 Sonia Benitez
Sonia Benitez Núria Puig1,†
Núria Puig1,† Pol Camps-Renom
Pol Camps-Renom José Luis Sánchez-Quesada
José Luis Sánchez-Quesada- 1Cardiovascular Biochemistry Group, Institut de Recerca Hospital de Sant Pau (IR Sant Pau), Barcelona, Spain
- 2CIBER-Diabetes and Metabolic Diseases (CIBERDEM), Madrid, Spain
- 3Stroke Unit, Department of Neurology, Hospital de La Santa Creu I Sant Pau, IR Sant Pau, Barcelona, Spain
The fundamental role of qualitative alterations of lipoproteins in the early development of atherosclerosis has been widely demonstrated. Modified low-density lipoproteins (LDL), such as oxidized LDL (oxLDL), small dense LDL (sdLDL), and electronegative LDL [LDL(-)], are capable of triggering the atherogenic process, favoring the subendothelial accumulation of cholesterol and promoting inflammatory, proliferative, and apoptotic processes characteristic of atherosclerotic lesions. In contrast, high-density lipoprotein (HDL) prevents and/or reverses these atherogenic effects. However, LDL's atherogenic and HDL's anti-atherogenic actions may result altered in certain pathological conditions. The molecular mechanisms underlying the impaired effects of altered lipoproteins have been studied in numerous in vitro and in vivo studies, and have been extensively analyzed in coronary atherosclerosis, especially in the context of pathologies such as dyslipidemia, diabetes, obesity, and metabolic syndrome. However, the corresponding studies are scarcer in the field of ischemic stroke, despite carotid arteriosclerosis progression underlies at least 20% of ischemic strokes. The present review relates qualitative alterations of LDL and HDL with the development of carotid arteriosclerosis and the occurrence of ischemic stroke.
1 Atherosclerotic cardiovascular disease
Cardiovascular (CV) diseases, responsible for more than 17.5 million deaths per year worldwide, represent a health and socioeconomic problem of huge magnitude. Increased CV disease prevalence in recent decades is largely due to the higher incidence of CV risk-associated pathologies, such as diabetes, dyslipidemia, obesity, and hypertension, with population ageing, unhealthy eating habits, and sedentary lifestyles undoubtedly acting as contributing factors (1).
Early development of atherosclerosis is a main underlying cause of CV disease, resulting in a condition known as atherosclerotic CV disease (ASCVD). Atherosclerosis is defined as a thickening and loss of elasticity in the arterial wall of large- and medium-sized arteries, and particularly in zones of curvature or bifurcation with turbulent flows, leading to a narrowing (stenosis) of the vascular lumen (2). Low-shear stress zones are susceptible to atherosclerotic plaque development, with rupture eventually leading to reduced blood flow and the onset of CV events (3). Vessels more prone to developing atherosclerosis are the abdominal aorta, and peripheral, coronary, and carotid arteries, leading, respectively, to the following forms of ASCVD: aortic atherosclerotic disease, peripheral artery disease (claudication), coronary heart disease (myocardial infarction, angina pectoris), and cerebrovascular disease (transient ischemic attack, ischemic stroke).
Atherosclerosis is a long-term process that is initially triggered by lipoprotein entry and retention in the artery wall, leading to intracellular and extracellular lipid accumulation in the subendothelial space. The progressive and slow deposition of lipids, occurring in parallel with an inflammatory response and monocyte recruitment from circulating blood to the arterial intima, together result in a narrowing of the artery wall, referred to as atherosclerotic plaque (2).
Low-density lipoprotein (LDL) plays a major role in different stages of the atherosclerotic plaque formation (Figure 1) (4). LDL entry to the microenvironment of the subendothelial space favors chemical modifications resulting from oxidative stress and the actions of lipolytic and proteolytic enzymes. Modified LDL, with the acquired pro-atherogenic properties, favors subendothelial retention and induces endothelial dysfunction, the recruitment of leukocytes with enhanced inflammatory response, the differentiation of monocytes into macrophages, the emergence of apoptotic processes, and the formation of lipid-loaded foam cells (5–7). To attract leukocytes to the lesioned area, modified LDL promotes the expression of adhesion molecules and chemokines by endothelial cells. In monocyte-derived macrophages, modified LDL promotes the release of more chemokines and cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1β), in addition to growth factors. This response contributes to the proliferation and activation of smooth muscle cells (SMCs), which together with macrophages, uptake modified LDL becoming lipid-loaded foam cells, a hallmark of atherosclerosis (8, 9).
