 Daniel A. Kasal1,2*†
Daniel A. Kasal1,2*† Viviane Sena1,†Grazielle Vilas Bôas Huguenin1,3,†Andrea De Lorenzo1,†
Viviane Sena1,†Grazielle Vilas Bôas Huguenin1,3,†Andrea De Lorenzo1,† Eduardo Tibirica1,†
Eduardo Tibirica1,†
- 1Research and Teaching Department, National Institute of Cardiology, Rio de Janeiro, Brazil
- 2Internal Medicine Department, State University of Rio de Janeiro, Rio de Janeiro, Brazil
- 3Nutrition and Dietetics Department, Fluminense Federal University, Rio de Janeiro, Brazil
Cardiovascular disease (CVD) is the main cause of morbidity and mortality in the adult and the elderly, with increasing prevalence worldwide. A growing body of research has focused on the earliest stage of vascular decline—endothelial dysfunction (ED)—which at the microvascular level can anticipate in decades the diagnosis of CVD. This review aims to provide a prospect of the literature regarding the development of ED as an indissociable feature of the aging of the cardiovascular system, highlighting the role of inflammation in the process. Vascular aging consists of a lifelong continuum, which starts with cell respiration and its inherent production of reactive oxygen species. Molecular imbalance is followed by cellular epigenetic changes, which modulate immune cells, such as macrophage and lymphocyte subtypes. These mechanisms are influenced by lifestyle habits, which affect inflammation hotspots in organism, such as visceral fat and gut microbiota. The process can ultimately lead to an environment committed to the loss of the physiological functions of endothelial cells. In addition, we discuss lifestyle changes targeting the connection between age-related inflammation and vascular dysfunction. Addressing microvascular ED represents a critical endeavor in order to prevent or delay vascular aging and associated diseases.
Introduction
The increasing proportion of older-age people in modern societies is considered to represent the outcome of long-lasting medical, scientific, and social achievements. However, aging is also typically associated with several changes in the cardiovascular system at different structural and functional levels (1). Cardiovascular disease (CVD) is the main cause of morbidity and mortality in adult and the elderly, with increasing prevalence worldwide (2). Whilst the pathophysiology underlying CVD has been described in detail in the last decades, chronic inflammation has been placed in the center of the molecular and cellular pathways, which ultimately lead to endothelial dysfunction (ED), the first identifiable stage of vascular decline. The mechanisms involved in vascular senescence have been recognized early, acting since the first developmental stages of cellular organisms. During mature life, the physiological processes inherent to growth and homeostasis of organic systems, including cardiovascular, eventually evolve to an unbalanced state, resulting in dysfunction and disease.
The age-related decline in vascular function occurs throughout the vascular tree in humans, ranging from large arteries, such as coronary, to peripheral conduit and resistance vessels, including the microcirculation (3). Accordingly, the vascular response induced by endothelium-dependent vasodilator substances display a progressive decline with age (4, 5). The same deterioration of endothelium-dependent vasodilation has been demonstrated in peripheral conduit vessels including brachial, femoral and popliteal arteries in the elderly (6–9). Predictably, the age-related decline in conduit artery endothelial function is reported to occur at an earlier stage in males than females (6). Moreover, at older age, the cutaneous microvascular endothelial function is also reduced in response to local and systemic physiological or pharmacological stimuli (10–12). Cutaneous microvascular reactivity has been shown to be correlated to microvascular function in different vascular beds, both in terms of intensity and regarding the underlying mechanisms (13).
Vascular aging is considered to be the deterioration in arterial structure and function over time, which ultimately leads to damage of the heart, brain, kidney, and other organs (14–16). Nevertheless, it is important to note that individual vascular age may be very different to their chronological age (17). A number of factors contribute to accelerate vascular aging. Demographic features, such as sex and gender, are well-known important modifiers of cardiovascular system pathophysiology and disease development via genetic, epigenetic, and hormonal pathways (18). Other factors, such as a sedentary lifestyle, poor diet, obesity, and smoking can also hasten vascular aging (14).
In fact, the development of premature vascular aging is influenced by different environmental and genetic factors. Exposure to traditional CVD risk factors, including smoking, obesity, hypertension, diabetes, and hypercholesterolemia promote the development and accumulation of sub-clinical vascular changes that undoubtedly contribute to early vascular aging (14). Genetic factors, represented by DNA damage, also play an important causal role in the dysfunction of endothelial and vascular smooth muscle cells (VSMC) during vascular aging (19, 20), which are associated with structural changes including the increase in vascular diameter and the thickening of arterial wall layers (mainly the intima) in large elastic arteries, throughout the lifespan (21). In this context, epidemiological studies suggest an inverse relationship between DNA integrity and age-related CVD (20). Moreover, extensive evidence supports a central role for DNA damage in the development and progression of macrovascular disease, which in turn support the concept that prolonged exposure to risk factors is a major stimulus for genomic instability within the vasculature (22). Also, epidemiological studies showed that intima-medial thickness of the carotid wall increases 2–3 folds between 20 and 90 years of age (23). Finally, epigenetic factors, including DNA methylation, histone-mediated transcriptional regulation and chromatin remodeling, are also involved in the pathophysiology of vascular aging (16).
In the present review, we define aging by a progressive loss of physiological integrity, leading to impaired function and increased vulnerability to death (24), meaning that aging is the overall process of decline in an organism, which involves the accumulation of senescent cells, among other factors. On the other hand, cellular senescence is characterized by cell-cycle arrest, which prevents the proliferation of damaged cells (25). Thus, while aging and senescence are interrelated, senescence is one of the cellular mechanisms contributing to the broader aging process.
This review aims to provide a prospect of the literature regarding the development of ED as an indissociable feature of the aging of the cardiovascular system, highlighting the role of inflammation in the process. In addition, we sought to provide insights to strategies aimed to slowing down vascular senescence.
Inflammation, homeostasis and the molecular origins of vascular senescence
Inflammation, while considered as an acute process comprising cellular and molecular phenomena in animals in response to infections or injury (26), is usually described as an advantageous response. On the other hand, a chronic inflammatory “sterile” state, is often view as a pathological process (27).
Systems homeostasis in animals can be defined as a set of mechanisms which preserve physiological processes within an optimal range, enabling maintenance of structure and function, besides the challenges imposed by changes in the environment (28). Accordingly, acute inflammation triggered against a pathogen or tissue damage is adaptive and aimed to restore organic homeostasis on the long term.
A rather dysfunctional, low grade and long-lasting inflammation is recognized in various chronic age-related diseases, including diabetes, hypertension, and CVD (29). At the molecular level, inflammation triggers are classified as pathogen-associated molecular patterns (PAMPs, which are microbial products) and their sterile counterparts, damage-associated molecular patterns (DAMPs, released through tissue damage). These molecules activate pattern recognition receptors (PRR), which are present mainly on innate immune cells, but are also expressed in endothelial cells (30), leading to leukocyte migration through the vessel wall and activation at the lesion site (29). PRR binding on target cells is followed by the production and release of proinflammatory cytokines [interleukin (IL)-1, tumor necrosis factor (TNF)-α], and dysregulation of cell redox equilibrium (31).
Different groups of highly reactive compounds are central to the understanding of the origins of vascular senescence and disease: reactive oxygen species [ROS, such as hydrogen peroxide (H2O2) and superoxide (O2.−)], reactive nitrogen species [RNS, including nitric oxide (NO) and peroxynitrite], and the less studied reactive sulfur species (RSS, comprising hydrogen sulfide and persulfides, among others). Theses unstable molecules are involved in the regulation of cell injury, death, cardiovascular pathologies, and inflammation (32, 33).
A key concept in the genesis of sterile chronic immune response is the activation against neoantigens, which are a vast array of molecules derived from healthy host tissues, when exposed to noxious stimuli. The mechanisms which generate neoantigens involve biochemical reactions with ROS. The production of ROS is inherent to physiological processes and immune function, being produced through cellular respiration in mitochondria and also by specific enzymes.
The molecular changes induced by ROS represent epigenetic (for DNA) or post-translational (for proteins) modulation, which are fundamental to physiological gene expression and cell signaling (34). However, depending of the ROS concentration, the cell types involved, the targets and chronicity of the stimulus, the result can be deregulation of homeostasis (35).
