 Patrick T. Wood
Patrick T. Wood Morgan M. Seffrood
Morgan M. Seffrood Brett A. Colson
Brett A. Colson Julian E. Stelzer
Julian E. Stelzer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med., 26 February 2025
Sec. General Cardiovascular Medicine
Volume 12 - 2025 | https://doi.org/10.3389/fcvm.2025.1550649
This article is part of the Research TopicDisparities, controversies and unmet needs in the management of cardiomyopathiesView all 3 articles
Hypertrophic cardiomyopathy (HCM) is a genetic disorder in the heart caused by variants in sarcomeric proteins that disrupt myocardial function, leading to hypercontractility, hypertrophy, and fibrosis. Optimal cardiac function relies on the precise coordination of thin and thick filament proteins that control the timing, magnitude of cellular force generation and relaxation, and in vivo systolic and diastolic function. Sarcomeric proteins, such as cardiac myosin binding protein C (cMyBP-C) play a crucial role in myocardial contractile function by modulating actomyosin interactions. Genetic variants in cMyBP-C are a frequent cause of HCM, highlighting its importance in cardiac health. This review explores the molecular mechanisms underpinning HCM and the rapidly advancing field of HCM translational research, including gene therapy and small-molecule interventions targeting sarcomere function. We will highlight novel approaches, including gene therapy using recombinant AAV vectors and small-molecule drugs targeting sarcomere function.
Heart disease is a major health challenge in the United States, contributing to millions of hospitalizations and accounts for 1 in every 5 deaths annually (1). Among the various forms of heart disease, hypertrophic cardiomyopathy (HCM) is the most common genetic cardiomyopathy, affecting approximately 1 in 500 people in the U.S (2). GWAS studies have shown that HCM is an autosomal dominant disease caused by one or more genetic variants in sarcomeric proteins (3–7). Alterations to sarcomere proteins result in either a gain or loss of function at the cellular level. This leads to dysregulation in the precise coordination of cardiac contraction and relaxation at the whole organ level.
Clinically, HCM presents as a heterogeneous phenotype that leads to variable degrees of cardiac remodeling (8–10). Common features of HCM include asymmetrical thickening of the left ventricular (LV) wall, hyperdynamic systolic performance, and a preserved or elevated left ventricular ejection fraction (LVEF) (10, 11). HCM is defined as either non-obstructive (nHCM), or obstructive (oHCM) depending on the severity of hypertrophy and the degree of left ventricular outflow tract obstruction (LVOTO), with the majority of patients with HCM experiencing LVOTO either at rest or during movement (10, 12).
Historically, therapeutic interventions such as beta-blockers (b-blockers), and non-dihydropyridine calcium channel blockers (CCBs) have been used to treat HCM symptoms by modulating adrenergic and calcium signaling pathways to improve patient quality of life (10, 13), but these therapies do not directly address the underlying problem of pathogenic variants in sarcomeric proteins (10, 14). Recent advances in therapeutic interventions are shifting toward directly targeting the primary defects found in altered sarcomere proteins to restore normal cardiac function at the molecular level (13, 15). This emerging paradigm highlights the growing interest in linking genotype to phenotype and connecting basic research to clinical practice.
Cardiac sarcomeric function is governed by the complex interaction of actin and myosin, in addition to several structural/regulatory proteins, such as the troponin complex, tropomyosin, and cardiac myosin binding protein C (cMyBP-C). These proteins are highly regulated and modulate contractile function in response to extracellular signaling (16). Numerous clinical studies demonstrate that pathogenic genetic variants of the myosin MYH7 gene or cMyBP-C MYBPC3 gene account for roughly 80% of all genetic variants that result in HCM (3, 9, 17), with MYBPC3 genetic variants being the most common (3, 18, 19).
Variants of MYBPC3 are a growing area of research due to its regulatory role in the sarcomere [see reviews, (19, 20)]. Localized in the C-zone of the A-band, cMyBP-C is anchored to the thick filament and arranged to create nine regularly spaced stripes ∼42 nm apart (21, 22). The 140-kDa protein consists of 11 globular immunoglobulin and fibronectin-like domains (C0–C10), with a flexible and phosphorylatable M-domain between C1 and C2, a key regulatory region (23–25).
Under the control of β-adrenergic stimulation, cMyBP-C's phosphorylation is regulated to control pressure development and relaxation in vivo (26). Unphosphorylated, cMyBP-C preferentially binds to myosin and acts as a mechanical tether that restrains the myosin heads of the thick filament (27–29). In this state, the myosin heads are spatially restricted, reducing their mobility and probability of interacting with actin, thus slowing the rate of force generation (20, 26, 30). The sequestration of the myosin heads is relieved via phosphorylation of the N-terminal domains (NTDs) of cMyBP-C, specifically at sites within the M-domain (20, 23, 31). The phosphorylation sites are targeted by various kinases, including protein kinase A (PKA), Ca2+/calmodulin-dependent protein kinase II, protein kinase C (PKC), and protein kinase G (PKG) allowing for precise adrenergic control over cardiac contraction (23, 32–34). In response to exercise or stress where increased cardiac output is necessary, phosphorylation of cMyBP-C relieves the restriction of myosin and reduces the tethering of cross-bridges via a reduction in cMyBP-C affinity for both myosin subfragment 1 and 2 (S1, S2) and actin (25, 35–37). The reduced constraint on myosin increases the probability of cross-bridge formation and enhances the transition of cross-bridges to force-bearing states, thereby accelerating contraction and increasing force generation (20, 26, 38, 39).
In vitro studies have also shown that specific regions of cMyBP-C, particularly the C0-C2 region, also modulate force generation (36, 40–42) by binding to the thin filament at low calcium concentrations to displace tropomyosin, and activate the thin filament (40, 43, 44). This way through interactions with myosin, actin, or both, cMyBP-C provides a high level of fine-tuning to cardiac contractility with a relatively low cMyBP-C to myosin ratio (1:8) and cMyBP-C to actin ratio (1:12) (36, 45).
