 Levon M. Khachigian
Levon M. Khachigian- Vascular Biology and Translational Research, Department of Pathology, Faculty of Medicine and Health, School of Biomedical Sciences, University of New South Wales, Sydney, NSW, Australia
The transcription factor, early growth response-1 (Egr-1) is the product of a prototypic immediate-early gene that plays an integral role in the pathogenesis of multiple cardiovascular diseases. Egr-1 has been linked with atherogenesis, myocardial ischemia-reperfusion injury, cardiac fibrosis and heart failure. Egr-1 expression is triggered by a host of factors including cytokines, hormones, growth factors, hyperglycaemia, biomechanical forces and oxygen deprivation. Egr-1 is a molecular conduit that links changes in the cellular environment with the inducible expression of genes whose products play a causative role in this inflammatory disease. It is rapidly synthesised, undergoes post-translational modification, interacts with a range of cofactors and drives gene expression. Studies in Egr-1 deficient mice, animal models using DNAzymes, RNA interference, oligodeoxynucleotide decoys, antisense oligonucleotides, and new insights provided by technologies such as single cell RNA sequencing, have shaped our understanding of the importance of Egr-1 in the initiation and progression of cardiovascular disease. This article describes Egr-1's role in various cardiovascular settings and discusses potential mechanisms of action. Given the range of conditions linked to Egr-1, this zinc finger protein may serve as a therapeutic target for intervention.
Introduction
Cardiovascular disease remains the leading cause of death worldwide (1). In 2020, approximately one-third of adults in the United States received care for a cardiovascular risk factor or condition (2). This public health burden is set to increase. For example, health care costs to manage cardiovascular risk factors are expected to rise dramatically, from $400 billion in 2020 to $1,344 billion in 2050 (2) and productivity losses are expected to rise by 54% from $234 billion to $361 billion.
Transcription factors regulate the expression of genes underpinning the pathogenesis of cardiovascular disease by binding to DNA regulatory elements, interacting with other regulatory proteins and undergoing modification themselves. Early growth response (Egr-1/EGR1) is a zinc finger transcription factor and product of an immediate-early gene which rapidly responds to changes in the cellular environment. Egr-1 has been linked with multiple cardiovascular disorders including reperfusion injury, myocardial fibrosis, no reflow and heart failure.
Egr-1 was discovered 3 decades ago and comprises N-terminal and C-terminal activation domains, several C2H2 type DNA-binding zinc fingers, nuclear localization signals and an inhibitory domain (3–8). Egr-1 functionally interacts with a broad range of partners, including protein kinases (e.g., MAPK3/ERK1), transcriptional activators [e.g., C/EBPβ (9), JUN (10)] and transcriptional repressors [e.g., NGFI-A binding protein 1 (NAB1) (11) and NAB2 (12)] (Figure 1). These cofactors have been found to play a causal role in the development of cardiovascular dysfunction. For example, ERK1 phosphorylates EGR1 (at Ser26), an amino acid in EGR1 critical for vascular endothelial cell proliferation and migration (13, 14). EGR1's interaction with C/EBPβ controls transcription of the human low-density lipoprotein (LDL) receptor gene (9), which is linked with hypercholesterolemia and cardiovascular disease (15). Cardiac-specific NAB1 overexpression regulates cardiomyocyte growth through interaction with Egr-1 and inhibits cardiac hypertrophy in response to pressure overload (16). Recent work indicates that EGR1 can also interact with chromatin remodeling proteins, including subunits of the nucleosome remodeling and deacetylation (NuRD) complex to repress inflammatory enhancers in macrophages and plays a gatekeeper role in monocytic commitment (17). Trizzino et al. correlated EGR1 binding with enhancer activation and repression during macrophage differentiation. On one hand, EGR1 transactivates through direct interactions with its DNA motif, while on the other, appears to act as a corepressor at myeloid enhancers enriched with AP1, PU.1, C/EBPα and other macrophage factors. EGR1 was found to be required for the repression of approximately 1,600 enhancers in the course of macrophage differentiation (17). Recent work by Ding et al. in cancer cells shows that EGR1 can directly bind HDAC9 (18), a histone deacetylase that promotes vascular inflammation and atherosclerosis (19).
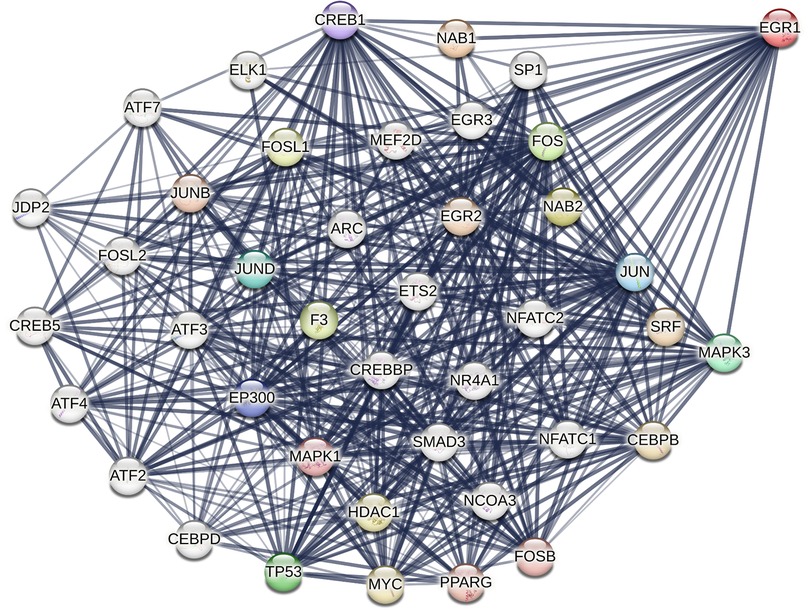
Figure 1. STRING representation of structural and/or functional interactions between human EGR1 with other proteins. Image was created in STRING (Protein-Protein Interaction Networks Functional Enrichment Analysis) using the full STRING network setting with edges indicating functional and physical protein associations in confidence mode, sourcing text, experiments and databases, and relative line thickness indicating strength of data support. Interactions limited to 40.
This article briefly reviews emerging evidence further implicating Egr-1 in the pathogenesis of cardiovascular disease.
