
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 04 April 2025
Sec. Cardiovascular Metabolism
Volume 12 - 2025 | https://doi.org/10.3389/fcvm.2025.1516279
 Chunling Wang1
Chunling Wang1 Jiashu Yang1Jianfang Yuan1Xuyong Wang1
Jiashu Yang1Jianfang Yuan1Xuyong Wang1 Qianrong Li1
Qianrong Li1 Chunzhen Ren1Xiaodong Zhi1,2
Chunzhen Ren1Xiaodong Zhi1,2 Xinfang Lv2
Xinfang Lv2 Kai Liu3Xinke Zhao1
Kai Liu3Xinke Zhao1 Yingdong Li1*
Yingdong Li1*
Circadian rhythm, or the biological clock, is an intrinsic timing system present in organisms that operates on a cycle of approximately 24 h. Nearly every cell in the human body adheres to a specific circadian rhythm, governing various biological processes essential for overall health. REV-ERB, a key circadian clock-regulating gene, plays a crucial role in maintaining the precision of these rhythms. This gene influences many downstream targets associated with diverse pathophysiological processes, including metabolism, autophagy, immunity, inflammation, and aging across multiple organs. REV-ERB specifically impacts cardiac systolic function by regulating myocardial energy metabolism. In contemporary society, health and well-being are increasingly challenged by disruptions to the biological clock, such as night shifts, late-night activities, and jet lag. These disruptions often lead to circadian rhythm disorders, which are now being increasingly linked to heart diseases. This review explored the potential role of REV-ERB in the cardiovascular system. Beyond its role in circadian rhythm regulation, REV-ERB could significantly influence physiological and pathological processes related to cardiovascular health, including atherosclerosis, myocardial ischemia/reperfusion injury, and heart failure. Mechanistically, REV-ERB could regulate glucose and lipid metabolism, inflammation, autophagy, ferroptosis, and mitochondrial function. The review highlighted the protective roles and underlying mechanisms of REV-ERB in cardiovascular diseases, suggesting that multidisciplinary research may provide a basis for breakthroughs in REV-ERB-targeted therapies for cardiovascular disorders.
Circadian rhythms are critical in regulating various physiological cardiovascular functions, e.g., heart rate and blood pressure, demonstrating rhythmic fluctuations. For instance, blood pressure typically peaks in the morning and declines at night (1, 2) Moreover, certain cardiovascular conditions, such as myocardial infarction (MI), arrhythmias, stroke, heart failure (HF), and sudden cardiac death, exhibit a close association with circadian rhythms, with a higher incidence of these events occurring in the early morning hours (3).
The rapid industrialization of society over the past century has significantly altered the human external environment. The clear distinction between day and night has been disrupted by artificial lighting and frequent travel across time zones. Shift work, in particular, has been linked to an increased risk of both non-cardiac and cardiovascular diseases (CVDs), including acute myocardial infarction (AMI). Among patients with acute coronary syndrome, shift work has been associated with a 15% higher risk of major cardiovascular events and worsened long-term cardiac outcomes (4, 5).
The biological clock is composed of a central clock, located in the suprachiasmatic nucleus of the hypothalamus, and peripheral clocks that function in nearly all tissues (6). The central clock receives optical signals from the retina, which synchronizes peripheral clocks through neurohumoral signals (7). In addition to central clock inputs, peripheral clocks respond to tissue-specific cues, such as food intake and physical activity (8–10).
The nuclear receptor subfamily 1, group D, member 1 (NR1D1, also known as REV-ERBα) was first identified in 1989 (11). In 1994, researchers discovered a related orphan receptor with high homology to REV-ERBα, named NR1D2 or REV-ERBβ REV-ERBα/β proteins (12), essential components of the circadian clock, are widely expressed across tissues and have become key therapeutic targets for heart diseases (13). REV-ERBs regulate glucose, lipids, energy metabolism, adipogenesis, and inflammation (Figure 1).
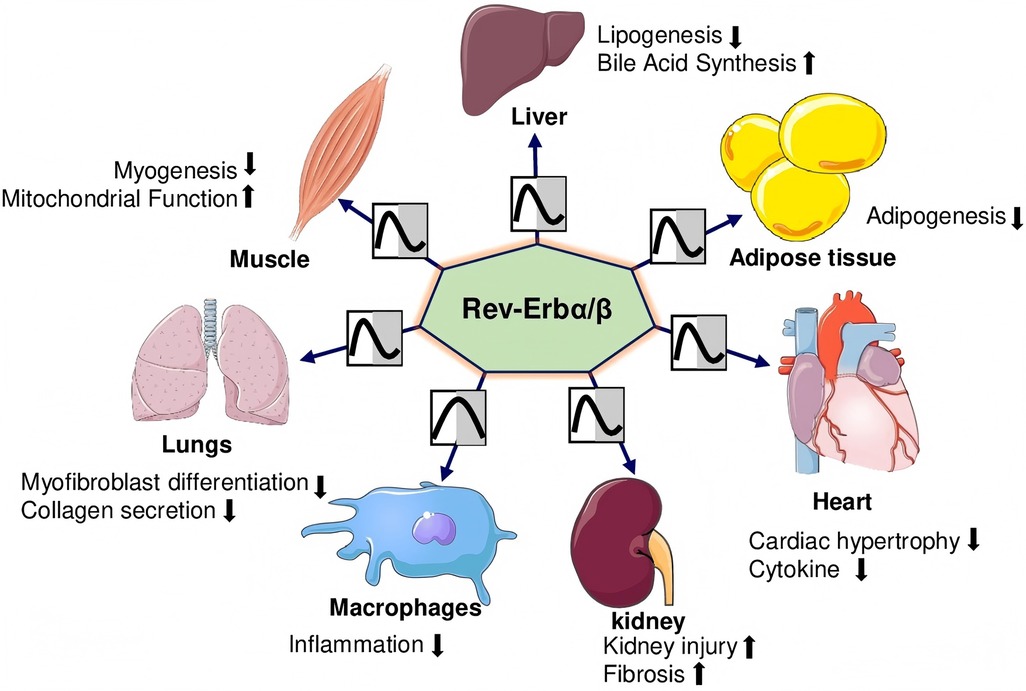
Figure 1. REV-ERBα/βs regulate the organs’ physiological functions. REV-ERBs suppress adipogenesis and lipogenesis while promoting bile acid synthesis in adipose tissue and the liver. In the heart, they mitigate cardiac hypertrophy and reduce inflammatory cytokine levels. In the lungs, they inhibit myofibroblast differentiation and collagen production. In skeletal muscles, REV-ERBs decrease myogenesis while enhancing mitochondrial function. However, in the kidneys, they contribute to increased injury and fibrosis.
This review discusses the role of REV-ERBα/β in circadian rhythm regulation and its association with processes, such as autophagy, inflammation, immunity, metabolism, fibrosis, and ferroptosis. It also examined the links between circadian rhythms and CVDs, emphasizing potential therapeutic strategies. Ligands targeting REV-ERBα/β or retinoic acid-related orphan receptor (RORα/γ), as well as lifestyle modifications, are noticeable for the prevention and treatment of CVDs.
In 1729, De Maran, a French astronomer, observed human physiological rhythms persisted without external cues, such as light. However, the concept of an intrinsic biological clock remained unaccepted until the 20th century (14). Circadian rhythm refers to behavioral, physiological, and metabolic cycles with a 24-h periodicity, evolved to synchronize biological functions with Earth's rotation. Derived from Latin, meaning “about a day”, this internal timing system ensures optimal adaptation (15). In 2017, Jeffrey C. Hall, Michael Rosbash, and Michael W. Young received the Nobel Prize in Physiology or Medicine for elucidating the molecular mechanisms of the biological clock. Since then, research has underscored its crucial role in human physiology and disease.
The biological clock is a conserved, endogenous timing system across animals, plants, fungi, and bacteria, operating on a 24-h cycle. It regulates physiological processes through rhythmic patterns, which are influenced by genetic, endocrine, behavioral factors, and external cues like light and temperature fluctuations (16). Despite external influences, the circadian system remains self-sustaining (17). Nearly all mammalian cells contain circadian rhythm molecules, with regulation centralized in the hypothalamic suprachiasmatic nucleus (SCN). This pacemaker, composed of ∼15,000 neurons, processes light signals received via the retinal hypothalamic tract, synchronizing peripheral oscillators through hormonal, autonomic, and temperature-mediated pathways (18–20). Strong intercellular coupling within the SCN maintains functional stability and coherence (18).
While the SCN orchestrates the circadian system, peripheral tissues can maintain independent rhythms influenced by cues like feeding schedules (18). Light exposure is essential in regulating circadian rhythms via SCN-mediated synchronization of peripheral clocks, including REV-ERB pathways. Disruptions in light-dark cycles, such as shift work, desynchronize these clocks, contributing to the progress of circadian rhythm disorders and CVDs. REV-ERB regulates clock genes and metabolic pathways, with light exposure modulating its activity through the SCN. Chronodisruption exacerbates conditions, including atherosclerosis, myocardial fibrosis, and heart failure (HF) (21–24). Harmonization of central and peripheral clocks supports stable physiological and behavioral responses (18).
Given the interaction among light exposure, circadian rhythms, and cardiovascular outcomes, targeting REV-ERB may serve as a therapeutic strategy to mitigate the adverse effects of chronodisruption. The pharmacological activation of REV-ERB has exhibited promise in reducing pathological gene expression and improving myocardial infarction and HF preclinical models, further emphasizing its relevance in light-exposure-related circadian dysfunction. Coordinating central and peripheral systems of circadian rhythm is necessary for optimal physiological functioning and maintaining physical and mental health.
The composition and regulation of circadian rhythms at the molecular level are as follows. The molecular oscillator of the clock system comprises two interlocking negative feedback loops of gene expression. This standard oscillator refers to the transcriptional translation feedback loop (TTFL). Two transcription factors named circadian locomotor output cycles kaput (CLOCK) and brain and muscle ARNT-like 1 (BMAL1) bind to the E-box DNA motif of the circadian clock-controlled genes (CCGs) and then recruit the co-activating proteins CBP/p300 (one group of histone acetyltransferases) along with Thyroid Hormone Receptor-associated protein 3 (THRAP3), steroid receptor coactivator 2 (SRC-2), and some other peptides, which then activate the period (PER1, PER2 and PER3) and cryptochrome (CRY1 and CRY2) genes in the primary feedback loop along with REV-ERBα and REV-ERBβ in the secondary feedback loop (25). The expression products of these genes include PER1, PER2, CRY1, CRY2, and several other peptides, such as casein kinases 1 ε (CK1 ε), which together form a repressor complex. After reaching the threshold activity level, this repressor complex weakens the transcriptional activity of CLOCK and BMAL1. Therefore, the PER and CRY repressor complex levels are reduced, lifting the inhibition of CLOCK and BMAL1 activity. This allows a new 24-h cycle of PER and CRY transcription and translation to begin, maintaining the circadian rhythm. In the secondary feedback loop, CLOCK–BMAL1 heterodimers bind to E-Box promoters and enhancers of REV-ERBα and REV-ERBβ, regulating their temporal expression as negative nuclear orphan receptors. The REV-ERBβ repressor protein competes with the ROR-Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1 α) activator/coactivator to bind to the ROR/REV-ERB-response elements (RORE) in BMAL1 and CLOCK. This leads to the recruitment of the nuclear receptor co-repressor 1 (NCoR1)-histone deacetylase 3 (HDAC3) co-repressor complex, inhibiting the transcription of Bmal1 and Clock genes. The transcription and translation feedback loop based on CLOCK-BMAL1 and ROR/RORE interaction operates in opposite phases aligned horizontally, i.e., at a time difference of 12 h. In addition, the phase difference is regulated by clock output regulators, such as the PAR-bZip (rich in proline and acidic amino groups) transcription factor and E4BP4/NFIL3 (E4BP4, also referred to as interleukin-3 nuclear gene NFIL3), the temporal expressions of which are regulated via primary and secondary feedback loops, respectively (26).
