 Marta Casula1,†
Marta Casula1,† Daniela Marchetti2,†
Daniela Marchetti2,† Lucia Trevisan3*
Lucia Trevisan3* Laura Pezzoli2
Laura Pezzoli2 Matteo Bellini2
Matteo Bellini2 Serena Patrone1
Serena Patrone1 Antonio Zingarelli4
Antonio Zingarelli4 Fabio Gotta3Maria Iascone2
Fabio Gotta3Maria Iascone2 Paola Mandich1,3
Paola Mandich1,3
- 1Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genoa, Genoa, Italy
- 2Laboratory of Medical Genetics, ASST Papa Giovanni XXIII, Bergamo, Italy
- 3Medical Genetics Unit, IRCCS Ospedale Policlinico San Martino, Genoa, Italy
- 4Cardiological Unit, Ospedale Policlinico IRCSS San Martino, Genoa, Italy
Spontaneous coronary artery dissection (SCAD) is a relevant non-atherosclerotic cause of acute coronary syndrome with a complex genetic architecture. Recent discoveries have highlighted the potential role of miRNAs and protein-coding genes involved in the processing of small RNAs in the pathogenesis of SCAD. Furthermore, there may be a connection between SCAD and the increased cardiovascular risk observed in fragile X premutation carriers as well as a correlation with pathogenetic variants in genes encoding for collagen and extracellular matrix, which are related to connective tissue disorders (CTDs). In our cohort of 15 Italian SCAD patients, a total of 37 rare variants were identified in 34 genes using whole exome sequencing (WES) and TRIO-WES analysis when both parents were available. Three likely pathogenic/pathogenetic variants were found in genes previously associated with SCAD and CTDs (COL3A1, COL1A2, and SMAD3) and 26 variants of uncertain significance in genes previously associated with SCAD and CTDs. TRIO-WES analysis revealed 7 de novo variants, 1 of which was found in a potential novel candidate gene (DROSHA). In addition, a premutation allele of 55 ± 2 CGG repeats in the promoter of the FMR1 gene was identified in two related SCAD patients by test for CGG-repeat expansions in the 5′-UTR of the FMR1 gene. Our findings suggest various potential mechanisms such as mRNA toxicity, miRNA regulation, alteration of collagen, and the extracellular matrix architecture, all of which could disrupt vascular homeostasis, and finally, WES and TRIO-WES have proven to be the most powerful approaches for characterizing the genetic background of SCAD.
1 Introduction
Spontaneous coronary artery dissection (SCAD) is a non-atherosclerotic cause of acute coronary syndrome (ACS) with a prevalence of 1%–4% (1, 2). It is one of the major causes of acute myocardial infarction in young to middle-aged women (3). Several different risk factors have been associated with SCAD including female sex, pregnancy-related factors, inflammation, emotional and/or physical stressors (4), hypothyroidism (5), migraine (6–8), and hypertension. Another frequently associated condition is fibromuscular dysplasia (FMD) which has been described, in some case series, in more than 50% of the patients (9–11). Although the complex genetic component of SCAD is still unexplained, increasing evidence suggests a multifactorial and polygenic basis with common genetic loci (about 16 associated with SCAD risk have been identified) contributing in the determination of the risk of SCAD (12). Furthermore, a growing number of reports associate SCAD with heritable connective tissue disorders (CTDs) such as Marfan syndrome, vascular Ehlers–Danlos syndrome (vEDs), and Loeys–Dietz syndrome (LDS) (13–17), suggesting an underestimated monogenic rare disease in SCAD (18). Moreover, the role of miRNAs and of genes encoded for proteins involved in processing of small RNAs is rapidly emerging, both in vascular biology and in possible pathogenic mechanisms of vascular diseases (19, 20) and a relationship between toxicity due to Fragile X premutation and the pathogenesis of SCAD has been hypothesized (21). Recently, the association of connective tissue involvement in females with intermediate or gray zone alleles was reported in a cohort of females with CTDs (22).
1.1 Aims of the research
Although genome-wide association studies (GWAS) have revealed several loci associated with increased risk of SCAD, the genetic causes of SCAD remain mostly unknown. For this purpose, we collected a homogeneous cohort of Italian patients, phenotypically well characterized and hospitalized for SCAD. This cohort underwent whole exome sequencing (WES) and, when both parents were available, TRIO-WES analysis. The goal of this study was to contribute to elucidate the role of genetic factors in pathological mechanisms underlying SCAD disease and the diagnostic route through genetic testing and clinical genetic evaluation.
2 Patients and methods
2.1 Study population
Data of SCAD patients evaluated at IRCCS Policlinico San Martino in Genoa between January 2010 and July 2023 were collected retrospectively. The inclusion criterion was SCAD diagnosis formulated in the catheterization laboratory. Fifteen patients out of 14 families meeting this criterion were enrolled. After the hospitalization for SCAD event and complete recovery, a genetic evaluation was offered to all patients. Clinical, therapeutic, electrocardiographic, echocardiographic, and angiographic data were gathered and analyzed. All patients underwent a regular clinical and instrumental follow-up (at 1, 6, and 12 months after SCAD and then every year, unless otherwise indicated), during which non-invasive imaging by coronary computed tomography angiography (CCTA) was employed to evaluate the spontaneous healing of the involved coronary vessels. Moreover, FMD was assessed by different imaging techniques. A detailed genetic visit was performed for all patients and their available relatives. A complete personal and three-generation family history was collected with a particular focus on cardiovascular disease and CTDs (see Family tree in the Supplementary Material). All patients and their parents underwent a specific physical evaluation for CTDs, including Beighton scale and Marfan scores. During genetic counseling, limits and implications of the study were discussed, and a signed informed written consent was collected. The local Ethical Committee approved the study (22 May 2022, n. 858).