One important aspect in the genesis of neoantigens and inflammation is the balance between oxidant and antioxidant producers in vasculature. On the pro-oxidant side, the main enzymes producing ROS are nicotinamide adenine dinucleotide phosphate oxidase (Nox), uncoupled endothelial nitric oxide synthase (eNOS), the mitochondrial respiratory system, and xanthine oxidase (XO) (36). Nox catalyzes the synthesis of superoxide and hydrogen peroxide (37), is involved in cell growth and immune activity, and has been extensively associated with ED. The Nox enzymes (isoforms 1–5), were initially characterized in phagocytes of the innate immune response, and later in VSMCs (38), endothelial cells and adventitial fibroblasts (39, 40). The O2.−-generating isoforms Nox 1, 2, and 5, are expressed in endothelial and VSMCs, and have been associated with ED, inflammation within the vessel wall, and intimal thickening (41). Nox4 is unique, since it synthetizes H2O2 and localizes to mitochondria (42).
This set of enzymes has emerged as a primary ROS source in vascular disease and aging. Nox activity is increased in hypertension patients (43) and is upregulated in perivascular tissue upon exposure to cardiovascular risk factors and conditions including smoking (44), diabetes and obesity (45). In a murine model, Nox4 expression in VSMCs increased almost two-fold with aging, which was associated with even higher increase in IL-6 expression (46).
Besides directly generating H2O2, Nox drives in the uncoupling of eNOS, (which starts producing O2.−, instead of the vasodilator NO) (47), and converts the antioxidant xanthine dehydrogenase in the O2.− and H2O2 producer XO (48).
On the other hand, important antioxidant mechanisms act to maintain cellular ROS levels under physiological concentrations in cardiovascular tissue, which have been demonstrated to be suppressed in CVD and aging. Special attention, including as a therapeutic target, has been devoted to the transcription factor nuclear factor erythroid 2-related factor 2 (Nfr2). This molecule upregulates a set of proteins with antioxidant actions, such as nicotinamide adenine dinucleotide phosphate quinone oxidoredutase-1, heme oxygenase 1 (HO-1), and glutathione peroxidase, among others (49). HO-1 degrades heme to produce carbon monoxide, capable of suppressing vasoconstriction through increased cyclic guanosine monophosphate (cGMP) (50).
Nfr2 is a downstream target of the renal protein klotho (KL). This hormone synthetized in the kidneys was originally described as an anti-aging molecule. Mice mutant for the KL gene displayed reduced lifespan and a senescent phenotype, including neurological, cutaneous and metabolic derangements. Accordingly, it was named by the original research authors after the Greek mythology deity Klotho, who spins the thread of life (51). The soluble KL protein was shown to attenuate Angiotensin II-mediated apoptosis and senescence in human aortic VSMCs, via activation of Nrf2 and HO-1 (52).
Both Nfr2 and HO-1 induction have been demonstrated to reduce senescence-related oxidative stress in human cell culture studies, in hyperglycemia and smoking conditions, respectively (53, 54). The cardiovascular protective effects through stimulus of the Nfr2 pathway have been studied with the employment of natural products, such as herbal medicines extracts enriched with flavonoids, terpenoids and phenols (55). Besides enzymatic systems reducing oxidative stress, a growing field of study and possible interventions include microRNAs. For instance, miR92-A, has been demonstrated to induce HO-1 and reduce oxidative stress in human endothelial cells (56).
When ROS react with lipids, a heterogeneous group of molecules, known as lipid peroxidation products (LPO) is generated. The degradation of LPO in turn results in reactive aldehydes, which have longer half-life and can diffuse to adjacent tissues (57). LPO-derived aldehydes can react with lipids, proteins and DNA, resulting in vascular pathology.
Both low-density lipopoproteins (LDL) and high-density lipoproteins, (HDL) traditionally recognized as displaying antagonistic actions in atherosclerosis, can react with LPO. The reaction with the former results in oxidized LDL (oxLDL) particles, which display new modified epitopes, capable of triggering macrophage and adaptive immune responses directed to the vessel wall (58).
OxLDL is capable of binding to a specific PRR in endothelial cells, the Toll-like receptor (TLR-4), which activates the nuclear factor kB (NFκB) transcription factor (59). This pathway in turn upregulates a series of proinflammatory mechanisms, including cytokine and mitochondrial ROS production. In addition, NFκB mediates the priming of the inflammasome NLRP3 (which encodes NOD-, LRR- and pyrin domain-containing protein 3). This inflammasome is of particular interest in CVD, since it can be also activated by cholesterol crystals and free fatty acids (60). The end result is the cleavage of pro-IL1β and pro-IL-18 into their mature forms, and the induction of pyroptosis, an inflammatory form of programmed cell death (61). In a murine model, NLRP3 has been demonstrated as upregulated in aging stem cells when compared with cells from young (62), highlighting the molecular link between chronic diseases and aging. The importance of NLRP3 chronic activation in vascular pathophysiology has prompted interest in the pharmacological inhibition of the pathway. Examples of therapeutic compounds with this purpose include glyburide derivatives, colchicine or specific synthetic inhibitors (63). Cytokines downstream from inflammasome activation have also been considered as direct targets in CVD treatment.
The possible use of anti-inflammatory pharmaceuticals for cardiovascular beneficial effects was highlighted by registry studies reporting lower cardiovascular event rates with patients receiving anti-TNF-α pharmaceuticals for autoimmune diseases (e.g., ertanecept or infliximab) (64). This concept was further explored in the first randomized trial with anti-IL1β designed for primary cardiovascular endpoints, showing a reduction in cardiovascular events in patients with previous myocardial infarction (65).
In contrast, HDL oxidation reverses the usual lipoprotein anti-inflammatory properties into proinflammatory, with oxHDL breakdown products being found in higher concentration in atherosclerotic plaques, when compared with serum levels (66). Besides triggering the recognition of host-derived modified molecules, neoantigens have also being implicated in suppressing self-tolerance to host antigens (67), leading to widespread low-grade persistent immune activation and amplification of oxidative stress. In addition to a wide set of modifications in gene expression, protein synthesis and processing, ROS inactivate NO, the main vasodilation mediator, with NO/ROS imbalance being a crucial mechanism to ED and CVD (68).
One important issue and still subject to debate is to determine in which extent these mechanisms are inherent to “healthy” aging or are unique pathophysiological processes. One perspective is that the senescent phenotype and inflammation are epiphenomena which coexist both in aging and age-related diseases, and act synergically, in a vicious cycle (69). Accordingly, the term “Inflammagin” was coined 25 years ago, to refer to the myriad of long-term chronic inflammatory processes, which occur through life (70).
The role of gut microbiota in vascular dysfunction and aging
The gut microbiota has been recognized as a key regulator of human physiology. The human gastrointestinal tract harbors a vast array of microorganisms (more than 1,000 species in the large intestine) that significantly interact with and affect nutrition, metabolic functions, the immune system, among other increasingly recognized relationships (71). Primarily, the gut microbiota takes part in food digestion and absorption; additionally, in the colon, complex carbohydrates are converted, mainly by bacteria from the Firmicutes phylum, into short chain fatty acids such as n-butyrate, acetate, and propionate, which regulate neutrophil function and migration, reduce colonic mucosal permeability, inhibit inflammatory cytokines, and control the redox environment in the cell (72). Akkermansia muciniphila is one of the short chain fatty acids producers, and its abundance in the gut is associated with several health benefits in humans. These include maintenance of the integrity of the intestinal barrier and decrease of the occurrence of diabetes, obesity, and inflammatory bowel disease, which are associated with epithelial gut damage and high permeability (73).
Direct and indirect roles of gut microbiota have been described on blood pressure regulation, vascular inflammation and stiffening. Dysbiosis, the change of its composition and function, has been linked to the development and progression of numerous disorders, including CVD (74), and is associated with in an increase of trimethylamine (TMA)-producing bacteria, such as Escherichia and Klebsiella. TMA is a compound obtained either directly from diet or formed by the metabolism of dietary choline, carnitine, betaine, and ergothioneine by intestinal microflora in the colon (75). TMA is absorbed from the intestine and oxidized to trimethylamine N-oxide (TMAO), in the liver (76). Both observational and experimental studies suggest a positive correlation between increased plasma TMAO concentrations and CVD, such as myocardial infarction, and stroke (77). TMAO was shown to activate NLRP3 in murine endothelial cells, resulting in vascular oxidative stress and endothelial dysfunction (78). This compound has also been associated to the induction of inflammatory markers in mice and in human aortic endothelial cells and VSMCs, through direct activation of the NF-κB signaling cascade, along with promoting the recruitment of activated leukocytes to endothelial cells (79).