Egr-1, atherosclerosis and acute coronary syndrome
Building on pioneering studies from a range of laboratories that established pro-inflammatory and pro-atherogenic roles for Egr-1 (20, 21), Manta and colleagues (22) studied links between Egr-1 and stabilin proteins in atherosclerosis using Apoe−/− mice interbred with stab1−/− and stab2−/− mice. Stab1 and stab2 are scavenger receptors expressed in the liver by sinusoidal endothelial cells implicated in atherogenesis. Low-density lipoprotein and ox-LDL uptake into caudal veins are reduced stab1−/−stab2−/− zebrafish (23). Manta et al. (22) found that atherosclerotic plaque burden was substantially lower in Apoe−/−stab1−/− and Apoe−/−stab2−/− animals, and that monoclonal antibodies targeting stab1 or stab2 reduced diet-associated atherosclerosis in Apoe−/−mice and Ldlr−/−mice. Single-cell RNA seq of circulating myeloid cells from Apoe−/−, Apoe−/−stab1−/− and Apoe−/−stab2−/− mice revealed transcriptional changes in patrolling (Ccr2–/Cx3cr1+/Ly6Clo) and inflammatory (Ccr2+/Cx3cr1+/Ly6Chi) monocytes, including reduced expression of Egr-1. Egr-1 was the only gene downregulated in both monocyte subtypes (22). scRNA-seq by Cochain et al. showed that EGR1 (along with PHLDA1, ATF3, NLRP3 and CCL2) is expressed in macrophages of both human and mice atherosclerotic lesions (24). Peripheral blood cytospin staining with anti-Cd11b and anti-Egr-1 antibodies confirmed downregulation of Egr-1 in Cd11b+ cells of Apoe−/−stab1−/− and Apoe−/−stab2−/− mice compared with those from Apoe−/− mice. Egr-1 attenuation may represent a common mechanism for lower proatherogenic activation of monocytes. Atheroprotection resulting from stab-deficiency is likely due to Egr-1 downregulation in monocytes by plasma proteins regulated by stabilin-reliant clearance.
Egr-1 is also thought to play a role in plaque thrombogenicity and instability, which underpin acute myocardial infarction. Egr-1 activates tissue factor expression in macrophages (25, 26). Tissue factor expression is inhibited by simvastatin in advanced atherosclerotic lesions of Apoe−/−mice (27). Recent studies by Severino et al. (28) investigated the effects of atorvastatin on CD4 + T-cells in statin-naïve non-ST elevation acute coronary syndrome patients. Atorvastatin reduced levels of EGR1 (−10.9 fold) (most profound inhibition) and FOS (−5.5 fold), and decreased the proportion of CD4+CD28−T-cells producing IFN-γ (from 44.1% to 15.0%). CD4+CD28−T-cells have been implicated in weakening of the fibrous cap and atherosclerotic plaque rupture (29). Interestingly, atorvastatin can reduce levels of tissue factor, an EGR1-dependent gene (30), in cholesterol-fed rabbits (31). Moreover, atorvastatin decreased levels of pro-inflammatory cytokines IL-6 and IL-18, chemokine (C-C motif) receptor-2 (CCR2) and Toll-like receptor-4 (TLR4). The authors surmised atorvastatin's inhibitory effects on EGR1 may account for why the statin reduces inflammation in acute coronary syndromes. Future studies should determine whether other statins have similar effects on EGR1 and tissue factor in this setting. Additionally, Wang et al. (32) reported the involvement of Egr-1 in coronary microembolization (CME), a thrombotic and microinfarction complication that can arise in acute coronary syndrome patients during percutaneous coronary intervention (PCI) and result in no reflow. In a rat model of CME, in which myocardial tissue underwent edema and inflammatory cell infiltration around injected microspheres, the authors found that Egr-1 shRNA reduced myocardial injury (serum cardiac troponin I levels) and microinfarct area. Moreover, qPCR after CME-induced myocardial injury revealed activation of the Egr-1/Bim/Beclin-1 pathway impacting autophagy and apoptosis. Egr-1 silencing increased LC3-II and Beclin-1 levels and ameliorated levels of cleaved caspase-3, p62 and Bim. Future studies using alternatives to plastic microspheres to cause a coronary microembolus would better reflect the clinical situation and build on these important proof-of-principle investigations.
Apoptosis underpins myocyte loss after acute myocardial infarction, which can lead to LV remodeling and heart failure (33). Recent studies by Zhou et al. revealed that EGR1 stimulates the expression of a range of apoptosis-related proteins (ATF2, ELK1, HAND2 and CTCF) to promote cardiomyocyte cell death. Mutation of Ser501 (to Ala) reduced EGR1 phosphorylation and impaired the pro-apoptotic effect of EGR1 in cardiomyocytes in a JNK-dependent manner (34).
Biomechanical forces impact atherosclerotic lesion formation and progression. Oscillatory shear stress (OSS) is an atheroprone biomechanical force that can induce the expression of endothelial cell surface adhesion molecules, such as VCAM-1 and ICAM-1, which facilitate leukocyte adhesion and invasion into the arterial wall (35). Egr-1 is activated in endothelial cells by fluid shear stress (36), a response inhibited by PD98059 indicating control via the MEK/ERK pathway (37). Bondareva et al. recently found that EGR1 was induced by OSS and its binding motif is among the most prominent enriched motifs (38). This followed work by Ajami et al. using pathway analysis to identify an SRF-EGR1-HIF1A regulatory axis for OSS up-regulation and that a range of mediators including cytokines, focal adhesions and eNOS activation are targets of EGR1 (39). Egr-1 modulation by biomechanical forces has recently been exploited in a bioreactor application using a non-endothelial system. Kwon et al. developed CHO-DG44 cells that express GFP in small-scale bioreactors secondary to EGR1 promoter activation by shear stress although the definition of an exact shear stress value was not achieved in their sensor design (40).
Egr-1, cardiomyopathy and fibrosis
The adult heart lacks the capacity to regenerate and heart failure can ensue after cardiac ischemia and fibrosis (41). Li et al. (42) sought to identify potential targets for vascular regeneration by investigating the transcriptomic dynamics of coronary vascular endothelial cells following ischemic injury in the heart through comprehensive meta-analysis of publicly available single-cell RNA-sequencing data. Egr-1/EGR1 was among the most significantly upregulated genes in human and murine vascular endothelial cells after ischemia (along with Jun, Zfp36, Fosb, and Hsp90aa1), findings supported by substantially greater EGR1+CD31+ double staining in coronary tissue from patients with ischemic cardiomyopathy compared with normal hearts (42). Cardiac ischemia and fibrosis are primary causes of end-stage heart failure (43). Egr-1 is induced by profibrotic stimuli such as TGF-β and can promote collagen synthesis, extracellular matrix production and fibrosis (44, 45).