In patients with MYBPC3-based HCM, the well-regulated control of the sarcomere is lost due to genetic variants in the protein. Pathogenic variants in MYBPC3 occur primarily in two forms: nonsense and missense, both of which play distinct roles in the development of HCM (9). The majority of pathogenic HCM-causing MYBPC3 variants are heterozygous frameshift, nonsense, or splice site variants (46, 47). These genetic variants result in the introduction of premature termination codons in one allele and theoretically result in the production of truncated cMyBP-C protein, however, truncated cMyBP-C proteins have not been observed in patient HCM cardiac tissue (47–49). Data from patient-derived, MYBPC3-variant induced pluripotent stem cell cardiomyocytes (iPSCMs) suggests that the MYBPC3 mRNA transcript is degraded through nonsense-mediated RNA decay resulting in protein haploinsufficiency (50). Reduction in cMyBP-C protein expression disrupts the stoichiometric balance between cMyBP-C, myosin, and actin in the sarcomere, leading to a loss of regulation over cross-bridge formation, and force generation (Figure 1) (19, 29, 48, 51). Patients with heterozygous MYBPC3 truncating variants can have a range of symptoms that develop over their life span, while patients with homozygous MYBPC3 truncating variants develop severe HF within the first year of life (52–54).
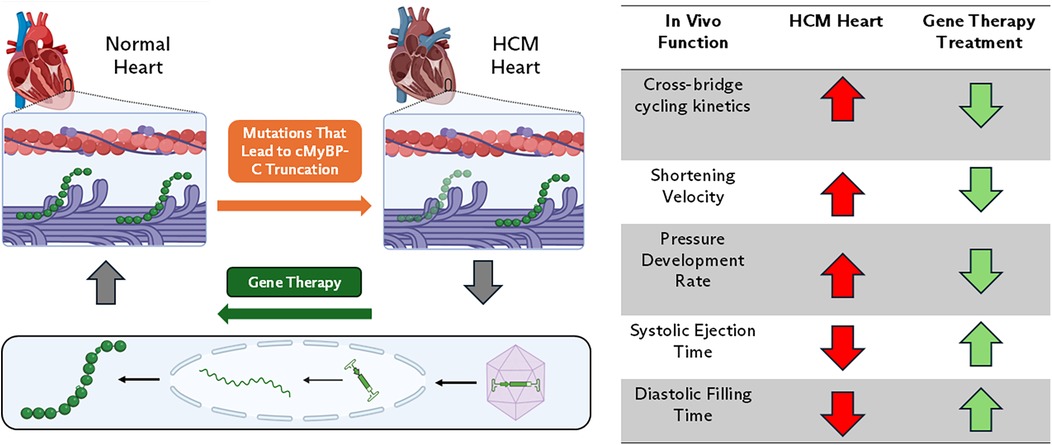
Figure 1. cMyBP-C haploinsufficiency-based HCM. In this form of disease, genetic variations in MYBPC3 result in prematurely truncated cMyBP-C protein. Reduction in cMyBP-C expression disrupts the stoichiometric balance between cMyBP-C, myosin, and actin in the sarcomere resulting in dysregulation of sarcomere contractility. Gene therapy using AAV9 aims to restore cMyBP-C levels to normal and mechanistically slow shortening velocity and increase thin filament drag. In vivo, this normalizes rates of pressure development and systolic ejection times, thereby improving systolic ejection and diastolic filling.
In contrast, pathogenic missense variants in MYBPC3 involve a single nucleotide substitution that results in the replacement of one amino acid with another in the protein (19, 55). Unlike truncating variants that result in haploinsufficiency, pathogenic missense variants typically lead to the production of full-length proteins that can be incorporated into the sarcomere, resulting in a dominant negative effect (19, 55). These variants impair cMyBP-C's ability to regulate actin/myosin interactions, however, the exact mechanism of impairment may vary depending on where in the protein the genetic variation is located (32, 56, 57). Some variants may alter cMyBP-C's structure to increase/decrease actin/myosin binding while others may affect how the protein is phosphorylated (32, 57–61). The example in Figure 2 highlights how a theoretical variation in the NTD can cause a change in protein structure, leading to increased cMyBP-C/actin binding and decreased cMyBP-C/myosin binding. Due to the low population frequency of missense variants, it is difficult to interpret how patients may develop disease and respond to treatments. This has led to a rise in novel molecular and computational screens that can be used to improve individualized patient care (58, 59, 62).
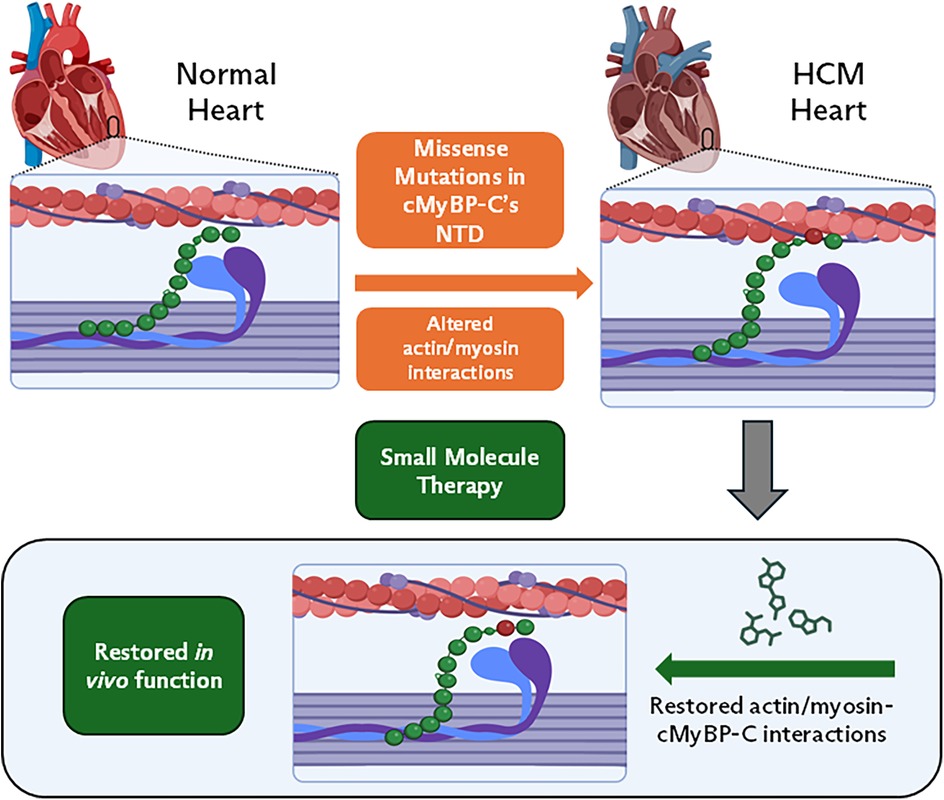
Figure 2. cMyBP-C missense variations in HCM. In this form of disease, genetic variations in MYBPC3 result in single nucleotide substitutions. These variations can have a wide range of effects on cMyBP-C's ability to regulate actin/myosin interactions. Small molecule (SM) therapy can revert hyper-contractility to normal, by restoring cMyBP-C's interactions with actin/myosin. This will have similar effects to in vivo function seen in Figure 1 table inset (right).
