 Zhong-He Zhang1,2,3,†
Zhong-He Zhang1,2,3,† Hector Barajas-Martinez1,4,5,†
Hector Barajas-Martinez1,4,5,† Hong-Yi Duan1,2,3
Hong-Yi Duan1,2,3 Guo-Hua Fan6
Guo-Hua Fan6 Hong Jiang1,2,3
Hong Jiang1,2,3 Charles Antzelevitch4,5
Charles Antzelevitch4,5 Hao Xia1,2,3*
Hao Xia1,2,3* Dan Hu1,2,3*‡
Dan Hu1,2,3*‡
- 1Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, China
- 2Cardiovascular Research Institute, Wuhan University, Wuhan, China
- 3Hubei Key Laboratory of Cardiology, Wuhan, China
- 4Cardiovascular Research Department, Lankenau Institute for Medical Research, Lankenau Heart Institute, Wynnwood, PA, United States
- 5Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, United States
- 6Department of Thoracic Surgery, Renmin Hospital of Wuhan University, Wuhan, China
Background: Previous studies suggested that SCN10A/Nav1.8 may influence cardiac electrophysiology and the susceptibility to cardiac arrhythmias. Notably, the expression of SCN10A is not uniform, showing variable expression in each cardiac chamber. The present study aims to explore the functional significance of Nav1.8 expression among different cell types present in the ventricular myocardium.
Methods: The effect of the specific Nav1.8 blocker, A-803467, on action potential was recorded from epicardial, mid-myocardial (M cells) and Purkinje tissue slices isolated from the canine left ventricle using standard microelectrode techniques and on late sodium current from Purkinje cells using patch-clamp techniques.
Results: A-803467 treatment did not significantly affect maximum diastolic potential, action potential amplitude or maximum rate of rise of the action potential upstroke in epicardial cells, M cells or Purkinje fibers. Action potential duration (APD) was also unaffected by A-803467 in epicardial cells. However, administration of 1,000 nmol/L A-803467 reduced APD30, APD50, and APD90 during relatively slow pacing rates of 0.2 and 0.5 Hz in M cells. In Purkinje fibers, A-803467 (100 and 1,000 nmol/L) substantially abbreviated APD50 and APD90 at slow pacing rates (0.2 and 0.5 Hz). Moreover, 100 nmol/L A-803467 significantly inhibited the development of early afterdepolarizations induced by 10 nmol/L ATX-II (7/8 vs. 2/8, p < 0.05) as well as the amplitude of late sodium current at 0.2 Hz in Purkinje cells.
Conclusions: The functional significance of Nav1.8 varies among different types of ventricular and conduction system cardiomyocytes. The reduction in INa,L and APD, as well as suppression of early afterdepolarizations by Nav1.8 block in Purkinje fibers suggests Nav1.8 as a potential therapeutic target for bradycardia-dependent arrhythmias.
Introduction
The voltage-gated sodium channel 1.5 (Nav1.5), encoded by SCN5A gene is recognized as the primary cardiac Nav isoform. Mutations in Nav1.5 have been associated with a variety of cardiac disorders, including long QT syndrome (LQTS), Brugada syndrome, cardiac conduction system disease and atrial fibrillation (AF) (1, 2). However, other sodium channels including Nav1.8 are also present in the heart. Nav1.8 is a tetrodotoxin-resistant voltage-gated sodium channel, initially identified as predominantly expressed in small-diameter sensory neurons of the dorsal root ganglia, where it plays a crucial role in pain perception and modulation (3).
Several genome-wide association studies have demonstrated that common single nucleotide polymorphisms and variants in SCN10A are associated with electrocardiographic indices, including PR, QRS and QT interval duration, as well as cardiac arrhythmia phenotypes, such as Brugada syndrome, AF, cardiac conduction system disease and sudden unexpected death syndrome (4–10). The expression of SCN10A mRNA and Nav1.8 protein has been detected in atrial and ventricular myocytes of mice, rabbits, and humans (4, 11–16). Moreover, pharmacological block of Na1.8 channels using the highly selective inhibitor A-803467 has been shown to prolong both PR interval and QRS duration in wild-type mice (4). Moreover, Yang et al. reported that application of A-803467 in ventricular cardiomyocytes of mice and rabbits results in abbreviated action potential duration (APD) as well as suppression of early afterdepolarizations (EADs) (15). These findings collectively suggest that SCN10A plays an important role in cardiac electrophysiology and arrhythmogenesis.
The distribution of SCN10A exhibits heterogeneity among atrial and ventricular tissues, as well as between the left and right ventricles. Yang et al. also reproted that Nav1.8-mediated late sodium currents (INa,L) varied among individual ventricular cardiomyocytes in mice. Furthermore, the APD-shortening effect of A-803467 demonstrated considerable cell-to-cell variability, with only approximately half of the tested cells showing significant reduction in APD (15). Accordingly, the physiological effects mediated by Nav1.8 may differ among the different cell types. The canine ventricular wall encompasses epicardial cells, M cells, and Purkinje cells, each exhibiting distinct electrophysiological and pharmacological properties. However, the electrophysiological characteristics of Nav1.8 across these cell subtypes remain unexplored. This study aims to elucidate the functional electrophysiological role of Nav1.8 in different cellular subtypes of the ventricular myocardium.
Materials and methods
Slice preparation and Purkinje fibers
All experiments involving animals were approved by the Animal Care and Use Committee of the Renmin Hospital of Wuhan University. All animal procedures conformed to the Guidelines for the Care and Use of Laboratory Animals outlined by the US National Institutes of Health. Adult mongrel dogs weighing 20–35 kg were anticoagulated with heparin and anesthetized with sodium pentobarbital (30 mg/kg, intravenous). The chest was opened via a left thoracotomy, and the heart was rapidly excised and placed in a cardioplegic solution (4℃ Tyrode solution containing 12 mmol/L KCl).
Epicardial (<1.5 mm from epicardial surface) and mid-myocardial (2–7 mm from epicardial surface) tissues were isolated from the left ventricle of the hearts by razor blade shavings (Davol Simon Dermatome, Cranston, RI, USA) made parallel to the surface of the ventricular free wall. Free running Purkinje fibers were dissected out from left ventricle. The preparations were placed in a tissue bath (volume 5 ml, flow rate 12 ml/min) and allowed to equilibrate for at least 3 h while superfused with Tyrode's solution bubbled with 95% O2-5% CO2 and maintained at (37 ± 0.5℃, pH = 7.35). The composition of the Tyrode's solution was (in mmol/L): NaCl 129, KCl 4, NaH2PO4 0.9, NaHCO3 20, CaCl2 1.8, MgSO4 0.5, and D-glucose 5.5.
Isolation of Purkinje cells
Purkinje cells were isolated via enzymatic dissociation as previously described (17). Purkinje fibers were dissected from the left ventricles and placed in a small dish. Purkinje fibers were then digested in a nominally Ca2+-free solution containing 1.0 mg/ml collagenase (type II, Worthington) and 30 mM 2,3-butanedione monoxime at 36°C. Gentle stirring of the enzyme solution with a small magnetic bar facilitated the release of cells from the fibers. The suspension containing dissociated Purkinje cells was periodically collected and transferred into a modified KB solution, while fresh enzyme solution was added to the remaining Purkinje fibers to maintain a volume of 2 ml. The enzymatic digestion process typically took 15–35 min to yield individual myocytes. Finally, the isolated Purkinje cells were stored in the modified KB solution at room temperature until further use.