Koenig et al. (46) used scRNA-seq and single-nucleus (sn)RNA-seq on left ventricular cardiac tissue from 17 individuals with dilated (non-ischemic) cardiomyopathy (DCM) and 28 non-diseased donors. EGR1 was one of the most significantly upregulated genes in vascular endothelial cells (alongside DUSP5/6, PDE4B/D, FGFR1, SMAD3/6, VEGF-A/C, APLNR) and macrophages, monocytes and dendritic cells (along with CCL3, NLRP3, NFKB2) in DCM material compared to non-diseased tissue. While this study sheds light on the transcriptional landscape of cells in healthy and diseased human heart, future experiments exploring signaling mechanisms or the potential role of chromatin modification and accessibility involving EGR1 in this context would provide added mechanistic insights on molecular control in the pathogenesis of heart failure.
Investigations by Shen et al. showed that inhibition of Egr1 expression in mice relieved the severity of myocardial fibrosis, whereas delivery of miR-150-5p antagomir exacerbated myocardial fibrosis, reversing the effect of sh-Egr1 (47). A recent study by Zhang et al. (48) showed that Egr-1 regulates regenerative senescence during neonatal heart regeneration as well as cardiac repair in adult mice. In neonatal hearts, Egr-1 mediates angiogenesis and cardiomyocyte replication, whereas in adult hearts, senescence and repair induced by the heparan sulfate proteoglycan, agrin, involves integrin–FAK–ERK–Akt1 signaling and Egr-1 activation in cardiac fibroblasts. The authors found increased fibrotic scar formation and impaired cardiac function in Egr-1 deficient mice treated with agrin compared with wild-type mice, demonstrating that Egr1 is needed for agrin-induced cardiac repair. Future work should delineate whether cellular localisation impacts Egr-1 function and use conditional Egr-1 deficient mice to better understand Egr-1 mediated senescence in cardiac fibroblasts during repair.
Egr-1 and vascular dysfunction
Tian et al. (49) reported EGR1 regulation of sushi, von Willebrand factor type A, EGF, and pentraxin domain-containing protein 1 (SVEP1), a large extracellular matrix protein associated with coronary artery disease (50). In Apoe−/− or Svep1+/−Apoe−/− mice fed a high fat diet for 8 weeks, Svep1+/−Apoe−/− mice had reduced atherosclerotic plaque burden. Moreover, Svep1 deficiency specifically in SMC reduced atherosclerotic and plaque size and complexity (51). Levels of SVEP1, which mediates SMC migration and proliferation in response to ox-LDL, are elevated in human atherosclerotic plaques compared to normal tissues. EGR1 binds to and transactivates the SVEP1 promoter and drives SVEP1 expression in SMCs treated with ox-LDL (49). Separate studies by Xie et al. (52) revealed that the splicing factor, serine/arginine-rich splicing factor 1 (SRSF1) promotes SMC proliferation and injury-induced neointima formation and involves induction of truncated form of p53, Δ133p53, which activates Krüppel-like factor 5 (KLF5), a process requiring the physical interaction of Δ133p53 and EGR1. These findings suggest that Δ133p53-EGR1 complex formation is needed for SRSF1-inducible KLF5 signalling and SRSF1-dependent proliferation (52). That Egr-1 promotes SMC hyperplasia has been demonstrated using a range of approaches (53–63). For example, intimal thickening is reduced when blood vessels from Egr-1-deficient mice are injured compared with wild type animals (64) and lumens of vein grafts from Egr-1−/− mice are wider than those of wild types (65). Tian et al. and Xie et al.'s data suggest that the growth-regulatory effects of EGR1 on SMCs are reliant on SVEP1, SRSF1 and/or other genes. For example, Mylonas et al. (66) reported in a rabbit vein graft model that Egr-1 oligodeoxynucleotide decoys that reduce the expression of pro-inflammatory transcription factors (NF-κB, KLF4, HIF1α), stem cell genes (NANOG and HOXA5), toll-like receptors (TLR2, TLR3, TLR4, TLR8), chemokines (CCL4, CCL20, CCR2), interleukins (IL1β, IL2, IL4, IL8, IL10, IL18), TNFα and interferons (IFNβ, IFNγ). These decoys reduce Egr-1 expression, ki-67+ proliferation and intimal hyperplasia (67). Egr-1 is thus an attractive target to control intimal thickening given the breadth and nature of genes it regulates.
Egr-1 in hypertension and preeclampsia
The direct relationship between hypertension and the risk of cardiovascular disease is well established (68, 69). Systolic blood pressure is a leading modifiable risk factor for premature cardiovascular death (70). EGR1 levels are elevated in advanced vascular lesions from individuals with late-stage pulmonary arterial hypertension (71). Laggner et al. recently implicated EGR1 as a driver of right ventricular remodeling in idiopathic pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension patients (72). Preeclampsia is a common complication in pregnancy and defined by onset of hypertension, proteinurea and organ failure after 20 weeks’ gestation. The pathogenesis of preeclampsia is incompletely understood but is associated with vasospasm and impaired placental angiogenesis. Zhao and colleagues (73) studied serum fibroblast growth factor 23 (FGF23) in healthy women or pregnant women with preeclampsia and examined its role mediating placental angiogenesis through ERK1/2-EGR1 signaling. They found that third trimester FGF23 levels were lower in women with preeclampsia. FGF23 stimulated endothelial cell migration, invasion and tubule formation in vitro. It also promoted placental VEGF-A expression through the ERK1/2-EGR-1 pathway since SCH772984 blocked both EGR1 and VEGF-A in these cells. Moreover, levels of p-ERK, EGR1 and VEGF-A in placental tissue were lower in the preeclampsia group than in controls. Whether Egr-1 directly regulates VEGF-A transcription in endothelial cells appears to be context-dependent since VEGF-A levels do not appear to change at least in retinas of mice deficient in Egr-1 (74) or when Egr-1 is knocked down (75) or overexpressed (76). While Zhao et al.'s studies suggest a potential mechanism in the development of preeclampsia, future studies should determine if FGF23 levels correlate with preeclampsia severity or if the FGF23-ERK1/2-EGR-1 pathway is causal for preeclampsia in animal models (73).
