
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Cardiovasc. Med. , 20 March 2025
Sec. Sex and Gender in Cardiovascular Medicine
Volume 12 - 2025 | https://doi.org/10.3389/fcvm.2025.1502985
This article is part of the Research Topic Sex-Specific Risk Factors and Cardiovascular Disease Risk in Women View all 9 articles
 Ava P. Aminbakhsh1,†
Ava P. Aminbakhsh1,† Emilie T. Théberge2,†
Emilie T. Théberge2,† Elizabeth Burden3,4Cindy Kalenga Adejumo3,4Annabel K. Gravely1Anna Lehman2,4
Elizabeth Burden3,4Cindy Kalenga Adejumo3,4Annabel K. Gravely1Anna Lehman2,4 Tara L. Sedlak4,5*
Tara L. Sedlak4,5*
Introduction: Coronary artery disease (CAD) is the leading cause of death around the world, with epidemiological sex and gender differences in prevalence, pathophysiology and outcomes. It has been hypothesized that sex steroids, like estrogen, may contribute to these sex differences. There is a relatively large genetic component to developing CAD, with heritability estimates ranging between 40%–60%. In the last two decades, genome-wide association studies (GWAS) have contributed substantially to advancing the understanding of genetic candidates contributing to CAD. The aim of this study was to determine if genes discovered in CAD GWASs are affected by estrogen via direct modulation or indirect down-stream targets.
Methods: A scoping review was conducted using MEDLINE and EMBASE for studies of atherosclerotic coronary artery disease and a genome-wide association study (GWAS) design. Analysis was limited to candidate genes with corresponding single nucleotide polymorphisms (SNPs) surpassing genome-wide significance and had been mapped to genes by study authors. The number of studies that conducted sex-stratified analyses with significant genes were quantified. A literature search of the final gene lists was done to examine any evidence suggesting estrogen may modulate the genes and/or gene products.
Results: There were 60 eligible CAD GWASs meeting inclusion criteria for data extraction. Of these 60, only 36 had genome-wide significant SNPs reported, and only 3 of these had significant SNPs from sex-stratified analyses mapped to genes. From these 36 studies, a total of 61 genes were curated, of which 26 genes (43%) were found to have modulation by estrogen. All 26 were discovered in studies that adjusted for sex. 12/26 genes were also discovered in studies that conducted sex-stratified analyses. 12/26 genes were classified as having a role in lipid synthesis, metabolism and/or lipoprotein mechanisms, while 11/26 were classified as having a role in vascular integrity, and 3/26 were classified as having a role in thrombosis.
Discussion: This study provides further evidence of the relationship between estrogen, genetic risk and the development of CAD. More sex-stratified research will need to be conducted to further characterize estrogen's relation to sex differences in the pathology and progression of CAD.
Coronary artery disease (CAD) is the leading cause of death around the world (1), with well-described epidemiological sex and gender differences in prevalence, pathophysiology, and management outcomes (2) (Box 1). CAD is attributed to atherosclerosis, where lesions (atheromas or atherosclerotic plaques) form in the luminal intima of coronary arteries. These lesions develop over time due to a complex interplay of risk factors within a state of chronic inflammation (3). Sex differences have been studied in the development and progression of atherosclerosis but are not completely understood (4). For instance, cisgender women have been found to have smaller coronary arteries and are more likely to experience plaque erosion, whereas men are more prone to plaque rupture (5).

Box 1. Distinguishing sex and gender definitions as outlined in the Canadian Institutes for Health Research Panel on Sex and Gender.
Sex differences in the presentation of CAD include the prevalence of traditional cardiovascular risk factors and a later average onset in women, approximately 10 years later than in men (6). Higher sex-specific risk in developing CAD has been observed in women who have diabetes, a history of smoking, depression and/or anxiety. Unique risk factors of women include sex-specific conditions such as premature menopause, gestational hypertension or preeclampsia, and polycystic ovarian syndrome (PCOS) (7–9). In pre- and peri-menopause, the risk of CAD is perceived to be lower due to the protective role of estrogens (10).
Cardiovascular health is modulated in part by the steroid sex hormones: androgens, estrogens and progestogens, among which the most abundant subtypes are testosterone, 17β -estradiol and progesterone, respectively (6, 11). These hormones bind to receptors expressed on the surface of most cell types in the cardiovascular system in both sexes, exerting physiological effects through regulation of genomic expression (“slow response”, spanning hours to days), or acting on extranuclear components in the cell (“fast non-genomic” response, spanning seconds to minutes) (12, 13). Estrogen has been shown to exert both direct and indirect regulatory effects on thousands of genes (14). Protective physiological effects include the promotion of vasodilation, anti-inflammatory cascades, and improvement of lipid profiles such as through decreased low-density lipoprotein (LDL) oxidation and binding (15–18). Theoretically, deleterious genetic variations in genes regulated by estrogen and with implications in cardiovascular health could increase the predisposition for CAD.
There are sex differences in physiologically normal ranges of androgen to estrogen ratios, which further differ by age (i.e., puberty, menopause, andropause) (19, 20). In women, premenopausal estradiol ranges from 30 to 400 pg/ml while postmenopausal estradiol ranges from 0 to 20 pg/ml (21, 22), while androgen levels mostly constant but gradually decrease over the course of a lifetime, and not to the rate or degree of estrogen decline (23). As such, the ratio of androgen to estrogens differs greatly between pre- and post-menopausal women (19, 20). When levels go outside normal ranges, variable effects are observed on cardiovascular health. Women with PCOS, a condition characterized by elevated androgen levels that disrupt the estrogen-to-testosterone ratios, have been shown to experience accelerated atherosclerosis compared to women without PCOS (24, 25). Further, a number of candidate gene studies have identified associations with increased risk of CAD or myocardial infarction (MI) in individuals with deleterious variants in the estrogen receptor 1 (ESR1 or ERα,) or 2 genes (ESR2 or ERβ) causing dysfunctional or null activity (26–29).
There is a relatively large genetic component to developing CAD, with heritability estimates ranging between 40%–60% (30, 31). Most of the heritability of CAD is polygenic, owing to the individually small but cumulatively large contribution of hundreds to thousands of genes considered in CAD risk (32). In the last two decades, the computational methods, capabilities and scalability of genome-wide association studies (GWAS) have contributed substantially to advancing the understanding of which genetic candidates contribute to CAD. Following the first CAD GWAS published in 2007 (33), novel or overlapping candidate genes have emerged through replication between studies, with differences between studies reflecting parameters such as sample size, genetic ancestry, male/female representation, and CAD definitions. Historically, GWASs were predominantly in populations of European genetic ancestry; with time, more diverse and multi-ethnic cohorts have emerged to broaden the generalizability and unique candidates from GWAS findings across different population groups. Furthermore, in addition to the small effects of many common genetic variants towards CAD risk, there are also genes that can contribute large independent risk towards CAD if an individual carries deleterious variants, such as in LDLR, APOB, PCSK9, and LPA (34, 35).
Investigating sex as a biological variable in GWAS studies is essential for accurately identifying sex-specific genetic associations and understanding their unique contributions to disease susceptibility through affected biological pathways. Studies that do not segregate by sex assume that the contributing genes to biological pathways of disease are shared between the sexes; this reduces trait specificity and inserts a level of bias into the results (36). The rationale may be from a fear of potential lowered statistical power when sample sizes are sex-stratified; however, this operates under the assumption that there are no sex differences. If there are indeed sex differences, sex-stratified analyses could in fact increase the power to detect these differences.
To our knowledge, there has not been a review conducted to date that quantifies CAD GWAS-identified gene candidates from both a sex-stratified lens and in assessment of associated modulation by estrogens. Therefore, the aims are twofold: first, we aim to quantify the number of CAD GWAS studies published to date that conducted any sex-stratified analyses. Second, we aim to identify gene candidates listed in both sex-stratified and non-stratified CAD GWAS publications that have functional evidence for direct or indirect regulation by estrogens.
A scoping review of the literature was conducted in accordance with PRISMA guidelines and consultation with a librarian by doing parallel electronic searches of MEDLINE (via Ovid, 1946—April 24, 2024) and EMBASE (via Ovid, 1974—April 24, 2024). The initial part of the search strategy included synonymous terms related to our desired atherosclerotic coronary artery disease phenotype: “Coronary Artery Disease” or “Coronary Heart Disease”. The second part of the search strategy included terms related to our desired study type: “Genome Wide Association Study” or “Genome-Wide Association Study”. The phenotype and study type were linked using the “AND” operator. In addition, manual searching was conducted through citations of relevant reviews and cross-referencing with studies published in the GWAS catalog (https://www.ebi.ac.uk/gwas/home).
To determine eligibility for full-text review, title and abstract screening was conducted by two independent reviewers (A.A., E.T.). All conflicts were resolved through discussion with investigators with the relevant domain expertise (A.L., T.S.). The same applied for screening full-text articles. Studies included were those describing: (1) CAD phenotype; and (2) original GWAS or a GWAS meta-analysis.
There is heterogeneity in how CAD is defined in the GWAS literature. In general, obstructive CAD refers to ≥50% stenosis in the left main coronary artery or ≥70% in a major epicardial vessel identified through coronary angiography (39). However, in studies where researchers do not include angiography results, a history of MI and/or percutaneous coronary intervention (PCI) and/or coronary artery bypass grafting (CABG) are often used as proxies for CAD. Therefore, we included papers with a definition of CAD as: (1) luminal stenosis more than 50% in a major coronary artery; (2) MI; (3) CABG and/or (4) PCI. An “original” GWAS refers to genome-wide association analyses conducted in a novel study population. This includes papers from consortia that published consecutive GWASs over time from cohorts that expanded in case sample size over time.
We excluded abstracts where no full text exists, non-English studies, non-GWAS study design (e.g., family studies, linkage analyses, candidate gene studies), CAD risk factors assessed as outcome (e.g., blood pressure, lipid traits), any non-CAD outcomes (e.g., heart failure), and any studies that were exclusively a secondary analysis using previously published GWAS data [e.g., Mendelian randomization, polygenic risk scores (PRS), causal pathway analysis].
Following abstract screening, full texts were examined for further exclusion. We limited our analysis to any candidate gene with a corresponding single nucleotide polymorphism (SNP) rsID that had a p-value below the widely accepted Bonferroni-corrected genome-wide significance value of p < 5*10−8. Data corresponding to these SNPs were extracted from all materials available, including any Supplementary Material.
For the studies that met the above criteria, full texts were reviewed for extraction of the following variables: Study identifiers (such as author, year of publication, title), study characteristics [sample size, CAD definition, genetic technology used to identify SNPs, population ethnicity(ies), sex ratio of included participants] and candidate gene information [SNP's rsID, mapped gene(s), chromosomal position, risk allele, odds ratio, p-value, minor/estimated allele frequency]. When sex/gender was regressed out as a covariate and not treated as an independent variable, these analyses were termed “sex-combined” analysis for our purposes. When quantifying ethnicity and ancestry representation, the following were grouped together: “White” and “Caucasian” as “European”; “Han Chinese”, “Japanese”, “Taiwanese” and “Korean” as “East Asian”; “Pakistani” and “Bangladeshi” as “South Asian”; “Saudi Arabian” and “Lebanese” as “Middle Eastern”; and “Black”, “Black British” and “African American” as “African descent”.
A summary of all genes mapped from SNPs was made using information provided by study authors of which SNPs were mapped to genes. If a SNP was not previously mapped to (a) gene(s) by study authors, they were excluded from further consideration; we did not infer mapping so as to not introduce bias in the interpretation (i.e., as SNPs can be mapped to multiple genes). Standardization of gene names was conducted, and the number of times each gene was listed by authors were quantified. If a SNP was mapped to multiple loci as explicitly described in the publication, all genes were included. The purpose of this review was not to interpret putatively causal genes from mapped SNP loci towards CAD risk. However, recognizing how many SNPs may be mapped to inconsequential loci (from functional points of view), for studies that conducted non-sex-stratified analyses, a cutoff of genes appearing in at least 5 studies was considered for literature review of evidence of the gene's relevant associations with CAD, and if there were any studies suggesting modulation by estrogens. Considering how few studies produced genome-wide significant sex-specific results, this minimum of 5 studies was not applied to the sex-stratified candidates.
A total of 2820 papers were initially identified (Embase n = 1,828; Medline n = 992). Manual search was conducted and discovered 4 additional papers through cross-referencing the GWAS Catalog (n = 3) and through citations of relevant reviews (n = 1). After the 580 duplicates were removed, the remaining 2,244 studies were screened for selection, after which 2,156 studies were excluded on initial abstract screening, leaving. From remaining articles, only 36 reported SNPs that reached genome-wide significance, including one paper that was included from manual searching (Figure 1).