SCN10A plasmid transfection in ND7/23 cells
The wild-type cDNA plasmid (2 µg) of SCN10A-encoded Nav1.8 channel and green fluorescent protein (0.5 µg) were co-transfected into ND7/23 cells with 10 µl FuGene6 in culture for 48 h. Then green fluorescent protein colored cells were selected as successful transfection for patching clamp experiments to verify the effect of the drug A-803467 (see below) on INa,L.
Action potential (AP) recordings
The preparations were stimulated at basic cycle lengths (BCLs) ranging from 500 to 5,000 ms. Standard techniques were used to deliver rectangular-wave pulses lasting 1–3 ms at 2.5 times diastolic threshold intensity through silver bipolar electrodes that were insulated except at the tips.
Transmembrane potentials were recorded using 3 mmol/L KCL-filled glass microelectrodes (tip resistances, 10–20 MΩ) connected to a high input-impedance amplification system (World Precision Instruments, New Haven, USA.) The signals were displayed on oscilloscopes and further amplified, digitized and analyzed (Spike 2, Cambridge Electronic Design, Cambridge, England). The following action potential (AP) parameters were evaluated before and after exposure to A-803467 (for 20–30 min): action potential amplitude (APA), maximum diastolic potential (MDP), and maximum rate of rise of the AP upstroke (Vmax), APD at 30%, 50% and 90% repolarization (APD30, APD50 and APD90). ATX-II (10 nmol/L, for 20–30 min) was used to induce EADs in canine Purkinje fibers. EADs, characterized by depolarizing oscillations during phase 2 or 3 of the action potential, were evaluated at BCLs ranging from 500 to 5,000 ms.
INa,L recordings
INa,L was recorded using the whole-cell patch-clamp technique. The bath solution used contained (in mmol/L): NaCl 140, CaCl2 2.0, MgCl2 1, glucose 10, and HEPES 10, pH adjusted to 7.4 with NaOH. 10 μmol/L nifedipine was added in bath solution to block calcium current. The pipette solution was composed of (in mmol/L): NaCl 10, aspartate 130, MgCl2 1, CsCl 10, HEPES 10, MgATP 5, and EGTA 10, pH adjusted to 7.2 with CsOH. INa,L was assessed by applying a 500-ms square depolarization pulse to −20 mV from a holding potential of −80 mV, repeated every 5 s. The amplitude of INa,L was measured at the termination of the testing depolarizing pulse. To record Nav1.8-mediated INa,L in ND7/23 cells, which inherently express tetrodotoxin-sensitive fast sodium currents, 30 nmol/L tetrodotoxin (for 20–30 min) was administered to selectively inhibit the endogenous currents.
Drugs
A-803467 and ATX-II were purchased from Sigma-Aldrich Co (St. Louis, Missouri, USA). A-803467 was dissolved in 5% dimethyl sulfoxide, stored at a concentration of 50 mmol/L, and diluted to the desired final concentration prior to use. ATX-II was dissolved in distilled water to create a stock solution of 200 nmol/L, and then diluted to a final concentration of 10 nmol/L for use.
Statistical analysis
All data are presented as mean ± standard error. The statistical analysis of the electrophysiological effects of A-803467 was conducted using one-way repeated measure analysis variance followed by Bonferroni's test or with a student t test. The incidence of EADs was assessed by using the Fisher exact test. The SPSS (version 26.0) and GraphPad Prism software (version 10.0) were used for data analysis and graphing. P < 0.05 was considered statistically significant.
Results
Effect of A-803467 on AP characteristics in epicardial cells
We first assessed the effects of the NaV1.8 specific blocker A-803467 on AP characteristics in canine left ventricular epicardial preparations at BCLs from 500 to 5,000 ms. A-803467 (3–1,000 nmol/L) had no significant effect on APA, MDP, and Vmax (Figure 1A–C) at any BCL tested. Treatment with 1,000 nmol/L A-803467 exhibited a trend towards abbreviation of APD30 at stimulation BCLs ranging from 500 to 5,000 ms. However, this reduction was not statistically significant (Figure 1D). Both APD50 and APD90 showed no substantial change under the same conditions (Figure 1E,F).
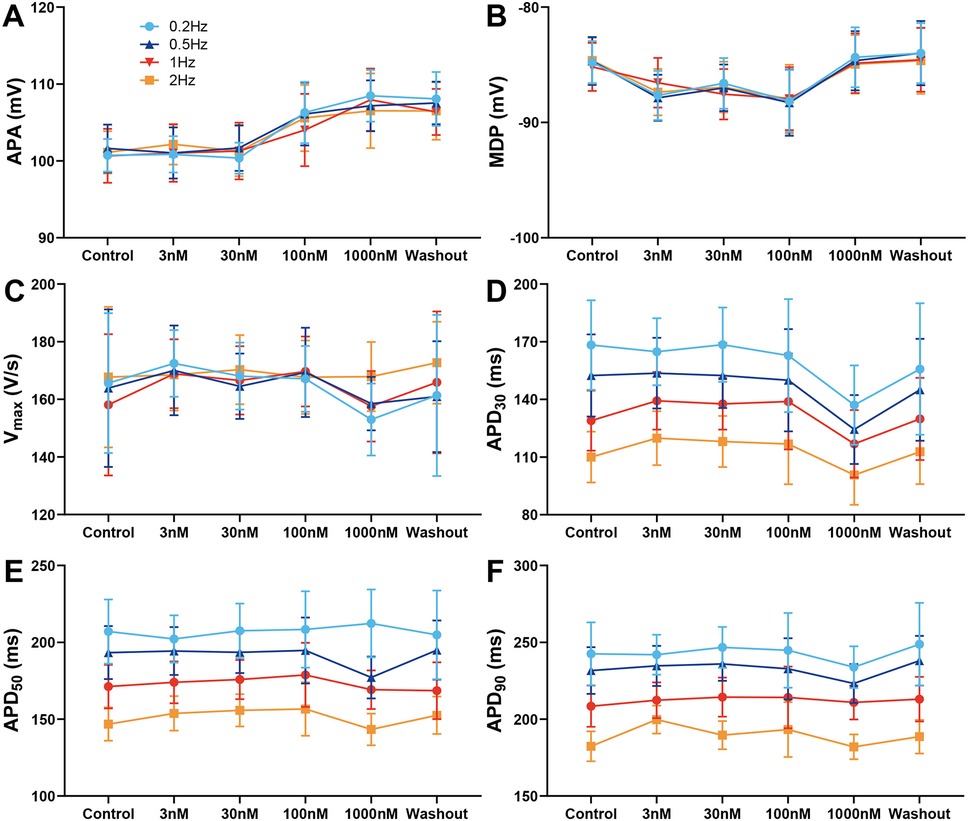
Figure 1. Effect of A-803467 (3–1,000 nmol/L) administration on action potential properties in canine left ventricular epicardial cardiomyocytes (N = number of dogs; n = number of tissue slices). (A–F) Average data for action potential amplitude (APA), maximum diastolic potential (MDP), maximum rate of rise of the action potential upstroke (Vmax), action potential duration at 30%, 50% and 90% repolarization (APD30, APD50 and APD90), before (control) and after administration and washout of A-803467 (N = 5 and n = 5, respectively).
