (mutant D32A/F101S/C290V of L-AmDH) (Franklin et al., 2020) and the mutant AmDH-M0 (mutant K68T/N261L of LfLeuAADH) (Chen et al., 2019). We recently described native AmDHs from biodiversity and provided a picture of their substrate scope (Mayol et al., 2019; Caparco et al., 2020). Their top tested substrates were mainly cyclohexanone and isobutyraldehyde, but some of them displayed activity toward alkyl linear ketones such as pentan-2-one enabling the biocatalytic synthesis of the corresponding (S)-amines on a millimolar scale (Mayol et al., 2019). The high enantiomeric excess (ee > 97%) in favor of the (S)-amines obtained in these cases prompted us to consider that high enantioselectivity could be achieved for other small chiral amines. In the present work, we sought to study in more detail the biocatalytic potential of some of these native AmDHs (CfusAmDH, MsmeAmDH, MicroAmDH, and MATOUAmDH2) for the synthesis of small chiral alkyl amines and small amino alcohols 2a–h (Figure 2).
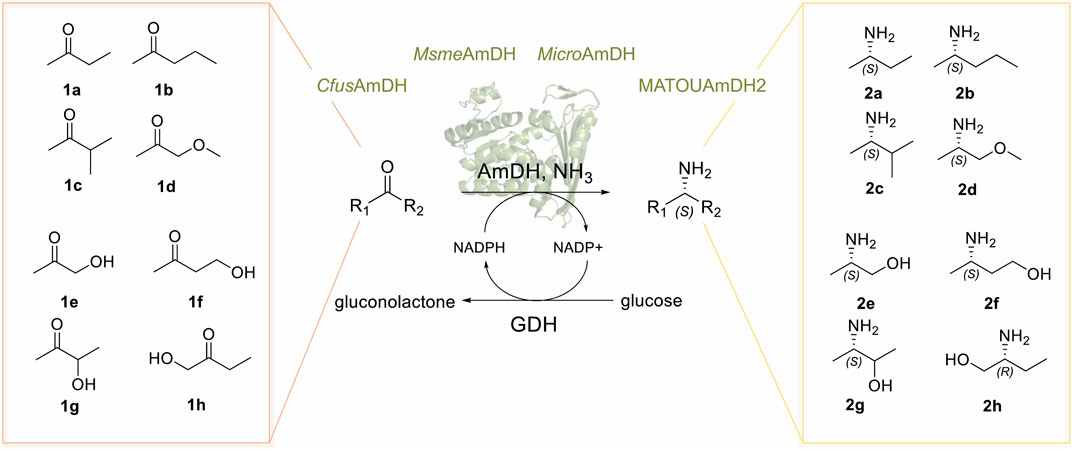
FIGURE 2. Reductive amination reactions catalyzed by native AmDHs detailed in this study. GDH = glucose dehydrogenase.
Materials and Methods
Chemicals and Materials
All reagents were purchased from commercial sources and used without additional purification, except in-house (S)-pentan-2-amine (S)-2b (Mayol et al., 2019). Butan-2-one (1a), pentan-2-one (1b), 1-methoxypropan-2-one (1d), 4-hydroxybutan-2-one (1f), 1-hydroxypropan-2-one (1e), 3-hydroxybutan-2-one (acetoin; 1g), 2-aminopropan-1-ol (alaninol; 2e), butan-2-amine (2a), pentan-2-amine (2b), 3-methylbutan-2-amine (2c), 1-methoxypropan-2-amine (2d), 2-aminobutan-1-ol (2h), (S)-butan-2-amine ((S)-2a), (R)-3-methylbutan-2-amine ((R)-2c), (S)-1-methoxypropan-2-amine ((S)-2d), (S)-2-aminopropan-1-ol (S-alaninol; (S)-2e), (S)-2-aminobutan-1-ol ((S)-2h), β-nicotinamide adenine dinucleotide phosphate disodium salt (NADP+), glucose dehydrogenase (GDH105), (S)-2-(5-fluoro-2,4-dinitrophenylamino)propanamide (FDAA), and benzoyl chloride (BzCl) were purchased from Merck/Sigma-Aldrich (St. Louis, MO, United States). 3-Methylbutan-2-one (1c), 1-hydroxybutan-2-one (1h), 3-aminobutan-1-ol (2f), 3-aminobutan-2-ol (2g), (R)-3-aminobutan-1-ol ((R)-2f), and (2R,3S)-3-aminobutan-2-ol ((2R,3S)-2g) were purchased from Enamine Ltd. (Kiev, Ukraine). Buffers were produced from substances purchased from Sigma–Aldrich and adjusted to the desired pH value with ammonium hydroxide 30%. UHPLC-UV analysis were performed on a UHPLC U3000 RS 1034 bar system (Thermo Fisher Scientific, Waltham, MA, United States) equipped with a UV detector, using a Kinetex F5 column (100 mm × 2.1 mm; 1.7 µm) (Phenomenex, California, United States). NMR spectra were recorded on a Bruker (Bruker, Billerica, MA, United States) 600-MHz spectrometer (Evry University, France) for 1H and 13C experiments. Chemical shifts (expressed in ppm) of 1H and 13C spectra were referenced to the solvent peak δ (H) = 4.79 for D2O.
Large-Scale Purification of Amine Dehydrogenases
Large-scale purification of MsmeAmDH, CfusAmDH, MicroAmDH, and MATOUAmDH2 was conducted using 2 × 500 ml culture by nickel affinity chromatography (His Trap FF 5 ml) in tandem with gel filtration (Hi Load 16/60 Superdex 200pg) as described elsewhere (Mayol et al., 2019). The storage buffer was 50 mM NaCl, 50 mM sodium phosphate pH 7.5–8.0, glycerol 10%–15%, and 1 mM DTT. Protein concentrations were determined by the Bradford method with bovine serum albumin as the standard (Bradford, 1976). The samples were analyzed by SDS-PAGE using the Invitrogen NuPAGE system. The purified proteins were stored at −80°C.
Screening of MsmeAmDH, CfusAmDH, MicroAmDH and MATOUAmDH2 for Conversion of Short Alkyl (Hydroxyl) Ketones
The selected AmDHs (CfusAmDH, MsmeAmDH, MicroAmDH, and MATOUAmDH2), were tested toward ketones 1a–1h in a 96-well plate with a cofactor regeneration system using UHPLC-UV monitoring. To a reaction mixture (total volume = 100 μl) containing 0.2 mM NADP+, 11 mM or 55 mM glucose (1.1 eq.), and 3 U ml−1 GDH105 in 2 M NH4HCO2 buffer, pH 9.0, was added 10 mM or 50 mM (final concentration) of substrate followed by 0.5 mg ml−1 of purified AmDH. The 96-well plate was covered with an aluminum thermowell sealer and a lid and left for 24 h at 30°C in a thermocontrolled oven. The 96-well plate contained wells with background reactions performed in the same manner but lacking the substrate or the purified AmDH, together with wells with calibration mixtures containing 2 M NH4HCO2 buffer pH 9.0 and various concentrations of amines (2, 4, 6, and 8 mM for 10 mM reactions and 10, 20, 30, and 40 mM for 50 mM reactions). All the wells were analyzed by UHPLC-UV after derivatization with BzCl in 96-well plates. The amounts of amines in reaction mixtures were deduced from calibration curves obtained from the UV-response of the calibration mixtures of racemic 2a–2h (Supplementary Figures S1, S2). Enantiomeric excess of products 2a–2h and diastereoisomeric excess for 2g were determined after derivatization with FDAA in 96-well plates for UHPLC-UV detection (Supplementary Figure S3). The reactions were performed in duplicate or triplicate at different days to ensure reliable reproducibility.