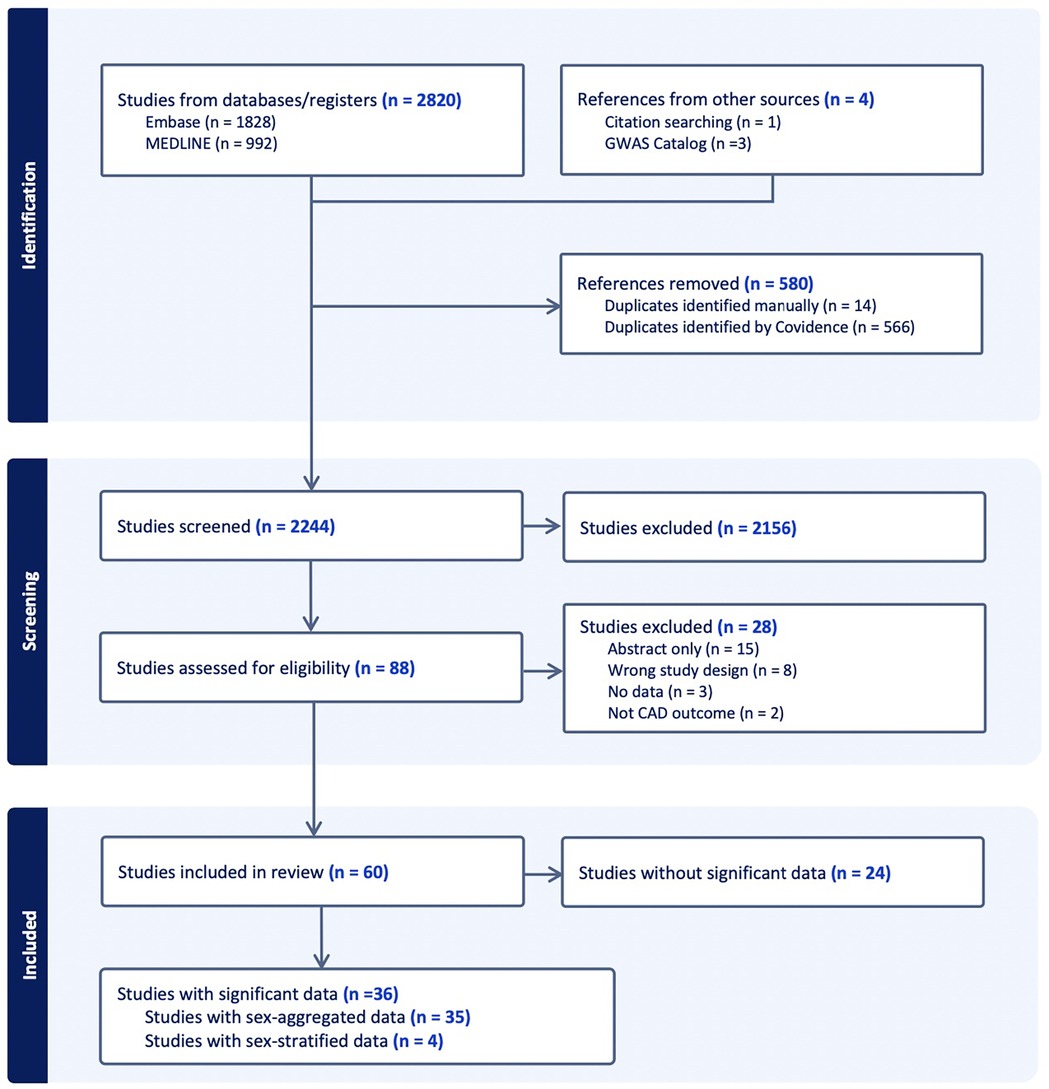
Figure 1. Abstract screening of CAD GWAS studies included shown through PRISMA flow diagram. “Significant” refers to genome-wide significance of p < 5 * 10−8 and “data” refer to SNPs.
Key cohort characteristics of the studies meeting inclusion criteria are summarized in Table 1. Studies that met inclusion criteria but did not contain genome-wide significant results are summarized in Supplementary Table S1.
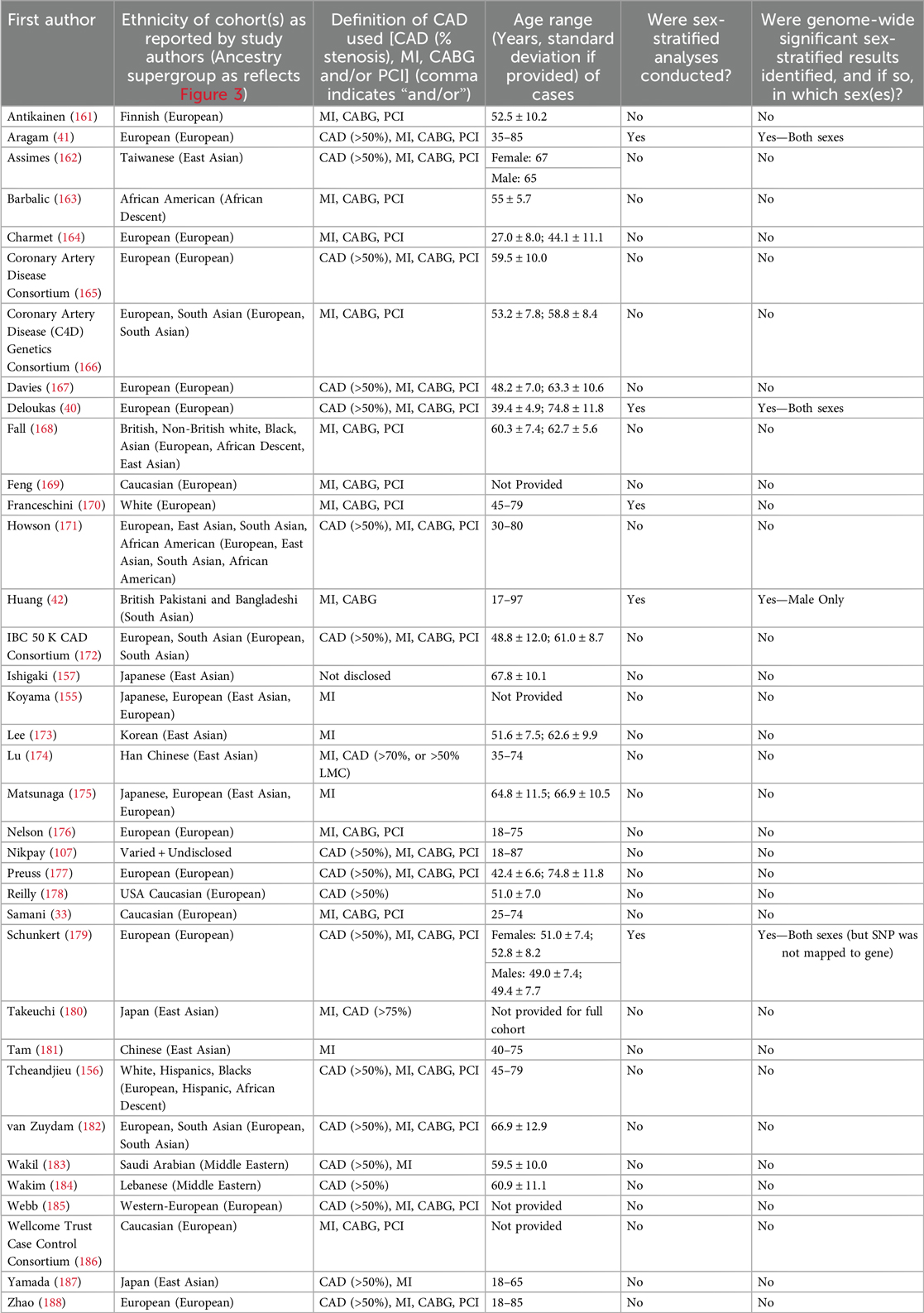
Table 1. Summary of GWAS studies included that had genome-wide significant data (n = 36).
Of the 36 studies included, only 4 reported any genome-wide significant sex-stratified results, from whom 3 had mapped their results to genes (Figure 2A) (40–42). More details of the studies and SNPs identified from sex-combined and sex-stratified analyses can be seen in Supplementary Tables S2 and S3, respectively. The size and clinical heterogeneity of the CAD presentation within and between cohorts are suspected to contribute to the under-representation and lack of sex-specific results. One of the four studies was a meta-analysis that reported the sex-specific age ranges, with female mean ages from the included studies ranging in the early 50 s (Table 1). Exploring the reasoning for why women were under-enrolled in the cohorts used for these GWAS analyses is beyond the scope of this paper.
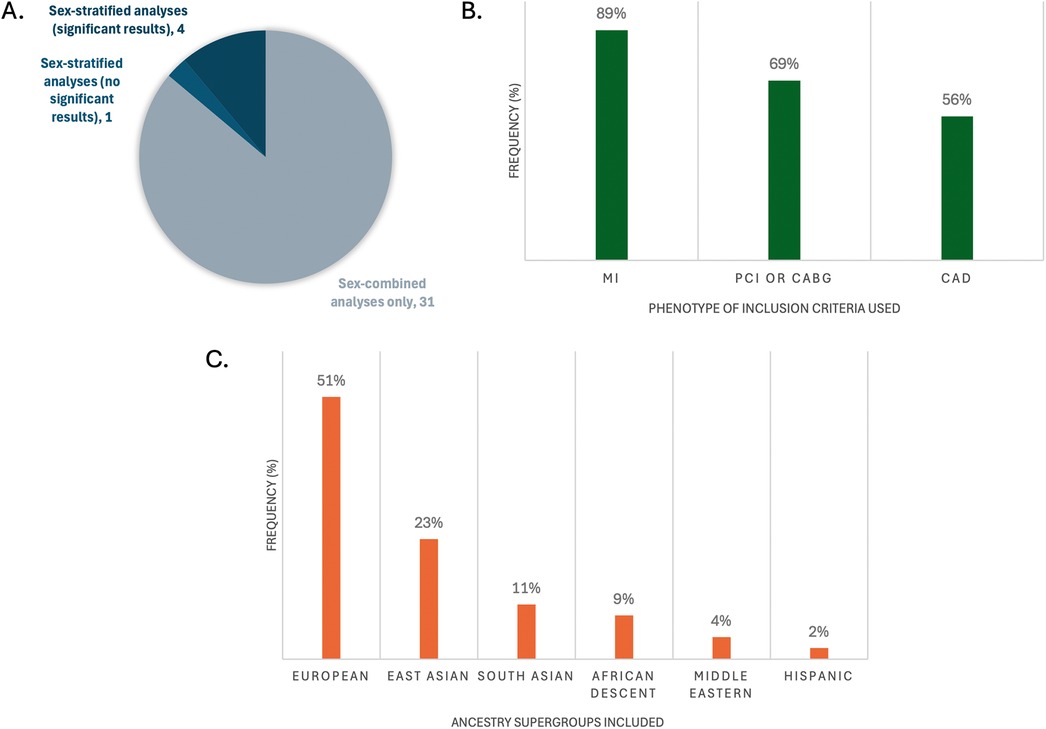
Figure 2. Representation of key characteristics of the GWASs that had genome-wide significant SNPs mapped to genes (n = 36). (A) Frequency of studies that did sex-stratified analyses and/or sex-combined analyses; (B) frequency of phenotype definitions used for inclusion criteria cases; (C) frequency of inclusion of the large ancestral supergroups of participants in the studies. Percents do not add up to 100% because multiple CAD definitions or ancestries may have been reported per study.
Most of the 36 studies included composite definitions that specified at least one of MI, PCI, CABG, and/or CAD with stenosis limits of >50%, >70% or >75%. In their definition of CAD, 33 studies included MI, 25 included CABG, 24 included PCI, and 20 included >50%, >70% or >75% stenosis limits (Figure 2B). An important limitation to underscore in any generalization of MI as CAD, in the absence of PCI, CABG or angiographic reporting of >50% epicardial stenosis, is the potential for confounding with non-atherosclerotic causes of MI. These causes are impossible to quantify without further individual-level information regarding the event. Specifically, MI with no obstructive coronary arteries (MINOCA) is a term encompassing coronary plaque disruption due to rupture, erosion, and calcific nodules (atherosclerotic causes) and non-atherosclerotic etiologies of MI, the most prevalent being epicardial vasospasm, coronary microvascular dysfunction, and spontaneous coronary artery dissection (SCAD). Further, non-ischemic causes of MI include MINOCA “mimickers”, such as Takotsubo cardiomyopathy, myocarditis and supply-demand mismatch (Type 2 MI) (43). MINOCA makes up about 6%–15% of all MIs (43), and is approximately 3 times more prevalent in women than men (44), which adds to clinical heterogeneity in CAD groups solely using MI as their inclusion criteria. Interestingly, individuals with a high PRS for SCAD have been shown to have a low PRS for atherosclerotic CAD and vice versa (45). Therefore, robust atherosclerotic phenotypes for CAD GWAS study designs are recommended so as to not risk accidental inclusion of individuals with non-CAD etiologies of MI, which could influence statistical significance of SNP candidates.
Most studies (26, 72%) reported only one ethnicity. The most commonly reported individuals were of self-reported European ethnicity (51%), followed by East Asian (23%), then South Asian (11%) (Figure 2C). There is a plethora of research demonstrating epidemiological differences in CAD prevalence in different racial/ethnic subgroups, for example with 2–4× higher rates observed in South Asian (46, 47), Hispanic, and Black (48) individuals compared to White individuals. There is an ongoing challenge by researchers to disentangle genetic risk (ancestry) from social (race/ethnicity) and environmental influences on this risk, such as differences in diet, exposure to stress and exercise, that increase CAD risk through variation in gene expression that strongly affect these observed ethnic differences (49) (Box 2). Further, the generalizability of results produced by SNP array technologies used in GWASs should be scrutinized based on the population for which the arrays were designed; for example, accuracy is greatly reduced when SNP arrays created from European-ancestry allele frequencies are applied on individuals of African ancestry (50). This technical bias is further perpetuated by analyses that include filtering of alleles of rare frequency (typically <5% or <1% prevalence in a referenced population). Importantly, there is the concept of “sex-influenced inheritance” (a.k.a. “sex-biased inheritance”) wherein there are more inter-sex similarities than inter-ethnicity similarities for a number of traits, reflected in sex-stratified analyses comparing the sexes within cohorts containing diverse ethnicities. This is exemplified in recent very large multi-ethnic sex-stratified GWASs of complex traits such as blood pressure (51) and lipid traits (52, 53) identifying sex-specific gene associations contributing to the traits. The more that risk genes are identified and replicated between GWAS studies with sex-stratified analyses of diverse genetic ancestries, the more robust these genes are for generalizability across population groups.

Box 2. Distinguishing race, ethnicity, and genetic ancestry definitions. Although these terms are sometimes used interchangeably, each carries distinct implications, particularly when assessing single nucleotide polymorphism (SNP) prevalence.
From the 36 studies included, 61 genes were identified for further literature review if modulated by estrogen. There were 52 genes found in sex-combined analyses with corresponding SNPs surpassing genome-wide significance in at least 5 studies, and 29 genes found in sex-stratified analyses whose corresponding SNPs also surpassed genome-wide significance (Table 2). Due to the small fraction of studies (n = 3) that had significant SNPs mapped to genes from sex-stratified analysis, all genes that surpassed genome-wide significance were retained, even if not replicated. The majority (n = 20/29) of these 29 sex-stratified gene associations were also identified in the sex-combined analyses replicated in at least 5 other studies (Figure 3).