Effect of A-803467 on AP characteristics in M cells
Figure 2 illustrates average APs recordings at baseline, during exposure to increasing concentrations of A-803467, and following drug wash-out. Similarly to its effects on epicardial cells, APA, MDP, and Vmax were not altered by A-803467 exposure at concentrations ranging from 3 to 1,000 nmol/L at different BCLs (Figure 2A–C). At BCLs of 500 and 1,000 ms, A-803467, even at concentrations up to 1,000 nmol/L, did not produce significant changes in APD30, APD50, or APD90 compared with control group (Figure 2D–F). However, at BCLs of 2,000 and 5,000 ms, 1,000 nmol/L A-803467 significantly decreased APD30 (BCL = 2,000 ms: 185.39 ± 14.21 ms vs. 147.34 ± 10.99 ms, p < 0.05; BCL = 5,000 ms: 214.29 ± 15.81 ms vs. 161.09 ± 11.76 ms, p < 0.01), APD50 (BCL = 2,000 ms: 261.84 ± 11.87 ms vs. 217.33 ± 7.61 ms, p < 0.01; BCL = 5,000 ms: 297.72 ± 13.07 ms vs. 237.07 ± 9.03 ms, p < 0.01), and APD90 (BCL = 2,000 ms: 318.62 ± 12.21 ms vs. 272.23 ± 8.11 ms, p < 0.01; BCL = 5,000 ms: 357.51 ± 13.58 ms vs. 294.29 ± 10.09 ms, p < 0.01) (Figure 2D–F). These effects were reversed following wash-out of the drug. At BCLs of 2,000 and 5,000 ms, the abbreviating effects on APD were not observed when A-803467 was administered at concentrations from 3 to 100 nmol/L.
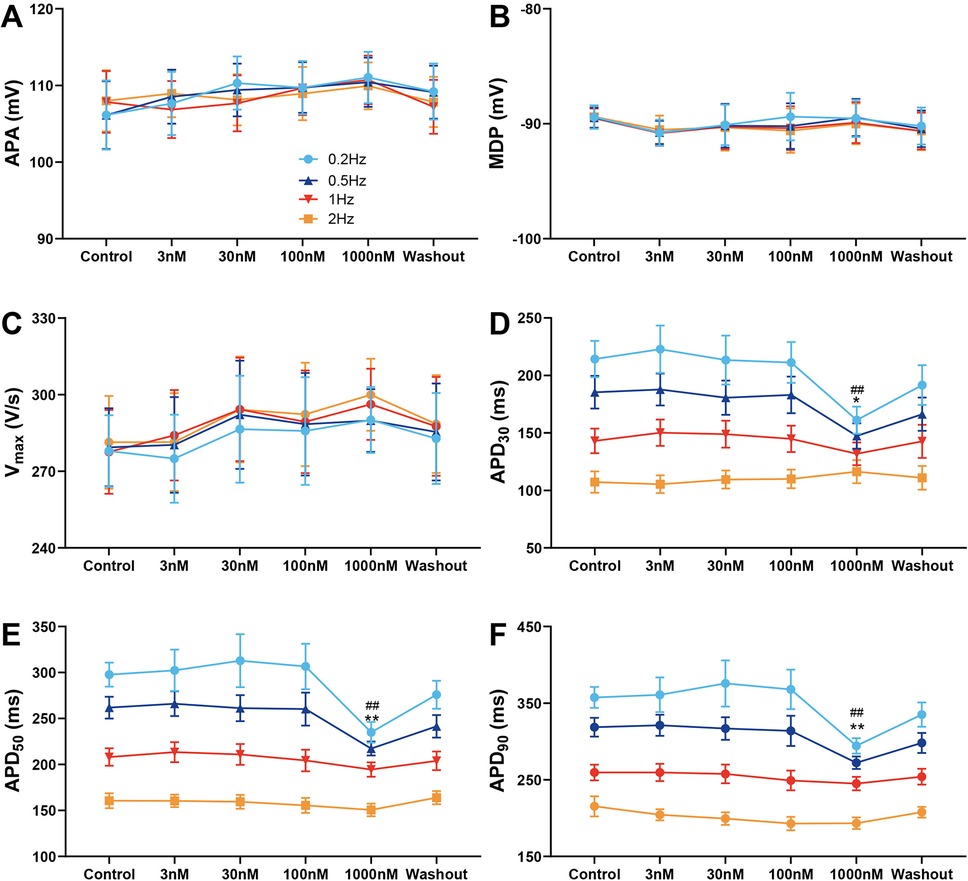
Figure 2. Effect of A-803467 (3–1,000 nmol/L) administration on action potential properties in canine left ventricular M cardiomyocytes (N = number of dogs; n = number of tissue slices). (A–F) Average data for action potential amplitude (APA), maximum diastolic potential (MDP), maximum rate of rise of the action potential upstroke (Vmax), action potential duration at 30%, 50% and 90% repolarization (APD30, APD50 and APD90), before (control) and after administration and washout of A-803467. *p < 0.05 vs. control at 0.5 Hz, **p < 0.01 vs. control at 0.5 Hz, #p < 0.05 vs. control at 0.2 Hz, ##p < 0.01 vs. control at 0.2 Hz (N = 10 and n = 10, respectively).
Effect of A-803467 on AP characteristics, EADs and INa,L in Purkinje fibers
Figure 3 shows the effects of various concentrations of A-803467 on APs recorded in Purkinje fibers. No significant changes in APA, MDP and Vmax were observed at any BCL or concentration studied (Figure 3A–C). At BCLs of 500 and 1,000 ms, treatment with A-803467 did not significantly reduce APD30, APD50, or APD90 (Figure 3D–F). However, at a BCL of 2,000 ms, 100 nmol/L A-803467 remarkably reduced APD50 (264.73 ± 10.92 ms vs. 220.20 ± 8.69 ms, p < 0.05) and APD90 (369.73 ± 14.67 ms vs. 319.34 ± 9.43 ms, p < 0.05). Additionally, 1,000 nmol/L A-803467 significantly decreased APD30 (94.64 ± 3.63 ms vs. 72.42 ± 6.29 ms, p < 0.05), as well as APD50 (264.73 ± 10.92 ms vs. 219.14 ± 8.79 ms, p < 0.05) and APD90 (369.73 ± 14.67 ms vs. 321.51 ± 13.25 ms, p < 0.05). At a BCL of 5,000 ms, both 100 and 1,000 nmol/L A-803467 diminished APD50 (301.69 ± 11.81 ms vs. 262.18 ± 9.74 ms, p < 0.05; 301.69 ± 11.81 ms vs. 256.98 ± 9.70 ms, p < 0.01) and APD90 (430.73 ± 16.64 ms vs. 385.82 ± 10.82 ms, p < 0.05; 430.73 ± 16.64 ms vs. 378.57 ± 12.01 ms, p < 0.01), while exerting no significant effect on APD30 (Figure 3D–F). The above-mentioned effect was reversed after the drug was washed out.
