 Alexander Burashnikov
Alexander Burashnikov- Lankenau Institute for Medical Research, Wynnewood, PA, United States
The primary electrophysiological mechanism of atrial fibrillation (AF) maintenance is poorly defined. AF mapping studies readily record focal activations (defining them as focal sources or breakthroughs) and “incomplete reentries” (defining them as reentries or would-be-reentries) but do not or rarely detect complete circular activations. Electrophysiological alterations induced by anti-AF drugs before AF cardioversion may help delineate the mechanism of AF maintenance. Cardioversion of AF by antiarrhythmic drugs is associated with prolongation of the AF cycle length and temporal excitable gap (t-EG), resulting in improvement in AF organization (AF-org), and with or without alterations in the refractory period, conduction velocity and wavelength. Such electrophysiological pattern is conceivable with termination of a single focal source but not a single reentry (Class III agents do not increase reentrant t-EG). Yet, a single focal source and multiple focal sources are plausible as the primary mechanism of AF maintenance prior drug administration. Improvement in AF-org caused by anti-AF agents before AF cardioversion is coherent with simultaneous multiple random reentries and wavelets. However, simultaneous multiple reentries are unlikely to occur regularly (most of the contemporary AF mapping studies report either a single reentry at a time or no reentry at all), and the ability of random wavelets to maintain AF is speculative. The conducted analysis inclines toward the focal source as the primary mechanism of AF maintenance.
Introduction
The primary electrophysiological mechanism sustaining atrial fibrillation (AF) remains unknown (1–3). The dominant role of reentry in the maintenance of AF, thought proven 1–3 decades ago (4–9), is the subject of debate these days (2, 10–12). Contemporary AF mapping studies either rarely or never detect full circular activations and readily record “incomplete reentries”, defining the latter as “reentries” or “would-be-reentries” depending on the chosen definition of reentry (1, 9–22). Practically all AF mapping studies readily detect focal activation patterns that, however, can reflect true focal sources and breakthroughs (1, 9–22). Generally, current AF mapping technologies do not usually permit the precise establishment of the AF mechanism. Electrophysiological alterations induced by antiarrhythmic drugs prior AF cardioversion may help in delineating the primary mechanism of AF maintenance.
Mechanisms of AF maintenance
Functional reentry was accepted as the primary mechanism of AF maintenance for decades (1, 4, 5, 8, 23–25). From the 1970s to 1990s, the leading circuit reentry was thought to be the main mechanism of AF (4, 5, 23, 24). According to the theory, the leading circuit is determined by the wavelength (WL) and characterized by the absence of the fully excitable gap (EG) and the presence of a refractory core (23, 26). The WL is the product of atrial effective refractory period (ERP) and conduction velocity (CV), which estimates the probability of the leading circuit reentry generation (the shorter the WL, the greater the probability, and vice versa) (26). By about 2000, due to significant inconsistencies of the leading circuit with cardiac fibrillation characteristics, the theory was essentially replaced with the “spiral wave/rotor” concept as the primary mechanism for cardiac fibrillation (1, 25, 27, 28). Spiral wave activity is determined by the sink-source relationship of the inner-curved wavefront tip with the tissue (25, 27, 28). Critical characteristics of the spiral wave are the presence of the EG and an excitable but not excited core (25, 27, 28).
At the beginning of the 20th century, Mines (29), and Lewis (30) suggested that reentry could sustain AF. Around mid-century, Scherf et al. (31) and Prinzmetal et al. (32) reasoned that AF could be maintained by focal source(s). In the 1950s to 1960s, Moe and colleagues proposed that AF was primarily maintained by multiple random wandering wavelets (33, 34). This theory was “upgraded” to the multiple random functional reentry paradigm in the 1980s–1990s (4–7). In fact, it was generally accepted at that point that the main mechanism of AF maintenance was simultaneous random multiple functional reentries (4–7, 24, 35–37). This acceptance seemed largely based on preconceived assumptions rather than reproducible data. In the 1980s–1990s, AF mapping studies detected “incomplete reentries” but not full-circuit reentries (4, 24, 38). After 2000, with improved mapping technologies, reproducible evidence for the existence of simultaneous multiple reentries during AF has not been obtained (10–13, 16, 17, 19–21, 39, 40), suggesting that such multiple reentries are either not detectable with available mapping technologies or do not exist. Recently, Lee et al., using a high-density biatrial epicardial mapping, reported that the maintenance of AF in Moe's original experimental AF model is associated with multiple simultaneous sustained and intermittent focal activation patterns without or only with rare full-circuit reentrant activations (13, 19).
Anatomical reentry may play a critical role in AF maintenance, particular in the setting of significant atrial structural remodeling (40, 41). However, how often anatomical reentries maintain AF is unknown. Most of the AF mapping studies utilizing simultaneous epicardial and endocardial mapping in the setting of significant atrial structural remodeling did not record any anatomical reentries (10, 39, 42, 43). Also, the causative role of atrial structural remodeling in AF generation is controversial (44–48).
In the past 30 years, mostly from the 1990s to early 2010s, some experimental and clinical mapping studies reported that AF could be maintained by a single rotor, causing fibrillatory conduction in the rest of the atria (8, 9, 49). In the last decade, however, the vast majority of AF mapping studies either have recorded sporadic short-lived circular activations (usually 1–3 rotations) or have not recorded any full circular activation at all (11–21), questioning a single stable rotor as the primary mechanism of AF maintenance.
The interpretation of AF mapping data is critical in determining reentry (1, 2, 22, 50–52). Most who consistently detected rotors during AF have used the phase mapping approach for rotor detection (9, 14, 53) [introduced by Gray et al. in the late 1990s (54) and preceded by failure of detecting full-circuit reentries during AF with “conventional” methodologies by this group (38)]. It is well recognized now that the phase mapping method often finds false reentries (12, 18, 55). False reentries (or would-be-reentries) are readily detected during AF (1, 11, 12, 20, 22, 50, 56). In fact, “reentries” are readily detected even at the absence of AF during atrial rapid pacing (51). One would intuitively expect that “incomplete reentries” are natural consequences of any rapid atrial activation(s) in the setting of anatomical and rate-dependent functional heterogeneities in the atrium. How many of these “incomplete reentries” are true reentries is unknown. Interestingly, ablation approaches targeting reentry substrate (i.e., CFAE, FIRM, and fibrosis), initially thought promising, essentially failed to reduce AF occurrence (48, 57–61).
It is well recognized that AF can be maintained by focal sources (11–13, 16, 17, 31, 32, 62–64). Practically all AF mapping studies readily record focal activations (9–13, 15, 16, 20, 21, 39, 62, 63, 65–69) and many AF mapping studies detect either focal activations without reentries or predominantly focal activations (10–13, 16, 17, 20, 66–69). Yet, a lot of the AF mapping investigators record only short-lasting focal activations (commonly ≤3 beats) (10, 15, 21, 39), with most of these focal patterns interpreted as breakthroughs (10, 15, 39). At the same time, many AF mapping studies consistently record both intermittent and sustained focal activations and many of these activations are thought to reflect true focal foci (11–13, 16, 17, 20, 62, 63, 70). Interestingly, electrically isolated conduction pathways, encountered in remodeled atria, may give rise to “trapped reentry” (71). “Trapped reentry” manifests as a focal source (71) that may complicate the discrimination of focal vs. reentrant mechanisms in AF. The contribution of focal sources to the maintenance of AF is poorly understood.
Driven by the absence of evidence for either reentry or focal sources as the primary underlying mechanism of long-standing persistent AF, De Groot et al. have suggested that long-standing persistent AF is maintained by constant aberrant multiwavelets propagating between epicardium and endocardium, secondary to atrial structural remodeling (i.e., the double-layer hypothesis) (10, 39). The basis for the double-layer AF mechanism is a major epi-endocardial electrical dissociation, mainly due to significant atrial structural remodeling. It was estimated that about 400 new wavelets are generated every second during longstanding persistent AF, sufficient to maintain AF (10). Note, while circular activation patterns were not or rarely observed in De Groot et al. studies, focal activation patterns were readily recorded; however, these focal activations were short-lived and the majority were deduced as breakthroughs (10, 39, 72). Interestingly, Lee et al. also did not or rarely detect full-circular activation pattens and consistently recorded focal activations during long-standing persistent AF (11, 12, 16). These foci, however, were both sustained (≥32 s) and intermittent (commonly firing simultaneously), and many of these focal activations were deduced as real focal sources maintaining AF (11, 12, 16).
There is some resemblance between the double-layer hypothesis (10, 39) and the original multiple wavelet hypothesis developed by Moe et al. in the 1950s and 1960s (34), i.e., in both theories AF is maintained by the multiple random wandering wavelets. It is practically impossible to prove or disprove the original multiple wavelet and double-layer hypotheses with current AF mapping technologies. Yet, while the ability of random multiple wavelets to maintain AF is speculative (1, 2), random multiple wavelets (or “fibrillatory conduction”) can be readily generated by a single stationary source (8, 13, 19, 49).
There are important limitations of the currently available AF mapping technologies, usually not permitting a precise discrimination of arrhythmic mechanisms maintaining AF (1). These limitations are 1) commonly insufficient resolution, 2) restricted atrial mapping area (usually a part of epicardial surface), 3) the absence or partial information on the intramural activation, 4) uncertainties in the interpretation of signals/electrograms, 5) short duration of uninterrupted mapping time (usually ≤30 s), etc (1, 2). For example, most of the small localized “reentries” detected by “conventional” mapping resolution appear to be pseudo-reentries when viewed in high resolution (22, 50). Yet, failure of detecting a full-circuit reentry or multiple simultaneous reentries does not exclude their presence in unmapped areas, particularly intramurally. Another example is that although “clean” focal activations on atrial surface are readily detected during AF with a high resolution, these focal activations can be breakthroughs from a remote source(s) (10). The presence or absence of focal and reentrant activations during AF must be confirmed with comprehensive and reliable 3-dimentional activation maps, not achievable currently (1, 2).
The autonomic nervous system (ANS) may importantly contribute to the generation of AF (most of the supporting data is related to the initiation of AF, with little data being related to the maintenance of AF) (73, 74). Pertinent data on the involvement of ANS in AF maintenance that may help distinguish reentry vs. focal source maintaining AF are scarce, indirect, and doubtful. For example, acetylcholine promotes AF maintenance by shortening atrial repolarization and hyperpolarizing atrial resting membrane potential (38, 49, 75), suggesting reentry but not focal sources as the underlying mechanism of AF maintainance (38, 76). However, during cholinergically-mediated AF mapping studies commonly either do not record complete reentry at all or observe it rarely while readily recording focal activation patterns (12, 13, 38, 77). The issue of AF cardioversion with IKACh blockers is controversial, i.e., IKACh blockers may or may not stop AF (78–81), and the specificity of IKACh blockers is a concern [e.g., XAF-1407 prolongs QRS (79), indicating a major block of the sodium channel]. While β-blockers are commonly inefficient in cardioversion of AF, the interpretation of these data in term of mechanisms maintaining AF is unclear. Sympathetic nervous system may or may not affect both focal and reentrant mechanisms (this issue is poorly understood) (73, 82, 83).
Thus, while reentry was considered proven as the primary mechanism of AF maintenance from at least the 1970s to the 2010s (4–9), its dominance now is debatable (2, 10–12). Contemporary AF mapping studies readily detect focal activations and “partial reentries” but not full circuit activations (10–13, 16, 17, 19, 20, 66, 68, 84). With available technologies, it is difficult or impossible to definitively prove reentrant, wavelet, and focal mechanisms of AF maintenance (1, 2). The primary mechanism of AF maintenance remains poorly defined.