2.2 Genetic analyses
Genomic DNA of patients and available parents was extracted from peripheral blood using commercial DNA extraction kits. WES analysis was performed at the Molecular Genetic Laboratory of ASST-Papa Giovanni XXIII of Bergamo.
The exonic and flanking splice junction regions were captured using the Clinical Research Exome v.2 kits (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed on a NovaSeq6000 Illumina system with 150 bp paired-end reads. Reads were aligned to human genome build GRCh38, and the minimum coverage of the samples was 10×. The variant call file, including single nucleotide polymorphism and indels, was annotated by querying population frequencies databases and mutation databases, including the Genome Aggregation Database (http://gnomad.broad institute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and Human Gene Mutation Database Professional (HGMD, Release 2023.3). To prioritize variants, a sequential filtering strategy was applied, retaining only variants with the following characteristics: (a) potential effect on protein and transcript (splicing, missense, nonsense, and frameshift); (b) consistency with the patient's phenotype according to a selected panel of genes (23) previously associated with CTD and vasculopathies or previously identified in SCAD patients (23); (c) consistency with the suspected inheritance model (X-linked, autosomal recessive or de novo) if parents were available. Variants were classified based on current guidelines (24, 25). The potential causative variants were subsequently confirmed by Sanger sequencing in the proband and parents. As previously described, two pipelines were used to identify the copy number variants (CNVs) based on ExomeDepth (26, 27). All patients were analyzed for CGG-repeat expansions in the 5′-UTR of the Fragile X messenger ribonucleoprotein 1 (FMR1) gene according to the manufacturer's instruction (FRAXA analysis, AmplidexTM kit, Asuragen, Austin, TX, USA) and evaluated following current guidelines (28, 29).
3 Results
3.1 Population characteristics
The cohort of 15 SCAD cases comprised 14 (93%) female Italian patients of European ancestry with an average age at the SCAD event of 46 years (range: 30–58 years). Two women in the cohort were sisters.
The clinical characteristics of SCAD patients are summarized in Table 1. Among all SCAD, two (13%) were pregnancy-related SCAD (P-SCAD), defined as SCAD occurring during pregnancy or within 12 months of delivery, and only one patient (6.7%) suffered a recurrence SCAD episode. Migraine was present in eight (53.3%) patients.
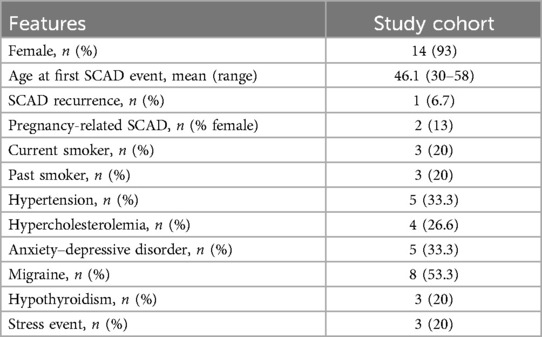
Table 1. SCAD patient characteristics.
Stressor factors at the time of SCAD event were present in three (20%) patients and five (33.3%) had anxiety–depressive disorders. The most common cardiovascular risk factor was hypertension (33.3%). The clinical presentation of SCAD was chest pain in all patients, but in three (20%) patients, the clinical onset was complicated by cardiac arrest. The tortuosity of the coronary vessels with a diameter ≥2 mm, defined as the presence of ≥3 consecutive curvatures of 90°–180° evaluated at end-diastole (30), was found in five patients (33.3%) during coronary angiography. Most patients 10 (66.6%) had a preserved anterograde coronary flow with initial thrombolysis in myocardial infarction (TIMI) 3. A percutaneous coronary intervention (PCI) was performed in three (20%) patients with initial absent antegrade flow (TIMI 0), ongoing chest pain, and persistent ST elevation on electrocardiogram. A conservative management with antiplatelet therapy was the prevalent strategy (80%) as suggested in recent ACS guidelines (31) proving to be the most effective as reported in some studies, in which a conservative approach is effective in the most part of patients (32–34) (Supplementary Table S1).
Clinical genetic evaluation excluded the presence of CTDs diagnosis in the probands, but two patients (13.3%) showed asymptomatic vascular abnormalities on imaging: one had a dissection of both internal carotid arteries and the other one had dissection of one of the anterior branches of the duplicated right vertebral artery. FMD was absent in our cohort. Beighton scale and Marfan scores were ≤2 in all patients.
3.2 Genetic results
Fifteen SCAD patients underwent WES analysis, first using a focused approach on SCAD-associated genes previously identified by different genetic approaches. All cases have read depth ≥10× for ≥98% of the consensus coding sequence of analyzed genes, suggesting adequate coverage to detect protein-coding single nucleotide variants and indels. TRIO-WES analysis was performed in five trios.
We found 37 rare variants in 34 genes. We found three likely pathogenic/pathogenetic (LP/P) variants in genes previously associated with SCAD and CTDs (COL3A1, COL1A2, and SMAD3) (Table 2).