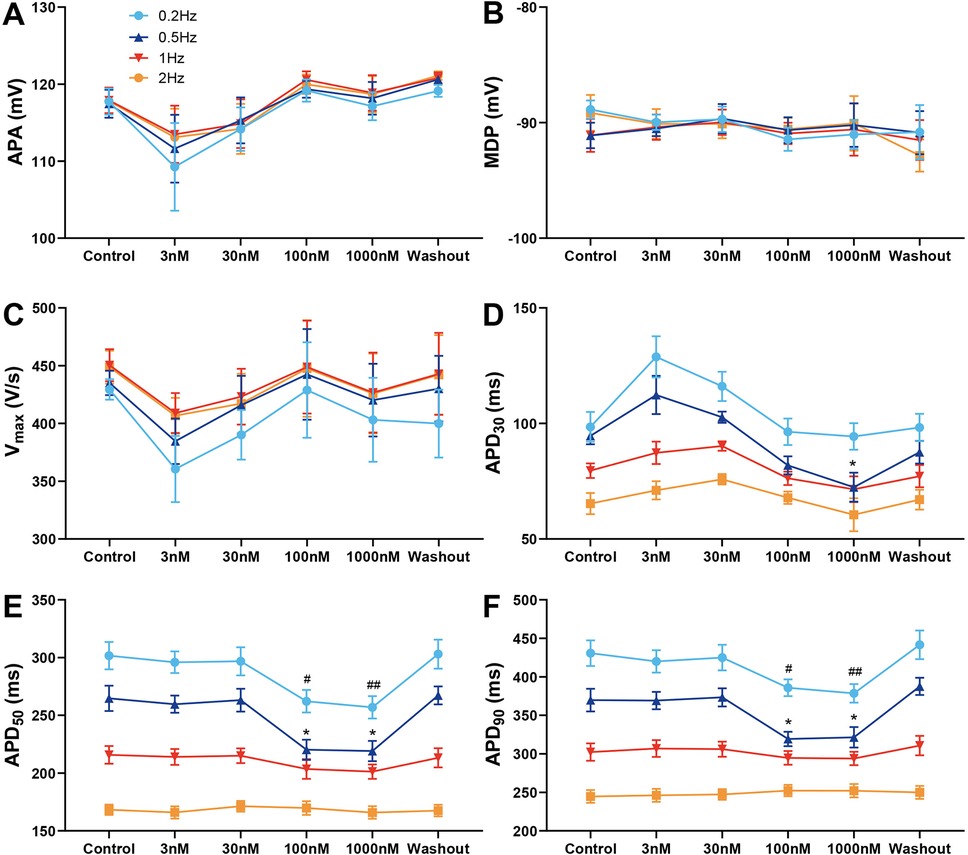
Figure 3. Effect of A-803467 (3–1,000 nmol/L) administration on action potential properties in canine left ventricular Purkinje fibers (N = number of dogs; n = number of tissue slices). (A–F) Average data for action potential amplitude (APA), maximum diastolic potential (MDP), maximum rate of rise of the action potential upstroke (Vmax), action potential duration at 30%, 50% and 90% repolarization (APD30, APD50 and APD90), before (control) and after administration and washout of A-803467. *p < 0.05 vs. control at 0.5 Hz, **p < 0.01 vs. control at 0.5 Hz, #p < 0.05 vs. control at 0.2 Hz, ##p < 0.01 vs. control at 0.2 Hz (N = 6 and n = 6, respectively).
The excessive prolongation of APs caused by the enhancement of INa,L can lead to EADs, which are associated with arrhythmogenesis in both congenital and acquired disease conditions, such as LQTS, heart failure, myocardial hypertrophy and ischemia-reperfusion. We examined the effects of 100 nmol/L A-803467 on EADs in Purkinje fibers triggered by the INa,L agonist ATX-II at a concentration of 10 nmol/L. As depicted in the inset of Figure 4A, ATX-II significantly prolonged APD and elicited EAD at a BCL of 2,000 ms. The administration of A-803467 attenuated APD prolongation induced by ATX-II and successfully suppressed the occurrence of EAD. Figure 4A summarizes the development of EADs in 8 Purkinje fibers exposed to 10 nmol/L ATX-II alone or together with 100 nmol/L A-803467 at different stimulation rates. At stimulation rates of 1 and 2 Hz, ATX-II barely failed to induce EADs. At a slower stimulation rate of 0.5 Hz, EADs developed in 2 of 8 (25%) Purkinje fibers following the addition of A-803467. In the absence of A-803467, EADs were induced by ATX-II in 4 of 8 (50%) Purkinje preparations. However, this reduction did not achieve the statistical significance. The incidence of EADs elicited by ATX-II was significantly reduced by A-803467 at a slower stimulation rate of 0.2 Hz (7/8 vs. 2/8, p < 0.05). To confirm that the underlying antiarrhythmic effects of A-803467 were mediated by inhibition of INa,L, we first examined its effects on SCN10A-encoded Nav1.8 channels heterologously expressed in ND7/23 cells. A-803467 (100 nmol/L) significantly inhibited INa,L in ND7/23 cells (Figure 4B). Furthermore, A-803467 also significantly reduced INa,L in Purkinje cells from 0.97 ± 0.08 pA/pF to 0.73 ± 0.06 pA/pF at 0.2 Hz (p < 0.01, Figure 4C).
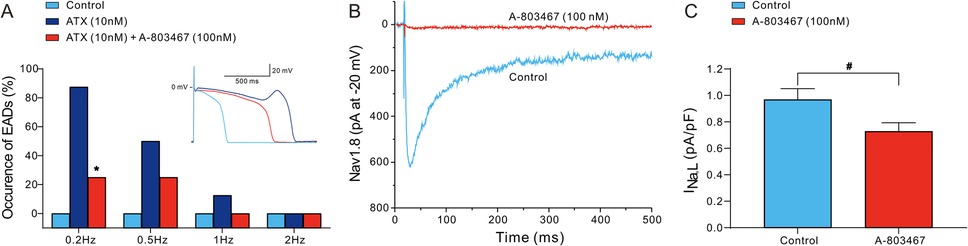
Figure 4. A-803467 inhibits early afterdepolarizations (EADs) and Nav1.8 induced INa,L in Purkinje fibers and cells (N = number of dogs; n = number of tissue slices or cells). (A) The incidence of EADs in Purkinje fibers exposed to ATX-II (10 nmol/L) alone or in combination with 100 nmol/L A-803467. The inset shows an example of A-803467 suppressing the EAD elicited by exposure to ATX-II at 0.5 Hz. *p < 0.05 vs. control + ATX (N = 8 and n = 8, respectively). (B) 100 nmol/L A-803467 inhibits SCN10A-mediated INa,L in transfected ND7/23 cells. (C) Summary data of relative INa,L at −20 mV in the absence (control) and presence of 100 nmol/L A-803467 in Purkinje cells. #p < 0.05 vs. control (N = 6 and n = 8, respectively).
