 Asad Nawaz
Asad Nawaz Zixian Sheng†
Zixian Sheng† Muhammad Junaid Akram
Muhammad Junaid Akram Lingjuan Liu
Lingjuan Liu Yuxing Yuan
Yuxing Yuan Jie Tian
Jie Tian- Department of Cardiology, National Clinical Research Center for Child Health and Disorders, Ministry of Education Key Laboratory of Child Development and Disorders, China International Science and Technology Cooperation Base of Child Development and Critical Disorders, Chongqing Key Laboratory of Pediatric Metabolism and Inflammatory Diseases, Key Laboratory of Children’s Important Organ Development and Diseases of Chongqing Municipal Health Commission, National Clinical Key Cardiovascular Specialty, Children’s Hospital of Chongqing Medical University, Chongqing, China
Background: Pediatric cardiomyopathies are rare but life-threatening conditions with high mortality. Limited data exists on their clinical features and risk factors, especially in Asian populations, highlighting the need for further research in this area.
Methods: This retrospective cohort study analyzed data from 212 pediatric patients diagnosed with hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), or restrictive phenotype hypertrophic cardiomyopathy (RP-HCM) at a single center in China from October 2012 to October 2023, with follow-up until October 31, 2024. Demographic, clinical, and diagnostic data, as well as follow-up outcomes, were reviewed. Logistic and Cox regression models identified risk factors for in-hospital and long-term mortality.
Results: Among the 212 patients, 79.72% (169/212) had HCM, 16.98% (36/212) had RCM, and 3.30% (7/212) had RP-HCM. Infection (75.47%, 160/212) and heart failure (51.42%, 109/212) were common comorbidities. In-hospital mortality was 5.19% (11/212), with follow-up mortality of 20.28% (43/212). The independent risk factors for mortality included left ventricular ejection fraction (LVEF), pulmonary hypertension, and low-density lipoprotein (LDL) levels (P < 0.05). Patients with RP-HCM showed the poorest outcomes, with a follow-up mortality rate of 42.86%. Only 10.4% (22/212) of patients underwent genetic testing, yet the positive detection rate was 63.7% (14/22).
Conclusions: This study underscores the importance of early diagnosis, genetic testing, and integrated management in pediatric cardiomyopathies. LVEF, pulmonary hypertension, and LDL levels are critical prognostic factors, offering insights for risk assessment and management in affected children.
1 Introduction
Pediatric cardiomyopathy is a rare but life-threatening condition with an estimated prevalence of approximately 1 in 100,000 (1, 2). Despite its rarity, it poses significant risks to affected children, leading to long-term influences on cardiac and pulmonary function, growth, development, and overall quality of life. Among the various types, dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) are more frequently observed and extensively studied. In contrast, restrictive cardiomyopathy (RCM), characterized by impaired ventricular filling and diastolic dysfunction, remains less understood due to its lower incidence (1–8). Recently, the emergence of a distinct subtype, hypertrophic cardiomyopathy with restrictive phenotype (RP-HCM), has garnered attention. RP-HCM presents with more severe clinical manifestations and a worse prognosis than typical HCM. However, due to its recent recognition and rarity, comprehensive data on RP-HCM remain limited, leaving significant gaps in our understanding of its clinical course and management strategies (9–13).
While recent studies have expanded the understanding of pediatric cardiomyopathy, there is still a lack of data specific to Asian populations, where genetic and environmental factors may influence disease characteristics and outcomes (1–15). To address this gap, we conducted a single-center retrospective cohort study in China to evaluate the clinical characteristics, outcomes, and mortality risk factors in children with HCM, RCM, and RP-HCM. By analyzing a multi-year dataset, this study aims to identify potential prognostic indicators to inform clinical decision-making and improve long-term outcomes for pediatric patients with these cardiomyopathies.
2 Methods
The observational study protocol was approved by the Ethical Committee of Chongqing Medical University (Date 2020/No 160-1). The parents of all participants provided informed consent. The data supporting this study's findings are available from the corresponding author upon reasonable request.
2.1 Patients
All hospitalized patients (aged <18 years) diagnosed with HCM, RCM, or RP-HCM from October 2012 to October 2023 were included in the study. Patients with comorbid malignant tumors or those with data missing for ≥20% were excluded. Follow-up records were established for each patient, and telephone follow-ups were conducted annually after discharge, with the follow-up period ending either upon the child's death or by October 31, 2024. Based on the follow-up results, the children were categorized as survival, death, or lost to follow-up.
2.2 Definition and diagnostic
The definition of CM in children is similar to that of adults. HCM refers to a thickening of the heart wall that cannot be explained by hemodynamic reasons and is not accompanied by ventricular dilation but physiological hypertrophy (such as that resulting from physical activity) and pathological hypertrophy (such as that resulting from hypertension, aortic valve stenosis, and other diseases) must be excluded. RCM is characterized by reduced ventricular compliance and is usually not associated with significant biventricular dilation, hypertrophy, or a decrease in systolic function. Their diagnostic criteria are based on pediatric guidelines (1, 2).
Research on RP-HCM in children is limited, and the diagnostic criteria refer to those for adults, including the following aspects: significant enlargement of both atria, mitral inflow E/A ratio of ≥2, and mitral deceleration time of ≤150 ms; absent or only mild left ventricular hypertrophy (LVH); ventricular cavity reduced or of normal size; left ventricular ejection fraction (LVEF) normal or mildly decreased (9–14).
2.3 Variables and outcomes
The primary outcome measure was all-cause mortality during the follow-up period, whereas the secondary outcome measure was in-hospital all-cause mortality. Data, including demographic characteristics, clinical features, auxiliary examinations, Genetic Report, diagnosis, and outcomes information, were collected for all patients based on their electronic medical records or clinical charts. All clinical characteristics were captured at the time of initial diagnosis/first evaluation at our center.