Drug-induced atrial electrophysiological alterations before AF termination
Atrial electrophysiological alterations induced by antiarrhythmic drugs before AF termination may shed light on electrophysiological mechanisms of AF maintenance. It is important to identify changes in electrophysiological parameters that consistently precede drug-induced cardioversion of AF as well as the value of these parameters for the differentiation of AF arrhythmic mechanisms. Alterations of the principal electrophysiological parameters related to AF (such as ERP) in the very last, or last several, AF beats before AF termination are practically unknown (e.g., ERP cannot be determined in the several last beats of AF) or inadequately defined [one study reported CV acceleration immediately before AF cardioversion (85)]. Therefore, the current analysis largely examines sustained drug-induced alterations of the electrophysiological parameters during AF maintenance before AF cardioversion [such alterations have been reported (85–95) or can be reasonably deducted, as discussed later]. To reduce uncertainties in interpretation, mostly data with Class I and III agents were analyzed.
Effective refractory period
Prolongation of atrial ERP by anti-AF agents is a surrogate for estimating anti-AF efficacy of the drugs (3, 96). IKr blockers increase ERP by prolongation of the action potential duration (APD) and INa blockers by induction of the post-repolarization refractoriness (97). When achievable, a significant drug-induced atrial ERP prolongation is likely to be associated with AF cardioversion or the AF rate slowing. However, prolongation of atrial ERP/APD may or may not be attainable with Class I and III agents during AF (86, 98, 99), and both classes can terminate AF without prolongation of atrial ERP (86, 100–102). Interestingly, IKr blockers are generally more efficient in cardioversion of persistent AF than INa blockers (86, 99, 103, 104), despite that Class III should not and Class I should significantly prolong atrial ERP at very rapid activation rates [due to their reverse and use dependency, respectively (3, 105, 106)]. Thus, atrial ERP prolongation cannot be considered a prerequisite for AF termination by anti-AF agents. Also, because atrial ERP prolongation acts to terminate, or at least slow down, all arrhythmic mechanisms, drug-induced ERP prolongation does not seem useful for discriminating among reentrant, wavelet, and focal mechanisms.
Conduction velocity
Slowing of atrial CV during AF generally occurs with inhibition of INa, but not with specific block of IKr (86). Whether INa block-induced CV slowing is involved in AF termination is not clear. In the case of reentry, INa block-induced CV slowing during AF may increase the chance of conduction block (that may lead to reentry termination; Figure 1), but slowing CV itself should promote reentry generation (a basic postulate for reentry) (107). Also, INa block-induced prolongation of the temporal excitable gap (t-EG) during AF (Figure 2; as discussed later) should reduce the chance of conduction block (Figure 1). Interestingly, CV consistently accelerates just before AF cardioversion with potassium and sodium channel blockers (85), likely due to a concomitant prolongation of the t-EG, as discussed later. In the case of a focal source, CV alterations by itself should not directly affect rapid focal source firing. Thus, the limited knowledge about the involvement of CV in drug-induced AF cardioversion is insufficient for the differentiation of arrhythmic mechanisms maintaining AF.
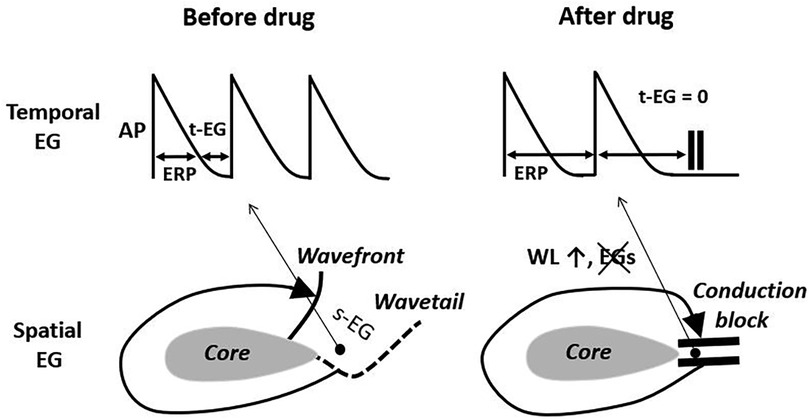
Figure 1. The elimination of the temporal and spatial excitable gaps (t-EG and s-EG, respectively) leading to conduction block is a well recognized mechanism of drug-induced reentry termination. The basis for this mechanism is wavelength (WL) prolongation, secondary to the effective refractory period (ERP) increase. AP, action potential. Pease see text for details.
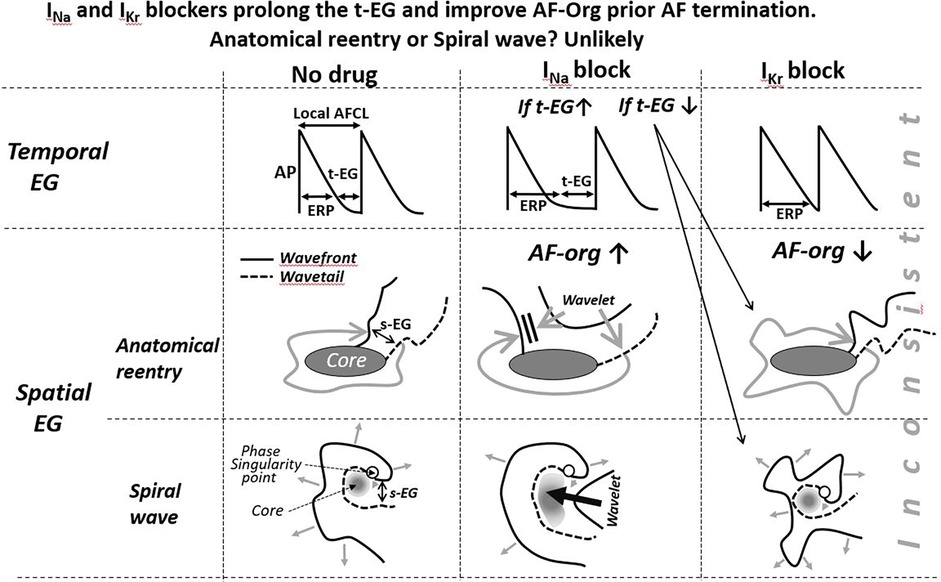
Figure 2. Schematic simplified illustrations of drug-induced alterations of the t-EG, s-EG, and AFCL in anatomical and spiral wave reentry. Maintenance and termination of reentries with prolongation of the EGs are not feasible with IKr blockers (IKr blockers decrease the EGs) and controversially conceivable with INa blockers. To be consistent with mother reentry being the main mechanism of AF, a reasonable explanation(s) for AF maintenance and termination associated with prolongation of the t-EG should be for both INa and IKr blockers. Pease see text for details.
Wavelength
In the 1980s and 1990s, wavelength (WL) was generally considered a predictable parameter for anti-AF efficacy of antiarrhythmic drugs based on numerous studies reporting that both INa and IKr blockers terminated and prevented AF by prolongation of the WL (Figure 1) (5, 108–110). It has become evident over the last two decades that anti-AF agents may effectively terminate AF with prolongation and shortening of the WL as well, without a change of the WL (3, 86, 100). Therefore, WL is not a reliable parameter for predicting drug-induced AF cardioversion. Note that drug-induced prolongation of WL is caused exclusively by ERP lengthening (CV slowing shortens the WL) (3, 5, 109). So, if drugs prolong WL, it is also invariably associated with ERP lengthening that alone may account for the termination of any arrhythmic mechanisms.
AF cycle length
Cardioversion of AF with INa and IKr blockers is quite consistently accompanied by prolongation of AF cycle length (AFCL) (85, 86, 89, 92, 94, 111–116). Yet, drug-induced prolongation of AFCL (i.e., slowing of AF activity) before AF termination is expected with any arrhythmic mechanisms, with apparent exception of anatomical reentry termination by IKr blockers (117). Therefore, drug-induced prolongation of AFCL by itself does not seem to be a reliable parameter for the differentiation of arrhythmic mechanisms.
The excitable gap and AF organization
There are temporal and spatial excitable gaps during AF (t-EG and s-EG, respectively) (86). The t-EG is defined as the difference between the AFCL and ERP at any given point, and the s-EG is defined as the excitable spatial distance between wavefront and wavetail at any given area (i.e., AFCL x CV minus ERP x CV). Spatiotemporal t-EG and s-EG during AF can be estimated but not measured nowadays (it is impossible to measure ERP simultaneously in multiple locations during AF, among many reasons).
Available data indicate that cardioversion of AF with antiarrhythmic agents is preceded by a sustained general prolongation of the t-EG (86, 88, 90) and improvement in AF organization (AF-org) (85–95, 113). The latter by itself demonstrates a sustained general prolongation of the t-EG. Indeed, improvement in AF-org is secondary to the prolongation of a general spatiotemporal t-EG that provides a longer recovery time for the activating wavefronts to become more organized. Drug-induced alteration of the s-EG prior AF termination is more difficult to estimate than the t-EG. Although drug-mediated improvement in AF-org is likely to be associated with a general increase in the s-EG, it is time (i.e., t-EG) and not space (i.e., s-EG) that determines atrial electrical recovery. According to the estimation of Wijffels et al., the s-EG before AF cardioversion (within minutes) was statistically significantly widened by d-sotalol, cibenzoline, and hydroquinidine, but not flecainide (86).
Directly comparing the effect of flecainide, d-sotalol, cibenzoline, and hydroquinidine on principal atrial electrophysiological parameters (ERP, WL, CV, s-EG, and t-EG) before termination of AF (within minutes) in goats, Wijffels et al. found that t-EG prolongation was the only parameter consistently associated with AF cardioversion (86). van Hunnik et al. (85) studied changes in AF organization, AFCL, and CV occurring immediately (within seconds) before spontaneous and drug (potassium and sodium channel blockers)-induced AF termination in goats. AF organization was consistently improved immediately before AF termination in all studied settings, coinciding with AFCL increase and CV acceleration (85). The improvement in AF organization and acceleration in CV were likely due to prolongation of a general t-EG.
In the case of AF sustained by a mother reentry, both the t-EG and s-EG around the reentry core are vital parts of the circuit (Figures 1–3). Drugs may decrease, increase, and not change the EGs around the reentry core (Figures 1–3) (25, 107, 117–119). The elimination of the t-EGs at any point around the reentry core (the t-EG cannot disappear without elimination of the s-EG and vice versa) should lead to conduction block at this point, which may result in termination of the circuit (Figure 1). Prolongation of a general t-EG along the reentrant pathway may make the circuit more stable (by reducing the chance of conduction block; Figure 1) or more vulnerable to termination. The latter may occur secondary to destabilization of the reentrant core by invading wandering wavelets (relevant to functional reentries; Figure 2) (120) or due to collision of the reentrant wavefront with some antidromic wavelets (relevant to anatomical and functional reentries; Figure 2) (119). Importantly, it is drug-induced termination of reentry with elimination of the EGs along the reentrant core that is well-recognized (Figure 1) (96, 107, 109) and drug-induced termination of reentry with prolongation of the EGs around the reentrant core is poorly defined and controversial (Figure 2; as discussed later).