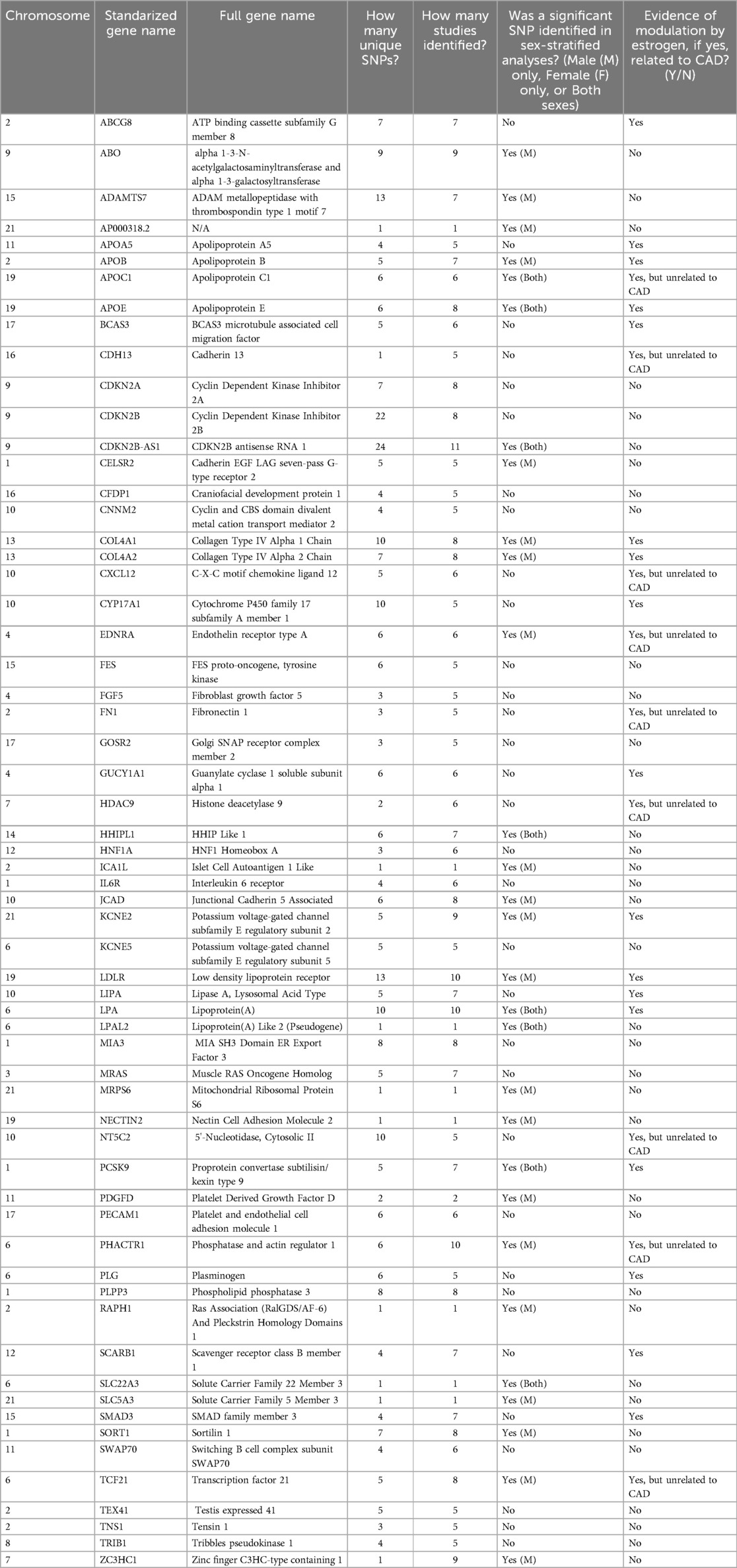
Table 2. Summary of all genes mapped from SNPs that met our inclusion criteria with a genome-wide significance value surpassing p < 5 × 10−8.
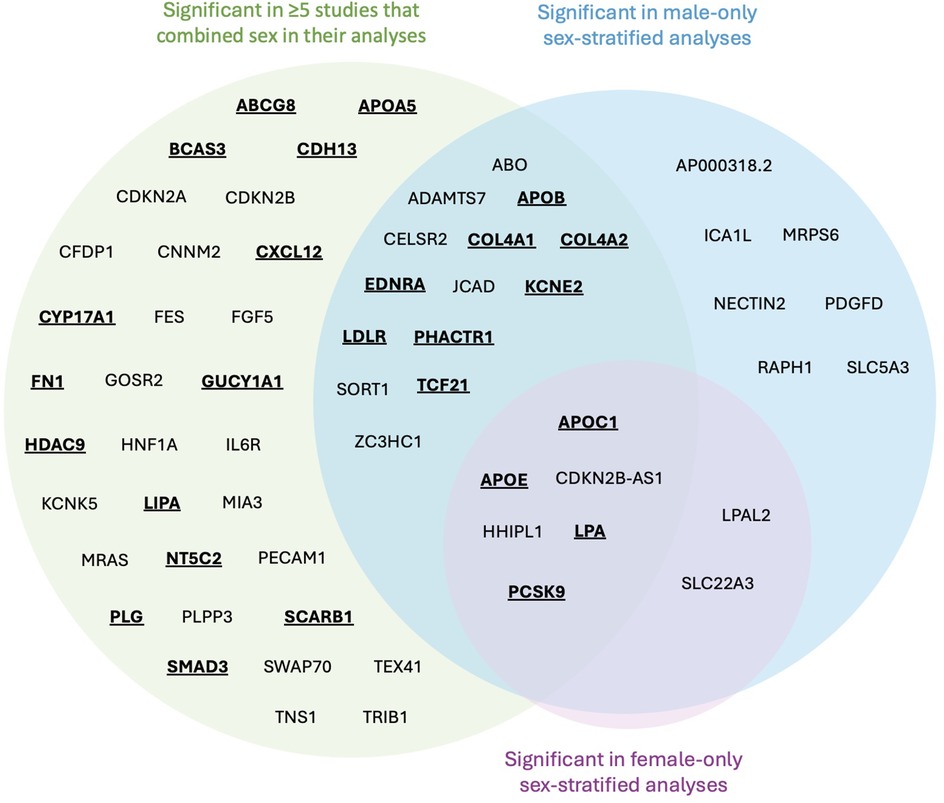
Figure 3. Venn diagram mapped genes from significant SNPs identified in CAD GWASs. Genes emphasized by bold and underscore were identified to have evidence of estrogen modulation of gene expression and/or translated protein activity. No SNPs were identified to be significant just in females.
Following literature review of the 61 genes, 26 (43%) were identified to be affected by estrogen (Figure 3, bold and underlined text). All 26 genes were identified in at least 5 papers with sex-combined analyses, while 12 of these 26 were also identified in at least 1 sex-stratified analysis. Of these 12, 8 were identified in male-stratified analyses in addition to the sex-combined analyses (APOB, COL4A1, COL4A2, EDNRA, KCNE2, LDLR, PHACTR1, TCF21), and 4 were identified in all three of the male-stratified, female-stratified, and sex-combined analyses (APOC1, APOE, LPA, PCSK9). Of note, all significant sex-stratified gene associations identified in females also overlapped with males. None of these 26 genes are on the X-chromosome. The remaining 35 genes did not have literature supporting any modulation of estrogen, directly or indirectly through relation to CAD or other diseases (e.g., breast cancer). These 35 genes were: ABO, ADAMTS7, AP000318.2, CDKN2A, CDKN2B, CDKN2B-AS1, CELSR2, CFDP1, CNNM2, FES, FGF5, GOSR2, HHIPL1, HNF1A, ICA1l, IL6R, JCAD, KCNE5, LPAL2, MIA3, MRAS, MRPS6, NECTIN2, PDGFD, PECAM1, PLPP3, RAPH1, SLC22A3, SLC5A3, SORT1, SWAP70, TEX41, TNS1, TRIB1, and ZC3HC1.
Of the 26 genes with evidence of estrogen modulation, the connection with CAD can be largely categorized by their involvement in lipid production/metabolism, arterial vascular properties, and/or thrombosis, and are expanded on below with relation to genetic variation towards CAD risk.
The following 12 genes were classified as having a role in lipid synthesis, metabolism and/or lipoprotein mechanisms: LDLR, APOB, PCSK9, LPA, LIPA, ABCG8, SCARB1, APOA5, APOE, APOC1, KCNE2, and HDAC9 (Figure 4). Of these, most had evidence of estrogen modulating some aspect of the CAD-related process, except KCNE2 and HDAC9, which had estrogen modulation evidence in other tissues (e.g., breast tissue, ovarian tissue).
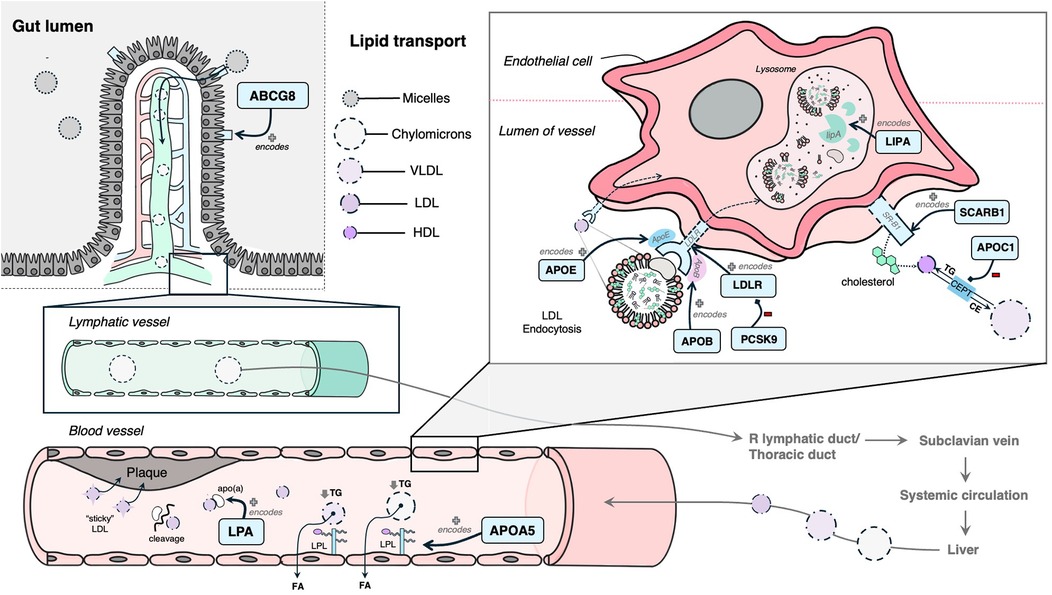
Figure 4. Mechanism diagram of the gene products identified in lipid synthesis, metabolism and/or lipoprotein mechanisms that have evidence for estrogen modulation. Genes described in text are in blue boxes with arrows identifying their gene product.
The following 11 genes were classified as having a role in vascular integrity through roles in the vascular endothelium and/or smooth muscle cells: BCAS3, COL4A1, COL4A2, SMAD3, CYP17A1, CDH13, CXCL12, EDNRA, NT5C2, TCF21, PHACTR1 (Figure 5). Of these, BCAS3, COL4A1, COL4A2, SMAD3 and CYP17A1 had evidence of estrogen modulating some aspect of CAD development, and the rest had estrogen modulation evidence in other tissues (e.g., breast tissue, uterine endothelium).
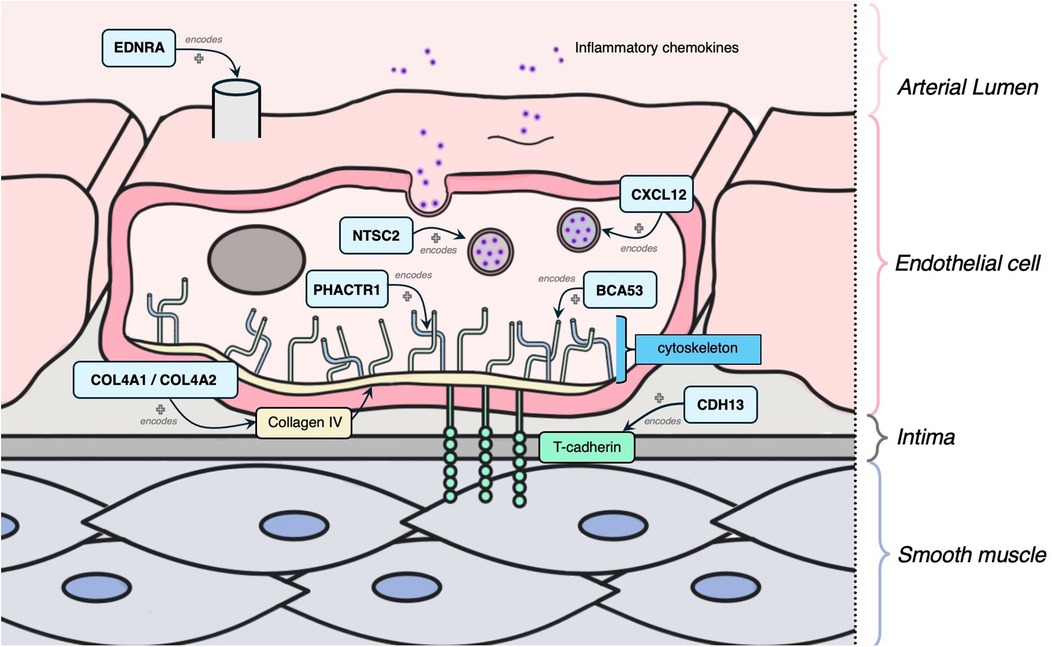
Figure 5. Mechanism diagram of the gene products identified in vascular integrity through roles in the vascular endothelium and/or smooth muscle cells that have evidence for estrogen modulation. Genes described in text are in blue boxes with arrows identifying their gene product.
Three genes were identified to have a role in thrombosis, and all had evidence of estrogen modulating some aspect of CAD development: GUCY1A1, PLG and FN1 (Figure 6).