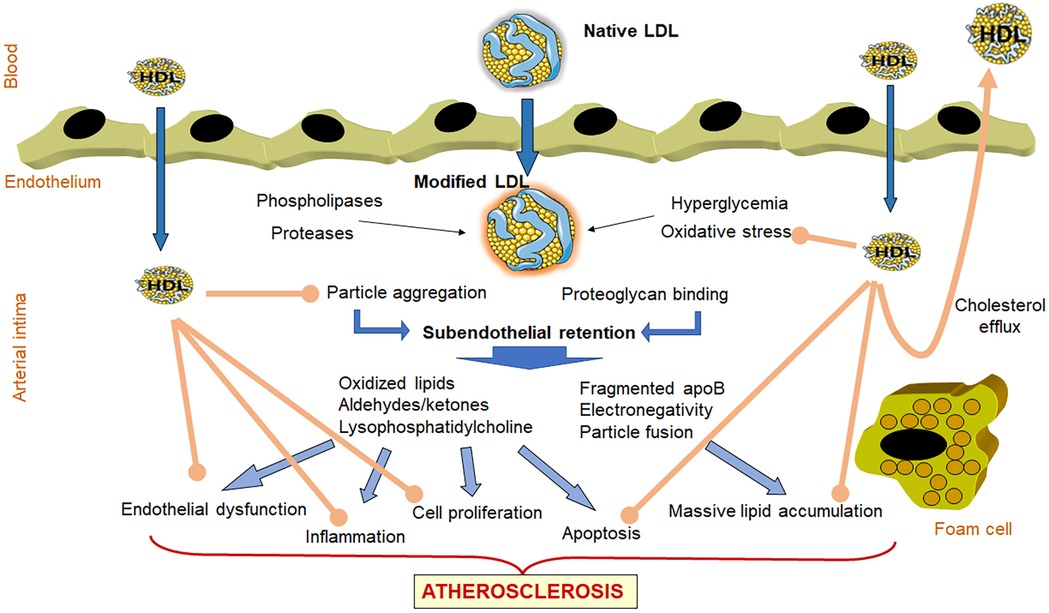
Figure 1. Role of LDL and HDL in atherogenesis. LDL entry to the microenvironment of the subendothelial space favors chemical modifications resulting from oxidative stress and the actions of lipolytic and proteolytic enzymes. These modifications favor LDL aggregation and increased binding to proteoglycans, facilitating its subendothelial retention. Retained modified LDL particles induce endothelial dysfunction, the recruitment of leukocytes with enhanced inflammatory response, the differentiation of monocytes into macrophages, the emergence of apoptotic processes, and the formation of lipid-loaded foam cells. Together, all of these events lead to the development of the atherosclerotic plaque. The atherogenic effects of LDL are indicated with blue arrows. In contrast, HDL particles that enter in the arterial wall can prevent LDL oxidation and aggregation, and display anti-inflammatory, antiproliferative and antiapoptotic properties (brown arrows). In addition, HDL induces the efflux of cholesterol from lipid-loaded foam cells and returning the excess of cholesterol to blood circulation for its metabolization in the liver.
Contrasting with LDL, high-density lipoprotein (HDL) is considered athero-protective (Figure 1), owing to its capacity to induce cholesterol efflux from tissues (10, 11) and to its anti-oxidant and anti-inflammatory properties (12). Of note, HDL's effects include the inhibition of LDL modification and mitigation of the atherogenic effects of modified LDL (12, 13).
The contribution of LDL cholesterol (LDL-C) to CV risk has been extensively studied, given its role in progression of atherosclerosis (14), which contrasts with the inverse association with vascular risk of HDL cholesterol (HDL-C) (15). However, the fact that therapies to regulate HDL-C or LDL-C levels do not eliminate vascular risk would suggest that, over and above concentration levels, the actual quality of lipoproteins is crucial to determining susceptibility to CV disease development.
2 Ischemic stroke and blood lipoprotein levels
Stroke represents the third cause of mortality in western countries and is a leading cause of disability and dementia worldwide. Furthermore, its impact is aggravated by the fact that around 20% of patients experience recurrence within 5 years of follow-up, increasing the risk of severe disability (16). Around 80% of strokes are ischemic in origin (17), and in approximately 20% of those cases, the cause is large-vessel atherosclerosis (18), referred to as atherothrombotic stroke (19). Plaques in the internal carotid artery are most frequently involved in this subtype (20), with stroke triggered by plaque rupture leading to thrombus formation and subsequent distal embolism (3), and/or by hemodynamic insufficiency, possibly attributable to progressive vessel occlusion caused by atherosclerotic plaque development (21).
The distribution of the modifiable vascular risk factors differs according to the type of stroke and the patient's sex and age (22). Stroke incidence is higher in women than in men for those younger than 30 years, whereas rates are higher in men during midlife. In subjects older than 80 years the incidence is slightly higher in women. Regarding carotid stenosis, women have a higher degree of stenosis, but less carotid plaque area (23) and less vulnerable plaques with lower risk than men (24, 25).
In atherothrombotic stroke, the recurrence risk of major vascular events is much higher than for other stroke subtypes (26), for which reason secondary prevention is essential. The main therapeutic approach is carotid endarterectomy, implemented according to the degree of stenosis (27). In addition to luminal narrowing, the inflammatory state and lipid content of the plaque are considered key factors in determining carotid plaque vulnerability. As explained above, lipoproteins play a key role in the progression of atherosclerosis, with LDL—most particularly when modified in the arterial wall—acting as a stimulus that strongly promotes inflammation and lipid accumulation (5).