2.4 Statistical analyses
Data processing was performed using RStudio 4.4.1 software. Differences with P < 0.05 were considered statistically significant. Impute missing values using the random forest algorithm. The Kolmogorov–Smirnov test was applied to verify whether our data were normally distributed. Means and standard deviations were calculated to describe the variables for normally distributed data, while medians and ranges were calculated for non-normally distributed data. Frequencies and percentages were used for categorical data. Significant differences between the two groups were measured using χ2 or Fischer exact tests for categorical variables and the t-test or Wilcoxon rank-sum test for continuous variables.
Additionally, we analyzed the factors affecting in-hospital mortality risk using a logistic regression model and the factors affecting mortality risk during the follow-up period using a Cox regression model. Performed inter-group difference analysis and univariate regression analysis to screen influencing factors, and then explored independent risk factors through multivariate stepwise regression analysis.
3 Results
3.1 General characteristics
A total of 212 patients were included in the study, including 169 (79.72%) for HCM, 36 (16.98%) for RP-HCM, and 7 (3.30%) for RP-HCM. The median age at admission was 0.83 (0.33, 7.00) years, the male-to-female ratio was about 1.9:1, 52.36% of patients were admitted through outpatient clinics, and 70.75% were diagnosed with cardiomyopathy for the first time. Infection (75.47%) was the most common co-diagnosis, followed by heart failure (51.42%) and arrhythmia (20.28%). The average length of hospitalization was 12.09 ± 11.75 days, and 11 cases (5.19%) died of all causes in hospital. Two patients (1 with HCM and 1 with RP-HCM) underwent pacemaker implantation during hospitalization. The duration of follow-up was 4.24 (1.87, 7.61) years, and the mean age at the end of follow-up was 7.56 (3.74, 12.51) years. During follow-up, 43 (20.28%) cases died, and 11 (5.19%) cases were lost to follow-up (Table 1).
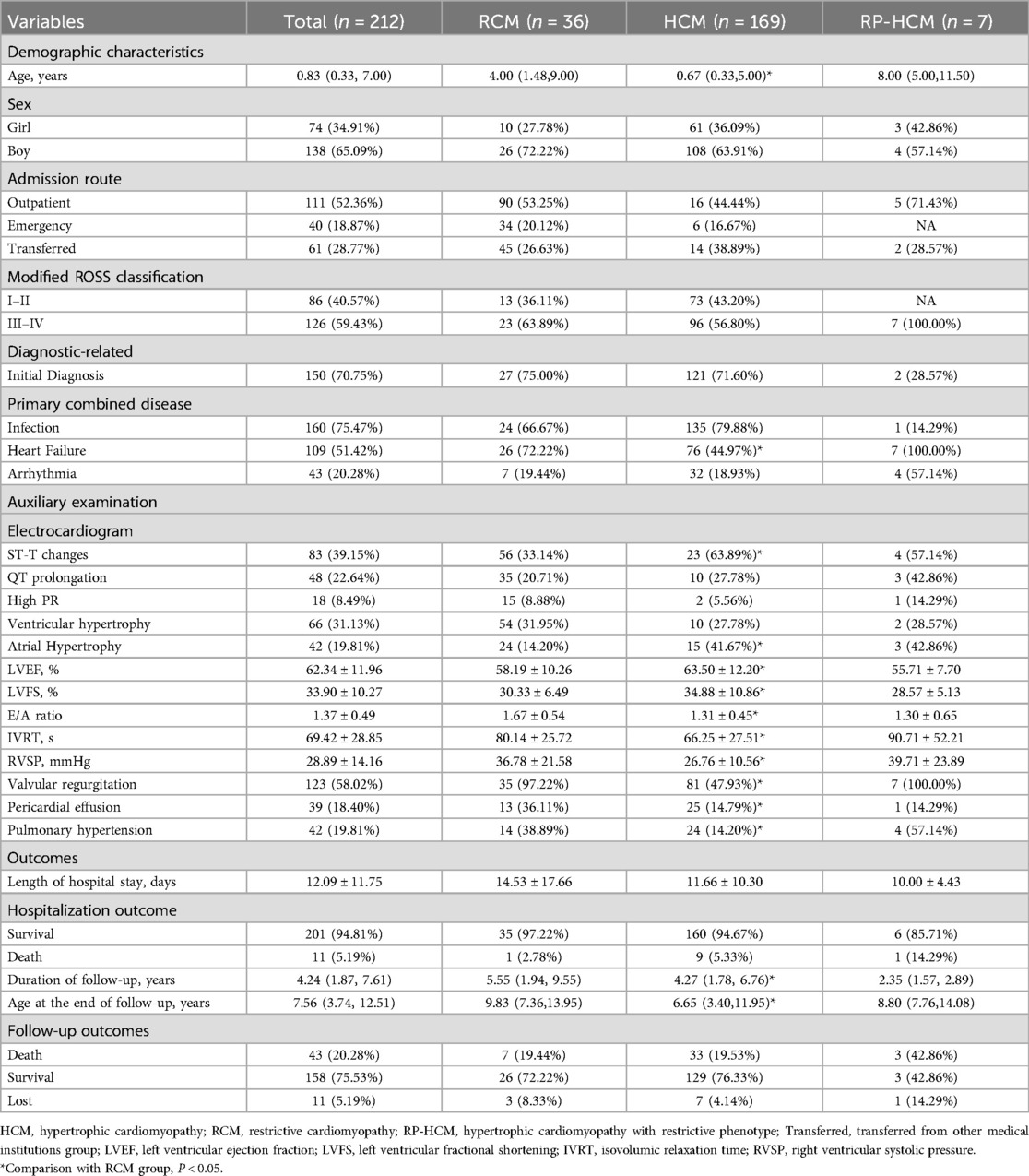
Table 1. Total sample characteristics and comparison of RCM, HCM, and RP-HCM.