On the other hand, the overgrowth of beneficial bacteria (mainly butyrate producers) is associated with lower arterial stiffness. Menni et al. found that gut microbiome diversity was inversely associated with arterial stiffness, evaluated by carotid-femoral pulse wave velocity (PWV). The abundance of Ruminococcaceae family bacteria, which are beneficial butyrate-producing bacteria linked to lower endotoxemia, was also associated with lower arterial stiffness (80). In a cross-sectional study, Biruete et al. (81) accessed the relationship between the gut microbiota composition and cardiometabolic risk factors in hemodialysis patients. In their study, bacterial abundance was inversely associated with age. The Firmicutes/Bacteroidetes (F/B) ratio was positively associated with traditional risk factors for CVD, such as arterial systolic blood pressure. Also, there was an inverse association between the genus Faecalibacterium and carotid-femoral PWV. The authors note that Faecalibacterium prausnitzii is a butyrate producer, and that might explain this finding, since there is a known inverse correlation between butyrate production and systemic inflammation (82). Finally, plasma concentration of lipopolysaccharide-binding protein, a marker of bacterial translocation, was negatively associated with butyrate-producing bacteria.
The modulation of vascular inflammatory response through aging
In the progression of vascular dysfunction which accompanies aging, the interaction between the immune system and vascular tissue plays a key role. A particular subset of CD4+T cells, known as T regulatory cells (Tregs), has received special interest in vascular pathology. This subpopulation of lymphocytes is important to the maintenance of self-antigen tolerance and has anti-inflammatory actions, through the production of transforming growth factor-β and IL-10 (83). Animal studies have shown that the venous infusion of Tregs prevents angiotensin-II and aldosterone-induced hypertension, oxidative stress and preserves microvascular vasodilation (84, 85). A subsequent human study has suggested that Treg populations are downregulated in hypertensive patients (86).
Remarkably, aging has been associated with Treg dysfunction and reduction in tissue and blood concentrations, a process associated with thymus involution (87). Thymic atrophy is also related to the expansion of senescent proinflammatory CD8+ T cells, which develop increased cytotoxic vascular activity (88).
Another important factor leading to increased inflammation through aging is represented by the adipose tissue, currently view as an endocrine organ regulating metabolic diseases and aging through hormones, known as adipokines. These are immunomodulating and metabolically active mediators produced in the adipose tissue, whose functions are not fully understood, but are regulators of local homeostasis as well as systemic inflammation, as they induce systemic elevation of cytokines (89). Chemerin, an adipokine, is associated with obesity, inflammation, angiogenesis, and metabolic syndrome. Adipose tissue expressing low levels of chemerin display a noninflammatory phenotype (90). Calprotectin is another upregulated adipokine in obesity, which is implicated in inflammation via enhancing adhesion of circulating monocytes or recruitment of macrophages (91). On the other hand, adiponectin is an adipokine with anti-inflammatory actions, which reduces adipose tissue inflammation and improves insulin sensitivity (92).
With aging, adipose tissue distribution and gene expression undergo important changes, with an increase in visceral vs. subcutaneous fat, and with the development of proinflammatory and coagulant gene expression. Dysregulated adipokines, such as very low leptin levels, along with increased TNF-α, are associated with subcutaneous lipoatrophy in the elderly and may determine poor prognosis (93). Conversely, upregulated adiponectin and increased insulin sensitivity are associated with longevity, since centenarians reportedly have increased adiponectin levels, which are inversely correlated with body mass index and body fat percentage (93, 94).
Microvascular endothelial dysfunction (MED) as a precursor of cardiovascular disease
It is currently well-recognized that both microvascular system alterations and subsequent tissue perfusion defects may precede and predict the development of cardiovascular and metabolic diseases, including arterial hypertension, diabetes and metabolic syndrome (95). The microcirculation is a comprehensive term used to describe the terminal vascular network with diameters of less than 300 µm categorized by a network of microvessels including small arteries, arterioles, capillaries, venules, and other cellular components all of which function to meet the oxygen and nutrient requirements of cells, and provide the large surface area needed for blood-tissue exchange (96, 97). Therefore, organs and tissues rely on adequate perfusion by the microcirculation, and suitable microvessel function is essential for the cardiovascular system to achieve this fundamental role (98). Thus, alterations in microvascular function resulting from aging have important consequences for organ function and cardiovascular health (98). Notwithstanding, endothelium-dependent vasodilatation declines with advancing age in humans independently of disease processes (99).
Early microvascular alterations and aging
Systemic microvascular dysfunction is considered to be a marker of CVD that manifests before the appearance of cardiovascular symptoms, and is a hallmark of the biological aging process (100). In the specific situation of coronary microvascular dysfunction, for instance, a systematic review and meta-analysis showed that coronary microvascular dysfunction is associated with a nearly 4-fold increase in mortality and a 5-fold increase in major adverse cardiac events (101).
The study of low birth weight (LBW) infants offers an insight of how early the determinants of CVD can occur. Diverse mechanisms by which LBW leads to the development of CVD have been proposed, and most involve ED. Several clinical studies have established a link between LBW and impaired endothelial function in the regulation of vascular tone in children and young adults (102). Indeed, birth weight has a significant positive correlation with flow-mediated dilation. There is evidence suggesting an association between LBW and dysfunction in the circulating number and functional capacity of endothelial progenitor cells (103).
Endothelium-derived microparticles (EMPs) are submicron anucleate vesicles that are released in response to apoptotic or activation stimuli. Once in circulation, EMPs bind, fuse and transfer microRNAs to target cells, thus acting as vectors that could mediate several biological processes with favorable or deleterious effects on vascular homeostasis (104). EMPs play an important role in vascular repair, degenerative process modulation, endothelial progenitor cell mobilization and differentiation, acting as crucial indicators of endothelial damage. The link between EMPs and endothelial function in children with LBW remains unclear, although evidence supports that early life events may have a negative impact on the endothelial cell. Children who had lower birth weight showed higher numbers of circulating CD31+/annexin V+ and CD144+ EMPs. In addition, LBW and high levels of both CD31+/annexin V+ and LDL-C were significant risk factors for the presence of microvascular ED (105). A possible mechanistic hypothesis is that changes in the epigenetic signatures occurring during fetal life would lead to an endothelial cell injury and trigger the release of EMPs.
Endothelial function declines with age, since aging is associated with senescence of the endothelium due to increased rate of apoptosis and reduced regenerative capacity (106). Endothelial cell senescence is an age-related process linked to ED, and denotes impairment in vasodilatation or reduction in the production of vasodilators (107). In fact, aging is associated with deficits in endothelial NO availability (108, 109) and NO synthase-dependent vasodilation of arterioles (110). ED in aging is also characterized by a reduced angiogenic capacity and increased expression of adhesion molecules, which amplify the interaction with circulating factors and inflammatory cells (111).
Genetic predisposition to microvascular dysfunction can constitute an important issue of the pathophysiology of cardiovascular diseases, including hypertension, and altered microvascular function might be an essential inherited phenotype in hypertensive patients. In this context, previous studies suggested that microvascular alterations can occur early in the development of hypertension (112). It has also been suggested that factors influencing microvascular growth and anatomy are potential candidate mechanisms in the pathogenesis of hypertension (113). In an elegant study by Noon et al., it has been demonstrated that hypertensive individuals who have hypertensive parents inherit attenuated microvascular function (113). In the offspring of parents with high blood pressure, the maximal skin blood flow response to physiological stimuli and capillary recruitment are blunted in those who have high blood pressure compared with those who have low blood pressure (113). Capillary rarefaction is considered to be a primary microvascular alteration in essential arterial hypertension, and also to be pathognomonic of this disease (114). In this context, it has been demonstrated that infants born at term to mothers with hypertensive disorders of pregnancy have significant structural capillary rarefaction at birth, when compared to term infants born to normotensive mothers (114).
The chronic and progressive increase in blood pressure that occur during aging, as a result of macrocirculatory changes, induce vasoconstriction within the microcirculation that promotes tissue hypoxia and decreases arteriolar and capillary density (115). This phenomena lead to further increase in peripheral vascular resistance, establishing a vicious cycle and finishing in both tissue injury and target organ damage (116) and is equally present in senescence and hypertension (117–119).