Treating HCM is difficult, in part, due to disease heterogeneity and long patient histories with several potential confounding variables. Current treatment strategies focus on relieving symptoms of HCM and improving patient quality of life through the use of pharmaceuticals with negative inotropic and chronotropic effects (10). Current AHA/ACC guidelines for the treatment of HCM suggest using non-vasodilating b-blockers first, followed by non-dihydropyridine CCBs (10). At the molecular level, b-blockers and CBCs decrease systolic function by blocking signaling pathways, inhibiting PKA, and decreasing intracellular calcium levels in cardiac myocytes to reduce contractility (63, 64). Due to their indirect mechanism of action, these drugs have several off-target side effects and do not specifically address genetic variants in sarcomeric proteins that a patient may have (65, 66).
Recent therapeutic advances are changing the paradigm in the treatment of HCM. The concept of direct modulation of the sarcomere provides an alternative approach to minimize off-target effects associated with indirect treatments (13, 15, 65). Unlike therapies that focus on symptom relief, direct sarcomere modulation addresses the regulation of contraction at the mechanistic level to reduce hypercontractility. Current direct modulation therapies for HCM treatment involve selective, allosteric, small molecules that specifically modulate cardiac myosin to directly affect the probability of actin-myosin cross-bridge formation (67–70). Drugs such as Mavacamten (MYK-461) and Aficamten (CK-274) are currently undergoing clinical trials to explore their effectiveness in treating and managing HCM (67, 68, 71). These novel therapeutics are first in class for the treatment of HCM, but like any new therapy, they have their caveats.
Clinical trials for Mavacamten, PIONEER-HCM, EXPLORER-HCM, and VALOR-HCM, were conducted on a limited patient population with oHCM (66, 71–73). These patients did not undergo genetic screening; leaving uncertainties about how Mavacamten affects patients with MYBPC3 genetic variants or other genetic variants. Studies using myocardium isolated from cMyBP-C−/− knockout (KO) mice, noted that higher concentrations of MYK-461 were required to achieve similar degrees of force depression compared with WT myocardium (74) suggesting genetic variants may play a role in the efficacy of MYK-461. Furthermore, it remains unclear how patients with other types of HCM, such as non-obstructive HCM (nHCM), may respond to Mavacamten treatment. These limitations ultimately reduce the utility of this drug to a smaller subset of the population. To address the critical need for the development of new therapies, there are currently two options under investigation: gene therapy or small molecule therapy (62, 75–80). Both therapy options allow for a more personalized and variant-specific approach to treatment.
The goal of gene therapy is to correct sarcomeric variants that drive the development of HCM by correcting haploinsufficiency, engineering therapeutic proteins, or modifying the expression of existing genes (81). There are several modalities of gene therapy for treating genetic-based HCM, including gene replacement, silencing, or editing.
For patients with truncation variants of MYBPC3, treatments that involve increasing cMyBP-C protein expression have shown promising results in small animal studies (82). Gene replacement therapies use cardiac targeting expression vectors, such as AAV9 vectors, with cardiac-specific promotors to increase the amount of cMyBP-C protein in the cardiomyocyte. This can be done with full-length peptides or therapeutic cMyBP-C protein fragments (Figure 1) (75, 77, 78, 83). Physiologically, in vivo reintroduction of cMyBP-C protein has been shown to slow shortening velocity in cMyBP-C −/− mice (83). At the molecular level, increasing the amount of cMyBP-C protein or protein fragments restricts myosin head availability, leading to the normalization of cross-bridge kinetics and cycling (75, 77, 83). Additionally, the reintroduction of protein or fragments has been shown to restore cMyBP-C phosphorylation via β-adrenergic stimulation (77). Restoration of cMyBP-C at the molecular level translates in vivo to a normalized rate of pressure development and systolic ejection time, thereby improving systolic ejection and diastolic filling (75, 77, 83).
The first MYBPC3-based clinical trial to treat HCM patients using a gene replacement technique is underway. The MyPeak-1 (Phase 1b) clinical trial started in 2023 to evaluate the safety and clinical efficacy of an AAV gene replacement therapy “TN-201” for pathogenic MYBPC3 variants related to HCM (NCT05836259). The therapy is a one-time treatment using an IV infusion of full-length MYBPC3 AAV. To be included in the trial, patients must be diagnosed with HCM (oHCM or nHCM) and have a LVEF >45%. Additionally, patients must undergo genetic testing to confirm the presence of pathogenic or likely pathogenic MYBPC3 truncating variants. During the 5-year study, clinicians will monitor changes in patient RV cMyBP-C protein levels, VO2-max, LV mass index, LV filling pressure, and New York Heart Association functional class changes (80). The goal is to halt disease progression or reverse symptoms over 5 years. Interim data from Cohort-1 showed that a low dose of TN-201 was well tolerated and the trial is currently recruiting patients for a higher dose trial, however, further public data is limited at the time of writing (80).