Recent studies (27–29) have highlighted the involvement of non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), in regulating circadian rhythms at the molecular level. Several miRNAs, such as miR-219 and miR-132, have been found to modulate the expression of core clock components, including CLOCK, BMAL1, PER, and CRY, thereby influencing circadian oscillations. Additionally, lncRNAs, such as Neat1 and locked nucleic acid, have been implicated in stabilizing rhythmic gene expression by interacting with chromatin modifiers and transcriptional regulators. These non-coding RNAs contribute to tuning the transcriptional-translational feedback loop well, reinforcing the robustness of circadian rhythms and linking them to metabolic and physiological processes. Their dysregulation has been associated with circadian misalignment and related disorders, including CVDs and metabolic syndromes. Integrating non-coding RNA regulation into the classical TTFL model expands the understanding of circadian control mechanisms and their broader implications for human health.
Several cardiovascular parameters, such as heart rate, heart rate variability, electrocardiogram (ECG) waveform, and blood pressure, fluctuate pronouncedly every day (30). Approximately 13% of genes and 8% of proteins involved in cardiac growth, metabolism, and molecular signaling follow rhythmic patterns. These fluctuations align with 24-h variations in cardiovascular neurohumoral factors, such as autonomic, sympathetic, and parasympathetic tone, renin-angiotensin-aldosterone system activity, and cortisol levels (31). Clock-regulated genes drive functional oscillations in various cardiac cell types, including endothelial cells, vascular smooth muscle cells, fibroblasts, cardiomyocytes, and cardiac progenitor-like cells (3).
Disruptions to normal circadian rhythms can significantly affect cardiovascular health due to inadequate sleep, shift work, or jet lag. Adverse cardiovascular events, such as myocardial infarction and stroke, occur more frequently in the early morning (6 a.m. to 2 p.m.), with elevated cardiac injury markers like creatine kinase detected in affected patients (32). Experimental studies show that circadian clock disruption in animal models leads to severe CVDs (23). For example, deletion of the circadian clock gene Bmal1 in mice alters cardiac myosin composition, impairs sarcomere structure, and accelerates age-related dilated cardiomyopathy (24).
Cardiomyocyte-specific Bmal1 knockout (CBK) mice exhibit reduced stroke volume and ejection fraction, progressing to early heart failure and premature death. Deleting Bmal1 from myocardial cells disrupts the circadian expression of Na+ and K+ channels, reducing heart rate and increasing arrhythmia risk, which may lead to sudden cardiac death (33, 34). Despite increasing knowledge of the molecular clock's role in cardiovascular physiology and disease, its potential for improving treatment for CVD remains largely untapped.
The REV-ERB gene is located on chromosome 17, and expresses in various cells throughout the human body, including those in the liver, heart, lung, adipose tissue, skeletal muscle, and brain. This gene is a critical regulator of metabolism, mitochondrial biogenesis, inflammatory responses, and fibrosis (35). The REV-ERBα protein, comprising 614 amino acids, consists of four distinct domains: A/B, C, D, and E, each with specialized functions. The A/B domain is a highly active nuclear receptor region capable of ligand-independent cis-activation, facilitating nucleic receptor interactions with other family members. The C domain, a conserved region, is responsible for DNA binding activity and determines the selection of chaperone nuclear receptors. The D domain, a flexible hinge region, contains nuclear localization signals. The E domain, characterized by proline- and acidic amino acid-rich regions (LBD), binds ligands, enabling dimerization and activation, thereby functioning as a transcription factor to regulate downstream target gene transcription. The LBD's primary structure includes ligand-binding pockets, especially the activation function 2 (AF-2) region, which features helices (H12), a co-regulatory factor-binding surface, and a dimerization surface. Notably, REV-ERBα lacks the AF-2 region, precluding transcriptional activation. Instead, REV-ERBα directly binds target gene promoters and enhancers, recruiting the nuclear receptor co-repressor (NCoR) and histone deacetylase 3 (HDAC3) complex to the REV-ERBα response element, thereby suppressing downstream gene transcription. Upon ligand binding, REV-ERBα undergoes a conformational change that enhances the recruitment of transcriptional co-regulatory proteins to receptor-specific gene promoter complexes, further inhibiting transcription (13). REV-ERBα also interacts with the retinoic acid-related orphan receptor α (RORα) to competitively regulate circadian rhythm oscillations (36). REV-ERBα inhibits the transcription and translation of the circadian clock components CLOCK and BMAL1 (also referred to as ARNTL), which form heterodimers that bind E-box elements to drive the transcription of core clock molecules and downstream targets (37). Conversely, RORα competes with REV-ERBα for ROR response element (RORE) binding, activating BMAL1 and CLOCK transcription (37). This interplay establishes a self-sustaining feedback loop involving REV-ERBα, ensuring robust and precise circadian clock regulation. Circadian rhythms orchestrate various physiological processes in a time-dependent manner to maintain homeostasis despite environmental factors, such as circadian circulation, food intake, and physical activity. Disruptions in circadian rhythm caused by gene mutations, shift work, exposure to artificial light, irregular eating patterns, or abnormal sleep cycles adversely affect human health (Figure 2).
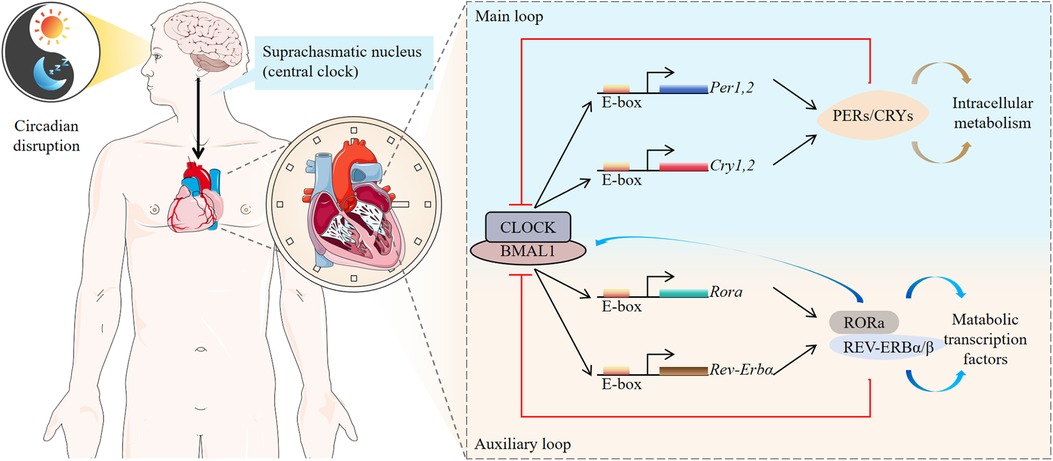
Figure 2. Molecular clocks in humans. The human molecular clock is driven by external cues, such as light and diet, which synchronize the central oscillator, or “master clock,” located in the suprachiasmatic nucleus (SCN). These cues reset peripheral oscillators found in nearly every cell of the body. The molecular clock mechanism consists of interconnected transcriptional feedback loops. Circadian clock-controlled genes (CCGs) are expressed under the regulation of these molecular clocks, driving physiological processes such as cardiac function throughout the day and night. REV-ERB and retinoid-related orphan receptor (ROR) stabilize and enhance the feedback loop mediated by BMAL1.
Autophagy, a highly conserved intracellular degradation system, is essential for maintaining cellular homeostasis under stress conditions. Eukaryotic autophagy involves the use of lysosomes for the degradation of cytoplasmic proteins and damaged organelles of cells under the regulation of the autophagy-related gene (Atg) (38–40). NR1D1 is involved in the autophagy process occurring within various organelles, including mitochondria and lysosomes. Autophagy failure reportedly promoted cell degeneration, age-related senescence, tumor formation, and infection in mice and has also been speculated to play a key role in human diseases (41).
REV-ERBα regulates the oxidative function of skeletal muscle by regulating the genes involved in the different steps of mitophagy (42). SR9009 interacts with the core autophagy gene Atg5, inhibits autophagy activity, and plays an anti-tumor role in SCLC (small cell lung cancer) (43). In hepatic stellate cells (HSCs) (44), SR9009 regulates the signaling pathways associated with growth and survival, such as protein kinase B (AKT)/mammalian target of rapamycin (mTOR)/P70S6K (p70 ribosomal protein S6 kinase) pathway, also inhibits AV (autophagosome) biogenesis. The expression levels of REV-ERBα and REV-ERBβ in the granulosa cells of patients with polycystic ovary syndrome (PCOS) are lower than those in healthy people. According to in vitro studies, the overexpression of REV-ERBα and REV-ERBβ promotes the expression of mitochondrial biosynthetic genes and inhibits autophagy in granulosa cells. REV-ERBα and REV-ERBβ inhibit granulosa cell apoptosis and promote proliferation. In a mouse PCOS model (45), the REV-ERB agonist SR9009 reportedly improved abnormal follicular development by promoting mitochondrial biosynthesis and inhibiting autophagy. A study (46) reported that the negative regulation of REV-ERBα provided partial protection against glucose toxicity and cytokine-induced β-cell apoptosis. Sulli et al. (47) reported that the agonists of REV-ERBs (SR9009/SR9011) specifically targeted cancer cell de novo fatty acid synthesis and early autophagy-related pathways in A172 Glioblastoma (GBM) cells by sharply reducing the expression levels of the key rate-limiting enzymes involved in de novo lipogenesis. The agonists of REV-ERBs (SR9009/SR9011) exert a specific damage effect on cancer cells and oncogene-induced senescent cells but not on the viability of normal cells or tissues. According to a study (48), the agonist of REV-ERBs named GSK4112 could stimulate the autophagy flow of human macrophages and the expressions of key genes involved in lysosomal biogenesis through the activation of NR1D1 via the transcription factor EB (TFEB)-related pathway. A recent study (49) demonstrated that the transcription factors TFEB and Transcription factor binding to IGHM enhancer (TFE3), which are the major regulators of autophagy and lysosomal biogenesis, do not regulate NR1D1 expression by forming a complex with BMAL1-CLOCK and rather than directly binding to the NR1D1 promoter to drive the expression of REV-ERBα. Overexpression of TFEB and TFE3 in cells and knockdown of endogenous NR1D1 (50) leads to the expression of autophagy genes and enhancement of the autophagy flux. Therefore, the opposite, although interrelated roles of TFEB and TFE3 along with NR1D1, determine the time of autophagy activation.
Circadian changes during inflammation and immune function have been observed in different physiological and pathological processes of humans and animals. However, the molecular mechanisms underlying these changes and the mediating cell types remain unknown. The biological clock controls several inflammatory processes, and the disturbed biological clock may lead to or aggravate inflammation. REV-ERB is vital in the inflammation process mediated by various cell types. REV-ERB regulates the inflammatory response by reducing the secretion of inflammatory cytokines, regulating gene transcription and the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome pathway, and inhibiting macrophage polarization.