It is important to mention that the evaluation of systemic microcirculatory function in human beings is only feasible in the skin or mucous membranes, including the sublingual microcirculation. In fact, the cutaneous microcirculation is considered to be not only an accessible vascular bed but also representative of other organ systems, while allowing the evaluation of mechanisms underlying vascular disease (13). Moreover, it is also noteworthy that not all microvessels are perfused under conditions of normal metabolic demand (120) and that the non-perfused portion of the microcirculation represents a reserve that may be recruited in response to varying metabolic requirements (119). The recruitment of this functional microvascular reserve appears to be more difficult in aged organs, due to impairments in the dynamic control of blood flow (121). The existence of an age-dependent microvascular rarefaction, which may be extended and amplified by hypertension, has already been reported in experimental studies (122). Moreover, this capillary rarefaction can be reversed by long-term effective anti-hypertensive treatment (122, 123), probably resulting in the improvement of target organ damage and slowing the development of hypertension (124).
The reduction of microvascular reactivity during aging can be explained by several mechanisms. For instance, endothelium-dependent vasodilatation pharmacologically induced by acetylcholine - a conventional test for endothelial function - decays with advancing age (99). Moreover, aging is associated with deficits in endothelial NO availability, including the human coronary arterioles (108), and NO synthase-dependent vasodilation of microcirculation, via an increase in superoxide produced by the activation of Nox (110). ROS signaling increases with age, through increased expression of Nox (125), and can impair endothelium-dependent vasodilation (126). The availability of collateral circulation also declines in the aging microvasculature. For example, collateral circulation increase by means of pial anastomoses after middle cerebral artery occlusion is reduced in an aged animal model (127).
Interestingly, vasoconstrictive responses are also altered during aging. In spite of the limitation in dynamic responses, age is associated with chronic vasoconstriction mediated by local activation of Rho-kinase signaling (128). Cutaneous vasoconstriction in response to low body temperature is also impaired in aging, a response that is largely dependent on adrenergic stimulation by norepinephrine (128). The adrenergic hypercontractility associated with aging is homogenous among vascular beds, starting at the same age in all the vascular territories without experiencing further significant impairments as age increases (129). Moreover, during aging, ED is due, at least in part, to the release of endothelium-derived contracting factors (EDCF) that counteract the vasodilator effect of NO. Endothelium-dependent contractions involve the activation of endothelial cyclooxygenases and the release of various prostanoids, which activate smooth muscle thromboxane prostanoid receptors of the underlying vascular smooth muscle (130–132). This type of vascular dysregulation is thought to contribute to additional age-related pathologies including hypertension and erectile dysfunction (133).
Lifestyle interventions targeting inflammation and vascular dysfunction
Various lifestyle interventions were studied aiming to modulate the inflammation-vasculature axis. Diet-induced changes of the gut microbiota are linked to changes in arterial stiffness. The concept was corroborated by recent studies, in which arterial stiffness was induced in murine models by fecal transplantation from CVD patients (134, 135). In addition, control mice fed with microbiota from obese mice develop changes in gut permeability and increased PWV (136). In a therapeutical experimental study in rats, soy supplementation shifted the cecal microbiota toward a lower F/B ratio and significantly improved aortic stiffness (137).
In humans, cocoa and chocolate, rich in flavonoids and proanthocyanidins, seem to reduce blood pressure levels and cardiovascular risk, with an improvement in measures of vascular health (arterial stiffness and endothelial function), possibly due to the increased production of NO, and antioxidant/anti-inflammatory properties (138). The impact of cocoa flavonoids on vascular health is mediated by metabolites which depend on gut microbiota metabolism (139). An interesting study of acute soy supplementation showed reduced arterial stiffness mediated by equol (a microbial-derived isoflavone metabolite, produced by gut microbiota after soy intake in some individuals of the Western population) (140).
Antibiotic treatment also induces changes in gut microbiota composition, which have been associated with parallel changes in arterial stiffness. In experimental studies, mice treated with antibiotics have displayed reversal of aortic stiffness induced by the Western diet (141). Surprisingly, antibiotic treatment restored arterial stiffness in old mice to normal levels (142). Nonetheless, the role of antibiotics in the modulation of human microbiota for the improvement of cardiovascular health is uncertain. Added to the fact that antibiotics impact the gut microbiota, reducing bacterial diversity and changing relative abundances, it cannot be recommended, at this time, that this class of drugs be used with that purpose (143).
An additional well studied strategy for improving ED and chronic inflammation that accompanies aging is physical activity. The study of Abd et al. compared the impact of six months of aerobic vs. resisted exercise training on inflammatory cytokines and endothelial activation markers among elderly. Both aerobic and resistance exercise training reduced plasma levels of TNF-α, IL-6 and C-reactive protein (CRP), in addition to elevation of IL-10. These findings suggested that exercise training could modulate systemic inflammation biomarkers in elderly individuals, with more significant changes following aerobic exercise training (144). The same pattern of cytokine modulation was found by Santos et al. who demonstrated that moderate aerobic exercise training for 60 min/day, 3 days/week for six months could improve sleep in elderly, with was associated with anti-inflammatory effects (145).
Hyperglycemia-induced ED and insulin resistance are potent risk factors for CVD, and likely contribute to multiple chronic disease complications associated with aging. Regular physical activity has been recommended as an effective approach, together with medications and dietary control, to improve endothelial function in type 2 diabetes (T2D). Skeletal muscle contraction during physical activity increases local blood flow and cardiac output, which results in increased shear stress on vascular endothelium and increased NO production. The systematic review and metanalysis by Lee et al. support the notion that in T2D patients (average age of 59 years), exercise has beneficial effects on endothelial function, with even better results in low/moderate intensity and aerobic training, when compared to moderate/high intensity and resistance subgroups (146). In a cohort study (average age of 40 years), Hong et al. showed that cardiorespiratory fitness is associated with lower levels of TNF-α and IL-1β. These findings were unrelated to body mass index, which could in part be mediated by enhancing the ability of immune cells to suppress inflammatory responses via adrenergic receptors (147).
Conclusions
Oxidative stress and inflammation are inherent to vascular aging, act through the lifespan of animals, and are probably set since intrauterine development. Mechanisms essential to homeostasis, depending on the local molecular environment and concentration, can become deleterious and induce ED. Both suppressing proinflammatory and enhancing anti-inflammatory pathways in vasculature are promising targets for treatments aiming to mitigate age-related vascular dysfunction and disease. Gut microbiota is an important milieu where a chronic inflammatory state can convey the progression of vascular decline, but also suitable to interventions through nutritional strategies. Regular physical activity has also been proved in multiple studies to reduce inflammation and ED in both young and the elderly, regardless of the intensity. The vast range of mechanisms coexisting in aging and CVD, and which initially cause ED, is summarized in Figure 1. There is extensive experimental and clinical evidence to support the concept that both microvascular network alterations and tissue perfusion defects may precede and predict the development of cardiovascular and metabolic diseases. Moreover, the functional and subsequent structural alterations in both microvascular reactivity and density, as well as the alterations in the macrocirculation characteristic of physiologic vascular aging, contribute to the development of target end-organ damage (148). Therefore, the microcirculation may be considered an essential target of both the pharmacological and the non-pharmacological treatment of arterial hypertension and other CVD (148).
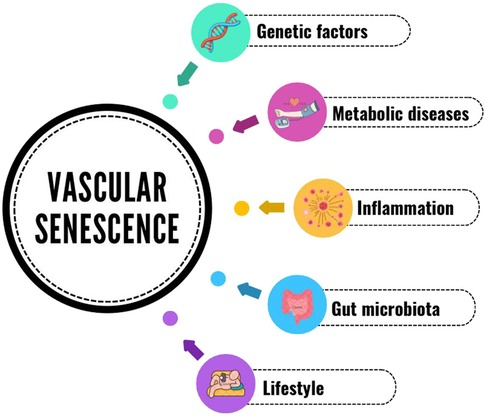
Figure 1. Vascular senescence & disease.