Egr-1 and complications of diabetes
Egr-1 plays a causal role in the development of type 2 diabetes. Shen and colleagues (77) found that glucagon, which increases blood glucose levels by regulating gluconeogenesis, stimulates Egr-1 which binds the promoter of the gluconeogenic transcription factor C/EBPα and activates the expression of hepatic gluconeogenic genes. Xu et al. reported that Egr-1 drives the development of renal tubulointerstitial fibrosis in the context of diabetic kidney disease. Egr-1 activates the expression of renal fibrosis markers (such as protease-activated receptor 1, TGF-β1, fibronectin and collagen I) via the TGF-β1/Smad pathway (78). Recent work comparing differential gene expression across multiple autoimmune disorder datasets, including type 1 diabetes, revealed that EGR1 was the only gene commonly expressed across four datasets (type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus, Crohn's disease) (79). Beyond type 2 diabetes, which mainly affects older individuals, EGR1 has been used as a marker gene for childhood-onset type 2 diabetes (80).
Diabetic retinopathy is a major cause of vision loss and disability. Ao et al. (81) found that Egr1 is highly expressed in retinas of hyperglycaemic streptozotocin (STZ)-treated diabetic rats. Intravitreal delivery of Egr-1 shRNA to STZ-induced diabetic rats reduced Egr-1 and p53 levels and inhibited apoptosis in the retina. Karthikkeyan et al. showed that EGR1 is also expressed human diabetic retina compared with non-diabetic retina (82). Moreover, El-Asrar et al. showed EGR1 expression in epiretinal membranes from individuals with proliferative diabetic retinopathy (mainly by vascular endothelial cells and stromal cells) and proliferative vitreoretinopathy (mainly myofibroblasts). EGR1 expression was coincident with HMGB1, RAGE, and OPN expression, suggesting its involvement in vitreoretinal angiogenic, inflammatory and fibrotic processes (83).
Future research directions
The studies above have expanded our appreciation of the regulatory role that Egr-1 plays in vascular disease. Several limitations have already been identified that need to be addressed to advance the field. Additionally, many articles have described Egr-1 function without clarifying direct molecular mechanisms in a specific cardiovascular disease. For example, while Egr-1 is a well-established prototypic transcription factor, does it functionally collaborate with other such factors and if so, which ones, how and in which cell types? Can single cell- or single nuclear-RNA-sequencing extend our appreciation of Egr-1 control in complex cardiovascular tissue? Does Egr-1 undergo post-translational modification(s), dynamic or otherwise, in the context of a specific cardiovascular disease? How can ERK-dependent Egr-1 promoter activation be studied separately from ERK-dependent Egr-1 phosphorylation in the context of cardiovascular disease when Egr-1 is expressed so rapidly? Moreover, it is vital that new EGR1-targeting agents be developed or existing approaches be refined, whether this be with natural, novel or repurposed pharmaceuticals or those that exploit nucleic acid strategies. In this regard, recent studies indicate that the naturally occurring agent astragaloside IV inhibits cardiac hypertrophy through ERK and PKCβII/Egr-1 activation in rodents born from mothers with intrauterine hypoxia (84). This can also include DNAzymes targeting EGR1, which have been used by ourselves and others in a range of local delivery models to reduce post-angioplasty restenosis (53, 54, 85), in-stent restenosis (54) and to lower infarct size following myocardial ischemia-reperfusion (86, 87). However, an important challenge has been overcoming low efficient tissue uptake after systemic delivery (88). Future studies could apply insights from other fields to models of cardiovascular disease. For example, Jiang et al. (89) encapsulated EGR1 DNAzymes (75) with poly(ethylenimine) and an amorphous Mn2+/Zn2+-coordinated inositol hexaphosphate (IP6) capsule modified with a cRGD targeting peptide shell then injected these intravenously into mice bearing tumors which accumulated the DNAzyme. cRGD peptides confer targeting ability via recognition of αvβ3 integrin receptors, mimicking integrin-mediated endocytic cell entry. Upon endocytosis, the Mn2+/Zn2+-IP6 shell is degraded in the cell's acidic lysosomal environment releasing Mn2+, Zn2+ and the DNAzyme. These biocapsulated DNAzymes suppressed EGR1 levels in tumors and induced tumor cell death. Delivered intravenously, biocapsule DNAzymes inhibited EGR1 levels in, and growth of, pulmonary metastatic tumors (89). While this approach demonstrates the clinical potential of biocapsules for targeted inhibition of breast cancer, the same approach may be useful in vascular settings, especially given the crucial role played by αvβ3 integrin in atherogenesis. For example, αvβ3 antibodies inhibit macrophage infiltration into atherosclerosis lesions and lesion formation in diabetic pigs fed a high fat diet (90). Moreover, αvβ3 mediates plaque angiogenesis, inflammation and SMC accumulation (91, 92) and positron emission tomography radiotracers (18F-fluciclatide) have targeted these as potential markers of atherosclerosis (93).
Concluding remarks
As an immediate-early gene activated by a range of environmental cues such as cytokines, hormones, growth factors and hypoxia, Egr-1 integrates changes outside the cell with programmatic inducible gene expression. The breadth of dependent genes and scope of proliferative, migratory, immune and inflammatory conditions that EGR1 controls suggest this factor represents a fertile therapeutic target for drug development for many types of cardiovascular disease. A key challenge, however, is devising effective, clinically viable, preferably systemically deliverable interventional strategies that can selectively target EGR1 and provide sustained inhibition in cardiovascular settings involving complex, interactive molecular networks and comorbidities.