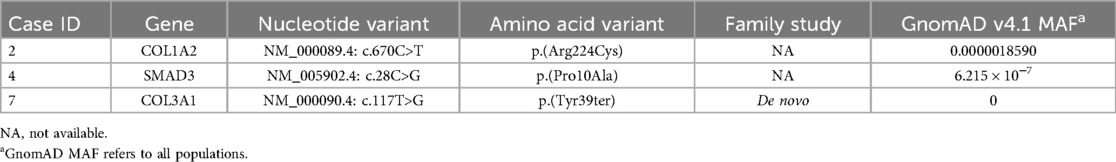
Table 2. Likely pathogenic and pathogenic variants (ACMG classification) in CTDs and SCAD-related genes.
Twenty-six variants were classified as variants of uncertain significance (VUS) in genes both previously associated with SCAD and CTDs or already described (30–33) (Supplementary Table S2). We found three variants in genes candidate for SCAD but also described in association with arterial dissection/aneurism (TLN1, SMAD6, and TSR1) (35–38).
Patient carrying the LP missense variant c.28C > G; p.(Pro10Ala) in SMAD3 gene associated with LDS type 3 showed coronary arteries tortuosity and scoliosis since puberty. During the follow-up, a progressive increase in the aortic root diameter was observed.
Patient with LP variant c.670C > T; p.(Arg224Cys) in COL1A2 present no involvement of other vascular districts and no signs of CTDs and/or osteogenesis imperfecta.
By TRIO-WES analysis, despite the small cohort of patients, we got seven de novo variants, one in a possible novel candidate gene (DROSHA), and four in genes not apparently correlated with SCAD or CTDs (SHANK2, SUPV3L1, SBNO1, and MCHR2) (Supplementary Table S3).
One variant [c.2277C > A; p.(Ser759Arg)] was found in COL4A1. The patient carrying this variant did not have any symptoms or signs associated with the COL4A1 syndrome.
A de novo truncating heterozygous LP/P variant was found in COL3A1 c.117T > G p.(Tyr39Ter), not previously reported in literature and absent in general population databases.
This variant was found in a patient with an asymptomatic dissection of both internal carotid arteries diagnosed after P-SCAD event.
The only male patient of the cohort had the DROSHA c.2317G > A p.(Val773Ile) variant. He presented a SCAD event at 45 years worsened by a cardiac arrest. He did not show any sign of CTDs and had a positive familial history for aortic aneurism (paternal uncle at 70 years) and P-SCAD (paternal cousin before 35 years).
The only two related SCAD patients of our cohort are two sisters who carried a premutation allele of 55 ± 2 CGG repeats in the promoter of FMR1 gene (Supplementary Table S4). One sister had a SCAD event at 37 years and the other one at 45 years. Both had subsequent multiple events of chest pain without new ACS events, but they did not present any involvement of other vascular districts nor any sign of CTDs.
4 Discussion
SCAD is no longer considered a rare condition. However, its true prevalence remains unknown largely due to underdiagnosis.
According to previous studies, we suggest three different mechanisms that could contribute to the production of this complex phenotype.
4.1 mRNA toxicity and miRNA regulation
The increased risk of cardiovascular disease in Fragile X premutation carriers is already well defined. Recently, SCAD has been reported to be associated with Fragile X premutation (22, 39). These reports suggest a possible role of the Fragile X premutation in SCAD pathogenesis through different mechanisms. First, Fragile X Messenger Ribonucleoprotein (FMRP) mild deficiency influences extracellular matrix (ECM) degradation contributing to vasal dissection. Its diminished level leads to abnormal connective tissue structure including shortened and fragmented elastin fibers (21). Moreover, the FMR1 premutation could affect the component of the elastin matrix in connective tissue. Second, the fragile X premutation causes an elevation of the amount of mRNA transcript, which is known to be linked to mRNA toxicity through the increase of RNA binding proteins bound to RNA. Among these, the sequestration of DROSHA/DGCR8 has the effect of downregulating miRNAs, essential for maintaining cellular normal functions (40–42). Endothelial cells may be more sensitive to miRNA dysregulation than other tissues (43). Furthermore, women with premutation may have many medical issues related to their condition, such as hormone replacement therapy for ovarian insufficiency (FXPOI), hypertension, and psychological stress, which overlap with known risk factors for SCAD (35, 44, 45).
In our study, we identified two sisters with FMR1 premutation at the lower limit of the range. The definition of the number of repeats associated with disease manifestations is traditionally related to intellectual disability. Historically, the limit between intermediate allele and premutation FMR1 allele was defined considering the risk of expansion to full mutation. Novel interest in other clinical aspects such as cardiovascular involvement is now emerging, and the border of these intervals could be reevaluated when these aspects will be taken into consideration. The role of the premutation FMR1 allele in the severity of our patient's clinical picture deserves further investigations. Recently, four candidate miRNAs have been identified as promising biomarkers for acute SCAD: miR-let-7f-5p, miR-146a-5p, miR-151a-3p, and miR-223-5p. These miRNAs were significantly expressed in the cohort of SCAD patients. The target genes of these four miRNAs are associated with vascular biology and TGF-beta signaling (20). To further support the role of miRNA dysregulation in SCAD pathogenesis, we found a de novo variant in DROSHA or ribonuclease III nuclear (RNASEN, OMIM*608828), an essential microRNA processing enzyme engaged in the process for maturing microRNA. miRNA biogenesis is initiated by transcription with RNA polymerase II (46) and their primary transcripts (pri-miRNAs) that harbor a local hairpin structure then cropped by a nuclear RNase III, DROSHA, into ∼70 nt precursor-miRNAs (pre-miRNAs). DROSHA works in a microcomplex nature in the nucleus and processes pre-miRNAs into precursor miRNA that contains a dsRNA-binding protein, DGCR8, required for vascular development (Figure 1). It has been shown that DGCR8 binds preferentially to expansion of CGG repeats in the 5′ UTR of FMR1 gene, thus showing a link between miRNAs regulation, Fragile X Syndrome, and vascular development. DROSHA, by processing miRNA, regulates vascular development and homeostasis (41). Therefore, the loss or reduction of ubiquitously expressed DROSHA may predispose to vascular damages. It is demonstrated that DROSHA is essential for fetal and postnatal endothelial development and promotes vascular development and homeostasis in vertebrates (43). This finding suggests the potential involvement of DROSHA in human vascular disorders. Interestingly, missense variants of DROSHA mediate primarily vascular abnormalities in humans. Since DROSHA is involved in vascular homeostasis, its role in determining SCAD events should be taken into consideration together with miRNA regulation. If confirmed, this may suggest experimenting treatments with miRNA mimics or antagonists to improve clinical features in patients with vascular abnormalities.