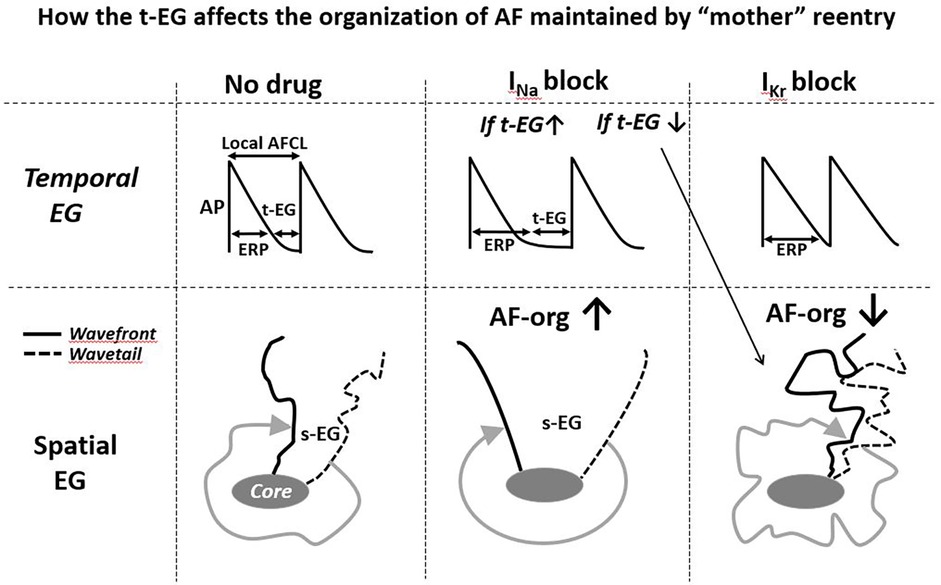
Figure 3. Simplified illustrations of how alterations in the t-EG affect the organization of AF (AF-org) sustianed by mother reentry. Prolongation of the t-EG improves and shortening of the t-EG aggravates AF-org (prolongation of the t-EG should be commonly associated with inrease of the s-EG). INa blockers may both increase and decrease the reentrant EGs. IKr blockers decrease the reentrant EGs. Please see text for details.
In a mother focal source sustaining AF the t-EG is an integral part of this source, affecting the rate of firing (by modulating the recovery of the ion channel currents involved in the firing) and being affected by the rate of firing (i.e., the slower the rate, the greater the t-EG, and vice versa). How and if the s-EGs surrounding a rapid focal source affect the generation of this focal source firing is unknown. Yet, source-sink relationship between focal source and surrounding tissue may change the s-EGs. It has been shown that autonomic influences and frequency of activation may alter source-sink relationship between focal source and surrounding tissue (121, 122), potentially changing the t-EG. How and if anti-AF drugs modify the s-EG surrounding focal source is unknown. Available data indicate that anti-AF agents reduce the firing rate of arrhythmic focal activity (123–126) that should prolong the t-EG in the spot of a rapid focal source (Figure 4). Cessation of rapid focal activity is commonly associated with its deceleration (Figure 4) (127–131).
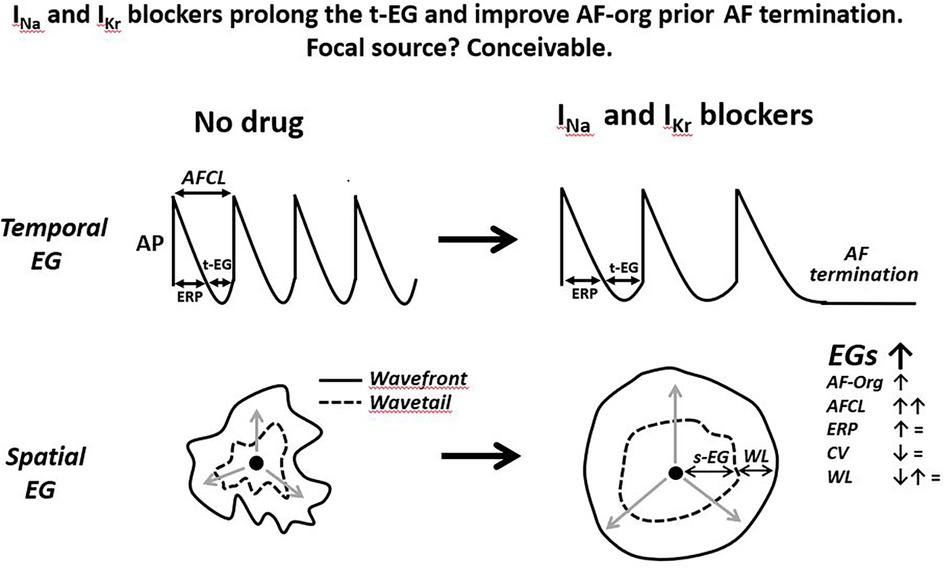
Figure 4. Schematic simplified illustrations of drug-induced alterations of the t-EG, s-EG, AF-org, and AFCL prior the termination of a single focal source firing. Anti-AF drugs prolong AFCL and t-EG, improving AF-org in AF maintained by a focal source. The slowing of rapid focal source firing is a plausible sign of its subsequent cessation. Drug-induced improvement in AF-org and termination of focal AF may or may not be associated with changes in atrial ERP, CV, and WL. The illustrated action potentials (AP) are from the focal source location, displaying diastolic depolarization. Please see text for details.
If AF is sustained by multiple simultaneous sources (reentrant, focal, or wavelet), prolongation of the spatiotemporal t-EG (i.e., improvement in AF-org) is likely to be associated with the reduction in the number of these sources, promoting the cessation of such AF. So, a general prolongation of the t-EG before AF termination appears to be consistent with all multisource AF mechanisms.
Thus, antiarrhythmic agents consistently induce prolongation of the t-EG and improvement in AF-org prior AF termination, and these parameters may help differentiate arrhythmic mechanisms.
Drug-induced prolongation of the t-EG and improvement in AF-org before AF termination: reentry, wavelets, or focal?
The following part of the analysis is an attempt to estimate how the drug-induced prolongation of the t-EG and improvement in AF-Org before AF termination correspond to reentrant, wavelet, and focal mechanisms.
A single anatomical reentry? unlikely
The classical mechanism of anatomical reentry termination by drugs is conduction block caused by the elimination of EGs in a critical area (secondary to prolongation of the WL; Figure 1) (107, 117, 118). This mechanism is inconsistent with prolongation of the t-EG induced by drugs prior AF cardioversion. With a general t-EG prolongation along the anatomical circuit, the probability of conduction block and, thus, termination of the reentry by this mechanism ought to be reduced (Figures 1, 2).
Class III agents shorten, not prolong, the t-EG in anatomical reentry [Figure 2; as it is logically conceivable and has been reported in atrial flutter studies (99, 117, 118)]. Therefore, lengthening of the t-EG with IKr blockers around the anatomical reentrant circuit is contradictory by itself. Class I agents may not only shorten and but also prolong the t-EG around the anatomical reentrant core (Figure 2) (132). In the case of such t-EG prolongation, anatomical reentry can be terminated by collision of the reentrant wavefront with antidromically propagating activation of any origin (Figure 2) (132). However, entrainment should readily occur also (Figure 2). To be coherent with a mother anatomical reentry, a reasonable explanation for its termination associated with the prolongation of the t-EG should be for both INa and IKr blockers. Considering this argument as well as the inconsistency with the classical mechanism of drug-induced termination of anatomical reentry with prolongation of the t-EG, anatomical reentry is unlikely as the primary mechanism of AF maintenance.
A mother functional reentry? Unlikely
There is little to no data and thoughts on termination of functional reentries by drugs associated with prolongation of the t-EG. In contrast, there are plenty of data and theories on termination of functional reentry by Class I and III agents associated with elimination of the t-EG, resulting to conduction block (secondary to prolongation of the WL; Figure 1) (107, 109, 133). This mechanism was directly or indirectly supported by many studies, largely in the 1980s and 1990s (96, 107, 109, 133). During that time, the primary interpretation of the functional reentry was based on the leading circuit reentry theory, and drug-induced termination of functional reentry due to elimination of the EGs leading to conduction block was consistent with this theory (Figure 1). By about 2000, due to significant inconsistencies of the leading circuit with cardiac fibrillation characteristics, the theory was essentially replaced with the “spiral wave/rotor” concept as the primary mechanism for functional reentry (1, 25, 27, 28). To what extent drug-induced termination of functional reentry via conduction block (Figure 1) is applicable to spiral waves is unclear.
The termination of mother rotor by anti-AF agents is poorly defined, perhaps due to the absence or rare detection of a sustained spiral wave during AF (10–13, 15–22, 38, 77, 94, 134, 135). All available data and theories suggest that IKr block (by prolonging ERP with no or little CV slowing) shortens the t-EG in spiral waves, as in all other reentrant mechanisms (25, 107, 117, 132, 136), that is inconsistent with t-EG prolongation induced by IKr blockers before AF termination (86). In the original study demonstrating that d-sotalol-induced termination of AF was associated with prolongation of the t-EG and s-EG, the only explanation for these EG prolongations, reconciling it with reentry, was that d-sotalol might inhibit INa (86) (high concentrations of d-sotalol block INa (137). However, d-sotalol did not affect atrial CV in this study (86), indicating that d-sotalol caused little to no inhibition of INa.
Experimental data indicate that INa blockers can shorten (109, 138, 139), prolong (120), or leave EGs unchanged (138) in functional reentrant circuits. There was one experimental study reporting that termination of vagally-mediated AF by pilsicainide (an INa blocker) was associated with prolongation of the t-EG and reduction of the WL (120). In this study, a mother rotor-based AF stopped when the spiral wave core was excited by a wavelet invader coming from widened spatiotemporal EGs (120) (as schematically illustrated in Figure 2). However, these results contradict to another study reporting that pilsicainide shortened the t-EG and prolonged the WL before cardioversion of vagally mediated AF (140).
IKr block-induced improvement of AF-org (86, 90, 95, 141) are discordant with the effects of IKr blockers to shorten the EGs of a mother spiral wave (Figures 2, 3) (25, 136). INa blockers may improve the AF-org of AF maintained by a mother rotor if INa blockers prolong a general t-EG and the degree of this prolongation is great enough to overwhelm the effect of INa blockers to worsen AF organization (Figure 3).
An apparent problem with the idea of mother spiral wave termination by antiarrhythmic drugs for the primary explanation of AF termination is the issue of sustainability of spiral waves. Spiral waves are generally unstable by nature (1, 27, 28), and the vast majority of AF mapping studies never recorded sustained rotors (i.e., rotors, when detected, commonly lasted for ≤3 rotations) (11, 15, 17, 18, 20, 21, 66, 94, 134, 135, 142, 143).
It is well recognized that the ERP, CV, WL, and EGs may vary significantly in space and time along function reentry pathway (25, 144). Yet, a spatiotemporal measurement of these parameters around the functional reentry core is technically unrealistic currently [putting aside that any chance of measuring these parameters is very low; the vast majority AF mapping studies never detect a sustained spiral wave (10–13, 15–22, 38, 77, 94, 134, 135)]. When selecting and manipulating the functional reentry-relevant parameters/conditions in mathematical models, there may be some explanations for drug-induced termination of reentry with the t-EG prolongation that are not considered in this review. Such theoretical explanations should reasonably substantiate the termination of functional reentry by both the INa and IKr blockers as well as in common and not specific conditions. Particularly, one should explain how IKr blockers prolong the t-EG in mother reentry-maintained AF.
Thus, drug-induced prolongation of the t-EG before AF cardioversion cannot be reasonably explained with the classical mechanism of drug-induced termination of functional reentry (Figure 1). Also, the termination of mother functional reentry with prolongation of the t-EG is not plausible with Class III blockers and only controversially conceivable with INa blockers. Therefore, mother functional reentry is unlikely to be the primary mechanism of AF maintenance.
Simultaneous random multiple reentries and wavelets? Speculative
Anti-AF drug-induced improvement in AF-org should reduce the appearance of random short-living reentries and, thus, is consistent with the termination of AF maintained by such reentries. However, contemporary AF mapping studies do not record simultaneous multiple reentries (10–13, 16, 17, 19, 39, 40). Moreover, increasingly more AF mapping studies either do not observe full circular activity at all or observe only a single rotational activity at a time (10–13, 16, 17, 19, 39, 40), with many or all of these rotational activities being would-be-reentries (12, 13, 19).
Drug-induced improvement in AF-org before AF cardioversion is also generally consistent with the random multiple wavelet theories (Moe's and double layer). The original Moe's hypothesis was largely abandoned in the 1980s to 1990s (in favor or multiple random reentries) (4–7, 24, 35–37), but still remains in circulation (134). The double-layer hypothesis seems limited, or largely limited, to long-standing persistent AF (10). It is impossible to definitively prove or disprove the multiple wavelet hypothesis(es) with available mapping technologies (1, 2). At the same time, random multiple wavelets (i.e., fibrillatory conduction) can be generated by a rapid stationary source (13, 19, 49), including by rapid pacing (51).