Author contributions
DK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. VS: Data curation, Investigation, Writing – original draft, Writing – review & editing. GH: Data curation, Validation, Visualization, Writing – review & editing. AD: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing. ET: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The publication of this article received financial support by a scientific budget from the Ministry of Health of Brazil, granted to the Professional Master's Program in Cardiovascular Sciences of the National Institute of Cardiology, Rio de Janeiro, Brazil.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Egan BM, Mattix-Kramer HJ, Basile JN, Sutherland SE. Managing hypertension in older adults. Curr Hypertens Rep. (2024) 26(4):157–67. doi: 10.1007/s11906-023-01289-7
2. Li Y, Cao GY, Jing WZ, Liu J, Liu M. Global trends and regional differences in incidence and mortality of cardiovascular disease, 1990–2019: findings from 2019 global burden of disease study. Eur J Prev Cardiol. (2023) 30(3):276–86. doi: 10.1093/eurjpc/zwac285
3. Thijssen DH, Carter SE, Green DJ. Arterial structure and function in vascular ageing: are you as old as your arteries? J Physiol. (2016) 594(8):2275–84. doi: 10.1113/JP270597
4. Vita JA, Treasure CB, Nabel EG, McLenachan JM, Fish RD, Yeung AC, et al. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation. (1990) 81(2):491–7. doi: 10.1161/01.CIR.81.2.491
5. Zeiher AM, Drexler H, Saurbier B, Just H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest. (1993) 92(2):652–62. doi: 10.1172/JCI116634
6. Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Coll Cardiol. (1994) 24(2):471–6. doi: 10.1016/0735-1097(94)90305-0
7. Parker BA, Ridout SJ, Proctor DN. Age and flow-mediated dilation: a comparison of dilatory responsiveness in the brachial and popliteal arteries. Am J Physiol Heart Circ Physiol. (2006) 291(6):H3043–9. doi: 10.1152/ajpheart.00190.2006
8. Black MA, Cable NT, Thijssen DH, Green DJ. Impact of age, sex, and exercise on brachial artery flow-mediated dilatation. Am J Physiol Heart Circ Physiol. (2009) 297(3):H1109–16. doi: 10.1152/ajpheart.00226.2009
9. Thijssen DH, de Groot P, Kooijman M, Smits P, Hopman MT. Sympathetic nervous system contributes to the age-related impairment of flow-mediated dilation of the superficial femoral artery. Am J Physiol Heart Circ Physiol. (2006) 291(6):H3122–9. doi: 10.1152/ajpheart.00240.2006
10. Holowatz LA, Houghton BL, Wong BJ, Wilkins BW, Harding AW, Kenney WL, et al. Nitric oxide and attenuated reflex cutaneous vasodilation in aged skin. Am J Physiol Heart Circ Physiol. (2003) 284(5):H1662–7. doi: 10.1152/ajpheart.00871.2002
11. Tew GA, Klonizakis M, Saxton JM. Effects of ageing and fitness on skin-microvessel vasodilator function in humans. Eur J Appl Physiol. (2010) 109(2):173–81. doi: 10.1007/s00421-009-1342-9
12. Black MA, Green DJ, Cable NT. Exercise prevents age-related decline in nitric-oxide-mediated vasodilator function in cutaneous microvessels. J Physiol. (2008) 586(14):3511–24. doi: 10.1113/jphysiol.2008.153742
13. Holowatz LA, Thompson-Torgerson CS, Kenney WL. The human cutaneous circulation as a model of generalized microvascular function. J Appl Physiol (1985). (2008) 105(1):370–2. doi: 10.1152/japplphysiol.00858.2007
14. Climie RE, Alastruey J, Mayer CC, Schwarz A, Laucyte-Cibulskiene A, Voicehovska J, et al. Vascular ageing: moving from bench towards bedside. Eur J Prev Cardiol. (2023) 30(11):1101–17. doi: 10.1093/eurjpc/zwad028
15. Wang Y, Wang J, Zheng XW, Du MF, Zhang X, Chu C, et al. Early-life cardiovascular risk factor trajectories and vascular aging in midlife: a 30-year prospective cohort study. Hypertension. (2023) 80(5):1057–66. doi: 10.1161/HYPERTENSIONAHA.122.20518
16. González LDM, Romero-Orjuela SP, Rabeya FJ, Del Castillo V, Echeverri D. Age and vascular aging: an unexplored frontier. Front Cardiovasc Med. (2023) 10:1278795. doi: 10.3389/fcvm.2023.1278795
17. Seeland U, Nemcsik J, Lønnebakken MT, Kublickiene K, Schluchter H, Park C, et al. Sex and gender aspects in vascular ageing—focus on epidemiology, pathophysiology, and outcomes. Heart Lung Circ. (2021) 30(11):1637–46. doi: 10.1016/j.hlc.2021.07.006
18. Mauvais-Jarvis F, Bairey Merz N, Barnes PJ, Brinton RD, Carrero JJ, DeMeo DL, et al. Sex and gender: modifiers of health, disease, and medicine. Lancet. (2020) 396(10250):565–82. doi: 10.1016/S0140-6736(20)31561-0
19. Zhang X, Zhao Q, Wang T, Long Q, Sun Y, Jiao L, et al. DNA damage response, a double-edged sword for vascular aging. Ageing Res Rev. (2023) 92:102137. doi: 10.1016/j.arr.2023.102137
20. Bautista-Niño PK, Portilla-Fernandez E, Vaughan DE, Danser AH, Roks AJ. DNA damage: a main determinant of vascular aging. Int J Mol Sci. (2016) 17(5):748–74. doi: 10.3390/ijms17050748
21. Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev. (1993) 73(2):413–67. doi: 10.1152/physrev.1993.73.2.413
22. Shah AV, Bennett MR. DNA damage-dependent mechanisms of ageing and disease in the macro- and microvasculature. Eur J Pharmacol. (2017) 816:116–28. doi: 10.1016/j.ejphar.2017.03.050
23. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. (2003) 107(1):139–46. doi: 10.1161/01.CIR.0000048892.83521.58
24. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
25. Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol. (2022) 18(10):611–27. doi: 10.1038/s41581-022-00601-z
26. Medzhitov R. Origin and physiological roles of inflammation. Nature. (2008) 454(7203):428–35. doi: 10.1038/nature07201
27. Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. (2010) 267(6):543–60. doi: 10.1111/j.1365-2796.2010.02218.x
28. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. (2015) 160(5):816–27. doi: 10.1016/j.cell.2015.02.010
29. Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation-nature’s way to efficiently respond to all types of challenges: implications for understanding and managing “the epidemic” of chronic diseases. Front Med (Lausanne). (2018) 5:316. doi: 10.3389/fmed.2018.00316
30. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell toll-like receptor pathways in sepsis. Innate Immun. (2015) 21(8):827–46. doi: 10.1177/1753425915606525
31. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
32. Kolluru GK, Shen X, Kevil CG. Reactive sulfur species: a new redox player in cardiovascular pathophysiology. Arterioscler Thromb Vasc Biol. (2020) 40(4):874–84. doi: 10.1161/ATVBAHA.120.314084
33. Weidinger A, Kozlov AV. Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules. (2015) 5(2):472–84. doi: 10.3390/biom5020472
34. Miyashita H, Chikazawa M, Otaki N, Hioki Y, Shimozu Y, Nakashima F, et al. Lysine pyrrolation is a naturally-occurring covalent modification involved in the production of DNA mimic proteins. Sci Rep. (2014) 4:5343. doi: 10.1038/srep05343
35. DeVallance E, Li Y, Jurczak MJ, Cifuentes-Pagano E, Pagano PJ. The role of NADPH oxidases in the etiology of obesity and metabolic syndrome: contribution of individual isoforms and cell biology. Antioxid Redox Signal. (2019) 31(10):687–709. doi: 10.1089/ars.2018.7674
36. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. (2000) 87(10):840–4. doi: 10.1161/01.RES.87.10.840
37. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. (2007) 87(1):245–313. doi: 10.1152/physrev.00044.2005
38. Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, et al. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res. (2002) 90(11):1205–13. doi: 10.1161/01.RES.0000020404.01971.2F
39. Petry A, Djordjevic T, Weitnauer M, Kietzmann T, Hess J, Görlach A. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal. (2006) 8(9-10):1473–84. doi: 10.1089/ars.2006.8.1473