Derivatization Procedures and UHPLC-UV Conditions
BzCl derivatization: 20 μl of each reaction mixture was mixed with 50 μl of a 200 mM Na2CO3/NaHCO3 aqueous solution, pH 9.5, and then mixed with 30 μl of a 50 mM BzCl solution in acetonitrile (MeCN). The derivatization reaction was left for 40 min at room temperature for reactions in a 96-well plate (or vortexed for 30 s in case of reactions in microtubes) and then quenched by the addition of 20 μl of a 1 M HCl aqueous solution and 30 μl of a 1/1 solution of H2O/ACN. After centrifugation (1,008 g, 5 min, 4°C) on a 0.22-μm filtration 96-well plate (or with 0.22-μm syringe filters in case of derivatization in microtubes), the filtrates were analyzed by UHPLC-UV (eluent MeCN/H2O + 0.1% formic acid with a linear gradient 20/80 during 1 min, then 20/80 to 70/30 in 3 min, then 70/30 during 2 min followed by re-equilibration time; flow 0.5 ml min−1; temperature 25°C; injection volume 3 μl; λ = 250 nm).
FDAA derivatization: 20 μl of each reaction mixture was mixed with 8 μl of a 1 M NaHCO3 aqueous solution, pH 8.0, and then mixed with 20 μl of a 15 mM FDAA solution in 1/1 acetone/EtOH. The derivatization reactions were left 2 h at 55°C for reactions in a 96-well plate (or stirred at 600 rpm for 2 h at 65°C in case of reactions in microtubes), and then quenched by the addition of 4 μl of a 2 M HCl aqueous solution and 80 μl of a 1/1 solution of H2O/MeOH. After centrifugation (1,008 g, 5 min, 5°C) on a 0.22-μm filtration 96-well plate (or with 0.22 μm syringe filters in case of derivatization in microtubes), the filtrate was analyzed by UHPLC-UV (eluent MeOH/H2O + 0.1% formic acid with a linear gradient 40/60 to 80/20 in 5 min, then 80/20 during 3 min followed by re-equilibration time; flow 0.3 ml min−1; temperature 25°C; injection volume 3 μl; λ = 340 nm). In the case of 2a, the eluent was as follows: a linear gradient 30/70 to 80/20 in 10 min, then 80/20 during 3 min followed by re-equilibration time.
Effect of Substrate Loadings on Conversion of 1-Methoxypropan-2-one and Butan-2-one
The tolerance of MsmeAmDH to higher substrate concentrations was studied by monitoring the conversion of 1-methoxypropan-2-one (1d) into (S)-1-methoxypropan-2-amine (2d) and butan-2-one (1a) to (S)-butan-2-amine (2a) for 24–48 h. Reaction mixtures (100 µl), containing 0.2 mM NADP+, 1.1 eq. of glucose, 3 U ml−1 GDH105 in 2 M NH4HCO2 buffer, pH 9.0, 0.5 mg ml−1 or 1 mg ml−1 of purified MsmeAmDH, and substrate at a concentration range of 50 to 200–300 mM, were incubated in microtubes at 30°C at 400 rpm. Aliquots were taken at 24 and 48 h and diluted in 2 M NH4HCO2 buffer, pH 9.0, to reach theoretically 50 mM (or 10 mM for ee value determination) product concentration. Conversion to (S)-amines and ee values was determined in 96-well plates by UHPLC-UV after derivatization with BzCl and FDAA, respectively.
Semi-Preparative Scale Reactions
1d to 2d: In a 50-ml Greiner tube equipped with a screw cap was poured 1-methoxypropan-2-one (1d) (7.50 ml of a 1 M stock solution in water, 1 eq., 7.5 mmol), distilled water (14.90 ml), NH4HCO2 buffer, pH 9.0 (10.00 ml of a 10 M stock solution), NADP+ (2.00 ml of a 5 mM stock solution, 0.001 eq., 0.01 mmol), glucose (9.27 ml of a 0.89 M stock solution, 1.1 eq., 8.25 mmol), GDH105 (1.5 ml of a 100 U ml−1 stock solution), and MsmeAmDH purified by gel filtration (4.83 ml of a 5.2 mg ml−1 stock solution). The reaction was shaken at 30°C, 300 rpm with UHPLC-UV monitoring after BzCl derivatization in microtubes (Supplementary Figures S4, S5). Isolation of the product 2d was not performed.
1a to 2a: In a 50-ml Greiner tube equipped with a screw cap was poured butan-2-one (7.50 ml of a 1 M stock solution in water, 1 eq., 7.5 mmol), distilled water (15.92 ml), NH4HCO2 buffer, pH 9.0 (10.00 ml of a 10 M stock solution), NADP+ (2.00 ml of a 5 mM stock solution, 0,001 eq., 0.01 mmol), glucose (8.25 ml of a 1 M stock solution, 1.1 eq., 8.25 mmol), GDH105 (1.5 ml of a 100 U ml−1 stock solution), and MsmeAmDH purified by gel filtration (4.83 ml of a 5.2 mg ml−1 stock solution). The reaction was shaken at 30°C, 300 rpm with UHPLC-UV monitoring after BzCl derivatization in microtubes. After 24 h, the reaction was acidified to pH 1–2 with concentrated HCl and concentrated in vacuo to remove unreacted ketone (water bath 45°C, p = 35 mbar). After addition of 10 ml of water, the mixture was basified with KOH 10 M under cooling by ice bath and distilled under atmospheric pressure. Three fractions were collected [fraction 1: boiling point (bp) 65–70°C, fraction 2: bp 70–80°C, fraction 3: bp 80–85°C]. The three fractions were analyzed by UHPLC-UV and NMR demonstrating the presence of the desired product with more or less water and ammonia salt. The three fractions were combined, acidified with concentrated HCl (approximatively 2 eq.), and lyophilized. The (2S)-butan-2-amine (2a) was obtained as monohydrochloride salt (421 mg, 51% yield, white solids) with 92.6% ee as determined by UHPLC-UV after FDAA derivatization (Supplementary Figures S6–S8). NMR spectra were identical to standard amine commercially available. 1H NMR (600 MHz, D2O) δ 3.31 (m, 1H), 1.57–1.73 (m, 2H), 1.29 (d, J = 6.74 Hz, 3H), 0.98 (t, J = 7.60 Hz, 3H). 13C NMR (150 MHz, D2O) δ 49.3, 27.2, 17.2, 9.0. (Supplementary Figures S9–S12).
Modeling and Docking Experiments
The products (R/S)-butan-2-amine (2a), (2R/S)-2-amino-1-propanol (2e), and (2R/S)-2-aminobutanol (2h) were docked into the structures MsmeAmDH and CfusAmDH, and into the models of MicroAmDH and MATOUAmDH2 to analyze the observed stereoselectivities. Each amine product was docked as their (R)- and (S)-enantiomers separately. The ligand PDB files were generated using the CORINA Molecular Online Tool. The docking was done in the RX structures of CfusAmDH (PDB: 6IAU) and MsmeAmDH (PDB: 6IAQ) and in the homology models generated for MicroAmDH (PDB template used: 6IAQ) and MATOUAmDH2 (PDB template used: 6IAU) using SWISS-MODEL. With AutoDockTool (Morris et al., 2009), the docking simulations were performed on rigid structures, with no flexibility given to any catalytic pocket residues. The number of Genetic Algorithm (GA) runs were fixed at 50 using the Lamarckian GA (4.2). The 50 ligand conformations obtained were then analyzed in PyMOL.
Results
Conversions of Various Small Alkyl and Hydroxylated Ketones
In the context of biocatalytic applications, we aim to explore the synthetic potential of native AmDHs already described mainly through determination of specific activities and kinetic parameters. The AmDHs MsmeAmDH, CfusAmDH, MicroAmDH, and MATOUAmDH2, selected according to their reported substrate profiles, were tested with a cofactor recycling system over 24 h to provide conversion data with the small alkyl ketones, butan-2-one (1a), pentan-2-one (1b), 3-methylbutan-2-one (1c), 1-methoxypropan-2-one (1d), 1-hydoxypropan-2-one (1e), 4-hydroxybutan-2-one (1f), 3-hydroxybutan-2-one (1g), and 1-hydroxybutan-2-one (1h). Each substrate was tested at 10 and 50 mM to provide preliminary data for biocatalytic syntheses at a more substantial substrate concentration, while using a reasonable amount of purified enzyme (0.5 mg ml−1). All reactions were carried out in 96-well plates at 30°C in ammonium formate buffer, pH 9.0, with a glucose–glucose dehydrogenase NADP+ recycling system. The conversions were calculated based on calibration curves obtained with commercialized standards after derivatization of the reaction mixtures with BzCl and UHPLC-UV analysis performed in 96-well plates. The enantiomeric excesses were calculated after UHPLC-UV analysis of the same reactions derivatized with FDAA in a 96-well plate.