There are additional issues with random multiple reentries and wavelets as the primary mechanisms of AF maintenance. First, there is substantial evidence for the usual presence of repetitive activation patterns and some spatiotemporal stability during paroxysmal and persistent AF (54, 145–154), arguing against a completely random nature of the primary mechanism of AF maintenance. Second, distinct and repetitive F waves displaying a narrow single or dominant power peak are usually recorded during paroxysmal and persistent AF in most patients (95, 111, 155–159) that does not appear to be consistent with random multiple reentries and wavelets. Third, if AF is sustained by simultaneous random multiple sources, the termination of such AF should be due to the cessation of the last random driver and not by a stable driver. However, available data indicate that anti-AF drugs by significantly slowing the rate of AF (86, 94, 100, 116, 159–161) commonly convert AF to a tachycardia-like arrhythmia before the arrhythmia termination (100, 113, 116, 125, 162, 163). Such tachycardia-like arrhythmias are likely to be maintained by a single stationary driver. All these data and arguments cast doubt on the primary role of random multiple reentries and wavelets in AF maintenance.
Focal source(s)? Conceivable
Drug-induced prolongation of the t-EG resulting to improvement in AF-org before AF termination, with or without changes in the ERP, CV, and WL, is conceivable with termination of a focal source (Figure 4). It is known that drug-induced prolongation of the t-EG is inevitably associated with increase of the AFCL (t-EG = AFCL-ERP while anti-AF drugs do not shorten ERP) and that anti-AF agents prolong AFCL before AF termination (86, 89, 92, 94, 111–116, 164). These facts can be explained with focal sources (Figure 4). Indeed, available data indicate that termination of arrhythmic focal source activity with or without drugs is preceded by slowing this activity (123–131) that should be associated with prolongation of the t-EG (Figure 4). Increase in the cycle length and t-EG in the location of focal source maintaining AF should spread throughout the atria (with spatiotemporal variations), leading to a general improvement in AF-org (Figure 4).
We know little about the mechanism(s) of rapid focal sources and mechanisms by which anti-AF drugs can slow down such focal activity. Mechanistically, drug-induced ERP prolongation, when it occurs, should slow down rapid focal source activity, promoting the termination of such activity. Abnormalities in intracellular calcium activity (Cai) and “funny” current (If) may be involved in the maintenance of AF (165–167) and many anti-AF agents are capable of reducing Cai (such as flecainide, ranolazine, amiodarone, etc.) (168, 169) and inhibiting If (such as amiodarone, flecainide, ibutilide, etc.) (170, 171).
Specific data and theories on focal AF termination by anti-AF drugs are scarce. AF termination by drugs are explained almost exclusively based on the reentry ideology (25, 96, 107, 109), even when the drugs do not prolong atrial ERP before AF termination (86). The investigation of pharmacological cardioversion of focal AF is problematic. In the whole atrium (in situ, in vivo, or in vitro), focal AF is commonly difficult to prove definitively (as discussed previously), and in isolated thin atrial slices or single cells (in which focal sources can be convincingly proved), sustained rapid focal activity with a rate equivalent to AF does not develop (127, 129, 172–174). In fact, focal arrhythmic activity in isolated thin atrial tissue slices and single cells is commonly induced artificially (i.e., by rapid pacing in the presence of a high concentration of β-receptor agonists), its rate of activation is typically slower than the rate of atrial tachycardia, and the duration of this focal activity is usually only in seconds (127, 129, 173). These data may argue against focal sources as important drive(s) for AF. However, the fact that focal activity from atrial tissue slices and single cells cannot account even for the rate and sustainability of focal atrial tachycardias refute this assumption. There are focal atrial tachycardias (131) and there is reasonable evidence for the existence of focal AF (11–13, 16, 17, 62–64). It appears that atrial arrhythmic focal sources are capable of generating sustained rapid activation only in natural or close to natural environments (i.e., in vivo, in situ, or in coronary-perfused atrial preparations) but not in superfused atrial tissue or isolated single cells (3).
Thus, available data indicate that the termination of atrial arrhythmic focal activity by drugs is preceded by the prolongation of the cycle length that should be naturally associated with the lengthening of the t-EG and improvement in AF-org (Figure 4). Also, drug-induced cardioversion of focal AF is plausible with or without alteration in ERP, CV, and WL. Therefore, the reported behaviors of atrial electrophysiological parameters prior AF termination in the presence of Class I and III agents are conceivable with a single focal source (Figure 4). Is this deduction pertinent to the mechanism of AF maintenance before drug application? The primary mechanism of AF maintenance could be a mother source before and after the drug application, with slower activation rate following the treatment. It also could be simultaneous multiple sources before the drug treatment, converting to slower activating fewer sources or a single source following the drug application. If AF is maintained by simultaneous multiple sources, drug-induced termination of AF is likely due to the slowing and cessation of the last source of the same mechanism that maintains AF before drug application (Figures 4, 5). Therefore, a mother focal source and multiple focal sources are conceivable as the primary mechanism of AF maintenance before drug application (Figure 5).
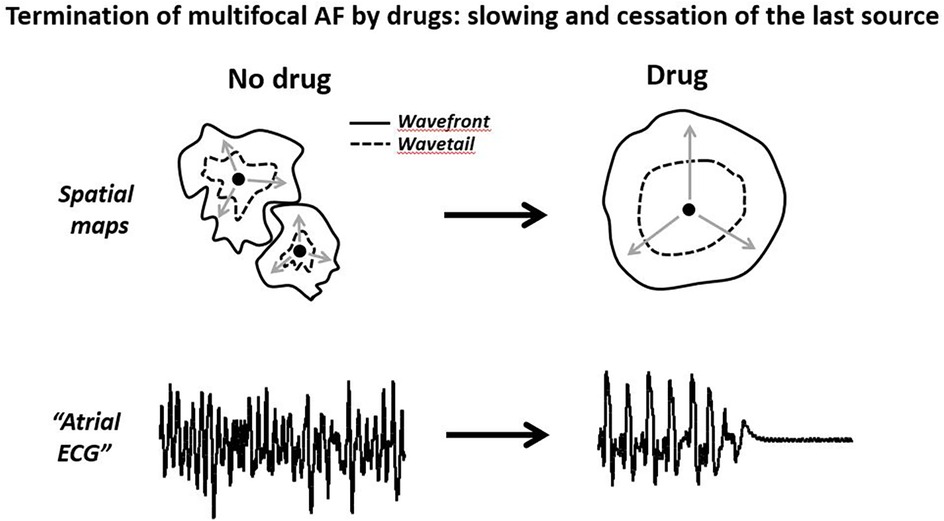
Figure 5. Schematic illustrations of cardioversion of a multifocal AF by anti-arrhythmic agents. The termination of multisource AF is likely due to the slowing and cessation of the last source of the same mechanism maintaining AF. Please see text for details.
Conclusions
(1) Drug-induced prolongation of the t-EG and improvement in AF-org prior AF termination, with or without changes in the ERP, CV, and WL, are consistent with a mother focal source and inconsistent with a mother reentry as the main mechanism of AF maintenance. Yet, a single and multiple focal sources are conceivable as the primary mechanism of AF maintenance prior drug administration.
(2) Drug-induced improvement in AF-org during AF maintenance before AF cardioversion is also coherent with simultaneous random multiple reentries and wavelets. However, AF mapping data indicate that simultaneous multiple reentries are unlikely to occur regularly, and the ability of random multiple wavelets to maintain AF is uncertain.
(3) The conducted “pharmacological” analysis in conjunction with AF mapping data supports the notion that focal sources are more likely to be the primary mechanism of AF maintenance than reentries.
Author contributions
AB: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Sharpe-Strumia Research Foundation (SSRF2023-06), Bryn Mawr, PA, USA
Acknowledgments
The author is grateful to Larry Hanover for editorial assistance.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. de Groot NMS, Shah D, Boyle PM, Anter E, Clifford GD, Deisenhofer I, et al. Critical appraisal of technologies to assess electrical activity during atrial fibrillation: a position paper from the European heart rhythm association and European Society of Cardiology Working Group on eCardiology in collaboration with the Heart Rhythm Society, Asia Pacific Heart Rhythm Society, Latin American Heart Rhythm Society and Computing in Cardiology. Europace. (2022) 24(2):313–30. doi: 10.1093/europace/euab254
2. Schotten U, Lee S, Zeemering S, Waldo AL. Paradigm shifts in electrophysiological mechanisms of atrial fibrillation. Europace. (2021) 23(23 Suppl 2):ii9–ii13. doi: 10.1093/europace/euaa384
3. Burashnikov A. Investigational anti-atrial fibrillation pharmacology and mechanisms by which antiarrhythmics terminate the arrhythmia: where are we in 2020? J Cardiovasc Pharmacol. (2020) 76(5):492–505. doi: 10.1097/FJC.0000000000000892
4. Allessie MA, Lammers WJEP, Bonke FIM, Hollen J. Experimental evaluation of moe’s multiple wavelet hypothesis of atrial fibrillation. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology and Arrhythmias. New York: Grune & Stratton (1985). p. 265–76.
