 Xinxin Shi1,2,†
Xinxin Shi1,2,† Liming He1,2,†Yucheng Wang1,2
Liming He1,2,†Yucheng Wang1,2 Yue Wu1,2Dongming Lin1Chao Chen3
Yue Wu1,2Dongming Lin1Chao Chen3 Ming Yang4*
Ming Yang4* Shuwei Huang1,2*
Shuwei Huang1,2*
- 1Department of Cardiology, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Hangzhou, China
- 2The First Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, China
- 3Department of Cardiology, Hangzhou TCM Hospital of Zhejiang Chinese Medical University, Hangzhou Hospital of Traditional Chinese Medicine, Hangzhou, China
- 4Department of Cardiology, The Second Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, China
Sick sinus syndrome (SSS) is a grave medical condition that can precipitate sudden death. The pathogenesis of SSS remains incompletely understood. Existing research postulates that the fundamental mechanism involves increased fibrosis of the sinoatrial node and its surrounding tissues, as well as disturbances in the coupled-clock system, comprising the membrane clock and the Ca2+ clock. Mitochondrial dysfunction exacerbates regional tissue fibrosis and disrupts the functioning of both the membrane and calcium clocks. This plays a crucial role in the underlying pathophysiology of SSS, including mitochondrial energy metabolism disorders, mitochondrial oxidative stress damage, calcium overload, and mitochondrial quality control disorders. Elucidating the mitochondrial mechanisms involved in the pathophysiology of SSS and further investigating the disease's mechanisms is of great significance.
1 Introduction
Sick sinus syndrome (SSS) encompasses a group of syndromes characterized by impaired pacing function and/or conduction of electrical impulses due to dysfunction of the sinoatrial node and its surrounding tissue (1, 2). The disease tends to occur in the elderly (3), with an overall annual incidence of nearly 1 per 1,000 individuals aged 45 years and above (4), and approximately 1 per 600 individuals aged 65 years and above (5). It is reported that the incidence of SSS is potentially associated with age and race (6). The onset of SSS is typically insidious and progresses slowly, often rendering early diagnosis challenging, as mild cases may remain asymptomatic. However, severe cases can manifest as sinus arrest, Adams-Stokes syndrome, or sudden death (7, 8). SSS is also one of the main indications for permanent pacemaker implantation (9, 10), but recent studies indicate that while pacemaker implantation ameliorates symptoms, it does not significantly enhance survival rates (11), and there are several main disadvantages such as high cost, unsatisfactory treatment effect and great intraoperative risk (12). The pathogenesis of SSS remains incompletely understood, so it is significant to delve into the core link of the disease mechanism. Mitochondrial dysfunction is implicated in the pathogenesis of various arrhythmia diseases (13), while the attention of SSS in arrhythmia-related diseases is far less than that of atrial fibrillation. There is a lack of comprehensive review and summary. This paper examines the pivotal role of mitochondrial dysfunction in the pathogenesis of SSS.
2 Basic mechanisms of SSS
2.1 Fibrosis of the sinoatrial node and its surrounding area
The cells in the sinoatrial node are composed of pacemaker cells (P cells), transition cells (T cells), cardiac fibroblast (CF), and atrial myocytes. There are 35%–55% fibrotic tissues in this region (14), and the necessary degree of fibrosis in the sinoatrial node tissues plays a role in maintaining the structural integrity and electrical insulation in the conduction process. Such structural characteristics ensure that the electrical signal is effectively transmitted to the myocardium via the sinoatrial conduction pathway (SACP) (15–17).
In general, the extent of degenerative fibrosis of the sinoatrial node exhibits a positive correlation with advancing age. As age increases, there is a notable decrease in sinoatrial node cells and/or an increase in fatty tissue infiltration (14, 18, 19), disrupting the continuity between the sinoatrial node and the surrounding atrial myocardium (20), which is considered the most significant intrinsic cause of SSS (21).As well as the atrial muscle tissue around the sinoatrial node, it's fibrotic process resulting in slowed conduction and atrial systolic dysfunction, which concomitantly increases the risk of atrial fibrillation (22). The two genetically engineered mice, ROSA-eGFP-DTA and HCN4-KiT Cre, cultured by Stefan Herrmann et al., accurately reflected the histopathological results of human with SSS, wherein tissue degenerative fibrosis in the sinoatrial node resulted in abnormal cardiac pacing (23).Pathological fibrosis of the sinoatrial node is also one of the etiologies of SSS (15, 24), CF within the sinoatrial node secrete substantial amounts of extracellular matrix (ECM) in response to angiotensin II (AngII), inflammatory injury, oxidative stress, overload, and an acidic environment, culminating in pathological fibrosis of the sinoatrial node (25–27). The TGF-β/SMAD signaling pathway, modulated by AngII, is a well-established mediator of interstitial fibrosis (28–30). The expression of TGF-β1 related genes is implicated in the pathogenesis of SSS (31), with elevated levels of TGF-β1 and Smad2 proteins observed in atrial muscle remodeling associated with SSS (32). The SSS mouse model developed by Chen et al. with a micro-osmotic pump to continuously administer AngII, further simulates tissue pathological fibrosis in the sinoatrial node stimulated by AngII (33).
2.2 Dysfunction of the coupled-clock system
Lakatta's laboratory proposed the coupled-clock system, comprising the integration of the “Ca2+ clock” and the “membrane clock”, which is regarded as a crucial mechanism for the functional operation of the sinoatrial node pacemaker (34, 35). The strict synchronization between the two clocks ensures that the sinoatrial node beats steadily and rhythmically, and the “membrane clock” is the periodic activity of the major ion currents on the cell membrane (Table 1), including the delayed rectifier potassium current (Ik), funny current (If), L-type voltage-gated calcium channel current (ICa,L), T-type voltage-gated calcium channel currents (ICa,T), and so forth (51). Among them, If controlled by hyperpolarization-activated cyclic-nucleotide gated (HCN) channels plays a dominant role (41, 52). The “Ca2+ clock” primarily involves the inward current generated by the sodium-calcium exchanger (NCX), which maintains intracellular Ca2+ concentrations during diastole. Ca2+ serves as a mediating signal between the membrane clock and the Ca2+ clock, facilitating current conduction within sinoatrial node cells (53). Changes in the initial membrane potential lead to the opening of L-type Ca2+ channels, and Ca2+ influx elevates intracellular Ca2+ concentration. This intracellular Ca2+ can be recycled from the cytoplasm into the sarcoplasmic reticulum (SR) via the sarcoplasmic reticulum Ca2+-ATPase (SERCA), which activates SR ryanodine receptors (RyRs) to release Ca2+, thereby generating local Ca2+ release (LCR). The LCR subsequently activates the NCX, and the resultant internal Sodium-calcium exchanger current (INCX) generates activates ICa,L again, and a new action potential is formed (54, 55). Any impairment within the coupling of the membrane clock and Ca2+ clock will disrupt the pacing currents of the sinoatrial node, adversely affecting the regular and stable rhythmicity of sinoatrial node contractions, thereby precipitating the occurrence of SSS (41, 56–59).
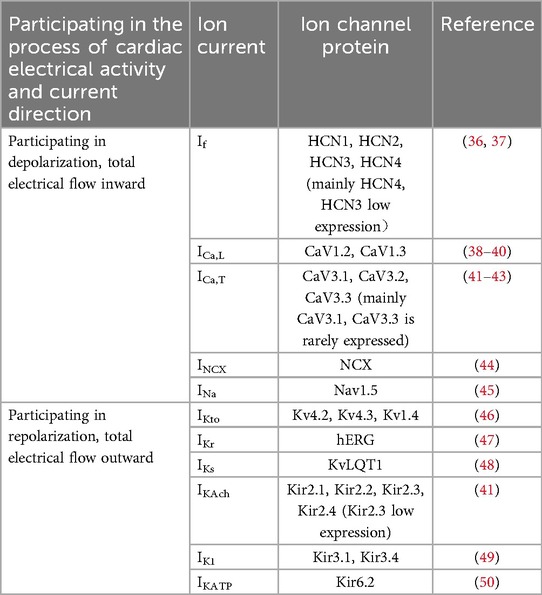
Table 1. Major ion currents and their regulatory proteins on the membrane clock.
In summary, the coupling mechanism of the coupled-clock system is essential for the sustained conduction activity of the sinoatrial node. Adequate levels of tissue fibrosis are crucial for maintaining the insulating properties of cardiac electrical conduction, while the interplay between conduction and insulation is vital for the normal functioning of the sinoatrial node. Remodeling of the sinoatrial node often results in fibrosis of both the sinoatrial node and the surrounding atrial muscle tissue (15, 60–65), induced by conditions such as atrial fibrillation, atrial flutter, heart failure, myocardial infarction. A small group of patients with SSS caused by family genetic factors are associated with regional tissue fibrosis, such as TGF-β1T869C gene (31), while the majority of cases are linked to mutations in ion channel-related genes, with SCN5A and HCN4 identified as two prominent pathogenic genes associated with SSS (66–72), and others include KCNG2 (73), SCN10A (74), GNB2 (75, 76). As well as endogenous metabolites like adenosine (77, 78)and adrenaline (79–81), are closely associated with sinoatrial node fibrosis and dysfunction of the coupled-clock system.
3 Mitochondria and mechanisms of SSS
3.1 Mitochondrial energy metabolism and mechanisms of SSS
Mitochondria, often known as the energy factories of cells, are responsible for the production of adenosine triphosphate (ATP) through oxidative phosphorylation, thereby supplying energy to the cell. In the context of cardiac physiology, sinoatrial node cells, functioning as the heart's natural pacemaker, must continuously generate electrical impulses, resulting in a heightened demand for energy. The mechanism of SSS and mitochondrial energy metabolism are reflected in the following aspects (Table 2).
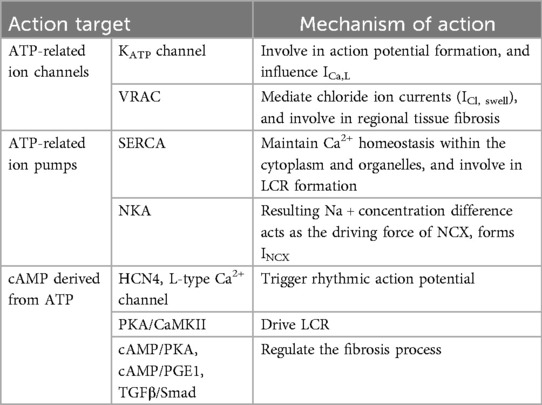
Table 2. Summary of mitochondrial energy metabolism and mechanisms of SSS.
3.1.1 ATP-related ion channels
3.1.1.1 ATP-sensitive potassium channel (KATP channel)
KATP channel comprises Kir6 main subunit and SuR auxiliary subunit (82), and participates in potassium ion current in sinoatrial node cells (83). Its activity is modulated by intracellular ATP concentrations; elevated ATP levels inhibit the channel, preventing its involvement in action potential formation and excitation-contraction coupling. Conversely, a decrease in ATP concentration leads to channel activation, facilitating K+ efflux, which accelerates repolarization and shortens the action potential plateau (84–86). In addition to KATP channels distributed on the cell membrane, which participate in the formation of membrane clock currents, KATP channels distributed on the inner mitochondrial membrane are closely related to mitochondrial energy metabolism, mitochondrial membrane potential maintenance, apoptosis inhibition, and Ca2+ overload alleviation, thereby sustaining the homeostatic state of the intracellular environment (87–89). The activity of KATP channel is regulated by mitochondrial energy metabolism and synchronously participates in mitochondrial energy metabolism and Ca2+ homeostasis, thereby influencing ICa,L (90), and repeatedly affecting the coupled-clock system mechanism which mediates the pacemaker currents of sinoatrial node cells.