Author contributions
LK: Writing – review & editing, Writing – original draft, Project administration, Investigation, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a National Heart Foundation (Vanguard Grant), New South Wales Ministry of Health (Senior Researcher Grant, NSW Cardiovascular Research Capacity Program) and a Cooperative Research Centre-Project (Department of Industry Science and Resources) (LMK).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Vaduganathan M, Mensah GA, Turco JV, Fuster V, Roth GA. The global burden of cardiovascular diseases and risk: a compass for future health. J Am Coll Cardiol. (2022) 80:2361–71. doi: 10.1016/j.jacc.2022.11.005
2. Kazi DS, Elkind MSV, Deutsch A, Dowd WN, Heidenreich P, Khavjou O, et al. Forecasting the economic burden of cardiovascular disease and stroke in the United States through 2050: a presidential advisory from the American Heart Association. Circulation. (2024) 150:e89–e101. doi: 10.1161/CIR.0000000000001258
3. Milbrandt J. A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science. (1987) 238:797–9. doi: 10.1126/science.3672127
4. Lemaire P, Revelant O, Bravo R, Charnay P. Two mouse genes encoding potential transcription factors with identical DNA-binding domains are activated by growth factors in cultured cells. Proc Natl Acad Sci U S A. (1988) 85:4691–5. doi: 10.1073/pnas.85.13.4691
5. Sukhatme VP, Cao X, Chang LL, Tsai-Morris C-H, Stamenkovich D, Ferreira PCP, et al. A zinc-finger encoding gene corregulated with c-fos during growth and differentiation and after depolarization. Cell. (1988) 53:37–43. doi: 10.1016/0092-8674(88)90485-0
6. Lemaire P, Vesque C, Schmitt J, Stunnenberg H, Frank R, Charnay P. The serum-inducible mouse gene Krox-24 encodes a sequence-specific transcriptional activator. Mol Cell Biol. (1990) 10:3456–67. doi: 10.1128/mcb.10.7.3456-3467.1990
7. Siderovski DP, Blum S, Forsdyke RE, Forsdyke DR. A set of human putative lymphocyte G0/G1 switch genes includes genes homologous to rodent cytokine and zinc finger protein-encoding genes. DNA Cell Biol. (1990) 9:579–87. doi: 10.1089/dna.1990.9.579
8. Chen J, Liu MY, Parish CR, Chong BH, Khachigian L. Nuclear import of early growth response-1 involves importin-7 and the novel nuclear localization signal serine-proline-serine. Int J Biochem Cell Biol. (2011) 43:905–12. doi: 10.1016/j.biocel.2011.03.004
9. Zhang F, Lin M, Abidi P, Thiel G, Liu J. Specific interaction of Egr1 and c/EBPbeta leads to the transcriptional activation of the human low density lipoprotein receptor gene. J Biol Chem. (2003) 278:44246–54. doi: 10.1074/jbc.M305564200
10. Jung E, Ou S, Ahn SS, Yeo H, Lee YH, Shin SY. The JNK-EGR1 signaling axis promotes TNF-alpha-induced endothelial differentiation of human mesenchymal stem cells via VEGFR2 expression. Cell Death Differ. (2023) 30:356–68. doi: 10.1038/s41418-022-01088-8
11. Swirnoff AH, Apel ED, Svaren J, Sevetson BR, Zimonjic DB, Popescu NC, et al. Nab1, a corepressor of NGFI-A (Egr-1), contains an active transcriptional repression domain. Mol Cell Biol. (1998) 18:512–24. doi: 10.1128/MCB.18.1.512
12. Svaren J, Sevetson BR, Apel ED, Zimonjic DB, Popescu NC, Milbrandt J. NAB2, a corepressor of NGFI-A (egr-1) and Krox20, is induced by proliferative and differentiative stimuli. Mol Cell Biol. (1996) 16:3545–53. doi: 10.1128/MCB.16.7.3545
13. Santiago FS, Sanchez-Guerrero E, Zhang G, Zhong L, Raftery M, Khachigian LM. Extracellular signal-regulated kinase-1 phosphorylates early growth response-1 at serine 26. Biochem Biophys Res Commun. (2019) 510:345–51. doi: 10.1016/j.bbrc.2019.01.019
14. Santiago FS, Li Y, Khachigian LM. Serine 26 in early growth response-1 is critical for endothelial proliferation, migration, and network formation. J Am Heart Assoc. (2021) 10:e020521. doi: 10.1161/JAHA.120.020521
15. Bjornsson E, Gunnarsdottir K, Halldorsson GH, Sigurdsson A, Arnadottir GA, Jonsson H, et al. Lifelong reduction in LDL (low-density lipoprotein) cholesterol due to a gain-of-function mutation in LDLR. Circ Genom Precis Med. (2021) 14:e003029. doi: 10.1161/CIRCGEN.120.003029
16. Buitrago M, Lorenz K, Maass M, Oberdorf-Maass S, Keller U, Schmitteckert EM, et al. The transcriptional repressor NAB1 is a specific regulator of pathological cardiac hypertrophy. Nature Med. (2005) 11:837–44. doi: 10.1038/nm1272
17. Trizzino M, Zucco A, Deliard S, Wang F, Barbieri E, Veglia F, et al. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci Adv. (2021) 7:eaaz8836. doi: 10.1126/sciadv.aaz8836
18. Ding S, Wang X, Lv D, Tao Y, Liu S, Chen C, et al. EBF3 reactivation by inhibiting the EGR1/EZH2/HDAC9 complex promotes metastasis via transcriptionally enhancing vimentin in nasopharyngeal carcinoma. Cancer Lett. (2022) 527:49–65. doi: 10.1016/j.canlet.2021.12.010
19. Das S, Natarajan R. HDAC9: an inflammatory link in atherosclerosis. Circ Res. (2020) 127:824–6. doi: 10.1161/CIRCRESAHA.120.317723
20. Mccaffrey TA, Fu C, Du C, Eskinar S, Kent KC, Bush H Jr, et al. High-level expression of egr-1 and egr-1-inducible genes in mouse and human atherosclerosis. J Clin Invest. (2000) 105:653–62. doi: 10.1172/JCI8592
21. Harja E, Bucciarelli LG, Lu Y, Stern DM, Zou YS, Schmidt AM, et al. Early growth response-1 promotes atherogenesis: mice deficient in early growth response-1 and apolipoprotein E display decreased atherosclerosis and vascular inflammation. Circ Res. (2004) 94:333–9. doi: 10.1161/01.RES.0000112405.61577.95