3.2 Types of cardiomyopathies
Because the sample size of PR-HCM was too small, we only compared the RCM group with the HCM group. The results showed that the median age of RCM children was significantly higher than that of the HCM group [4.00 (1.48, 9.00) years vs. 0.67 (0.33, 5.00) years], and there was no statistical difference in the sex ratio among the groups. More men than women. The proportion of children with heart failure in the RCM group was significantly higher than that in the HCM group (72.22% vs. 44.97%), and the proportion of children with co-infection in both groups was higher than 2/3 (RCM 66.67%; HCM 79.88%), about 1/5 of the patients with arrhythmia (RCM 19.44%; HCM 18.93%). The results of cardiac ultrasound showed that LVEF (58.19 ± 10.26% vs. 63.50 ± 12.20%) and LVFS (30.33 ± 6.49% vs. 34.88 ± 10.86%) in the RCM group were significantly lower than those in the HCM group. The E/A ratio of RCM group was (1.67 ± 0.54 vs. 1.31 ± 0.45), IVRT (80.14 ± 25.72 s vs. 66.25 ± 27.51 s) and RVSP (36.78 ± 21.58 mmHg vs. 26.76) ± 10.56 mmHg), pulmonary hypertension (97.22% vs. 47.93%), pericardial effusion (36.11% vs. 14.79%) and moderate to severe valve regurgitation (38.89% vs. 14.20%) were significantly higher than those in HCM group (Table 1). The results of electrocardiogram, cardiac biomarkers, complete blood count, and biochemical tests were different in only some indicators (Supplementary Table 1). All the above differences were statistically significant (P < 0.05).
There was no significant difference in outcome indicators between the HCM and RCM groups. The in-hospital all-cause mortality was 2.78% (1/36) in the RCM group and 19.44% (7/36) in the follow-up period. In-hospital all-cause mortality was 5.33% (9/169) in the HCM group and 19.53% (33/169) during follow-up (Table 1).
We also described the characteristics of the PR-HCM group, and the results showed that the median age of 7 children at admission was 8.00 (5.00, 11.50) years, all with heart failure, and the modified cardiac function grade was III to IV. Four patients (57.14%) had arrhythmia, but only one (14.29) had infection. LVEF (55.71 ± 7.70%) and LVFS (28.57 ± 5.13%) of these children were lower than the mean values of HCM and RCM groups, and the results of IVRT (90.71 ± 52.21) and RVSP (39.71 ± 23.89) were more similar to those of RCM group than HCM group. All seven patients had moderate to severe valve regurgitation, and four (57.14%) had pulmonary hypertension. Although only one (14.29%) person died during hospitalization in the PR-HCM group, all-cause mortality was 42.86% (3/7) during follow-up (Table 1).
3.3 Comparison of patients with different admission routes
111, 40 and 61 patients were transferred to outpatient, emergency and other medical institutions, respectively. There were no significant differences in age and sex ratio among the three groups. However, the modified ROSS classification of Transferred group and emergency group was III–IV (Transferred group 86.89%; 75% in the emergency group) and the first diagnosis of cardiomyopathy (85.25% in the Transferred group; The proportion of patients in the emergency group (77.50%) was significantly higher than that in the outpatient group (38.74% with modified ROSS grade III to IV, 60.36% with first test diagnosis). The proportion of patients with infection in the emergency department group was the highest (87.50%), followed by the Transferred group (80.33%) and the outpatient group (68.47%) (the above differences were statistically significant (P < 0.05, Table 2). No significant differences were observed in heart failure, arrhythmia, and laboratory test results among the three groups (Supplementary Material 3).
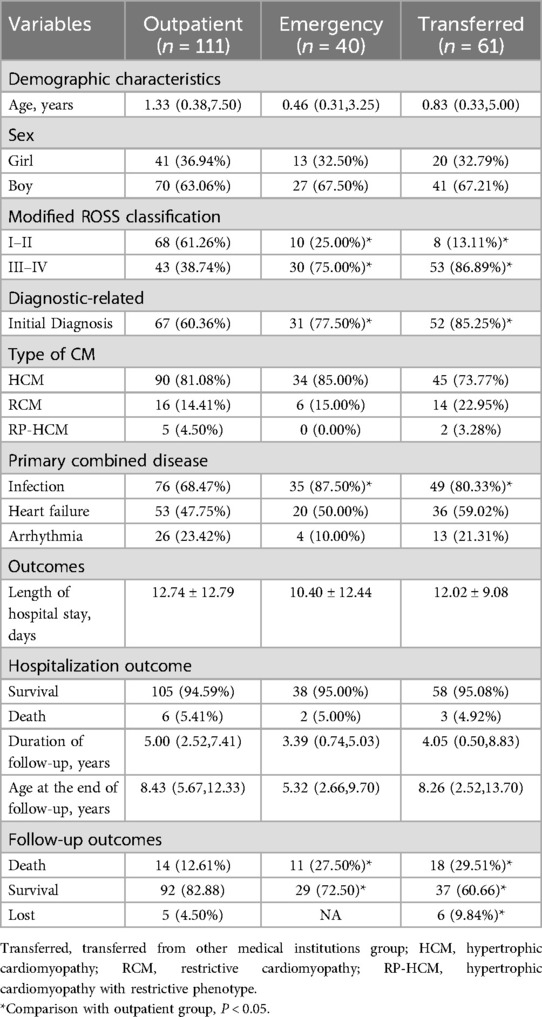
Table 2. Comparison of patients admitted through different modes of admission.