Other therapeutic options may be better suited for patients with missense variants that result in full-length proteins with negative effects. Under these conditions, gene replacement therapy might not fully correct the HCM phenotype as the missense variant is still transcribed. To correct genetic variants that result in cMyBP-C structural changes causing hypercontractility, gene silencing techniques could use rAAV to deliver small interfering RNAs (siRNAs) to stop transcription, or silence, pathogenic genes (84). This technique successfully prevented HCM pathology for at least 6 months in mice carrying a pathogenic variant in the β-myosin heavy chain gene (76) and could be adapted for MYBPC3 variants. However, SiRNA therapy could be difficult to implement in MYBPC3-HCM patients, as this therapy could result in a haploinsufficient phenotype.
Alternatively, missense variants can be corrected or edited using a genome editing technique instead of silenced. By delivering an RNA-guided Cas9 nuclease via AAV9, the variant gene can be edited back to its non-variant state (79). Genetic editing of β-myosin heavy chain gene R403Q variant mice corrected 70% of variant transcripts and prevented the associated HCM phenotype (79). While promising, gene therapy is still a novel technique and faces barriers like manufacturing ability, scalability, and cost (85). Additionally, some patients may also have strong immunity to AAV-based therapies that prevent delivery and transcription of the treatment, thus these patients may require higher doses (85) or alternative therapies, such as small molecules.
Small-molecule drugs that bind to cMyBP-C and alter its interaction with the thick and thin filaments could be used to improve cardiac function in HCM patients with genetic variants of MYBPC3. By acting to inhibit or enhance cMyBP-C binding with actin and myosin, small molecules directly target the dysfunctional protein interactions at the sarcomeric level to normalize cardiac muscle performance at the whole heart level (Figure 2).
To treat HCM patients with hyper-contractility and incomplete relaxation, the mechanisms of drug action on cMyBP-C function can either increase or decrease interactions with actin and myosin to result in reduced contractility and enhanced relaxation. In one scenario, several HCM-linked missense variants located in the M-domain of cMyBP-C exhibit enhanced interactions with actin and myosin (30, 61). Thus, compounds designed to modulate actin-cMyBP-C interactions and reduce myocardial ATPase activity (58, 62) could result in therapies that directly normalize affinities and conformations of variant cMyBP-C with the myofilaments, improving heart function. Contrary, in the case of cMyBP-C haploinsufficiency (i.e., less cMyBP-C protein) or missense variants in non-cMyBP-C sarcomeric proteins (i.e., beta-myosin) that result in hyper-contractility due to an increased probability of cross-bridges, drugs designed to modulate cMyBP-C interactions with myosin to decrease actin-myosin interactions in favor of relaxed myocardium and reduced contractility (27–29, 86) could mitigate hyper-contractility in these patients. Alternatively, drugs that target cMyBP-C to modulate its binding to actin and tropomyosin to lessen cMyBP-C-induced thin filament activation that sustains force generation (43, 44) could reduce hyper-contractility and enhance relaxation. Thus, cMyBP-C targeting modulators for the treatment of HCM would be expected to impact in vivo cardiac performance by suppressing systolic contractility while enhancing diastolic relaxation to ameliorate symptoms.
There are currently no known cMyBP-C binding drugs despite the desirability of such modulators of cMyBP-C. This is due to the lack of high-throughput screening (HTS) assays to identify drugs that bind specifically to cMyBP-C and alter its function. Current industry screening methods mostly rely on myosin ATPase assays which have been used to identify small molecule treatments for HCM such as Mavacamten and Omecamtiv mecarbil (69, 87) which target cardiac myosin. It is possible that these myosin ATPase assays (activated by actin, Ca2+, cMyBP-C NTDs), could be used with the addition of cMyBP-C or its fragments (e.g., C0-C2); however, these methods are not ideally suited for identifying cMyBP-C-specific binding compounds (without actin or myosin binding properties). Other approaches such as using genetically altered or iPSC-derived cardiomyocytes in contractility assays, are time-consuming, expensive, and labor-intensive and still have their caveats. Testing for small molecules that specifically target and bind to cMyBP-C and modulate its interactions with actin or myosin is the critical first step in the drug development process. Using HTS assays expedites this process and accelerates the identification of drugs for treating HCM.
Fluorescence lifetime (FLT) and FLT-detected FRET assays using probes on human cMyBP-C C0-C2, and actin (or myosin) have recently been developed (58, 60–62). Combined with a plate reader, drug screens of ∼1,300–2,800 compounds have been performed (58, 62). Pharmacologically active compounds identified with changes in lifetime or FRET indicated a change in cMyBP-C binding to actin. Further testing of some of the compounds identified from the HTS in vitro assays on function revealed effects on modulating myosin ATPase activity measured by in situ assays using skinned cardiac or skeletal muscle (88). Thus, the HTS assays developed can quickly and accurately detect valid cMyBP-C binding compounds to be further validated in downstream functional assays of muscle function (62).
Currently, small-molecule modulators binding to C0-C2 have been identified to either inhibit or enhance interactions with actin (58, 62). A subset of these modulators affect the Ca2+-sensitivity ATPase activity in cardiac myofibrils (62). For example, Pneumocandin B0 increased ATPase activity in cardiac myofibrils and was identified to increase FRET between cMyBP-C C0-C2 and actin in the screen (62), suggesting it may be an activator of cMyBP-C. In another example, Enoxaparin sodium decreased ATPase activity in cardiac myofibrils and was identified to decrease FRET between C0-C2 and actin in the screen (62), suggesting it could be an inhibitor of cMyBP-C. A cMyBP-C C0-C2-myosin assay has also been developed (61). In addition to primary HTS for compounds affecting myosin-cMyBP-C interactions, these assays are also useful for deprioritizing actin and myosin-binding drugs (to prioritize cMyBP-C binding drugs). Future work will establish the efficacy of screened compounds in vitro and in vivo models of HCM.