Discussion
SCN10A is located on human chromosome 3p22, adjacent to SCN5A. Nav1.8 shares 70.4% protein sequence similarity with Nav1.5 and plays a crucial role in driving the upstroke of APs and facilitating conduction in peripheral nerves (18). The role of SCN10A in the heart began to attract attention following initial genome-wide association studies that revealed its association with cardiac conduction. SCN10A, like its high expression in peripheral neurons, is also readily detectable in intracardiac neurons, primarily cholinergic neurons (16). Chambers et al. detected SCN10A mRNA expression in both the atria and ventricles of mouse cardiac tissue, a finding further corroborated in isolated atrial and ventricular myocytes from wild-type mice (11). Subsequent studies documented SCN10A mRNA expression in ventricular myocytes and Purkinje fiber of mice, humans, and rabbits (4, 5, 12–15, 19–22). However, some studies also exhibited divergent results, failing to detect mRNA or full-length mRNA in ventricles or ventricular myocytes (7, 23, 24). This discrepancy may be attributed to variations in experimental conditions, expression levels below detection thresholds, high rates of alternative splicing, or methodological differences (primer position in quantitative polymerase chain reaction etc.) (14, 19, 23, 25). More recently, Man et al. reported that the full-length SCN10A transcript was undetectable in the ventricles of both mouse and human heart using quantitative reverse transcription polymerase chain reaction, although short SCN10A transcripts containing the final seven exons was detected (25). Similar findings were reported via RNA sequencing (7). By designing primers for individual exons to address alternative splicing, Dybkova et al. successfully detected SCN10A mRNA expression in both human left ventricular myocardium and isolated failing cardiomyocytes (22). Nav1.8 protein expression has been observed in ventricular myocardium or cardiomyocytes from mice, rats, humans, and canines via immunohistochemistry and/or Western blot techniques (12–14, 22, 26–28). In line with SCN10A mRNA and protein expression, Nav1.8-mediated INa,L in ventricular myocytes is well recognized under both physiological conditions and pharmacological provocation (13, 15, 22, 23, 29, 30). Moreover, polymorphic variants of SCN10A have been linked to the QT interval, with rare variants identified in patients with LQTS (6, 31). Collectively, these findings substantiate the presence of Nav1.8 in ventricular myocardium, although at significantly lower expression levels than Nav1.5, both at the mRNA and protein levels.
Our findings demonstrated that A-803467 did not exert significant effects on the APA, Vmax, and MDP in epicardial cells, M cells, or Purkinje cells. Considering that Vmax and APA serve as indicators of rapid Na+ influx and peak Na+ current, our experimental data suggest that NaV1.8 makes a negligible contribution to the peak Na+ current in ventricular cardiomyocytes. This observation concurs with prior studies showing that neither specific pharmacological inhibition nor genetic ablation of Nav1.8 affects peak Na+ current or its kinetics in either atrial or ventricular cardiomyocytes (13, 15, 23, 30). Our data revealed that non-cardiac sodium channels contribute approximately 10% of the peak Na+ current in canine cardiomyocytes, implying that Nav1.8's contribution to the overall peak sodium current is likely minimal (32). Peak Na+ current is a major determinant of cardiac conduction velocity. The lack of significant effect on Vmax upon Nav1.8 inhibition suggests that Nav1.8 may not directly influence cardiac conduction through its electrophysiological effects in cardiomyocytes. However, it is noteworthy that Macri et al. identified a negative correlation between Nav1.8-dependent INa,L and PR intervals in a large-scale genetic screen (5). Additionally, Nav1.8 may modulate cardiac conduction indirectly by modulating Nav1.5 or via effects on intracardiac neurons (33).
INa,L persists long after peak Na+ current is inactivated, substantially influencing the shape and duration of the AP (34). Our group previously demonstrated that non-cardiac Nav isoforms contribute approximately 50% of INa,L in canine ventricular cardiomyocytes under physiological conditions (32). Nav1.8 manifests a more depolarized voltage dependence of inactivation and slower inactivation kinetics compared to Nav1.5 (7). Furthermore, INa,L mediated by Nav1.8 is considerably greater than that mediated by Nav1.5 in heterologous system (15, 35). These outcomes align with numerous studies indicating that Nav1.8 mainly contributes to the INa,L (5, 13, 15, 22, 23, 29, 36). We observed that A-803467 had no effect on APD in epicardial cells. In contrast, it significantly reduced APD50 and APD90 in M cells and Purkinje fiber. This discrepancy suggests that the distribution of Nav1.8 and the associated INa,L is heterogeneous across different cardiac cell types. Indeed, canine M cells and Purkinje fibers exhibit a larger INa,L amplitude compared to epicardial cells, and this more prominent INa,L contributes to their longer APD and steeper rate dependence of APD (34, 37, 38) Previous studies have demonstrated enrichment of SCN10A mRNA in the cardiac conduction system and Purkinje fibers during both developmental stages and in the adult murine heart (4, 20, 25). Vassalle et al. discovered a slowly inactivating sodium current in canine Purkinje cells that contributes to APD prolongation, while being minimally present or absent in ventricular cardiomyocytes. Remarkably, about 30% of the slowly inactivating sodium current remained even in the presence of 30 µmol/L tetrodotoxin, indicating that Nav1.8 might be responsible for this current (39). Yang et al. discovered that Nav1.8-mediated INa,L varies among different ventricular cardiomyocytes, leading to inconsistent APD shortening effects of A-803467, which was observed in only 50% of cells (15). Casini et al. observed that treatment with A-803467 induced a slight but statistically significant reduction in both APD50 and APD90 in rabbit ventricular cardiomyocytes, although this effect was only partially reversible in a minority of cells after washout. Of note, A-803467 treatment significantly reduced APD50 in human-induced pluripotent stem cell-derived cardiomyocytes, with the effect being reversible upon washout in the majority of cells, although the blocker did not alter APD in every cell (19). Verkert et al. reported that the expression of Nav1.8 was also heterogeneous in intracardiac neurons (16). Similarly, other ionic channels such as Iks and Ito are also not uniformly distributed across the ventricular wall (40, 41).
Some researchers were unable to detect functional Nav1.8-mediated INa,L (16, 19). The discrepancy potentially arises from cell-specific variations in Nav1.8 expression. As we have described, Nav1.8 expression levels differ among cell types, and the low expression in certain cells might preclude the detection of Nav1.8-mediated INa,L. Additionally, this discrepancy could also stem from variations in experimental conditions, such as temperature differences, the use of perforated patch vs. ruptured patch techniques, and variations in recording solutions. A previous study demonstrated that INa,L was markedly elevated in human right atrial cardiomyocytes from AF patients compared to those from patients in sinus rhythm at room temperature. However, this disparity in INa,L amplitudes between AF and sinus rhythm cells diminished and statistically insignificant at physiological temperature (42). Similarly, INa,L in human-induced pluripotent stem cell-derived cardiomyocytes decreased with increasing temperature (43). We found that A-803467 shortened APD in M cells and Purkinje cells only at slow pacing rates, which aligns with previous studies. Notably, Yang et al. and Bengel et al. reported similar rate-dependent APD shortening by A-803467 in mouse ventricular myocytes, with the latter specifically demonstrating this effect in ATX-II stimulated preparations. Importantly, this phenomenon was abolished in SCN10A knockout models (15, 29, 33). Li et al. reported that INa,L was frequency-dependent, with its amplitude increasing as the pacing frequency decreased. Notably, Purkinje cells displayed a significantly larger rate dependence of INa,L compared to ventricular myocytes (44). This characteristic may explain why A-803467 shortens APD only under slow pacing rates.