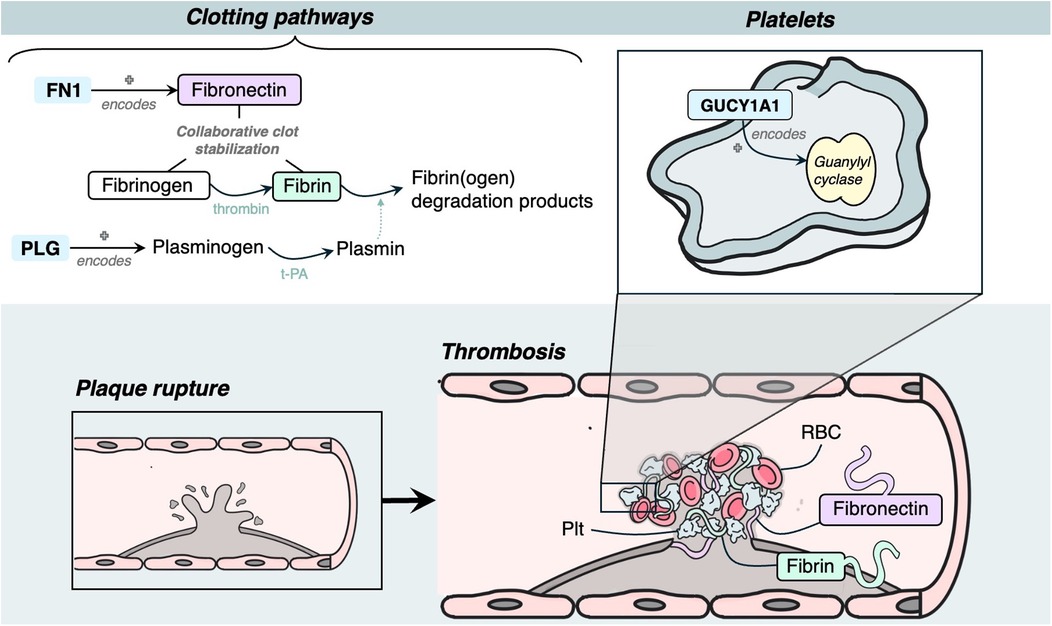
Figure 6. Mechanism diagram of the gene products identified in thrombosis roles that have evidence for estrogen modulation. Genes described in text are in blue boxes with arrows identifying their gene product.
Upon review of gene candidates replicated across the CAD GWAS literature, we found that a large proportion of gene candidates (26/61, 43%) had evidence of modulation by estrogen through direct or indirect mechanisms. Genes identified through GWAS SNP-gene mapping indicate that the SNP in that gene was more frequently present in the cases (individuals with CAD) compared to controls (individuals without CAD). However, a gene associated with CAD in a male-only GWAS may still be influenced by estrogen. We posit this is because estrogen may exert a protective effect against the pathological variant in women, thereby preventing it from reaching statistical significance in female GWAS. In men, the absence of this protective effect could contribute to a pathological phenotype. As we demonstrate later in our discussion, many of estrogen's effects on these genes are through receptor signalling and downstream pathways. Thus, it is reasonable to conclude that estrogen may be cardioprotective for women through its downstream effects on the proteins these genes produce. Mechanisms whereby estrogen affects the gene expression and/or functioning of the protein product are described below.
LDLR encodes a cell surface receptor predominantly expressed in the liver involved in the endocytosis of LDL particles, where they are further metabolized and degraded in hepatocytes (55, 56). Missense or loss of function (LoF) mutations in the LDLR gene results in reduced affinity or inability of the LDLR to bind LDL, resulting in higher plasma lipid levels and thus increased risk of hypercholesterolemia (57). Estrogen influences plasma lipid concentration through LDLR-dependent and LDLR-independent pathways (58). It has been shown in rabbit and rat models that LDLR expression increases up to 10-fold when treated with estrogen, resulting in increased clearance of plasma LDL (59, 60).
APOB encodes for a major apolipoprotein ApoB, which is a high affinity ligand for LDLR attached to LDL particles (61). Missense or LoF variants in APOB can result in decreased binding affinity of LDL to bind to LDLR, resulting in elevated plasma lipid levels (62). Estrogen can reduce ApoB concentrations as demonstrated by experiments in mice via LDLR-independent mechanisms (63).
PCSK9 encodes proprotein convertase subtilisin/kexin type 9, which plays a crucial role in cholesterol metabolism by promoting LDLR degradation (64). PCSK9 gain of function (GoF) mutations further increase LDLR degradation, thus increasing plasma lipid levels. Estradiol reduces PCSK9-mediated LDLR degradation through a mechanism involving activation of the G-protein-coupled estrogen receptor (65). Moreover, circulating levels of PCSK9 are generally higher in women than in men, and this difference is more pronounced post-menopause, which has been suggested to be due to lower circulating estrogen (66). However, in premenopausal women, PCSK9 levels vary with the menstrual cycle, showing an inverse relationship with estradiol (67).
Several studies of the SLC22A3-LPAL2-LPA gene cluster have suggested that polymorphisms in this region are associated with an increased risk of CAD (68, 69). From our review, among genes with evidence to be modulated by estrogen, the SNPs mapped to genes in this cluster had the highest odds ratios, with the highest in LPA of 1.51 (1.33–1.70) (Refer to Supplementary Table S2).
LPA encodes a highly polymorphic glycoprotein, apolipoprotein-a (“apo-a”), which attaches to LDL and is, together, referred to as lipoprotein-a [“Lp(a)”]. Lp(a) is similar to LDL but an independent risk factor of CAD that does not lower with statins (70). Copy number variations in the kringle-iv repeat region of LPA are the main genetic determinants of circulating Lp(a) concentrations (71). When apo(a) is cleaved, it results in fragments that are prone to attaching to atherosclerotic lesions and promoting thrombogenesis through plasminogen activation (70, 71). There is evidence that estrogen decreases Lp(a) plasma levels by increasing its uptake via LDLR (72). In postmenopausal women, taking hormonal therapy has been associated with lower Lp(a) values and subsequent lower risk of CAD (72).
ABCG8 encodes for jejunal and ileal sterol efflux transporters (73). LoF mutations of ABCG8 have been shown to lead to elevated plasma sitosterol and LDL levels (74). ABCG8 has been shown to significantly increase sitosterolemia and potentially accelerate progression of CAD (75). Estrogen upregulates intestinal ABCG8 activity through the intestinal ERa pathway, thus leading to increased cholesterol absorption (73). Interestingly, this upregulation can be completely counteracted by use of an estrogen antagonist (73).
APOA5 encodes a minor apolipoprotein that is an important component of high-density lipoprotein (HDL) and very lower density lipoprotein (VLDL) (76). It directly regulates triglycerides by increasing lipoprotein lipase (LPL), which stimulates the breakdown of triglyceride-rich lipoproteins resulting in lower plasma triglycerides levels (77). Knockout mouse models of APOA5 have shown 4-fold increases in triglyceride levels, and as such it has been linked as a CAD risk gene (78). Oral administration of estrogen has been shown to increase triglyceride concentration, suggested to be due to increased hepatic production of triglycerides due to reduced ApoB production and decreased triglyceride clearance through inhibition of LPL (79). In addition, APOA5 expression has been shown to be higher in women than in men, potentially suggesting that there may be sex differences in its regulation (80).
APOE encodes a plasma protein that is involved in transport and metabolism of cholesterol and triglycerides (81). It is a major ligand for LDLR (82) and is known for having 3 major alleles impacting disease risk: APOE2 (“E2”), APOE3 (“E3”), and APOE4 (“E4”) (82). Expression of these alleles result in higher circulating plasma LDL levels: the E2 and E3 alleles demonstrate poorer binding affinity to LDLR receptors, and E4 alleles bind preferentially to very low density lipoprotein particles (VLDLs), resulting in decreased lipid uptake by LDLR on hepatocytes (82, 83). While these variants have similar background population frequencies between sexes, there have been sex differences observed in associated cardiovascular risk. For example, the E4 allele is associated with higher risk in men, while the E2 has a protective effect observed in women only (84). APOE has been demonstrated to be upregulated by estrogen through the estrogen-receptor alpha pathway (85), although the effects are variable based on the APOE alleles carried. For example, the E4 allele has shown to have higher expression following menopausal hormone therapy than E2 and E3 variants (84). Many studies use APOE-knockout murine models to study accelerated atherosclerosis and effects of estrogen, underscoring the importance of this ligand in atherosclerotic pathology (86).
SCARB1 encodes an HDL receptor that mediates the cholesterol transfer to and from HDL (87). LoF or missense alleles increased dimerization and decreased hepatocellular uptake of HDL, resulting in higher risk of atherosclerosis (87). Estradiol has been shown to be an indirect modulator of the SCARB1 receptor in rat models (88). Estradiol does not directly decrease expression of SCARB1 in the liver, however it has been demonstrated to decrease SCARB1 expression levels secondary to the estrogen-induced increase in LDLR activity and ACTH presence (88).
LIPA encodes lipase A, which functions in lysosomes to hydrolyze cholesteryl esters and triglycerides to generate free cholesterol and free fatty acids following LDLR-mediated LDL endocytosis in hepatocytes (89). LoF variants in LIPA have been shown to result in accumulation of triglycerides and cholesterol esters that contribute to foam cell development and premature atherosclerotic plaque formation (89, 90). Deficient LIPA activity may have an indirect effect on CAD risk through its downstream effects on estrogen production. Since LIPA functions in hydrolysis of triglycerides and cholesterol, it provides energy while also influencing the synthesis and secretion of sex hormones including estrogen (91).
APOC1 encodes an apolipoprotein C1 family member that plays a role in HDL and VLDL metabolism through highly selective inhibition of cholesteryl ester transfer protein (CETP) in plasma (92, 93). Decreased expression of APOC1 from LoF or missense variants, leads to increased plasma triglycerides, which confers increased CAD risk (94). One study suggested that APOC1 could promote the estrogen receptor expression in the context of ovarian cancer (95). In addition, there is marked APOC1 elevation in women with PCOS; however it is unclear how much of the altered lipid metabolism is due to estrogen and/or androgen metabolism or because of insulin resistance (96).
KCNE2 encodes a voltage-gated potassium channel involved in regulating heart rate, neurotransmitter release and smooth muscle contraction (97). Demonstrated by mouse knockout models, causal links between KCNE2 and atherosclerosis have been suggested to be due to raising serum LDL and impairing glucose tolerance (98, 99). Estrogen action alters KCNE2 expression directly through binding of estrogen receptor alpha to the estrogen-responsive element in the KCNE2 regulatory domain (100). Estrogen was observed to be less responsive in the presence of variants altering affinity of DNA-binding domains in either the estrogen receptor or KCNE2 estrogen-responsive element (100). Further, the nuclear estrogen-related receptor has also been shown to have a crucial role in modulating KCNE2 by binding to its promoter (101).
HDAC9 encodes enzyme histone deacetylase 9, and has been implicated in progression of atherosclerosis through histone acetylation and subsequent expression of specific genes related to lipid metabolism and macrophage polarization (102). Missense and LoF HDAC9 mutations result in downregulation of inflammatory genes and increased Apolipoprotein-A1 and HDL-mediated cholesterol efflux, resulting in decreased plasma cholesterol levels (103). As such, it is thought that upregulation of HDAC9 in macrophages has increased atherosclerotic risk through suppression of cholesterol efflux and proinflammatory actions (103). A variant has also been shown to directly modulate the expression of TWIST1, a gene that regulates arterial wall proliferation and calcification (104). Increased expression of HDAC9 has been shown to be associated with decreased expression and activity of estrogen receptor alpha in MCF-7 cells in breast cancer studies (105).
BCAS3 encodes for a cytoskeletal protein that functions in angiogenesis and related processes like TGFβ signaling, cell adhesion, peptidase activity and matrix organization (106). It is thought to do this by activating Cdc42 which in turn affects actin organization, cell polarity and cell motility in endothelial cells (107). Endothelial BCAS3 knockout mice survive to only embryonic day 11.5 and have diffuse vascular patterning defects (106). However, despite numerous studies citing BCAS3 association with CAD, functional studies are still lacking. Estrogen directly induces BCAS3 transcript expression by binding to estrogen receptor alpha and subsequently inducing MTA1, which is a transcriptional factor of BCAS3 (108).
COL4A1 and COL4A2 encode subunits of type IV collagen, which is an essential structural component of the basement membrane (109). LoF alleles in these genes result in lower collagen IV abundance and thinner fibrous caps, thus creating very unstable plaques and contributing to smooth muscle cell pathology (110). SMAD3 encodes the SMAD family member 3, a signaling molecule involved in the transforming growth factor-beta (TGF-β) signaling pathway (111), which is a cell growth inhibitor crucial in the regulation of inflammation and fibrosis (112). SMAD3-knockout mice have decreased fibrotic response, resulting in thin, unstable fibrous caps (113). SMAD3 induces TGF-β which is necessary for TGFβ-stimulated expression of both COL4A1 and COL4A2 (113). Estrogen inhibits TGF-β signaling by binding to estrogen receptor alpha, thus decreasing SMAD3 levels, which subsequently decreases COL4A1 and COL4A2 expression (114, 115).
CYP17A1 encodes a member of the CYP450 superfamily of enzymes, which contain a heme cofactor and mostly function as monooxygenases (116). CYP17A1 is specifically localized in the endoplasmic reticulum and involved in the steroidogenic pathway that produces mineralocorticoids, glucocorticoids, androgens, progestins and estrogens (117). One study showed that CYP17A1 knockout mice develop atherosclerotic lesions at a higher rate compared to WT (118). The functional evidence linking CYP17A1 and CAD is not entirely understood, however it is thought to involve glucose homeostasis regulation by promoting glucose uptake and utilization (118). Further, there is evidence of genetic variants that are associated with severe hypertension, a well-known risk factor for CAD (119, 120). CYP17A1 has a role in estrogen production; it is highly expressed in granulosa cells, and can catalyze pregnenolone and progesterone to form androstenedione, which CYP19A1 later converts to estrogen (121). Missense/LoF variants in CYP17A1 have been shown to cause fertility impairments and related cancers (119, 122).
CDH13 encodes a T-cadherin that is expressed on endothelial and smooth muscle cells (123). Increased CDH13 activity results in increased migration and proliferation of endothelial cells and therefore effects vascular remodeling and atherosclerosis development (124). Knockout T-cadherin and adiponectin mice had increased neointimal thickness following carotid artery ligation (125). A variant within this gene was found to be associated with estrogen signaling metabolism in breast cancer, menstruation patterns and pregnancy (126).