Regarding outcomes, although there was no significant difference in in-hospital all-cause mortality among the three groups, all-cause mortality during follow-up was significantly higher in both the Transferred (29.51%) and emergency (27.50%) groups than in the outpatient group (12.61%). All the above differences were statistically significant (P < 0.05, Table 2).
3.4 Risk factors for death from cardiomyopathy in children
Logistic stepwise regression analysis showed that LDL (OR 2.15; 95% CI 1.28–3.61) and RVSP (OR 1.04; 95% CI 1.01–1.07) were positively correlated with the risk of hospital death in children with cardiomyopathy, LVEF (OR 0.95; 95% CI 0.90–0.99) was negatively associated with the risk of death in hospital (Figure 1A, Supplementary Material 5). COX stepped-regression analysis showed Pulmonary hypertension (OR 2.40; 95% CI 1.24–4.64), LDL (OR 1.48; 95% CI 1.11–1.96), CKMB (OR 1.01; 95% CI 1.01–1.01) was positively associated with the risk of death during follow-up in children with cardiomyopathy, while Weight (OR 0.92; 95% CI 0.88–0.96), ALB (OR 0.94; 95% CI 0.90–0.98), TG (OR 0.52; 95% CI 0.31–0.87), LVEF (OR 0.96; 95% CI 0.93–0.98) was negatively associated with the risk of death during follow-up (Figure 1B, Supplementary Material 6).
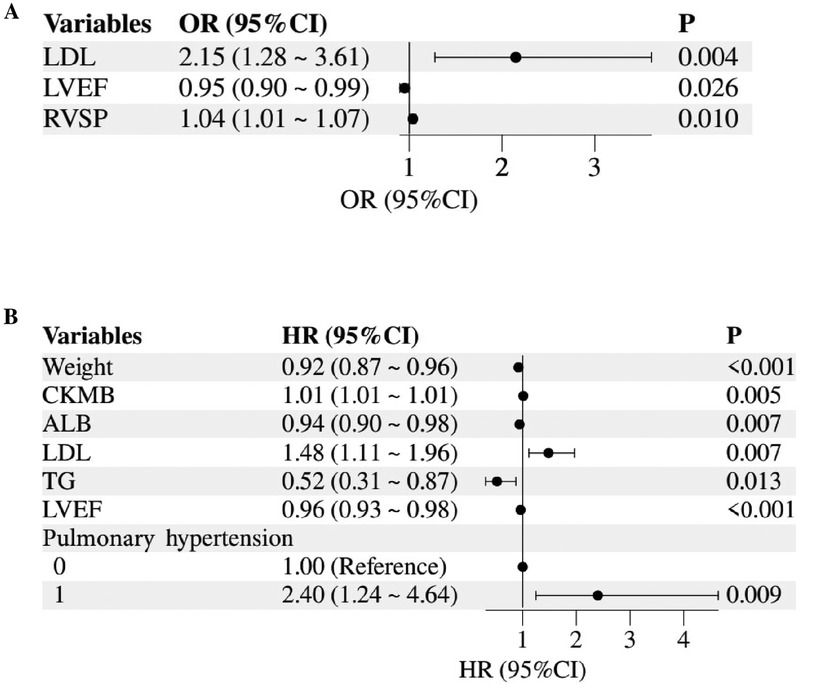
Figure 1. Forest plot of mortality risk factors in children with HCM or RCM. (A) Logistic regression for in-hospital mortality risk; (B) cox regression for mortality risk during follow-up. LDL, low-density lipoprotein; LVEF, left ventricular ejection fraction; RVSP, right ventricular systolic pressure; CKMB, creatine kinase MB; ALB, albumin; TG, triglycerides.
3.5 Genetic analysis
In our sample, only 10.4% (22/212) of the children were genetically tested. Of these 22, 68.2% (15/22) were tested after 5 years of age, and the positive detection rate was 63.7% (14/22). Among them, 40.1% (9/22) were pathogenic mutations, 27.3% (5/22) were likely pathogenic mutations. At the same time, 31.8% (7/22) had multiple gene mutations, and 40.1% (9/22) had mutations from their parents. The most frequently mutated gene was MYH7 (5 HCM, 1 RCM and 3 RP-HCM), followed by TNNI3 (1 HCM and 2 RCM) and GAA (2 HCM). Five children died during follow-up, two of whom had polygenic mutations, and the pathogenic or suspected mutations detected included PTPN11 (1), MYH7 (2), TNNI3 (1), KCNE3 (1), and HCN4 (1) (Table 3).