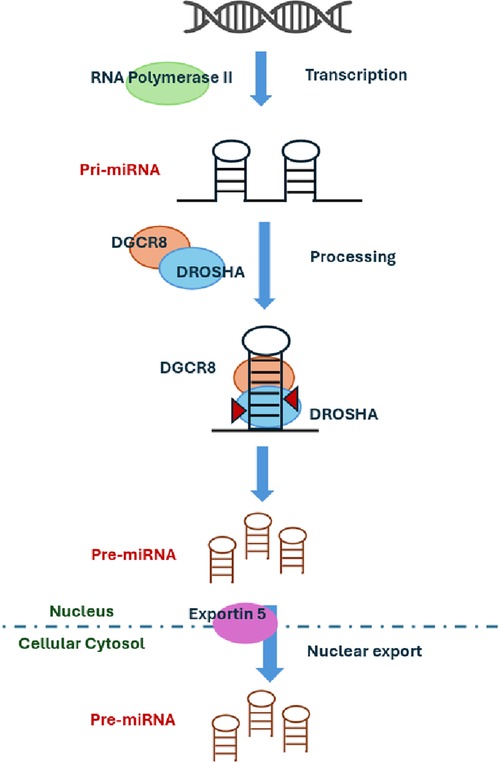
Figure 1. The role of DROSHA in miRNA production.
4.2 Role of collagen
Robust evidence showed the involvement of 10 collagen genes among the ECM structural constituent: the most common genes with interesting variants were COL3A1, COL5A1, and COL4A1 (14, 23, 47, 48).
In our cohort we identified 11 rare variants (18% LP/P) in collagen genes. Pathogenetic variants were found in COL3A1 and COL1A2 genes (Table 2).
The role of collagen genes (especially COL3A1 and COL5A1) in arterial dissection and FMD has been described already (47, 48), and spontaneous dissection has been demonstrated in a murine model (47).
COL3A1 associated with vEDs encodes for proα1chain of type 3 collagen, and it is the most associated with arterial dissections and/or ruptures of variable severity (47, 49). Our patient with a COL3A1 variant presents only vascular signs. In fact, not only the vEDs but also a less severe phenotypic spectrum has been described in the last few years in association with COL3A1 variants (50–53).
The COL1A2 gene, classically linked with osteogenesis imperfecta and Ehler-Danlos syndrome (EDS), has been recently associated with hereditary thoracic aortic aneurysm and dissection disease complex (TAAD), suggesting a genetic overlap between SCAD and TAAD (54).
Moreover, a VUS variant was observed in COL4A1 gene. COL4A1 was already described as associated with a wide spectrum of small-vessel brain disease including porencephaly, variably associated with eye defects and systemic findings (kidney involvement, muscle cramps, cerebral aneurysms, Raynaud phenomenon, cardiac arrhythmia, and hemolytic anemia). Our patients did not show any of these signs.
The role of collagen genes is significant even in our smaller cohort (detection rate 2/15, 13%), highlighting the indication to analyze collagen pathways in SCAD patients for the important implications in their surveillance.
Therefore, our findings confirm the role of collagen genes in SCAD pathogenesis even in patients not fulfilling the clinical diagnostic criteria.
4.3 Extracellular matrix and role of cell-to-cell interaction
ECM is a complex and dynamic structure involved in the physiological and pathological processes of the cardiovascular system. The cardiomyocytes are interconnected by the ECM to form a multicellular syncytium that allows the coordinated myocardial excitation and contraction. Collagen is the main component of heart ECM and its quantitative/qualitative alterations have effects on myocardial contraction, relaxation, diastolic stiffness, and electrical conduction. ECM composes the vascular structure and provides endothelial functions such as the expression of adhesion molecules, the formation of tight junctions, and the endothelial metabolism. During pregnancy, ECM is an important component of blood vessel remodeling with a potential role in cardiovascular disease, such as P-SCAD. Moreover, the ECM plays a significant role in the inflammatory system, contributing to vascular pathologies, as observed in SCAD (7, 55, 56).
An LP variant in SMAD3 was observed in one patient in our cohort. The SMAD family proteins are a group of intracellular signal transducer proteins like the gene products of the Drosophila gene “mothers against decapentaplegic” (Mad) and the Caenorhabditis elegans gene Sma. SMAD3 functions in the transforming growth factor-beta signaling pathway and transmits signals from the cell surface to the nucleus, regulating gene activity and cell proliferation. This protein forms a complex with other SMAD proteins and binds DNA, functioning both as a transcription factor and tumor suppressor. Variants in this gene are associated with aneurysms-osteoarthritis syndrome and LDS3. Our patient with SMAD3 variant presented, at follow-up, mild signs of LDS, as described above.