The results are shown in Figure 3 and detailed in Supplementary Figure S13. At 10 mM, low conversions (6.8%–18.5%) were obtained for small alkyl ketones devoid of any functionality, but interestingly, high ee values (96.0 and 98.1% respectively) were still recorded in the case of the smallest amine (S)-butan-2-amine (2a) with MsmeAmDH and CfusAmDH, despite the high similarity between the size of each substituent. We note that MATOUAmDH2 and especially MicroAmDH gave lower ee values (88.9% and 69.1%, respectively). Conversions and enantioselectivity results for (S)-pentan-2-amine (2b) were similar to that previously described for CfusAmDH, despite lower conversions observed experimentally in plates compared to closed microtubes, presumably due to undesired evaporation of the substrate. Very interestingly, good to high conversions of 1-methoxypropan-2-one (1d) to the corresponding amine (S)-MOIPA (2d) were achieved with CfusAmDH (63.9%), MsmeAmDH (56.5%), and MicroAmDH (78.4%). Lower conversions were obtained with MATOUAmDH2 (30.0%), together with slightly lower ee values (90.4% against 97.4–98.1) for the three other enzymes. Even more remarkable were the moderate to high conversions obtained with non-protected alcohols. With 10 mM as the starting substrate concentration, the small vicinal amino alcohol (3S)-3-aminobutan-2-ol (2g) was obtained with 64.3%–73.3% conversion with the four tested enzymes. For ketones 1e, 1f, and 1h with a terminal alcohol, the conversions were much more dependent on the enzyme. (S)-2-aminopropan-1-ol (2e) and (S)-2-aminobutan-1-ol (2h) were formed with low to moderate conversions with CfusAmDH and MATOUAmDH2 (26.1%–31.6% and 37.2%–13.4%, respectively) whereas good conversions were reached with MsmeAmDH and MicroAmDH (67.4%–57.6% and 60.9%–91.5%, respectively). (S)-3-aminobutan-1-ol (2f) was formed with only 9.6% with MATOUAmDH2, but with 48.2%–65.7% with the other three AmDHs. High stereoselectivity in favor of the (S)-enantiomer were confirmed for all these substrates with ee values generally above 96%, except for 2-aminobutan-1-ol (2h) for which an (R)- preference was determined in the case of CfusAmDH, even if the enantiomeric excess was quite low (48.1%). Taking into account Cahn-Ingold-Prelog rules, this is in accordance with the same positioning of the longest carbon chain of the substrate in the active site. For MsmeAmDH and MicroAmDH, the (S)-amines were formed with 67.0%–84.8% ee, whereas only 14.9% ee was measured with MATOUAmDH2, in addition to low conversion. Excellent ee values (98.8%–99.4%) were particularly obtained for (3S)-3-aminobutan-2-ol (2g) with CfusAmDH, MsmeAmDH and MATOUAmDH2 (98.2% with MicroAmDH). In case of this chiral substrate, bad diasteroisomeric excess was obtained with the four enzymes (10.4%–13.0%, Supplementary Figure S13) showing that they accept both enantiomers at the alcohol position. Overall, MicroAmDH displayed lower enantioselectivities compared to the three other enzymes, particularly for alkylketones 2a–2c and for 2-aminopropan-1-ol (2e). For the former, the not quantified (R)-enantiomer was present in larger amounts with MicroAmDH than with other enzymes, for which only traces could be detected. We noted that the four enzymes did not differentiate the stereochemistry at the vicinal OH position, with the trans/cis ratio ranging from 55.2% to 56.5%.
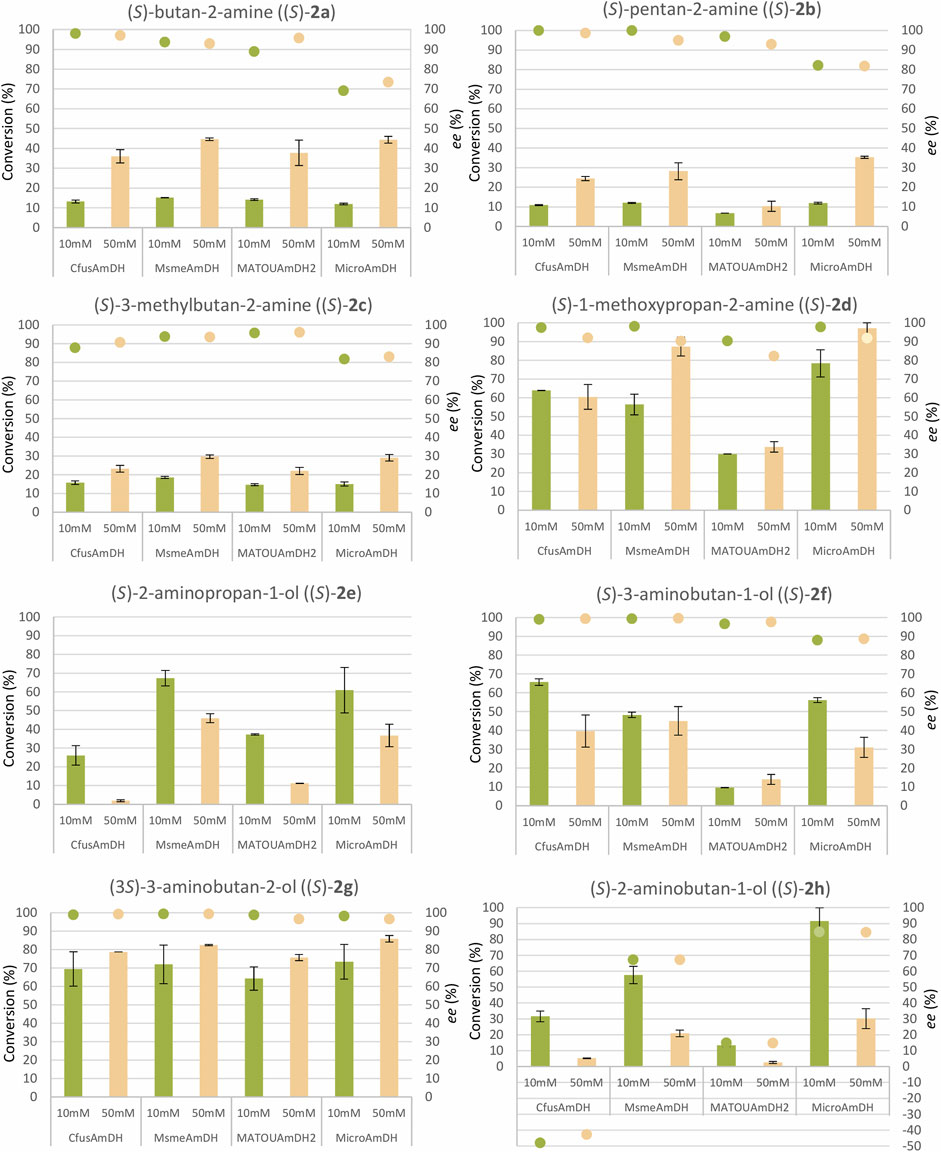
FIGURE 3. Conversions and enantiomeric excess results with CfusAmDH, MsmeAmDH, MATOUAmDH2, and MicroAmDH. Reactions conditions: 10 mM or 50 mM substrate, 2 M NH4HCO2 buffer, pH 9.0, 0.2 mM NADP+, 1.1 eq. glucose, 3 U ml−1 GDH105, 0.5 mg ml−1 purified AmDH, 24 h, 30°C. Error bars represent standard deviations of two or three independent experiments. Due to coelution with FDAA-derivatized co-product, the enantiomeric excess of (S)-2-aminopropan-1-ol (2e) could not be calculated, but the (S)-enantiomer was the major peak (Supplementary Figure 3E). The data are given in Supplementary Figure S13.