3.1.1.2 Volume-regulated anion channel (VRAC)
VRAC is expressed in atrial myocytes, ventricular myocytes and P cells, and is involved in cardiac physiology and pathophysiological processes (91, 92). VRAC activation occurs in response to cellular swelling, hence it is also referred to as the swelling-activated chloride channel. The primary component of VRAC is leucine-rich repeat protein 8 (LRRC8)A, commonly known as Swell1 (93), is essential for regulating cellular volume reduction, maintaining cell volume homeostasis, and mediating chloride ion currents (ICl, swell) (94). Activation of VRAC requires the participation of ATP (95, 96), and the function is reversibly inhibited in hypoxic environment and under the action of mitochondrial inhibitors (97). Interestingly, VRAC is regulated by mitochondrial energy metabolism; inhibition of VRAC consequently affects the mitochondrial electron transport chain, thereby impairing ATP production (98). On the other hand, normally functioning VRAC channels also play an important role in CF (99). Given their role as energy-demanding ion channel proteins, it is plausible to suggest that metabolic disorders arising from mitochondrial dysfunction may provoke inflammatory responses in the sinoatrial node or contribute to increased extracellular matrix deposition, thereby exacerbating regional tissue fibrosis.
3.1.2 ATP-related ion pumps
3.1.2.1 SERCA
SERCA is a crucial ion pump integral to the Ca2+ clock mechanism of the sinoatrial node. Its primary function is to restore intracellular Ca2+ levels and maintain calcium homeostasis within the cytoplasm and organelles, such as mitochondria and the SR. The stable release of Ca2+ in the SR ensures the stable LCR of the Ca2+ clock. SERCA has a high affinity with ATP (100), while ATP consumption is accompanied by Ca2+ recycling (101, 102), which can negatively regulate SERCA activity by restricting ATP produced in mitochondria (103). Meanwhile, Claudia Rodriguez et al. also demonstrated that AMP-activated protein kinase (AMPK), which is involved in mitochondrial protection, can stimulate the ATP-generating pathway and restore homeostasis to activate SERCA activity (104). This indicates that mitochondria play a significant role in cytoplasmic Ca2+ recovery through SERCA based on energy metabolism, and further participate in the coupled-clock system's coupling mechanism. The ability of sinoatrial node cells to generate larger and rhythmic LCRs should be linked with increased abundance of SERCA (55), while the expression level and activity of SERCA tend to decline with age (105), which also proves that mitochondria-driven attenuation of SERCA during aging is one of the mechanisms of SSS.
3.1.2.2 Na+/K+-ATPase (NKA)
NKA is an ion pump that consumes ATP to maintain the balance of sodium (Na+) and potassium (K+) ions within the cell (106). It works in concert with other ion channels and pumps to regulate calcium (Ca2+) homeostasis within the SR, which is crucial for both SR Ca2+ equilibrium and membrane depolarization in sinoatrial node cells. This regulation ensures the maintenance of the resting membrane potential and the rhythmic function of the sinoatrial node (107, 108). When the concentration of cytoplasmic Ca2+ is low, SERCA is in a low activity state, while NKA, which is in a high activity state, can account for 30% of all ATPases (109), and participate in the transport of Na+ and K+. The resulting Na+ concentration difference acts as the driving force of NCX (110–112). The whole process is regulated by intracellular Ca2+ signaling (111), and eventually INCX is formed (113), which is vital to the electrical function of the heart (114). Gvantsa Chkadua et al. noted that NKA activity is regulated not only by mitochondrial ATP but also by cytochrome c (Cyt C) released from mitochondria, which mediates non-apoptotic effects. NKA is activated by low Cyt C concentrations and inhibited at higher concentrations (115). Norbert A. Dencher et al. observed impaired mitochondrial function and significantly reduced NKA expression in aged rats or those with age-related diseases (116). Therefore, NKA is intricately linked with mitochondrial function and is a potential target for mitochondrial-mediated cardiac protection (117).
3.1.3 Cyclic adenosine monophosphate (cAMP) derived from ATP is involved in the cardiac electrical activity of sinoatrial node cells
cAMP, mainly derived from ATP, is formed by the cyclization reaction of ATP after the removal of one pyrophosphate (two phosphorus atoms) under the catalysis of adenylyl cyclase (AC) (118). It is widely present in cells and plays a crucial role in various physiological processes, notably acting as an intracellular “second messenger” in the sinoatrial node (81, 119). cAMP/protein kinase A (PKA) signal is an important mechanism driving the coupled-clock system of sinoatrial node cell membrane to trigger rhythmic action potential (120), and the efficacy of this mechanism diminishes with age due to a reduction in cAMP levels (80). Therefore, many iron channels on the sinoatrial node cell membrane are regulated by cAMP, with the most dependent being HCN4 (121–124), L-type Ca2+ channels (125–127). Regarding the regulation of the Ca2+ clock, the occurrence of LCR is dependent on the phosphorylation of cAMP downstream PKA and Ca2+/calmodulin-dependent protein kinase II (CaMKII) (127–129), and cAMP also plays a regulatory role in inhibiting tissue fibrosis (130). Age-related cardiac contraction decline is partly attributed to the desensitization of β-adrenergic/cAMP signaling. Enhancing cardiac cAMP bioavailability and PKA activity has been shown to improve contractile function in mice, potentially alleviating fibrosis and cardiac tissue remodeling (131). Besides that, cAMP-elevating receptor agonist prostaglandin E1 (PGE1) can inhibit cardiac fibroblast proliferation (132). Alternatively, cAMP inhibits the downstream TGFβ/Smad signaling pathway, reduces the expression of α-SMA, alleviates ECM deposition, thus regulating the fibrosis process (133–137).
In addition, although the cystic fibrosis transmembrane conductance regulator (CFTR) has not been reported to be related to the SSS mechanism, it holds potential as a target protein for future investigations into its role in the electrophysiological processes and fibrosis progression within the sinoatrial node and adjacent tissues. CFTR is extensively expressed in the heart (138), which requires ATP hydrolysis for energy (139–141), and is also shown as cAMP-dependent (142), and accompanied by current signals ICl,cAMP during chloride ion transport (143, 144). What is more interesting is that gene mutations resulting in the mistranslation of CFTR can directly cause cystic fibrosis (139), suggesting a subtle connection between the fundamental mechanisms of SSS and cystic fibrosis, whether by coincidence or inevitability.
3.2 Mitochondrial oxidative stress and SSS
Aging and a variety of cardiovascular diseases are believed to be related to mitochondrial oxidative stress. For instance, mice with a germline deletion of the Ndufs4 subunit of respiratory complex I exhibit chronic arrhythmias, atrioventricular block, and other sinoatrial node dysfunctions. Targeted anti-oxidative stress therapy has been shown to ameliorate chronic arrhythmias and prolong their lifespan (145). The role of mitochondrial oxidative stress in the pathogenesis of SSS may be related to the following aspects (Table 3).
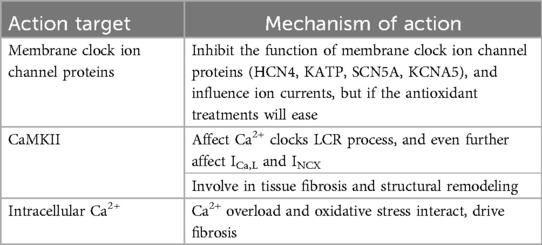
Table 3. Summary of mitochondrial oxidative stress and SSS.
3.2.1 Ion channels dysfunction is mediated by mitochondrial oxidative stress injury
Thioredoxin 2 (Trx2) serves as a rate-limiting enzyme in the mitochondrial thioredoxin system, which is one of the principal pathways for scavenging reactive oxygen species (ROS). Trx2 can bind to apoptosis signal-regulated kinase-1 (ASK1), inhibiting its activity and thereby suppressing apoptosis induced by the pro-apoptotic factor cysteine-aspartic acid protease-3 (Caspase3) (146, 147). This mechanism plays a pivotal role in maintaining cellular survival, reducing oxidative stress, and regulating mitochondrial apoptosis signal transduction (148–150). Bicheng Yan et al. discovered that Trx2 is essential for maintaining HCN4-mediated normal heart rate; the absence of Trx2 results in significant ROS accumulation, leading to the dysfunction of HCN4, a critical factor in the development of SSS (151). Carlos H. Pereira et al. found that p21-activated kinase 1 (Pak1) enhances NADPH oxidase 2-dependent ROS production, thereby reducing HCN expression and inhibiting sinoatrial node activity. The sinoatrial node dysfunction, associated with a decreased heart rate following oxidative stress injury controlled by Pak1, is linked to membrane clock dysfunction rather than Ca2+ clock dysfunction. Additionally, the application of TEMPOL, a ROS scavenger, can clear ROS and reverse such sinoatrial node function impairment (152). It is evident that HCN4 is an ion channel significantly affected by mitochondrial oxidative stress (152). A large amount of ROS accumulated by oxidative stress damage can affect the function of KATP (153), the process of tachypacing-induced mitochondrial dysfunction is often accompanied by oxidative stress damage and Ca2+ overload, targeted use of antioxidants can reverse associated mitochondrial dysfunction, which reduced ADP and increased ATP production in cells, meanwhile, KATP expression increased (154). The cardiac sodium channel NaV1.5 (SCN5A) has a fundamental role in excitability and conduction, peroxisome proliferator activated receptor-γ (PPARγ) coactivator-1 (Pgc-1)-deficient murine cardiac models simulate mitochondrial dysfunction, a decrease in Nav1.5 channel protein expression was found in these models (155). Upon tetrodotoxin (TTX) exposure, it is showed that ROS increase, mitochondrial function decreases, SCN5A expression decreased (156). As we can see, mitochondrial oxidative stress injury is an important factor affecting the function and expression of SCN5A, meliorate mitochondrial oxidative stress and preserve bioenergetics can improve mitochondrial dysfunction and protect the function of SCN5A (157). Angiotensin II (Ang II) enhances the expression of KV1.5 (KCNA5) by activating ROS-dependent phosphorylation of Smad2/3 (forming P-Smad2/3) and ERK 1/2 (forming P-ERK1/2), antioxidant can diminish Ang II-induced reactive oxygen species (ROS) generation, inhibit Ang II-induced expression of P-Smad2/3, phosphorylated ERK (P-ERK1/2), which maintains the normal function of KV1.5 (158–160).Trx2 is regulated by nuclear factor-erythroid 2-related factor 2 (Nrf2), which together constitute a crucial component of mitochondrial oxidative stress (161, 162). Heng Zhang et al. found that SSS is associated with oxidative stress damage, which may be related to the loss of HCN4 and the weakening of If in the process of oxidative stress, and the anti-oxidative stress effect of regulating the Nrf-2/HO-1 axis can improve SSS (163).
3.2.2 Mitochondrial oxidative stress and Ca2+/CaMKII activation participate in Ca2+ clock and structural remodeling
During mitochondrial oxidative stress, a substantial accumulation of ROS participates in the calcium release process of RyRs on the SR (164). This phenomenon is related to ROS-mediated phosphorylation of CaMKII, where aberrant activation of CaMKII phosphorylation results in Ca2+ leakage of RyRs and abnormal LCR (165, 166). LCR affects the discharge rhythm and the recycling and release of cytoplasmic Ca2+ by SR, ICa,L and INCX (128, 167). Duanyang Xie et al. identified that mitochondrial excitatory amino acid transporter 1 (EAAT1)-dependent mitochondrial glutamate input enhances ROS production, leading to the oxidation of CaMKII protein, ultimately augmenting LCR (168). Jian-Bin Xue et al. observed that sinoatrial node dysfunction following heart failure manifested as decreased CaMKII phosphorylation, reduced RyRs protein expression, diminished SERCA function, lowered SR Ca2+ content, attenuated LCR, and inhibited Ca2+ clock. Furthermore, CaMKII was implicated in subsequent tissue fibrosis and structural remodeling during this process (169, 170). Despite appearing contradictory, the findings of the two scholars are reconcilable. Duanyang Xie's observations pertain to glutamate-mediated ROS generation within the controlled oxidative stress damage range of sinoatrial node cells, whereby sufficient ROS and substrates required by CaMKII can enhance LCR. Under severe oxidative stress, CaMKII often mediates apoptosis, exacerbating the initial injury (171, 172). Jian-Bin Xue's findings are predicated on sinoatrial node dysfunction post-heart failure, where CaMKII contributes more significantly to tissue fibrosis and structural remodeling as part of compensatory mechanisms. Both accelerated and decelerated heart rates mediated by LCR enhancement or attenuation align with clinical manifestations of SSS, including fast-slow syndrome and slow-fast syndrome. Our inferences, based on clinical presentations, suggest that CaMKII activation mediated by mitochondrial oxidative stress injury and the abnormality of the sinoatrial node Ca2+ clock LCR are experimentally demonstrated, also involving fibrosis and remodeling.