The distinctive properties of Purkinje fibers-including their complex anatomical structure, excitation-contraction coupling modes, and unique electrophysiological characteristics such as AP morphology, APD, and the intricate interplay of membrane currents render them particularly susceptible to afterdepolarizations and ventricular arrhythmias (40, 45). Accumulating evidence has demonstrated that ablation of Purkinje fiber-originated triggers effectively prevents or reduces the recurrence of ventricular tachycardia/fibrillation across a spectrum of cardiac conditions, encompassing both inherited disorders (Brugada syndrome, early repolarization syndrome, LQTS and idiopathic ventricular fibrillation) as well as acquired conditions such as ischemic heart disease (46, 47). Enhanced INa,L underlies the generation of the afterdepolarizations and triggered activity in Purkinje cells (44, 48). ATX-II can enhances the INa,L generated by multiple voltage-gated sodium channels, including Nav1.8, resulting in APD prolongation that can trigger EADs. We reported that in Purkinje fibers, A-803467 showed a tendency to inhibit ATX-II induced EADs at a pacing frequency of 0.5 Hz, with significant suppression of EADs observed at a lower frequency of 0.2 Hz via reduction of INa,L. These findings provide compelling evidence that Nav1.8-mediated INa,L represents a promising therapeutic target for the pharmacological management of bradycardia associated arrhythmias. Yang et al. illustrated A-803467 effectively eliminated ATX-II induced EADs and trigged activity in mouse and rabbit ventricular cardiomyocytes (15). Furthermore, an enhanced INa,L can precipitate intracellular Ca2+ overload by activating the reverse mode of the Na+/Ca2+ exchanger. This may lead to diastolic sarcoplasmic reticulum Ca2+ leak (Ca2+ sparks) and delayed afterdepolarizations due to increased Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent ryanodine receptor 2 phosphorylation. Ca2+ sparks are considered as a crucial substrate for delayed afterdepolarizations and triggered arrhythmias (13, 48). In mouse ventricular cardiomyocytes stimulated by ATX-II, pharmacological inhibition of Nav1.8 significantly reduced INa,L, thereby diminishing reverse mode Na+/Ca2+ exchanger current, diastolic sarcoplasmic reticulum Ca2+ leak and CaMKII-dependent phosphorylation of ryanodine receptor 2 (29). In human ventricular cardiomyocytes from hearts with failure and hypertrophy, both mRNA and protein expression levels of Nav1.8, as well as the INa,L mediated by Nav1.8, were found to be significantly increased. Inhibition of Nav1.8 notably reduced INa,L and diastolic sarcoplasmic reticulum Ca2+ leak (22, 49). Moreover, in a chronic CaMKIIδc overexpression heart failure mouse model, genetic ablation of SCN10A significantly suppressed INa,L, reduced Ca2+-dependent proarrhythmic triggers and delayed afterdepolarizations in ventricular myocytes, and decreased in vivo arrhythmia events (13). Wu et al. revealed endogenous INa,L underlies the reverse rate-dependent prolongation of APD induced by IKr inhibitors, enhances beat-to-beat repolarization variability, and promotes bradycardia-associated ventricular arrhythmias (50). Our findings further illustrated that A-803467 significantly reduced APD and suppressed EADs through INa,L inhibition under slow pacing conditions. During bradycardia, particularly in pathological conditions, such as myocardial infarction, heart failure, and ventricular hypertrophy, the pathological upregulation of Nav1.8-mediated INa,L significantly elevates the risk of arrhythmias at slow heart rates, with this effect being most prominent in M cells and Purkinje fibers. This electrophysiological paradigm elucidates Nav1.8 channels as a compelling therapeutic target in the pharmacological management of bradycardia-associated arrhythmogenesis.
The underlying mechanisms by which SCN10A modulates cardiac electrophysiology in ventricular myocytes are not yet fully elucidated. Beyond the direct electrophysiological effects of Nav1.8 on ventricular cardiomyocyte, van den Boogaard et al. proposed that intronic variants in SCN10A altered the transcription factor binding site for TBX5 and TBX3, thereby regulating the expression of SCN5A (51). Additionally, our group previously demonstrated that in a heterologous expression system, the co-expression of SCN10A with SCN3B yielded a negligible sodium current. In contrast, co-expressing SCN10A with both SCN5A and SCN3B nearly doubled the mediated sodium current compared to co-expression of SCN5A and SCN3B. Co-immunoprecipitation assays further revealed an association between Nav1.5 and Nav1.8 when co-expressed (3, 52). Moreover, when short SCN10A was co-expressed with SCN5A in heterologous expression system, nearly identical results were observed (25). These findings support the premise that Nav1.8 may affect the electrophysiology of cardiomyocytes either through direct physical interactions or indirect interactions with Nav1.5 as a protein complex. Additional evidence further supports this hypothesis. in situ hybridization analysis revealed that SCN10A and SCN5A exhibited overlapping spatial and temporal expression patterns within the mouse heart (21). Besides, CaMKIIδc co-immunoprecipitated with Nav1.8 in human ventricular myocardium from both non-failing and heart failure samples. Immunofluorescence staining further corroborated co-localization of CaMKIIδc and Nav1.8 in isolated human ventricular cardiomyocytes (13). Moreover, it is also well established that CaMKIIδc interacts with Nav1.5 as part of a macro complex. However, evidence for such a protein complex in vivo is currently lacking, and further studies are needed to confirm this hypothesis. What's more, SCN10A can also indirectly modulate the electrophysiology of cardiomyocytes through intracardiac neurons (33).
Limitations
The experimental data were derived from canine tissue, and the cell type-specific results obtained from our particular canine model may limit the generalizability of our findings to other species or humans. Therefore, clinical extrapolation should be approached with caution. Further validation in more physiologically relevant models, including arterially perfused ventricular wedge preparations and in vivo studies is warranted. Additionally, given the known heterogeneous distribution of SCN10A between ventricles, the applicability of our results, obtained from left ventricular tissue, to the right ventricle and whole heart requires further exploration. Although A-803467 is currently the most selective Nav1.8 blocker available, the possibility of off-target effects on other sodium channels cannot be entirely ruled out.
Conclusion
Nav1.8 exhibits a heterogeneous role in cardiac electrophysiology across the ventricular wall. Nav1.8 inhibition in Purkinje fibers attenuates INa,L, shortens APD, and suppresses EADs, highlighting its potential as a therapeutic target for arrhythmias associated with bradycardia.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by Animal Care and Use Committee of Renmin Hospital of Wuhan University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
Z-HZ: Writing – original draft, Conceptualization, Data curation, Formal analysis, Investigation, Visualization, Writing – review & editing, Validation. HB-M: Writing – original draft, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Visualization, Writing – review & editing, Validation. H-YD: Writing – original draft, Data curation, Investigation, Writing – review & editing, Validation. G-HF: Writing – original draft, Data curation, Investigation, Writing – review & editing, Validation. HJ: Writing – original draft, Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Validation. CA: Writing – original draft, Conceptualization, Funding acquisition, Writing – review & editing, Validation. HX: Writing – original draft, Conceptualization, Data curation, Investigation, Project administration, Supervision, Writing – review & editing, Validation. DH: Data curation, Investigation, Writing – original draft, Writing – review & editing, Conceptualization, Formal analysis, Funding acquisition, Project administration, Supervision, Visualization, Methodology, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Grants from National Key R&D Program of China (grant no. 2023YFE0118300); National Natural Science Foundation Project of China (grant nos. 82270332, 81670304 and 82370286); Fundamental Research Funds for the Central Universities of China (grant no. 2042022kf1217); National Institutes of Health of USA, NIH R56 (grant no. HL47678); National Institutes of Health of USA, NIH R01 (grant nos. HL138103 and HL152201); Woman Board Foundation Grant from Main Line Health, USA (grant no. 25401); W.W. Smith Charitable Trust (grant no. 06417-5222); and the Wistar and Martha Morris Funds and Sharpe-Strumia Research Foundation (grant no. 06417-5221/5524), USA.