22. Manta CP, Leibing T, Friedrich M, Nolte H, Adrian M, Schledzewski K, et al. Targeting of scavenger receptors stabilin-1 and stabilin-2 ameliorates atherosclerosis by a plasma proteome switch mediating monocyte/macrophage suppression. Circulation. (2022) 146:1783–99. doi: 10.1161/CIRCULATIONAHA.121.058615
23. Verwilligen RaF, Mulder L, Rodenburg FJ, Van Dijke A, Hoekstra M, Bussmann J, et al. Stabilin 1 and 2 are important regulators for cellular uptake of apolipoprotein B-containing lipoproteins in zebrafish. Atherosclerosis. (2022) 346:18–25. doi: 10.1016/j.atherosclerosis.2022.02.018
24. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-Cell RNA-Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
25. Bea F, Puolakkainen MH, Mcmillen T, Hudson FN, Mackman N, Kuo CC, et al. Chlamydia pneumoniae induces tissue factor expression in mouse macrophages via activation of Egr-1 and the MEK-ERK1/2 pathway. Circ Res. (2003) 92:394–401. doi: 10.1161/01.RES.0000059982.43865.75
26. Albrecht C, Preusch MR, Hofmann G, Morris-Rosenfeld S, Blessing E, Rosenfeld ME, et al. Egr-1 deficiency in bone marrow-derived cells reduces atherosclerotic lesion formation in a hyperlipidaemic mouse model. Cardiovasc Res. (2010) 86:321–9. doi: 10.1093/cvr/cvq032
27. Bea F, Blessing E, Shelley MI, Shultz JM, Rosenfeld ME. Simvastatin inhibits expression of tissue factor in advanced atherosclerotic lesions of apolipoprotein E deficient mice independently of lipid lowering: potential role of simvastatin-mediated inhibition of Egr-1 expression and activation. Atherosclerosis. (2003) 167:187–94. doi: 10.1016/S0021-9150(02)00387-8
28. Severino A, Zara C, Campioni M, Flego D, Angelini G, Pedicino D, et al. Atorvastatin inhibits the immediate-early response gene EGR1 and improves the functional profile of CD4+ T-lymphocytes in acute coronary syndromes. Oncotarget. (2017) 8:17529–50. doi: 10.18632/oncotarget.15420
29. Dumitriu IE, Araguas ET, Baboonian C, Kaski JC. CD4+ CD28 null T cells in coronary artery disease: when helpers become killers. Cardiovasc Res. (2009) 81:11–9. doi: 10.1093/cvr/cvn248
30. Mackman N. Regulation of the tissue factor gene. Thromb Haemost. (1997) 78:747–54. doi: 10.1055/s-0038-1657623
31. Li JQ, Zhao SP, Li QZ, Cai YC, Wu LR, Fang Y, et al. Atorvastatin reduces tissue factor expression in adipose tissue of atherosclerotic rabbits. Int J Cardiol. (2007) 115:229–34. doi: 10.1016/j.ijcard.2006.02.007
32. Wang X, Wu X, Lu Y, Sun Y, Zhu H, Liang J, et al. Egr-1 is involved in coronary microembolization-induced myocardial injury via Bim/Beclin-1 pathway-mediated autophagy inhibition and apoptosis activation. Aging. (2018) 10:3136–47. doi: 10.18632/aging.101616
33. Abbate A, Biondi-Zoccai GG, Bussani R, Dobrina A, Camilot D, Feroce F, et al. Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. J Am Coll Cardiol. (2003) 41:753–60. doi: 10.1016/S0735-1097(02)02959-5
34. Zhou J, Yao Y, Zhang J, Wang Z, Zheng T, Lu Y, et al. JNK-dependent phosphorylation and nuclear translocation of EGR-1 promotes cardiomyocyte apoptosis. Apoptosis. (2022) 27:246–60. doi: 10.1007/s10495-022-01714-3
35. Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ Res. (1998) 82:532–9. doi: 10.1161/01.RES.82.5.532
36. Khachigian LM, Anderson KA, Halnon NJ, Resnick N, Gimbrone MA Jr, Collins T. Egr-1 is activated in endothelial cells exposed to fluid shear stress and interacts with a novel shear-stress response element in the PDGF A-chain promoter. Arterioscl Thromb Vasc Biol. (1997) 17:2280–6. doi: 10.1161/01.ATV.17.10.2280
37. Schwachtgen J-L, Houston P, Campbell C, Sukhatme V, Braddock M. Fluid shear stress activation of egr-1 transcription in cultured human endothelial and epithelial cells is mediated via the extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway. J Clin Invest. (1998) 101:2540–9. doi: 10.1172/JCI1404
38. Bondareva O, Tsaryk R, Bojovic V, Odenthal-Schnittler M, Siekmann AF, Schnittler HJ. Identification of atheroprone shear stress responsive regulatory elements in endothelial cells. Cardiovasc Res. (2019) 115:1487–99. doi: 10.1093/cvr/cvz027
39. Ajami NE, Gupta S, Maurya MR, Nguyen P, Li JY, Shyy JY, et al. Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc Natl Acad Sci U S A. (2017) 114:10990–5. doi: 10.1073/pnas.1707517114
40. Kwon T, Leroux AC, Zang H, Pollard D, Zehe C, Akbari S. Cell-based shear stress sensor for bioprocessing. J Biotechnol. (2024) 390:71–9. doi: 10.1016/j.jbiotec.2024.04.016
41. Hinderer S, Schenke-Layland K. Cardiac fibrosis - a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. (2019) 146:77–82. doi: 10.1016/j.addr.2019.05.011
42. Li Z, Solomonidis EG, Berkeley B, Tang MNH, Stewart KR, Perez-Vicencio D, et al. Multi-species meta-analysis identifies transcriptional signatures associated with cardiac endothelial responses in the ischaemic heart. Cardiovasc Res. (2023) 119:136–54. doi: 10.1093/cvr/cvac151
43. Ravassa S, Lopez B, Treibel TA, San Jose G, Losada-Fuentenebro B, Tapia L, et al. Cardiac fibrosis in heart failure: focus on non-invasive diagnosis and emerging therapeutic strategies. Mol Aspects Med. (2023) 93:101194. doi: 10.1016/j.mam.2023.101194
44. Bhattacharyya S, Wu M, Fang F, Tourtellotte W, Feghali-Bostwick C, Varga J. Early growth response transcription factors: key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix Biol. (2011) 30:235–42. doi: 10.1016/j.matbio.2011.03.005
45. Bhattacharyya S, Fang F, Tourtellotte W, Varga J. Egr-1: new conductor for the tissue repair orchestra directs harmony (regeneration) or cacophony (fibrosis). J Pathol. (2013) 229:286–97. doi: 10.1002/path.4131
46. Koenig AL, Shchukina I, Amrute J, Andhey PS, Zaitsev K, Lai L, et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat Cardiovasc Res. (2022) 1:263–80. doi: 10.1038/s44161-022-00028-6