Ideal compounds would only bind to cardiac MyBP-C and not skeletal MyBP-C (62). Additionally, they may also bind to the interface of cMyBP-C in complex with actin or myosin. The drugs should have good ADMET properties, such as cell permeability, be nontoxic, and have minimal side effects. Additionally, ideal compounds should effectively treat certain HCM subtypes (obstructive or non-obstructive, cMyBP-C or non-cMyBP-C variants).
Several avenues for future investigation are in development using the techniques described above to treat HCM and other cardiomyopathies. Gene therapies could introduce designer cMyBP-C proteins that mimic different phosphorylation states, thereby providing more nuanced and targeted treatment for different cardiomyopathies (75). The engineered cMyBP-C variants would be designed to express phospho-mimetic variants that can modulate contractility in specific HCM subtypes or other cardiomyopathies where increased or decreased contractility might be beneficial. Similarly, drugs that mimic cMyBP-C phosphorylation by affecting cMyBP-C structure (30, 60, 62, 89) and function (90, 91) can enhance myocardial contraction and relaxation. Importantly, these perturbations would be independent of adrenergic stimuli or neurohormonal signaling, which are often dysregulated in heart failure.
cMyBP-C-targeted small molecules could also treat other cardiac disorders, such as dilated cardiomyopathy (DCM) by reducing myosin and increasing actin interactions to enhance contractility. Likewise, non-genetic HF (HFrEF/HFpEF) by similar mechanisms of action to change cMyBP-C interactions as in HCM/DCM for alleviating dysfunction of hypo- or hyper-contractility could provide effective new intervening therapies.
HCM is a diverse and complicated disease that can have multiple genetic aspects in different sarcomere and potentially non-sarcomere proteins (6, 7). By working closely with clinicians and developing genetic screens and patient-specific treatment regimes, it may be possible to combine multiple therapeutic approaches to deliver a personalized approach to HCM treatment. Additionally, investigating the safety and efficacy profiles of these novel interventions in pediatric populations may offer unprecedented opportunities for preventing or delaying the onset of HCM in genetically susceptible individuals (52). These novel therapies will need to be tested against current standard-of-care treatments to establish cMyBP-C-targeted therapies within the broader therapeutic landscape.
PW: Writing – original draft, Writing – review & editing. MS: Writing – original draft, Writing – review & editing. BC: Writing – original draft, Writing – review & editing. JS: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants awarded by the NIH National Heart, Lung, and Blood Institute (NHLBI) grants R01HL146676 (JS), R01HL153236 (JS), R01HL141564 (BC), the American Heart Association (AHA) grants 2022TPA961478 (JS), National Institute of Arthritis and Musculoskeletal and Skin Diseases grants R01AR079435 (BC), R01AR079435-S1 (BC, MS).
Figures 1, 2 were created with a licensed version of Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Martin SS, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. 2024 heart disease and stroke statistics: a report of US and global data from the American Heart Association. Circulation. (2024) 149(8):e347–913. doi: 10.1161/CIR.0000000000001209
2. Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. (2015) 65(12):1249–54. doi: 10.1016/j.jacc.2015.01.019
3. Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. (2015) 17(11):880–8. doi: 10.1038/gim.2014.205
4. Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. (2011) 364(17):1643–56. doi: 10.1056/NEJMra0902923
5. Biddinger KJ, Jurgens SJ, Maamari D, Gaziano L, Choi SH, Morrill VN, et al. Rare and common genetic variation underlying the risk of hypertrophic cardiomyopathy in a national biobank. JAMA Cardiol. (2022) 7(7):715–22. doi: 10.1001/jamacardio.2022.1061
6. Tadros R, Zheng SL, Grace C, Jorda P, Francis C, Jurgens SJ, et al. Large scale genome-wide association analyses identify novel genetic loci and mechanisms in hypertrophic cardiomyopathy. medRxiv [Preprint]. (2023). doi: 10.1101/2023.01.28.23285147
7. Tadros R, Francis C, Xu X, Vermeer AMC, Harper AR, Huurman R, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. (2021) 53(2):128–34. doi: 10.1038/s41588-020-00762-2
8. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. (2002) 287(10):1308–20. doi: 10.1001/jama.287.10.1308
9. Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. (2001) 104(4):557–67. doi: 10.1016/S0092-8674(01)00242-2
10. Ommen SR, Ho CY, Asif IM, Balaji S, Burke MA, Day SM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology joint committee on clinical practice guidelines. Circulation. (2024) 149(23):e1239–311. doi: 10.1161/CIR.0000000000001250
11. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. (2017) 121(7):749–70. doi: 10.1161/CIRCRESAHA.117.311059
12. Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, Kuvin JT, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. (2006) 114(21):2232–9. doi: 10.1161/CIRCULATIONAHA.106.644682
13. Kawana M, Spudich JA, Ruppel KM. Hypertrophic cardiomyopathy: mutations to mechanisms to therapies. Front Physiol. (2022) 13:975076. doi: 10.3389/fphys.2022.975076