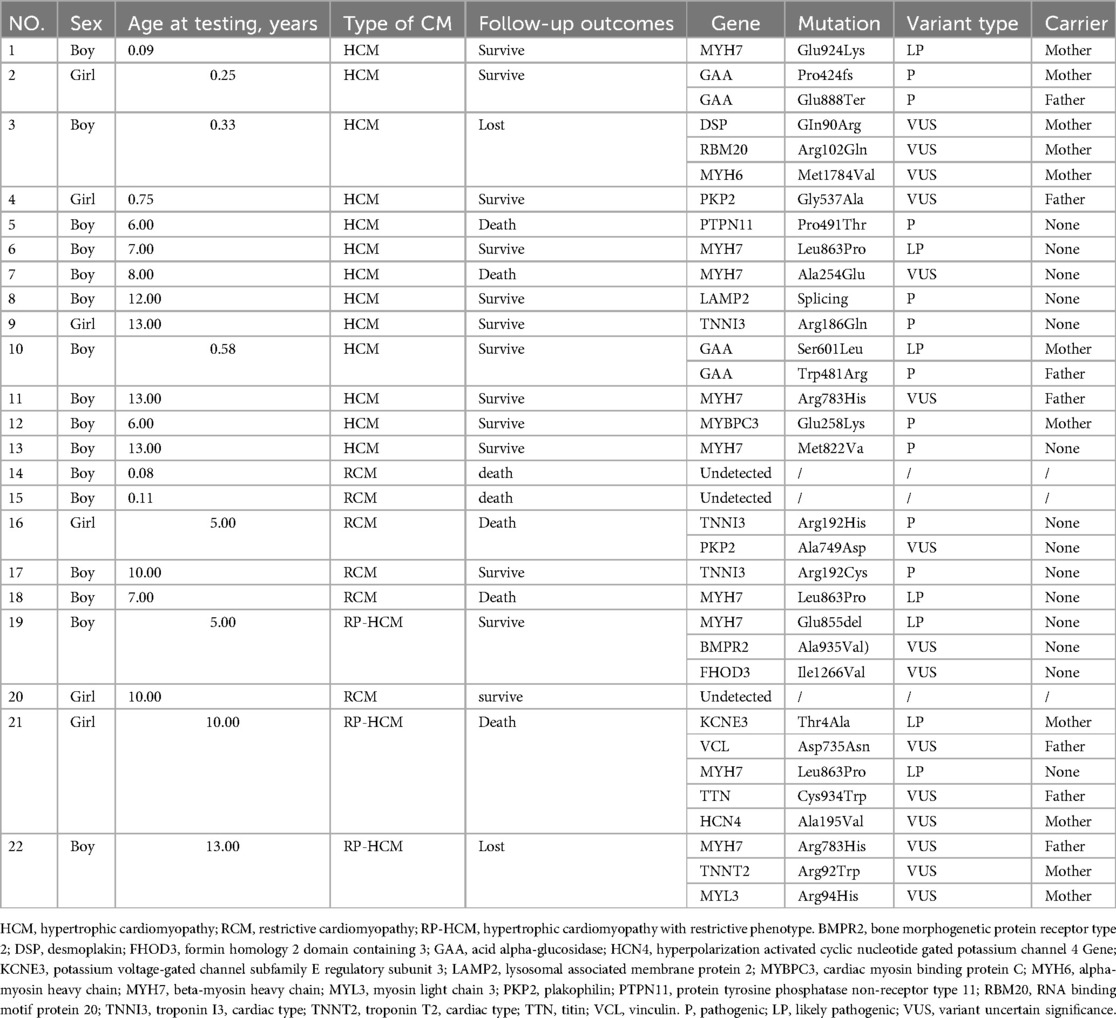
Table 3. Genetic analysis.
4 Discussion
This study conducted long-term follow-up of hypertrophic cardiomyopathy (HCM), restricted cardiomyopathy (RP-HCM), and restricted phenotype hypertrophic cardiomyopathy (RP-HCM) in China, and comprehensively evaluated clinical features, outcomes, and mortality risk factors, emphasizing the importance of genetic testing, early diagnosis, and integrated management to improve prognosis. It enhances our current understanding of childhood cardiomyopathy.
Consistent with previous studies, far more children are diagnosed with HCM than RCM, and patients with HCM are also diagnosed at a younger age (3–8). Notably, the median age at HCM diagnosis in our cohort is younger than that reported in European/North American cohorts (7, 8), likely reflecting regional differences in early screening practices. Both cardiomyopathy phenotypes are more common in boys than in girls. Previous studies have suggested that RCM is rare in childhood, but the prognosis is poor (16). The results of our analysis showed that the heart function of children with RCM was significantly worse than that of HCM, specifically as follows: Children with RCM had lower left ventricular ejection fraction (LVEF) and left ventricular shortening fraction (LVFS) than children with HCM, and higher rates of early/atrial (E/A) ratio, isovolumic diastolic time (IVRT), right ventricular systolic blood pressure (RVSP), pulmonary hypertension, and moderate to severe valve regurgitation. Therefore, the incidence of heart failure on admission in the RCM group was significantly higher than that in the HCM group. Interestingly, despite these severe functional impairments, the cumulative mortality of RCM in our cohort (19.5%) was lower than historical Western data (5-year survival rate 51.1%–64.6%) (3, 17–19). This may be related to the fact that RCM children in our study were diagnosed at a younger age than previously reported, allowing timely intervention to delay disease progression. Additionally, the high infection comorbidity rate in our cohort [75.47% vs. <30% in Western studies (7)] underscores the need for region-specific strategies to address infectious triggers. In addition to the differences in the pathological mechanisms of the two phenotypes, this result may also be influenced by the generally late age of diagnosis of RCM. In addition, the progress of the medical level and the update of guidelines also provided great help to improve the clinical outcome of children with CM.
Of course, as we expected, children with this particular phenotype of RP-HCM had a very bad outcome. This is why the clinical characteristics and outcomes of children with RP-HCM are described separately (9–13). RP-HCM was first proposed by Kubo et al. (9), and restrictive filling pattern (10, 11) and restrictive physiology (12) were also named in other literature. Previous studies have mainly been carried out in adults. As one of the adverse remodeling types of HCM, its incidence in HCM ranges from 1.5% to 16% (9–11). Compared with typical HCM, RP-HCM has a more severe clinical phenotype, higher rates of atrial fibrillation, stroke, and heart failure, and a relatively poor clinical prognosis, with 22.6%–43.6% of patients dying or having a heart transplant during 5–10 years of follow-up (9–11). Of our patients, all seven had heart failure and moderate to severe valvular regurgitation, 57.14% had arrhythmias, and all of the cardiac ultrasound assessments were worse than those in the typical HCM group. Although only one person died during hospitalization, the all-cause mortality rate for RP-HCM was 42.86% during 2.35 (95% CI 1.57–2.89) years of follow-up. Although we only have seven samples, these results suggest that we need to pay attention to this particular phenotype and conduct higher-quality clinical and mechanistic studies in children to inform the development of treatment strategies in this population.