The study revealed that aged mice exhibit time-dependent differences in controlling Streptococcus pneumoniae infection. Transcriptomic analysis of pulmonary circadian rhythms demonstrated significant alterations in the rhythmic expression of the core clock gene REV-ERBα and the lung's apelin/apelin receptor pathway. Further mechanistic investigations indicated that REV-ERBα mediates the host defense function of alveolar macrophages by regulating the apelinergic signaling axis. Pharmacological inhibition of REV-ERBα enhanced the resistance of aged mice to pneumococcal infection (51). In a study (52), the expression patterns of the biological clock-related genes in the thyroid of patients with autoimmune thyroiditis (AIT) were altered. Animal experiments have demonstrated that chronic circadian rhythm disorders cause significant oscillations in the biological clock-related genes, such as Bmal1, Clock, Per2, Cry1, Ror, and REV-ERB, in addition to causing increases in the secretion of tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and anti-thyroglobulin antibodies, thereby aggravating the inflammatory response of AIT. GSK4112 or SR9011 (NR1D1 agonist) attenuates microglia-mediated neuroinflammation by blocking the nuclear translocation of the NF-κB subunit p65 and inhibiting the expressions and secretion of pro-inflammatory cytokines [such as interleukin 6 (IL-6) and TNFα] in a dose-dependent manner (53). The pretreatment of primary chondrocytes with SR9009 (REV-ERB agonist) reportedly blocked the secretion of inflammatory molecules (matrix metalloproteinase, MMP3, MMP9, and MMP13) and cytokines (interleukin-1 β and tumor necrosis factor) in the cells induced by lipopolysaccharide. The repeated intra-articular treatment of SR9009 could prevent sodium iodoacetate-induced mechanical hypersensitivity and partially reduce knee joint injury in mice (54). A study of the peripheral blood mononuclear cells (PBMC) of young people after mild-to-moderate COVID-19 infection revealed that the expression of REV-ERBα in PBMC after COVID-19 infection was decreased upon LPS stimulation, indicating that REV-ERBα plays a role in both circadian rhythm control and inflammatory pathway (55). Studies have demonstrated that REV-ERBs inactivate the transcription of matrix metalloproteinase 9 (MMP9) and chemokine receptor (CX3CR1) in macrophages by inhibiting the transcription of enhancer-derived RNA (eRNA) (56). TH17 is a pro-inflammatory immune cell that prevents bacterial and fungal infections on the mucosal surface. REV-ERB α links the cellular development of TH17 to the circadian clock network, and its deletion increases the expressions of TH17-mediated pro-inflammatory cytokines and enhances the inflammatory response (57, 58). Dysregulation of the NLRP3 signaling cascade is associated with several inflammatory and metabolic diseases, including rheumatoid arthritis, gout, atherosclerosis, or type-2 diabetes. The biological clock controls several inflammatory processes, and the interrupted clock may lead to or aggravate inflammation. The circadian clock controls the expression and activation of NLRP3, thereby controlling the secretion of interleukin IL-1 β and IL-18 in different tissues and immune cells, particularly in macrophages. The circadian rhythm oscillation of the NLRP3 signal reportedly disappeared in the circadian clock disorder model, leading to the generation of peritonitis, hepatitis, or colitis (59). Morioka et al. demonstrated that REV-ERBα and REV-ERBβ mRNAs were expressed in cultured rat spinal cord microglia. Treatment of a culture of rat spinal microglia with SR9009 significantly blocked the lipopolysaccharide-induced increases in the expression levels of IL-1 β and IL-6 mRNA. Treatment of cultured rat spinal microglia with SR9009 significantly blocked lipopolysaccharide-induced increases in IL-1 β and IL-6 mRNA expression levels. Activation of REV-ERB exhibited analgesic effects in the spinal dorsal horn by negatively regulating spinal microglial activity (60). The findings demonstrate that ischemic stroke during the sleep phase (ZT06) significantly reduces the expression level of Rev-Erbα and exacerbates neuroinflammatory responses as well as stroke severity. Pharmacological intervention experiments reveal that the Rev-Erbα agonist SR9009, administered at ZT06, significantly alleviates neuroinflammation, reduces cerebral infarct volume, and downregulates the expression of the NLRP3 inflammasome in monocytes and neutrophils, as well as in brain tissue. Furthermore, SR9009 treatment markedly downregulates the expression of the pro-inflammatory cytokine TNFα while upregulating the expression of the anti-inflammatory cytokine IL-10. However, slight changes were found under the same intervention during the awake phase (ZT18) (61).
Obesity and circadian rhythm disruption represent global health challenges, significantly elevating the risk of metabolic disorders. Adipose tissue and circadian rhythms are integral to regulating energy homeostasis, and their dysregulation is intricately associated with the pathogenesis of obesity. Circadian rhythm is closely related to metabolic regulation. The experimental study of the circadian rhythm gene expression profile revealed that approximately 3%–10% of the transcriptome is regulated by circadian rhythm, with the highest involvement in regulating basic metabolic processes (62). In the liver, the circadian rhythm regulates the basic metabolic processes, such as glycolysis, fatty acid metabolism, cholesterol biosynthesis, xenobiotics, and intermediate metabolism, which is realized through control of the rate-limiting steps of these processes. Studies in mice have demonstrated that genetic damage to the core clock gene leads to abnormal metabolic phenotypes, such as obesity, dyslipidemia, diabetes, hypertension, and HF (13). Circadian rhythm disorders due to shift work or lack of sleep are closely related to metabolic syndrome (63). Numerous studies have confirmed that ROR and REV-ERB coordinate the core circadian oscillators and clock-controlled genes to regulate metabolic pathways.
REV-ERB is expressed highly in the adipose tissue and exhibits a circadian rhythm, which is necessary for adipogenesis. A study (64) reported that REV-ERB inhibits adipogenesis by inhibiting the expression of peroxisome proliferator-activated receptor γ2, (PPARγ2), which is the primary transcriptional regulator of adipogenesis, and the dynamic change in the REV-ERB protein levels involved an initial increase followed by a decrease, leading to adipocyte differentiation (64). Studies have demonstrated that the double knockout of REV-ERBα/β caused a significant increase in the levels of TAG in the liver along with hepatic steatosis (65). An imbalance in the cholesterol metabolism plays a critical role in atherosclerosis. Cholesterol is a major part of the cell membrane and a significant metabolic precursor of the biosynthetic pathway, including the synthesis pathway for steroid hormones, vitamin D, and bile acids. The rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) and sterol regulatory element binding protein 2 (SREBP2) are the key regulators of cholesterol homeostasis (66). REV-ERBα participates in the circadian rhythm of sterol regulatory element binding protein (SREBP) activity regulation, and further the target genes related to cholesterol and lipid metabolism. REV-ERBα also participates in rhythmic bile acid metabolism regulation via regulating the expression of cholesterol 7 αhydroxylase (CYP7A1) (67). Studies have demonstrated that treatment with synthetic REV-ERB agonists decreases the plasma levels of cholesterol and the liver levels of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl coenzyme a reductase in the biosynthesis reaction of cholesterol in mice. The REV-ERB agonist SR9009 reportedly reduced the plasma levels of cholesterol in wild-type mice and low-density lipoprotein receptor (LDLR)-deficient mice, while reducing the expressions of the related genes in the cholesterol biosynthesis pathway (66). REV-ERB ligands significantly regulate circadian behaviors and the rhythmic expression of core clock genes in the hypothalamus of mice. They also modulate the circadian expression patterns of metabolic genes in the liver, skeletal muscle, and adipose tissue, thereby promoting increased energy expenditure. Intervention with REV-ERB agonists in diet-induced obese mouse models effectively reduces fat mass and markedly improves dyslipidemia and hyperglycemia, thereby alleviating obesity (68).
REV-ERB agonists could effectively inhibit the expressions of fatty acid synthase (FASN), stearoyl-CoA desaturase 1 (SCD1), and PPAR-γ coactivator 1 β (PGC-1 β) in diet-induced obese mice, in addition to decreasing fat mass, improving dyslipidemia and hyperglycemia, and reducing obesity. Following eight weeks of continuous light exposure, mice exhibited increased body weight, insulin resistance, elevated white fat mass, and altered expression of circadian clock genes. Chronic administration of SR9009 restored the expression of REV-ERBα and REV-ERBβ in mice subjected to either a 24-h light cycle or constant light while suppressing the expression of Bmal1 in both white adipose tissue (WAT) and brown adipose tissue (BAT). By inhibiting adipogenesis, SR9009 effectively reduced weight gain, improved insulin resistance, and decreased white fat mass in mice under these lighting conditions (69). REV-ERBα (−/−) mice exhibited obesity and mild hyperglycemia under a regular diet. Moreover, the REV-ERBα (−/−) mice with overexpressed lipoprotein lipase (Lp1) gene under a high-fat diet exhibited promoted utilization of muscle fatty acids and fat overload (70). Heme reportedly inhibits hepatic gluconeogenic gene expression and glucose output via REV-ERBα-mediated genes (71). According to a study (72), the nuclear receptor REV-ERBs in GABAergic (γ-aminobutyric acid production) neurons (SCNGABA neurons) in the suprachiasmatic nucleus (SCN) control the rhythmic expressions of genes involved in neurotransmission within the SCN, regulate insulin secretion, and inhibit the circadian rhythm of hepatic glucose production in mice, while exerting no effects on the circadian rhythms of eating or regular light and dark cycle movement behavior. A clinical observational study demonstrated (73) that the novel pan-PPAR agonist Chiglitazar significantly reduces low-density lipoprotein cholesterol (LDL-C), free fatty acids (FFA), 3:00 a.m. blood glucose, and fasting blood glucose levels. Notably, its glucose-lowering effect is independent of lipid metabolism regulation mechanisms and may be modulated by the REV-ERB nuclear receptor pathway. This finding provides new theoretical evidence for the glucose-lowering mechanism of Chiglitazar, suggesting its potential role in glycemic regulation via targeting molecular pathways associated with circadian rhythms.
REV-ERB is primarily expressed in the liver, heart, lungs, adipose tissue, skeletal muscle, and brain, serving as a key regulator of fibrosis (35). Transforming growth factor-β (TGF-β), the central mediator of tissue fibrosis plays a critical role in the fibrotic response to injury. Chronic inflammation is increasingly recognized as a major contributor to morbidity and mortality (74). TGF-β has been shown to induce the expression of Bmal1 and Clock while suppressing PER1/2, REV-ERBα, and RORα, thereby influencing circadian clock genes (75).
In a study by Cunningham et al. (76), REV-ERBα inhibited differentiation and collagen secretion in cultured embryonic lung fibroblasts and pulmonary myofibroblasts derived from patients with pulmonary fibrosis. Conversely, in REV-ERBα-deficient (REV-ERB −/−) mice, pulmonary myofibroblasts exhibited increased collagen-1 and α-smooth muscle actin (α-SMA) secretion. The same study observed that the mRNA and protein levels of BMAL1, PER2, CRY1, and REV-ERBα were reduced in the peripheral blood mononuclear cells (PBMCs), lung tissue, and sputum cells of smokers and patients with chronic obstructive pulmonary disease (COPD) compared to non-smokers (77). Studies have demonstrated that REV-ERBα agonists, such as SR9009 and GSK4112 inhibit fibroblast differentiation in human fetal lung fibroblasts (HFL-1) induced by TGF-β or cigarette smoke (CS) (78). These agonists also attenuate the inflammatory response and pulmonary fibrosis in airway epithelial cells and mouse lung fibroblasts exposed to lipopolysaccharide (LPS) or cigarette smoke (79).
Hepatic stellate cells (HSCs), mesenchymal cells located in the space of Disse between hepatocyte basements and sinusoidal endothelial cells, play a vital role in liver fibrosis (80). In their resting state, HSCs primarily store vitamin A and retinol. However, liver damage activates HSCs, transforming them into myofibroblasts with contractile, migratory, and fibrogenic properties. Chronic liver injury leads to repeated HSC activation, resulting in cirrhosis, hepatocellular carcinoma (HCC), and fibrous tissue accumulation that disrupts liver structure, increases portal vein pressure, and impairs liver function. Research by Li et al. (81) indicated that REV-ERBα protein expression was upregulated in activated HSCs and damaged liver tissue. The REV-ERB ligand SR6452 improved liver fibrosis and portal hypertension (PH) in rats by inhibiting cytoplasmic REV-ERBα expression, reducing α-SMA and TGF-β levels, and suppressing HSC activation. In a mouse model of non-alcoholic steatohepatitis (NASH), SR9009 was reported to reduce glucose levels, improve glucose tolerance, and inhibit the expression of fibrotic markers, such as collagen α1(III) (COLl3A1), α-SMA, STAT1, MMP13, TIMP1, and TGF-β. These effects reduced liver fibrosis and inflammation and improved liver function (82). In another study using a CCl4-induced liver fibrosis mouse model, the REV-ERB agonist SR9009 inhibited autophagosome biogenesis, fibrosis-related gene expression, and HSC proliferation through the regulation of P70S6K (44). These findings underscore the potential therapeutic value of REV-ERBα agonists in fibrosis and associated pathologies treatments.