CXCL12 encodes for a chemokine that is produced in endothelial cells and as such has an important role in angiogenesis, hematopoiesis, and tissue regeneration (127, 128). CXCL12-knockout mice models demonstrated compromised artery coverage, suggesting a key role in arterial development and regulation (129). CXCL12 has also been shown to consistently be upregulated in individuals with calcific aortic valve disease (130). However, the underlying mechanism between poor prognosis of CAD and high levels of CXCL12 is not understood (127). Research has shown estradiol regulation of the CXCL12 axis in the growth of breast cancer cells (131). Estradiol directly induces transcription of CXCR4 and CXCR7, which are both receptors of CXCL12, through control of their promoters (131).
EDNRA encodes endothelin receptor A, which mediates cell proliferation and long-lasting vasoconstriction (132). In coordination with EDNRB, EDNRA can mediate contraction post-relaxation of intact endothelium vessels (132). Increased EDNRA expression has shown to play an important role in hypertension and thus progression of vascular proliferation (132, 133). One study showed that EDNRA transcript in endometrial stromal cells increases in the proliferative phase when given estradiol in rhesus macaques (134). Estrogen binding to estrogen receptor alpha directly results in increased epithelial EDN3 expression, which is thought to act via EDNRA to further stimulate cell proliferation (134).
NT5C2 encodes for a 5′ nucleotidase that functions in purine metabolism (135), and has a suggested role in type 2 diabetes and hypertension (136). NT5C2 is present on vascular endothelium cells and likely has an inflammatory process relation to CAD (136, 137). Knockdown NT5C2 zebrafish had higher blood flow and elevated linear velocity, in addition to increased inflammatory markers such as angiotensin-converting enzyme and C-reactive protein (138). There is some evidence to suggest that NT5C2 may be involved in the estradiol regulation of fibroblasts (137). Expression levels of NT5C2 were increased 2 h after estradiol administration in primary uterine endometrial epithelial cells (137).
TCF21 encodes a transcription factor that has been identified as a “master regulator” for smooth muscle cell gene expression (139). Increased TCF21 expression has been suggested to be associated with decreased risk of CAD by influencing smooth muscle cell behavior in developing lesions, contributing to a protective fibrous cap (140). It achieves this by disrupting the MYOCD-SRF pathway, which is crucial for SMC differentiation (140), as shown by in vitro experiments of variants causing overexpression of TCF21 and TCF21-knockout mouse models (141). TCF21 interacts with USF2 in endometriotic stromal cells and activates the promoters of SF-1 and estrogen receptor beta, thereby influencing the estrogen pathway in endometriosis (142). While it is unknown if this estrogen activation is widespread throughout the body, it is interesting to note that TCF21 also regulates fibrosis in endometriosis (142).
PHACTR1 encodes for a member of the phosphatase and actin regulator family (143). It has a crucial role in binding to actin to regulate the organization of the actin cytoskeleton and important roles in tubule formation, and thus in endothelial cell survival (143). PHACTR1 deficiency from knockout mice or LoF experiments demonstrated accelerated foam cell formation and thus increased atherosclerosis (144). In ovarian granulosa cells, PHACTR1 was identified to be a regulatory target by estrogen through experiments in estrogen receptor 2-depleted mice (145); however this connection has not been replicated in vascular endothelial cells.
GUCY1A1 encodes the alpha subunit of the guanylate cyclase enzyme, which is an essential enzyme in platelets (146). LoF alleles result in lack of guanylate cyclase enzyme in platelets, which have been shown experimentally to result in vascular inflammation through increased leukocyte recruitment and endothelial cell activation, and thus atherosclerotic plaque progression (146). Estradiol has been shown to highly up-regulate both expression and activity of the a1 subunit of guanylate cyclase through estrogen receptor activation (147).
PLG encodes a plasminogen protein that is converted to active plasmin by plasminogen activators such as tissue plasminogen activator (148). Plasmin acts as an antithrombotic agent, and is responsible for degrading fibrin-containing blood clots (148), in addition to cleaving fibronectin and von Willebrand factor (149). It is hypothesized that increased binding of plasminogen kringle domains to tissue plasminogen activator can lead to increased activator activity, thus increasing the conversion of plasminogen to plasmin, and as such cause unstable plaque formations to occur (148). Estrogen directly increases PLG expression by binding to 5′-region-flanking enhancers (150).
FN1 encodes fibronectin, a glycoprotein involved in the cell adhesion and migration processes of thrombosis and coagulation (151). Fibronectin works with fibrin and fibrinogen in clot formation, contributing to thrombus stability (151). Fibronectin has also shown to play a role in mediating platelet adhesion (151). In knockout mouse models, a splice variant of fibronectin containing extra domain A has been shown to have twice the amount of atherosclerotic lesions and macrophage content in plaques (152). Inhibition of the estrogen receptor is suggested to significantly decrease FN1 expression (153), consistent with studies demonstrating increased expression in breast cancer cells through G-protein coupled transmembrane receptors (154).
There are several limitations from the methods used in our study. It must be acknowledged that due to the lack of sex-stratified data available, sex-stratified gene associations were only required to be discovered once. This means that there is less credibility to these sex-stratified genes. This is in comparison to sex-combined genes analysis where a gene had to be replicated in at least 5 studies to be included. This cutoff was chosen so as to increase confidence in the SNPs included in the discussion, as several identified papers listed all potential genes that could be mapped to a SNP (41, 155–157), without secondary analysis that would result in one high confidence causal gene.
Age ranges varied significantly by study, with many reporting aggregate ranges (i.e., 18–85 years old) that encompasses both cases and controls. The lack of specificity in these studies to disentangle ranges of the cases calls into question if the younger participants included as controls were in fact true negatives, considering that they are not at the age range where CAD may generally occur, such that their SNPs may have confounded analyses and subsequent gene associations with CAD. Furthermore, menopause status cannot be inferred from the reported age ranges, including the two studies that reported sex-stratified age ranges. Even if the age at study data capture was after menopause, it is still the case that the health effects of past higher relative levels of estrogen pre-menopause could remain a health risk modifier for CAD later in life. This principle is analogously supported by evidence that the duration of estrogen exposure during the lifetime has been shown to influence the risk of breast cancer in later years (158).
Additionally, due to the nature of our search for estrogen, it is possible that there are connections between estrogen and genes that were missed. For example, any downstream metabolites or effectors of the estrogen system were not included in this review. In addition, estrogen is not the only sex hormone. There are androgens that may have a role in CAD that were not evaluated. Androgen regulation has demonstrated potential involvement in the CAD pathway (159), and is an area that continues to grow with more research.
Moreover, there are flaws innate to the study design of GWASs that limit the conclusions that can be made. To increase power in a GWAS, meta-analyses will incorporate many cohorts to increase sample size. However, this causes heterogeneity and leads to decreased confidence as many of these cohorts use varying inclusion/exclusion criteria, technology to identify SNPs, and have varying amounts of male to female participants. In fact, every study that reported case/control sex ratios had a larger percentage of men in the cases and more women being included as controls (data not shown). We speculate that this may be due to the observation that women tend to develop CAD later in life than men by about 10 years (160) and/or assumptions that there are no genetic sex differences that will influence results. This may thus confound the validity of the results. It is thus an important consideration for future studies to include equal proportions of the sexes and conduct sex-stratified analyses to be able to identify candidates with sex-specific effects in the pathogenesis of CAD.
Our scoping review identified 26 genes through CAD GWASs that are modulated by estrogen. Some of these genes are well-studied contributors to the development of CAD, such as LDLR and PCSK9. This study provides further evidence of the relationship between the actions of estrogen and the development of CAD. More research will need to be conducted to establish estrogen's relation to sex differences in the pathology and progression of CAD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
AA: Writing – original draft, Writing – review & editing. ET: Writing – original draft, Writing – review & editing. EB: Writing – review & editing. CA: Writing – review & editing. AG: Writing – review & editing. AL: Writing – review & editing. TS: Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
We would like to thank UBC Faculty of Medicine librarian Aubrey Geyer for providing support during the design and execution of scoping review study methodology in the context of our article. The authors confirm that ethics protocol and patient consent forms were not required for this manuscript.
The authors declare that there are no commercial or financial relationships that could be perceived as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1502985/full#supplementary-material
Supplemental Table 1 | These 24 studies had sex-combined analyses that met all other inclusion criteria, but contained no SNPs that surpassed genome-wide statistical significance of p<5*10^-8.
Supplemental Table 2 | This table contains the 1658 SNPs that achieved genome-wide significance (p<5*10^-8) from the included GWASs in sex-combined analyses.
Supplemental Table 3 | This table contains the 597 SNPs from sex-stratified analyses that had achieved genome-wide significance (p<5*10^-8) from the 4 papers that met our criteria. 413 SNPs were mapped from female-stratified data while 184 SNPS were mapped from male-stratified data.
1. Stark B, Johnson C, Roth GA. Global prevalence of coronary artery disease: an update from the global burden of disease study. J Am Coll Cardiol. (2024) 83(13_Supplement):2320–2320. doi: 10.1016/S0735-1097(24)04310-9
2. Regitz-Zagrosek V, Gebhard C. Gender medicine: effects of sex and gender on cardiovascular disease manifestation and outcomes. Nat Rev Cardiol. (2023) 20(4):236–47. doi: 10.1038/s41569-022-00797-4
3. Lorenzatti AJ, Retzlaff BM. Unmet needs in the management of atherosclerotic cardiovascular disease: is there a role for emerging anti-inflammatory interventions? Int J Cardiol. (2016) 221:581–6. doi: 10.1016/j.ijcard.2016.07.061
4. Sato Y, Kawakami R, Sakamoto A, Cornelissen A, Mori M, Kawai K, et al. Sex differences in coronary atherosclerosis. Curr Atheroscler Rep. (2022) 24(1):23–32. doi: 10.1007/s11883-022-00980-5
5. Pacheco C, Mullen KA, Coutinho T, Jaffer S, Parry M, Van Spall HGC, et al. The Canadian women’s heart health alliance atlas on the epidemiology, diagnosis, and management of cardiovascular disease in women—chapter 5: sex- and gender-unique manifestations of cardiovascular disease. CJC Open. (2022) 4(3):243–62. doi: 10.1016/j.cjco.2021.11.006
6. Mulvagh SL, Mullen KA, Nerenberg KA, Kirkham AA, Green CR, Dhukai AR, et al. The Canadian women’s heart health alliance atlas on the epidemiology, diagnosis, and management of cardiovascular disease in women—chapter 4: sex- and gender-unique disparities: CVD across the lifespan of a woman. CJC Open. (2022) 4(2):115–32. doi: 10.1016/j.cjco.2021.09.013
7. Anand SS, Islam S, Rosengren A, Franzosi MG, Steyn K, Yusufali AH, et al. Risk factors for myocardial infarction in women and men: insights from the INTERHEART study. Eur Heart J. (2008) 29(7):932–40. doi: 10.1093/eurheartj/ehn018
8. Manfrini O, Yoon J, van der Schaar M, Kedev S, Vavlukis M, Stankovic G, et al. Sex differences in modifiable risk factors and severity of coronary artery disease. J Am Heart Assoc. (2020) 9(19):e017235. doi: 10.1161/JAHA.120.017235
9. Norris CM, Yip CYY, Nerenberg KA, Clavel MA, Pacheco C, Foulds HJA, et al. State of the science in women’s cardiovascular disease: a Canadian perspective on the influence of sex and gender. J Am Heart Assoc. (2020) 9(4):e015634. doi: 10.1161/JAHA.119.015634
10. Barrett-Connor E. Menopause, atherosclerosis, and coronary artery disease. Curr Opin Pharmacol. (2013) 13(2):186–91. doi: 10.1016/j.coph.2013.01.005
11. Regitz-Zagrosek V, Kararigas G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev. (2017) 97(1):1–37. doi: 10.1152/physrev.00021.2015
12. Yaşar P, Ayaz G, User SD, Güpür G, Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol. (2017) 16(1):4–20. doi: 10.1002/rmb2.12006
13. Vaura F, Palmu J, Aittokallio J, Kauko A, Niiranen T. Genetic, molecular, and cellular determinants of sex-specific cardiovascular traits. Circ Res. (2022) 130(4):611–31. doi: 10.1161/CIRCRESAHA.121.319891
14. Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
15. Ouyang P, Michos ED, Karas RH. Hormone replacement therapy and the cardiovascular system lessons learned and unanswered questions. J Am Coll Cardiol. (2006) 47(9):1741–53. doi: 10.1016/j.jacc.2005.10.076
16. Medina D, Mehay D, Arnold AC. Sex differences in cardiovascular actions of the renin-angiotensin system. Clin Auton Res. (2020) 30(5):393–408. doi: 10.1007/s10286-020-00720-2
17. Jones TH, Kelly DM. Randomized controlled trials—mechanistic studies of testosterone and the cardiovascular system. Asian J Androl. (2018) 20(2):120–30. doi: 10.4103/aja.aja_6_18
18. Dama A, Baggio C, Boscaro C, Albiero M, Cignarella A. Estrogen receptor functions and pathways at the vascular immune interface. Int J Mol Sci. (2021) 22(8):4254. doi: 10.3390/ijms22084254
19. Zhao D, Guallar E, Ouyang P, Subramanya V, Vaidya D, Ndumele CE, et al. Endogenous sex hormones and incident cardiovascular disease in post-menopausal women. J Am Coll Cardiol. (2018) 71(22):2555–66. doi: 10.1016/j.jacc.2018.01.083
20. van Koeverden ID, de Bakker M, Haitjema S, van der Laan SW, de Vries JPPM, Hoefer IE, et al. Testosterone to oestradiol ratio reflects systemic and plaque inflammation and predicts future cardiovascular events in men with severe atherosclerosis. Cardiovasc Res. (2019) 115(2):453–62. doi: 10.1093/cvr/cvy188
21. Malik R, Pokeria C, Singh S. Correlation of menopausal symptoms with Serum estradiol: a study in urban Indian postmenopausal women. J Obstet Gynaecol India. (2022) 72(4):322–9. doi: 10.1007/s13224-021-01518-6
22. Peacock K, Carlson K, Ketvertis KM. Menopause. In: StatPearls. Treasure Island (FL): StatPearls Publishing (2024). Available at: http://www.ncbi.nlm.nih.gov/books/NBK507826/ (Accessed August 22, 2024).