At 50 mM substrate, all the conversions were similar or even much higher compared with 10 mM for substrates 1a–1d without any free hydroxyl group, at equivalent concentration of biocatalyst. The conversion of butan-2-one (1a) was 2.7–3.7 times higher at 50 mM than at 10 mM. For the one and two more carbon substrates 1b and 1c, this behavior was also observed but to a lesser extent (1.5–3 times for 1b and 1.5–1.9 times for 1c). With free hydroxylated ketones 1e–1h, the increase in substrate concentration was tolerated by the four enzymes for 4-hydroxybutan-2-one (1f) and 3-hydroxybutan-2-one (1g) but not for 1-hydoxypropan-2-one (1e) and 1-hydroxybutan-2-one (1h), where significant decreases in conversion were observed. Among the four tested enzymes, CfusAmDH seemed to be the most affected. In terms of enantiomeric excess, overall, the ratio between enzymes for all the tested substrates was approximately conserved.
Increase of Butan-2-one and 1-Methoxypropan-2-one Concentration
Based on the screening results, we were prompted to test an increase in substrate concentration above 50 mM towards the small ketone butan-2-one (1a), which displayed a clear increase in yield between 10 and 50 mM. This study was conducted on MsmeAmDH and the reaction was monitored at 24 h (Figure 4A). The already high ammonia concentration was retained at 2 M, the same ratio NH4+/substrate could not be maintained for reasons of solubility and ionic strenghts. The results obtained after UHPLC-UV analysis showed that 150 mM of 1a was still converted in good yield (58.3%) to the corresponding (S)-amine 2a with 0.5 mg ml−1 of MsmeAmDH in 24 h. Even if the conversion dropped at 300 mM of substrate, the amount of amine formed was similar to that formed at 150 and 200 mM. The calculated ee values were similar for the amine 2a whatever the initial substrate concentration, i.e., approximately, 91.8%–92.0%.
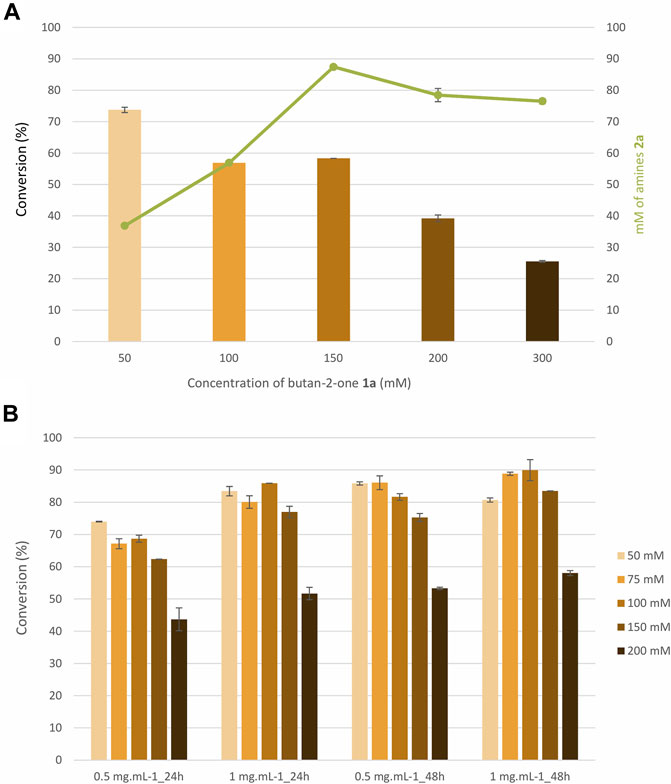
FIGURE 4. (A) and (B) Effect of concentration in butan-2-one 1a (A) and 1-methoxypropan-2-one 1d (B) on the conversion with MsmeAmDH. Reactions conditions: 10–200 mM substrate, 2 M NH4HCO2 buffer, pH 9.0, 0.2 mM NADP+, 1.1 eq. glucose, 3 U ml−1 GDH105, 0.5 mg ml−1, or 1.0 mg ml−1 purified AmDH, 24 h or 48 h, 30°C. Error bars represent standard deviations of two independent experiments.
A similar study was performed for 1-methoxypropan-2-one (1d) at two different enzyme loadings (Figure 4B). The conversions were similar at 50–150 mM and decreased 1.4–1.7 times at 200 mM depending on the reaction duration and enzyme concentration. Running the reaction for 48 h with 0.5 mg ml−1 or 24 h with twice the amount of enzyme enabled 81.7%–85.9% conversion of 100 mM of ketone, which were roughly the conversions obtained at 50 mM (74.0–85.9%). At 200 mM, the conversions did not exceed 58% whatever the conditions used, which were still similar to that obtained at 10 mM during the screening in 96-well plates (56.5%). At 200 mM, the space time yields were 5.2–7.9 g L−1 day−1, depending on the reaction time and enzyme loading.
Semi-preparative Synthesis of (S)-MOIPA ((S)-2d) and (S)-Butan-2-amine ((S)-2a)
In view of the tolerance to high loadings of 1-methoxypropan-2-one 1d and butan-2-one 1a, the semi-preparative syntheses of (S)-MOIPA ((S)-2d) and (S)-2a were performed to highlight the potential of these enzymes, and more particularly of wild-type MsmeAmDH. Therefore, the synthesis of (S)-MOIPA was carried out at 7.5 mmol scale at 150 mM substrate concentration over 48 h with 0.5 mg ml−1 of MsmeAmDH. Based on calibration curves, 6.6 mmol of (S)-MOIPA was formed (88.3% conversion, ee = 98.6%), superior to the result on the 100-µl scale, probably due to a better control of the reaction conditions at larger scale (Figure 5). Isolation of the product was not performed due to high water solubility and volatility of the product but the established industrial synthesis of this compound is evidence for the feasibility of this process step.
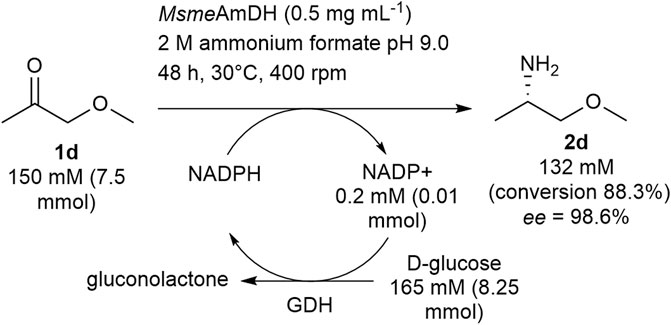
FIGURE 5. Semi-preparative synthesis of (S)-1-methoxypropan-2-amine (S)-2d.
The synthesis of 2a was also carried out at the 7.5 mmol scale at 150 mM substrate concentration over 24 h with 0.5 mg ml−1 of MsmeAmDH. The resulting conversion (>99%) was again higher than the one obtained at the 100-µl scale. Following a similar protocol to the one described by Heckmann et al., the desired (S)-butan-2-amine (2a) was isolated thanks to a distillation procedure with 51% yield and 92.6% ee after added lyophilization step (Figure 6).
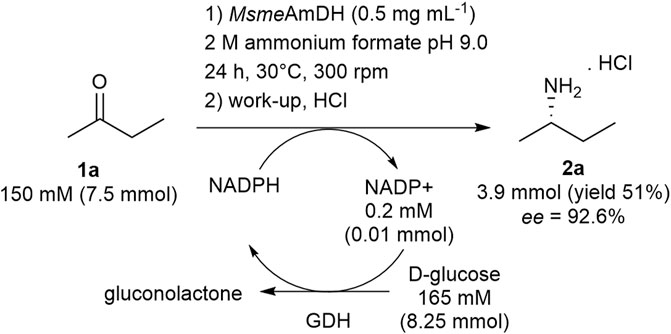
FIGURE 6. Semi-preparative synthesis of (S)-butan-2-amine (2a).