47. Shen J, Xing W, Gong F, Wang W, Yan Y, Zhang Y, et al. MiR-150-5p retards the progression of myocardial fibrosis by targeting EGR1. Cell Cycle. (2019) 18:1335–48. doi: 10.1080/15384101.2019.1617614
48. Zhang L, Elkahal J, Wang T, Rimmer R, Genzelinakh A, Bassat E, et al. Egr1 regulates regenerative senescence and cardiac repair. Nat Cardiovasc Res. (2024) 3:915–32. doi: 10.1038/s44161-024-00493-1
49. Tian Q, Chen JH, Ding Y, Wang XY, Qiu JY, Cao Q, et al. EGR1 Transcriptionally regulates SVEP1 to promote proliferation and migration in human coronary artery smooth muscle cells. Mol Biol Rep. (2024) 51:365. doi: 10.1007/s11033-024-09322-x
50. Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, Konig IR, et al. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. (2016) 374:1134–44. doi: 10.1056/NEJMoa1507652
51. Jung IH, Elenbaas JS, Alisio A, Santana K, Young EP, Kang CJ, et al. SVEP1 is a human coronary artery disease locus that promotes atherosclerosis. Sci Transl Med. (2021) 13:eabe0357. doi: 10.1126/scitranslmed.abe0357
52. Xie N, Chen M, Dai R, Zhang Y, Zhao H, Song Z, et al. SRSF1 Promotes vascular smooth muscle cell proliferation through a Delta133p53/EGR1/KLF5 pathway. Nat Commun. (2017) 8:16016. doi: 10.1038/ncomms16016
53. Santiago FS, Lowe HC, Kavurma MM, Chesterman CN, Baker A, Atkins DG, et al. New DNA enzyme targeting Egr-1 mRNA inhibits vascular smooth muscle proliferation and regrowth factor injury. Nature Med. (1999) 5:1264–9. doi: 10.1038/15215
54. Lowe HC, Fahmy RG, Kavurma MM, Baker A, Chesterman CN, Khachigian LM. Catalytic oligodeoxynucleotides define a key regulatory role for early growth response factor-1 in the porcine model of coronary in-stent restenosis. Circ Res. (2001) 89:670–7. doi: 10.1161/hh2001.097867
55. Fahmy RG, Khachigian LM. Antisense Egr-1 RNA driven by the CMV promoter is an inhibitor of vascular smooth muscle cell proliferation and regrowth after injury. J Cell Biochem. (2002) 84:575–82. doi: 10.1002/jcb.10057
56. Wada Y, Fujimori M, Suzuki J, Tsukioka K, Ito K, Sawa Y, et al. Egr-1 in vascular smooth muscle cell proliferation in response to allo-antigen. J Surg Res. (2003) 115:294–302. doi: 10.1016/S0022-4804(03)00213-0
57. Fahmy RG, Khachigian LM. Locked nucleic acid-modified DNA enzymes targeting early growth response-1 inhibit vascular smooth muscle cell growth. Nucleic Acids Res. (2004) 32:2281–5. doi: 10.1093/nar/gkh543
58. Ohtani K, Egashira K, Usui M, Ishibashi M, Hiasa KI, Zhao Q, et al. Inhibition of neointimal hyperplasia after balloon injury by cis-element ‘decoy’ of early growth response gene-1 in hypercholesterolemic rabbits. Gene Ther. (2004) 11:126–32. doi: 10.1038/sj.gt.3302153
59. Liu GN, Teng YX, Yan W. Transfected synthetic DNA enzyme gene specifically inhibits Egr-1 gene expression and reduces neointimal hyperplasia following balloon injury in rats. Int J Cardiol. (2008) 129:118–24. doi: 10.1016/j.ijcard.2007.11.066
60. Han W, Liu GN. EGR-1 decoy ODNs inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia of balloon-injured arteries in rat. Life Sci. (2010) 86:234–43. doi: 10.1016/j.lfs.2009.12.005
61. Vazquez-Padron RI, Mateu D, Rodriguez-Menocal L, Wei Y, Webster KA, Pham SM. Novel role of egr-1 in nicotine-related neointimal formation. Cardiovasc Res. (2010) 88:296–303. doi: 10.1093/cvr/cvq213
62. Zhang J, Guo C, Wang R, Huang L, Liang W, Liu R, et al. An Egr-1-specific DNAzyme regulates Egr-1 and proliferating cell nuclear antigen expression in rat vascular smooth muscle cells. Exp Ther Med. (2013a) 5:1371–4. doi: 10.3892/etm.2013.1013
63. Li Y, Mcrobb LS, Khachigian LM. MicroRNA miR-191 targets the zinc finger transcription factor Egr-1 and suppresses intimal thickening after carotid injury. Int J Cardiol. (2016) 212:229–302. doi: 10.1016/j.ijcard.2016.03.037
64. Zhang K, Cao J, Dong R, Du J. Early growth response protein 1 promotes restenosis by upregulating intercellular adhesion molecule-1 in vein graft. Oxid Med Cell Longev. (2013b) 2013:432409.24386503
65. Dong R, Zhang K, Wang YL, Zhang F, Cao J, Zheng JB, et al. MiR-551b-5p contributes to pathogenesis of vein graft failure via upregulating early growth response-1 expression. Chin Med J (Engl). (2017) 130:1578–85. doi: 10.4103/0366-6999.208246
66. Mylonas KS, Peroulis M, Kapelouzou A. Transfection of vein grafts with early growth response factor-1 oligodeoxynucleotide decoy: effects on stem-cell genes and toll-like receptor-mediated inflammation. Int J Mol Sci. (2023) 24:5866. doi: 10.3390/ijms242115866
67. Peroulis M, Kakisis J, Kapelouzou A, Giagini A, Giaglis S, Mantziaras G, et al. The role of ex-vivo gene therapy of vein grafts with Egr-1 decoy in the suppression of intimal hyperplasia. Eur J Vasc Endovasc Surg. (2010) 40:216–23. doi: 10.1016/j.ejvs.2010.04.013
68. Whelton SP, Mcevoy JW, Shaw L, Psaty BM, Lima JC, Budoff M, et al. Association of normal systolic blood pressure level with cardiovascular disease in the absence of risk factors. JAMA Cardiol. (2020) 5:1011–8. doi: 10.1001/jamacardio.2020.1731
69. Liu J, Li Y, Ge J, Yan X, Zhang H, Zheng X, et al. Lowering systolic blood pressure to less than 120 mm hg versus less than 140 mm hg in patients with high cardiovascular risk with and without diabetes or previous stroke: an open-label, blinded-outcome, randomised trial. Lancet. (2024) 404:245–55. doi: 10.1016/S0140-6736(24)01028-6
70. Razo C, Welgan CA, Johnson CO, Mclaughlin SA, Iannucci V, Rodgers A, et al. Effects of elevated systolic blood pressure on ischemic heart disease: a burden of proof study. Nat Med. (2022) 28:2056–65. doi: 10.1038/s41591-022-01974-1