14. Rossignol P, Hernandez AF, Solomon SD, Zannad F. Heart failure drug treatment. Lancet. (2019) 393(10175):1034–44. doi: 10.1016/S0140-6736(18)31808-7
15. Holmes JB, Doh CY, Mamidi R, Li J, Stelzer JE. Strategies for targeting the cardiac sarcomere: avenues for novel drug discovery. Expert Opin Drug Discov. (2020) 15(4):457–69. doi: 10.1080/17460441.2020.1722637
16. van der Velden J, Stienen GJM. Cardiac disorders and pathophysiology of sarcomeric proteins. Physiol Rev. (2019) 99(1):381–426. doi: 10.1152/physrev.00040.2017
17. Weissler-Snir A, Hindieh W, Gruner C, Fourey D, Appelbaum E, Rowin E, et al. Lack of phenotypic differences by cardiovascular magnetic resonance imaging in MYH7 (β-myosin heavy chain)- versus MYBPC3 (myosin-binding protein C)-related hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. (2017) 10(2):e005311. doi: 10.1161/CIRCIMAGING.116.005311
18. Jacques A, Hoskins AC, Kentish JC, Marston SB. From genotype to phenotype: a longitudinal study of a patient with hypertrophic cardiomyopathy due to a mutation in the MYBPC3 gene. J Muscle Res Cell Motil. (2008) 29(6):239–46. doi: 10.1007/s10974-009-9174-0
19. Suay-Corredera C, Alegre-Cebollada J. The mechanics of the heart: zooming in on hypertrophic cardiomyopathy and cMyBP-C. FEBS Lett. (2022) 596(6):703–46. doi: 10.1002/1873-3468.14301
20. Moss RL, Fitzsimons DP, Ralphe JC. Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium cardiac myosin binding protein C. Circ Res. (2015) 116(1):183–92. doi: 10.1161/CIRCRESAHA.116.300561
21. Craig R, Offer G, Boycott BB. The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond B Biol Sci. (1997) 192(1109):451–61. doi: 10.1098/rspb.1976.0023
22. Tonino P, Kiss B, Gohlke J, Smith JE, Granzier H. Fine mapping Titin’s C-zone: matching cardiac myosin-binding protein C stripes with titin’s super-repeats. J Mol Cell Cardiol. (2019) 133:47–56. doi: 10.1016/j.yjmcc.2019.05.026
23. Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. (1995) 14(9):1952–60. doi: 10.1002/j.1460-2075.1995.tb07187.x
24. Gresham KS, Mamidi R, Stelzer JE. The contribution of cardiac myosin binding protein-c Ser282 phosphorylation to the rate of force generation and in vivo cardiac contractility. J Physiol. (2014) 592(17):3747–65. doi: 10.1113/jphysiol.2014.276022
25. Mamidi R, Gresham KS, Li J, Stelzer JE. Cardiac myosin binding protein-C Ser302 phosphorylation regulates cardiac β-adrenergic reserve. Sci Adv. (2017) 3(3):e1602445. doi: 10.1126/sciadv.1602445
26. Gresham KS, Stelzer JE. The contributions of cardiac myosin binding protein C and troponin I phosphorylation to β-adrenergic enhancement of in vivo cardiac function. J Physiol. (2016) 594(3):669–86. doi: 10.1113/JP270959
27. Colson BA, Bekyarova T, Fitzsimons DP, Irving TC, Moss RL. Radial displacement of myosin cross-bridges in mouse myocardium due to ablation of myosin binding protein-C. J Mol Biol. (2007) 367(1):36–41. doi: 10.1016/j.jmb.2006.12.063
28. McNamara JW, Li A, Smith NJ, Lal S, Graham RM, Kooiker KB, et al. Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J Mol Cell Cardiol. (2016) 94:65–71. doi: 10.1016/j.yjmcc.2016.03.009
29. Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ Res. (2006) 98(9):1212–8. doi: 10.1161/01.RES.0000219863.94390.ce
30. Previs MJ, Mun JY, Michalek AJ, Previs SB, Gulick J, Robbins J, et al. Phosphorylation and calcium antagonistically tune myosin-binding protein c’s structure and function. Proc Natl Acad Sci. (2016) 113(12):3239–44. doi: 10.1073/pnas.1522236113
31. Heling LWHJ, Geeves MA, Kad NM. MyBP-C: one protein to govern them all. J Muscle Res Cell Motil. (2020) 41(1):91–101. doi: 10.1007/s10974-019-09567-1
32. Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. (2010) 48(5):866–75. doi: 10.1016/j.yjmcc.2009.11.014
33. Kuster DWD, Bawazeer AC, Zaremba R, Goebel M, Boontje NM, van der Velden J. Cardiac myosin binding protein C phosphorylation in cardiac disease. J Muscle Res Cell Motil. (2012) 33(1):43–52. doi: 10.1007/s10974-011-9280-7
34. Thoonen R, Giovanni S, Govindan S, Lee DI, Wang GR, Calamaras TD, et al. Molecular screen identifies cardiac myosin–binding protein-C as a protein kinase G-iα substrate. Circ Heart Fail. (2015) 8(6):1115–22. doi: 10.1161/CIRCHEARTFAILURE.115.002308
35. Colson BA, Bekyarova T, Locher MR, Fitzsimons DP, Irving TC, Moss RL. Protein kinase A–mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ Res. (2008) 103(3):244–51. doi: 10.1161/CIRCRESAHA.108.178996
36. Kampourakis T, Yan Z, Gautel M, Sun YB, Irving M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc Natl Acad Sci. (2014) 111(52):18763–8. doi: 10.1073/pnas.1413922112
37. Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J Gen Physiol. (2006) 128(3):261–72. doi: 10.1085/jgp.200609547
38. Sadayappan S, de Tombe PP. Cardiac myosin binding protein-C: redefining its structure and function. Biophys Rev. (2012) 4(2):93. doi: 10.1007/s12551-012-0067-x
39. Kensler RW, Craig R, Moss RL. Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc Natl Acad Sci U S A. (2017) 114(8):E1355–64. doi: 10.1073/pnas.1614020114
40. Craig R, Lee KH, Mun JY, Torre I, Luther PK. Structure, sarcomeric organization, and thin filament binding of cardiac myosin-binding protein-C. Pflüg Arch - Eur J Physiol. (2014) 466(3):425–31. doi: 10.1007/s00424-013-1426-6
41. Razumova MV, Bezold KL, Tu AY, Regnier M, Harris SP. Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae. J Gen Physiol. (2008) 132(5):575. doi: 10.1085/jgp.200810013