3.2.3 Mitochondrial Ca2+ overload and oxidative stress damage mediated fibrosis
Mitochondria affect intracellular Ca2+ concentration by modulating Ca2+ absorption and release. When Ca2+ overload occurs in mitochondria, mitochondrial membrane potential changes will cause oxidative stress or aggravate mitochondrial oxidative stress damage. ROS generated by oxidative stress can further damage mitochondrial membrane structure, affecting mitochondrial Ca2+ uniporter (MCU) and mitochondrial permeability transition pore (mPTP), thereby aggravatingCa2+ overload (173–175). Mitochondrial Ca2+ overload and oxidative stress often interact (176). Xing Chang et al. demonstrated that hypoxia/reoxygenation (H/R) injury disrupts the equilibrium of “Ca2+ release” and “Ca2+ cycling” in rat sinoatrial node cells, and abnormal Ca2+ concentration or Ca2+ overload in sinoatrial node cells further aggravated mitochondrial oxidative stress injury (177). Oxidative stress and Ca2+ overload are frequently implicated in the pathogenesis of fibrosis (178–180). An imbalance in Ca2+ homeostasis is a critical factor in sinoatrial node dysfunction and regional tissue fibrosis. Mingjie Zheng et al. found that Hippo-Yap pathway plays a role in maintaining sinoatrial node homeostasis by regulating Ca2+ homeostasis, and the inactivation of Lats1/2, pathologically associated with the Hippo-Yap signaling pathway, results in severe dysfunction of the sinoatrial node, which is manifested by Ca2+ homeostasis imbalance and increased fibrosis in the sinoatrial node region (181). Impulse conduction disorders in the atrial region are still an important part of SSS, and mitochondrial oxidative stress in atrial myocytes is a significant cause of atrial fibrosis (182).
3.3 Mitochondrial quality control (MQC) and SSS mechanism
MQC refers to the mechanisms within the cell that regulate the number, morphology and function of mitochondria to ensure their normal functioning and cell health, and involves mitochondrial protein homeostasis, mitochondrial autophagy, and mitochondrial dynamics and biogenesis (183–185). MQC defects often play a crucial role in the pathogenesis of cardiovascular diseases (186), especially degenerative diseases correlated with aging (187). MQC defects associated with the pathogenesis of SSS have been reported mainly in fusion protein-mediated mitochondria-SR coupling and dynamin-related protein mediated mitochondria fission and autophagy (Table 4).
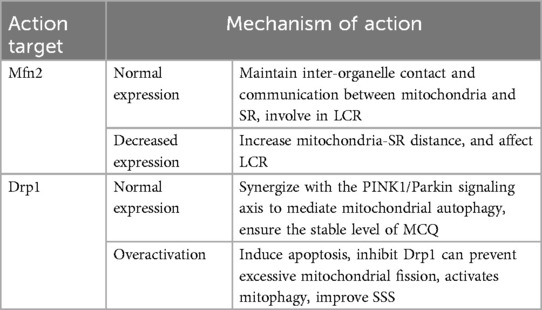
Table 4. Summary of MQC and SSS mechanism.
3.3.1 Mitofusin 2(Mfn2) mediated mitochondria-SR coupling
Mfn2 is a transmembrane guanosine triphosphatase (GTPase) located in the outer membrane of mitochondria, and a key factor in regulating mitochondrial fusion and maintaining mitochondrial structure (188). Mfn2 is also expressed on SR, and the proximity between mitochondria and SR (189), facilitates a hub for crosstalk between them (190, 191). Inter-organelle contact and communication between mitochondria and SR maintain cellular homeostasis (192), especially in the Ca2+ cycle involved in LCR (189, 190, 193, 194). Under normal conditions, mitochondrial ATP is vital to power SR Ca2+ cycling that drives phasic contraction/relaxation, and changes in SR Ca2+ release are sensed by mitochondria and used to modulate oxidative phosphorylation based on metabolic need (195). Disruption of the physical link between SR and mitochondria mediated by Mfn2 impairs the mitochondria-SR metabolic feedback mechanism. This results in diminished SERCA activity, obstructed Ca2+ cycling, impaired mitochondrial ATP production, disrupted energy metabolism, and potentially programmed cell death (195, 196). Lu Ren et al. identified that sinoatrial node dysfunction is associated with Mfn2-mediated alterations in mitochondria-SR connectomics. In a mouse model of sinoatrial node dysfunction induced by heart failure, electron microscopy (EM) tomography revealed mitochondrial structural abnormalities and increased mitochondria-SR distance. This results in abnormal mitochondrial Ca2+ processing, altered local PKA activity, and impaired mitochondrial function in sinoatrial node cells (197).
3.3.2 Dynamin-related protein 1 (Drp1) mediates mitochondrial fission and autophagy
Drp1, a GTPase widely distributed in the cytoplasm, is a major regulatory factor in the process of mitochondrial fission (198, 199). When the mitochondrial fission process is activated, Drp1 is transported to the mitochondrial surface, where it binds to related receptors to form helical oligomers. These helical Drp1 structures facilitate its GTP hydrolysis (200, 201), encircle the mitochondrial outer membrane, and mediate its scission (202, 203). Drp1 plays a major role in the entire process of mitochondrial fission (204), DRP1-mediated mitochondrial fission promotes the occurrence of mitochondrial autophagy, and the dysfunctional mitochondria produced by fission depend on the clearance of mitochondrial autophagy (205, 206). Under normal cellular conditions, Drp1 synergizes with the PINK1/Parkin signaling axis to mediate mitochondrial autophagy, ensuring the stable mitochondrial quality level and function (207). Excessive activation of Drp1 leads to excessive mitochondrial fission beyond the scope of mitochondrial autophagy, the increase of Drp1 level promotes excessive opening of mPTP, oligomerization of BCL2-associated X protein (Bax) and release of Cyt C, and mitochondrial ROS accumulation, and loss of mitochondrial membrane potential, ultimately inducing apoptosis (208, 209). Conversely, the application of Drp1 inhibitors can ameliorate these adverse effects (210, 211). Rebecca Z Fan et al. found that a partial Drp1 knockout improves autophagy (212). Mitochondrial fission is integral to fibrosis, as evidenced by Ching-Yi Chen et al., who found that Drp1 inhibition elevated ATP levels and reduced mitochondrial fission and apoptosis, thereby mitigating fibrosis (213). Enhanced mitochondrial autophagy also contributed to the reduction of ECM in the sinoatrial node (33), the mechanism of fibrosis in SSS is linked to Drp1-mediated mitochondrial fission and autophagy. Xing Chang et al. proved that Tongyang Huoxue decoction (TYHX) inhibits Drp1 translocation to mitochondria, prevents excessive mitochondrial fission, activates mitophagy, and enhances mitochondrial membrane potential, demonstrating TYHX's efficacy in improving SSS (177).
The foregoing is the process of mitochondria related mechanisms participating in the pathological mechanism of SSS, which is mainly reflected in mitochondrial energy metabolism, mitochondrial oxidative stress damage, MCQ's involvement in the basic pathological mechanism of SSS regarding the exacerbation of regional tissue fibrosis and the dysfunction of the coupled-clock system mechanism. For the summary, please refer to the mechanism diagram (Figure 1).
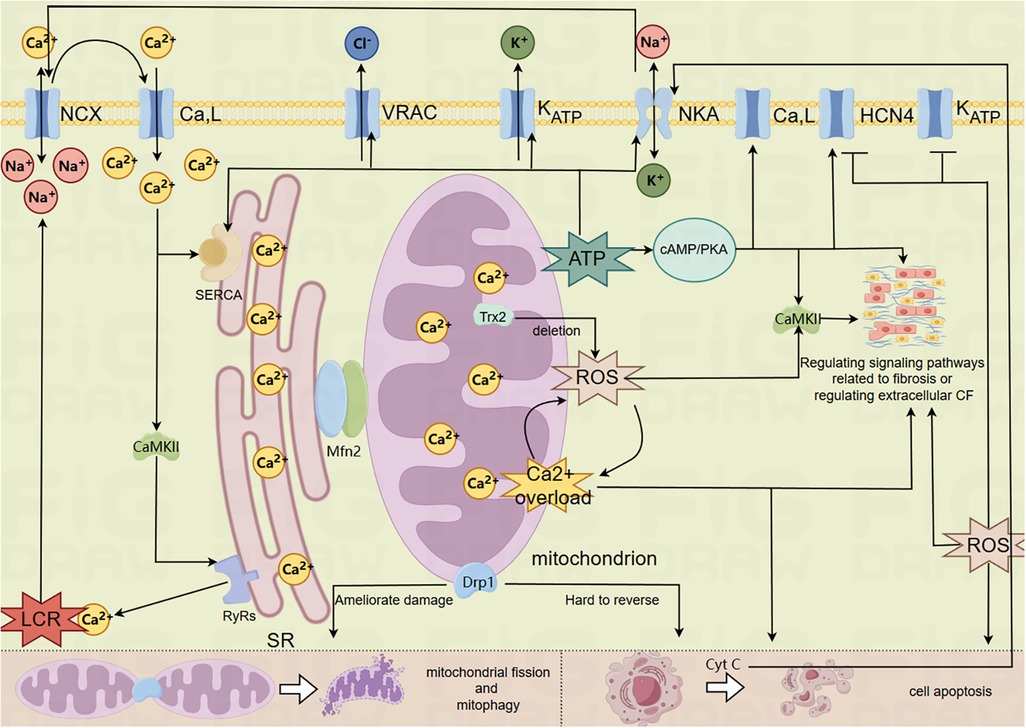
Figure 1. Mechanism of mitochondrial involvement in SSS. (The model membrane clock ion channel proteins in the figure are closely related to mitochondria and have been mentioned in relevant literature, so they cannot represent all membrane clock ion channel proteins in the figure).
4 Summary and conclusion
The rhythmically beating heart is an organ with high energy demand (214). Mitochondrial dysfunction mechanisms are frequently implicated in the physiological and pathological processes of various cardiovascular diseases, including myocardial ischemia-reperfusion injury (215), atrial fibrillation (216), heart failure (217) and recovery after myocardial infarction (218). There is no doubt that SSS cannot be excluded, particularly as this class of diseases is clearly associated with aging, which has been shown to correlate with mitochondrial dysfunction (219, 220). Disruption of the coupled-clock system mechanism and severe regional tissue fibrosis are the fundamental mechanisms of SSS. ATP and its derivative, cAMP, produced via mitochondrial energy metabolism, are involved in regulating membrane clock-related ion channel proteins and Ca2+ clock-related ion pumps, and in modulating signaling pathways that mediate fibrosis or extracellular CF. Additionally, mitochondria-mediated oxidative stress damage, Ca2+ overload, and MCU dysfunction contribute to the development and progression of these mechanisms in SSS.