From the perspective of admission routes, nearly 50% of hospitalized children with cardiomyopathy are still from the emergency department or transferred hospitals (transferred from lower-level hospitals), and these children have worse heart function. The proportion of patients with first diagnosis of cardiomyopathy (77.5%–85.3%), co-infection (80.3%–87.5%), and mortality during follow-up (27.5%–29.5%) were higher than those admitted through outpatient clinics. Combined with the patient records of these children, we found that many children are still treated for heart failure caused by infection or infection, and then diagnosed with cardiomyopathy, but most of the children are at high risk of worsening heart function. This may be related to the lack of awareness of childhood cardiomyopathy and the imperfect management of early screening and diagnosis of childhood cardiomyopathy. Secondly, the high proportion of co-infection is also very worthy of attention, not only for cardiomyopathy. In all cases, infection is an important risk factor leading to the deterioration of children's cardiopulmonary function, so for children with cardiomyopathy, it is very important to do a good job of infection prevention and standardized treatment. Finally, for children with confirmed cardiomyopathy, regular outpatient follow-up and daily management are very important for delaying the course of the disease and improving the prognosis of the disease.
In addition, in our data, the median age of hospitalized children is less than 1 year old, and they can get reasonable diagnosis and treatment earlier, which reflects the progress of medical treatment level and is a very good phenomenon. However, the bad thing is that only 10.4% of the children had genetic testing, and among the children who had genetic testing, more than 2/3 of the sick children were older than 5 years old, which suggests that the coverage of genetic testing for children with cardiomyopathy may be insufficient, and the detection time is lagging. Whether in the study of adult or childhood cardiomyopathy, scholars have repeatedly emphasized the important value of genetic testing in cardiomyopathy (1–3, 20–23). Results of a multi-centre study conducted at 14 institutions in North America showed that 81 children tested for clinical cardiomyopathy gene combinations (21), 39 (48%) were positive and 19 (24%) were VUS of unknown significance, and the positive rate of testing was higher in children with HCM (68%) than in children with DCM. Therefore, genetic testing can help a large proportion of children with cardiomyopathy to make a definitive molecular genetic diagnosis. In addition to assisting in diagnosing and classifying cardiomyopathy, genetic testing is also an important basis for assessing prognosis and developing individualized treatment plans. For example, in HCM, patients with MYH7 gene mutation may have more obvious clinical symptoms, a younger age of onset, and a worse prognosis. Patients with MYBPC3 gene mutation showed more benign disease progression without serious clinical symptoms, and most of the first onset age was 40–50 years old (24). However, the clinical practice of genetic detection of childhood cardiomyopathy and the mechanism exploration of abnormal genes and clinical phenotypes still have many shortcomings that need improvement. Therefore, in clinical work for children with cardiomyopathy and children with first-degree family members with cardiomyopathy, we suggest actively improving genetic testing and genetic counselling (1, 2, 23), and we need to refer to the results of genetic testing to carry out more in-depth mechanism research to provide a reference for the precise treatment of cardiomyopathy.
Finally, we believe that LVEF, pulmonary hypertension, and LDL are all high-risk factors for death in children with HCM and RCM (25–30). LVEF is closely related to the heart's pumping function, especially in cardiomyopathy, and the decline of LVEF may mean the progressive deterioration of cardiac remodeling, accompanied by the progression of heart failure, resulting in adverse outcomes. RVSP is a measure of pulmonary hypertension, which may be caused by abnormalities in the structure and function of the heart and may aggravate the process of heart failure and ventricular remodeling; and is also considered to be an important factor indicating poor prognosis in children with cardiomyopathy (31). The effect of LDL on the prognosis of children with cardiomyopathy may be related to lipid metabolism, but its specific mechanism of action remains to be explored. We speculate that elevated LDL may reflect impaired myocardial energy metabolism or genetic defects in lipid regulation, as seen in other pediatric metabolic disorders. For example, a 2007 trial reported that omega-3 supplementation improved ventricular function in pediatric dilated cardiomyopathy, though LDL was not analyzed in that study (32). This suggests potential interactions between lipid homeostasis and myocardial repair. Previous scholars have suggested that cardiomyopathy and heart failure with ejection fraction retention can be regarded as metabolic diseases. Sodium-dependent glucose transporters 2 (SGLT-2) also improved cardiac function in these patients (33). On the other hand, recent studies in adults have found that either too low or too high LDL is strongly associated with an increased risk of death from cardiovascular disease. In our study, LVEF, pulmonary hypertension, and LDL are closely related to the risk of death in children with cardiomyopathy during hospitalization and post-discharge follow-up. These variables are easily accessible in clinical practice, and their combined use may enhance risk stratification. Future large-scale cohort studies are needed to validate their prognostic value and explore the underlying mechanisms through basic research.
The main limitation of this study was its retrospective, single-center design with a small sample size and limited genetic testing data, which may not have fully represented the characteristics of pediatric HCM and RCM. Despite this, long-term follow-up was conducted, and the clinical outcomes of RP-HCM were specifically described. Non-medical factors, including maternal history and education of guardians, showed no significant correlation with outcomes (SM1-4). Future multicenter studies with larger cohorts are warranted to advance the understanding of pediatric cardiomyopathy, particularly focusing on distinct subtypes such as HCM, RCM, and RP-HCM.