23. Brzozowska M, Lewiński A. Changes of androgens levels in menopausal women. Prz Menopauzalny. (2020) 19(4):151–4. doi: 10.5114/pm.2020.101941
24. Vryonidou A, Papatheodorou A, Tavridou A, Terzi T, Loi V, Vatalas IA, et al. Association of hyperandrogenemic and metabolic phenotype with carotid intima-media thickness in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. (2005) 90(5):2740–6. doi: 10.1210/jc.2004-2363
25. Gao L, Zhao Y, Wu H, Lin X, Guo F, Li J, et al. Polycystic ovary syndrome fuels cardiovascular inflammation and aggravates ischemic cardiac injury. Circulation. (2023) 148(24):1958–73. doi: 10.1161/CIRCULATIONAHA.123.065827
26. Shearman AM, Cupples LA, Demissie S, Peter I, Schmid CH, Karas RH, et al. Association between estrogen receptor alpha gene variation and cardiovascular disease. JAMA. (2003) 290(17):2263–70. doi: 10.1001/jama.290.17.2263
27. Shearman AM, Cooper JA, Kotwinski PJ, Miller GJ, Humphries SE, Ardlie KG, et al. Estrogen receptor alpha gene variation is associated with risk of myocardial infarction in more than seven thousand men from five cohorts. Circ Res. (2006) 98(5):590–2. doi: 10.1161/01.RES.0000210578.62102.a6
28. Schuit SCE, Oei HHS, Witteman JCM, Geurts van Kessel CH, van Meurs JBJ, Nijhuis RL, et al. Estrogen receptor alpha gene polymorphisms and risk of myocardial infarction. JAMA. (2004) 291(24):2969–77. doi: 10.1001/jama.291.24.2969
29. Henttonen AT, Kortelainen ML, Kunnas TA, Nikkari ST. Estrogen receptor-1 genotype is related to coronary intima thickness in young to middle-aged women. Scand J Clin Lab Invest. (2007) 67(4):380–6. doi: 10.1080/00365510601085951
30. Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, De Faire U. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med. (2002) 252(3):247–54. doi: 10.1046/j.1365-2796.2002.01029.x
31. McPherson R, Tybjaerg-Hansen A. Genetics of coronary artery disease. Circ Res. (2016) 118(4):564–78. doi: 10.1161/CIRCRESAHA.115.306566
32. Klarin D, Natarajan P. Clinical utility of polygenic risk scores for coronary artery disease. Nat Rev Cardiol. (2022) 19(5):291–301. doi: 10.1038/s41569-021-00638-w
33. Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. (2007) 357(5):443–53. doi: 10.1056/NEJMoa072366
34. Watts GF, Gidding S, Wierzbicki AS, Toth PP, Alonso R, Brown WV, et al. Integrated guidance on the care of familial hypercholesterolaemia from the international FH foundation. Int J Cardiol. (2014) 171(3):309–25. doi: 10.1016/j.ijcard.2013.11.025
35. Reyes-Soffer G, Ginsberg HN, Berglund L, Duell PB, Heffron SP, Kamstrup PR, et al. Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American Heart Association. Arterioscler, Thromb, Vasc Biol. (2022) 42(1):e48–60. doi: 10.1161/ATV.0000000000000147
36. Shapiro JR, Klein SL, Morgan R. Stop “controlling” for sex and gender in global health research. BMJ Glob Health. (2021) 6(4):e005714. doi: 10.1136/bmjgh-2021-005714
37. Government of Canada CI of HR. How to integrate sex and gender into research—CIHR. (2018). Available at: https://www.cihr-irsc.gc.ca/e/50836.html (Accessed August 22, 2024).
38. Connelly PJ, Azizi Z, Alipour P, Delles C, Pilote L, Raparelli V. The importance of gender to understand sex differences in cardiovascular disease. Can J Cardiol. (2021) 37(5):699–710. doi: 10.1016/j.cjca.2021.02.005
39. Writing Committee Members, Virani SS, Newby LK, Arnold SV, Bittner V, Brewer LC, et al. 2023 AHA/ACC/ACCP/ASPC/NLA/PCNA guideline for the management of patients with chronic coronary disease: a report of the American Heart Association/American College of Cardiology joint committee on clinical practice guidelines. J Am Coll Cardiol. (2023) 82(9):833–955. doi: 10.1016/j.jacc.2023.04.003
40. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. (2013) 45(1):25–33. doi: 10.1038/ng.2480
41. Aragam KG, Jiang T, Goel A, Kanoni S, Wolford BN, Atri DS, et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat Genet. (2022) 54(12):1803–15. doi: 10.1038/s41588-022-01233-6
42. Huang QQ, Sallah N, Dunca D, Trivedi B, Hunt KA, Hodgson S, et al. Transferability of genetic loci and polygenic scores for cardiometabolic traits in British Pakistani and Bangladeshi individuals. Nat Commun. (2022) 13(1):4664. doi: 10.1038/s41467-022-32095-5
43. Pacheco C, Coutinho T, Bastiany A, Beanlands R, Boczar KE, Gulati M, et al. Canadian Cardiovascular society/Canadian women’s heart health alliance clinical practice update on myocardial infarction with No obstructive coronary artery disease (MINOCA). Can J Cardiol. (2024) 40(6):953–68. doi: 10.1016/j.cjca.2024.02.032
44. Mahajan AM, Gandhi H, Smilowitz NR, Roe MT, Hellkamp AS, Chiswell K, et al. Seasonal and circadian patterns of myocardial infarction by coronary artery disease status and sex in the ACTION registry-GWTG. Int J Cardiol. (2019) 274:16–20. doi: 10.1016/j.ijcard.2018.08.103
45. Adlam D, Berrandou TE, Georges A, Nelson CP, Giannoulatou E, Henry J, et al. Genome-wide association meta-analysis of spontaneous coronary artery dissection identifies risk variants and genes related to artery integrity and tissue-mediated coagulation. Nat Genet. (2023) 55(6):964–72. doi: 10.1038/s41588-023-01410-1
46. Gupta M, Brister S. Is south Asian ethnicity an independent cardiovascular risk factor? Can J Cardiol. (2006) 22(3):193–7. doi: 10.1016/S0828-282X(06)70895-9
47. Sucato V, Coppola G, Manno G, Vadalà G, Novo G, Corrado E, et al. Coronary artery disease in south Asian patients: cardiovascular risk factors, pathogenesis and treatments. Curr Probl Cardiol. (2023) 48(8):101228. doi: 10.1016/j.cpcardiol.2022.101228
48. Lima Dos Santos CC, Matharoo AS, Pinzón Cueva E, Amin U, Perez Ramos AA, Mann NK, et al. The influence of sex, age, and race on coronary artery disease: a narrative review. Cureus. (2023) 15(10):e47799. doi: 10.7759/cureus.47799
49. Sumi MP, Mahajan B, Sattar RSA, Nimisha , Apurva , Kumar A, et al. Elucidation of epigenetic landscape in coronary artery disease: a review on basic concept to personalized medicine. Epigenet Insights. (2021) 14:2516865720988567. doi: 10.1177/2516865720988567
50. Bentley AR, Callier SL, Rotimi CN. Evaluating the promise of inclusion of African ancestry populations in genomics. NPJ Genom Med. (2020) 5:5. doi: 10.1038/s41525-019-0111-x
51. Shetty NS, Pampana A, Patel N, Li P, Yerabolu K, Gaonkar M, et al. Sex differences in the association of genome-wide systolic blood pressure polygenic risk score with hypertension. Circ Genom Precis Med. (2023) 16(6):e004259. doi: 10.1161/CIRCGEN.123.004259
52. Hoffmann TJ, Theusch E, Haldar T, Ranatunga DK, Jorgenson E, Medina MW, et al. A large electronic health record-based genome-wide study of serum lipids. Nat Genet. (2018) 50(3):401–13. doi: 10.1038/s41588-018-0064-5
53. Tabassum R, Widén E, Ripatti S. Effect of biological sex on human circulating lipidome: an overview of the literature. Atherosclerosis. (2023) 384:117274. doi: 10.1016/j.atherosclerosis.2023.117274
54. Hynes G, Riley D. Guidance on the Use of Standards for Race-Based and Indigenous Identity Data Collection and Health Reporting in Canada. Ottawa, ON: CIHI (2022).
55. Defesche JC. Low-density lipoprotein receptor–its structure, function, and mutations. Semin Vasc Med. (2004) 4(1):5–11. doi: 10.1055/s-2004-822993
56. van de Sluis B, Wijers M, Herz J. News on the molecular regulation and function of hepatic LDLR and LRP1. Curr Opin Lipidol. (2017) 28(3):241–7. doi: 10.1097/MOL.0000000000000411
57. Srivastava RAK. A review of progress on targeting LDL receptor-dependent and -independent pathways for the treatment of hypercholesterolemia, a Major risk factor of ASCVD. Cells. (2023) 12(12):1648. doi: 10.3390/cells12121648
58. Palmisano BT, Zhu L, Stafford JM. Estrogens in the regulation of liver lipid metabolism. Adv Exp Med Biol. (2017) 1043:227–56. doi: 10.1007/978-3-319-70178-3_12
59. Ma PT, Yamamoto T, Goldstein JL, Brown MS. Increased mRNA for low density lipoprotein receptor in livers of rabbits treated with 17 alpha-ethinyl estradiol. Proc Natl Acad Sci U S A. (1986) 83(3):792–6. doi: 10.1073/pnas.83.3.792
60. Chao YS, Windler EE, Chen GC, Havel RJ. Hepatic catabolism of rat and human lipoproteins in rats treated with 17 alpha-ethinyl estradiol. J Biol Chem. (1979) 254(22):11360–6. doi: 10.1016/S0021-9258(19)86494-3
61. Devaraj S, Semaan JR, Jialal I. Biochemistry, apolipoprotein B. In: StatPearls. Treasure Island (FL): StatPearls Publishing (2024). Available at: http://www.ncbi.nlm.nih.gov/books/NBK538139/ (Accessed August 22, 2024).
62. Coto E, Lorca R, Rodríguez-Reguero J, Martín M, Pascual I, Avanzas P, et al. The APOB polymorphism rs1801701 A/G (p.R3638Q) is an independent risk factor for early-onset coronary artery disease: data from a Spanish cohort. Nutr Metab Cardiovasc Dis. (2021) 31(5):1564–8. doi: 10.1016/j.numecd.2021.02.010
63. Colvin PL. Estrogen increases low-density lipoprotein receptor-independent catabolism of apolipoprotein B in hyperlipidemic rabbits. Metab Clin Exp. (1996) 45(7):889–96. doi: 10.1016/S0026-0495(96)90165-1
64. Puteri MU, Azmi NU, Kato M, Saputri FC. PCSK9 Promotes cardiovascular diseases: recent evidence about its association with platelet activation-induced myocardial infarction. Life (Basel). (2022) 12(2):190. doi: 10.3390/life12020190
65. Fu W, Gao XP, Zhang S, Dai YP, Zou WJ, Yue LM. 17β-Estradiol Inhibits PCSK9-mediated LDLR degradation through GPER/PLC activation in HepG2 cells. Front Endocrinol. (2019) 10:930. doi: 10.3389/fendo.2019.00930
66. Ghosh M, Gälman C, Rudling M, Angelin B. Influence of physiological changes in endogenous estrogen on circulating PCSK9 and LDL cholesterol. J Lipid Res. (2015) 56(2):463–9. doi: 10.1194/jlr.M055780
67. Guo W, Fu J, Chen X, Gao B, Fu Z, Fan H, et al. The effects of estrogen on Serum level and hepatocyte expression of PCSK9. Metab Clin Exp. (2015) 64(4):554–60. doi: 10.1016/j.metabol.2015.01.009
68. Wang L, Chen J, Zeng Y, Wei J, Jing J, Li G, et al. Functional variant in the SLC22A3-LPAL2-LPA gene cluster contributes to the severity of coronary artery disease. Arterioscler, Thromb, Vasc Biol. (2016) 36(9):1989–96. doi: 10.1161/ATVBAHA.116.307311
69. Trégouët DA, König IR, Erdmann J, Munteanu A, Braund PS, Hall AS, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. (2009) 41(3):283–5. doi: 10.1038/ng.314
70. Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. (2016) 57(8):1339–59. doi: 10.1194/jlr.R067314
71. Wu Z, Sheng H, Chen Y, Tang J, Liu Y, Chen Q, et al. Copy number variation of the lipoprotein(a) (LPA) gene is associated with coronary artery disease in a southern han Chinese population. Int J Clin Exp Med. (2014) 7(10):3669–77.25419416
72. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy and risk of future cardiovascular events. J Am Coll Cardiol. (2008) 52(2):124–31. doi: 10.1016/j.jacc.2008.04.009
73. Duan LP, Wang HH, Ohashi A, Wang DQH. Role of intestinal sterol transporters ABCG5, ABCG8, and NPC1L1 in cholesterol absorption in mice: gender and age effects. Am J Physiol Gastrointest Liver Physiol. (2006) 290(2):G269–276. doi: 10.1152/ajpgi.00172.2005
74. Miwa K, Inazu A, Kobayashi J, Higashikata T, Nohara A, Kawashiri M, et al. ATP-binding cassette transporter G8 M429V polymorphism as a novel genetic marker of higher cholesterol absorption in hypercholesterolaemic Japanese subjects. Clin Sci. (2005) 109(2):183–8. doi: 10.1042/CS20050030
75. Tada H, Nohara A, Inazu A, Sakuma N, Mabuchi H, Kawashiri M. Sitosterolemia, hypercholesterolemia, and coronary artery disease. J Atheroscler Thromb. (2018) 25(9):783–9. doi: 10.5551/jat.RV17024
76. Jakel H, Nowak M, Helleboid-Chapman A, Fruchart-Najib J, Fruchart J. Is apolipoprotein A5 a novel regulator of triglyceride-rich lipoproteins? Ann Med. (2006) 38(1):2–10. doi: 10.1080/07853890500407488
77. Wang F, Wang I, Ellis S, Archacki S, Barnard J, Hubbard C, et al. Analysis of causal effect of APOA5 variants on premature coronary artery disease. Ann Hum Genet. (2018) 82(6):437–47. doi: 10.1111/ahg.12273
78. Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. (2001) 294(5540):169–73. doi: 10.1126/science.1064852
79. Wójcik C, Fazio S, McIntyre AD, Hegele RA. Co-occurrence of heterozygous CREB3L3 and APOA5 nonsense variants and polygenic risk in a patient with severe hypertriglyceridemia exacerbated by estrogen administration. J Clin Lipidol. (2018) 12(5):1146–50. doi: 10.1016/j.jacl.2018.05.014
80. Luo F, Guo Y, Ruan Gy, Peng R, Li Xp. Estrogen lowers triglyceride via regulating hepatic APOA5 expression. Lipids Health Dis. (2017) 16:72. doi: 10.1186/s12944-017-0463-0
81. Ma W, Ren X, Zhang L, Dong H, Lu X, Feng W. Apolipoprotein E gene polymorphism and coronary artery disease risk among patients in northwest China. Pharmgenomics Pers Med. (2021) 14:1591–9. doi: 10.2147/PGPM.S338285
82. Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med. (2016) 94(7):739–46. doi: 10.1007/s00109-016-1427-y
83. Alagarsamy J, Jaeschke A, Hui DY. Apolipoprotein E in cardiometabolic and neurological health and diseases. Int J Mol Sci. (2022) 23(17):9892. doi: 10.3390/ijms23179892
84. Valencia-Olvera AC, Maldonado Weng J, Christensen A, LaDu MJ, Pike CJ. Role of estrogen in women’s Alzheimer’s disease risk as modified by APOE. J Neuroendocrinol. (2023) 35(2):e13209. doi: 10.1111/jne.13209
85. Srivastava RAK, Srivastava N, Averna M, Lin RC, Korach KS, Lubahn DB, et al. Estrogen up-regulates apolipoprotein E (ApoE) gene expression by increasing ApoE mRNA in the translating pool via the estrogen receptor α-mediated pathway*. J Biol Chem. (1997) 272(52):33360–6. doi: 10.1074/jbc.272.52.33360
86. Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein E–deficient mouse. Arterioscler, Thromb, Vasc Biol. (2004) 24(6):1006–14. doi: 10.1161/01.ATV.0000128849.12617.f4
87. Koenig SN, Sucharski HC, Jose EM, Dudley EK, Madiai F, Cavus O, et al. Inherited variants in SCARB1 cause severe early-onset coronary artery disease. Circ Res. (2021) 129(2):296–307. doi: 10.1161/CIRCRESAHA.120.318793
88. Stangl H, Graf GA, Yu L, Cao G, Wyne K. Effect of estrogen on scavenger receptor BI expression in the rat. (2002). Available at: https://joe.bioscientifica.com/view/journals/joe/175/3/663.xml (Accessed August 22, 2024).