Conflict of interest
CA serves as a consultant and received grant funds from Novartis and Trevena Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang ZH, Barajas-Martínez H, Xia H, Li B, Capra JA, Clatot J, et al. Distinct features of probands with early repolarization and Brugada syndromes carrying SCN5A pathogenic variants. J Am Coll Cardiol. (2021) 78(16):1603–17. doi: 10.1016/j.jacc.2021.08.024
2. Walsh R, Mauleekoonphairoj J, Mengarelli I, Bosada FM, Verkerk AO, van Duijvenboden K, et al. A rare noncoding enhancer variant in SCN5A contributes to the high prevalence of Brugada syndrome in Thailand. Circulation. (2025) 151(1):31–44. doi: 10.1161/circulationaha.124.069041
3. Hu D, Barajas-Martínez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. (2014) 64(1):66–79. doi: 10.1016/j.jacc.2014.04.032
4. Sotoodehnia N, Isaacs A, de Bakker PI, Dörr M, Newton-Cheh C, Nolte IM, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. (2010) 42(12):1068–76. doi: 10.1038/ng.716
5. Macri V, Brody JA, Arking DE, Hucker WJ, Yin X, Lin H, et al. Common coding variants in SCN10A are associated with the Nav1.8 late current and cardiac conduction. Circ Genom Precis Med. (2018) 11(5):e001663. doi: 10.1161/circgen.116.001663
6. Avery CL, Wassel CL, Richard MA, Highland HM, Bien S, Zubair N, et al. Fine mapping of QT interval regions in global populations refines previously identified QT interval loci and identifies signals unique to African and Hispanic descent populations. Heart Rhythm. (2017) 14(4):572–80. doi: 10.1016/j.hrthm.2016.12.021
7. Gando I, Williams N, Fishman GI, Sampson BA, Tang Y, Coetzee WA. Functional characterization of SCN10A variants in several cases of sudden unexplained death. Forensic Sci Int. (2019) 301:289–98. doi: 10.1016/j.forsciint.2019.05.042
8. Huang Y, Chen XM, Barajas-Martinez H, Jiang H, Antzelevitch C, Hu D. Common variants in SCN10A gene associated with Brugada syndrome. Hum Mol Genet. (2021) 31(2):157–65. doi: 10.1093/hmg/ddab217
9. Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. (2010) 90(2):755–96. doi: 10.1152/physrev.00020.2009
10. Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, et al. Common variants at SCN5A-SCN10A and Hey2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. (2013) 45(9):1044–9. doi: 10.1038/ng.2712
11. Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. (2010) 42(2):149–52. doi: 10.1038/ng.516
12. Huang Y, Wang LL, Liu ZB, Chen C, Ren X, Luo AT, et al. Underlying mechanism of atrial fibrillation-associated Nppa-I137T mutation and cardiac effect of potential drug therapy. Heart Rhythm. (2024) 21(2):184–96. doi: 10.1016/j.hrthm.2023.10.025
13. Bengel P, Dybkova N, Tirilomis P, Ahmad S, Hartmann N, Mohamed BA, et al. Detrimental proarrhythmogenic interaction of Ca(2+)/calmodulin-dependent protein kinase II and Na(V)1.8 in heart failure. Nat Commun. (2021) 12(1):6586. doi: 10.1038/s41467-021-26690-1
14. Pabel S, Ahmad S, Tirilomis P, Stehle T, Mustroph J, Knierim M, et al. Inhibition of Na(V)1.8 prevents atrial arrhythmogenesis in human and mice. Basic Res Cardiol. (2020) 115(2):20. doi: 10.1007/s00395-020-0780-8
15. Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking SCN10A channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. (2012) 111(3):322–32. doi: 10.1161/circresaha.112.265173
16. Verkerk AO, Remme CA, Schumacher CA, Scicluna BP, Wolswinkel R, de Jonge B, et al. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. (2012) 111(3):333–43. doi: 10.1161/circresaha.112.274035
17. Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, et al. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. (2005) 65(1):117–27. doi: 10.1016/j.cardiores.2004.08.017
18. Zimmer T, Haufe V, Blechschmidt S. Voltage-gated sodium channels in the mammalian heart. Glob Cardiol Sci Pract. (2014) 2014(4):449–63. doi: 10.5339/gcsp.2014.58
19. Casini S, Marchal GA, Kawasaki M, Nariswari FA, Portero V, van den Berg NWE, et al. Absence of functional Na(V)1.8 channels in non-diseased atrial and ventricular cardiomyocytes. Cardiovasc Drugs Ther. (2019) 33(6):649–60. doi: 10.1007/s10557-019-06925-6
20. Pallante BA, Giovannone S, Fang-Yu L, Zhang J, Liu N, Kang G, et al. Contactin-2 expression in the cardiac Purkinje fiber network. Circ Arrhythm Electrophysiol. (2010) 3(2):186–94. doi: 10.1161/circep.109.928820
21. van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. (2012) 122(7):2519–30. doi: 10.1172/jci62613
22. Dybkova N, Ahmad S, Pabel S, Tirilomis P, Hartmann N, Fischer TH, et al. Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc Res. (2018) 114(13):1728–37. doi: 10.1093/cvr/cvy152
23. Stroud DM, Yang T, Bersell K, Kryshtal DO, Nagao S, Shaffer C, et al. Contrasting Nav1.8 activity in SCN10A-/- ventricular myocytes and the intact heart. J Am Heart Assoc. (2016) 5(11):e002946. doi: 10.1161/jaha.115.002946
24. Verweij N, Mateo Leach I, van den Boogaard M, van Veldhuisen DJ, Christoffels VM, Hillege HL, et al. Genetic determinants of P wave duration and PR segment. Circ Cardiovasc Genet. (2014) 7(4):475–81. doi: 10.1161/circgenetics.113.000373
25. Man JCK, Bosada FM, Scholman KT, Offerhaus JA, Walsh R, van Duijvenboden K, et al. Variant intronic enhancer controls SCN10A-short expression and heart conduction. Circulation. (2021) 144(3):229–42. doi: 10.1161/circulationaha.121.054083
26. Burel S, Coyan FC, Lorenzini M, Meyer MR, Lichti CF, Brown JH, et al. C-Terminal phosphorylation of Na(V)1.5 impairs FGF13-dependent regulation of channel inactivation. J Biol Chem. (2017) 292(42):17431–48. doi: 10.1074/jbc.M117.787788
27. Facer P, Punjabi PP, Abrari A, Kaba RA, Severs NJ, Chambers J, et al. Localisation of SCN10A gene product Na(V)1.8 and novel pain-related Ion channels in human heart. Int Heart J. (2011) 52(3):146–52. doi: 10.1536/ihj.52.146