40. Chamseddine AH, Miller FJ. Gp91phox contributes to NADPH oxidase activity in aortic fibroblasts but not smooth muscle cells. Am J Physiol Heart Circ Physiol. (2003) 285(6):H2284–9. doi: 10.1152/ajpheart.00459.2003
41. Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. (2012) 110(10):1364–90. doi: 10.1161/CIRCRESAHA.111.243972
42. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. (2009) 106(34):14385–90. doi: 10.1073/pnas.0906805106
43. Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens. (2001) 19(7):1245–54. doi: 10.1097/00004872-200107000-00009
44. Chang KH, Park JM, Lee CH, Kim B, Choi KC, Choi SJ, et al. NADPH oxidase (NOX) 1 mediates cigarette smoke-induced superoxide generation in rat vascular smooth muscle cells. Toxicol in Vitro. (2017) 38:49–58. doi: 10.1016/j.tiv.2016.10.013
45. Antonopoulos AS, Margaritis M, Coutinho P, Shirodaria C, Psarros C, Herdman L, et al. Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: the regulatory role of perivascular adipose tissue. Diabetes. (2015) 64(6):2207–19. doi: 10.2337/db14-1011
46. Lozhkin A, Vendrov AE, Pan H, Wickline SA, Madamanchi NR, Runge MS. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J Mol Cell Cardiol. (2017) 102:10–21. doi: 10.1016/j.yjmcc.2016.12.004
47. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. (2003) 111(8):1201–9. doi: 10.1172/JCI200314172
48. McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, et al. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. (2003) 285(6):H2290–7. doi: 10.1152/ajpheart.00515.2003
49. Zhu L, He S, Huang L, Ren D, Nie T, Tao K, et al. Chaperone-mediated autophagy degrades Keap1 and promotes Nrf2-mediated antioxidative response. Aging Cell. (2022) 21(6):e13616. doi: 10.1111/acel.13616
50. Álvarez-Maestro M, Eguibar A, Chanca P, Klett-Mingo M, Gómez Rivas J, Buño-Soto A, et al. Androgen deprivation therapy in patients with prostate cancer increases serum levels of thromboxane A2: cardiovascular implications. Front Cardiovasc Med. (2021) 8:653126. doi: 10.3389/fcvm.2021.653126
51. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. (1997) 390(6655):45–51. doi: 10.1038/36285
52. Maltese G, Psefteli PM, Rizzo B, Srivastava S, Gnudi L, Mann GE, et al. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J Cell Mol Med. (2017) 21(3):621–7. doi: 10.1111/jcmm.12996
53. Even B, Fayad-Kobeissi S, Gagliolo JM, Motterlini R, Boczkowski J, Foresti R, et al. Heme oxygenase-1 induction attenuates senescence in chronic obstructive pulmonary disease lung fibroblasts by protecting against mitochondria dysfunction. Aging Cell. (2018) 17(6):e12837. doi: 10.1111/acel.12837
54. Senthil KKJ, Gokila VM, Wang SY. Activation of Nrf2-mediated anti-oxidant genes by antrodin C prevents hyperglycemia-induced senescence and apoptosis in human endothelial cells. Oncotarget. (2017) 8(57):96568–87. doi: 10.18632/oncotarget.19951
55. Wu X, Wei J, Yi Y, Gong Q, Gao J. Activation of Nrf2 signaling: a key molecular mechanism of protection against cardiovascular diseases by natural products. Front Pharmacol. (2022) 13:1057918. doi: 10.3389/fphar.2022.1057918
56. Gou L, Zhao L, Song W, Wang L, Liu J, Zhang H, et al. Inhibition of miR-92a suppresses oxidative stress and improves endothelial function by upregulating heme oxygenase-1 in db/db mice. Antioxid Redox Signal. (2018) 28(5):358–70. doi: 10.1089/ars.2017.7005
57. Gentile F, Arcaro A, Pizzimenti S, Daga M, Cetrangolo GP, Dianzani C, et al. DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genet. (2017) 4(2):103–37. doi: 10.3934/genet.2017.2.103
58. Hartley A, Haskard D, Khamis R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis—novel insights and future directions in diagnosis and therapy. Trends Cardiovasc Med. (2019) 29(1):22–6. doi: 10.1016/j.tcm.2018.05.010
59. Masters SL, Latz E, O'Neill LA. The inflammasome in atherosclerosis and type 2 diabetes. Sci Transl Med. (2011) 3(81):81ps17. doi: 10.1126/scitranslmed.3001902
60. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12(5):408–15. doi: 10.1038/ni.2022
61. Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. (2012) 150(3):606–19. doi: 10.1016/j.cell.2012.07.007
62. Luo H, Mu WC, Karki R, Chiang HH, Mohrin M, Shin JJ, et al. Mitochondrial stress-initiated aberrant activation of the NLRP3 inflammasome regulates the functional deterioration of hematopoietic stem cell aging. Cell Rep. (2019) 26(4):945–954.e4. doi: 10.1016/j.celrep.2018.12.101
63. Toldo S, Mezzaroma E, Buckley LF, Potere N, Di Nisio M, Biondi-Zoccai G, et al. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol Ther. (2022) 236:108053. doi: 10.1016/j.pharmthera.2021.108053
64. Greenberg JD, Kremer JM, Curtis JR, Hochberg MC, Reed G, Tsao P, et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. (2011) 70(4):576–82. doi: 10.1136/ard.2010.129916
65. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Anti-inflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
66. Bergt C, Pennathur S, Fu X, Byun J, O'Brien K, McDonald TO, et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci U S A. (2004) 101(35):13032–7. doi: 10.1073/pnas.0405292101
67. Mustelin T, Andrade F. Autoimmunity: the neoantigen hypothesis. Front Immunol. (2024) 15:1432985. doi: 10.3389/fimmu.2024.1432985
68. Tejero J, Shiva S, Gladwin MT. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol Rev. (2019) 99(1):311–79. doi: 10.1152/physrev.00036.2017
69. Li X, Li C, Zhang W, Wang Y, Qian P, Huang H. Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct Target Ther. (2023) 8(1):239. doi: 10.1038/s41392-023-01502-8
70. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
71. Lozupone C, Faust K, Raes J, Faith JJ, Frank DN, Zaneveld J, et al. Identifying genomic and metabolic features that can underlie early successional and opportunistic lifestyles of human gut symbionts. Genome Res. (2012) 22(10):1974–84. doi: 10.1101/gr.138198.112
72. Levine UY, Looft T, Allen HK, Stanton TB. Butyrate-producing bacteria, including mucin degraders, from the swine intestinal tract. Appl Environ Microbiol. (2013) 79(12):3879–81. doi: 10.1128/AEM.00589-13
73. Ghaffari S, Abbasi A, Somi MH, Moaddab SY, Nikniaz L, Kafil HS, et al. From its critical role in human health to strategies for promoting its abundance in human gut microbiome. Crit Rev Food Sci Nutr. (2023) 63(25):7357–77. doi: 10.1080/10408398.2022.2045894
74. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. (2018) 24(4):392–400. doi: 10.1038/nm.4517
75. Zeisel SH, Mar MH, Howe JC, Holden JM. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. (2003) 133(5):1302–7. doi: 10.1093/jn/133.5.1302
76. Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. (2013) 17(1):49–60. doi: 10.1016/j.cmet.2012.12.011
77. Zeisel SH, Warrier M. Trimethylamine N-oxide, the microbiome, and heart and kidney disease. Annu Rev Nutr. (2017) 37:157–81. doi: 10.1146/annurev-nutr-071816-064732
78. Boini KM, Hussain T, Li PL, Koka S. Trimethylamine-N-oxide instigates NLRP3 inflammasome activation and endothelial dysfunction. Cell Physiol Biochem. (2017) 44(1):152–62. doi: 10.1159/000484623
79. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc. (2016) 5(2):e002767–779. doi: 10.1161/JAHA.115.002767
80. Menni C, Lin C, Cecelja M, Mangino M, Matey-Hernandez ML, Keehn L, et al. Gut microbial diversity is associated with lower arterial stiffness in women. Eur Heart J. (2018) 39(25):2390–7. doi: 10.1093/eurheartj/ehy226