5 Conclusion
This study describes the clinical features, outcomes, and influencing factors of children with HCM, RCM, and RP-HCM in China. It emphasizes the importance of early diagnosis, genetic testing, and integrated management to improve prognosis. LVEF, pulmonary hypertension, and LDL levels are suggested as key risk factors for poor prognosis in children with cardiomyopathy, providing new insights for risk assessment and treatment management of pediatric cardiomyopathy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethical Committee of Chongqing Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
AN: Data curation, Formal analysis, Software, Writing – original draft, Writing – review & editing. ZS: Formal analysis, Writing – original draft. MA: Writing – original draft, Writing – review & editing. JL: Formal analysis, Writing – review & editing. LL: Formal analysis, Writing – review & editing. YY: Formal analysis, Project administration, Software, Writing – review & editing. JT: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Clinical Research Center for Child Health and Disorders (Grant No: NCRCCHD-2021-KP-01 to JT).
Acknowledgments
We appreciate the participation of all patients in the present study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1541651/full#supplementary-material
References
1. Bogle C, Colan SD, Miyamoto SD, Choudhry S, Baez-Hernandez N, Brickler MM, et al. Treatment strategies for cardiomyopathy in children: a scientific statement from the American heart association. Circulation. (2023) 148(2):174–95. doi: 10.1161/CIR.0000000000001151
2. Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American heart association. Circulation. (2019) 140:e9–68. doi: 10.1161/CIR.0000000000000682
3. Yuan W, Jia Z, Li J, Liu L, Tian J, Huang X, et al. The clinical profile, genetic basis and survival of childhood cardiomyopathy: a single-center retrospective study. Eur J Pediatr. (2024) 183:1389–401. doi: 10.1007/s00431-023-05358-6
4. Rath A, Weintraub R. Overview of cardiomyopathies in childhood. Front Pediatr. (2021) 9:708732. doi: 10.3389/FPED.2021.708732
5. Ciarambino T, Menna G, Sansone G, Giordano M. Cardiomyopathies: an overview. Int J Mol Sci. (2021) 22:7722. doi: 10.3390/ijms22147722
6. Sarnecki J, Paszkowska A, Petryka-Mazurkiewicz J, Kubik A, Feber J, Jurkiewicz E, et al. Left and right ventricular morphology, function and myocardial deformation in children with left ventricular non-compaction cardiomyopathy: a case-control cardiovascular magnetic resonance study. J Clin Med. (2022) 11:1104. doi: 10.3390/jcm11041104
7. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. (2003) 348:1639–46. doi: 10.1056/NEJMoa021737
8. Elmasry OA, Kamel TB, El-Feki NF. Pediatric cardiomyopathies over the last decade: a retrospective observational epidemiology study in a tertiary institute, Egypt. J Egypt Public Health Assoc. (2011) 86:63–7. doi: 10.1097/01.EPX.0000399140.68151.6a
9. Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, Osman E, et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol. (2007) 49(25):2419–26. doi: 10.1016/j.jacc.2007.02.061
10. Pinamonti B, Di Lenarda A, Nucifora G, Gregori D, Perkan A, Sinagra G. Incremental prognostic value of restrictive filling pattern in hypertrophic cardiomyopathy: a Doppler echocardiography study. Eur J Echocardiogr. (2008) 9(4):466–71. doi: 10.1016/j.euje.2007.06.008
11. Biagini E, Spirito P, Rocchi G, Ferlito M, Rosmini S, Lai F, et al. Prognostic implication of the Doppler restrictive filling pattern in hypertrophic cardiomyopathy. Am J Cardiol. (2009) 104(12):1727–31. doi: 10.1016/j.amjcard.2009.07.057
12. De Bortoli M, Vio R, Basso C, Calore M, Minervini G, Angelini A, et al. Novel missense variant in MYL2 gene associated with hypertrophic cardiomyopathy showing high incidence of restrictive physiology. Circ Genom Precis Med. (2020) 13(2):e002824. doi: 10.1161/CIRCGEN.119.002824
13. Li S, Wu B, Yin G, Song L, Jiang Y, Huang J, et al. MRI characteristics, prevalence, and outcomes of hypertrophic cardiomyopathy with restrictive phenotype. Radiol Cardiothorac Imaging. (2020) 2(4):e190158. doi: 10.1148/ryct.2020190158
14. Priotto F, Albani S, Mabritto B, Luceri S, Pizzuti A, Bongioanni S, et al. C48 prognostic impact of echocardiographic restrictive filling pattern in patients affected by hypertrophic cardiomyopathy and preserved systolic ejection fraction. Eur Heart J Suppl. (2022) 24(Supplement_C):suac011-047. doi: 10.1093/eurheartj/suac011.047
15. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic evaluation of cardiomyopathy-a heart failure society of America practice guideline. J Card Fail. (2018) 24:281–302. doi: 10.1016/j.cardfail.2018.03.004
16. Ditaranto R, Caponetti AG, Ferrara V, Parisi V, Minnucci M, Chiti C, et al. Pediatric restrictive cardiomyopathies. Front Pediatr. (2022) 9:745365. doi: 10.3389/fped.2021.745365