28. Qi B, Dai S, Song Y, Shen D, Li F, Wei L, et al. Blockade of Na(V)1.8 increases the susceptibility to ventricular arrhythmias during acute myocardial infarction. Front Cardiovasc Med. (2021) 8:708279. doi: 10.3389/fcvm.2021.708279
29. Bengel P, Ahmad S, Tirilomis P, Trum M, Dybkova N, Wagner S, et al. Contribution of the neuronal sodium channel Na(V)1.8 to sodium- and calcium-dependent cellular proarrhythmia. J Mol Cell Cardiol. (2020) 144:35–46. doi: 10.1016/j.yjmcc.2020.05.002
30. Hartmann N, Knierim M, Maurer W, Dybkova N, Zeman F, Hasenfuß G, et al. Na(V)1.8 as proarrhythmic target in a ventricular cardiac stem cell model. Int J Mol Sci. (2024) 25(11):6144. doi: 10.3390/ijms25116144
31. Abou Ziki MD, Seidelmann SB, Smith E, Atteya G, Jiang Y, Fernandes RG, et al. Deleterious protein-altering mutations in the SCN10A voltage-gated sodium channel gene are associated with prolonged QT. Clin Genet. (2018) 93(4):741–51. doi: 10.1111/cge.13036
32. Biet M, Barajas-Martínez H, Ton AT, Delabre JF, Morin N, Dumaine R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na(+) channels. J Mol Cell Cardiol. (2012) 53(5):593–8. doi: 10.1016/j.yjmcc.2012.06.012
33. Park DS, Fishman GI. Nav-Igating through a complex landscape: SCN10A and cardiac conduction. J Clin Invest. (2014) 124(4):1460–2. doi: 10.1172/jci75240
34. Antzelevitch C, Nesterenko V, Shryock JC, Rajamani S, Song Y, Belardinelli L. The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol. (2014) 221:137–68. doi: 10.1007/978-3-642-41588-3_7
35. Jabbari J, Olesen MS, Yuan L, Nielsen JB, Liang B, Macri V, et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ Cardiovasc Genet. (2015) 8(1):64–73. doi: 10.1161/HCG.0000000000000022
36. El-Battrawy I, Albers S, Cyganek L, Zhao Z, Lan H, Li X, et al. A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. Europace. (2019) 21(9):1410–21. doi: 10.1093/europace/euz122
37. Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell. Circ Res. (1991) 68(6):1729–41. doi: 10.1161/01.res.68.6.1729
38. Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol. (2001) 281(2):H689–97. doi: 10.1152/ajpheart.2001.281.2.H689
39. Vassalle M, Bocchi L, Du F. A slowly inactivating sodium current (INa2) in the plateau range in canine cardiac Purkinje single cells. Exp Physiol. (2007) 92(1):161–73. doi: 10.1113/expphysiol.2006.035279
40. Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, et al. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. (1999) 10(8):1124–52. doi: 10.1111/j.1540-8167.1999.tb00287.x
41. Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res. (1995) 76(3):351–65. doi: 10.1161/01.res.76.3.351
42. Poulet C, Wettwer E, Grunnet M, Jespersen T, Fabritz L, Matschke K, et al. Late sodium current in human atrial cardiomyocytes from patients in sinus rhythm and atrial fibrillation. PLoS One. (2015) 10(6):e0131432. doi: 10.1371/journal.pone.0131432
43. El-Battrawy I, Lang S, Zhao Z, Akin I, Yücel G, Meister S, et al. Hyperthermia influences the effects of sodium channel blocking drugs in human-induced pluripotent stem cell-derived cardiomyocytes. PLoS One. (2016) 11(11):e0166143. doi: 10.1371/journal.pone.0166143
44. Li W, Yu Y, Hou JW, Zhou ZW, Guo K, Zhang PP, et al. Larger rate dependence of late sodium current in cardiac Purkinje cells: a potential link to arrhythmogenesis. Heart Rhythm. (2017) 14(3):422–31. doi: 10.1016/j.hrthm.2016.11.036
45. Sicouri S, Belardinelli L, Antzelevitch C. Antiarrhythmic effects of the highly selective late sodium channel current blocker gs-458967. Heart Rhythm. (2013) 10(7):1036–43. doi: 10.1016/j.hrthm.2013.03.023
46. Haïssaguerre M, Extramiana F, Hocini M, Cauchemez B, Jaïs P, Cabrera JA, et al. Mapping and ablation of ventricular fibrillation associated with long-QT and Brugada syndromes. Circulation. (2003) 108(8):925–8. doi: 10.1161/01.Cir.0000088781.99943.95
47. Haissaguerre M, Vigmond E, Stuyvers B, Hocini M, Bernus O. Ventricular arrhythmias and the his-Purkinje system. Nat Rev Cardiol. (2016) 13(3):155–66. doi: 10.1038/nrcardio.2015.193
48. Shryock JC, Song Y, Rajamani S, Antzelevitch C, Belardinelli L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res. (2013) 99(4):600–11. doi: 10.1093/cvr/cvt145
49. Ahmad S, Tirilomis P, Pabel S, Dybkova N, Hartmann N, Molina CE, et al. The functional consequences of sodium channel Na(V) 1.8 in human left ventricular hypertrophy. ESC Heart Fail. (2019) 6(1):154–63. doi: 10.1002/ehf2.12378
50. Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, et al. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation. (2011) 123(16):1713–20. doi: 10.1161/circulationaha.110.000661
51. van den Boogaard M, Smemo S, Burnicka-Turek O, Arnolds DE, van de Werken HJ, Klous P, et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest. (2014) 124(4):1844–52. doi: 10.1172/jci73140
Keywords: SCN10A, pharmacology, arrhythmia, Purkinje fibers, cardiac electrophysiology
Citation: Zhang Z-H, Barajas-Martinez H, Duan H-Y, Fan G-H, Jiang H, Antzelevitch C, Xia H and Hu D (2025) Characterization of novel arrhythmogenic patterns arising secondary to heterogeneous expression and activation of Nav1.8. Front. Cardiovasc. Med. 12:1546803. doi: 10.3389/fcvm.2025.1546803
Received: 17 December 2024; Accepted: 7 March 2025;
Published: 20 March 2025.
Edited by:
Konstantinos Letsas, Onassis Cardiac Surgery Center, GreeceReviewed by:
Jerome Montnach, INSERM U1087 Institut du Thorax, FranceDimitra Antonakaki, St Bartholomew's Hospital, United Kingdom
Copyright: © 2025 Zhang, Barajas-Martinez, Duan, Fan, Jiang, Antzelevitch, Xia and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Xia, eGlhaGFvMTk2NkAxNjMuY29t; Dan Hu, aHVkYW4wNzE2QGhvdG1haWwuY29t; cm0wMDI2NDZAd2h1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
‡ORCID:
Dan Hu
orcid.org/0000-0002-0994-8434