71. Van Der Feen DE, Dickinson MG, Bartelds B, Borgdorff MA, Sietsma H, Levy M, et al. Egr-1 identifies neointimal remodeling and relates to progression in human pulmonary arterial hypertension. J Heart Lung Transplant. (2016) 35:481–90. doi: 10.1016/j.healun.2015.12.004
72. Laggner M, Oberndorfer F, Golabi B, Bauer J, Zuckermann A, Hacker P, et al. EGR1 Is implicated in right ventricular cardiac remodeling associated with pulmonary hypertension. Biology (Basel). (2022) 11:677. doi: 10.3390/biology11050677
73. Zhao S, Zhou J, Chen R, Zhou W, Geng H, Huang Y, et al. Decreased FGF23 inhibits placental angiogenesis via the ERK1/2-EGR-1 signaling pathway in preeclampsia. Cytokine. (2024) 176:156508. doi: 10.1016/j.cyto.2024.156508
74. Schippert R, Schaeffel F, Feldkaemper MP. Microarray analysis of retinal gene expression in Egr-1 knockout mice. Mol Vis. (2009) 15:2720–39.20019881
75. Fahmy RG, Dass CR, Sun LQ, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nature Med. (2003) 9:1026–32. doi: 10.1038/nm905
76. Gille J, Swerlick RA, Caughman SW. Transforming growth factor-alpha-induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2-dependent DNA binding and transactivation. EMBO J. (1997) 16:750–9. doi: 10.1093/emboj/16.4.750
77. Shen N, Jiang S, Lu JM, Yu X, Lai SS, Zhang JZ, et al. The constitutive activation of egr-1/C/EBPa mediates the development of type 2 diabetes mellitus by enhancing hepatic gluconeogenesis. Am J Pathol. (2015) 185:513–23. doi: 10.1016/j.ajpath.2014.10.016
78. Xu P, Zhan H, Zhang R, Xu XJ, Zhang Y, Le Y, et al. Early growth response factor 1 upregulates pro-fibrotic genes through activation of TGF-beta1/Smad pathway via transcriptional regulation of PAR1 in high-glucose treated HK-2 cells. Mol Cell Endocrinol. (2023) 572:111953. doi: 10.1016/j.mce.2023.111953
79. Rajalingam A, Ganjiwale A. Identification of common genetic factors and immune-related pathways associating more than two autoimmune disorders: implications on risk, diagnosis, and treatment. Genomics Inform. (2024) 22:10. doi: 10.1186/s44342-024-00004-5
80. Jia K, Wu Y, Ju J, Wang L, Shi L, Wu H, et al. The identification of gene signature and critical pathway associated with childhood-onset type 2 diabetes. PeerJ. (2019) 7:e6343. doi: 10.7717/peerj.6343
81. Ao H, Liu B, Li H, Lu L. Egr1 mediates retinal vascular dysfunction in diabetes mellitus via promoting p53 transcription. J Cell Mol Med. (2019) 23:3345–56. doi: 10.1111/jcmm.14225
82. Karthikkeyan G, Nareshkumar RN, Aberami S, Sulochana KN, Vedantham S, Coral K. Hyperglycemia induced early growth response-1 regulates vascular dysfunction in human retinal endothelial cells. Microvasc Res. (2018) 117:37–43. doi: 10.1016/j.mvr.2018.01.002
83. El-Asrar AM, Missotten L, Geboes K. Expression of high-mobility groups box-1/receptor for advanced glycation end products/osteopontin/early growth response-1 pathway in proliferative vitreoretinal epiretinal membranes. Mol Vis. (2011) 17:508–18.21365018
84. Zhang Y, Wu M, Deng Y, He B, Li H. Astragaloside IV attenuates cardiac hypertrophy in rats born from mothers with intrauterine hypoxia through the PKCbetaII/Egr-1 pathway. Exp Ther Med. (2023) 26:365. doi: 10.3892/etm.2023.12064
85. Wang TR, Yang G, Liu GN. DNA Enzyme ED5 depletes egr-1 and inhibits neointimal hyperplasia in rats. Cardiology. (2013) 125:192–200. doi: 10.1159/000350364
86. Bhindi R, Khachigian LM, Lowe HC. DNAzymes targeting the transcription factor Egr-1 reduce myocardial infarct size following ischemia-reperfusion in rats. J Thromb Haemost. (2006) 4:1479–83. doi: 10.1111/j.1538-7836.2006.02022.x
87. Bhindi R, Fahmy RG, Mcmahon AC, Khachigian LM, Lowe HC. Intracoronary delivery of DNAzymes targeting human EGR-1 reduces infarct size following myocardial ischaemia reperfusion. J Pathol. (2012) 227:157–64. doi: 10.1002/path.2991
88. Khachigian LM. Deoxyribozymes as catalytic nanotherapeutic agents. Cancer Res. (2019) 79:879–88. doi: 10.1158/0008-5472.CAN-18-2474
89. Jiang R, Li L, Li M. Biomimetic construction of degradable DNAzyme-loaded nanocapsules for self-sufficient gene therapy of pulmonary metastatic breast cancer. ACS Nano. (2023) 17:22129–44. doi: 10.1021/acsnano.3c09581
90. Maile LA, Busby WH, Xi G, Gollahan KP, Flowers W, Gafbacik N, et al. An anti-alphaVbeta3 antibody inhibits coronary artery atherosclerosis in diabetic pigs. Atherosclerosis. (2017) 258:40–50. doi: 10.1016/j.atherosclerosis.2017.01.030
91. Hoshiga M, Alpers CE, Smith LL, Giachelli CM, Schwartz SM. Alpha-v beta-3 integrin expression in normal and atherosclerotic artery. Circ Res. (1995) 77:1129–35. doi: 10.1161/01.RES.77.6.1129
92. Dietz M, Kamani CH, Deshayes E, Dunet V, Mitsakis P, Coukos G, et al. Imaging angiogenesis in atherosclerosis in large arteries with (68)Ga-NODAGA-RGD PET/CT: relationship with clinical atherosclerotic cardiovascular disease. EJNMMI Res. (2021) 11:71. doi: 10.1186/s13550-021-00815-5
Keywords: cardiovascular disease, early growth response-1, EGR1, acute coronary syndrome, vascular biology
Citation: Khachigian LM (2024) Early growth response-1, a dynamic conduit in cardiovascular disease. Front. Cardiovasc. Med. 11:1487668. doi: 10.3389/fcvm.2024.1487668
Received: 28 August 2024; Accepted: 14 October 2024;
Published: 15 November 2024.
Edited by:
Ilia Fishbein, University of Pennsylvania, United StatesReviewed by:
Shunchang Li, Chengdu Sport University, ChinaCopyright: © 2024 Khachigian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Levon M. Khachigian, bC5raGFjaGlnaWFuQHVuc3cuZWR1LmF1