Several studies found enrichment in rare variants in PKD1 gene, both PV and VUS, in SCAD patients with and without kidney involvement (14, 17, 27, 47, 57, 58). PKD1 encodes a membrane protein, polycystin-1, involved in cell-to-cell or cell-to-matrix interactions. This protein, besides its well-known role in kidney disease, is implicated in the structural integrity of blood vessels with a potential role in SCAD pathogenesis (17): VUS in PKD1 were found in heterozygous form in 14% of our patients, without any kidney involvement.
MYH11 is one of the four genes associated with the dysregulation of smooth muscle cells contractile function (ACTA2, MYH11, MYLK, and PRKG1) (59, 60).
MYH11 encodes the smooth muscle–specific myosin heavy chain, and its genetic variants in patients with arterial dissections demonstrate its role in inducing structural vascular fragility due to defects in focal adhesion, intercellular adhesion, and the actomyosin network (61, 62).
One of our patients presented a VUS in MYH11. Albeit this finding is intriguing, the role of this gene in SCAD pathogenesis remains to be proven.
TLN1 is a large gene highly expressed in the vasculature, including aorta, tibial, and coronary arteries. It encodes for a protein engaged in focal adhesion and essential for integrin activation. Alterations in the expression of TLN1 in smooth muscle cells have been associated with major risk of human arterial diseases, confirming the importance of talin 1 in vascular integrity (63). A study demonstrated that TLN1 was significantly downregulated in aortic tissue of patients with aortic dissection (64), and another case report showed rare missense variants in TLN1 associated with familial and sporadic SCAD cases (65). Furthermore, a murine model showed that the inactivation of talin 1 leads to defects in angiogenesis (66). A rare variant of TLN1 was found in one of our cases with SCAD, dissection of one of the anterior branches of the duplicated right vertebral artery and positive paternal family history. Therefore, our findings support the role of ECM and the need of investigating this group of genes in SCAD (diagnostic rate 1/15, 6.7%).
5 Conclusion
In our study, we collected data from a small cohort of SCAD patients and, although this is a limited case series, the detection rate of LP/P variants has been higher than expected and previously reported (20%) (17, 47).
This discrepancy can be due to the small sample of our cohort and the inclusion criteria used; however, our results underline the importance of studying the genetics of these patients for the possible diagnosis of unexpected systemic disorders.
These findings, together with the increasing evidence from recent literature, support the idea that nowadays, accurate genetic diagnosis and multidisciplinary assessment are important to improve the management in SCAD patients.
The characterization of the genetic background could change in the future the complex working diagnosis and could personalize patient care, familial ascertainment, and management and enable access to specific treatments. Although in a limited series, our data showed that, due to the large number of the genes involved in SCAD, WES-TRIO is the most powerful approach to identify variants in genes already associated with SCAD and to discover new pathways that can contribute to elucidate the complex genetic architecture of SCAD.
Data availability statement
The datasets presented in this article are not readily available because due to ethical/privacy restrictions. Italian law does not allow the deposit of exome data in a public repository. Furthermore, patients have not authorized us to deposit genetic data. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Comitato Etico Territorale (CET) Liguria. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
MC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. DM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LT: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LP: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – review & editing. MB: Data curation, Formal Analysis, Methodology, Project administration, Software, Writing – review & editing. SP: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. AZ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. FG: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MI: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. PM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1486273/full#supplementary-material
References
1. Matta A, Levai L, Elbaz M, Nader V, Parada FC, Carrié D, et al. Spontaneous coronary artery dissection: a review of epidemiology, pathophysiology and principles of management. Curr Probl Cardiol. (2023) 48:101682. doi: 10.1016/j.cpcardiol.2023.101682