89. Vargas-Alarcón G, Posadas-Romero C, Villarreal-Molina T, Alvarez-León E, Angeles J, Vallejo M, et al. Single nucleotide polymorphisms within LIPA (lysosomal acid lipase A) gene are associated with susceptibility to premature coronary artery disease. a replication in the genetic of atherosclerotic disease (GEA) Mexican study. PLoS One. (2013) 8(9):e74703. doi: 10.1371/journal.pone.0074703
90. Pasta A, Borro P, Cremonini AL, Formisano E, Tozzi G, Cecchi S, et al. Effect of a common missense variant in LIPA gene on fatty liver disease and lipid phenotype: new perspectives from a single-center observational study. Pharmacol Res Perspect. (2021) 9(5):e00820. doi: 10.1002/prp2.820
91. Cai P, Zhang W, Jiang S, Xiong Y, Qiao H, Yuan H, et al. Role of mn-LIPA in sex hormone regulation and gonadal development in the oriental river prawn, macrobrachium nipponense. Int J Mol Sci. (2024) 25(3):1399. doi: 10.3390/ijms25031399
92. Li W, Wang Y, Huang R, Lian F, Xu G, Wang W, et al. Association of lipid metabolism-related gene promoter methylation with risk of coronary artery disease. Mol Biol Rep. (2022) 49(10):9373–8. doi: 10.1007/s11033-022-07789-0
93. Gautier T, Masson D, Jong MC, Duverneuil L, Le Guern N, Deckert V, et al. Apolipoprotein CI deficiency markedly augments plasma lipoprotein changes mediated by human cholesteryl ester transfer protein (CETP) in CETP transgenic/ApoCI-knocked out mice. J Biol Chem. (2002) 277(35):31354–63. doi: 10.1074/jbc.M203151200
94. Rouland A, Masson D, Lagrost L, Vergès B, Gautier T, Bouillet B. Role of apolipoprotein C1 in lipoprotein metabolism, atherosclerosis and diabetes: a systematic review. Cardiovasc Diabetol. (2022) 21(1):272. doi: 10.1186/s12933-022-01703-5
95. Yang S, Du J, Wang W, Zhou D, Xi X. APOC1 Is a prognostic biomarker associated with M2 macrophages in ovarian cancer. BMC Cancer. (2024) 24(1):364. doi: 10.1186/s12885-024-12105-z
96. Wild RA. Dyslipidemia in PCOS. Steroids. (2012) 77(4):295–9. doi: 10.1016/j.steroids.2011.12.002
97. KCNE2 potassium voltage-gated channel subfamily E regulatory subunit 2 [Homo sapiens (human)]—Gene—NCBI. Available at: https://www.ncbi.nlm.nih.gov/gene/9992 (Accessed August 22, 2024).
98. Lee SM, Nguyen D, Hu Z, Abbott GW. KCNE2 deletion promotes atherosclerosis and diet-dependent sudden death. J Mol Cell Cardiol. (2015) 87:148–51. doi: 10.1016/j.yjmcc.2015.08.013
99. Lisewski U, Köhncke C, Schleussner L, Purfürst B, Lee SM, De Silva A, et al. Hypochlorhydria reduces mortality in heart failure caused by KCNE2 gene deletion. FASEB J. (2020) 34(8):10699–719. doi: 10.1096/fj.202000013RR
100. Kundu P, Ciobotaru A, Foroughi S, Toro L, Stefani E, Eghbali M. Hormonal regulation of cardiac KCNE2 gene expression. Mol Cell Endocrinol. (2008) 292(1–2):50–62. doi: 10.1016/j.mce.2008.06.003
101. Alaynick WA, Way JM, Wilson SA, Benson WG, Pei L, Downes M, et al. ERRγ regulates cardiac, gastric, and renal potassium homeostasis. Mol Endocrinol. (2010) 24(2):299–309. doi: 10.1210/me.2009-0114
103. Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler, Thromb, Vasc Biol. (2014) 34(9):1871–9. doi: 10.1161/ATVBAHA.114.303393
104. Ma L, Bryce NS, Turner AW, Di Narzo AF, Rahman K, Xu Y, et al. The HDAC9-associated risk locus promotes coronary artery disease by governing TWIST1. PLoS Genet. (2022) 18(6):e1010261. doi: 10.1371/journal.pgen.1010261
105. Yang C, Croteau S, Hardy P. Histone deacetylase (HDAC) 9: versatile biological functions and emerging roles in human cancer. Cell Oncol. (2021) 44(5):997–1017. doi: 10.1007/s13402-021-00626-9
106. Shetty R, Joshi D, Jain M, Vasudevan M, Paul JC, Bhat G, et al. Rudhira/BCAS3 is essential for mouse development and cardiovascular patterning. Sci Rep. (2018) 8(1):5632. doi: 10.1038/s41598-018-24014-w
107. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. (2015) 47(10):1121–30. doi: 10.1038/ng.3396
108. Gururaj AE, Singh RR, Rayala SK, Holm C, den Hollander P, Zhang H, et al. MTA1, A transcriptional activator of breast cancer amplified sequence 3. Proc Natl Acad Sci U S A. (2006) 103(17):6670–5. doi: 10.1073/pnas.0601989103
109. Plaisier E, Ronco P. COL4A1-Related Disorders. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle (1993). Available at: http://www.ncbi.nlm.nih.gov/books/NBK7046/ (Accessed August 22, 2024).
110. Yang W, Ng FL, Chan K, Pu X, Poston RN, Ren M, et al. Coronary-Heart-Disease-Associated genetic variant at the COL4A1/COL4A2 locus affects COL4A1/COL4A2 expression, vascular cell survival, atherosclerotic plaque stability and risk of myocardial infarction. PLoS Genet. (2016) 12(7):e1006127. doi: 10.1371/journal.pgen.1006127
111. Derynck R, Zhang YE. Smad-dependent and smad-independent pathways in TGF-β family signalling. Nature. (2003) 425(6958):577–84. doi: 10.1038/nature02006
112. Meng Xm, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. (2016) 12(6):325–38. doi: 10.1038/nrneph.2016.48
113. Turner AW, Nikpay M, Silva A, Lau P, Martinuk A, Linseman TA, et al. Functional interaction between COL4A1/COL4A2 and SMAD3 risk loci for coronary artery disease. Atherosclerosis. (2015) 242(2):543–52. doi: 10.1016/j.atherosclerosis.2015.08.008
114. Ito I, Hanyu A, Wayama M, Goto N, Katsuno Y, Kawasaki S, et al. Estrogen inhibits transforming growth factor β signaling by promoting SMAD2/3 degradation. J Biol Chem. (2010) 285(19):14747–55. doi: 10.1074/jbc.M109.093039
115. Silbiger S, Lei J, Ziyadeh FN, Neugarten J. Estradiol reverses TGF-beta1-stimulated type IV collagen gene transcription in murine mesangial cells. Am J Physiol. (1998) 274(6):F1113–1118. doi: 10.1152/ajprenal.1998.274.6.F1113
116. InterPro. Available at: https://www.ebi.ac.uk/interpro/entry/InterPro/IPR001128/ (Accessed August 22, 2024).
117. PubChem. CYP17A1—cytochrome P450 family 17 subfamily A member 1 (human). Available at: https://pubchem.ncbi.nlm.nih.gov/gene/CYP17A1/human (Accessed August 22, 2024).
118. Wu TT, Zheng YY, Ma X, Xiu WJ, Yang HT, Hou XG, et al. Mutated CYP17A1 promotes atherosclerosis and early-onset coronary artery disease. Cell Commun Signal. (2023) 21(1):155. doi: 10.1186/s12964-023-01061-z
119. Marsh CA, Auchus RJ. Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17,20-lyase deficiency and isolated 17,20-lyase deficiency. Fertil Steril. (2014) 101(2):317–22. doi: 10.1016/j.fertnstert.2013.11.011
120. Camats N, Üstyol A, Atabek ME, Dick B, Flück CE. A novel CYP17A1 deletion causes a functional knockout of the steroid enzyme 17-hydroxylase and 17,20-lyase in a turkish family and illustrates the precise role of the CYP17A1 gene. Clin Case Rep. (2015) 3(10):793–7. doi: 10.1002/ccr3.343
121. Zhao D, Fan Y, Xiong X, Yin S, Fu W, Ma Y, et al. Molecular characterization of TRIB1 gene and its role in regulation of steroidogenesis in bos grunniens granulosa cells. Theriogenology. (2022) 191:1–9. doi: 10.1016/j.theriogenology.2022.07.012
122. Yoshimoto FK, Auchus RJ. The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J Steroid Biochem Mol Biol. (2015) 151:52–65. doi: 10.1016/j.jsbmb.2014.11.026
123. Lee JH, Shin DJ, Park S, Kang SM, Jang Y, Lee SH. Association between CDH13 variants and cardiometabolic and vascular phenotypes in a Korean population. Yonsei Med J. (2013) 54(6):1305–12. doi: 10.3349/ymj.2013.54.6.1305
124. Putku M, Kals M, Inno R, Kasela S, Org E, Kožich V, et al. CDH13 Promoter SNPs with pleiotropic effect on cardiometabolic parameters represent methylation QTLs. Hum Genet. (2015) 134(3):291–303. doi: 10.1007/s00439-014-1521-6
125. Fujishima Y, Maeda N, Matsuda K, Masuda S, Mori T, Fukuda S, et al. Adiponectin association with T-cadherin protects against neointima proliferation and atherosclerosis. FASEB J. (2017) 31(4):1571–83. doi: 10.1096/fj.201601064R
126. Song SS, Kang S, Park S. Association of estrogen-related polygenetic risk scores with breast cancer and interactions with alcohol intake, early menarche, and nulligravida. Asian Pac J Cancer Prev. (2022) 23(1):13–24. doi: 10.31557/APJCP.2022.23.1.13
127. Zhang S, Ding Y, Feng F, Gao Y. The role of blood CXCL12 level in prognosis of coronary artery disease: a meta-analysis. Front Cardiovasc Med. (2022) 9:938540. doi: 10.3389/fcvm.2022.938540
128. Döring Y, van der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, et al. CXCL12 Derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation. (2019) 139(10):1338–40. doi: 10.1161/CIRCULATIONAHA.118.037953
129. Rios Coronado PE, Zanetti D, Zhou J, Naftaly JA, Prabala P, Martínez Jaimes AM, et al. CXCL12 drives natural variation in coronary artery anatomy across diverse populations. medRxiv (2024 July 5);2023.10.27.23297507.
130. Hu T, Jiang Y, Yang JS, Hu FJ, Yuan Y, Liu JC, et al. Investigation of autophagy-related genes and immune infiltration in calcific aortic valve disease: a bioinformatics analysis and experimental validation. Exp Ther Med. (2024) 27(5):233. doi: 10.3892/etm.2024.12521
131. Boudot A, Kerdivel G, Habauzit D, Eeckhoute J, Le Dily F, Flouriot G, et al. Differential estrogen-regulation of CXCL12 chemokine receptors, CXCR4 and CXCR7, contributes to the growth effect of estrogens in breast cancer cells. PLoS One. (2011) 6(6):e20898. doi: 10.1371/journal.pone.0020898
132. Zhang L, Sui R. Effect of SNP polymorphisms of EDN1, EDNRA, and EDNRB gene on ischemic stroke. Cell Biochem Biophys. (2014) 70(1):233–9. doi: 10.1007/s12013-014-9887-6
133. Hasegawa K, Fujiwara H, Doyama K, Inada T, Ohtani S, Fujiwara T, et al. Endothelin-1-selective receptor in the arterial intima of patients with hypertension. Hypertension. (1994) 23(3):288–93. doi: 10.1161/01.HYP.23.3.288
134. Keator CS, Mah K, Ohm L, Slayden OD. Estrogen and progesterone regulate expression of the endothelins in the rhesus macaque endometrium. Hum Reprod. (2011) 26(7):1715–28. doi: 10.1093/humrep/der115
135. NT5C2 5′-nucleotidase, cytosolic II [Homo sapiens (human)]—Gene—NCBI. Available at: https://www.ncbi.nlm.nih.gov/gene/22978 (Accessed August 23, 2024).