42. Shaffer JF, Kensler RW, Harris SP. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner *. J Biol Chem. (2009) 284(18):12318–27. doi: 10.1074/jbc.M808850200
43. Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, et al. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc Natl Acad Sci. (2014) 111(6):2170–5. doi: 10.1073/pnas.1316001111
44. Risi C, Belknap B, Forgacs-Lonart E, Harris SP, Schröder GF, White HD, et al. N-Terminal domains of cardiac myosin binding protein C cooperatively activate the thin filament. Structure. (2018) 26(12):1604–1611.e4. doi: 10.1016/j.str.2018.08.007
45. Morimoto K, Harrington WF. Isolation and composition of thick filaments from rabbit skeletal muscle. J Mol Biol. (1973) 77(1):165–75. doi: 10.1016/0022-2836(73)90370-7
46. Carrier L. Targeting the population for gene therapy with MYBPC3. J Mol Cell Cardiol. (2021) 150:101–8. doi: 10.1016/j.yjmcc.2020.10.003
47. Suay-Corredera C, Pricolo MR, Herrero-Galán E, Velázquez-Carreras D, Sánchez-Ortiz D, García-Giustiniani D, et al. Protein haploinsufficiency drivers identify MYBPC3 variants that cause hypertrophic cardiomyopathy. J Biol Chem. (2021) 297(1):100854. doi: 10.1016/j.jbc.2021.100854
48. Glazier AA, Thompson A, Day SM. Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy. Pflugers Arch. (2019) 471(5):781–93. doi: 10.1007/s00424-018-2226-9
49. Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. (2009) 105(3):219–22. doi: 10.1161/CIRCRESAHA.109.202440
50. Helms AS, Tang VT, O’Leary TS, Friedline S, Wauchope M, Arora A, et al. Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight. (2020) 5(2):e133782. doi: 10.1172/jci.insight.133782
51. Carrier L, Bonne G, Bahrend E, Yu B, Richard P, Niel F, et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res. (1997) 80(3):427–34. doi: 10.1161/01.res.0000435859.24609.b3
52. Zahka K, Kalidas K, Simpson MA, Cross H, Keller BB, Galambos C, et al. Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the Amish. Heart. (2008) 94(10):1326–30. doi: 10.1136/hrt.2007.127241
53. Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy. Circulation. (2018) 138(14):1387–98. doi: 10.1161/CIRCULATIONAHA.117.033200
54. Xin B, Puffenberger E, Tumbush J, Bockoven JR, Wang H. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am J Med Genet A. (2007) 143A(22):2662–7. doi: 10.1002/ajmg.a.31981
55. Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. (2015) 573(2):188–97. doi: 10.1016/j.gene.2015.09.008
56. van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JMJ, Winegrad S, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy. Circulation. (2009) 119(11):1473–83. doi: 10.1161/CIRCULATIONAHA.108.838672
57. Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy. Circulation. (2003) 107(17):2227–32. doi: 10.1161/01.CIR.0000066323.15244.54
58. Bunch TA, Lepak VC, Bortz KM, Colson BA. A high-throughput fluorescence lifetime-based assay to detect binding of myosin-binding protein C to F-actin. J Gen Physiol. (2021) 153(3):e202012707. doi: 10.1085/jgp.202012707
59. Doh CY, Kampourakis T, Campbell K, Stelzer J. Basic science methods for the characterization of variants of uncertain significance in hypertrophic cardiomyopathy. Front Cardiovasc Med. (2023) 10:1238515. doi: 10.3389/fcvm.2023.1238515
60. Kanassatega RS, Bunch TA, Lepak VC, Wang C, Colson BA. Human cardiac myosin-binding protein C phosphorylation- and mutation-dependent structural dynamics monitored by time-resolved FRET. J Mol Cell Cardiol. (2022) 166:116–26. doi: 10.1016/j.yjmcc.2022.02.005
61. Wong FL, Bunch TA, Lepak VC, Steedman AL, Colson BA. Cardiac myosin-binding protein C N-terminal interactions with myosin and actin filaments: opposite effects of phosphorylation and M-domain mutations. J Mol Cell Cardiol. (2024) 186:125–37. doi: 10.1016/j.yjmcc.2023.11.010
62. Bunch TA, Guhathakurta P, Thompson AR, Lepak VC, Carter AL, Thomas JJ, et al. Drug discovery for heart failure targeting myosin-binding protein C. J Biol Chem. (2023) 299(12):105369. doi: 10.1016/j.jbc.2023.105369
63. Bers DM. Cardiac excitation–contraction coupling. Nature. (2002) 415(6868):198–205. doi: 10.1038/415198a
64. Wang J, Gareri C, Rockman HA. G-protein–coupled receptors in heart disease. Circ Res. (2018) 123(6):716–35. doi: 10.1161/CIRCRESAHA.118.311403
65. Ammirati E, Contri R, Coppini R, Cecchi F, Frigerio M, Olivotto I. Pharmacological treatment of hypertrophic cardiomyopathy: current practice and novel perspectives. Eur J Heart Fail. (2016) 18(9):1106–18. doi: 10.1002/ejhf.541
66. Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2020) 396(10253):759–69. doi: 10.1016/S0140-6736(20)31792-X
67. Braunwald E, Saberi S, Abraham TP, Elliott PM, Olivotto I. Mavacamten: a first-in-class myosin inhibitor for obstructive hypertrophic cardiomyopathy. Eur Heart J. (2023) 44(44):4622–33. doi: 10.1093/eurheartj/ehad637
68. Sebastian SA, Padda I, Lehr EJ, Johal G. Aficamten: a breakthrough therapy for symptomatic obstructive hypertrophic cardiomyopathy. Am J Cardiovasc Drugs. (2023) 23(5):519–32. doi: 10.1007/s40256-023-00599-0
69. Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. (2016) 351(6273):617–21. doi: 10.1126/science.aad3456
70. Day SM, Tardiff JC, Ostap EM. Myosin modulators: emerging approaches for the treatment of cardiomyopathies and heart failure. J Clin Invest. (2022) 132(5):e148557. doi: 10.1172/JCI148557