Modeling Studies
Taking advantage of the crystallographic structures of MsmeAmDH and CfusAmDH with their cofactor NADP+ (PDB ID: 6IAQ and 6IAU, respectively), molecular docking using some tested amines and the closed form of MsmeAmDH-NADP+ or CfusAmDH-NADP+ were performed using Autodock software (Morris et al., 2009). In addition, MicroAmDH was modeled by homology using the closed form of MsmeAmDH (PDB ID: 6IAQ) as a template, as already described for MATOUAmDH2 with CfusAmDH as a template (Caparco et al., 2020). Molecular docking was performed on these models following the same procedure as on MsmeAmDH or CfusAmDH. Among the fifty docking conformations obtained from the docking simulations (docking protocol detailed in Materials and Methods), the ratio between correct conformations harboring (S)-stereochemistry and those having (R)-stereochemistry was analyzed (Figure 7). As the final step of the reductive amination mechanism is the transfer of the hydride from C4 of the nicotinamide ring of the NAD(P)H cofactor to the C atom of the iminium intermediate to give respectively the oxidized NAD(P)+ and the amine product, only conformations having the correct orientation of the hydrogen toward the cofactor were selected.
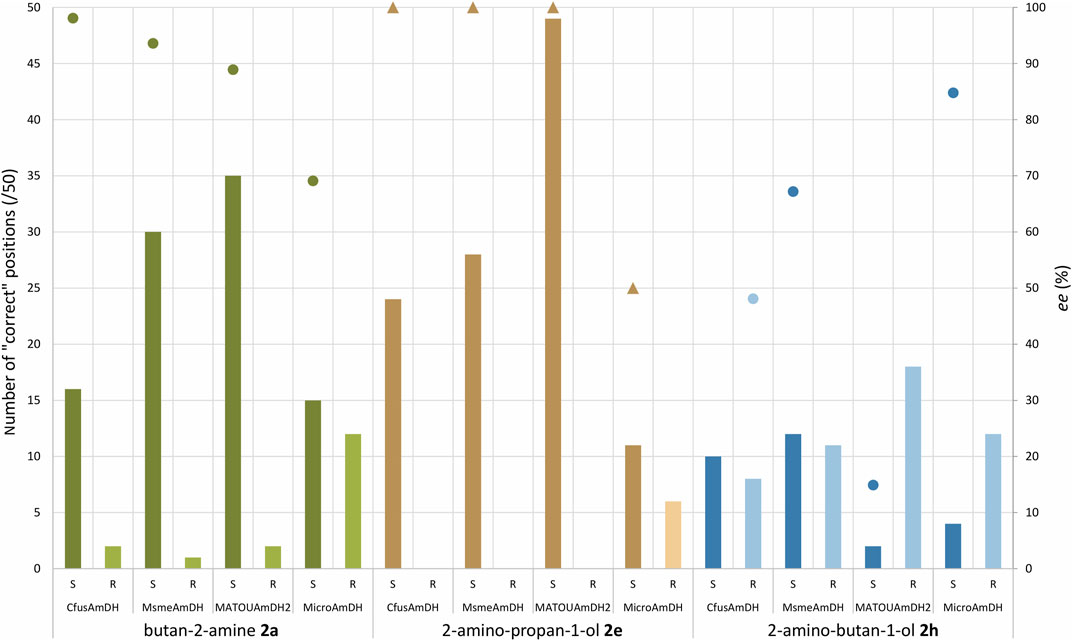
FIGURE 7. Docking simulation of (S)- and (R)-amines 2a, 2e, and 2h. Bars represent the number of “correct” positions obtained out of 50 generated conformations while the scatter plot represents the enantiomeric excess obtained from the corresponding reaction (circles: calculated experimental data, triangles: estimated experimental data). ee values are indicated for (S)-amines except for 2h with CfusAmDH which is for (R)-2h. For 2e ligand, ee values could not be determined but (R)-2e was only found in traces with CfusAmDH, MsmeAmDH, and MATOUAmDH2 (ee values arbitrarily indicated at 100%) but was obtained in larger proportion with MicroAmDH (ee arbitrarily indicated at 50%).
As detailed in Figure 7, for the butan-2-amine (2a) ligand, a majority of (S)-conformations were generated with the correct orientation in CfusAmDH, MsmeAmDH, and MATOUAmDH2, respectively 16, 30, and 35 out of 50 poses while the (R)-conformations gave respectively only 2, 1, and 2 correct positions out of 50 poses. On the other hand, for MicroAmDH, the difference is much lower with 15 out of 50 poses of (S)-conformations and 12 out of 50 poses of (R)-conformations. These in silico results are in accordance with the experimental data that showed good ee values for the first three enzymes (88.9%–98.1%) while MicroAmDH reached only a 69.1% ee with this substrate.
Based on the docking results, the different (R)/(S) ratios are difficult to explain as the positioning of the docked amines in their (R) and (S) conformations is almost identical, as illustrated in Figure 8 in the case of MsmeAmDH. No clear correlation between residues surrounding the substrate, or residues of the second layer, and the docking results with each enzyme could be established. Possibly, the features of the docking algorithm, such as the assumption of rigid protein and ligand, can obscure the reasons for these differences in enantioselectivity.
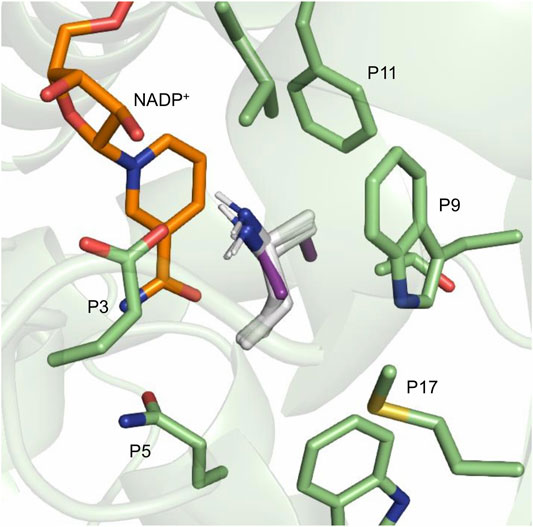
FIGURE 8. MsmeAmDH RX structure (PDB: 6IAQ) and NADP+ cofactor, in orange, with docked structures of (S)-2a, in white, and (R)-2a, in purple. All the (S)-2a docked structures belong to the 20 conformations displaying the lowest binding energy.
A good correlation between in silico and in vitro results was observed with the 2-amino-propan-1-ol (2e) ligand, for which only (S)-enantiomers were obtained both from these docking studies and from the biocatalytic reactions with CfusAmDH, MsmeAmDH, and MATOUAmDH2. Without being quantified, some (R)-2e was only experimentally identified with MicroAmDH while also being docked in 35% correct conformations [11/50 for (S)-conformation against 6/50 for (R)-conformation]. This different enantioselectivity and (R)-/(S)-ratios obtained with MicroAmDH with 2e can be due to a coordination by hydrogen bonding of the hydroxyl group of the ligand with the hydroxyl group of Y169 (P11), which can favor the positioning of the ketone for the formation of the corresponding (R)-amine (Figure 9). If we use the nomenclature already published (Mayol et al., 2019), where the residues composing the active site pocket were renamed as their spatial positions P1 to P20, this key residue is P11. Both mechanistic and structural roles have been postulated for P3 as the catalytic residue while P11 was hypothesized to be essential for the closing of the active site (Mayol et al., 2019). As a reminder, alignment of these P1–P20 residues in the studied AmDHs is presented in Figure 10.