81. Biruete A, Allen JM, Kistler BM, Jeong JH, Fitschen PJ, Swanson KS, et al. Gut microbiota and cardiometabolic risk factors in hemodialysis patients: a pilot study. Top Clin Nutr. (2019) 34(2):153–60. doi: 10.1097/TIN.0000000000000170
82. Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE, et al. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. (2016) 48(11):826–34. doi: 10.1152/physiolgenomics.00089.2016
83. Mallat Z, Ait-Oufella H, Tedgui A. Regulatory T-cell immunity in atherosclerosis. Trends Cardiovasc Med. (2007) 17(4):113–8. doi: 10.1016/j.tcm.2007.03.001
84. Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. (2011) 57(3):469–76. doi: 10.1161/HYPERTENSIONAHA.110.162941
85. Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. (2012) 59(2):324–30. doi: 10.1161/HYPERTENSIONAHA.111.181123
86. Chen ZY, Chen F, Wang YG, Wang DH, Jang LL, Cheng LX. Down-regulation of helios expression in tregs from patients with hypertension. Curr Med Sci. (2018) 38(1):58–63. doi: 10.1007/s11596-018-1846-9
87. Palatella M, Guillaume SM, Linterman MA, Huehn J. The dark side of tregs during aging. Front Immunol. (2022) 13:940705. doi: 10.3389/fimmu.2022.940705
88. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. (2015) 116(6):1022–33. doi: 10.1161/CIRCRESAHA.116.303697
89. Lago F, Dieguez C, Gómez-Reino J, Gualillo O. Adipokines as emerging mediators of immune response and inflammation. Nat Clin Pract Rheumatol. (2007) 3(12):716–24. doi: 10.1038/ncprheum0674
90. Bozaoglu K, Curran JE, Stocker CJ, Zaibi MS, Segal D, Konstantopoulos N, et al. Chemerin, a novel adipokine in the regulation of angiogenesis. J Clin Endocrinol Metab. (2010) 95(5):2476–85. doi: 10.1210/jc.2010-0042
91. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, et al. Increased levels of calprotectin in obesity are related to macrophage content: impact on inflammation and effect of weight loss. Mol Med. (2011) 17(11-12):1157–67. doi: 10.2119/molmed.2011.00144
92. Engeli S, Feldpausch M, Gorzelniak K, Hartwig F, Heintze U, Janke J, et al. Association between adiponectin and mediators of inflammation in obese women. Diabetes. (2003) 52(4):942–7. doi: 10.2337/diabetes.52.4.942
93. Arai Y, Takayama M, Abe Y, Hirose N. Adipokines and aging. J Atheroscler Thromb. (2011) 18(7):545–50. doi: 10.5551/jat.7039
94. Atzmon G, Pollin TI, Crandall J, Tanner K, Schechter CB, Scherer PE, et al. Adiponectin levels and genotype: a potential regulator of life span in humans. J Gerontol A Biol Sci Med Sci. (2008) 63(5):447–53. doi: 10.1093/gerona/63.5.447
95. Kovacic JC, Moreno P, Nabel EG, Hachinski V, Fuster V. Cellular senescence, vascular disease, and aging: part 2 of a 2-part review: clinical vascular disease in the elderly. Circulation. (2011) 123(17):1900–10. doi: 10.1161/CIRCULATIONAHA.110.009118
96. Guven G, Hilty MP, Ince C. Microcirculation: physiology, pathophysiology, and clinical application. Blood Purif. (2020) 49(1-2):143–50. doi: 10.1159/000503775
97. Secomb TW, Pries AR. The microcirculation: physiology at the mesoscale. J Physiol. (2011) 589(Pt 5):1047–52. doi: 10.1113/jphysiol.2010.201541
98. Gates PE, Strain WD, Shore AC. Human endothelial function and microvascular ageing. Exp Physiol. (2009) 94(3):311–6. doi: 10.1113/expphysiol.2008.043349
99. DeSouza CA, Clevenger CM, Greiner JJ, Smith DT, Hoetzer GL, Shapiro LF, et al. Evidence for agonist-specific endothelial vasodilator dysfunction with ageing in healthy humans. J Physiol. (2002) 542(Pt 1):255–62. doi: 10.1113/jphysiol.2002.019166
100. SenthilKumar G, Gutierrez-Huerta CA, Freed JK, Beyer AM, Fancher IS, LeBlanc AJ. New developments in translational microcirculatory research. Am J Physiol Heart Circ Physiol. (2022) 323(6):H1167–H75. doi: 10.1152/ajpheart.00566.2022
101. Gdowski MA, Murthy VL, Doering M, Monroy-Gonzalez AG, Slart R, Brown DL. Association of isolated coronary microvascular dysfunction with mortality and Major adverse cardiac events: a systematic review and meta-analysis of aggregate data. J Am Heart Assoc. (2020) 9(9):e014954. doi: 10.1161/JAHA.119.014954
102. Joo Turoni C, Chaila Z, Chahla R, Bazán de Casella MC, Peral de Bruno M. Vascular function in children with low birthweight and its relationship with early markers of cardiovascular risk. Horm Res Paediatr. (2016) 85(6):396–405. doi: 10.1159/000445949
103. Souza LV, De Meneck F, Oliveira V, Higa EM, Akamine EH, Franco MDC. Detrimental impact of low birth weight on circulating number and functional capacity of endothelial progenitor cells in healthy children: role of angiogenic factors. J Pediatr. (2019) 206:72–77.e1. doi: 10.1016/j.jpeds.2018.10.040
104. Shu Z, Tan J, Miao Y, Zhang Q. The role of microvesicles containing microRNAs in vascular endothelial dysfunction. J Cell Mol Med. (2019) 23(12):7933–45. doi: 10.1111/jcmm.14716
105. Parizotto GP, de Souza LV, Thomazini F, Prado MS, Agudelo JSH, de Almeida DC, et al. Birth weight and its relationship with endothelial function and pattern of endothelium-derived microparticles during childhood: new insight about early vascular damage. Life Sci. (2022) 298:120517. doi: 10.1016/j.lfs.2022.120517
106. Savoia C, Battistoni A, Calvez V, Cesario V, Montefusco G, Filippini A. Microvascular alterations in hypertension and vascular aging. Curr Hypertens Rev. (2017) 13(1):16–23. doi: 10.2174/1573402113666170505115010
107. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. (2018) 123(7):825–48. doi: 10.1161/CIRCRESAHA.118.312563
108. Feher A, Broskova Z, Bagi Z. Age-related impairment of conducted dilation in human coronary arterioles. Am J Physiol Heart Circ Physiol. (2014) 306(12):H1595–601. doi: 10.1152/ajpheart.00179.2014
109. Behringer EJ. Impact of aging on vascular ion channels: perspectives and knowledge gaps across major organ systems. Am J Physiol Heart Circ Physiol. (2023) 325(5):H1012–38. doi: 10.1152/ajpheart.00288.2023
110. Mayhan WG, Arrick DM, Sharpe GM, Sun H. Age-related alterations in reactivity of cerebral arterioles: role of oxidative stress. Microcirculation. (2008) 15(3):225–36. doi: 10.1080/10739680701641421
111. Scioli MG, Bielli A, Arcuri G, Ferlosio A, Orlandi A. Ageing and microvasculature. Vasc Cell. (2014) 6:19. doi: 10.1186/2045-824X-6-19
112. Sullivan JM, Prewitt RL, Josephs JA. Attenuation of the microcirculation in young patients with high-output borderline hypertension. Hypertension. (1983) 5(6):844–51. doi: 10.1161/01.HYP.5.6.844
113. Noon JP, Walker BR, Webb DJ, Shore AC, Holton DW, Edwards HV, et al. Impaired microvascular dilatation and capillary rarefaction in young adults with a predisposition to high blood pressure. J Clin Invest. (1997) 99(8):1873–9. doi: 10.1172/JCI119354
114. Antonios TF, Raghuraman RP, D’Souza R, Nathan P, Wang D, Manyonda IT. Capillary remodeling in infants born to hypertensive pregnancy: pilot study. Am J Hypertens. (2012) 25(8):848–53. doi: 10.1038/ajh.2012.51
115. Feihl F, Liaudet L, Levy BI, Waeber B. Hypertension and microvascular remodelling. Cardiovasc Res. (2008) 78(2):274–85. doi: 10.1093/cvr/cvn022
116. Yannoutsos A, Levy BI, Safar ME, Slama G, Blacher J. Pathophysiology of hypertension. J Hypertens. (2014) 32(2):216–24. doi: 10.1097/HJH.0000000000000021
117. Paneni F, Costantino S, Cosentino F. Molecular pathways of arterial aging. Clin Sci (Lond). (2015) 128(2):69–79. doi: 10.1042/CS20140302
118. Muris DM, Houben AJ, Kroon AA, Henry RM, van der Kallen CJ, Sep SJ, et al. Age, waist circumference, and blood pressure are associated with skin microvascular flow motion: the Maastricht study. J Hypertens. (2014) 32(12):2439–49. discussion 49. doi: 10.1097/HJH.0000000000000348
119. Levy BI, Schiffrin EL, Mourad JJ, Agostini D, Vicaut E, Safar ME, et al. Impaired tissue perfusion. Circulation. (2008) 118(9):968–76. doi: 10.1161/CIRCULATIONAHA.107.763730