2. Van Damme A, McDermott S, McMurtry S, Kung JY, Gyenes G, Norris C. Secondary prevention and rehabilitation for spontaneous coronary artery dissection: a systematic review. Can J Cardiol. (2023) 39:S395–411. doi: 10.1016/j.cjca.2023.08.013
3. Gulati R, Behfar A, Narula J, Kanwar A, Lerman A, Cooper L, et al. Acute myocardial infarction in young individuals. Mayo Clin Proc. (2020) 95:136–56. doi: 10.1016/j.mayocp.2019.05.001
4. Gurgoglione FL, Rizzello D, Giacalone R, Ferretti M, Vezzani A, Pfleiderer B, et al. Precipitating factors in patients with spontaneous coronary artery dissection: clinical, laboratoristic and prognostic implications. Int J Cardiol. (2023) 385:1–7. doi: 10.1016/j.ijcard.2023.05.027
5. Camacho Freire SJ, Díaz Fernández JF, Gheorghe LL, Gómez Menchero AE, León Jiménez J, Roa Garrido J, et al. Spontaneous coronary artery dissection and hypothyroidism. Revista Española de Cardiología. (2019) 72:625–33. doi: 10.1016/j.rec.2018.06.031
6. Stanojevic D, Apostolovic S, Kostic T, Mitov V, Kutlesic-Kurtovic D, Kovacevic M, et al. A review of the risk and precipitating factors for spontaneous coronary artery dissection. Front Cardiovasc Med. (2023) 10:1273301. doi: 10.3389/fcvm.2023.1273301
7. Hayes SN, Kim ESH, Saw J, Adlam D, Arslanian-Engoren C, Economy KE, et al. Spontaneous coronary artery dissection: current state of the science: a scientific statement from the American Heart Association. Circulation. (2018) 137:e523–57. doi: 10.1161/CIR.0000000000000564
8. Kok SN, Hayes SN, Cutrer FM, Raphael CE, Gulati R, Best PJM, et al. Prevalence and clinical factors of migraine in patients with spontaneous coronary artery dissection. J Am Heart Assoc. (2018) 7:e010140. doi: 10.1161/JAHA.118.010140
9. Tweet MS, Gulati R, Hayes SN. What clinicians should know about spontaneous coronary artery dissection. Mayo Clin Proc. (2015) 90:1125–30. doi: 10.1016/j.mayocp.2015.05.010
10. Combaret N, Gerbaud E, Dérimay F, Souteyrand G, Cassagnes L, Bouajila S, et al. National French registry of spontaneous coronary artery dissections: prevalence of fibromuscular dysplasia and genetic analyses. EuroIntervention. (2021) 17:508–15. doi: 10.4244/EIJ-D-20-01046
11. Hayes SN, Tweet MS, Adlam D, Kim ESH, Gulati R, Price JE, et al. Spontaneous coronary artery dissection. J Am Coll Cardiol. (2020) 76:961–84. doi: 10.1016/j.jacc.2020.05.084
12. Adlam D, Berrandou T-E, Georges A, Nelson CP, Giannoulatou E, Henry J, et al. Genome-wide association meta-analysis of spontaneous coronary artery dissection identifies risk variants and genes related to artery integrity and tissue-mediated coagulation. Nat Genet. (2023) 55:964–72. doi: 10.1038/s41588-023-01410-1
13. Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers–Danlos syndrome (EDS type IV). Genet Med. (2014) 16:881–8. doi: 10.1038/gim.2014.72
14. Kaadan MI, MacDonald C, Ponzini F, Duran J, Newell K, Pitler L, et al. Prospective cardiovascular genetics evaluation in spontaneous coronary artery dissection. Circ Genom Precis Med. (2018) 11:e001933. doi: 10.1161/CIRCGENETICS.117.001933
15. Krittanawong C, Kumar A, Johnson KW, Luo Y, Yue B, Wang Z, et al. Conditions and factors associated with spontaneous coronary artery dissection (from a national population-based cohort study). Am J Cardiol. (2019) 123:249–53. doi: 10.1016/j.amjcard.2018.10.012
16. Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers–Danlos syndrome type IV, the vascular type. N Engl J Med. (2000) 342:673–80. doi: 10.1056/NEJM200003093421001
17. Carss KJ, Baranowska AA, Armisen J, Webb TR, Hamby SE, Premawardhana D, et al. Spontaneous coronary artery dissection. Circ Genom Precis Med. (2020) 13(6). doi: 10.1161/CIRCGEN.120.003030
18. Weldy CS, Murtha R, Kim JB. Dissecting the genomics of spontaneous coronary artery dissection. Circ Genom Precis Med. (2022) 15:e003867. doi: 10.1161/CIRCGEN.122.003867
19. Bauersachs J, Thum T. Biogenesis and regulation of cardiovascular MicroRNAs. Circ Res. (2011) 109:334–47. doi: 10.1161/CIRCRESAHA.110.228676
20. Lozano-Prieto M, Adlam D, García-Guimaraes M, Sanz-García A, Vera-Tomé P, Rivero F, et al. Differential miRNAs in acute spontaneous coronary artery dissection: pathophysiological insights from a potential biomarker. EBioMedicine. (2021) 66:103338. doi: 10.1016/j.ebiom.2021.103338
21. Tassanakijpanich N, Cohen J, Cohen R, Srivatsa UN, Hagerman RJ. Cardiovascular problems in the fragile X premutation. Front Genet. (2020) 11:586910. doi: 10.3389/fgene.2020.586910
22. Butler MG, Hossain WA, Steinle J, Gao H, Cox E, Niu Y, et al. Connective tissue disorders and fragile X molecular status in females: a case series and review. Int J Mol Sci. (2022) 23:9090. doi: 10.3390/ijms23169090
23. Tarr I, Hesselson S, Iismaa SE, Rath E, Monger S, Troup M, et al. Exploring the genetic architecture of spontaneous coronary artery dissection using whole-genome sequencing. Circ Genom Precis Med. (2022) 15:e003527. doi: 10.1161/CIRCGEN.121.003527
24. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
25. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho Y-Y, et al. Sherloc: a comprehensive refinement of the ACMG–AMP variant classification criteria. Genet Med. (2017) 19:1105–17. doi: 10.1038/gim.2017.37
26. Pezzoli L, Pezzani L, Bonanomi E, Marrone C, Scatigno A, Cereda A, et al. Not only diagnostic yield: whole-exome sequencing in infantile cardiomyopathies impacts on clinical and family management. J Cardiovasc Dev Dis. (2022) 9:2. doi: 10.3390/jcdd9010002
27. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. (2012) 28:2747–54. doi: 10.1093/bioinformatics/bts526
28. Biancalana V, Glaeser D, McQuaid S, Steinbach P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur J Hum Genet. (2015) 23:417–25. doi: 10.1038/ejhg.2014.185
29. Spector E, Behlmann A, Kronquist K, Rose NC, Lyon E, Reddi HV. Laboratory testing for fragile X, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2021) 23:799–812. doi: 10.1038/s41436-021-01115-y
30. Jakob M, Spasojevic D, Krogmann ON, Wiher H, Hug R, Hess OM. Tortuosity of coronary arteries in chronic pressure and volume overload. Cathet Cardiovasc Diagn. (1996) 38:25–31. doi: 10.1002/(SICI)1097-0304(199605)38:1%3C25::AID-CCD7%3E3.0.CO;2-5
31. Byrne RA, Rossello X, Coughlan JJ, Barbato E, Berry C, Chieffo A, et al. 2023 ESC guidelines for the management of acute coronary syndromes. Eur Heart J. (2023) 44:3720–826. doi: 10.1093/eurheartj/ehad191
32. Tweet MS, Eleid MF, Best PJM, Lennon RJ, Lerman A, Rihal CS, et al. Spontaneous coronary artery dissection. Circ Cardiovasc Interv. (2014) 7:777–86. doi: 10.1161/CIRCINTERVENTIONS.114.001659
33. Bocchino PP, Angelini F, Franchin L, D’Ascenzo F, Fortuni F, De Filippo O, et al. Invasive versus conservative management in spontaneous coronary artery dissection: a meta-analysis and meta-regression study. Hellenic J Cardiol. (2021) 62:297–303. doi: 10.1016/j.hjc.2021.02.013
34. Lettieri C, Zavalloni D, Rossini R, Morici N, Ettori F, Leonzi O, et al. Management and long-term prognosis of spontaneous coronary artery dissection. Am J Cardiol. (2015) 116:66–73. doi: 10.1016/j.amjcard.2015.03.039
35. Turley TN, O’Byrne MM, Kosel ML, de Andrade M, Gulati R, Hayes SN, et al. Identification of susceptibility loci for spontaneous coronary artery dissection. JAMA Cardiol. (2020) 5:929. doi: 10.1001/jamacardio.2020.0872
36. Sun Y, Chen Y, Li Y, Li Z, Li C, Yu T, et al. Association of TSR1 variants and spontaneous coronary artery dissection. J Am Coll Cardiol. (2019) 74:167–76. doi: 10.1016/j.jacc.2019.04.062
37. Grond-Ginsbach C, Böckler D, Newton-Cheh C. Pathogenic TSR1 gene variants in patients with spontaneous coronary artery dissection. J Am Coll Cardiol. (2019) 74:177–8. doi: 10.1016/j.jacc.2019.06.005
38. Li Y, Fang M, Yang J, Yu C, Kuang J, Sun T, et al. Analysis of the contribution of 129 candidate genes to thoracic aortic aneurysm or dissection of a mixed cohort of sporadic and familial cases in South China. Am J Transl Res. (2021) 13(5):4281–95.34150014
39. McKenzie FJ, Tassanakijpanich N, Epps KC, March SK, Hagerman RJ. Spontaneous coronary artery dissection in females with the fragile X FMR1 premutation. JACC Case Rep. (2020) 2:40–4. doi: 10.1016/j.jaccas.2019.11.058
40. Garcia-Arocena D, Hagerman PJ. Advances in understanding the molecular basis of FXTAS. Hum Mol Genet. (2010) 19:R83–9. doi: 10.1093/hmg/ddq166
41. Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. (2013) 3:869–80. doi: 10.1016/j.celrep.2013.02.004
42. Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. (2013) 12:786–98. doi: 10.1016/S1474-4422(13)70125-X
43. Jiang X, Wooderchak-Donahue WL, McDonald J, Ghatpande P, Baalbaki M, Sandoval M, et al. Inactivating mutations in Drosha mediate vascular abnormalities similar to hereditary hemorrhagic telangiectasia. Sci Signal. (2018) 11:513. doi: 10.1126/scisignal.aan6831
44. Verstraeten A, Perik MHAM, Baranowska AA, Meester JAN, Van Den Heuvel L, Bastianen J, et al. Enrichment of rare variants in Loeys–Dietz syndrome genes in spontaneous coronary artery dissection but not in severe fibromuscular dysplasia. Circulation. (2020) 142:1021–4. doi: 10.1161/CIRCULATIONAHA.120.045946
45. MacKenzie J, Sumargo I, Taylor S. A cryptic full mutation in a male with a classical fragile X phenotype. Clin Genet. (2006) 70:39–42. doi: 10.1111/j.1399-0004.2006.00634.x
46. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. (2003) 425:415–9. doi: 10.1038/nature01957
47. Zekavat SM, Chou EL, Zekavat M, Pampana A, Paruchuri K, Lino Cardenas CL, et al. Fibrillar collagen variants in spontaneous coronary artery dissection. JAMA Cardiol. (2022) 7:396. doi: 10.1001/jamacardio.2022.0001
48. Richer J, Hill HL, Wang Y, Yang M-L, Hunker KL, Lane J, et al. A novel recurrent COL5A1 genetic variant is associated with a dysplasia-associated arterial disease exhibiting dissections and fibromuscular dysplasia. Arterioscler Thromb Vasc Biol. (2020) 40:2686–99. doi: 10.1161/ATVBAHA.119.313885
49. Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, et al. Diagnosis, natural history, and management in vascular Ehlers–Danlos syndrome. Am J Med Genet C Semin Med Genet. (2017) 175:40–7. doi: 10.1002/ajmg.c.31553
50. Ghali N, Baker D, Brady AF, Burrows N, Cervi E, Cilliers D, et al. Atypical COL3A1 variants (glutamic acid to lysine) cause vascular Ehlers–Danlos syndrome with a consistent phenotype of tissue fragility and skin hyperextensibility. Genet Med. (2019) 21:2081–91. doi: 10.1038/s41436-019-0470-9
51. Ruscitti F, Trevisan L, Rosti G, Gotta F, Cianflone A, Geroldi A, et al. A novel mutation in COL3A1 associates to vascular Ehlers–Danlos syndrome with predominant musculoskeletal involvement. Mol Genet Genomic Med. (2021) 9:e1753. doi: 10.1002/mgg3.1753
52. Subramaniam SR, Yeung LF, Choy LYL, Kwok JSS. Vascular Ehlers-Danlos syndrome: a null COL3A1 variant found in a patient with loin pain without marked cutaneous features (case report). Clin Case Rep. (2024) 12:e8517. doi: 10.1002/ccr3.8517
53. Valencia-Cifuentes V, Sinisterra-Díaz SE, Quintana-Peña V, Folleco E, Nastasi-Catanese JA, Pachajoa H, et al. Case report: a novel COL3A1 variant in a Colombian patient with isolated cerebrovascular involvement in vascular Ehlers-Danlos syndrome. Front Med (Lausanne). (2024) 11:1304168. doi: 10.3389/fmed.2024.1304168
54. Jeoffrey SMH, Kalyanasundaram A, Zafar MA, Ziganshin BA, Elefteriades JA. Genetic overlap of spontaneous dissection of either the thoracic aorta or the coronary arteries. Am J Cardiol. (2023) 205:69–74. doi: 10.1016/j.amjcard.2023.07.046
55. Grandoch M, Bollyky PL, Fischer JW. Hyaluronan. Circ Res. (2018) 122:1341–3. doi: 10.1161/CIRCRESAHA.118.312522
56. Çanga Y. Systemic inflammatory activation in patients with acute coronary syndrome secondary to nonatherosclerotic spontaneous coronary artery dissection. North Clin Istanb. (2017) 5(3):186–94. doi: 10.14744/nci.2017.59244
57. Klingenberg-Salachova F, Limburg S, Boereboom F. Spontaneous coronary artery dissection in polycystic kidney disease. Clin Kidney J. (2012) 5:44–6. doi: 10.1093/ndtplus/sfr158
58. Saw J, Yang M-L, Trinder M, Tcheandjieu C, Xu C, Starovoytov A, et al. Chromosome 1q21.2 and additional loci influence risk of spontaneous coronary artery dissection and myocardial infarction. Nat Commun. (2020) 11:4432. doi: 10.1038/s41467-020-17558-x
59. Zhu L, Vranckx R, Van Kien PK, Lalande A, Boisset N, Mathieu F, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. (2006) 38:343–9. doi: 10.1038/ng1721
60. Wang L, Guo D, Cao J, Gong L, Kamm KE, Regalado E, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. (2010) 87:701–7. doi: 10.1016/j.ajhg.2010.10.006
61. Erhart P, Körfer D, Dihlmann S, Qiao J-L, Hausser I, Ringleb P, et al. Multiple arterial dissections and connective tissue abnormalities. J Clin Med. (2022) 11:3264. doi: 10.3390/jcm11123264
62. Negishi K, Aizawa K, Shindo T, Suzuki T, Sakurai T, Saito Y, et al. An Myh11 single lysine deletion causes aortic dissection by reducing aortic structural integrity and contractility. Sci Rep. (2022) 12:8844. doi: 10.1038/s41598-022-12418-8
63. Li Y, Gao S, Han Y, Song L, Kong Y, Jiao Y, et al. Variants of focal adhesion scaffold genes cause thoracic aortic aneurysm. Circ Res. (2021) 128:8–23. doi: 10.1161/CIRCRESAHA.120.317361
64. Wei X, Sun Y, Wu Y, Zhu J, Gao B, Yan H, et al. Downregulation of Talin-1 expression associates with increased proliferation and migration of vascular smooth muscle cells in aortic dissection. BMC Cardiovasc Disord. (2017) 17:162. doi: 10.1186/s12872-017-0588-0
65. Turley TN, Theis JL, Sundsbak RS, Evans JM, O’Byrne MM, Gulati R, et al. Rare missense variants in TLN1 are associated with familial and sporadic spontaneous coronary artery dissection. Circ Genom Precis Med. (2019) 12:e002437. doi: 10.1161/CIRCGEN.118.002437
Keywords: whole exome sequencing (WES), DROSHA, connective tissue disorder, miRNA, spontaneous coronary artery dissection (SCAD), Fragile X premutation
Citation: Casula M, Marchetti D, Trevisan L, Pezzoli L, Bellini M, Patrone S, Zingarelli A, Gotta F, Iascone M and Mandich P (2024) Genetics architecture of spontaneous coronary artery dissection in an Italian cohort. Front. Cardiovasc. Med. 11:1486273. doi: 10.3389/fcvm.2024.1486273
Received: 25 August 2024; Accepted: 1 November 2024;
Published: 25 November 2024.
Edited by:
Tommaso Gori, Johannes Gutenberg University Mainz, GermanyReviewed by:
Pavol Tomasov, Liberec Regional Hospital, CzechiaCopyright: © 2024 Casula, Marchetti, Trevisan, Pezzoli, Bellini, Patrone, Zingarelli, Gotta, Iascone and Mandich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucia Trevisan, bHVjaWEudHJldmlzYW5AaHNhbm1hcnRpbm8uaXQ=
†These authors share first authorship