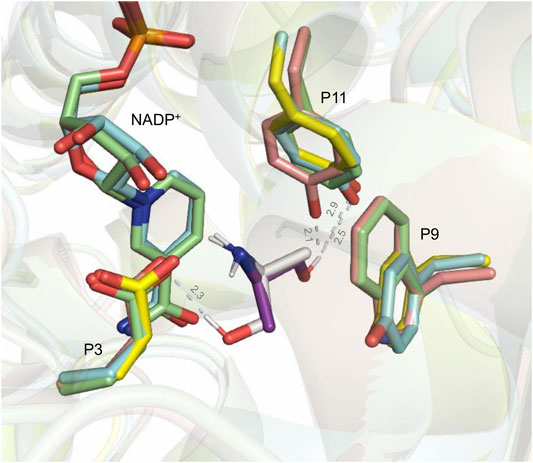
FIGURE 9. Structural alignment of CfusAmDH RX structure (PDB: 6IAU; blue), MicroAmDH homology model (PDB template: 6IAQ; pink), and MATOUAmDH2 homology model (PDB template: 6IAU; yellow) to MsmeAmDH RX structure (PDB: 6IAQ; green). The corresponding NADP+ cofactors are shown in the same color as the enzyme to which they belong. Docked structures of (S)-2e and (R)-2e in MicroAmDH are respectively shown in white and purple. The distance between the hydrogen atom of the hydroxyl group of (S)-2e and the oxygen atom of the carboxylate group of E104 (P3) in MicroAmDH, and between the hydrogen atom of the hydroxyl group of (R)-2e and the oxygen atom of the hydroxyl group of Y169, Y173, and Y176 (P11), respectively, of MicroAmDH, CfusAmDH, and MATOUAmDH2, are given in Å.
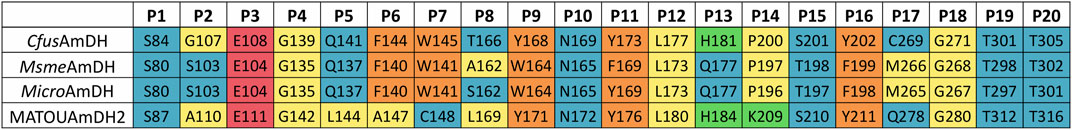
FIGURE 10. Comparison of the active site residues P1–P20 of the selected AmDHs. All the P1 to P20 residues listed come from an alignment with CfusAmDH (PDB: 6IAU). The color code used refers to the polarity and charge of the corresponding residue [blue: polar residues, yellow: hydrophobic residues, orange: aromatic residues, red: negatively charged residues, and green: positively charged residues (charges at physiological pH)].
In CfusAmDH and MATOUAmDH2, P11 is also occupied by Y, respectively Y173 and Y176, but with a slightly different position that moves the hydroxyl group further away from the ligand (∆ = 0.4 Å between Y169 of MicroAmDH and Y173 of CfusAmDH, and ∆ = 0.8 Å between Y169 of MicroAmDH and Y176 of MATOUAmDH2). In addition, the more hindered W164 (P9) in MicroAmDH compared to Y168 and Y171 in CfusAmDH and MATOUAmDH2 can also impart less flexibility to the alkylhydroxyl chain. In the docking poses, the latter is oriented more towards Y169 in MicroAmDH and its hydroxyl group, increasing access to (R)-configurations. In MsmeAmDH, P11 is occupied by a phenylalanine that cannot provide a hydrogen bond with the substrate to favor the formation of the (R)-amine. In that case, only (S)-2e can be docked with the hydroxyl group orientated towards the carboxylate function of the glutamate in P3, with which a hydrogen bond can occur (Figure 8).
For 2-aminobutanol (2h) ligand, overall, the in silico analysis revealed much more (R)-poses with all the enzymes compared to the other ligands, which is in accordance with the in vitro analysis displaying the lowest ee values for this amine. For example, in MsmeAmDH, approximately the same number of (S)- and (R)- correct poses were obtained, and the ee value for the (S)-2h was experimentally 67%. Nevertheless, the higher ees obtained experimentally for MicroAmDH and MsmeAmDH compared to MATOUAmDH2 and CfusAmDH are not directly related to the respective ratios of correct (R)- and (S)-conformations in this modeling study. For example, in MATOUAmDH2, the ratio of (R)/(S) poses was higher than with CfusAmDH, whereas CfusAmDH facilitated the formation of (R)-2h with an ee value of 48% and MATOUAmDH2 facilitated the formation of (S)-2h with an ee value of 15%. For CfusAmDH and MsmeAmDH, this ratio was similar (respectively 8/10 and 11/12), whereas MsmeAmDH displayed a clear preference for the formation of (S)-2h (ee = 67%). For CfusAmDH, many (R)-poses seemed stabilized with the hydroxyl group of Y173 (Figure 11A), thus explaining the greatest rate of formation of (R)-amines compared to other substrates, even if the ee value remained low (ee(R) = 48.1%). The more hindered ethyl moiety of (R)-2h, compared to (R)-2e, might “push” the rotation of the substrate, still keeping the chiral center close to the NADP+ hydride carbon, and make the hydroxyl group closer to the tyrosine even in CfusAmDH (2 Å) and MATOUAmDH2 (2.9 Å). It is noticeable that for MATOUAmDH2 and MsmeAmDH, the different conformations displayed much more variability of positions, explained structurally by the presence of other residues (P5, P8, P9, P16, and P17) surrounding these key positions, thus enabling other interactions and positioning of the key residues potentially less visible in these models (Figure 11B).
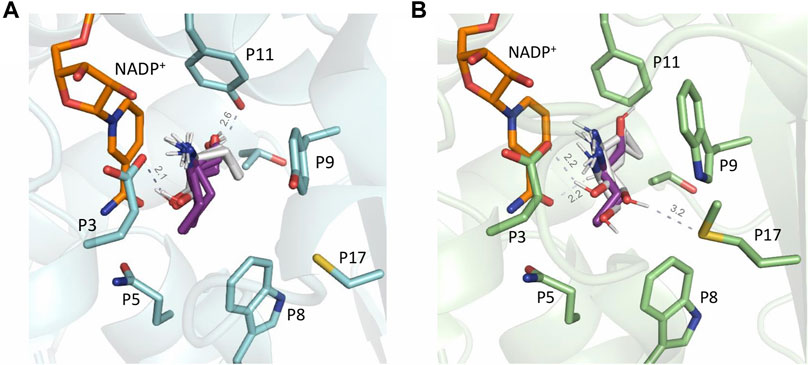
FIGURE 11. (A) CfusAmDH RX structure (PDB: 6IAU) and NADP+ cofactor, in orange, with docked structures of (S)-2e, in white, and (R)-2e, in purple; (B) MsmeAmDH RX structure (PDB: 6IAQ) and NADP+ cofactor, in orange, with docked structures of (S)-2e, in white, and (R)-2e, in purple.