71. Dong T, Alencherry B, Ospina S, Desai MY. Review of mavacamten for obstructive hypertrophic cardiomyopathy and future directions. Drug Des Devel Ther. (2023) 17:1097–106. doi: 10.2147/DDDT.S368590
72. Desai MY, Owens A, Wolski K, Geske JB, Saberi S, Wang A, et al. Mavacamten in patients with hypertrophic cardiomyopathy referred for septal reduction: week 56 results from the VALOR-HCM randomized clinical trial. JAMA Cardiol. (2023) 8(10):968–77. doi: 10.1001/jamacardio.2023.3342
73. Heitner SB, Jacoby D, Lester SJ, Owens A, Wang A, Zhang D, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy. Ann Intern Med. (2019) 170(11):741–8. doi: 10.7326/M18-3016
74. Mamidi R, Li J, Doh CY, Verma S, Stelzer JE. Impact of the myosin modulator mavacamten on force generation and cross-bridge behavior in a murine model of hypercontractility. J Am Heart Assoc Cardiovasc Cerebrovasc Dis. (2018) 7(17):e009627. doi: 10.1161/JAHA.118.009627
75. Dominic KL, Choi J, Holmes JB, Singh M, Majcher MJ, Stelzer JE. The contribution of N-terminal truncated cMyBPC to in vivo cardiac function. J Gen Physiol. (2023) 155(6):e202213318. doi: 10.1085/jgp.202213318
76. Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. (2013) 342(6154):111–4. doi: 10.1126/science.1236921
77. Li J, Mamidi R, Doh CY, Holmes JB, Bharambe N, Ramachandran R, et al. AAV9 gene transfer of cMyBPC N-terminal domains ameliorates cardiomyopathy in cMyBPC-deficient mice. JCI Insight. (2020) 5(17):e130182. doi: 10.1172/jci.insight.130182
78. Mearini G, Stimpel D, Geertz B, Weinberger F, Krämer E, Schlossarek S, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun. (2014) 5(1):5515. doi: 10.1038/ncomms6515
79. Reichart D, Newby GA, Wakimoto H, Lun M, Gorham JM, Curran JJ, et al. Efficient in vivo genome editing prevents hypertrophic cardiomyopathy in mice. Nat Med. (2023) 29(2):412–21. doi: 10.1038/s41591-022-02190-7
80. Tenaya Therapeutics. First-in-Human, Open-Label, Safety, Tolerability, Dose-Finding, Pharmacodynamic and Cardiac Transgene Expression Study of TN-201, a Recombinant Adenoassociated Virus Serotype 9 (AAV9) Containing Myosin Binding Protein C Transgene, in Adults With MYBPC3 Mutation-Associated Hypertrophic Cardiomyopathy (HCM). (2024) Report No.: NCT05836259. Available online at: https://clinicaltrials.gov/study/NCT05836259 (cited December 31, 2024).
81. Chancellor D, Barrett D, Nguyen-Jatkoe L, Millington S, Eckhardt F. The state of cell and gene therapy in 2023. Mol Ther. (2023) 31(12):3376–88. doi: 10.1016/j.ymthe.2023.11.001
82. Merkulov S, Chen X, Chandler MP, Stelzer JE. In vivo cardiac myosin binding protein C gene transfer rescues myofilament contractile dysfunction in cardiac myosin binding protein C null mice. Circ Heart Fail. (2012) 5(5):635–44. doi: 10.1161/CIRCHEARTFAILURE.112.968941
83. Li J, Gresham KS, Mamidi R, Doh CY, Wan X, Deschenes I, et al. Sarcomere-based genetic enhancement of systolic cardiac function in a murine model of dilated cardiomyopathy. Int J Cardiol. (2018) 273:168–76. doi: 10.1016/j.ijcard.2018.09.073
84. Zhang J, Chen B, Gan C, Sun H, Zhang J, Feng L. A comprehensive review of small interfering RNAs (siRNAs): mechanism, therapeutic targets, and delivery strategies for cancer therapy. Int J Nanomedicine. (2023) 18:7605–35. doi: 10.2147/IJN.S436038
85. Kohn DB, Chen YY, Spencer MJ. Successes and challenges in clinical gene therapy. Gene Ther. (2023) 30(10):738–46. doi: 10.1038/s41434-023-00390-5
86. Nelson S, Beck-Previs S, Sadayappan S, Tong C, Warshaw DM. Myosin-binding protein C stabilizes, but is not the sole determinant of SRX myosin in cardiac muscle. J Gen Physiol. (2023) 155(4):e202213276. doi: 10.1085/jgp.202213276
87. Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. (2011) 331(6023):1439–43. doi: 10.1126/science.1200113
88. Chandler J, Treacy C, Ameer-Beg S, Ehler E, Irving M, Kampourakis T. in situ FRET-based localization of the N terminus of myosin binding protein-C in heart muscle cells. Proc Natl Acad Sci U S A. (2023) 120(12):e2222005120. doi: 10.1073/pnas.2222005120
89. Colson BA, Thompson AR, Espinoza-Fonseca LM, Thomas DD. Site-directed spectroscopy of cardiac myosin-binding protein C reveals effects of phosphorylation on protein structural dynamics. Proc Natl Acad Sci. (2016) 113(12):3233–8. doi: 10.1073/pnas.1521281113
90. Rosas PC, Liu Y, Abdalla MI, Thomas CM, Kidwell DT, Dusio GF, et al. Phosphorylation of cardiac myosin-binding protein-C is a critical mediator of diastolic function. Circ Heart Fail. (2015) 8(3):582–94. doi: 10.1161/CIRCHEARTFAILURE.114.001550
Keywords: hypertrophic cardiomyopathy, cMyBP-C, myosin binding protein C, gene therapy, AAV9 gene transfer, small-molecule, high-throughput screening, small molecule therapy
Citation: Wood PT, Seffrood MM, Colson BA and Stelzer JE (2025) cMyBP-C in hypertrophic cardiomyopathy: gene therapy and small-molecule innovations. Front. Cardiovasc. Med. 12:1550649. doi: 10.3389/fcvm.2025.1550649
Received: 23 December 2024; Accepted: 13 February 2025;
Published: 26 February 2025.
Edited by:
Nicola Mumoli, ASST Ovest Milanese, ItalyReviewed by:
Kristina Bezold Kooiker, University of Washington, United StatesCopyright: © 2025 Wood, Seffrood, Colson and Stelzer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick T. Wood, cGF0cmljay53b29kQGNhc2UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.