136. Chen YT, Lin WD, Liao WL, Tsai YC, Liao JW, Tsai FJ. NT5C2 Methylation regulatory interplay between DNMT1 and insulin receptor in type 2 diabetes. Sci Rep. (2020) 10(1):16087. doi: 10.1038/s41598-020-71336-9
137. Shen Z, Fahey JV, Bodwell JE, Rodriguez-Garcia M, Rossoll RM, Crist SG, et al. Estradiol regulation of nucleotidases in female reproductive tract epithelial cells and fibroblasts. PLoS One. (2013) 8(7):e69854. doi: 10.1371/journal.pone.0069854
138. Vishnolia KK, Hoene C, Tarhbalouti K, Revenstorff J, Aherrahrou Z, Erdmann J. Studies in zebrafish demonstrate that CNNM2 and NT5C2 are most likely the causal genes at the blood pressure-associated locus on human chromosome 10q24.32. Front Cardiovasc Med. (2020) 7:135. doi: 10.3389/fcvm.2020.00135
139. Xie Y, Martin KA. TCF21—flipping The phenotypic switch in SMC. Circ Res. (2020) 126(4):530–2. doi: 10.1161/CIRCRESAHA.120.316533
140. Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T, et al. Coronary disease associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-Serum response factor pathway. Circ Res. (2020) 126(4):517–29. doi: 10.1161/CIRCRESAHA.119.315968
141. Kim JB, Pjanic M, Nguyen T, Miller CL, Iyer D, Liu B, et al. TCF21 And the environmental sensor aryl-hydrocarbon receptor cooperate to activate a pro-inflammatory gene expression program in coronary artery smooth muscle cells. PLoS Genet. (2017) 13(5):e1006750. doi: 10.1371/journal.pgen.1006750
142. Zhu J, Wu P, Zeng C, Xue Q. Increased SUMOylation of TCF21 improves its stability and function in human endometriotic stromal cells†. Biol Reprod. (2021) 105(1):128–36. doi: 10.1093/biolre/ioab038
143. PHACTR1 phosphatase and actin regulator 1 [Homo sapiens (human)]—Gene—NCBI. Available at: https://www.ncbi.nlm.nih.gov/gene/221692 (Accessed August 22, 2024).
144. Rezvan A. PHACTR1 And atherosclerosis: it’s complicated. Arterioscler, Thromb, Vasc Biol. (2023) 43(8):1409–11. doi: 10.1161/ATVBAHA.123.319545
145. Herman L, Legois B, Todeschini AL, Veitia RA. Genomic exploration of the targets of FOXL2 and ESR2 unveils their implication in cell migration, invasion, and adhesion. FASEB J. (2021) 35(4):e21355. doi: 10.1096/fj.202002444R
146. Mauersberger C, Sager HB, Wobst J, Dang TA, Lambrecht L, Koplev S, et al. Loss of soluble guanylyl cyclase in platelets contributes to atherosclerotic plaque formation and vascular inflammation. Nat Cardiovasc Res. (2022) 1(12):1174–86. doi: 10.1038/s44161-022-00175-w
147. Pino MTL, Ronchetti SA, Cordeiro G, Bollani S, Duvilanski BH, Cabilla JP. Soluble guanylyl cyclase Alpha1 subunit: a new marker for estrogenicity of endocrine disruptor compounds. Environ Toxicol Chem. (2019) 38(12):2719–28. doi: 10.1002/etc.4591
148. Drinane MC, Sherman JA, Hall AE, Simons M, Mulligan-Kehoe MJ. Plasminogen and plasmin activity in patients with coronary artery disease. J Thromb Haemost. (2006) 4(6):1288–95. doi: 10.1111/j.1538-7836.2006.01979.x
149. PLG plasminogen [Homo sapiens (human)]—Gene—NCBI. Available at: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=5340 (Accessed August 22, 2024).
150. Kobelt L, Klammt J, Tefs K, Schuster V. Estrogen modulates plasminogen promoter activity. Biochem Biophys Res Commun. (2013) 438(1):110–5. doi: 10.1016/j.bbrc.2013.07.035
151. Maurer LM, Tomasini-Johansson BR, Mosher DF. Emerging roles of fibronectin in thrombosis. Thromb Res. (2010) 125(4):287–91. doi: 10.1016/j.thromres.2009.12.017
152. Doddapattar P, Gandhi C, Prakash P, Dhanesha N, Grumbach IM, Dailey ME, et al. Fibronectin splicing variants containing extra domain A promote atherosclerosis in mice through toll-like receptor 4. Arterioscler, Thromb, Vasc Biol. (2015) 35(11):2391–400. doi: 10.1161/ATVBAHA.115.306474
153. Schiavi J, Fodera DM, Brennan MA, McNamara LM. Estrogen depletion alters osteogenic differentiation and matrix production by osteoblasts in vitro. Exp Cell Res. (2021) 408(1):112814. doi: 10.1016/j.yexcr.2021.112814
154. Quinn JA, Graeber CT, Frackelton AR, Kim M, Schwarzbauer JE, Filardo EJ. Coordinate regulation of estrogen-mediated fibronectin matrix assembly and epidermal growth factor receptor transactivation by the G protein-coupled receptor, GPR30. Mol Endocrinol. (2009) 23(7):1052–64. doi: 10.1210/me.2008-0262
155. Koyama S, Ito K, Terao C, Akiyama M, Horikoshi M, Momozawa Y, et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet. (2020) 52(11):1169–77. doi: 10.1038/s41588-020-0705-3
156. Tcheandjieu C, Zhu X, Hilliard AT, Clarke SL, Napolioni V, Ma S, et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat Med. (2022) 28(8):1679–92. doi: 10.1038/s41591-022-01891-3
157. Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat Genet. (2020) 52(7):669–79. doi: 10.1038/s41588-020-0640-3
158. Travis RC, Key TJ. Oestrogen exposure and breast cancer risk. Breast Cancer Res. (2003) 5(5):239–47. doi: 10.1186/bcr628
159. Pott J, Bae YJ, Horn K, Teren A, Kühnapfel A, Kirsten H, et al. Genetic association study of eight steroid hormones and implications for sexual dimorphism of coronary artery disease. J Clin Endocrinol Metab. (2019) 104(11):5008–23. doi: 10.1210/jc.2019-00757
160. Maas AHEM, Appelman YEA. Gender differences in coronary heart disease. Neth Heart J. (2010) 18(12):598–602. doi: 10.1007/s12471-010-0841-y
161. Antikainen AAV, Sandholm N, Trégouët DA, Charmet R, McKnight AJ, Ahluwalia TS, et al. Genome-wide association study on coronary artery disease in type 1 diabetes suggests beta-defensin 127 as a risk locus. Cardiovasc Res. (2020) 117(2):600–12. doi: 10.1093/cvr/cvaa045
162. Assimes TL, Lee IT, Juang JM, Guo X, Wang TD, Kim ET, et al. Genetics of coronary artery disease in Taiwan: a cardiometabochip study by the taichi consortium. PLoS One. (2016) 11(3):e0138014. doi: 10.1371/journal.pone.0138014
163. Barbalic M, Reiner AP, Wu C, Hixson JE, Franceschini N, Eaton CB, et al. Genome-wide association analysis of incident coronary heart disease (CHD) in African Americans: a short report. PLoS Genet. (2011) 7(8):e1002199. doi: 10.1371/journal.pgen.1002199
164. Charmet R, Duffy S, Keshavarzi S, Gyorgy B, Marre M, Rossing P, et al. Novel risk genes identified in a genome-wide association study for coronary artery disease in patients with type 1 diabetes. Cardiovasc Diabetol. (2018) 17(1):61. doi: 10.1186/s12933-018-0705-0
165. Coronary Artery Disease Consortium, Samani NJ, Deloukas P, Erdmann J, Hengstenberg C, Kuulasmaa K, et al. Large scale association analysis of novel genetic loci for coronary artery disease. Arterioscler, Thromb, Vasc Biol. (2009) 29(5):774–80. doi: 10.1161/ATVBAHA.108.181388
166. Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in europeans and south asians identifies five new loci for coronary artery disease. Nat Genet. (2011) 43(4):339–44. doi: 10.1038/ng.782
167. Davies RW, Wells GA, Stewart AFR, Erdmann J, Shah SH, Ferguson JF, et al. A genome-wide association study for coronary artery disease identifies a novel susceptibility locus in the major histocompatibility complex. Circ Cardiovasc Genet. (2012) 5(2):217–25. doi: 10.1161/CIRCGENETICS.111.961243
168. Fall T, Gustafsson S, Orho-Melander M, Ingelsson E. Genome-wide association study of coronary artery disease among individuals with diabetes: the UK biobank. Diabetologia. (2018) 61(10):2174–9. doi: 10.1007/s00125-018-4686-z
169. Feng T, Zhu X. Genome-wide searching of rare genetic variants in WTCCC data. Hum Genet. (2010) 128(3):269–80. doi: 10.1007/s00439-010-0849-9
170. Franceschini N, Carty C, Bůzková P, Reiner AP, Garrett T, Lin Y, et al. Association of genetic variants and incident coronary heart disease in multiethnic cohorts: the PAGE study. Circ Cardiovasc Genet. (2011) 4(6):661–72. doi: 10.1161/CIRCGENETICS.111.960096
171. Howson JMM, Zhao W, Barnes DR, Ho WK, Young R, Paul DS, et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. (2017) 49(7):1113–9. doi: 10.1038/ng.3874
172. IBC 50K CAD Consortium. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. (2011) 7(9):e1002260. doi: 10.1371/journal.pgen.1002260
173. Lee JY, Lee BS, Shin DJ, Woo Park K, Shin YA, Joong Kim K, et al. A genome-wide association study of a coronary artery disease risk variant. J Hum Genet. (2013) 58(3):120–6. doi: 10.1038/jhg.2012.124
174. Lu X, Wang L, Chen S, He L, Yang X, Shi Y, et al. Genome-wide association study in han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. (2012) 44(8):890–4. doi: 10.1038/ng.2337
175. Matsunaga H, Ito K, Akiyama M, Takahashi A, Koyama S, Nomura S, et al. Transethnic meta-analysis of genome-wide association studies identifies three new loci and characterizes population-specific differences for coronary artery disease. Circ Genom Precis Med. (2020) 13(3):e002670. doi: 10.1161/CIRCGEN.119.002670
176. Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. (2017) 49(9):1385–91. doi: 10.1038/ng.3913
177. Preuss M, König IR, Thompson JR, Erdmann J, Absher D, Assimes TL, et al. Design of the coronary ARtery DIsease genome-wide replication and meta-analysis (CARDIoGRAM) study: a genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet. (2010) 3(5):475–83. doi: 10.1161/CIRCGENETICS.109.899443
178. Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. (2011) 377(9763):383–92. doi: 10.1016/S0140-6736(10)61996-4
179. Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. (2011) 43(4):333–8. doi: 10.1038/ng.784
180. Takeuchi F, Yokota M, Yamamoto K, Nakashima E, Katsuya T, Asano H, et al. Genome-wide association study of coronary artery disease in the Japanese. Eur J Hum Genet. (2012) 20(3):333–40. doi: 10.1038/ejhg.2011.184
181. Tam CHT, Lim CKP, Luk AOY, Shi M, Man Cheung H, Ng ACW, et al. Identification of a common variant for coronary heart disease at PDE1A contributes to individualized treatment goals and risk stratification of cardiovascular complications in Chinese patients with type 2 diabetes. Diabetes Care. (2023) 46(6):1271–81. doi: 10.2337/dc22-2331
182. van Zuydam NR, Ladenvall C, Voight BF, Strawbridge RJ, Fernandez-Tajes J, Rayner NW, et al. Genetic predisposition to coronary artery disease in type 2 diabetes Mellitus. Circ Genom Precis Med. (2020) 13(6):e002769. doi: 10.1161/CIRCGEN.119.002769
183. Wakil SM, Ram R, Muiya NP, Mehta M, Andres E, Mazhar N, et al. A genome-wide association study reveals susceptibility loci for myocardial infarction/coronary artery disease in Saudi arabs. Atherosclerosis. (2016) 245:62–70. doi: 10.1016/j.atherosclerosis.2015.11.019
184. Wakim V, Abi Khalil E, Salloum AK, Khazen G, Ghassibe-Sabbagh M, Zalloua PA. New susceptibility alleles associated with severe coronary artery stenosis in the Lebanese population. BMC Med Genomics. (2021) 14(1):90. doi: 10.1186/s12920-021-00942-x
185. Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NGD, Jansen H, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. (2017) 69(7):823–36. doi: 10.1016/j.jacc.2016.11.056
186. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447(7145):661–78. doi: 10.1038/nature05911
187. Yamada Y, Yasukochi Y, Kato K, Oguri M, Horibe H, Fujimaki T, et al. Identification of 26 novel loci that confer susceptibility to early-onset coronary artery disease in a Japanese population. Biomed Rep. (2018) 9(5):383–404. doi: 10.3892/br.2018.1152
Keywords: coronary artery disease, genome wide association study, estrogen, gene candidates, sex differences
Citation: Aminbakhsh AP, Théberge ET, Burden E, Adejumo CK, Gravely AK, Lehman A and Sedlak TL (2025) Exploring associations between estrogen and gene candidates identified by coronary artery disease genome-wide association studies. Front. Cardiovasc. Med. 12:1502985. doi: 10.3389/fcvm.2025.1502985
Received: 27 September 2024; Accepted: 4 March 2025;
Published: 20 March 2025.
Edited by:
Anita Cote, Trinity Western University, CanadaReviewed by:
Emma Louise Robinson, Edwards Lifesciences, United StatesCopyright: © 2025 Aminbakhsh, Théberge, Burden, Adejumo, Gravely, Lehman and Sedlak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tara L. Sedlak, VGFyYS5TZWRsYWtAdmNoLmNh
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.