Discussion
In addition to the protein engineering work that can be performed to access valuable biocatalysts, the deep characterization of wild-type enzymes selected from biodiversity is also a very efficient strategy. This study showed that recently described native AmDHs (CfusAmDH, MsmeAmDH, MicroAmDH, and MATOUAmDH2) appear as suitable biocatalysts for sustainable access to very small chiral amines. This was exemplified by moderate to high conversion rates obtained with 2-amino C3-C5 alkanes at high substrate loadings, and with hydroxylated 2-aminoalkanes such as (3S)-3-aminobutan-2-ol (2g) and (S)-1-methoxypropan-2-amine (2d), (S)-MOIPA. Despite its modeled larger catalytic pocket, MATOUAmDH2 displayed similar conversion rates and very high ee values, even if it seemed less well adapted for hydroxy ketones. The studied amines, challenging to obtain at high ee values due to inherent similarly sized substituents, are obtained with good to very good ee values with these enzymes. They are therefore positioned perfectly as a complement to the only few enzymes performing chemical reductive amination already described on these small ketones. Particularly, they differ from the imine reductase studied by Johnson-Matthey and Turner’s group reported to be active toward 1-methoxypropan-2-one (1d) but with an alkylated amine and not ammonia as amine source, and without further published details (Montgomery et al., 2020). In view of their previously described substrate scope, these native AmDHs clearly displayed preference for small substrates, and particularly these 2-aminoketones with C3–C5 carbon chain length, whereas RedAms such as NfRedAm, AtRedAm, or AdRedAm have been shown to be active toward carbon chain of C6 length or above and again mostly with larger amines than ammonia (Sharma et al., 2018; González-Martínez et al., 2020; Mangas-Sanchez et al., 2020). Compared to the engineered enzymes, only the modified AADH L-AmDH-TV was reported to be active toward the short C4 aliphatic ketones butan-2-one (1b). This modified leucine DH derived from Bacillus stearothermophilus displayed interesting specific activities toward pentan-2-one (1c) (1,303.6 mU mg−1), and, in a lesser extent, toward butan-2-one (1b) (225.5 mU mg−1) (Franklin et al., 2020). The absence of conversion data with these substrates does not allow to compare this enzyme more accurately with our native AmDHs. For the higher homolog 1c, both L-AmDH-TV and the mutant LE-AmDH-v1 from the ɛ-deaminating L-lysine dehydrogenase from Geobacillus stearothermophilus appeared to be more efficient biocatalysts (Tseliou et al., 2019). Nevertheless, their opposite enantioselectivity renders them complementary enzymes to CfusAmDH, MsmeAmDH, and MATOUAmDH2, but not alternative ones. Regarding the hydroxyl-functionalized tested ketones, the studied native AmDHs seemed competitive compared to the mutant AmDH-M0 displaying very interesting biocatalytic capabilities for the synthesis of different chiral vicinal amino alcohols. Even if no conversion data were reported by Chen et al. for the C3 amine 2e and the C4 amine 2f, the commonly studied amine (2S)-aminobutan-1-ol (2h) was obtained after a 24 h reaction with AmDH-M0 with 91% conversion and ee > 99% at 30°C starting from 50 mM substrate concentration (Chen et al., 2019). Taking into account that the data provided here for hydroxylated ketones may be underestimated due to a study carried out in 96-well plates, and that the work of Chen et al. was performed with a triple amount of purified enzyme, similar conversions (70%–90%) may be reached with MicroAmDH for 2h, a precursor of the drug ethambutol. Further improvement should permit higher tolerance to higher substrate concentrations and higher ee values for this substrate. Interestingly, the authors suggested a favorable coordination of the hydroxyl group of the ligand with some key residues, as we postulated here with Y in P11 in some native AmDHs. In their case, the hydroxyl group of 1-hydroxypentan-2-one may interact with the mutated residue T68, the same as for the carboxylic moiety of the amino acid in the AADH native enzyme. To access amines (S)-2e and (S)-2f via reductive amination, especially with high enantioselectivity, MsmeAmDH and CfusAmDH seemed to be the only described dehydrogenases.
Concerning enzymes not performing reductive amination, many data have been reported with TAs. For example, the (S)-1-methoxypropan-2-one (2d) was synthesized with a ω-TA (Höhne and Bornscheuer, 2009). Despite the completely different mechanism of action between TAs and reductive aminases such as AmDHs, we discussed here some results obtained with TAs to underline the key role of AmDHs in the biocatalytic synthesis of such compounds. Native AmDHs and more particularly MsmeAmDH seemed complementary to HewT or *RTA-X43 used by Heckmann et al. for the continuous-flow synthesis of butan-2-amine (2a). If we refer to the data provided in batch conditions without immobilization for more accurate comparison, MsmeAmDH behaves similarly to HewT or *RTA-X43 in terms of conversion increase with higher substrate loadings in the range 10–100 mM, but not above. Both TAs displayed similar or higher conversions at 300 mM compared to 100 mM, whereas conversions with MsmeAmDH drastically dropped above 150 mM. This behavior tends to suggest that these TAs have higher affinity for higher homologs than 1a whereas MsmeAmDH seemed more designed for small substrates such as 1a–1h, but limited by very high substrate loadings. MsmeAmDH favored the formation of the (S)-amine like HewT but with higher ee values (91% at 150 mM vs. 45% at 100 mM). Further exploration of AmDH homologs may enable the selection of (R)-selective AmDHs for 1a, to provide an alternative to *RTA-X43, highly stereoselective for the (R)-amine formation. Again, protein engineering on MsmeAmDH may also help to improve from the currently good ee value to excellent ee value for this target. As successfully demonstrated by the authors with TAs, scale-up synthesis of 2a in flow with these AmDHs may provide alternative process systems, also potentially extendable to at least 2b–2d, especially as these AmDHs do not require the use of a sacrificial amine donor such as isopropylamine (IPA), which clearly complicates the purification of the target amine. As exemplified in this work, the distillation protocol greatly elaborated by Paradisi et al. for 2a has been facilitated here by the absence of IPA.
CfusAmDH and MsmeAmDH are also complementary to RbTA, a (R)-selective TA applied by Li et al. for the synthesis of various (R)-hydroxyl amines such as (R)-ABOL (2f). CfusAmDH and MsmeAmDH harbor a less narrow substrate scope, as RbTA can convert C4-C7 ketones, but furnish the opposite (S)-enantiomer, thus widening the biocatalytic solutions for accessing such chiral building blocks. Obtained here in non-optimal condition in a 96-well plate with 48%–66% yield with excellent (S)-stereoselectivity (ee > 99%), the opposite enantiomer (S)-2f ((S)-ABOL) could be synthesized with MsmeAmDH or CfusAmDH without requiring high loading of IPA as it was done by Li et al. with 1 M IPA for 100 mM substrate 1f and 30 g L−1 of whole cells in 60 h (Li et al., 2021). Again, Y125 in RbTA was postulated to play a key role in substrate hydroxy ketone recognition by forming a hydrogen bond with the hydroxyl functionality according to docking results and loss of activity with the mutation Y125A, thus supporting our hypothesis of key role of P11 in our case.
The structurally based preliminary explanation of the slight differences in (S)-enantioselectivities observed experimentally for 2a–2g, together with the important variability for 2h, helps to better understand the enzyme-substrate interactions within the catalytic pockets of these four enzymes. It would be interesting to perform some deeper computational studies, including molecular dynamics simulations, as done by Tseliou et al., to explain the substrate-dependent stereo-switchable selectivity in the case of the mutant LE-AmDH-v1 (Tseliou et al., 2019). In silico studies with the iminium intermediates or carbinolamines, instead of the amine products, may provide additional explanations for the different rates of enantioselectivity observed depending on AmDHs and substrates.
This study highlights the potential of native AmDHs, especially CfusAmDH, MsmeAmDH, MicroAmDH and MATOUAmDH2, for the biocatalytic synthesis of short aliphatic amines and hydroxy amines. For the latter, in one simple step, high conversion rates and ee values can be reached, which is remarkable compared to conventional synthesis. Due to high (S)-enantioselectivity, they are perfectly complementary to the few reductive amination enzymes described for these substrates, which are mostly (R)-selective. They are also positioned as very good alternatives to TAs such as *RTA-X43. (S)-MOIPA currently produced by the ChiPros™ process at more than 5,000 tons per year is a nice example of what can be achieved with these native enzymes (non-optimized STY 7.9 g L−1 day−1 with MsmeAmDH). Protein engineering work coupled with the structural analysis provided should improve their efficiency and use in chemical synthesis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
CV-V conceived the project. LD, CV-V designed and conducted the experiments, with input of AC and MB. LD, GG, and CV-V analyzed data with input from AZ, AB, and JC. LD and CV-V wrote the manuscript with input from GG, AZ, AC, and AB. All authors read and approved the final manuscript.