Mitochondrial biosynthesis is a complex process involving the growth and division of mitochondria to meet increased energy demands (83). This process relies on coordinated signaling pathways to ensure the production of new mitochondria. Studies have established that REV-ERBs are essential in mitochondrial biogenesis (84). For instance, Woldt et al. (42). demonstrated that REV-ERBα knockout in skeletal muscle inactivated the Lkb1-Ampk-Sirt1-Ppargc-1α signaling pathway. This inactivation reduced mitochondrial content, impaired mitochondrial oxidative function, increased autophagy, and diminished exercise capacity collectively. In contrast, overexpression of REV-ERBα increased mitochondrial numbers and improved mitochondrial respiration. In patients with polycystic ovary syndrome (PCOS), the expression levels of REV-ERBα and REV-ERBβ in ovarian granulosa cells were significantly lower than in healthy controls (45). In vitro experiments with human ovarian granulosa cells (KGN) showed that overexpression of REV-ERBα and REV-ERBβ upregulated the mitochondrial biosynthesis genes, such as PGC-1α, NRF1, and TFAM, while reducing mitophagy. Furthermore, treatment with the REV-ERB agonist SR9009 promoted mitochondrial biosynthesis, alleviating follicular dysplasia. Mitochondrial dynamics involves the processes, such as fission, fusion, and subcellular translocation, which are crucial for maintaining mitochondrial DNA integrity and respiratory function (85). In many cell types, mitochondria exist as dynamic networks, with fission and fusion regulating their function and adaptation. In a mouse model of Parkinson's disease (PD) induced by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and in SH-SY5Y neuronal cells, sinapic acid was observed to exert protective effects against MPTP-induced PD. This protection involved increased expression of REV-ERBα protein and decreased levels of mitochondrial fission proteins, including dynein-related protein 1 (DRP1) and phosphorylated DRP1 at SER616 (86). These findings emphasize the essential roles of REV-ERBs in mitochondrial biosynthesis and dynamics, with significant implications for understanding and managing metabolic and neurodegenerative disorders.
Ferroptosis is an iron-dependent, non-apoptotic form of cell death, characterized by increased lipid peroxidation (87). Type 2 diabetes was induced by a high-fat diet (HFD) and an intraperitoneal injection of streptozotocin (STZ) in mice with cardiac-specific knockout of the REV-ERBα gene (88). Meanwhile, high glucose (HG) levels and high palmitic acid (PA) levels were administered to induce glycolipid toxicity in H9C2 cardiomyocytes in vitro. It was observed that after REV-ERBα was knocked out, the disorder of iron metabolism in the myocardium was aggravated, the expression of glutathione peroxidase 4 (GPX4) was decreased, the cardiac function significantly deteriorated in the diabetic mice, and the indicators of myocardial inflammation, myocardial fibrosis, and oxidative stress were significantly increased. Contrary to the view that REV-ERBα/β deficiency is associated with poor health conditions, such as cancer, metabolic disorders, and severe inflammation, a study (89) demonstrated that REV-ERB deficiency inhibits ferroptosis and improves folic acid-induced acute kidney injury. Another study (90) on renal injury induced by aristolochic acid I (AAI, a typical AA) reported that the kidney-specific knockout of REV-ERBα in mice decreased the sensitivity of these mice to AAI-induced ferroptosis and renal injury. Meanwhile, treatment with siRNA or SR8278 (a REV-ERBα antagonist) in vitro reduced the aristolochic lactam I (ALI)-induced ferroptosis in mouse renal tubular epithelial cells (mRTECs). In addition, the REV-ERBα antagonistic effect of SR8278 alleviated the AAI-induced ferroptosis and renal injury in mice. ArBu exhibits a broad spectrum of anti-tumor activity (91). Treating human gastric cancer cells with ArBu increases the expression of REV-ERBα and causes ferroptosis. Numerous studies have demonstrated that REV-ERB inhibition could be harmful or beneficial, depending on the type of disease (Figure 3).
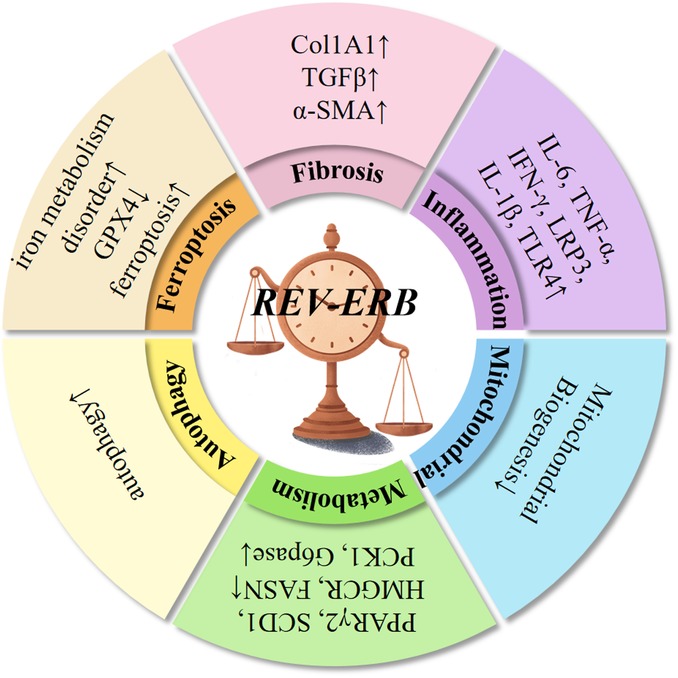
Figure 3. REV-ERBα/β proteins exhibit robust 24-h oscillations and regulate various physiological functions. These proteins inhibit autophagy, suppress gluconeogenesis, regulate insulin and glucagon levels, and maintain rhythmic glucose oscillations. In the liver, REV-ERBα/β regulate lipid synthesis, transport, bile acid metabolism, and control adipocyte differentiation and adipose tissue expansion. They also contribute to fatty acid oxidation in skeletal muscle and the heart. As inflammatory regulators, REV-ERBα/β are involved in NF-κB signaling, NLRP3 inflammasome activation, transcription of inflammation-related genes, macrophage polarization, and immune cell development. Additionally, REV-ERBα/β inhibit fibrosis across multiple organs, regulate mitochondrial biogenesis, prevent intracellular iron metabolism disorders, and suppress ferroptosis.
CVDs present high morbidity and mortality and are associated with tremendous healthcare costs that are ever-increasing. According to the 2019 Global Burden of Disease (GBD) study, the total number of cases of cardiovascular diseases increased from 271 million in 1990 to 523 million in 2019. The number of deaths due to CVDs has been on a steady rise, from 12.1 million deaths reported in the year 1990 to 18.6 million deaths reported in 2019, with the latter accounting for 33% of all deaths worldwide (92). Atherosclerosis is a vital risk factor for CVDs. Its pathogenesis is characterized by cholesterol deposition, smooth muscle hyperplasia, inflammatory cell infiltration, and connective tissue accumulation, manifesting as plaque formation (atherosclerosis) in the intima of the arterial wall. Changes in the expressions of biological clock-related genes contribute to the pathogenesis of atherosclerosis. Aortic aneurysm, rupture, and dissection are among the most life-threatening arterial diseases and are closely associated with atherosclerosis. Plaque deposition, inflammation, and arterial wall remodeling driven by atherosclerosis contribute to the weakening of vessel walls, increasing the risk of these conditions. Studies have demonstrated that the circadian rhythms influencing blood pressure, heart rate, vascular tone, and vasoconstrictor hormone levels also affect the incidence of aortic aneurysms and dissections. The early morning hours are particularly critical, as physiological processes, such as plaque rupture, hypercoagulability, and coronary vasoconstriction are often aggravated during this time, heightening the risk of cardiovascular events (93, 94). In contrast to ischemic events, which are primarily influenced by acute occlusion of coronary arteries, aortic diseases emerge as chronic outcomes of arterial wall deterioration linked to prolonged exposure to atherosclerotic factors. While circadian rhythm disorders exacerbate the risk, these conditions should be distinctly categorized as arterial diseases mediated by structural and inflammatory mechanisms rather than I/R injury (95).
The severity of atherosclerotic lesions is determined by the balance of monocyte recruitment, macrophage excretion, proliferation, survival, and arterial wall apoptosis (96). Apolipoprotein CIII (apoCIII) functions critically in plasma triglycerides and residual lipoproteins. The presence of triglyceride-rich residual lipoproteins in plasma is closely related to atherosclerosis. In the human liver HepG2 cells expressing apoCIII, silencing the REV-ERBα gene could specifically inhibit the apoCIII gene promoter activity (97). A key step before the formation of atherosclerotic lesions is the migration of monocytes to the arterial wall and their differentiation into macrophages (98). Macrophages are the primary organizers of plaque inflammation (98, 99). Sato et al. reported that REV-ERBα inhibits the expression of the IL-6 gene in macrophages, either directly or indirectly, via RORE and nuclear factor-κB (NF-κB) response elements in the IL-6 promoter region, and the expression of the IL-6 gene was increased in the peritoneal macrophages of the mice lacking REV-ERBα (100). Several NRs are associated with developing CVDs and atherosclerosis (101). A study demonstrated that shRNA-mediated REV-ERBα deficiency, especially in hematopoietic cells, exacerbated the development of atherosclerotic lesions in LDLR (−/−) mice. At the cellular level, REV-ERBα knockdown in bone marrow-derived monocytes promoted the shift of pro-inflammatory macrophages to the anti-inflammatory phenotype (102). In the same study, treating LDL receptor-deficient mice with the REV-ERBα agonist SR9009 reduced the polarization of mouse macrophages (BMDMs) to pro-inflammatory M1 macrophages which increased the polarization of BMDMs to anti-inflammatory M2 macrophages. After seven weeks of this treatment, the size of the atherosclerotic plaques in the treated mice was significantly reduced (103). In another study (104), REV-ERBα expression in primary human macrophages could be induced using the synthetic ligands of the liver X receptor (LXR). Overexpression of REV-ERBα inhibited the induction of toll-like receptor TLR-4 by LXR agonists. However, its knockdown increased the expression of TLR-4, indicating that this was the molecular mechanism underlying the regulatory effect of REV-ERBα on cholesterol homeostasis. Vulnerable plaque rupture is the main trigger for most acute cardiovascular events. The study of vulnerable plaque models in hypercholesterolemia ApoE (−/−) mice and NR1D1 (−/−) ApoE (−/−) mice revealed that NR1D1 deficiency could significantly increase the vulnerability/rupture of plaques (105). The incidence of intraplaque hemorrhage and spontaneous plaque rupture with intravascular thrombosis, together with the findings of in vitro experiments conducted using mouse bone marrow-derived macrophages (BMDMs), confirmed that NR1D1 plays a protective role by inhibiting macrophage pyroptosis. Table 1 summarizes studies involving REV-ERB in CVDs, detailing the type of CVDs, the animal/cell model used, and pathophysiological implications.