In fact, SSS is a kind of syndrome of pacing function and (or) impulse conduction dysfunction caused by dysfunction of sinoatrial node and surrounding tissue lesions. This syndrome encompasses lesions in the sinoatrial node region as well as in the adjacent atrial and atrioventricular junction areas, complicating both clinical and experimental research efforts. Clinical diagnosis primarily relies on electrocardiographic evaluations, yet the diverse and complex electrocardiographic manifestations across different heart regions further complicate the classification of disease types. At the same time, various animal modeling methods of SSS often fail to reflect the complexity of clinical diseases, such as gene regulation (23), inducing ischemia-reperfusion injury on the sinoatrial node area, and injecting sodium hydroxide to the sinoatrial node area through internal jugular vein (221), among others. On the other hand, mitochondria are ingenious and magical cellular organelles with powerful functions. They can regulate energy metabolism, oxidative stress and Ca2+ level, communicate with other neighboring organelles, participate in intracellular communication between organelles and mitochondrial autophagy to clear dysfunctional mitochondria and maintain intracellular environmental homeostasis. At the same time, mitochondrial dysfunction to the point of inability to stabilize the entire cell function can mediate the occurrence of apoptosis. The multifaceted roles of mitochondria are interdependent; for instance, oxidative stress damage can be exacerbated by abnormal energy metabolism or Ca2+ overload in mitochondria. On the contrary, energy metabolism is influenced by mitochondrial oxidative stress and Ca2+ concentrations, with oxidative stress and Ca2+ overload mutually interacting. Mitochondrial dynamics, such as fission, fusion, and autophagy, are integral to maintaining complete mitochondrial functionality, thus adding to the complexity and challenges of related studies.
Based on the published clinical and experimental findings concerning the mechanisms of mitochondrial dysfunction and SSS, it is evident that further investigation is warranted. Specifically, it remains unclear whether other ion channel proteins associated with the membrane clock are directly regulated by mitochondrial mechanisms and contribute to the fundamental pathology of SSS, or if they exert no significant regulatory effects. Additionally, the role of mitochondria in the fibrosis process mediated by certain ion channel proteins, which may be central to the basic mechanism of SSS, requires further elucidation. Therefore, current research in this area is insufficient. Further in-depth and comprehensive mechanism studies will help clarify the involvement of mitochondrial mechanism in the pathological mechanism of SSS and identify the core link of disease pathogenesis, which is conducive to the development of relevant new effective drugs to prevent and treat SSS.
Author contributions
XS: Writing – original draft. LH: Writing – original draft. YWa: Writing – review & editing. YWu: Writing – review & editing. DL: Writing – review & editing. CC: Writing – review & editing. MY: Writing – original draft. SH: Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Key Project of Traditional Chinese Medicine Science and Technology Program of Zhejiang Province (Grant No.2022ZZ015) and the Project of Traditional Chinese Medicine Science and Technology Program of Zhejiang Province (Grant No.2024ZL117).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Liang J, Luo S, Huang B. Case report: sCN5A mutations in three young patients with sick sinus syndrome. Front Cardiovasc Med. (2023) 10:1294197. doi: 10.3389/fcvm.2023.1294197
2. Manoj P, Kim JA, Kim S, Li T, Sewani M, Chelu MG, et al. Sinus node dysfunction: current understanding and future directions. Am J Physiol Heart Circ Physiol. (2023) 324(3):H259–h78. doi: 10.1152/ajpheart.00618.2022
3. Ewy GA. Sick sinus syndrome. J Am Coll Cardiol. (2014) 64(6):539–40. doi: 10.1016/j.jacc.2014.05.029
4. Jensen PN, Gronroos NN, Chen LY, Folsom AR, deFilippi C, Heckbert SR, et al. Incidence of and risk factors for sick sinus syndrome in the general population. J Am Coll Cardiol. (2014) 64(6):531–8. doi: 10.1016/j.jacc.2014.03.056
5. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation. (2022) 145(8):e153–639. doi: 10.1161/cir.0000000000001052
6. Mond HG, Proclemer A. The 11th world survey of cardiac pacing and implantable cardioverter-defibrillators: calendar year 2009–a world society of arrhythmia's project. Pacing Clin Electrophysiol. (2011) 34(8):1013–27. doi: 10.1111/j.1540-8159.2011.03150.x
7. Bodin A, Bisson A, Gaborit C, Herbert J, Clementy N, Babuty D, et al. Ischemic stroke in patients with sinus node disease, atrial fibrillation, and other cardiac conditions. Stroke. (2020) 51(6):1674–81. doi: 10.1161/STROKEAHA.120.029048
8. De Ponti R, Marazzato J, Bagliani G, Leonelli FM, Padeletti L. Sick sinus syndrome. Card Electrophysiol Clin. (2018) 10(2):183–95. doi: 10.1016/j.ccep.2018.02.002
9. Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomström-Lundqvist C, et al. 2020 ESC guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European association for cardio-thoracic surgery (EACTS). Eur Heart J. (2021) 42(5):373–498. doi: 10.1093/eurheartj/ehaa612
10. Pourshams IA, Lin B, Wang PJ, Stafford RS. Decision-making experiences and decisional regret in patients receiving implanted cardioverter-defibrillators. Heart Mind. (2022) 6(1):32–5. doi: 10.4103/hm.hm_51_21
11. Dang D, Galand V, Loirat A, Auffret V, Behar N, Mabo P, et al. Procedural safety and long-term follow-up after pacemaker implantation in nonagenarians. Clin Cardiol. (2018) 41(10):1315–21. doi: 10.1002/clc.23083
12. Poole JE, Gleva MJ, Mela T, Chung MK, Uslan DZ, Borge R, et al. Complication rates associated with pacemaker or implantable cardioverter-defibrillator generator replacements and upgrade procedures. Circulation. (2010) 122(16):1553–61. doi: 10.1161/CIRCULATIONAHA.110.976076
13. Brown DA, O'Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res. (2010) 88(2):241–9. doi: 10.1093/cvr/cvq231
14. Shiraishi I, Takamatsu T, Minamikawa T, Onouchi Z, Fujita S. Quantitative histological analysis of the human sinoatrial node during growth and aging. Circulation. (1992) 85(6):2176–84. doi: 10.1161/01.CIR.85.6.2176
15. Csepe TA, Kalyanasundaram A, Hansen BJ, Zhao J, Fedorov VV. Fibrosis: a structural modulator of sinoatrial node physiology and dysfunction. Front Physiol. (2015) 6:37. doi: 10.3389/fphys.2015.00037
16. Fedorov VV, Glukhov AV, Chang R, Kostecki G, Aferol H, Hucker WJ, et al. Optical mapping of the isolated coronary-perfused human sinus node. J Am Coll Cardiol. (2010) 56(17):1386–94. doi: 10.1016/j.jacc.2010.03.098
17. Oren RV, Clancy CE. Determinants of heterogeneity, excitation and conduction in the sinoatrial node: a model study. PLoS Comput Biol. (2010) 6(12):e1001041. doi: 10.1371/journal.pcbi.1001041
18. Yan J, Li H, Bu H, Jiao K, Zhang AX, Le T, et al. Aging-associated sinus arrest and sick sinus syndrome in adult zebrafish. PLoS One. (2020) 15(5):e0232457. doi: 10.1371/journal.pone.0232457
19. Sugiura M, Ohkawa S, Hiraoka K, Okimoto T, Ueda K. A clinicopathological study on the sick sinus syndrome. Jpn Heart J. (1976) 17(6):731–41. doi: 10.1536/ihj.17.731
20. Machida N, Hirakawa A. The anatomical substrate for sick sinus syndrome in dogs. J Comp Pathol. (2021) 189:125–34. doi: 10.1016/j.jcpa.2021.10.007
21. Demoulin JC, Kulbertus HE. Histopathological correlates of sinoatrial disease. Br Heart J. (1978) 40(12):1384–9. doi: 10.1136/hrt.40.12.1384
22. Chang W, Li G. Clinical review of sick sinus syndrome and atrial fibrillation. Herz. (2022) 47(3):244–50. doi: 10.1007/s00059-021-05046-x
23. Herrmann S, Fabritz L, Layh B, Kirchhof P, Ludwig A. Insights into sick sinus syndrome from an inducible mouse model. Cardiovasc Res. (2011) 90(1):38–48. doi: 10.1093/cvr/cvq390
24. Chen H, Chew G, Devapragash N, Loh JZ, Huang KY, Guo J, et al. The E3 ubiquitin ligase WWP2 regulates pro-fibrogenic monocyte infiltration and activity in heart fibrosis. Nat Commun. (2022) 13(1):7375. doi: 10.1038/s41467-022-34971-6
25. Hof T, Chaigne S, Récalde A, Sallé L, Brette F, Guinamard R. Transient receptor potential channels in cardiac health and disease. Nat Rev Cardiol. (2019) 16(6):344–60. doi: 10.1038/s41569-018-0145-2
26. Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med. (2009) 19(8):247–52. doi: 10.1016/j.tcm.2010.02.005
27. Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, et al. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest. (2011) 121(8):3277–88. doi: 10.1172/JCI57833
28. Rupérez M, Lorenzo O, Blanco-Colio LM, Esteban V, Egido J, Ruiz-Ortega M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation. (2003) 108(12):1499–505. doi: 10.1161/01.CIR.0000089129.51288.BA
29. Bujak M, Frangogiannis NG. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. (2007) 74(2):184–95. doi: 10.1016/j.cardiores.2006.10.002
30. Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. (2020) 577(7791):566–71. doi: 10.1038/s41586-019-1897-5
31. Chen JY, Liu JH, Wu HD, Lin KH, Chang KC, Liou YM. Transforming growth factor-β1 T869C gene polymorphism is associated with acquired sick Sinus syndrome via linking a higher Serum protein level. PLoS One. (2016) 11(7):e0158676. doi: 10.1371/journal.pone.0158676
32. Xiao M, Zhang M, Bie M, Wang X, Guo J, Xiao H. Galectin-3 induces atrial fibrosis by activating the TGF-β1/smad pathway in patients with atrial fibrillation. Cardiology. (2020) 145(7):446–55. doi: 10.1159/000506072
33. Chen C, Zhang H, Hou S, Liu Y, Ren L, Hao M, et al. Shenxian-Shengmai oral liquid evoke autophagy of fibroblast to attenuate sinoatrial node fibrosis in sick Sinus syndrome mice via the AKT/mTOR pathway. Evid Based Complement Alternat Med. (2022) 2022:1. doi: 10.1155/2022/5219277
34. Maltsev VA, Lakatta EG. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am J Physiol Heart Circ Physiol. (2009) 296(3):H594–615. doi: 10.1152/ajpheart.01118.2008