Funding
This study was supported by the Commissariat à l’Énergie Atomique et aux Énergies Alternatives (CEA), the CNRS and the University of Evry Val d’Essonne.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank P. Wincker and V. de Berardinis for supporting the project, P. Sirvain and A. Perret for large-scale purification of the described enzymes, O. Maciejak (University Val d’Essonne) for NMR assistance, and the Region Ile de France for financial support of the 600-MHz spectrometer.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2021.781284/full#supplementary-material
References
Ager, D. J., Prakash, I., and Schaad, D. R. (1996). 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev. 96, 835–876. doi:10.1021/cr9500038
Bradford, M. M. (1976). A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 72, 248–254. doi:10.1016/0003-2697(76)90527-3
Caparco, A. A., Pelletier, E., Petit, J. L., Jouenne, A., Bommarius, B. R., Berardinis, V., et al. (2020). Metagenomic Mining for Amine Dehydrogenase Discovery. Adv. Synth. Catal. 362, 2427–2436. doi:10.1002/adsc.202000094
Chen, F.-F., Cosgrove, S. C., Birmingham, W. R., Mangas-Sanchez, J., Citoler, J., Thompson, M. P., et al. (2019). Enantioselective Synthesis of Chiral Vicinal Amino Alcohols Using Amine Dehydrogenases. ACS Catal. 9, 11813–11818. doi:10.1021/acscatal.9b03889
Cheng, F., Li, Q., Li, H., and Xue, Y. (2020). NAD(P)H-dependent Oxidoreductases for Synthesis of Chiral Amines by Asymmetric Reductive Amination of Ketones. Sheng Wu Gong Cheng Xue Bao 36, 1794–1816. doi:10.13345/j.cjb.190582
Cosgrove, S. C., Ramsden, J. I., Mangas‐Sanchez, J., and Turner, N. J. (2021). “Biocatalytic Synthesis of Chiral Amines Using Oxidoreductases,” in Methodologies in Amine Synthesis: Challenges and Applications. Editors A. Ricci, and L. Bernardi (Weinheim: Wiley VCH), 243–283. doi:10.1002/9783527826186.ch7
Ditrich, K. (2020). “Optically Active Amines on an Industrial Scale,” in Amine Biocatalysis (Stuttgart, Germany), 4.
Ducrot, L., Bennett, M., Grogan, G., and Vergne‐Vaxelaire, C. (2020). NAD(P)H‐Dependent Enzymes for Reductive Amination: Active Site Description and Carbonyl‐Containing Compound Spectrum. Adv. Synth. Catal. 363, 328–351. doi:10.1002/adsc.202000870
Franklin, R. D., Mount, C. J., Bommarius, B. R., and Bommarius, A. S. (2020). Separate Sets of Mutations Enhance Activity and Substrate Scope of Amine Dehydrogenase. ChemCatChem 12, 2436–2439. doi:10.1002/cctc.201902364
Gomm, A., and O’Reilly, E. (2018). Transaminases for Chiral Amine Synthesis. Curr. Opin. Chem. Biol. 43, 106–112. doi:10.1016/j.cbpa.2017.12.007
González‐Martínez, D., Cuetos, A., Sharma, M., García‐Ramos, M., Lavandera, I., Gotor‐Fernández, V., et al. (2020). Asymmetric Synthesis of Primary and Secondary β‐Fluoro‐arylamines Using Reductive Aminases from Fungi. ChemCatChem 12, 2421–2425. doi:10.1002/cctc.201901999
Heckmann, C. M., Dominguez, B., and Paradisi, F. (2021). Enantio-Complementary Continuous-Flow Synthesis of 2-Aminobutane Using Covalently Immobilized Transaminases. ACS Sustain. Chem. Eng. 9, 4122–4129. doi:10.1021/acssuschemeng.0c09075
Höhne, M., and Bornscheuer, U. T. (2009). Biocatalytic Routes to Optically Active Amines. ChemCatChem 1, 42–51. doi:10.1002/cctc.200900110
Koszelewski, D., Pressnitz, D., Clay, D., and Kroutil, W. (2009). Deracemization of Mexiletine Biocatalyzed by ω-Transaminases. Org. Lett. 11, 4810–4812. doi:10.1021/ol901834x
Li, F., Liang, Y., Wei, Y., Zheng, Y., Du, Y., and Yu, H. (2021). Biochemical and Structural Characterization of an ( R )‐Selective Transaminase in the Asymmetric Synthesis of Chiral Hydroxy Amines. Adv. Synth. Catal. 363, 4582–4589. doi:10.1002/adsc.202100636
Mangas-Sanchez, J., Sharma, M., Cosgrove, S. C., Ramsden, J. I., Marshall, J. R., Thorpe, T. W., et al. (2020). Asymmetric Synthesis of Primary Amines Catalyzed by Thermotolerant Fungal Reductive Aminases. Chem. Sci. 11, 5052–5057. doi:10.1039/d0sc02253e
Mayol, O., Bastard, K., Beloti, L., Frese, A., Turkenburg, J. P., Petit, J.-L., et al. (2019). A Family of Native Amine Dehydrogenases for the Asymmetric Reductive Amination of Ketones. Nat. Catal. 2, 324–333. doi:10.1038/s41929-019-0249-z
Montgomery, S. L., Pushpanath, A., Heath, R. S., Marshall, J. R., Klemstein, U., Galman, J. L., et al. (2020). Characterization of Imine Reductases in Reductive Amination for the Exploration of Structure-Activity Relationships. Sci. Adv. 6, eaay9320. doi:10.1126/sciadv.aay9320
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 30, 2785–2791. doi:10.1002/jcc.21256
Mutti, F. G., and Knaus, T. (2021). “Enzymes Applied to the Synthesis of Amines,” in Biocatalysis for Practitioners: Techniques, Reactions and Applications. Editors D. G. Gonzalo, and I. Lavandera (Weinheim: Wiley VCH), 143–180. doi:10.1002/9783527824465.ch6
Patil, M. D., Grogan, G., Bommarius, A., and Yun, H. (2018a). Oxidoreductase-Catalyzed Synthesis of Chiral Amines. ACS Catal. 8, 10985–11015. doi:10.1021/acscatal.8b02924
Patil, M. D., Grogan, G., Bommarius, A., and Yun, H. (2018b). Recent Advances in ω-Transaminase-Mediated Biocatalysis for the Enantioselective Synthesis of Chiral Amines. Catalysts 8, 254. doi:10.3390/catal8070254
Rehn, G., Ayres, B., Adlercreutz, P., and Grey, C. (2016). An Improved Process for Biocatalytic Asymmetric Amine Synthesis by In Situ Product Removal Using a Supported Liquid Membrane. J. Mol. Catal. B: Enzymatic 123, 1–7. doi:10.1016/j.molcatb.2015.10.010
Sharma, M., Mangas-Sanchez, J., France, S. P., Aleku, G. A., Montgomery, S. L., Ramsden, J. I., et al. (2018). A Mechanism for Reductive Amination Catalyzed by Fungal Reductive Aminases. ACS Catal. 8, 11534–11541. doi:10.1021/acscatal.8b03491
Slabu, I., Galman, J. L., Lloyd, R. C., and Turner, N. J. (2017). Discovery, Engineering, and Synthetic Application of Transaminase Biocatalysts. ACS Catal. 7, 8263–8284. doi:10.1021/acscatal.7b02686
Tseliou, V., Knaus, T., Masman, M. F., Corrado, M. L., and Mutti, F. G. (2019). Generation of Amine Dehydrogenases with Increased Catalytic Performance and Substrate Scope from ε-deaminating L-Lysine Dehydrogenase. Nat. Commun. 10, 3717. doi:10.1038/s41467-019-11509-x
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K., and Bornscheuer, U. T. (2021). Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 60, 88–119. doi:10.1002/anie.202006648
Keywords: native amine dehydrogenases, reductive amination, short chiral amines, amino alcohols, (S)-MOIPA, (S)-butan-2-amine
Citation: Ducrot L, Bennett M, Caparco AA, Champion JA, Bommarius AS, Zaparucha A, Grogan G and Vergne-Vaxelaire C (2021) Biocatalytic Reductive Amination by Native Amine Dehydrogenases to Access Short Chiral Alkyl Amines and Amino Alcohols. Front. Catal. 1:781284. doi: 10.3389/fctls.2021.781284
Received: 22 September 2021; Accepted: 29 October 2021;
Published: 26 November 2021.
Edited by:
Francesca Paradisi, University of Bern, SwitzerlandReviewed by:
Elaine O'Reilly, University College Dublin, IrelandJan von Langermann, University of Rostock, Germany
Copyright © 2021 Ducrot, Bennett, Caparco, Champion, Bommarius, Zaparucha, Grogan and Vergne-Vaxelaire. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carine Vergne-Vaxelaire, Y2FyaW5lLnZlcmduZUBnZW5vc2NvcGUuY25zLmZy