17. Tsuda E, Ito Y, Kato Y, Sakaguchi H, Ohuchi H, Kurosaki K. Thirty-year outcome in children with hypertrophic cardiomyopathy based on the type. J Cardiol. (2022) 80:557–62. doi: 10.1016/j.jjcc.2022.07.016
18. Chan W, Yang S, Wang J, Tong S, Lin M, Lu P, et al. Clinical characteristics and survival of children with hypertrophic cardiomyopathy in China: a multicentre retrospective cohort study. EClinicalMedicine. (2022) 49:101466. doi: 10.1016/j.eclinm.2022.101466
19. Hong JA, Kim MS, Cho MS, Choi HI, Kang DH, Lee SE, et al. Clinical features of idiopathic restrictive cardiomyopathy: a retrospective multicenter cohort study over 2 decades. Medicine. (2017) 96:e7886. doi: 10.1097/MD.0000000000007886
20. Vio R, Angelini A, Basso C, Cipriani A, Zorzi A, Melacini P, et al. Hypertrophic cardiomyopathy and primary restrictive cardiomyopathy: similarities, differences and phenocopies. J Clin Med. (2021) 10(9):1954. doi: 10.3390/JCM10091954
21. Ware SM, Wilkinson JD, Tariq M, Schubert JA, Lipshultz SE. Genetic causes of cardiomyopathy in children: first results from the pediatric cardiomyopathy genes study. J Am Heart Assoc. (2021) 10(9):e017731. doi: 10.1161/JAHA.120.017731
22. Girolami F, Gozzini A, Passantino S, Calabri GB, Spaziani G, Porcedda G, et al. 263 Genetic causes of cardiomyopathies in children. Eur Heart J Suppl. (2022) 24(Supplement_K):suac121-409. doi: 10.1093/eurheartjsupp/suac121.409
23. Pidaparti M, Geddes GC, Durbin MD. Clinical genetic and genomic testing in congenital heart disease and cardiomyopathy. J Clin Med. (2024) 3(9):2544. doi: 10.3390/jcm13092544
24. Section of Precision Cardiovascular Medicine of Chinese Society of Cardiology; Precision Cardiovascular Medicine Branch of China International Exchange; Promotive Association for Medical; Health Care Editorial Board of Chinese Journal of Cardiology. Guideline for the genetic diagnosis of monogenic cardiovascular diseases. Zhonghua Xin Xue Guan Bing Za Zhi. (2019) 47(3):175–96. doi: 10.3760/cma.j.issn.0253-3758.2019.03.003
25. Nasermoaddeli A, Miura K, Matsumori A, Soyama Y, Morikawa Y, Kitabatake A, et al. Prognosis and prognostic factors in patients with hypertrophic cardiomyopathy in Japan: results from a nationwide study. Heart. (2007) 93:711–5. doi: 10.1136/hrt.2006.095232
26. Xia K, Sun D, Wang R, Zhang Y. Factors associated with the risk of cardiac death in children with hypertrophic cardiomyopathy: a systematic review and meta-analysis. Heart Lung. (2022) 52:26–36. doi: 10.1016/j.hrtlng.2021.11.006
27. Norrish G, Qu C, Field E, Cervi E, Khraiche D, Klaassen S, et al. External validation of the HCM risk-kids model for predicting sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur J Prev Cardiol. (2022) 29:678–86. doi: 10.1093/eurjpc/zwab181
28. Anderson HN, Cetta F, Driscoll DJ, Olson TM, Ackerman MJ, Johnson JN. Idiopathic restrictive cardiomyopathy in children and young adults. Am J Cardiol. (2018) 121:1266–70. doi: 10.1016/j.amjcard.2018.01.045
29. Ammash NM, Seward JB, Bailey KR, Edwards WD, Tajik AJ. Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation. (2000) 101:2490–6. doi: 10.1161/01.cir.101.21.2490
30. Sherazi S, McNitt S, Choudhary N, Shah AH, Aktas MK, Asgher A, et al. Predictors of mortality in patients hospitalized for congestive heart failure with left ventricular ejection fraction ≥ 40. Cardiol J. (2015) 22:382–90. doi: 10.5603/CJ.a2015.0003
31. Morell E, Gaies M, Fineman JR, Charpie J, Rao R, Sasaki J, et al. Mortality from pulmonary hypertension in the pediatric cardiac ICU. Am J Respir Crit Care Med. (2021) 204(4):454–61. doi: 10.1164/RCCM.202011-4183OC
32. Olgar S, Ertugrul T, Nisli K, Omeroglu RE, Dindar A, Aydogan U. Fish oil supplementation improves left ventricular function in children with idiopathic dilated cardiomyopathy. Congest Heart Fail. (2007) 13(6):308–12. doi: 10.1111/j.1527-5299.2007.07135.x
Keywords: hypertrophic cardiomyopathy, restrictive cardiomyopathy, rapidly progressive hypertrophic cardiomyopathy, mortality risk factors, genetic testing
Citation: Nawaz A, Sheng Z, Akram MJ, Li J, Liu L, Yuan Y and Tian J (2025) Clinical characteristics and mortality risk factors in pediatric hypertrophic, restrictive, and rapidly progressive hypertrophic cardiomyopathy: a retrospective cohort study with follow-up. Front. Cardiovasc. Med. 12:1541651. doi: 10.3389/fcvm.2025.1541651
Received: 8 December 2024; Accepted: 19 March 2025;
Published: 31 March 2025.
Edited by:
Xupei Huang, Florida Atlantic University, United StatesReviewed by:
Howard Prentice, Florida Atlantic University, United StatesGabrielle Norrish, Great Ormond Street Hospital Children's Charity, United Kingdom
Copyright: © 2025 Nawaz, Sheng, Akram, Li, Liu, Yuan and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuxing Yuan, eXVhbnl1eGluZ0BzdHUuY3FtdS5lZHUuY24=; Jie Tian, amlldGlhbkBjcW11LmVkdS5jbg==
†These authors share first authorship
‡ORCID:
Yuxing Yuan
orcid.org/0000-0003-3867-9779
Jie Tian
orcid.org/0000-0002-9006-6710