35. Lakatta EG, Maltsev VA, Vinogradova TM. A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res. (2010) 106(4):659–73. doi: 10.1161/CIRCRESAHA.109.206078
36. Boyett MR, Yanni J, Tellez J, Bucchi A, Mesirca P, Cai X, et al. Regulation of sinus node pacemaking and atrioventricular node conduction by HCN channels in health and disease. Prog Biophys Mol Biol. (2021) 166:61–85. doi: 10.1016/j.pbiomolbio.2021.06.008
37. Wang Y, Anderson C, Dobrzynski H, Hart G, D'Souza A, Boyett MR. RNAseq shows an all-pervasive day-night rhythm in the transcriptome of the pacemaker of the heart. Sci Rep. (2021) 11(1):3565. doi: 10.1038/s41598-021-82202-7
38. Logantha S, Yamanushi TT, Absi M, Temple IP, Kabuto H, Hirakawa E, et al. Remodelling and dysfunction of the sinus node in pulmonary arterial hypertension. Philos Trans R Soc Lond B Biol Sci. (2023) 378(1879):20220178. doi: 10.1098/rstb.2022.0178
39. Mesirca P, Bidaud I, Briec F, Evain S, Torrente AG, Le Quang K, et al. G protein-gated IKACh channels as therapeutic targets for treatment of sick sinus syndrome and heart block. Proc Natl Acad Sci U S A. (2016) 113(7):E932–41. doi: 10.1073/pnas.1517181113
40. Zhang J, Wei F, Ding L, Wang L, Zhang X, Yu L, et al. MicroRNA-1976 regulates degeneration of the sinoatrial node by targeting Cav1.2 and Cav1.3 ion channels. J Mol Cell Cardiol. (2019) 134:74–85. doi: 10.1016/j.yjmcc.2019.06.018
41. Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, et al. Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation. (2009) 119(12):1562–75. doi: 10.1161/CIRCULATIONAHA.108.804369
42. Li Y, Zhang X, Zhang C, Zhang X, Li Y, Qi Z, et al. Increasing T-type calcium channel activity by β-adrenergic stimulation contributes to β-adrenergic regulation of heart rates. J Physiol. (2018) 596(7):1137–51. doi: 10.1113/JP274756
43. Mesirca P, Torrente AG, Mangoni ME. T-type channels in the sino-atrial and atrioventricular pacemaker mechanism. Pflugers Arch. (2014) 466(4):791–9. doi: 10.1007/s00424-014-1482-6
44. Kohajda Z, Loewe A, Tóth N, Varró A, Nagy N. The cardiac pacemaker story—fundamental role of the Na+/Ca2+ exchanger in spontaneous automaticity. Front Pharmacol. (2020) 11:516. doi: 10.3389/fphar.2020.00516
45. Baruscotti M, Difrancesco D, Robinson RB. A TTX-sensitive inward sodium current contributes to spontaneous activity in newborn rabbit sino-atrial node cells. J Physiol. (1996) 492(Pt 1):21–30. doi: 10.1113/jphysiol.1996.sp021285
46. Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. (2010) 48(1):12–25. doi: 10.1016/j.yjmcc.2009.07.013
47. Massaeli H, Guo J, Xu J, Zhang S. Extracellular K+ is a prerequisite for the function and plasma membrane stability of HERG channels. Circ Res. (2010) 106(6):1072–82. doi: 10.1161/CIRCRESAHA.109.215970
48. Verkerk AO, Wilders R. Human sinoatrial node pacemaker activity: role of the slow component of the delayed rectifier K+ current, IKs. Int J Mol Sci. (2023) 24(8):7264. doi: 10.3390/ijms24087264
49. Yamada N, Asano Y, Fujita M, Yamazaki S, Inanobe A, Matsuura N, et al. Mutant KCNJ3 and KCNJ5 potassium channels as novel molecular targets in bradyarrhythmias and atrial fibrillation. Circulation. (2019) 139(18):2157–69. doi: 10.1161/CIRCULATIONAHA.118.036761
50. Inagaki N, Gonoi T, Clement JPT, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. (1995) 270(5239):1166–70. doi: 10.1126/science.270.5239.1166
51. Pan Z, Yamaguchi R, Doi S. Bifurcation analysis and effects of changing ionic conductances on pacemaker rhythm in a sinoatrial node cell model. Biosystems. (2011) 106(1):9–18. doi: 10.1016/j.biosystems.2011.06.001
52. Darche FF, Rivinius R, Köllensperger E, Leimer U, Germann G, Seckinger A, et al. Pacemaker cell characteristics of differentiated and HCN4-transduced human mesenchymal stem cells. Life Sci. (2019) 232:116620. doi: 10.1016/j.lfs.2019.116620
53. Joung B, Chen PS, Lin SF. The role of the calcium and the voltage clocks in sinoatrial node dysfunction. Yonsei Med J. (2011) 52(2):211–9. doi: 10.3349/ymj.2011.52.2.211
54. Lyashkov AE, Juhaszova M, Dobrzynski H, Vinogradova TM, Maltsev VA, Juhasz O, et al. Calcium cycling protein density and functional importance to automaticity of isolated sinoatrial nodal cells are independent of cell size. Circ Res. (2007) 100(12):1723–31. doi: 10.1161/CIRCRESAHA.107.153676
55. Vinogradova TM, Tagirova Sirenko S, Lakatta EG. Unique Ca2+-cycling protein abundance and regulation sustains local Ca2+ releases and spontaneous firing of rabbit sinoatrial node cells. Int J Mol Sci. (2018) 19(8):2173. doi: 10.3390/ijms19082173
56. Alghamdi AM, Boyett MR, Hancox JC, Zhang H. Cardiac pacemaker dysfunction arising from different studies of Ion channel remodeling in the aging rat heart. Front Physiol. (2020) 11:546508. doi: 10.3389/fphys.2020.546508
57. Mesirca P, Bidaud I, Mangoni ME. Rescuing cardiac automaticity in L-type Cav1.3 channelopathies and beyond. J Physiol. (2016) 594(20):5869–79. doi: 10.1113/JP270678
58. Bernardi J, Aromolaran KA, Zhu H, Aromolaran AS. Circadian mechanisms: cardiac Ion channel remodeling and arrhythmias. Front Physiol. (2021) 11:611860. doi: 10.3389/fphys.2020.611860
59. Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, et al. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation. (2005) 112(4):471–81. doi: 10.1161/CIRCULATIONAHA.104.506857
60. Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation. (2007) 115(14):1921–32. doi: 10.1161/CIRCULATIONAHA.106.616011
61. Thiyagarajah A, Lau DH, Sanders P. Atrial fibrillation and conduction system disease: the roles of catheter ablation and permanent pacing. J Interv Card Electrophysiol. (2018) 52(3):395–402. doi: 10.1007/s10840-018-0429-9
62. Elvan A, Wylie K, Zipes DP. Pacing-induced chronic atrial fibrillation impairs sinus node function in dogs. Circulation. (1996) 94(11):2953–60. doi: 10.1161/01.CIR.94.11.2953
63. Glukhov AV, Hage LT, Hansen BJ, Pedraza-Toscano A, Vargas-Pinto P, Hamlin RL, et al. Sinoatrial node reentry in a canine chronic left ventricular infarct model. Circ Arrhythm Electrophysiol. (2013) 6(5):984–94. doi: 10.1161/CIRCEP.113.000404
64. Hansen BJ, Zhao J, Csepe TA, Moore BT, Li N, Jayne LA, et al. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur Heart J. (2015) 36(35):2390–401. doi: 10.1093/eurheartj/ehv233
65. Kalyanasundaram A, Li N, Hansen BJ, Zhao J, Fedorov VV. Canine and human sinoatrial node: differences and similarities in the structure, function, molecular profiles, and arrhythmia. J Vet Cardiol. (2019) 22:2–19. doi: 10.1016/j.jvc.2018.10.004
66. Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. (2003) 112(7):1019–28. doi: 10.1172/JCI200318062
67. Verkerk AO, Wilders R. Pacemaker activity of the human sinoatrial node: an update on the effects of mutations in HCN4 on the hyperpolarization-activated current. Int J Mol Sci. (2015) 16(2):3071–94. doi: 10.3390/ijms16023071
68. Chiang DY, Kim JJ, Valdes SO, de la Uz C, Fan Y, Orcutt J, et al. Loss-of-function SCN5A mutations associated with sinus node dysfunction, atrial arrhythmias, and poor pacemaker capture. Circ Arrhythm Electrophysiol. (2015) 8(5):1105–12. doi: 10.1161/CIRCEP.115.003098
69. Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, et al. Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest. (2003) 111(10):1537–45. doi: 10.1172/JCI200316387
70. Raucci FJ Jr, Shoemaker MB, Knollmann BC. Clinical phenotype of HCN4-related sick sinus syndrome. Heart Rhythm. (2017) 14(5):725–6. doi: 10.1016/j.hrthm.2017.02.006
71. Lee YS, Olaopa MA, Jung BC, Lee SH, Shin DG, Park HS, et al. Genetic variation of SCN5A in Korean patients with sick sinus syndrome. Korean Circ J. (2016) 46(1):63–71. doi: 10.4070/kcj.2016.46.1.63
72. Butters TD, Aslanidi OV, Inada S, Boyett MR, Hancox JC, Lei M, et al. Mechanistic links between Na+ channel (SCN5A) mutations and impaired cardiac pacemaking in sick sinus syndrome. Circ Res. (2010) 107(1):126–37. doi: 10.1161/CIRCRESAHA.110.219949
73. Sun C, Li N, Wang QQ, Yan LY, Ba SK, Zhang SS, et al. Whole genome sequencing identifies a deletion mutation in the unknown-functional KCNG2 from familial sick sinus syndrome. Physiol Genomics. (2022) 54(4):141–52. doi: 10.1152/physiolgenomics.00132.2021
74. Thorolfsdottir RB, Sveinbjornsson G, Aegisdottir HM, Benonisdottir S, Stefansdottir L, Ivarsdottir EV, et al. Genetic insight into sick sinus syndrome. Eur Heart J. (2021) 42(20):1959–71. doi: 10.1093/eurheartj/ehaa1108
75. Stallmeyer B, Kuß J, Kotthoff S, Zumhagen S, Vowinkel K, Rinné S, et al. A mutation in the G-protein gene GNB2 causes familial Sinus node and atrioventricular conduction dysfunction. Circ Res. (2017) 120(10):e33–44. doi: 10.1161/CIRCRESAHA.116.310112
76. Kuß J, Stallmeyer B, Goldstein M, Rinné S, Pees C, Zumhagen S, et al. Familial sinus node disease caused by a gain of GIRK (G-protein activated inwardly rectifying K+ channel) channel function. Circ Genom Precis Med. (2019) 12(1):e002238. doi: 10.1161/CIRCGEN.118.002238
77. Lou Q, Hansen BJ, Fedorenko O, Csepe TA, Kalyanasundaram A, Li N, et al. Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual-sided intramural optical mapping. Circulation. (2014) 130(4):315–24. doi: 10.1161/CIRCULATIONAHA.113.007086
78. Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. (1987) 325(6102):321–6. doi: 10.1038/325321a0
79. Yang D, Morrell CH, Lyashkov AE, Tagirova Sirenko S, Zahanich I, Yaniv Y, et al. Ca2+ and membrane potential transitions during action potentials are self-similar to each other and to variability of AP firing intervals across the broad physiologic range of AP intervals during autonomic receptor stimulation. Front Physiol. (2021) 12:612770. doi: 10.3389/fphys.2021.612770
80. Segal S, Shemla O, Shapira R, Peretz NK, Lukyanenko Y, Brosh I, et al. cAMP signaling affects age-associated deterioration of pacemaker beating interval dynamics. Geroscience. (2023) 45(4):2589–600. doi: 10.1007/s11357-023-00787-5
81. Difrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. (1991) 351(6322):145–7. doi: 10.1038/351145a0
82. Wang M, Wu JX, Ding D, Chen L. Structural insights into the mechanism of pancreatic KATP channel regulation by nucleotides. Nat Commun. (2022) 13(1):2770. doi: 10.1038/s41467-022-30430-4