5. Wang Z, Page P, Nattel S. Mechanism of flecainide’s antiarrhythmic action in experimental atrial fibrillation. Circ Res. (1992) 71(2):271–87. doi: 10.1161/01.RES.71.2.271
6. Nattel S, Bourne G, Talajic M. Insights into mechanisms of antiarrhythmic drug action from experimental models of atrial fibrillation. J Cardiovasc Electrophysiol. (1997) 8(4):469–80. doi: 10.1111/j.1540-8167.1997.tb00813.x
7. Cox JL, Canavan TE, Schuessler RB, Cain ME, Lindsay BD, Stone C, et al. The surgical treatment of atrial fibrillation. II. Intraoperative electrophysiologic mapping and description of the electrophysiologic basis of atrial flutter and atrial fibrillation. J Thorac Cardiovasc Surg. (1991) 101(3):406–26. doi: 10.1016/S0022-5223(19)36723-6
8. Jalife J. Deja vu in the theories of atrial fibrillation dynamics. Cardiovasc Res. (2011) 89(4):766–75. doi: 10.1093/cvr/cvq364
9. Narayan SM, Krummen DE, Shivkumar K, Clopton P, Rappel WJ, Miller JM. Treatment of atrial fibrillation by the ablation of localized sources: CONFIRM (conventional ablation for atrial fibrillation with or without focal impulse and rotor modulation) trial. J Am Coll Cardiol. (2012) 60(7):628–36. doi: 10.1016/j.jacc.2012.05.022
10. de Groot NMS, Allessie MA. Pathophysiology of atrial fibrillation: focal patterns of activation. Pacing Clin Electrophysiol. (2019) 42(10):1312–9. doi: 10.1111/pace.13777
11. Lee S, Sahadevan J, Khrestian CM, Cakulev I, Markowitz A, Waldo AL. Simultaneous biatrial high-density (510–512 electrodes) epicardial mapping of persistent and long-standing persistent atrial fibrillation in patients: new insights into the mechanism of its maintenance. Circulation. (2015) 132(22):2108–17. doi: 10.1161/CIRCULATIONAHA.115.017007
12. Lee S, Khrestian CM, Sahadevan J, Markowitz A, Waldo AL. New insights into understanding rotor versus focal activation in patients with persistent atrial fibrillation. JACC Clinical Electrophysiol. (2021) 7(7):909–19. doi: 10.1016/j.jacep.2020.12.010
13. Lee S, Sahadevan J, Khrestian CM, Durand DM, Waldo AL. High density mapping of atrial fibrillation during vagal nerve stimulation in the canine heart: restudying the moe hypothesis. J Cardiovasc Electrophysiol. (2013) 24(3):328–35. doi: 10.1111/jce.12032
14. Haissaguerre M, Hocini M, Denis A, Shah AJ, Komatsu Y, Yamashita S, et al. Driver domains in persistent atrial fibrillation. Circulation. (2014) 130(7):530–8. doi: 10.1161/CIRCULATIONAHA.113.005421
15. Lee G, Kumar S, Teh A, Madry A, Spence S, Larobina M, et al. Epicardial wave mapping in human long-lasting persistent atrial fibrillation: transient rotational circuits, complex wavefronts, and disorganized activity. Eur Heart J. (2014) 35(2):86–97. doi: 10.1093/eurheartj/eht267
16. Lee S, Sahadevan J, Khrestian CM, Markowitz A, Waldo AL. Characterization of foci and breakthrough sites during persistent and long-standing persistent atrial fibrillation in patients: studies using high-density (510–512 electrodes) biatrial epicardial mapping. J Am Heart Assoc. (2017) 6(3):e005274. doi: 10.1161/JAHA.116.005274
17. Takahashi Y, Akiyoshi K, Sekigawa M, Yagishita A, Yamamoto T, Maeda S, et al. Endocardial contact mapping of the left atrial appendage in persistent atrial fibrillation. J Cardiovasc Electrophysiol. (2020) 31(1):112–8. doi: 10.1111/jce.14278
18. Podziemski P, Zeemering S, Kuklik P, van Hunnik A, Maesen B, Maessen J, et al. Rotors detected by phase analysis of filtered, epicardial atrial fibrillation electrograms colocalize with regions of conduction block. Circ Arrhythm Electrophysiol. (2018) 11(10):e005858. doi: 10.1161/CIRCEP.117.005858
19. Lee S, Khrestian CM, Sahadevan J, Waldo AL. Reconsidering the multiple wavelet hypothesis of atrial fibrillation. Heart Rhythm. (2020) 17(11):1976–83. doi: 10.1016/j.hrthm.2020.06.017
20. Nayyar S, Ha ACT, Timmerman N, Suszko A, Ragot D, Chauhan VS. Focal and pseudo/rotational activations in human atrial fibrillation defined with automated periodicity mapping. J Cardiovasc Electrophysiol. (2021) 32(2):212–23. doi: 10.1111/jce.14812
21. Parameswaran R, Kalman JM, Royse A, Goldblatt J, Larobina M, Watts T, et al. Endocardial-epicardial phase mapping of prolonged persistent atrial fibrillation recordings: high prevalence of dissociated activation patterns. Circ Arrhythm Electrophysiol. (2020) 13(8):e008512. doi: 10.1161/CIRCEP.120.008512
22. Luther V, Sikkel M, Bennett N, Guerrero F, Leong K, Qureshi N, et al. Visualizing localized reentry with ultra-high density mapping in iatrogenic atrial tachycardia: beware Pseudo-reentry. Circ Arrhythm Electrophysiol. (2017) 10(4):e004724. doi: 10.1161/CIRCEP.116.004724
23. Allessie MA, Bonke FIM, Schopman JG. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. CircRes. (1973) 33:54–62.
24. Konings KT, Kirchhof CJ, Smeets JR, Wellens HJ, Penn OC, Allessie MA. High-density mapping of electrically induced atrial fibrillation in humans. Circulation. (1994) 89(4):1665–80. doi: 10.1161/01.CIR.89.4.1665
25. Nattel S, Xiong F, Aguilar M. Demystifying rotors and their place in clinical translation of atrial fibrillation mechanisms. Nat Rev Cardiol. (2017) 14(9):509–20. doi: 10.1038/nrcardio.2017.37
26. Rensma PL, Allessie MA, Lammers WJEP, Bonke FIM, Schalij MJ. Length of excitation wave and susceptibility to reentrant atrial arrhythmias in normal conscious dogs. Circ Res. (1988) 62:395–410. doi: 10.1161/01.RES.62.2.395
27. Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. CircRes. (1993) 72:631–50. doi: 10.1161/01.res.72.3.631
28. Pandit SV, Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ Res. (2013) 112(5):849–62. doi: 10.1161/CIRCRESAHA.111.300158
29. Mines GR. On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. TransRSocCan. (1914) 8:43–52.
30. Lewis T. Oliver-sharpey lectures ON THE NATURE OF FLUTTER AND FIBRILLATION OF THE AURICLE. Br Med J. (1921) 1(3146):551–5. doi: 10.1136/bmj.1.3146.551
31. Scherf D, Romano FJ, Terranova R. Experimental studies on auricular flutter and auricular fibrillation. Am Heart J. (1948) 36:241–51. doi: 10.1016/0002-8703(48)90403-7
32. Prinzmetal M, Rakata L, Borduas JL, Flamm E, Goldman L. The nuture of spontanous auricular fibrillation in man. JAMA. (1955) 157:1175–82. doi: 10.1001/jama.1955.02950310001001
33. Moe GK, Abildskov JA. Atrial fibrillation as a self-sustaining arrhythmia independent of focal discharge. Am Heart J. (1959) 58(1):59–70. doi: 10.1016/0002-8703(59)90274-1
34. Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J. (1964) 67:200–20. doi: 10.1016/0002-8703(64)90371-0
35. Asano Y, Saito J, Matsumoto K, Kaneko K, Yamamoto T, Uchida M. On the mechanism of termination and perpetuation of atrial fibrillation. Am J Cardiol. (1992) 69(12):1033–8. doi: 10.1016/0002-9149(92)90859-W
36. Sih HJ, Berbari EJ, Zipes DP. Epicardial maps of atrial fibrillation after linear ablation lesions [see comments]. J Cardiovasc Electrophysiol. (1997) 8(9):1046–54. doi: 10.1111/j.1540-8167.1997.tb00628.x
37. Kumagai K, Khrestian C, Waldo AL. Simultaneous multisite mapping studies during induced atrial fibrillation in the sterile pericarditis model. Insights into the mechanism of its maintenance. Circulation. (1997) 95(2):511–21. doi: 10.1161/01.CIR.95.2.511
38. Gray RA, Pertsov AM, Jalife J. Incomplete reentry and epicardail breakthrough patterns during atrial fibrillation in the sheep heart. Circulation. (1996) 94:2649–61. doi: 10.1161/01.CIR.94.10.2649
39. de Groot N, van der Does L, Yaksh A, Lanters E, Teuwen C, Knops P, et al. Direct proof of endo-epicardial asynchrony of the atrial wall during atrial fibrillation in humans. Circ Arrhythm Electrophysiol. (2016) 9(5):e003648. doi: 10.1161/CIRCEP.115.003648
40. Hansen BJ, Zhao J, Csepe TA, Moore BT, Li N, Jayne LA, et al. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur Heart J. (2015) 36(35):2390–401. doi: 10.1093/eurheartj/ehv233
41. Haissaguerre M, Shah AJ, Cochet H, Hocini M, Dubois R, Efimov I, et al. Intermittent drivers anchoring to structural heterogeneities as a major pathophysiological mechanism of human persistent atrial fibrillation. J Physiol. (2016) 594(9):2387–98. doi: 10.1113/JP270617
42. Eckstein J, Zeemering S, Linz D, Maesen B, Verheule S, van Hunnik A, et al. Transmural conduction is the predominant mechanism of breakthrough during atrial fibrillation: evidence from simultaneous endo-epicardial high-density activation mapping. Circ Arrhythm Electrophysiol. (2013) 6(2):334–41. doi: 10.1161/CIRCEP.113.000342
43. Verheule S, Eckstein J, Linz D, Maesen B, Bidar E, Gharaviri A, et al. Role of endo-epicardial dissociation of electrical activity and transmural conduction in the development of persistent atrial fibrillation. Prog Biophys Mol Biol. (2014) 115(2-3):173–85. doi: 10.1016/j.pbiomolbio.2014.07.007
44. Verheule S, Schotten U. Electrophysiological consequences of cardiac fibrosis. Cells. (2021) 10(11):3220. doi: 10.3390/cells10113220
45. Burashnikov A, Antzelevitch C. Is extensive atrial fibrosis in the setting of heart failure associated with a reduced atrial fibrillation burden? Pacing Clin Electrophysiol. (2018) 41(10):1289–97. doi: 10.1111/pace.13474
46. Ramos K, Pool L, van Schie MS, Wijdeleld L, an der Does WFB, Baks L, et al. Degree of fibrosis in human atrial tissue is not the hallmark driving AF. Cells. (2022) 11(3):427. doi: 10.3390/cells11030427
47. Ballatore A, Negrello E, Gatti M, Matta M, Desalvo P, Marcialis L, et al. Evaluation of pulmonary vein fibrosis following cryoballoon ablation of atrial fibrillation: a semi-automatic MRI analysis. J Cardiovasc Dev Dis. (2023) 10(9):396. doi: 10.3390/jcdd10090396.37754825
48. Marrouche NF, Wazni O, McGann C, Green T, Dean JM, Dagher K, et al. Effect of MRI-guided fibrosis ablation vs conventional catheter ablation on atrial arrhythmia recurrence in patients with persistent atrial fibrillation: the DECAAF II randomized clinical trial. JAMA. (2022) 327(23):2296–305. doi: 10.1001/jama.2022.8831
49. Schuessler RB, Grayson TM, Bromberg BI, Cox JL, Boineau JP. Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ Res. (1992) 71:1254–67. doi: 10.1161/01.RES.71.5.1254
50. Roney CH, Cantwell CD, Bayer JD, Qureshi NA, Lim PB, Tweedy JH, et al. Spatial resolution requirements for accurate identification of drivers of atrial fibrillation. Circ Arrhythm Electrophysiol. (2017) 10(5):e004899. doi: 10.1161/CIRCEP.116.004899
51. Hemam ME, Dave AS, Rodríguez-Mañero M, Valderrábano M. Epiphenomenal re-entry and spurious focal activation detection by atrial fibrillation mapping algorithms. JACC Clinical Electrophysiology. (2021) 7(7):923–32. doi: 10.1016/j.jacep.2020.12.005