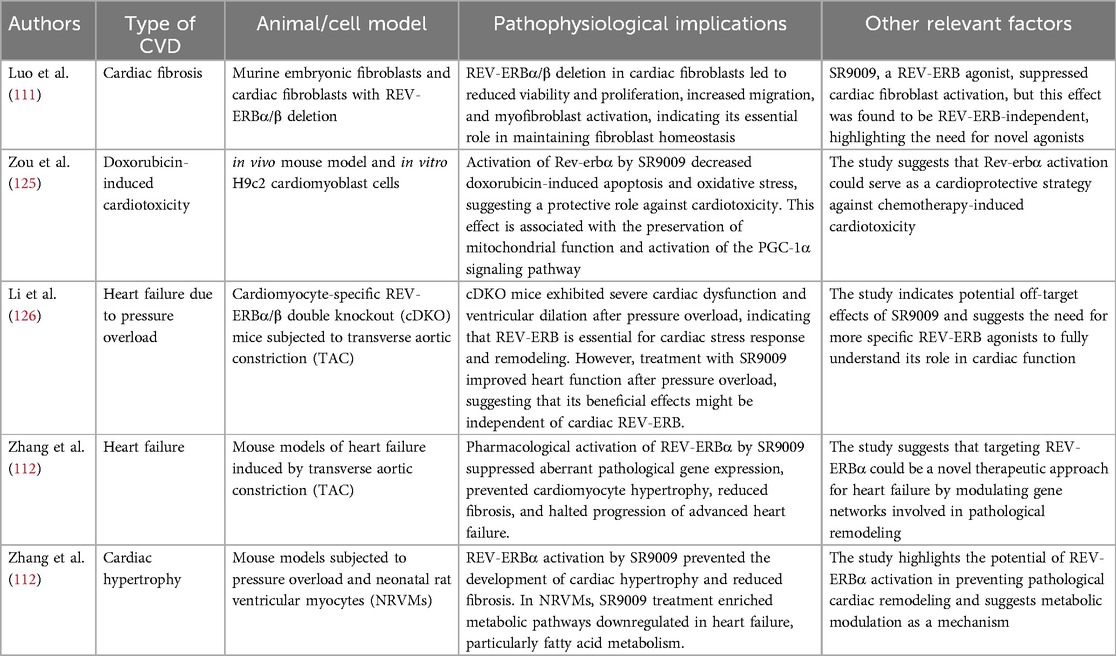
Table 1. Summarizing studies involving REV-ERB in cardiovascular diseases.
The prevalence of heart disease is increasing with time, with approximately 64.3 million cases of HF reported worldwide (106). Heart disease is associated with severe morbidity and mortality, along with a poor quality of life (107). The prevalence of all types of HF in patients aged 65 and above is approximately 11.8% in developed nations (108). Pathologically, HF is characterized by interstitial fibrosis, ventricular remodeling, and decreased ventricular compliance (109). In the heart of adult mammals, cardiomyocytes account for approximately 75% of the myocardial volume and are organized into 2–5 cell-thick layers. These myocardial cell layers are surrounded by interstitial extracellular matrix (ECM), which is mainly composed of fibrous collagen (110). As a mechanical scaffold, the epicardium is vital for contractile force transmission. In addition to type I collagen (the most abundant protein in the cardiac ECM) and type III collagen, the cardiac ECM consists of various glycoproteins, glycosaminoglycans, and proteoglycans (111). Moreover, it has a reservoir of stored potential growth factors and proteases, which are rapidly activated upon injury to stimulate repair. Several epidemiological studies have demonstrated that shift workers are at an increased risk of developing CVDs (112–115). Various non-standard schedules required by shift workers force sudden changes in their sleep time and light and dark exposure periods. These changes lead to a disordered endogenous circadian rhythm system, and disbalance the external body environment. The circadian rhythm system disorder caused by night shift work, besides causing an imbalance between the circadian rhythm system and the external light-dark cycle, also leads to an internal asynchrony among the different levels of the circadian rhythm system (116). Genome-wide gene expression analyses have revealed that shift work reprogrammed the cardiac circulatory transcriptome and led to cardiac fibrosis. REV-ERBα/β double deletion decreased the viability and proliferation of cardiac fibroblasts while increasing the migration of cardiac fibroblasts and myofibroblast activation. REV-ERBα/β are reported to maintain cardiac fibroblasts in a healthy resting state (117). The REV-ERB agonist SR9009 selectively inhibits abnormal pathological gene expression and prevents cardiomyocyte hypertrophy (118). The activation of REV-ERBα prevented the development of cardiac hypertrophy in mouse models, reduced the degree of fibrosis, and delayed the progression to advanced HF.
Early studies conducted on humans have revealed circadian rhythm patterns in the onset of acute myocardial ischemia (MI). The incidence of MI is higher in the morning than in the evening, aligning with the early-morning onset of other adverse cardiovascular events, such as unstable angina pectoris, sudden death, stroke, ventricular arrhythmia, cardiogenic shock, stent thrombosis, and transient MI. When the plaque narrows the arterial lumen, blood flow is limited, leading to ischemia of the distal tissue. When the lumen is completely blocked, blood flow is interrupted further, which may lead to non-fatal or fatal coronary artery disease or cerebrovascular events. Changes in the expressions of biological clock-related genes contribute to the pathogenesis of ischemic events. Studies have reported that in the morning, several physiological processes that might lead to plaque rupture, hypercoagulability, or coronary vasoconstriction are aggravated (94). Clinical studies have shown that the severity of MI is also heightened in the morning. Furthermore, the incidence of ischemic heart disease is significantly higher among shift workers compared to daytime workers, independent of lifestyle factors, such as smoking or age. Maintaining normal circadian rhythms or healthy sleep patterns is thus crucial for cardiovascular health (119).
Ischemic/reperfusion (I/R) injury studies in animal models provide insight into the circadian regulation of cardiac damage (120). For instance, rats subjected to left coronary artery occlusion followed by reperfusion showed altered circadian gene expression in ischemic regions (121). These changes included diminished peak expressions of critical clock genes, such as Npas2, Per1-3, Cry1-2, and REV-ERBα, and reduced amplitude of Bmal1, Clock, and other circadian regulators (122). This rapid inactivation of the biological clock in ischemic heart regions disrupts synchronization with the environment and neighboring heart regions (123).
Time-of-day studies in a mouse model of left anterior descending coronary artery occlusion demonstrated that ischemia during the sleep-to-awake transition (ZT12) resulted in larger infarct size, increased fibrosis, and impaired remodeling compared to ischemia during the awake-to-sleep transition (ZT0). Zhao et al. observed that shift workers with circadian rhythm disruption exhibited larger infarct sizes (5), reduced left ventricular ejection fraction (LVEF), and a higher risk of major adverse cardiac events. Mechanistic studies identified reduced Nr1d1 expression as a key driver of exacerbated MI outcomes in human and animal models. Cardiomyocyte-specific NR1D1 knockout mice showed increased infarct size, cardiomyocyte death, and worsened LVEF after MI and reperfusion injury. Therapeutic interventions targeting REV-ERB, such as intraperitoneal injections of the REV-ERB agonist SR9009, demonstrated significant benefits. These included improved survival rates, enhanced left ventricular function, reduced brain natriuretic peptide (BNP) levels, and lower inflammatory markers (e.g., IL-6, Mcp1, Ly6g). SR9009 also mitigated the activation of NF-κB and MAPK signaling pathways (124). Furthermore, SR9009 modulated cardiac fibroblast function, weakening the NLRP3 inflammasome, reducing immune cell recruitment, and accelerating myocardial repair (125).
HF is characterized by many clinical symptoms resulting from various pathogenic factors impairing the heart structure and/or function. It has become one of the most serious threats to human health. Notably, approximately 13% of cardiac genes and nearly 8% of related proteins exhibit rhythmic patterns, and disruptions in these rhythms can contribute to the development of HF. To further investigate circadian rhythm-related genes that may aid in identifying and treating HF, a study (126) analyzed expression data from ischemic and dilated cardiomyopathy patients with or without HF, using datasets from the GEO database. The study identified 723 differentially expressed circadian rhythm-related genes (DEGs) in HF patients compared to healthy controls. Among these, the relative mRNA expression levels of CRY2 and BHLHE41 were significantly elevated in the HF group, while ARNTL and NPAS2 expression levels were reduced. The cardiomyocyte circadian clock is crucial in myocardial systolic regulation, metabolism, and gene expression (34). Studies on Bmal1 (−/−) mice have demonstrated that Bmal1 deficiency leads to progressive myocardial pathological changes, first appearing at 36 weeks of age (24). These changes initially manifest as a transient increase in myocardial mass, followed by gradual ventricular dilation, ultimately resulting in HF and death. An in vitro working heart perfusion test revealed that systolic ventricular dysfunction coincided with the onset of dilation and failure. This was accompanied by the downregulation of two myosin heavy chain subtype mRNAs, sarcomeric structural abnormalities, and a shift in annexin isomer composition toward the stiffer N2B isomer.
The nuclear receptors REV-ERBα/β, critical to the circadian clock, have emerged as promising pharmacological targets for cardiac diseases. Pieterjan Dierickx et al. discovered that mice with cardiomyocyte-specific deletion of Rev-erb exhibit premature death due to dilated cardiomyopathy. Further mechanistic studies revealed that the absence of REV-ERB downregulates the expression of fatty acid oxidation genes through its direct target, E4BP4, and disrupts NAD+ biosynthesis, impairing cardiac metabolism and function (127). A study (128) using a mouse model of myocardial cell REV-ERB gene knockout (KO) compared the cardiac function of KO and WT mice. KO mice exhibited impaired systolic function, left ventricular enlargement, and a decreased ejection fraction starting at 4.5 months. By 6–8 months, most KO mice succumbed to cardiac dysfunction. Mechanistic studies revealed that heart REV-ERB plays a crucial role in enhancing the expression of fatty acid metabolism-related genes (FAO genes) during the light cycle while counteracting diet-induced activation of glucose metabolism genes in the dark cycle. REV-ERB knockout impaired oxidative lipid metabolism, leading to systolic and diastolic dysfunction. Fat reduction may serve as a predictor of adverse outcomes in advanced heart failure (HF), with circadian clock disruption identified as a key driver of lipid metabolism disorders. To further investigate the role of circadian rhythm disruption in lipid metabolism disorders associated with HF, a study (129) established an HF model and divided it into different groups: normal rhythm (LD), inverted rhythm (DL), a lentiviral vector carrying Bmal1 short hairpin RNA (LV-Bmal1 shRNA), and empty lentiviral vector control (LV-Control shRNA). Monitoring lipid metabolism levels revealed that BMAL1 protein levels in the adipose tissue of the LD group were lower than in the control group. In the normal-rhythm HF model, REV-ERBα protein levels increased, indicating circadian rhythm disruption. Additionally, HF rats exhibited decreased fat mass, increased ectopic lipid deposition, smaller adipocytes with lower lipid content, and fibrotic adipose tissue. Mechanistic studies further demonstrated that the disruption of the BMAL1/REV-ERBα circadian rhythm loop contributed to increased fat consumption in HF. Treatment with the REV-ERB agonist SR9009 reduced cardiac remodeling by decreasing AKT activity in aged WT mice (130). MEF2a and MEF2c are key regulators of the cardiac hypertrophy gene program. Studies have shown that ectopic expression of MEF2a or MEF2c in the heart can lead to dilated cardiomyopathy and HF (131, 132). A study (118) found that REV-ERBα, upon co-localization with MEF2a and MEF2c, could specifically inhibit MEF2a/MEF2c-driven cardiac hypertrophy and the aberrant activation of gene programs in HF, thereby playing a critical role in preventing ventricular remodeling.