120. Chade AR. Renal vascular structure and rarefaction. Compr Physiol. (2013) 3(2):817–31. doi: 10.1002/cphy.c120012
121. Jackson DN, Moore AW, Segal SS. Blunting of rapid onset vasodilatation and blood flow restriction in arterioles of exercising skeletal muscle with ageing in male mice. J Physiol. (2010) 588(Pt 12):2269–82. doi: 10.1113/jphysiol.2010.189811
122. Henrich HA, Geyer P, Conrad F, Silber RE. Age-related and pathophysiologic significance of capillary bed rarefaction of the hypertensive vascular system. Z Gerontol. (1990) 23(3):147–51.2392871
123. Sabino B, Lessa MA, Nascimento AR, Rodrigues CA, Henriques M, Garzoni LR, et al. Effects of antihypertensive drugs on capillary rarefaction in spontaneously hypertensive rats: intravital microscopy and histologic analysis. J Cardiovasc Pharmacol. (2008) 51(4):402–9. doi: 10.1097/FJC.0b013e3181673bc5
124. Battegay EJ, de Miguel LS, Petrimpol M, Humar R. Effects of anti-hypertensive drugs on vessel rarefaction. Curr Opin Pharmacol. (2007) 7(2):151–7. doi: 10.1016/j.coph.2006.09.007
125. Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, et al. Direct evidence of endothelial oxidative stress with aging in humans. Circ Res. (2007) 100(11):1659–66. doi: 10.1161/01.RES.0000269183.13937.e8
126. Sindler AL, Reyes R, Chen B, Ghosh P, Gurovich AN, Kang LS, et al. Age and exercise training alter signaling through reactive oxygen species in the endothelium of skeletal muscle arterioles. J Appl Physiol (1985). (2013) 114(5):681–93. doi: 10.1152/japplphysiol.00341.2012
127. Yamaguchi S, Kobayashi S, Murata A, Yamashita K, Tsunematsu T. Effect of aging on collateral circulation via pial anastomoses in cats. Gerontology. (1988) 34(4):157–64. doi: 10.1159/000212946
128. Thompson CS, Kenney WL. Altered neurotransmitter control of reflex vasoconstriction in aged human skin. J Physiol. (2004) 558(Pt 2):697–704. doi: 10.1113/jphysiol.2004.065714
129. Assar ME, Angulo J, García-Rojo E, Sevilleja-Ortiz A, García-Gómez B, Fernández A, et al. Early manifestation of aging-related vascular dysfunction in human penile vasculature-A potential explanation for the role of erectile dysfunction as a harbinger of systemic vascular disease. Geroscience. (2022) 44(1):485–501. doi: 10.1007/s11357-021-00507-x
130. Félétou M, Huang Y, Vanhoutte PM. Vasoconstrictor prostanoids. Pflugers Arch. (2010) 459(6):941–50. doi: 10.1007/s00424-010-0812-6
131. Félétou M, Vanhoutte PM, Verbeuren TJ. The thromboxane/endoperoxide receptor (TP): the common villain. J Cardiovasc Pharmacol. (2010) 55(4):317–32. doi: 10.1097/FJC.0b013e3181d8bc8a
132. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease—a 30th anniversary update. Acta Physiol (Oxf). (2017) 219(1):22–96. doi: 10.1111/apha.12646
133. Nunes KP, Rigsby CS, Webb RC. Rhoa/rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. (2010) 67(22):3823–36. doi: 10.1007/s00018-010-0460-1
134. Trikha SRJ, Lee DM, Ecton KE, Wrigley SD, Vazquez AR, Litwin NS, et al. Transplantation of an obesity-associated human gut microbiota to mice induces vascular dysfunction and glucose intolerance. Gut Microbes. (2021) 13(1):1940791. doi: 10.1080/19490976.2021.1940791
135. Liu H, Tian R, Wang H, Feng S, Li H, Xiao Y, et al. Gut microbiota from coronary artery disease patients contributes to vascular dysfunction in mice by regulating bile acid metabolism and immune activation. J Transl Med. (2020) 18(1):382. doi: 10.1186/s12967-020-02539-x
136. Battson ML, Lee DM, Li Puma LC, Ecton KE, Thomas KN, Febvre HP, et al. Gut microbiota regulates cardiac ischemic tolerance and aortic stiffness in obesity. Am J Physiol Heart Circ Physiol. (2019) 317(6):H1210–H20. doi: 10.1152/ajpheart.00346.2019
137. Cross TL, Zidon TM, Welly RJ, Park YM, Britton SL, Koch LG, et al. Soy improves cardiometabolic health and cecal Microbiota in female low-fit rats. Sci Rep. (2017) 7(1):9261. doi: 10.1038/s41598-017-08965-0
138. Ludovici V, Barthelmes J, Nägele MP, Enseleit F, Ferri C, Flammer AJ, et al. Cocoa, blood pressure, and vascular function. Front Nutr. (2017) 4:36. doi: 10.3389/fnut.2017.00036
139. Rodriguez-Mateos A, Weber T, Skene SS, Ottaviani JI, Crozier A, Kelm M, et al. Assessing the respective contributions of dietary flavanol monomers and procyanidins in mediating cardiovascular effects in humans: randomized, controlled, double-masked intervention trial. Am J Clin Nutr. (2018) 108(6):1229–37. doi: 10.1093/ajcn/nqy229
140. Hazim S, Curtis PJ, Schär MY, Ostertag LM, Kay CD, Minihane AM, et al. Acute benefits of the microbial-derived isoflavone metabolite equol on arterial stiffness in men prospectively recruited according to equol producer phenotype: a double-blind randomized controlled trial. Am J Clin Nutr. (2016) 103(3):694–702. doi: 10.3945/ajcn.115.125690
141. Battson ML, Lee DM, Jarrell DK, Hou S, Ecton KE, Weir TL, et al. Suppression of gut dysbiosis reverses western diet-induced vascular dysfunction. Am J Physiol Endocrinol Metab. (2018) 314(5):E468–E77. doi: 10.1152/ajpendo.00187.2017
142. Brunt VE, Gioscia-Ryan RA, Richey JJ, Zigler MC, Cuevas LM, Gonzalez A, et al. Suppression of the gut microbiome ameliorates age-related arterial dysfunction and oxidative stress in mice. J Physiol. (2019) 597(9):2361–78. doi: 10.1113/JP277336
143. Elvers KT, Wilson VJ, Hammond A, Duncan L, Huntley AL, Hay AD, et al. Antibiotic-induced changes in the human gut microbiota for the most commonly prescribed antibiotics in primary care in the UK: a systematic review. BMJ Open. (2020) 10(9):e035677. doi: 10.1136/bmjopen-2019-035677
144. Abd El-Kader SM, Al-Shreef FM, Al-Jiffri OH. Impact of aerobic exercise versus resisted exercise on endothelial activation markers and inflammatory cytokines among elderly. Afr Health Sci. (2019) 19(4):2874–80. doi: 10.4314/ahs.v19i4.9
145. Santos RV, Viana VA, Boscolo RA, Marques VG, Santana MG, Lira FS, et al. Moderate exercise training modulates cytokine profile and sleep in elderly people. Cytokine. (2012) 60(3):731–5. doi: 10.1016/j.cyto.2012.07.028
146. Lee JH, Lee R, Hwang MH, Hamilton MT, Park Y. The effects of exercise on vascular endothelial function in type 2 diabetes: a systematic review and meta-analysis. Diabetol Metab Syndr. (2018) 10:15. doi: 10.1186/s13098-018-0316-7
147. Hong S, Dimitrov S, Pruitt C, Shaikh F, Beg N. Benefit of physical fitness against inflammation in obesity: role of beta adrenergic receptors. Brain Behav Immun. (2014) 39:113–20. doi: 10.1016/j.bbi.2013.12.009
Keywords: microvascular endothelial dysfunction, inflammation, aging, cardiovascular disease, oxidative stress
Citation: Kasal DA, Sena V, Huguenin GVB, De Lorenzo A and Tibirica E (2025) Microvascular endothelial dysfunction in vascular senescence and disease. Front. Cardiovasc. Med. 12:1505516. doi: 10.3389/fcvm.2025.1505516
Received: 3 October 2024; Accepted: 21 January 2025;
Published: 18 February 2025.
Edited by:
Tlili Barhoumi, King Abdullah International Medical Research Center (KAIMRC), Saudi ArabiaReviewed by:
Tiago J. Costa, University of São Paulo, BrazilMingyi Wang, National Institute on Aging (NIH), United States
Copyright: © 2025 Kasal, Sena, Huguenin, De Lorenzo and Tibirica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel A. Kasal, ZGFuaWVsLmthc2FsQGluYy5zYXVkZS5nb3YuYnI=
†ORCID:
Daniel A. Kasal
orcid.org/0000-0002-6931-0950
Viviane Sena
orcid.org/0009-0003-2340-9239
Grazielle Vilas Bôas Huguenin
orcid.org/0000-0002-0553-4939
Andrea De Lorenzo
orcid.org/0000-0001-8522-6612
Eduardo Tibirica
orcid.org/0000-0002-3406-9300