83. Brahmajothi MV, Morales MJ, Campbell DL, Steenbergen C, Strauss HC. Expression and distribution of voltage-gated ion channels in ferret sinoatrial node. Physiol Genomics. (2010) 42a(2):131–40. doi: 10.1152/physiolgenomics.00049.2010
84. Terzic A, Alekseev AE, Yamada S, Reyes S, Olson TM. Advances in cardiac ATP-sensitive K+ channelopathies from molecules to populations. Circ Arrhythm Electrophysiol. (2011) 4(4):577–85. doi: 10.1161/CIRCEP.110.957662
85. Han X, Light PE, Giles WR, French RJ. Identification and properties of an ATP-sensitive K+ current in rabbit sino-atrial node pacemaker cells. J Physiol. (1996) 490(Pt 2):337–50. doi: 10.1113/jphysiol.1996.sp021148
86. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. (2006) 440(7083):470–6. doi: 10.1038/nature04711
87. Corradi J, Thompson B, Fletcher PA, Bertram R, Sherman AS, Satin LS. KATP channel activity and slow oscillations in pancreatic beta cells are regulated by mitochondrial ATP production. J Physiol. (2023) 601(24):5655–67. doi: 10.1113/JP284982
88. Foster HR, Ho T, Potapenko E, Sdao SM, Huang SM, Lewandowski SL, et al. β-cell deletion of the PKm1 and PKm2 isoforms of pyruvate kinase in mice reveals their essential role as nutrient sensors for the KATP channel. Elife. (2022) 11:e79422. doi: 10.7554/eLife.79422
89. Han Y, Li C, Zhang P, Yang X, Min J, Wu Q, et al. Protective effects of 5(S)-5-carboxystrictosidine on myocardial ischemia-reperfusion injury through activation of mitochondrial KATP channels. Eur J Pharmacol. (2022) 920:174811. doi: 10.1016/j.ejphar.2022.174811
90. Michailova A, Saucerman J, Belik ME, McCulloch AD. Modeling regulation of cardiac KATP and L-type Ca2+ currents by ATP, ADP, and Mg2+. Biophys J. (2005) 88(3):2234–49. doi: 10.1529/biophysj.104.046284
91. Sato R, Koumi S. Characterization of the stretch-activated chloride channel in isolated human atrial myocytes. J Membr Biol. (1998) 163(1):67–76. doi: 10.1007/s002329900371
92. Baumgarten CM, Clemo HF. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Prog Biophys Mol Biol. (2003) 82(1-3):25–42. doi: 10.1016/S0079-6107(03)00003-8
93. Bertelli S, Remigante A, Zuccolini P, Barbieri R, Ferrera L, Picco C, et al. Mechanisms of activation of LRRC8 volume regulated anion channels. Cell Physiol Biochem. (2021) 55(S1):41–56. doi: 10.33594/000000329
94. Sawicka M, Dutzler R. Regulators of cell volume: the structural and functional properties of anion channels of the LRRC8 family. Curr Opin Struct Biol. (2022) 74:102382. doi: 10.1016/j.sbi.2022.102382
95. Liu GX, Vepa S, Artman M, Coetzee WA. Modulation of human cardiovascular outward rectifying chloride channel by intra- and extracellular ATP. Am J Physiol Heart Circ Physiol. (2007) 293(6):H3471–9. doi: 10.1152/ajpheart.00357.2007
96. Kimelberg HK. Increased release of excitatory amino acids by the actions of ATP and peroxynitrite on volume-regulated anion channels (VRACs) in astrocytes. Neurochem Int. (2004) 45(4):511–9. doi: 10.1016/j.neuint.2003.11.002
97. Patel AJ, Lauritzen I, Lazdunski M, Honoré E. Disruption of mitochondrial respiration inhibits volume-regulated anion channels and provokes neuronal cell swelling. J Neurosci. (1998) 18(9):3117–23. doi: 10.1523/JNEUROSCI.18-09-03117.1998
98. Afzal A, Figueroa EE, Kharade SV, Bittman K, Matlock BK, Flaherty DK, et al. The LRRC8 volume-regulated anion channel inhibitor, DCPIB, inhibits mitochondrial respiration independently of the channel. Physiol Rep. (2019) 7(23):e14303. doi: 10.14814/phy2.14303
99. Bryan-Sisneros A, Sabanov V, Thoroed SM, Doroshenko P. Dual role of ATP in supporting volume-regulated chloride channels in mouse fibroblasts. Biochim Biophys Acta. (2000) 1468(1-2):63–72. doi: 10.1016/S0005-2736(00)00243-1
100. Kabashima Y, Ogawa H, Nakajima R, Toyoshima C. What ATP binding does to the Ca2+ pump and how nonproductive phosphoryl transfer is prevented in the absence of Ca 2+. Proc Natl Acad Sci U S A. (2020) 117(31):18448–58. doi: 10.1073/pnas.2006027117
101. Thirman J, Rui H, Roux B. Elusive Intermediate State Key in the Conversion of ATP hydrolysis into useful work driving the Ca2+ pump SERCA. J Phys Chem B. (2021) 125(11):2921–8. doi: 10.1021/acs.jpcb.1c00558
102. Mueller B, Zhao M, Negrashov IV, Bennett R, Thomas DD. SERCA structural dynamics induced by ATP and calcium. Biochemistry. (2004) 43(40):12846–54. doi: 10.1021/bi0489457
103. De Marchi U, Castelbou C, Demaurex N. Uncoupling protein 3 (UCP3) modulates the activity of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J Biol Chem. (2011) 286(37):32533–41. doi: 10.1074/jbc.M110.216044
104. Rodríguez C, Contreras C, Sáenz-Medina J, Muñoz M, Corbacho C, Carballido J, et al. Activation of the AMP-related kinase (AMPK) induces renal vasodilatation and downregulates nox-derived reactive oxygen species (ROS) generation. Redox Biol. (2020) 34:101575. doi: 10.1016/j.redox.2020.101575
105. Liu J, Sirenko S, Juhaszova M, Sollott SJ, Shukla S, Yaniv Y, et al. Age-associated abnormalities of intrinsic automaticity of sinoatrial nodal cells are linked to deficient cAMP-PKA-Ca2+ signaling. Am J Physiol Heart Circ Physiol. (2014) 306(10):H1385–97. doi: 10.1152/ajpheart.00088.2014
106. Huang S, Dong W, Lin X, Bian J. Na+/K+-ATPase: ion pump, signal transducer, or cytoprotective protein, and novel biological functions. Neural Regen Res. (2024) 19(12):2684–97. doi: 10.4103/NRR.NRR-D-23-01175
107. Morth JP, Pedersen BP, Buch-Pedersen MJ, Andersen JP, Vilsen B, Palmgren MG, et al. A structural overview of the plasma membrane Na+, K+-ATPase and H+-ATPase ion pumps. Nat Rev Mol Cell Biol. (2011) 12(1):60–70. doi: 10.1038/nrm3031
108. Hund TJ, Mohler PJ. Ankyrin-based targeting pathway regulates human sinoatrial node automaticity. Channels (Austin). (2008) 2(6):404–6. doi: 10.4161/chan.2.6.7220
109. Sepp M, Sokolova N, Jugai S, Mandel M, Peterson P, Vendelin M. Tight coupling of Na+/K+-ATPase with glycolysis demonstrated in permeabilized rat cardiomyocytes. PLoS One. (2014) 9(6):e99413. doi: 10.1371/journal.pone.0099413
110. Namekata I, Jitsukata K, Fukuda A, Odaka R, Hamaguchi S, Tanaka H. Intracellular Ca2+-mediated mechanisms for the pacemaker depolarization of the mouse and guinea pig sinus node tissue. Biomolecules. (2022) 12(3):377. doi: 10.3390/biom12030377
111. Morotti S, Ni H, Peters CH, Rickert C, Asgari-Targhi A, Sato D, et al. Intracellular Na(+) modulates pacemaking activity in murine sinoatrial node myocytes: an in silico analysis. Int J Mol Sci. (2021) 22(11):5645. doi: 10.3390/ijms22115645
112. Skogestad J, Aronsen JM, Tovsrud N, Wanichawan P, Hougen K, Stokke MK, et al. Coupling of the Na+/K+-ATPase to Ankyrin B controls Na+/Ca2+ exchanger activity in cardiomyocytes. Cardiovasc Res. (2020) 116(1):78–90. doi: 10.1093/cvr/cvz087
113. Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. (2012) 125(17):2059–70. doi: 10.1161/CIRCULATIONAHA.111.067306
114. O'Brien SE, Apkon M, Berul CI, Patel HT, Saupe K, Spindler M, et al. Phenotypical features of long Q-T syndrome in transgenic mice expressing human Na-K-ATPase α3-isoform in hearts. Am J Physiol Heart Circ Physiol. (2000) 279(5):H2133–42. doi: 10.1152/ajpheart.2000.279.5.H2133
115. Chkadua G, Nozadze E, Tsakadze L, Shioshvili L, Arutinova N, Leladze M, et al. The effect of cytochrome C on Na, K-ATPase. J Bioenerg Biomembr. (2024) 56(3):221–34. doi: 10.1007/s10863-024-10012-3
116. Dencher NA, Frenzel M, Reifschneider NH, Sugawa M, Krause F. Proteome alterations in rat mitochondria caused by aging. Ann N Y Acad Sci. (2007) 1100:291–8. doi: 10.1196/annals.1395.030
117. Garlid KD, Costa AD, Quinlan CL, Pierre SV, Dos Santos P. Cardioprotective signaling to mitochondria. J Mol Cell Cardiol. (2009) 46(6):858–66. doi: 10.1016/j.yjmcc.2008.11.019
118. Zhang Y, Chen S, Luo L, Greenly S, Shi H, Xu JJ, et al. Role of cAMP in cardiomyocyte viability: beneficial or detrimental? Circ Res. (2023) 133(11):902–23. doi: 10.1161/CIRCRESAHA.123.322652
119. Brand T, Poon KL, Simrick S, Schindler RF. The popeye domain containing genes and cAMP signaling. J Cardiovasc Dev Dis. (2014) 1(1):121–33. doi: 10.3390/jcdd1010121
120. Segal S, Arbel-Ganon L, Mazgaoker S, Davoodi M, Yaniv Y. Increase in Ca2+-activated cAMP/PKA signaling prevents hydroxychloroquine-induced bradycardia of the cardiac pacemaker. Front Physiol. (2022) 13:839140. doi: 10.3389/fphys.2022.839140
121. Baruscotti M, Bucchi A, Milanesi R, Paina M, Barbuti A, Gnecchi-Ruscone T, et al. A gain-of-function mutation in the cardiac pacemaker HCN4 channel increasing cAMP sensitivity is associated with familial inappropriate Sinus tachycardia. Eur Heart J. (2017) 38(4):280–8. doi: 10.1093/eurheartj/ehv582
122. Fenske S, Hennis K, Rötzer RD, Brox VF, Becirovic E, Scharr A, et al. cAMP-dependent regulation of HCN4 controls the tonic entrainment process in sinoatrial node pacemaker cells. Nat Commun. (2020) 11(1):5555. doi: 10.1038/s41467-020-19304-9
123. Cámara-Checa A, Perin F, Rubio-Alarcón M, Dago M, Crespo-García T, Rapún J, et al. A gain-of-function HCN4 mutant in the HCN domain is responsible for inappropriate sinus tachycardia in a Spanish family. Proc Natl Acad Sci U S A. (2023) 120(49):e2305135120. doi: 10.1073/pnas.2305135120
124. Boulton S, Akimoto M, Akbarizadeh S, Melacini G. Free energy landscape remodeling of the cardiac pacemaker channel explains the molecular basis of familial sinus bradycardia. J Biol Chem. (2017) 292(15):6414–28. doi: 10.1074/jbc.M116.773697
125. Grammatika Pavlidou N, Dobrev S, Beneke K, Reinhardt F, Pecha S, Jacquet E, et al. Phosphodiesterase 8 governs cAMP/PKA-dependent reduction of L-type calcium current in human atrial fibrillation: a novel arrhythmogenic mechanism. Eur Heart J. (2023) 44(27):2483–94. doi: 10.1093/eurheartj/ehad086
126. Dorey TW, Liu Y, Jansen HJ, Bohne LJ, Mackasey M, Atkinson L, et al. Natriuretic peptide receptor B protects against atrial fibrillation by controlling atrial cAMP via phosphodiesterase 2. Circ Arrhythm Electrophysiol. (2023) 16(11):e012199. doi: 10.1161/CIRCEP.123.012199
127. Reinhardt F, Beneke K, Pavlidou NG, Conradi L, Reichenspurner H, Hove-Madsen L, et al. Abnormal calcium handling in atrial fibrillation is linked to changes in cyclic AMP dependent signaling. Cells. (2021) 10(11):3042. doi: 10.3390/cells10113042