The pathological remodeling of the myocardium is associated with typical gene expression programs and had long been considered the result of the disease, until recently, when it was indicated to be the driver of the disease. Numerous studies have demonstrated that the pharmacological activation of REV-ERB selectively inhibits abnormal pathological gene expression and prevents cardiomyocyte hypertrophy (Figure 4). Therefore, regulating the gene network by targeting REV-ERBα could be a novel strategy for curing HF (118).
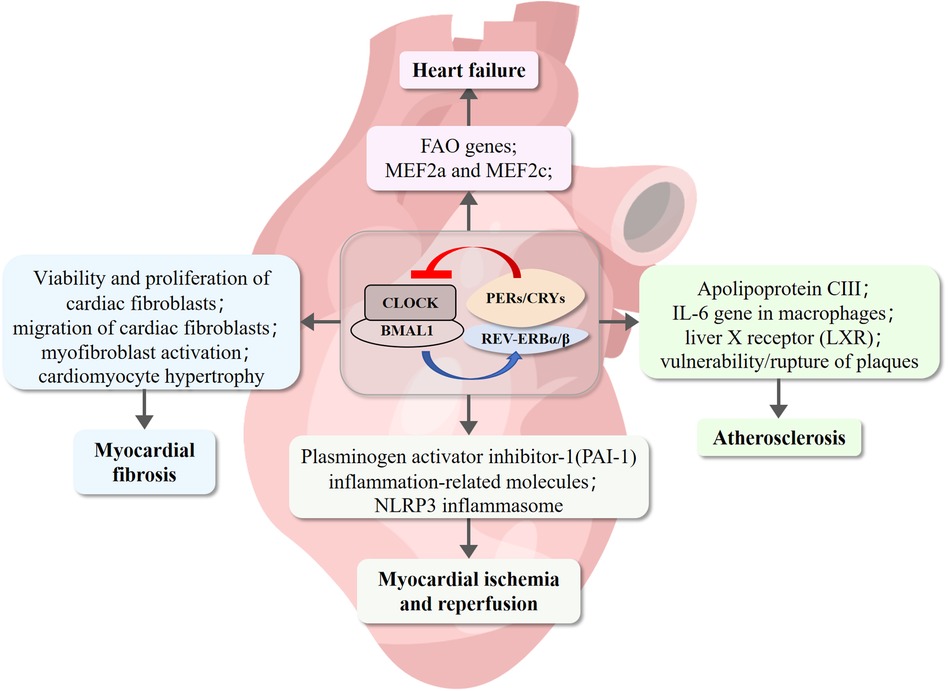
Figure 4. REV-ERB involvement of cardiovascular diseases pathogenesis. REV-ERB is implicated in the pathogenesis of cardiovascular diseases. Deficiency in REV-ERB increases the vulnerability and rupture of plaques. Its expression is significantly reduced in myocardial infarction and reperfusion injury. REV-ERB plays a crucial role in maintaining cardiac fibroblasts in a healthy resting state, preventing pathological gene expression, and inhibiting cardiomyocyte hypertrophy.
Circadian rhythms, intrinsic 24-h cycles regulating human physiology, play a fundamental role in maintaining health. Disruptions caused by modern lifestyles, work schedules, and increased screen time have led to widespread chronodisruption, contributing to adverse effects on sleep, physical health, and mental well-being. The regulation of circadian rhythms is crucial for cardiovascular health. Molecular mechanisms involving REV-ERB, a key circadian regulator, have shown protective roles against CVDs, such as atherosclerosis, MI/reperfusion injury, and HF. These findings highlight the importance of maintaining circadian rhythm stability to prevent and manage CVDs. Future research should continue to explore the interplay between circadian rhythm regulators and pathophysiological processes, aiming to develop effective therapeutic strategies for combating circadian rhythm-related disorders and enhancing overall health.
CW: Investigation, Methodology, Writing – original draft. JYa: Investigation, Writing – original draft. JYu: Methodology, Writing – original draft. XW: Investigation, Writing – original draft. QL: Investigation, Writing – original draft. CR: Writing – original draft, Visualization. XZhi: Visualization, Writing – original draft. XL: Visualization, Writing – original draft. KL: Conceptualization, Writing – review & editing. XZha: Conceptualization, Writing – review & editing. YL: Conceptualization, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the 2021 Gansu Jiaoyu Jiebang Guashuai Project (No. 2021 JYJBGS-03), 2022 Gansu Province Natural Sciences Fund (22JR11RA128) and National Natural Science Foundation of China (NO. 82260869), National Natural Science Foundation of China (NO. 82360926) and Lanzhou Youth Science and Technology Talents Innovation Project (2023-QN-191).
The authors declare that the research was conducted with no commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hermida R, Crespo JJ, Otero A, Dominguez-Sardina M, Moya A, Rios M, et al. Asleep blood pressure: significant prognostic marker of vascular risk and therapeutic target for prevention. Eur Heart J. (2018) 39(47):4159–71. doi: 10.1093/eurheartj/ehy475
2. Zhao Y, Liu Y, Sun Q, Han J, Wei Y, Lu Y, et al. Effects of blood pressure and heart rate circadian rhythms on left atrial function. J Hypertens. (2021) 39(11):2318–24. doi: 10.1097/HJH.0000000000002923
3. Crnko S, Du Pre BC, Sluijter JPG, Van Laake LW. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat Rev Cardiol. (2019) 16(7):437–47. doi: 10.1038/s41569-019-0167-4
4. Bochaton T, Ovize M. Circadian rhythm and ischaemia-reperfusion injury. Lancet. (2018) 391(10115):8–9. doi: 10.1016/S0140-6736(17)32177-3
5. Zhao Y, Lu X, Wan F, Gao L, Lin N, He J, et al. Disruption of circadian rhythms by shift work exacerbates reperfusion injury in myocardial infarction. J Am Coll Cardiol. (2022) 79(21):2097–115. doi: 10.1016/j.jacc.2022.03.370
6. Brown TM, Piggins HD. Electrophysiology of the suprachiasmatic circadian clock. Prog Neurobiol. (2007) 82(5):229–55. doi: 10.1016/j.pneurobio.2007.05.002
7. Cajochen C, Krauchi K, Wirz-Justice A. Role of melatonin in the regulation of human circadian rhythms and sleep. J Neuroendocrinol. (2003) 15(4):432–7. doi: 10.1046/j.1365-2826.2003.00989.x
8. Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the circadian clock in the liver by feeding. Science. (2001) 291(5503):490–3. doi: 10.1126/science.291.5503.490
9. Mistlberger RE, Skene DJ. Social influences on mammalian circadian rhythms: animal and human studies. Biol Rev Camb Philos Soc. (2004) 79(3):533–56. doi: 10.1017/s1464793103006353
10. Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. (2000) 14(23):2950–61. doi: 10.1101/gad.183500
11. Lazar MA, Hodin RA, Darling DS, Chin WW. A novel member of the thyroid/steroid hormone receptor family is encoded by the opposite strand of the rat c-erbA alpha transcriptional unit. Mol Cell Biol. (1989) 9(3):1128–36. doi: 10.1128/mcb.9.3.1128-1136.1989
12. Dumas B, Harding HP, Choi HS, Lehmann KA, Chung M, Lazar MA, et al. A new orphan member of the nuclear hormone receptor superfamily closely related to rev-erb. Mol Endocrinol. (1994) 8(8):996–1005. doi: 10.1210/mend.8.8.7997240
13. Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discov. (2014) 13(3):197–216. doi: 10.1038/nrd4100
14. Van Laake LW, Luscher TF, Young ME. The circadian clock in cardiovascular regulation and disease: lessons from the nobel prize in physiology or medicine 2017. Eur Heart J. (2018) 39(24):2326–9. doi: 10.1093/eurheartj/ehx775
15. Sehgal A. Physiology flies with time. Cell. (2017) 171(6):1232–5. doi: 10.1016/j.cell.2017.11.028
16. Cederroth CR, Albrecht U, Bass J, Brown SA, Dyhrfjeld-Johnsen J, Gachon F, et al. Medicine in the fourth dimension. Cell Metab. (2019) 30(2):238–50. doi: 10.1016/j.cmet.2019.06.019
17. Astiz M, Heyde I, Oster H. Mechanisms of communication in the mammalian circadian timing system. Int J Mol Sci. (2019) 20(2):343. doi: 10.3390/ijms20020343
18. Sollars PJ, Pickard GE. The neurobiology of circadian rhythms. Psychiatr Clin North Am. (2015) 38(4):645–65. doi: 10.1016/j.psc.2015.07.003
19. Hughes S, Jagannath A, Hankins MW, Foster RG, Peirson SN. Photic regulation of clock systems. Methods Enzymol. (2015) 552:125–43. doi: 10.1016/bs.mie.2014.10.018
20. Schibler U, Gotic I, Saini C, Gos P, Curie T, Emmenegger Y, et al. Clock-talk: interactions between central and peripheral circadian oscillators in mammals. Cold Spring Harb Symp Quant Biol. (2015) 80:223–32. doi: 10.1101/sqb.2015.80.027490
21. Fodor DM, Marta MM, Perju-Dumbrava L. Implications of circadian rhythm in stroke occurrence: certainties and possibilities. Brain Sci. (2021) 11(7):865. doi: 10.3390/brainsci11070865
22. De Luca G, Suryapranata H, Ottervanger JP, van ‘t Hof AW, Hoorntje JC, Gosselink AT, et al. Circadian variation in myocardial perfusion and mortality in patients with ST-segment elevation myocardial infarction treated by primary angioplasty. Am Heart J. (2005) 150(6):1185–9. doi: 10.1016/j.ahj.2005.01.057
23. Martino TA, Oudit GY, Herzenberg AM, Tata N, Koletar MM, Kabir GM, et al. Circadian rhythm disorganization produces profound cardiovascular and renal disease in hamsters. Am J Physiol Regul Integr Comp Physiol. (2008) 294(5):R1675–83. doi: 10.1152/ajpregu.00829.2007
24. Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol. (2012) 303(4):H475–85. doi: 10.1152/ajpheart.00238.2012
25. Ozturk N, Ozturk D, Kavakli IH, Okyar A. Molecular aspects of circadian pharmacology and relevance for cancer chronotherapy. Int J Mol Sci. (2017) 18(10):2168. doi: 10.3390/ijms18102168
26. Dibner C, Schibler U. Circadian timing of metabolism in animal models and humans. J Intern Med. (2015) 277(5):513–27. doi: 10.1111/joim.12347
27. Mosig RA, Kojima S. Timing without coding: how do long non-coding RNAs regulate circadian rhythms? Semin Cell Dev Biol. (2022) 126:79–86. doi: 10.1016/j.semcdb.2021.04.020
28. Zeng Y, Wu N, Zhang Z, Zhong L, Li G, Li Y. Non-coding RNA and arrhythmias: expression, function, and molecular mechanism. Europace. (2023) 25(4):1296–308. doi: 10.1093/europace/euad047
29. Zhang TN, Wang W, Yang N, Huang XM, Liu CF. Regulation of glucose and lipid metabolism by long non-coding RNAs: facts and research progress. Front Endocrinol (Lausanne). (2020) 11:457. doi: 10.3389/fendo.2020.00457
30. Portaluppi F, Hermida RC. Circadian rhythms in cardiac arrhythmias and opportunities for their chronotherapy. Adv Drug Deliv Rev. (2007) 59(9–10):940–51. doi: 10.1016/j.addr.2006.10.011
31. Young ME. The cardiac circadian clock: implications for cardiovascular disease and its treatment. JACC Basic Transl Sci. (2023) 8(12):1613–28. doi: 10.1016/j.jacbts.2023.03.024
32. Thosar SS, Butler MP, Shea SA. Role of the circadian system in cardiovascular disease. J Clin Invest. (2018) 128(6):2157–67. doi: 10.1172/JCI80590
33. Young ME, Brewer RA, Peliciari-Garcia RA, Collins HE, He L, Birky TL, et al. Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms. (2014) 29(4):257–76. doi: 10.1177/0748730414543141
34. Bray MS, Shaw CA, Moore MW, Garcia RA, Zanquetta MM, Durgan DJ, et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol. (2008) 294(2):H1036–47. doi: 10.1152/ajpheart.01291.2007
35. Raza GS, Sodum N, Kaya Y, Herzig KH. Role of circadian transcription factor rev-erb in metabolism and tissue fibrosis. Int J Mol Sci. (2022) 23(21):12954. doi: 10.3390/ijms232112954
36. Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. (2012) 74(2):246–60. doi: 10.1016/j.neuron.2012.04.006
37. Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. (2012) 35:445–62. doi: 10.1146/annurev-neuro-060909-153128
38. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147(4):728–41. doi: 10.1016/j.cell.2011.10.026
39. Deretic V. Links between autophagy, innate immunity, inflammation and crohn’s disease. Dig Dis. (2009) 27(3):246–51. doi: 10.1159/000228557
40. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. (2019) 176(1–2):11–42. doi: 10.1016/j.cell.2018.09.048
41. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. (2014) 24(1):69–79. doi: 10.1038/cr.2013.161
42. Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, et al. Rev-erb-alpha modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med. (2013) 19(8):1039–46. doi: 10.1038/nm.3213
43. Shen W, Zhang W, Ye W, Wang H, Zhang Q, Shen J, et al. SR9009 induces a REV-ERB dependent anti-small-cell lung cancer effect through inhibition of autophagy. Theranostics. (2020) 10(10):4466–80. doi: 10.7150/thno.42478
44. Thomes PG, Brandon-Warner E, Li T, Donohue TM Jr, Schrum LW. Rev-erb agonist and TGF-beta similarly affect autophagy but differentially regulate hepatic stellate cell fibrogenic phenotype. Int J Biochem Cell Biol. (2016) 81(Pt A):137–47. doi: 10.1016/j.biocel.2016.11.007
45. Sun L, Tian H, Xue S, Ye H, Xue X, Wang R, et al. Circadian clock genes REV-ERBs inhibits Granulosa cells apoptosis by regulating mitochondrial biogenesis and autophagy in polycystic ovary syndrome. Front Cell Dev Biol. (2021) 9:658112. doi: 10.3389/fcell.2021.658112
46. Brown MR, Laouteouet D, Delobel M, Villard O, Broca C, Bertrand G, et al. The nuclear receptor REV-ERBalpha is implicated in the alteration of beta-cell autophagy and survival under diabetogenic conditions. Cell Death Dis. (2022) 13(4):353. doi: 10.1038/s41419-022-04767-z
47. Sulli G, Rommel A, Wang X, Kolar MJ, Puca F, Saghatelian A, et al. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature. (2018) 553(7688):351–5. doi: 10.1038/nature25170
48. Chandra V, Bhagyaraj E, Nanduri R, Ahuja N, Gupta P. NR1D1 ameliorates mycobacterium tuberculosis clearance through regulation of autophagy. Autophagy. (2015) 11(11):1987–97. doi: 10.1080/15548627.2015.1091140
49. Pastore N, Ballabio A. Keeping the autophagy tempo. Autophagy. (2019) 15(10):1854–6. doi: 10.1080/15548627.2019.1645545
50. Pastore N, Vainshtein A, Herz NJ, Huynh T, Brunetti L, Klisch TJ, et al. Nutrient-sensitive transcription factors TFEB and TFE3 couple autophagy and metabolism to the peripheral clock. EMBO J. (2019) 38(12):e101347. doi: 10.15252/embj.2018101347
51. Silva Angulo F, Joseph CV, Delval L, Deruyter L, Heumel S, Bicharel M, et al. Rev-erb-alpha antagonism in alveolar macrophages protects against pneumococcal infection in elderly mice. Cell Rep. (2025) 44(2):115273. doi: 10.1016/j.celrep.2025.115273
52. Fu J, Fan Z, He L, Liu Q, Liu H, Li Y, et al. Circadian clock disruption in autoimmune thyroiditis. Eur Thyroid J. (2023) 12(5):e230035. doi: 10.1530/ETJ-23-0035
53. Guo DK, Zhu Y, Sun HY, Xu XY, Zhang S, Hao ZB, et al. Pharmacological activation of REV-ERBalpha represses LPS-induced microglial activation through the NF-kappaB pathway. Acta Pharmacol Sin. (2019) 40(1):26–34. doi: 10.1038/s41401-018-0064-0
54. Hashizume H, Motonari H, Yamamoto K, Nakamura Y, Hisaoka-Nakashima K, Morioka N. Stimulation of nuclear receptor REV-ERBs alleviates monosodium iodoacetate-induced osteoarthritis pathology of mice and the induction of inflammatory molecules expression in primary cultured chondrocytes. Int Immunopharmacol. (2024) 127:111349. doi: 10.1016/j.intimp.2023.111349
55. Silva BSA, Pereira T, Minuzzi LG, Padilha CS, Figueiredo C, Olean-Oliveira T, et al. Mild to moderate post-COVID-19 alters markers of lymphocyte activation, exhaustion, and immunometabolic responses that can be partially associated by physical activity level- an observational sub-analysis fit- COVID study. Front Immunol. (2023) 14:1212745. doi: 10.3389/fimmu.2023.1212745
56. Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. (2013) 498(7455):511–5. doi: 10.1038/nature12209
57. Amir M, Chaudhari S, Wang R, Campbell S, Mosure SA, Chopp LB, et al. REV-ERBalpha regulates T(H)17 cell development and autoimmunity. Cell Rep. (2018) 25(13):3733–49.e8. doi: 10.1016/j.celrep.2018.11.101
58. Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, Kashiwada M, et al. TH17 cell differentiation is regulated by the circadian clock. Science. (2013) 342(6159):727–30. doi: 10.1126/science.1243884
59. Pourcet B, Duez H. Circadian control of inflammasome pathways: implications for circadian medicine. Front Immunol. (2020) 11:1630. doi: 10.3389/fimmu.2020.01630
60. Morioka N, Kodama K, Tsuruta M, Hashizume H, Kochi T, Nakamura Y, et al. Stimulation of nuclear receptor REV-ERBs suppresses inflammatory responses in spinal microglia. Neurochem Int. (2021) 151:105216. doi: 10.1016/j.neuint.2021.105216
61. Kamat PK, Khan MB, Siddiqui S, Hattaway TG, Anas A, Rudic RD, et al. Time of day dependent reduction in stroke infarct volume by the reverb agonist SR9009 in mice. Exp Neurol. (2025) 384:115067. doi: 10.1016/j.expneurol.2024.115067
62. Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. (2002) 109(3):307–20. doi: 10.1016/s0092-8674(02)00722-5
63. Markwald RR, Melanson EL, Smith MR, Higgins J, Perreault L, Eckel RH, et al. Impact of insufficient sleep on total daily energy expenditure, food intake, and weight gain. Proc Natl Acad Sci U S A. (2013) 110(14):5695–700. doi: 10.1073/pnas.1216951110
64. Wang J, Lazar MA. Bifunctional role of rev-erbalpha in adipocyte differentiation. Mol Cell Biol. (2008) 28(7):2213–20. doi: 10.1128/MCB.01608-07
65. Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, et al. Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature. (2012) 485(7396):123–7. doi: 10.1038/nature11048
66. Sitaula S, Zhang J, Ruiz F, Burris TP. Rev-erb regulation of cholesterologenesis. Biochem Pharmacol. (2017) 131:68–77. doi: 10.1016/j.bcp.2017.02.006
67. Le Martelot G, Claudel T, Gatfield D, Schaad O, Kornmann B, Lo Sasso G, et al. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. (2009) 7(9):e1000181. doi: 10.1371/journal.pbio.1000181
68. Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. (2012) 485(7396):62–8. doi: 10.1038/nature11030
69. Yang MY, Lin HY, Chen YM, Hu ML, Chen IY, Yang CH. Chronic low-dose REV-ERBs agonist SR9009 mitigates constant light-induced weight gain and insulin resistance via adipogenesis modulation. Biomed J. (2025):100830. doi: 10.1016/j.bj.2025.100830
70. Delezie J, Dumont S, Dardente H, Oudart H, Grechez-Cassiau A, Klosen P, et al. The nuclear receptor REV-ERBalpha is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. (2012) 26(8):3321–35. doi: 10.1096/fj.12-208751
71. Yin L, Wu N, Lazar MA. Nuclear receptor rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl Recept Signal. (2010) 8:e001. doi: 10.1621/nrs.08001
72. Ding G, Li X, Hou X, Zhou W, Gong Y, Liu F, et al. REV-ERB in GABAergic neurons controls diurnal hepatic insulin sensitivity. Nature. (2021) 592(7856):763–7. doi: 10.1038/s41586-021-03358-w
73. Li W, Wang Y, Liu C, Yu Y, Xu L, Dong B. Evaluation of the regulatory effect of the pan-PPAR agonist chiglitazar on the dawn phenomenon. Diabetes Ther. (2025) 10:731–748. doi: 10.1007/s13300-025-01708-9
74. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. (2014) 69(Suppl 1):S4–9. doi: 10.1093/gerona/glu057
75. Lopez M, Meier D, Muller A, Franken P, Fujita J, Fontana A. Tumor necrosis factor and transforming growth factor beta regulate clock genes by controlling the expression of the cold inducible RNA-binding protein (CIRBP). J Biol Chem. (2014) 289(5):2736–44. doi: 10.1074/jbc.M113.508200
76. Cunningham PS, Meijer P, Nazgiewicz A, Anderson SG, Borthwick LA, Bagnall J, et al. The circadian clock protein REVERBalpha inhibits pulmonary fibrosis development. Proc Natl Acad Sci U S A. (2020) 117(2):1139–47. doi: 10.1073/pnas.1912109117
77. Yao H, Sundar IK, Huang Y, Gerloff J, Sellix MT, Sime PJ, et al. Disruption of sirtuin 1-mediated control of circadian molecular clock and inflammation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2015) 53(6):782–92. doi: 10.1165/rcmb.2014-0474OC
78. Wang Q, Sundar IK, Lucas JH, Muthumalage T, Rahman I. Molecular clock REV-ERBalpha regulates cigarette smoke-induced pulmonary inflammation and epithelial-mesenchymal transition. JCI Insight. (2021) 6(12):e145200. doi: 10.1172/jci.insight.145200
79. Sundar IK, Rashid K, Sellix MT, Rahman I. The nuclear receptor and clock gene REV-ERBalpha regulates cigarette smoke-induced lung inflammation. Biochem Biophys Res Commun. (2017) 493(4):1390–5. doi: 10.1016/j.bbrc.2017.09.157
80. Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
81. Li T, Eheim AL, Klein S, Uschner FE, Smith AC, Brandon-Warner E, et al. Novel role of nuclear receptor rev-erbalpha in hepatic stellate cell activation: potential therapeutic target for liver injury. Hepatology. (2014) 59(6):2383–96. doi: 10.1002/hep.27049
82. Griffett K, Bedia-Diaz G, Elgendy B, Burris TP. REV-ERB agonism improves liver pathology in a mouse model of NASH. PLoS One. (2020) 15(10):e0236000. doi: 10.1371/journal.pone.0236000
83. Li H, Cai Z. SIRT3 regulates mitochondrial biogenesis in aging-related diseases. J Biomed Res. (2022) 37(2):77–88. doi: 10.7555/JBR.36.20220078
84. Amador A, Campbell S, Kazantzis M, Lan G, Burris TP, Solt LA. Distinct roles for REV-ERBalpha and REV-ERBbeta in oxidative capacity and mitochondrial biogenesis in skeletal muscle. PLoS One. (2018) 13(5):e0196787. doi: 10.1371/journal.pone.0196787
85. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. (2014) 15(10):634–46. doi: 10.1038/nrm3877