52. Roney CH, Wit AL, Peters NS. Challenges associated with interpreting mechanisms of AF. Arrhythm Electrophysiol Rev. (2020) 8(4):273–84. doi: 10.15420/aer.2019.08
53. Zaman JAB, Sauer WH, Alhusseini MI, Baykaner T, Borne RT, Kowalwski CAB, et al. Identification and characterization of sites where persistent atrial fibrillation is terminated by localized ablation. Circ Arrhythm Electrophysiol. (2018) 11(1):e005258. doi: 10.1161/CIRCEP.117.005258
54. Gray RA, Pertsov AM, Jalife J. Spatial and temporal organization during cardiac fibrillation. Nature. (1998) 392(6671):75–8. doi: 10.1038/32164
55. Lee S, Khrestian CM, Sahadevan J, Waldo AL. An evaluation of phase analysis to interpret atrial activation patterns during persistent atrial fibrillation for targeted ablation. J Clin Med. (2022) 11(19):5807. doi: 10.3390/jcm11195807
56. Pathik B, Kalman JM, Walters T, Kuklik P, Zhao J, Madry A, et al. Absence of rotational activity detected using 2-dimensional phase mapping in the corresponding 3-dimensional phase maps in human persistent atrial fibrillation. Heart Rhythm. (2018) 15(2):182–92. doi: 10.1016/j.hrthm.2017.09.010
57. Sohal M, Choudhury R, Taghji P, Louw R, Wolf M, Fedida J, et al. Is mapping of complex fractionated electrograms obsolete? Arrhythm Electrophysiol Rev. (2015) 4(2):109–15. doi: 10.15420/AER.2015.04.02.109
58. Tilz RR, Lenz C, Sommer P, Roza MS, Sarver AE, Williams CG, et al. Focal impulse and rotor modulation ablation vs. Pulmonary vein isolation for the treatment of paroxysmal atrial fibrillation: results from the FIRMAP AF study. Europace. (2021) 23(5):722–30. doi: 10.1093/europace/euaa378
59. Spitzer SG, Miller JM, Sommer P, Szili-Torok T, Reddy VY, Nolker G, et al. Randomized evaluation of redo ablation procedures of atrial fibrillation with focal impulse and rotor modulation-guided procedures: the REDO-FIRM study. Europace. (2023) 25(1):74–82. doi: 10.1093/europace/euac122
60. Verma A, Jiang CY, Betts TR, Chen J, Deisenhofer I, Mantovan R, et al. Approaches to catheter ablation for persistent atrial fibrillation. N Engl J Med. (2015) 372(19):1812–22. doi: 10.1056/NEJMoa1408288
61. McCauley MD, Iacobellis G, Li N, Nattel S, Goldberger JJ. Targeting the substrate for atrial fibrillation: JACC review topic of the week. J Am Coll Cardiol. (2024) 83(20):2015–27. doi: 10.1016/j.jacc.2024.02.050
62. Zhou S, Chang CM, Wu TJ, Miyauchi Y, Okuyama Y, Park AM, et al. Nonreentrant focal activations in pulmonary veins in canine model of sustained atrial fibrillation. Am J Physiol Heart Circ Physiol. (2002) 283(3):H1244–52. doi: 10.1152/ajpheart.01109.2001
63. Nitta T, Ishii Y, Miyagi Y, Ohmori H, Sakamoto S, Tanaka S. Concurrent multiple left atrial focal activations with fibrillatory conduction and right atrial focal or reentrant activation as the mechanism in atrial fibrillation. J Thorac Cardiovasc Surg. (2004) 127(3):770–8. doi: 10.1016/j.jtcvs.2003.05.001
64. Knecht S, O'Neill MD, Matsuo S, Lim KT, Arantes L, Derval N, et al. Focal arrhythmia confined within the coronary sinus and maintaining atrial fibrillation. J Cardiovasc Electrophysiol. (2007) 18(11):1140–6. doi: 10.1111/j.1540-8167.2007.00927.x
65. Holm M, Johansson R, Brandt J, Lührs C, Olsson SB. Epicardial right atrial free wall mapping in chronic atrial fibrillation. Documentation of repetitive activation with a focal spread–a hitherto unrecognised phenomenon in man. Eur Heart J. (1997) 18(2):290–310. doi: 10.1093/oxfordjournals.eurheartj.a015233
66. Wolf M, Tavernier R, Zeidan Z, El Haddad M, Vandekerckhove Y, Pooter J, et al. Identification of repetitive atrial activation patterns in persistent atrial fibrillation by direct contact high-density electrogram mapping. J Cardiovasc Electrophysiol. (2019) 30(12):2704–12. doi: 10.1111/jce.14214
67. Nagase T, Kato R, Asano S, Fukunaga H, Yoshida Y, Hayashi Y, et al. Spatial relationship of localized sources of persistent atrial fibrillation identified by a unipolar-based automated algorithm to complex fractionated atrial electrocardiograms and atrial low voltage areas. J Cardiovasc Electrophysiol. (2023) 34(2):337–44. doi: 10.1111/jce.15755
68. Chu GS, Li X, Stafford PJ, Vanheusden FJ, Salinet JL, Almeida TP, et al. Simultaneous whole-chamber non-contact mapping of highest dominant frequency sites during persistent atrial fibrillation: a prospective ablation study. Front Physiol. (2022) 13:826449. doi: 10.3389/fphys.2022.826449
69. Unland R, Bergau L, El Hamriti M, Guckel D, Piran M, Fink T, et al. Find me if you can: first clinical experience using the novel CARTOFINDER algorithm in a routine workflow for atrial fibrillation ablation. J Clin Med. (2021) 10(13):2979. doi: 10.3390/jcm10132979
70. Fenelon G, Shepard RK, Stambler BS. Focal origin of atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J Cardiovasc Electrophysiol. (2003) 14(10):1093–102. doi: 10.1046/j.1540-8167.2003.03110.x
71. de Coster T, Teplenin AS, Feola I, Bart CI, Ramkisoensing AA, den Ouden BL, et al. ‘Trapped reentry’ as source of acute focal arrhythmias. Cardiovasc Res. (2024) 120(3):249–61. doi: 10.1093/cvr/cvad179
72. de Groot NM, Houben RP, Smeets JL, Boersma E, Schotten U, Schalij MJ, et al. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation. (2010) 122(17):1674–82. doi: 10.1161/CIRCULATIONAHA.109.910901
73. Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. (2014) 114(9):1500–15. doi: 10.1161/CIRCRESAHA.114.303772
74. Karatela MF, Fudim M, Mathew JP, Piccini JP. Neuromodulation therapy for atrial fibrillation. Heart Rhythm. (2023) 20(1):100–11. doi: 10.1016/j.hrthm.2022.08.011
75. Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. (2003) 107(18):2355–60. doi: 10.1161/01.CIR.0000065578.00869.7C
76. Burashnikov A, Antzelevitch C. Mechanisms of cardiac arrhythmias & conduction disturbances. In: O'Rourke RA, Walsh RA, Fuster V, editors. Hurst’s the Heart Manual of Cardiology. 12th ed. New York, NY: McGraw-Hill Professional; 2009:95–103.
77. Hirose M, Leatmanoratn Z, Laurita KR, Carlson MD. Partial vagal denervation increases vulnerability to vagally induced atrial fibrillation. J Cardiovasc Electrophysiol. (2002) 13(12):1272–9. doi: 10.1046/j.1540-8167.2002.01272.x
78. Hashimoto N, Yamashita T, Tsuruzoe N. Tertiapin, a selective IK,ACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol Res. (2006) 54(2):136–41. doi: 10.1016/j.phrs.2006.03.021
79. Sobota V, Gatta G, van Hunnik A, van Tuijn I, Kuiper M, Milnes J, et al. The acetylcholine-activated potassium current inhibitor XAF-1407 terminates persistent atrial fibrillation in goats. Front Pharmacol. (2020) 11:608410. doi: 10.3389/fphar.2020.608410
80. Walfridsson H, Anfinsen OG, Berggren A, Frison L, Jensen S, Linhardt G, et al. Is the acetylcholine-regulated inwardly rectifying potassium current a viable antiarrhythmic target? Translational discrepancies of AZD2927 and A7071 in dogs and humans. Europace. (2015) 17(3):473–82. doi: 10.1093/europace/euu192
81. Podd SJ, Freemantle N, Furniss SS, Sulke N. First clinical trial of specific IKACh blocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: pacemaker assessment of BMS 914392 in patients with paroxysmal atrial fibrillation. Europace. (2016) 18(3):340–6. doi: 10.1093/europace/euv263
82. Workman AJ. Cardiac adrenergic control and atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. (2010) 381(3):235–49. doi: 10.1007/s00210-009-0474-0
83. Liu T, Xiong F, Qi XY, Xiao J, Villeneuve L, Abu-Taha I, et al. Altered calcium handling produces reentry-promoting action potential alternans in atrial fibrillation-remodeled hearts. JCI Insight. (2020) 5(8):e133754. doi: 10.1172/jci.insight.133754
84. Honarbakhsh S, Roney C, Wharmby A, Vidal Horrach C, Hunter RJ. Spatial and temporal relationship between focal and rotational activations and their relationship to structural remodeling in patients with persistent atrial fibrillation. Heart Rhythm. (2024) (6):752–61. doi: 10.1016/j.hrthm.2024.01.039
85. van Hunnik A, Sobota V, Zeemering S, Opacic D, Scaf B, D’Alessandro E, et al. Analysis of drug-induced and spontaneous cardioversions reveals similar patterns leading to termination of atrial fibrillation. Front Physiol. (2024) 15:1399037. doi: 10.3389/fphys.2024.1399037
86. Wijffels MC, Dorland R, Mast F, Allessie MA. Widening of the excitable gap during pharmacological cardioversion of atrial fibrillation in the goat: effects of cibenzoline, hydroquinidine, flecainide, and d-sotalol. Circulation. (2000) 102(2):260–7. doi: 10.1161/01.CIR.102.2.260
87. Kakugawa H, Shimizu A, Yamagata T, Esato M, Ueyama T, Yoshiga Y, et al. Decrease in the spatial dispersion at the termination of atrial fibrillation by intravenous cibenzoline. Circ J. (2003) 67(10):810–5. doi: 10.1253/circj.67.810
88. Shan Z, Van Der Voort PH, Blaauw Y, Duytschaever M, Allessie MA. Fractionation of electrograms and linking of activation during pharmacologic cardioversion of persistent atrial fibrillation in the goat. J Cardiovasc Electrophysiol. (2004) 15(5):572–80. doi: 10.1046/j.1540-8167.2004.03402.x
89. Fujiki A, Sakabe M, Nishida K, Sugao M, Tsuneda T, Iwamoto J, et al. Drug-induced changes in fibrillation cycle length and organization index can predict chemical cardioversion of long-lasting atrial fibrillation with bepridil alone or in combination with aprindine. Circ J. (2004) 68(12):1139–45. doi: 10.1253/circj.68.1139
90. Santos PE, Duytschaever M, Allessie MA. Low-frequency oscillations of atrial fibrillation cycle length in goats: characterization and potentiation by class III antiarrhythmic almokalant. J Electrocardiol. (2008) 41(6):711–23. doi: 10.1016/j.jelectrocard.2008.03.006
91. Aunes-Jansson M, Edvardsson N, Stridh M, Sornmo L, Frison L, Berggren A. Decrease of the atrial fibrillatory rate, increased organization of the atrial rhythm and termination of atrial fibrillation by AZD7009. J Electrocardiol. (2013) 46(1):29–35. doi: 10.1016/j.jelectrocard.2012.09.002
92. Husser D, Stridh M, Sornmo L, Geller C, Klein HU, Olsson SB, et al. Time-frequency analysis of the surface electrocardiogram for monitoring antiarrhythmic drug effects in atrial fibrillation. Am J Cardiol. (2005) 95(4):526–8. doi: 10.1016/j.amjcard.2004.10.025
93. Tuan J, Osman F, Jeilan M, Kundu S, Mantravadi R, Stafford PJ, et al. Increase in organization index predicts atrial fibrillation termination with flecainide post-ablation: spectral analysis of intracardiac electrograms. Europace. (2010) 12(4):488–93. doi: 10.1093/europace/eup405