128. Sirenko ST, Zahanich I, Li Y, Lukyanenko YO, Lyashkov AE, Ziman BD, et al. Phosphoprotein phosphatase 1 but not 2A activity modulates coupled-clock mechanisms to impact on intrinsic automaticity of sinoatrial nodal pacemaker cells. Cells. (2021) 10(11):3106. doi: 10.3390/cells10113106
129. Wirth AN, Tsutsui K, Maltsev VA, Lakatta EG. Adenosine reduces sinoatrial node cell action potential firing rate by uncoupling its membrane and calcium clocks. Front Physiol. (2022) 13:977807. doi: 10.3389/fphys.2022.977807
130. Sassi Y, Ahles A, Truong DJ, Baqi Y, Lee SY, Husse B, et al. Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J Clin Invest. (2014) 124(12):5385–97. doi: 10.1172/JCI74349
131. Mougenot N, Mika D, Czibik G, Marcos E, Abid S, Houssaini A, et al. Cardiac adenylyl cyclase overexpression precipitates and aggravates age-related myocardial dysfunction. Cardiovasc Res. (2019) 115(12):1778–90. doi: 10.1093/cvr/cvy306
132. Marienfeld U, Walter U, Simm A. Inhibition of rat cardiac fibroblast growth by cAMP–but not by cGMP-dependent protein kinase. Basic Res Cardiol. (2001) 96(2):184–91. doi: 10.1007/s003950170069
133. Chakraborty D, Zhu H, Jüngel A, Summa L, Li YN, Matei AE, et al. Fibroblast growth factor receptor 3 activates a network of profibrotic signaling pathways to promote fibrosis in systemic sclerosis. Sci Transl Med. (2020) 12(563):eaaz5506. doi: 10.1126/scitranslmed.aaz5506
134. Mallano T, Palumbo-Zerr K, Zerr P, Ramming A, Zeller B, Beyer C, et al. Activating transcription factor 3 regulates canonical TGFβ signalling in systemic sclerosis. Ann Rheum Dis. (2016) 75(3):586–92. doi: 10.1136/annrheumdis-2014-206214
135. Meng L, Lu Y, Wang X, Cheng C, Xue F, Xie L, et al. NPRC deletion attenuates cardiac fibrosis in diabetic mice by activating PKA/PKG and inhibiting TGF-β1/smad pathways. Sci Adv. (2023) 9(31):eadd4222. doi: 10.1126/sciadv.add4222
136. Penke LR, Speth JM, Dommeti VL, White ES, Bergin IL, Peters-Golden M. FOXM1 is a critical driver of lung fibroblast activation and fibrogenesis. J Clin Invest. (2018) 128(6):2389–405. doi: 10.1172/JCI87631
137. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci U S A. (2005) 102(2):437–42. doi: 10.1073/pnas.0408704102
138. Wang Y, Zhao J, Cai Y, Ballard HJ. Cystic fibrosis transmembrane conductance regulator-dependent bicarbonate entry controls rat cardiomyocyte ATP release via pannexin1 through mitochondrial signalling and caspase activation. Acta Physiol (Oxf). (2020) 230(1):e13495. doi: 10.1111/apha.13495
139. Zhang Z, Chen J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell. (2016) 167(6):1586–97.e9. doi: 10.1016/j.cell.2016.11.014
140. Yamamoto S, Ichishima K, Ehara T. Regulation of extracellular UTP-activated Cl− current by P2Y-PLC-PKC signaling and ATP hydrolysis in mouse ventricular myocytes. J Physiol Sci. (2007) 57(2):85–94. doi: 10.2170/physiolsci.RP011406
141. Csanády L, Vergani P, Gadsby DC. Structure, gating, and regulation of the cftr anion channel. Physiol Rev. (2019) 99(1):707–38. doi: 10.1152/physrev.00007.2018
142. Hart P, Warth JD, Levesque PC, Collier ML, Geary Y, Horowitz B, et al. Cystic fibrosis gene encodes a cAMP-dependent chloride channel in heart. Proc Natl Acad Sci U S A. (1996) 93(13):6343–8. doi: 10.1073/pnas.93.13.6343
143. Gao Z, Sun HY, Lau CP, Chin-Wan Fung P, Li GR. Evidence for cystic fibrosis transmembrane conductance regulator chloride current in swine ventricular myocytes. J Mol Cell Cardiol. (2007) 42(1):98–105. doi: 10.1016/j.yjmcc.2006.10.002
144. Goodstadt L, Powell T, Figtree GA. 17β-estradiol potentiates the cardiac cystic fibrosis transmembrane conductance regulator chloride current in guinea-pig ventricular myocytes. J Physiol Sci. (2006) 56(1):29–37. doi: 10.2170/physiolsci.R2131
145. Chen B, Daneshgar N, Lee HC, Song LS, Dai DF. Mitochondrial oxidative stress mediates bradyarrhythmia in leigh syndrome mitochondrial disease mice. Antioxidants (Basel). (2023) 12(5):1001. doi: 10.3390/antiox12051001
146. Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. (2015) 131(12):1082–97. doi: 10.1161/CIRCULATIONAHA.114.012725
147. Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem. (2010) 285(6):3997–4005. doi: 10.1074/jbc.M109.034421
148. Chen C, Wang K, Zhang H, Zhou HJ, Chen Y, Min W. A unique SUMO-interacting motif of Trx2 is critical for its mitochondrial presequence processing and anti-oxidant activity. Front Physiol. (2019) 10:1089. doi: 10.3389/fphys.2019.01089
149. Hu C, Zhang H, Qiao Z, Wang Y, Zhang P, Yang D. Loss of thioredoxin 2 alters mitochondrial respiratory function and induces cardiomyocyte hypertrophy. Exp Cell Res. (2018) 372(1):61–72. doi: 10.1016/j.yexcr.2018.09.010
150. Holzerova E, Danhauser K, Haack TB, Kremer LS, Melcher M, Ingold I, et al. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain. (2016) 139(Pt 2):346–54. doi: 10.1093/brain/awv350
151. Yang B, Huang Y, Zhang H, Huang Y, Zhou HJ, Young L, et al. Mitochondrial thioredoxin-2 maintains HCN4 expression and prevents oxidative stress-mediated sick sinus syndrome. J Mol Cell Cardiol. (2020) 138:291–303. doi: 10.1016/j.yjmcc.2019.10.009
152. Pereira CH, Bare DJ, Rosas PC, Dias FAL, Banach K. The role of P21-activated kinase (Pak1) in sinus node function. J Mol Cell Cardiol. (2023) 179:90–101. doi: 10.1016/j.yjmcc.2023.04.004
153. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. (2014) 114(3):524–37. doi: 10.1161/CIRCRESAHA.114.300559
154. Lkhagva B, Lin YK, Chen YC, Cheng WL, Higa S, Kao YH, et al. ZFHX3 knockdown dysregulates mitochondrial adaptations to tachypacing in atrial myocytes through enhanced oxidative stress and calcium overload. Acta Physiol (Oxf). (2021) 231(4):e13604. doi: 10.1111/apha.13604
155. Chadda KR, Edling CE, Valli H, Ahmad S, Huang CL, Jeevaratnam K. Gene and protein expression profile of selected molecular targets mediating electrophysiological function in pgc-1α deficient murine atria. Int J Mol Sci. (2018) 19(11):3450. doi: 10.3390/ijms19113450
156. Pérez-Hernández M, Leo-Macias A, Keegan S, Jouni M, Kim JC, Agullo-Pascual E, et al. Structural and functional characterization of a Na(v)1.5-mitochondrial couplon. Circ Res. (2021) 128(3):419–32. doi: 10.1161/CIRCRESAHA.120.318239
157. Hu YF, Wu CH, Lai TC, Chang YC, Hwang MJ, Chang TY, et al. ALDH2 deficiency induces atrial fibrillation through dysregulated cardiac sodium channel and mitochondrial bioenergetics: a multi-omics analysis. Biochim Biophys Acta Mol Basis Dis. (2021) 1867(5):166088. doi: 10.1016/j.bbadis.2021.166088
158. Lu G, Xu S, Peng L, Huang Z, Wang Y, Gao X. Angiotensin II upregulates Kv1.5 expression through ROS-dependent transforming growth factor-beta1 and extracellular signal-regulated kinase 1/2 signalings in neonatal rat atrial myocytes. Biochem Biophys Res Commun. (2014) 454(3):410–6. doi: 10.1016/j.bbrc.2014.10.088
159. Lu G, Xu C, Tang K, Zhang J, Li Q, Peng L, et al. H 2 S inhibits angiotensin II-induced atrial Kv1.5 upregulation by attenuating Nox4-mediated ROS generation during atrial fibrillation. Biochem Biophys Res Commun. (2017) 483(1):534–40. doi: 10.1016/j.bbrc.2016.12.110
160. Lu G, Li J, Zhai Y, Li Q, Xie D, Zhang J, et al. Spironolactone suppresses aldosterone-induced Kv1.5 expression by attenuating mineralocorticoid receptor-Nox1/2/4-mediated ROS generation in neonatal rat atrial myocytes. Biochem Biophys Res Commun. (2019) 520(2):379–84. doi: 10.1016/j.bbrc.2019.10.039
161. Cano M, Datta S, Wang L, Liu T, Flores-Bellver M, Sachdeva M, et al. Nrf2 deficiency decreases NADPH from impaired IDH shuttle and pentose phosphate pathway in retinal pigmented epithelial cells to magnify oxidative stress-induced mitochondrial dysfunction. Aging Cell. (2021) 20(8):e13444. doi: 10.1111/acel.13444
162. Imhoff BR, Hansen JM. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem J. (2009) 424(3):491–500. doi: 10.1042/BJ20091286
163. Zhang H, Li L, Hao M, Chen K, Lu Y, Qi J, et al. Yixin-Fumai granules improve sick sinus syndrome in aging mice through nrf-2/HO-1 pathway: a new target for sick sinus syndrome. J Ethnopharmacol. (2021) 277:114254. doi: 10.1016/j.jep.2021.114254
164. Yang Z, Song T, Truong L, Reyes-García J, Wang L, Zheng YM, et al. Important Role of Sarcoplasmic Reticulum Ca2+ release via ryanodine receptor-2 channel in hypoxia-induced rieske iron–sulfur protein-mediated mitochondrial reactive oxygen species generation in pulmonary artery smooth muscle cells. Antioxid Redox Signal. (2020) 32(7):447–62. doi: 10.1089/ars.2018.7652
165. El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, et al. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. (2006) 114(7):670–80. doi: 10.1161/CIRCULATIONAHA.106.636845
166. Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. (2010) 106(6):1134–44. doi: 10.1161/CIRCRESAHA.109.203836
167. Lyashkov AE, Behar J, Lakatta EG, Yaniv Y, Maltsev VA. Positive Feedback mechanisms among local Ca releases, NCX, and ICaL ignite pacemaker action potentials. Biophys J. (2018) 114(5):1176–89. doi: 10.1016/j.bpj.2017.12.043
168. Xie D, Xiong K, Su X, Wang G, Zou Q, Wang L, et al. Glutamate drives ‘local Ca2+ release’ in cardiac pacemaker cells. Cell Res. (2022) 32(9):843–54. doi: 10.1038/s41422-022-00693-z
169. Xue J-B, Val-Blasco A, Davoodi M, Gómez S, Yaniv Y, Benitah J-P, et al. Sinus node dysfunction in heart failure is characterized by reduced CaMKII signaling. J Gen Physiol. (2021) 154(9):e2021ecc1. doi: 10.1085/jgp.2021ecc1
170. Xue JB, Val-Blasco A, Davoodi M, Gómez S, Yaniv Y, Benitah JP, et al. Heart failure in mice induces a dysfunction of the sinus node associated with reduced CaMKII signaling. J Gen Physiol. (2022) 154(9):e202112895. doi: 10.1085/jgp.202112895
171. Qi Y, Xu H, Li X, Zhao X, Li Y, Zhou X, et al. Silica nanoparticles induce cardiac injury and dysfunction via ROS/Ca2+/CaMKII signaling. Sci Total Environ. (2022) 837:155733. doi: 10.1016/j.scitotenv.2022.155733
172. Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. (2016) 22(2):175–82. doi: 10.1038/nm.4017
173. Dridi H, Santulli G, Bahlouli L, Miotto MC, Weninger G, Marks AR. Mitochondrial calcium overload plays a causal role in oxidative stress in the failing heart. Biomolecules. (2023) 13(9):1409. doi: 10.3390/biom13091409