86. Lee SB, Yang HO. Sinapic acid ameliorates REV-ERB alpha modulated mitochondrial fission against MPTP-induced Parkinson’s disease model. Biomol Ther (Seoul). (2022) 30(5):409–17. doi: 10.4062/biomolther.2022.020
87. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
88. Tian H, Huang Q, Cheng J, Xiong Y, Xia Z. Rev-erbalpha attenuates diabetic myocardial injury through regulation of ferroptosis. Cell Signal. (2024) 114:111006. doi: 10.1016/j.cellsig.2023.111006
89. Guo L, Zhang T, Wang F, Chen X, Xu H, Zhou C, et al. Targeted inhibition of rev-erb-alpha/beta limits ferroptosis to ameliorate folic acid-induced acute kidney injury. Br J Pharmacol. (2021) 178(2):328–45. doi: 10.1111/bph.15283
90. Wang Y, Wang Z, Wu Z, Chen M, Dong D, Yu P, et al. Involvement of REV-ERBalpha dysregulation and ferroptosis in aristolochic acid I-induced renal injury. Biochem Pharmacol. (2021) 193:114807. doi: 10.1016/j.bcp.2021.114807
91. Chen K, Li A, Wang J, Li D, Wang X, Liu C, et al. Arenobufagin causes ferroptosis in human gastric cancer cells by increasing rev-erbalpha expression. J Tradit Complement Med. (2023) 13(1):72–80. doi: 10.1016/j.jtcme.2022.10.007
92. Collaborators GBDRF. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet. (2020) 396(10258):1223–49. doi: 10.1016/S0140-6736(20)30752-2
93. Portaluppi F, Manfredini R, Fersini C. From a static to a dynamic concept of risk: the circadian epidemiology of cardiovascular events. Chronobiol Int. (1999) 16(1):33–49. doi: 10.3109/07420529908998710
94. Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. (1989) 79(4):733–43. doi: 10.1161/01.cir.79.4.733
95. Gui T, Shimokado A, Sun Y, Akasaka T, Muragaki Y. Diverse roles of macrophages in atherosclerosis: from inflammatory biology to biomarker discovery. Mediators Inflamm. (2012) 2012:693083. doi: 10.1155/2012/693083
96. Galkina E, Ley K. Leukocyte influx in atherosclerosis. Curr Drug Targets. (2007) 8(12):1239–48. doi: 10.2174/138945007783220650
97. Coste H, Rodriguez JC. Orphan nuclear hormone receptor rev-erbalpha regulates the human apolipoprotein CIII promoter. J Biol Chem. (2002) 277(30):27120–9. doi: 10.1074/jbc.M203421200
98. Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. (2012) 18(28):4266–88. doi: 10.2174/138161212802481237
99. Chinetti-Gbaguidi G, Staels B. Macrophage polarization in metabolic disorders: functions and regulation. Curr Opin Lipidol. (2011) 22(5):365–72. doi: 10.1097/MOL.0b013e32834a77b4
100. Sato S, Sakurai T, Ogasawara J, Shirato K, Ishibashi Y, Oh-ishi S, et al. Direct and indirect suppression of interleukin-6 gene expression in murine macrophages by nuclear orphan receptor REV-ERBalpha. ScientificWorldJournal. (2014) 2014:685854. doi: 10.1155/2014/685854
101. Neels JG, Hassen-Khodja R, Chinetti G. Nuclear receptors in abdominal aortic aneurysms. Atherosclerosis. (2020) 297:87–95. doi: 10.1016/j.atherosclerosis.2020.02.009
102. Ma H, Zhong W, Jiang Y, Fontaine C, Li S, Fu J, et al. Increased atherosclerotic lesions in LDL receptor deficient mice with hematopoietic nuclear receptor rev-erbalpha knock- down. J Am Heart Assoc. (2013) 2(4):e000235. doi: 10.1161/JAHA.113.000235
103. Sitaula S, Billon C, Kamenecka TM, Solt LA, Burris TP. Suppression of atherosclerosis by synthetic REV-ERB agonist. Biochem Biophys Res Commun. (2015) 460(3):566–71. doi: 10.1016/j.bbrc.2015.03.070
104. Fontaine C, Rigamonti E, Pourcet B, Duez H, Duhem C, Fruchart JC, et al. The nuclear receptor rev-erbalpha is a liver X receptor (LXR) target gene driving a negative feedback loop on select LXR-induced pathways in human macrophages. Mol Endocrinol. (2008) 22(8):1797–811. doi: 10.1210/me.2007-0439
105. Wu Z, Liao F, Luo G, Qian Y, He X, Xu W, et al. NR1D1 deletion induces rupture-prone vulnerable plaques by regulating macrophage pyroptosis via the NF-kappaB/NLRP3 inflammasome pathway. Oxid Med Cell Longev. (2021) 2021:5217572. doi: 10.1155/2021/5217572
106. Disease GBD, Injury I, Prevalence C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392(10159):1789–858. doi: 10.1016/S0140-6736(18)32279-7
107. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. (2023) 118(17):3272–87. doi: 10.1093/cvr/cvac013
108. Orso F, Fabbri G, Maggioni AP. Epidemiology of heart failure. Handb Exp Pharmacol. (2017) 243:15–33. doi: 10.1007/164_2016_74
109. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. (2021) 117(6):1450–88. doi: 10.1093/cvr/cvaa324
110. Bowers SL, Banerjee I, Baudino TA. The extracellular matrix: at the center of it all. J Mol Cell Cardiol. (2010) 48(3):474–82. doi: 10.1016/j.yjmcc.2009.08.024
111. Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. (2003) 108(11):1395–403. doi: 10.1161/01.CIR.0000085658.98621.49
112. Vyas MV, Garg AX, Iansavichus AV, Costella J, Donner A, Laugsand LE, et al. Shift work and vascular events: systematic review and meta-analysis. Br Med J. (2012) 345:e4800. doi: 10.1136/bmj.e4800
113. Torquati L, Mielke GI, Brown WJ, Kolbe-Alexander T. Shift work and the risk of cardiovascular disease. A systematic review and meta-analysis including dose-response relationship. Scand J Work Environ Health. (2018) 44(3):229–38. doi: 10.5271/sjweh.3700
114. Knutsson A. Health disorders of shift workers. Occup Med (Lond). (2003) 53(2):103–8. doi: 10.1093/occmed/kqg048
115. Boggild H, Knutsson A. Shift work, risk factors and cardiovascular disease. Scand J Work Environ Health. (1999) 25(2):85–99. doi: 10.5271/sjweh.410
116. Boivin DB, Boudreau P, Kosmadopoulos A. Disturbance of the circadian system in shift work and its health impact. J Biol Rhythms. (2022) 37(1):3–28. doi: 10.1177/07487304211064218
117. Luo X, Song S, Qi L, Tien CL, Li H, Xu W, et al. REV-ERB is essential in cardiac fibroblasts homeostasis. Front Pharmacol. (2022) 13:899628. doi: 10.3389/fphar.2022.899628
118. Zhang L, Zhang R, Tien CL, Chan RE, Sugi K, Fu C, et al. REV-ERBalpha ameliorates heart failure through transcription repression. JCI Insight. (2017) 2(17):e95177. doi: 10.1172/jci.insight.95177
119. Eckle T, Bertazzo J, Khatua TN, Fatemi Tabatabaei SR, Moori Bakhtiari N, Walker LA, et al. Circadian influences on myocardial ischemia-reperfusion injury and heart failure. Circ Res. (2024) 134(6):675–94. doi: 10.1161/CIRCRESAHA.123.323522
120. Manfredini R, Boari B, Gallerani M, Salmi R, Bossone E, Distante A, et al. Chronobiology of rupture and dissection of aortic aneurysms. J Vasc Surg. (2004) 40(2):382–8. doi: 10.1016/j.jvs.2004.04.019
121. Mukamal KJ, Muller JE, Maclure M, Sherwood JB, Mittleman MA. Increased risk of congestive heart failure among infarctions with nighttime onset. Am Heart J. (2000) 140(3):438–42. doi: 10.1067/mhj.2000.108830
122. Maemura K, Layne MD, Watanabe M, Perrell MA, Nagai R, Lee ME. Molecular mechanisms of morning onset of myocardial infarction. Ann N Y Acad Sci. (2001) 947:398–402. doi: 10.1111/j.1749-6632.2001.tb03972.x
123. Kung TA, Egbejimi O, Cui J, Ha NP, Durgan DJ, Essop MF, et al. Rapid attenuation of circadian clock gene oscillations in the rat heart following ischemia-reperfusion. J Mol Cell Cardiol. (2007) 43(6):744–53. doi: 10.1016/j.yjmcc.2007.08.018
124. Stujanna EN, Murakoshi N, Tajiri K, Xu D, Kimura T, Qin R, et al. Rev-erb agonist improves adverse cardiac remodeling and survival in myocardial infarction through an anti-inflammatory mechanism. PLoS One. (2017) 12(12):e0189330. doi: 10.1371/journal.pone.0189330
125. Reitz CJ, Alibhai FJ, Khatua TN, Rasouli M, Bridle BW, Burris TP, et al. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun Biol. (2019) 2:353. doi: 10.1038/s42003-019-0595-z
126. Sun Q, Zhao J, Liu L, Wang X, Gu X. Identification of the potential biomarkers associated with circadian rhythms in heart failure. PeerJ. (2023) 11:e14734. doi: 10.7717/peerj.14734
127. Dierickx P, Zhu K, Carpenter BJ, Jiang C, Vermunt MW, Xiao Y, et al. Circadian REV-ERBs repress E4bp4 to activate NAMPT-dependent NAD(+) biosynthesis and sustain cardiac function. Nat Cardiovasc Res. (2022) 1(1):45–58. doi: 10.1038/s44161-021-00001-9
128. Song S, Tien CL, Cui H, Basil P, Zhu N, Gong Y, et al. Myocardial rev-erb-mediated diurnal metabolic rhythm and obesity paradox. Circulation. (2022) 145(6):448–64. doi: 10.1161/CIRCULATIONAHA.121.056076
129. Ma D, Qu Y, Wu T, Liu X, Cai L, Wang Y. Excessive fat expenditure in MCT-induced heart failure rats is associated with BMAL1/REV-ERBalpha circadian rhythmic loop disruption. Sci Rep. (2024) 14(1):8128. doi: 10.1038/s41598-024-58577-8
130. Alibhai FJ, LaMarre J, Reitz CJ, Tsimakouridze EV, Kroetsch JT, Bolz SS, et al. Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. J Mol Cell Cardiol. (2017) 105:24–37. doi: 10.1016/j.yjmcc.2017.01.008
131. van Oort RJ, van Rooij E, Bourajjaj M, Schimmel J, Jansen MA, van der Nagel R, et al. MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. (2006) 114(4):298–308. doi: 10.1161/CIRCULATIONAHA.105.608968
Keywords: circadian rhythm, transcription factor, REV-ERB, cardiovascular diseases, biological clock regulation
Citation: Wang C, Yang J, Yuan J, Wang X, Li Q, Ren C, Zhi X, Lv X, Liu K, Zhao X and Li Y (2025) Role of circadian transcription factor REV-ERB in cardiovascular diseases: a review. Front. Cardiovasc. Med. 12:1516279. doi: 10.3389/fcvm.2025.1516279
Received: 24 October 2024; Accepted: 24 March 2025;
Published: 4 April 2025.
Edited by:
Tatsuya Sato, Sapporo Medical University, JapanReviewed by:
Gobinath Shanmugam, University of Alabama at Birmingham, United StatesCopyright: © 2025 Wang, Yang, Yuan, Wang, Li, Ren, Zhi, Lv, Liu, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingdong Li, d2Nsd2NsenNAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.