94. van Hunnik A, Zeemering S, Podziemski P, Kuklik P, Kuiper M, Verheule S, et al. Bi-atrial high-density mapping reveals inhibition of wavefront turning and reduction of complex propagation patterns as main antiarrhythmic mechanisms of vernakalant. Europace. (2021) 23(7):1114–23. doi: 10.1093/europace/euab026
95. Raygor VP, Ng J, Goldberger JJ. Surface ECG f wave analysis of dofetilide drug effect in the atrium. J Cardiovasc Electrophysiol. (2015) 26(6):644–8. doi: 10.1111/jce.12645
96. Workman AJ, Smith GL, Rankin AC. Mechanisms of termination and prevention of atrial fibrillation by drug therapy. Pharmacol Ther. (2011) 131(2):221–41. doi: 10.1016/j.pharmthera.2011.02.002
97. Burashnikov A, Antzelevitch C. New development in atrial antiarrhythmic drug therapy. Nat RevCardiol. (2010) 7(3):139–48. doi: 10.1038/nrcardio.2009.245
98. Kirchhof P, Engelen M, Franz MR, Ribbing M, Wasmer K, Breithardt G, et al. Electrophysiological effects of flecainide and sotalol in the human atrium during persistent atrial fibrillation. Basic Res Cardiol. (2005) 100(2):112–21. doi: 10.1007/s00395-005-0513-4
99. Stambler BS, Wood MA, Ellenbogen KA. Antiarrhythmic actions of intravenous ibutilide compared with procainamide during human atrial flutter and fibrillation: electrophysiological determinants of enhanced conversion efficacy. Circulation. (1997) 96(12):4298–306. doi: 10.1161/01.CIR.96.12.4298
100. Wijffels MC, Dorland R, Allessie MA. Pharmacologic cardioversion of chronic atrial fibrillation in the goat by class IA, IC, and III drugs: a comparison between hydroquinidine, cibenzoline, flecainide, and d-sotalol. J Cardiovasc Electrophysiol. (1999) 10(2):178–93. doi: 10.1111/j.1540-8167.1999.tb00660.x
101. Haugaard MM, Pehrson S, Carstensen H, Flethoj M, Hesselkilde EZ, Praestegaard KF, et al. Antiarrhythmic and electrophysiologic effects of flecainide on acutely induced atrial fibrillation in healthy horses. J Vet Intern Med. (2015) 29(1):339–47. doi: 10.1111/jvim.12496
102. Carstensen H, Kjær L, Haugaard MM, Flethoj M, Hesselkilde EZ, Kanters JK, et al. Antiarrhythmic effects of combining dofetilide and ranolazine in a model of acutely induced atrial fibrillation in horses. J Cardiovasc Pharmacol. (2018) 71(1):26–35. doi: 10.1097/FJC.0000000000000541
103. Volgman AS, Carberry PA, Stambler B, Lewis WR, Dunn GH, Perry KT, et al. Conversion efficacy and safety of intravenous ibutilide compared with intravenous procainamide in patients with atrial flutter or fibrillation. J Am Coll Cardiol. (1998) 31(6):1414–9. doi: 10.1016/S0735-1097(98)00078-3
104. Wolbrette DL, Hussain S, Maraj I, Naccarelli GV. A quarter of a century later: what is dofetilide’s clinical role today? J Cardiovasc Pharmacol Ther. (2019) 24(1):3–10. doi: 10.1177/1074248418784288
105. Colatsky TJ, Follmer CH, Starmer CF. Channel specificity in antiarrhythmic drug action. Mechanism of potassium channel block and its role in suppressing and aggravating cardiac arrhythmias. Circulation. (1990) 82:2235–42. doi: 10.1161/01.CIR.82.6.2235
106. Pallandi RT, Lovell NH, Campbell TJ. Class III antiarrhythmic effects of dofetilide in rabbit atrial myocardium. J Cardiovasc Pharmacol Ther. (1996) 1(3):229–34. doi: 10.1177/107424849600100306
107. Wit AL, Coromilas J. Role of alterations in refractoriness and conduction in the genesis of reentrant arrhythmias. Implications for antiarrhythmic effects of class III agents. Am J Cardiol. (1993) 72:3F–12F. doi: 10.1016/0002-9149(93)90958-F
108. Kirchhof C, Wijffels M, Brugada J, Planellas J, Allessie M. Mode of action of a new class IC drug (ORG 7797) against atrial fibrillation in conscious dogs. J Cardiovasc Pharmacol. (1991) 17(1):116–24. doi: 10.1097/00005344-199101000-00017
109. Wang JJ, Bourne GW, Wang ZG, Villemaire C, Talajic M, Nattel S. Comparative mechanisms of antiarrhythmic drug action in experimental atrial fibrillation—importance of use-dependent effects on refractoriness. Circulation. (1993) 88:1030–44. doi: 10.1161/01.CIR.88.3.1030
110. Wang J, Feng J, Nattel S. Class III antiarrhythmic drug action in experimental atrial fibrillation. Differences in reverse use dependence and effectiveness between d-sotalol and the new antiarrhythmic drug ambasilide. Circulation. (1994) 90(4):2032–40. doi: 10.1161/01.CIR.90.4.2032
111. Bollmann A, Kanuru NK, McTeague KK, Walter PF, DeLurgio DB, Langberg JJ. Frequency analysis of human atrial fibrillation using the surface electrocardiogram and its response to ibutilide. Am J Cardiol. (1998) 81(12):1439–45. doi: 10.1016/S0002-9149(98)00210-0
112. Husser D, Stridh M, Sornmo L, Platonov P, Olsson SB, Bollmann A. Analysis of the surface electrocardiogram for monitoring and predicting antiarrhythmic drug effects in atrial fibrillation. Cardiovasc Drugs Ther. (2004) 18(5):377–86. doi: 10.1007/s10557-005-5062-z
113. Brugada J, Gursoy S, Brugada P, Atie J, Guiraudon G, Andries E. Cibenzoline transforms random re-entry into ordered re-entry in the atria. Eur Heart J. (1993) 14(2):267–72. doi: 10.1093/eurheartj/14.2.267
114. Aunes M, Egstrup K, Frison L, Berggren A, Stridh M, Sornmo L, et al. Rapid slowing of the atrial fibrillatory rate after administration of AZD7009 predicts conversion of atrial fibrillation. J Electrocardiol. (2014) 47(3):316–23. doi: 10.1016/j.jelectrocard.2013.12.008
115. John S, Salmas J, Kornej J, Lobe S, Stridh M, Sornmo L, et al. Effects of dronedarone and amiodarone on atrial fibrillatory rate in patients with persistent atrial fibrillation. Int J Cardiol. (2013) 167(5):2354–6. doi: 10.1016/j.ijcard.2012.11.042
116. Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. (2007) 116(13):1449–57. doi: 10.1161/CIRCULATIONAHA.107.704890
117. Yamabe H, Tanaka Y, Morihisa K, Uemura T, Koyama J, Kanazawa H, et al. Electrophysiologic mechanism of typical atrial flutter termination by nifekalant: effect of a pure IKr -selective blocking agent. Pacing Clin Electrophysiol. (2013) 36(9):1123–31. doi: 10.1111/pace.12147
118. Inoue H, Yamashita T, Nozaki A, Sugimoto T. Effects of antiarrhythmic drugs on canine atrial flutter due to reentry: role of prolongation of refractory period and depression of conduction to excitable gap. J Am Coll Cardiol. (1991) 18(4):1098–104. doi: 10.1016/0735-1097(91)90773-3
119. Boyden PA, Graziano JN. Activation mapping of reentry around an anatomical barrier in the canine atrium: observations during the action of the class III agent, d-sotalol. J Cardiovasc Electrophysiol. (1993) 4:266–79. doi: 10.1111/j.1540-8167.1993.tb01230.x
120. Kawase A, Ikeda T, Nakazawa K, Ashihara T, Namba T, Kubota T, et al. Widening of the excitable gap and enlargement of the core of reentry during atrial fibrillation with a pure sodium channel blocker in canine atria. Circulation. (2003) 107(6):905–10. doi: 10.1161/01.CIR.0000050148.72502.3A
121. Ashton JL, Trew ML, LeGrice IJ, Paterson DJ, Paton JF, Gillis AM, et al. Shift of leading pacemaker site during reflex vagal stimulation and altered electrical source-to-sink balance. J Physiol. (2019) 597(13):3297–313. doi: 10.1113/JP276876
122. Cacciani F, Zaniboni M. Chronotropic modulation of the source-sink relationship of sinoatrial-atrial impulse conduction and its significance to initiation of AF: a one-dimensional model study. Biomed Res Int. (2015) 2015:496418. doi: 10.1155/2015/496418
123. Kunze KP, Kuck KH, Schlüter M, Bleifeld W. Effect of encainide and flecainide on chronic ectopic atrial tachycardia. J Am Coll Cardiol. (1986) 7(5):1121–6. doi: 10.1016/S0735-1097(86)80233-9
124. Eidher U, Freihoff F, Kaltenbrunner W, Steinbach K. Efficacy and safety of ibutilide for the conversion of monomorphic atrial tachycardia. Pacing Clin Electrophysiol. (2006) 29(4):358–62. doi: 10.1111/j.1540-8159.2006.00353.x
125. Chou CC, Zhou S, Miyauchi Y, Pak NH, Okuyama Y, Fishbein MC, et al. Effects of procainamide on electrical activity in thoracic veins and atria in canine model of sustained atrial fibrillation. Am J Physiol Heart Circ Physiol. (2004) 286(5):H1936–45. doi: 10.1152/ajpheart.00754.2003
126. Chou CC, Zhou S, Tan AY, Hayashi H, Nihei M, Chen PS. High-density mapping of pulmonary veins and left atrium during ibutilide administration in a canine model of sustained atrial fibrillation. Am J Physiol Heart Circ Physiol. (2005) 289(6):H2704–13. doi: 10.1152/ajpheart.00537.2005
127. Johnson N, Danilo P Jr, Wit AL, Rosen MR. Characteristics of initiation and termination of catecholamine- induced triggered activity in atrial fibers of the coronary sinus. Circulation. (1986) 74:1168–79. doi: 10.1161/01.CIR.74.5.1168
128. Tanaka Y, Obata K, Ohmori T, Ishiwata K, Abe M, Hamaguchi S, et al. Angiotensin II induces automatic activity of the isolated Guinea pig pulmonary vein myocardium through activation of the IP(3) receptor and the na(+)-ca(2+) exchanger. Int J Mol Sci. (2019) 20(7):1768. doi: 10.3390/ijms20071768
129. Burashnikov A, Sicouri S, Di Diego JM, Belardinelli L, Antzelevitch C. Synergistic effect of the combination of dronedarone and ranolazine to suppress atrial fibrillation. J Am Coll Cardiol. (2010) 56(15):1216–24. doi: 10.1016/j.jacc.2010.08.600
130. Laszlo A, Polic S, Kampine JP, Turner LA, Atlee JL 3rd, Bosnjak ZJ. Halothane, enflurane and isoflurane on abnormal automaticity and triggered rhythmic activity of Purkinje fibers from 24-hour-old infarcted canine hearts. Anesthesiology. (1991) 75(5):847–53. doi: 10.1097/00000542-199111000-00017
131. Page RL, Joglar JA, Caldwell MA, Calkins H, Conti JB, Deal BJ, et al. 2015 ACC/AHA/HRS guideline for the management of adult patients with supraventricular tachycardia: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the heart rhythm society. Circulation. (2016) 133(14):e471–505. doi: 10.1161/CIR.0000000000000310
132. Tai CT, Chen SA. Mechanisms of antiarrhythmic drug action on termination of atrial flutter. Pacing Clin Electrophysiol. (2001) 24(5):824–34. doi: 10.1046/j.1460-9592.2001.00824.x
133. Wang ZG, Fermini B, Nattel S. Mechanism of flecainide’s rate-dependent actions on action potential duration in canine atrial tissue. J Pharmacol Exp Ther. (1993) 267:575–81.8246130
134. Child N, Clayton RH, Roney CH, Laughner JI, Shuros A, Neuzil P, et al. Unraveling the underlying arrhythmia mechanism in persistent atrial fibrillation: results from the STARLIGHT study. Circ Arrhythm Electrophysiol. (2018) 11(6):e005897. doi: 10.1161/CIRCEP.117.005897
135. Pathik B, Kalman JM, Walters T, Kuklik P, Zhao J, Madry A, et al. Transient rotor activity during prolonged 3-dimensional phase mapping in human persistent atrial fibrillation. JACC Clinical Electrophysiology. (2018) 4(1):72–83. doi: 10.1016/j.jacep.2017.06.005