174. Li S, Ma Y, Ye S, Tang S, Liang N, Liang Y, et al. Polystyrene microplastics trigger hepatocyte apoptosis and abnormal glycolytic flux via ROS-driven calcium overload. J Hazard Mater. (2021) 417:126025. doi: 10.1016/j.jhazmat.2021.126025
175. Demaurex N, Rosselin M. Redox control of mitochondrial calcium uptake. Mol Cell. (2017) 65(6):961–2. doi: 10.1016/j.molcel.2017.02.029
176. Liu Y, Wang Y, Song S, Zhang H. Cascade-responsive nanobomb with domino effect for anti-tumor synergistic therapies. Natl Sci Rev. (2022) 9(3):nwab139. doi: 10.1093/nsr/nwab139
177. Chang X, Li Y, Liu J, Wang Y, Guan X, Wu Q, et al. ß-tubulin contributes to tongyang huoxue decoction-induced protection against hypoxia/reoxygenation-induced injury of sinoatrial node cells through SIRT1-mediated regulation of mitochondrial quality surveillance. Phytomedicine. (2023) 108:154502. doi: 10.1016/j.phymed.2022.154502
178. Lu W, Zhu H, Wu J, Liao S, Cheng G, Li X. Rhein attenuates angiotensin II-induced cardiac remodeling by modulating AMPK-FGF23 signaling. J Transl Med. (2022) 20(1):305. doi: 10.1186/s12967-022-03482-9
179. Casado-Barragán F, Lazcano-Páez G, Larenas PE, Aguirre-Delgadillo M, Olivares-Aravena F, Witto-Oyarce D, et al. Increased renal medullary NOX-4 in female but not male mice during the early phase of type 1 diabetes: potential role of ROS in upregulation of TGF-β1 and fibronectin in collecting duct cells. Antioxidants (Basel). (2023) 12(3):729. doi: 10.3390/antiox12030729
180. Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res. (2011) 64(4):381–92. doi: 10.1016/j.phrs.2011.06.018
181. Zheng M, Li RG, Song J, Zhao X, Tang L, Erhardt S, et al. Hippo-Yap signaling maintains sinoatrial node homeostasis. Circulation. (2022) 146(22):1694–711. doi: 10.1161/CIRCULATIONAHA.121.058777
182. Cui C, Jiang X, Ju W, Wang J, Wang D, Sun Z, et al. Atrial remodeling and metabolic dysfunction in idiopathic isolated fibrotic atrial cardiomyopathy. Int J Cardiol. (2018) 265:155–61. doi: 10.1016/j.ijcard.2018.04.080
183. Baumann K. Quality control: triaging mitochondrial membrane proteins. Nat Rev Mol Cell Biol. (2016) 17(8):463. doi: 10.1038/nrm.2016.103
184. Li B, Liu F, Chen X, Chen T, Zhang J, Liu Y, et al. FARS2 deficiency causes cardiomyopathy by disrupting mitochondrial homeostasis and the mitochondrial quality control system. Circulation. (2024) 149(16):1268–84. doi: 10.1161/CIRCULATIONAHA.123.064489
185. Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. (2018) 28(4):R170–r85. doi: 10.1016/j.cub.2018.01.004
186. Wu L, Wang L, Du Y, Zhang Y, Ren J. Mitochondrial quality control mechanisms as therapeutic targets in doxorubicin-induced cardiotoxicity. Trends Pharmacol Sci. (2023) 44(1):34–49. doi: 10.1016/j.tips.2022.10.003
187. Picca A, Mankowski RT, Burman JL, Donisi L, Kim JS, Marzetti E, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. (2018) 15(9):543–54. doi: 10.1038/s41569-018-0059-z
188. Zacharioudakis E, Agianian B, Kumar Mv V, Biris N, Garner TP, Rabinovich-Nikitin I, et al. Modulating mitofusins to control mitochondrial function and signaling. Nat Commun. (2022) 13(1):3775. doi: 10.1038/s41467-022-31324-1
189. Li Q, Su D, O'Rourke B, Pogwizd SM, Zhou L. Mitochondria-derived ROS bursts disturb Ca2+ cycling and induce abnormal automaticity in guinea pig cardiomyocytes: a theoretical study. Am J Physiol Heart Circ Physiol. (2015) 308(6):H623–36. doi: 10.1152/ajpheart.00493.2014
190. Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. (2016) 529(7587):528–31. doi: 10.1038/nature16500
191. Joaquim M, Escobar-Henriques M. Role of mitofusins and mitophagy in life or death decisions. Front Cell Dev Biol. (2020) 8:572182. doi: 10.3389/fcell.2020.572182
192. Yepuri G, Ramirez LM, Theophall GG, Reverdatto SV, Quadri N, Hasan SN, et al. DIAPH1-MFN2 interaction regulates mitochondria-SR/ER contact and modulates ischemic/hypoxic stress. Nat Commun. (2023) 14(1):6900. doi: 10.1038/s41467-023-42521-x
193. Ernst P, Chen K, Tang Y, Kim S, Guan J, He J, et al. Investigation into the difference in mitochondrial-cytosolic calcium coupling between adult cardiomyocyte and hiPSC-CM using a novel multifunctional genetic probe. Pflugers Arch. (2021) 473(3):447–59. doi: 10.1007/s00424-021-02524-3
194. Qiu Y, Cheng R, Liang C, Yao Y, Zhang W, Zhang J, et al. MicroRNA-20b promotes cardiac hypertrophy by the inhibition of mitofusin 2-mediated inter-organelle Ca2+ cross-talk. Mol Ther Nucleic Acids. (2020) 19:1343–56. doi: 10.1016/j.omtn.2020.01.017
195. Dorn GW 2nd, Song M, Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardiol. (2015) 78:123–8. doi: 10.1016/j.yjmcc.2014.09.015
196. Seidlmayer LK, Mages C, Berbner A, Eder-Negrin P, Arias-Loza PA, Kaspar M, et al. Mitofusin 2 is essential for IP3-mediated SR/Mitochondria metabolic feedback in ventricular myocytes. Front Physiol. (2019) 10:733. doi: 10.3389/fphys.2019.00733
197. Ren L, Gopireddy RR, Perkins G, Zhang H, Timofeyev V, Lyu Y, et al. Disruption of mitochondria-sarcoplasmic reticulum microdomain connectomics contributes to sinus node dysfunction in heart failure. Proc Natl Acad Sci U S A. (2022) 119(36):e2206708119. doi: 10.1073/pnas.2206708119
198. Zhang P, Hinshaw JE. Three-dimensional reconstruction of dynamin in the constricted state. Nat Cell Biol. (2001) 3(10):922–6. doi: 10.1038/ncb1001-922
199. Kraus F, Roy K, Pucadyil TJ, Ryan MT. Function and regulation of the divisome for mitochondrial fission. Nature. (2021) 590(7844):57–66. doi: 10.1038/s41586-021-03214-x
200. Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ, Ryan MT. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat Commun. (2018) 9(1):5239. doi: 10.1038/s41467-018-07543-w
201. Francy CA, Alvarez FJ, Zhou L, Ramachandran R, Mears JA. The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. J Biol Chem. (2015) 290(18):11692–703. doi: 10.1074/jbc.M114.610881
202. Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature. (2018) 558(7710):401–5. doi: 10.1038/s41586-018-0211-2
203. Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife. (2015) 4:e11553. doi: 10.7554/eLife.11553
204. Fonseca TB, Sánchez-Guerrero Á, Milosevic I, Raimundo N. Mitochondrial fission requires DRP1 but not dynamins. Nature. (2019) 570(7761):E34–e42. doi: 10.1038/s41586-019-1296-y
205. Tong M, Mukai R, Mareedu S, Zhai P, Oka SI, Huang CY, et al. Distinct roles of DRP1 in conventional and alternative mitophagy in obesity cardiomyopathy. Circ Res. (2023) 133(1):6–21. doi: 10.1161/CIRCRESAHA.123.322512
206. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. (2021) 593(7859):435–9. doi: 10.1038/s41586-021-03510-6
207. Lee Y, Lee HY, Hanna RA, Gustafsson ÅB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. (2011) 301(5):H1924–31. doi: 10.1152/ajpheart.00368.2011
208. Duan C, Kuang L, Hong C, Xiang X, Liu J, Li Q, et al. Mitochondrial Drp1 recognizes and induces excessive mPTP opening after hypoxia through BAX-PiC and LRRK2-HK2. Cell Death Dis. (2021) 12(11):1050. doi: 10.1038/s41419-021-04343-x
209. Lim JR, Lee HJ, Jung YH, Kim JS, Chae CW, Kim SY, et al. Ethanol-activated CaMKII signaling induces neuronal apoptosis through Drp1-mediated excessive mitochondrial fission and JNK1-dependent NLRP3 inflammasome activation. Cell Commun Signal. (2020) 18(1):123. doi: 10.1186/s12964-020-00572-3
210. Kalkhoran SB, Kriston-Vizi J, Hernandez-Resendiz S, Crespo-Avilan GE, Rosdah AA, Lees JG, et al. Hydralazine protects the heart against acute ischaemia/reperfusion injury by inhibiting Drp1-mediated mitochondrial fission. Cardiovasc Res. (2022) 118(1):282–94. doi: 10.1093/cvr/cvaa343
211. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. (2010) 121(18):2012–22. doi: 10.1161/CIRCULATIONAHA.109.906610
212. Fan RZ, Sportelli C, Lai Y, Salehe SS, Pinnell JR, Brown HJ, et al. A partial Drp1 knockout improves autophagy flux independent of mitochondrial function. Mol Neurodegener. (2024) 19(1):26. doi: 10.1186/s13024-024-00708-w
213. Chen CY, Li SJ, Wang CY, Mersmann HJ, Ding ST. The impact of DRP1 on myocardial fibrosis in the obese minipig. Eur J Clin Invest. (2020) 50(3):e13204. doi: 10.1111/eci.13204
214. Zuccolotto Dos Reis FH. Mitochondria and the heart. Eur Heart J. (2024) 45(22):1963–4. doi: 10.1093/eurheartj/ehae031
215. Deng J, Liu Q, Ye L, Wang S, Song Z, Zhu M, et al. The Janus face of mitophagy in myocardial ischemia/reperfusion injury and recovery. Biomed Pharmacother. (2024) 173:116337. doi: 10.1016/j.biopha.2024.116337
216. Wiersma M, van Marion DMS, Wüst RCI, Houtkooper RH, Zhang D, Groot NMS, et al. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells. (2019) 8(10):1202. doi: 10.3390/cells8101202
217. Schwemmlein J, Maack C, Bertero E. Mitochondria as therapeutic targets in heart failure. Curr Heart Fail Rep. (2022) 19(2):27–37. doi: 10.1007/s11897-022-00539-0
218. Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. (2020) 57:102884. doi: 10.1016/j.ebiom.2020.102884
219. Harrington JS, Ryter SW, Plataki M, Price DR, Choi AMK. Mitochondria in health, disease, and aging. Physiol Rev. (2023) 103(4):2349–422. doi: 10.1152/physrev.00058.2021
220. Guarente L. Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell. (2008) 132(2):171–6. doi: 10.1016/j.cell.2008.01.007
Keywords: Ca2+ clock, fibrosis, ion channel protein, membrane clock, mitochondria, review, sick sinus syndrome
Citation: Shi X, He L, Wang Y, Wu Y, Lin D, Chen C, Yang M and Huang S (2024) Mitochondrial dysfunction is a key link involved in the pathogenesis of sick sinus syndrome: a review. Front. Cardiovasc. Med. 11:1488207. doi: 10.3389/fcvm.2024.1488207
Received: 29 August 2024; Accepted: 15 October 2024;
Published: 29 October 2024.
Edited by:
Rui Providencia, University College London, United KingdomReviewed by:
Di Lang, University of California, San Francisco, United StatesJiang Hong, Shanghai First People’s Hospital, China
Copyright: © 2024 Shi, He, Wang, Wu, Lin, Chen, Yang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuwei Huang, aHN3MTEwNEAxMjYuY29t; Ming Yang, MjAyMTExMDExNTExMDQyQHpjbXUuZWR1LmNu
†These authors have contributed equally to this work