136. Honjo H, Yamazaki M, Kamiya K, Kodama I. Modulation of spiral wave reentry by K(+) channel blockade. Circ J. (2007) 71 Suppl A:A26–31. doi: 10.1253/circj.71.A26
137. Carmeliet E. Electrophysiologic and voltage clamp analysis of the effects of sotalol on isolated cardiac muscle and Purkinje fibers. J Pharmacol Exp Ther. (1985) 232:817–25.3973830
138. Spinelli W, Hoffman BF. Mechanisms of termination of reentrant atrial arrhythmias by class I and class III antiarrhythmic agents. Circ Res. (1989) 65:1565–79. doi: 10.1161/01.RES.65.6.1565
139. Li D, Benardeau A, Nattel S. Contrasting efficacy of dofetilide in differing experimental models of atrial fibrillation. Circulation. (2000) 102(1):104–12. doi: 10.1161/01.CIR.102.1.104
140. Shinagawa K, Mitamura H, Takeshita A, Sato T, Kanki H, Takatsuki S, et al. Determination of refractory periods and conduction velocity during atrial fibrillation using atrial capture in dogs: direct assessment of the wavelength and its modulation by a sodium channel blocker, pilsicainide. J Am Coll Cardiol. (2000) 35(1):246–53. doi: 10.1016/S0735-1097(99)00488-X
141. Singh SM, D'Avila A, Kim SJ, Houghtaling C, Dukkipati SR, Reddy VY. Intraprocedural use of ibutilide to organize and guide ablation of complex fractionated atrial electrograms: preliminary assessment of a modified step-wise approach to ablation of persistent atrial fibrillation. J Cardiovasc Electrophysiol. (2010) 21(6):608–16. doi: 10.1111/j.1540-8167.2009.01671.x
142. Benharash P, Buch E, Frank P, Share M, Tung R, Shivkumar K, et al. Quantitative analysis of localized sources identified by focal impulse and rotor modulation mapping in atrial fibrillation. Circ Arrhythm Electrophysiol. (2015) 8(3):554–61. doi: 10.1161/CIRCEP.115.002721
143. Walters TE, Lee G, Lee A, Sievers R, Kalman JM, Gerstenfeld EP. Site-specific epicardium-to-endocardium dissociation of electrical activation in a swine model of atrial fibrillation. JACC Clin Electrophysiol. (2020) 6(7):830–45. doi: 10.1016/j.jacep.2020.04.015
144. Peters NS, Coromilas J, Hanna MS, Josephson ME, Costeas C, Wit AL. Characteristics of the temporal and spatial excitable gap in anisotropic reentrant circuits causing sustained ventricular tachycardia. CircRes. (1998) 82:279–93. doi: 10.1161/01.res.82.2.279
145. Ryu K, Sahadevan J, Khrestian CM, Stambler BS, Waldo AL. Frequency analysis of atrial electrograms identifies conduction pathways from the left to the right atrium during atrial fibrillation-studies in two canine models. J Cardiovasc Electrophysiol. (2009) 20(6):667–74. doi: 10.1111/j.1540-8167.2008.01403.x
146. Gerstenfeld EP, Sahakian AV, Swiryn S. Evidence for transient linking of atrial excitation during atrial fibrillation in humans. Circulation. (1992) 86(2):375–82. doi: 10.1161/01.CIR.86.2.375
147. Harada A, Sasaki K, Fukushima T, Ikeshita M, Asano T, Yamauchi S, et al. Atrial activation during chronic atrial fibrillation in patients with isolated mitral valve disease. Ann Thorac Surg. (1996) 61(1):104–11. discussion 111-102. doi: 10.1016/0003-4975(95)00824-1
148. Roithinger FX, Sippensgroenewegen A, Karch MR, Steiner PR, Ellis WS, Lesh MD. Organized activation during atrial fibrillation in man: endocardial and electrocardiographic manifestations. J Cardiovasc Electrophysiol. (1998) 9(5):451–61. doi: 10.1111/j.1540-8167.1998.tb01836.x
149. Jarman JWE, Wong T, Kojodjojo P, Spohr H, Davies JER, Roughton M, et al. Organizational index mapping to identify focal sources during persistent atrial fibrillation. J Cardiovasc Electrophysiol. (2014) 25(4):355–63. doi: 10.1111/jce.12352
150. Zeemering S, van Hunnik A, van Rosmalen F, Bonizzi P, Scaf B, Delhaas T, et al. A novel tool for the identification and characterization of repetitive patterns in high-density contact mapping of atrial fibrillation. Front Physiol. (2020) 11:570118. doi: 10.3389/fphys.2020.570118
151. van Rosmalen F, Maesen B, van Hunnik A, Hermans BJM, Bonizzi P, Bidar E, et al. Incidence, prevalence, and trajectories of repetitive conduction patterns in human atrial fibrillation. Europace. (2021) 23(23 Suppl 1):i123–32. doi: 10.1093/europace/euaa403
152. Roney CH, Child N, Porter B, Sim I, Whitaker J, Clayton RH, et al. Time-averaged wavefront analysis demonstrates preferential pathways of atrial fibrillation, predicting pulmonary vein isolation acute response. Front Physiol. (2021) 12:707189. doi: 10.3389/fphys.2021.707189
153. Mann I, Linton NWF, Coyle C, Howard JP, Fudge M, Lim E, et al. RETRO-MAPPING: a new approach to activation mapping in persistent atrial fibrillation reveals evidence of spatiotemporal stability. Circ Arrhythm Electrophysiol. (2021) 14(6):e009602. doi: 10.1161/CIRCEP.121.009602
154. Ryu K, Shroff SC, Sahadevan J, Martovitz NL, Khrestian CM, Stambler BS. Mapping of atrial activation during sustained atrial fibrillation in dogs with rapid ventricular pacing induced heart failure: evidence for a role of driver regions. J Cardiovasc Electrophysiol. (2005) 16(12):1348–58. doi: 10.1111/j.1540-8167.2005.00266.x
155. Holm M, Pehrson S, Ingemansson M, Sornmo L, Johansson R, Sandhall L, et al. Non-invasive assessment of the atrial cycle length during atrial fibrillation in man: introducing, validating and illustrating a new ECG method. Cardiovasc Res. (1998) 38(1):69–81. doi: 10.1016/S0008-6363(97)00289-7
156. Pehrson S, Holm M, Meurling C, Ingemansson M, Smideberg B, Sornmo L, et al. Non-invasive assessment of magnitude and dispersion of atrial cycle length during chronic atrial fibrillation in man. Eur Heart J. (1998) 19(12):1836–44. doi: 10.1053/euhj.1998.1200
157. Xi Q, Sahakian AV, Frohlich TG, Ng J, Swiryn S. Relationship between pattern of occurrence of atrial fibrillation and surface electrocardiographic fibrillatory wave characteristics. Heart Rhythm. (2004) 1(6):656–63. doi: 10.1016/j.hrthm.2004.09.010
158. Husser D, Stridh M, Cannom DS, Bhandari AK, Girsky MJ, Kang S, et al. Validation and clinical application of time-frequency analysis of atrial fibrillation electrocardiograms. J Cardiovasc Electrophysiol. (2007) 18(1):41–6. doi: 10.1111/j.1540-8167.2006.00683.x
159. Platonov PG, Corino VD, Seifert M, Holmqvist F, Sornmo L. Atrial fibrillatory rate in the clinical context: natural course and prediction of intervention outcome. Europace. (2014) 16 Suppl 4:iv110–9. doi: 10.1093/europace/euu249
160. Eijsbouts S, Ausma J, Blaauw Y, Schotten U, Duytschaever M, Allessie MA. Serial cardioversion by class IC drugs during 4 months of persistent atrial fibrillation in the goat. J Cardiovasc Electrophysiol. (2006) 17(6):648–54. doi: 10.1111/j.1540-8167.2006.00407.x
161. Blaauw Y, Schotten U, van HA, Neuberger HR, Allessie MA. Cardioversion of persistent atrial fibrillation by a combination of atrial specific and non-specific class III drugs in the goat. Cardiovasc Res. (2007) 75(1):89–98. doi: 10.1016/j.cardiores.2007.03.021
162. Burashnikov A, Barajas-Martinez H, Hu D, Nof E, Blazek J, Antzelevitch C. Atrial-selective prolongation of refractory period with AVE0118 is due principally to inhibition of sodium channel activity. J Cardiovasc Pharmacol. (2012) 59(6):539–46. doi: 10.1097/FJC.0b013e31824e1b93
163. Burashnikov A, Antzelevitch C. Mild elevation of extracellular potassium greatly potentiates the effect of sodium channel block to cardiovert atrial fibrillation: the Lankenau approach. Heart Rhythm. (2023) 20(9):1257–64. doi: 10.1016/j.hrthm.2023.05.005
164. Burashnikov A, Di Diego JM, Patocskai B, Echt DS, Belardinelli L, Antzelevitch C. Effect of flecainide and ibutilide alone and in combination to terminate and prevent recurrence of atrial fibrillation. Circ Arrhythm Electrophysiol. (2024) 17(1):e012454. doi: 10.1161/CIRCEP.123.012454
165. Dobrev D, Wehrens XHT. Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol. (2017) 595(12):4001–8. doi: 10.1113/JP273048
166. Stillitano F, Lonardo G, Giunti G, Del Lungo M, Coppini R, Spinelli V, et al. Chronic atrial fibrillation alters the functional properties of if in the human atrium. J Cardiovasc Electrophysiol. (2013) 24(12):1391–400. doi: 10.1111/jce.12212
167. Lai LP, Su MJ, Lin JL, Tsai CH, Lin FY, Chen YS, et al. Measurement of funny current (I(f)) channel mRNA in human atrial tissue: correlation with left atrial filling pressure and atrial fibrillation. J Cardiovasc Electrophysiol. (1999) 10(7):947–53. doi: 10.1111/j.1540-8167.1999.tb01265.x
168. Salvage SC, Chandrasekharan KH, Jeevaratnam K, Dulhunty AF, Thompson AJ, Jackson AP, et al. Multiple targets for flecainide action: implications for cardiac arrhythmogenesis. Br J Pharmacol. (2018) 175(8):1260–78. doi: 10.1111/bph.13807
169. Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiological basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. (2011) 8(8):1281–90. doi: 10.1016/j.hrthm.2011.03.045
170. Tamura A, Ogura T, Uemura H, Reien Y, Kishimoto T, Nagai T, et al. Effects of antiarrhythmic drugs on the hyperpolarization-activated cyclic nucleotide-gated channel current. J Pharmacol Sci. (2009) 110(2):150–9. doi: 10.1254/jphs.08312FP
171. Sato N, Tanaka H, Habuchi Y, Giles WR. Electrophysiological effects of ibutilide on the delayed rectifier K(+) current in rabbit sinoatrial and atrioventricular node cells. Eur J Pharmacol. (2000) 404(3):281–8. doi: 10.1016/S0014-2999(00)00603-8
172. Cha TJ, Ehrlich JR, Zhang L, Chartier D, Leung TK, Nattel S. Atrial tachycardia remodeling of pulmonary vein cardiomyocytes: comparison with left atrium and potential relation to arrhythmogenesis. Circulation. (2005) 111(6):728–35. doi: 10.1161/01.CIR.0000155240.05251.D0
173. Wang TM, Luk HN, Sheu JR, Wu HP, Chiang CE. Inducibility of abnormal automaticity and triggered activity in myocardial sleeves of canine pulmonary veins. IntJ Cardiol. (2005) 104(1):59–66. doi: 10.1016/j.ijcard.2004.10.016
Keywords: atrial fibrillation, reentry, spiral wave, rotor, focal source, excitable gap, antiarrhythmic agents
Citation: Burashnikov A (2025) “Pharmacological” analysis of atrial fibrillation maintenance mechanism: reentry, wavelets, or focal?. Front. Cardiovasc. Med. 12:1447542. doi: 10.3389/fcvm.2025.1447542
Received: 11 June 2024; Accepted: 6 January 2025;
Published: 24 January 2025.
Edited by:
Frank Heinzel, Städtisches Klinikum Dresden, GermanyReviewed by:
Jerome Montnach, INSERM U1087 Institut du Thorax, FranceRohit Kharbanda, Leiden University Medical Center (LUMC), Netherlands
Copyright: © 2025 Burashnikov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Burashnikov, YnVyYXNobmlrb3ZhQG1saHMub3Jn