Although elevated serum lipids and lipoproteins have been extensively associated with CV disease, there is little information regarding cerebrovascular disease. However, coronary patients are reported to have an increased risk of stroke (28), and the relationship between lipids/lipoproteins and ischemic stroke, and particularly atherothrombotic stroke, has been addressed in several epidemiological studies.
High plasma levels of total cholesterol, and particularly of LDL-C, have been associated with atherothrombotic stroke (29, 30) and progressive carotid stenosis (31). In line with those findings, lipid-lowering therapy to decrease total cholesterol and LDL-C levels has been shown to mitigate the progression of carotid atherosclerosis (32, 33). According to the SPARCL study (Stroke Prevention by Aggressive Reduction in Cholesterol Levels), high-dose statins reduce the overall occurrence of strokes and CV events, with only a slight increase in the rate of hemorrhagic strokes (34). Likewise, PCSK9 inhibitors have been reported to reduce the incidence of stroke by 25% (35). Although some studies have pointed to an association between aggressive LDL-C-lowering therapies and an increased risk of hemorrhagic stroke, this association has not been conclusively demonstrated (36).
Notably, Sniderman et al. (37) argued that actual quantification of LDL particles or of apolipoprotein (apo) B was more important than LDL-C plasma levels in coronary artery disease. For ischemic stroke, the risk attributable to elevated apoB or non-HDL-C has been reported to be higher than the risk attributable to elevated LDL-C (38).
Elevated levels of lipoprotein(a) [Lp(a)] are postulated to contribute to so-called residual CV risk, i.e., risk not directly derived from elevated LDL-C (39, 40). Lp(a) consists of an LDL molecule that also contains the apolipoprotein (a) [apo(a)], covalently linked to apoB-100. Apo(a) is formed by multiple repetitions of structures called kringles, and depending on the number of repetitions differ in the CV risk, having small apo(a) isoforms a greater CV risk compared to larger isoforms (41). Higher Lp(a) concentrations are independently associated with long-term ASCVD risk and may amplify CV risk when concomitant with carotid plaque (42). Referring specifically to ischemic stroke, Lp(a) may play an especially relevant role, since its antifibrinolytic action may be determinant in the surge of thrombotic processes leading to an ischemic event (43, 44). Besides this antifibrinolytic action, the atherogenic effect of Lp(a) seems to be related to its ability to bind highly inflammatory oxidized phospholipids. These phospholipids include a soluble fraction in the surface of the lipoprotein particle and a fraction of these phospholipids covalently bound to apo(a) (45, 46). Indeed, Lp(a) is the main plasma transporter of oxidized phospholipids (47). Consequently, Lp(a) could play a role in the oxidative stress associated with the development of arteriosclerosis (48) and, therefore, be related to the oxidative modifications of LDL described in the following sections, which play a prominent role in the early appearance of ischemic events. Elevated Lp(a) levels have been associated with a high incidence of ischemic stroke in most (49–51)—but not all (52)—studies, and have also been associated with a greater vascular event recurrence risk in patients with acute first-ever ischemic stroke (53) and with vulnerable carotid plaque and plaque development (54, 55). The association of Lp(a) with carotid plaque vulnerability is sex-specific, since it was associated with presence of intraplaque hemorrhage in women and with stenosis degree in men (54). A recent meta-analysis concluded that increased Lp(a) concentrations could be considered a predictive marker for identifying individuals at risk of developing ischemic stroke (56).
Triglycerides may be independently associated with the risk of ischemic stroke (57), while elevated remnant cholesterol, i.e., the cholesterol transported in triglyceride-rich lipoproteins, has likewise been associated with a higher risk of ischemic stroke in the general population (58). A recent genome-wide association study (GWAS), using Mendelian randomization, has suggested that remnant cholesterol is causally associated with large-artery atherothrombotic stroke (59). Moreover, remnant cholesterol levels in patients with ischemic stroke have been positively associated with carotid artery intima-media thickness (60), with the concentration of triglyceride-rich particles predicting the presence of vulnerable carotid plaque independently of LDL-C (61, 62).
Despite the well-known inverse association between HDL-C levels and coronary heart disease, findings have not been so consistent for stroke. HDL-C levels are weakly or not at all associated with the incidence or prevalence of ischemic events, and do not predict recovery from stroke. However, some studies have shown an inverse association in the specific case of atherothrombotic stroke (63, 64). As occurs with LDL, the HDL particle count is a promising risk prediction parameter. While most studies to date have been single-cohort studies assessing only coronary disease or composite vascular outcomes, a study by Singh et al. (65) has reported that HDL particles is a robust marker for ischemic stroke in the overall population, and also that it inversely correlates with both myocardial infarction and stroke, albeit displaying racial disparities.
Conflicting results regarding the predictive power of lipid and lipoprotein blood levels for stroke may be partly due to the time of blood collection in patients, because, as already described in the 1980s (28), stroke itself exerts a lipid-lowering effect. Growing evidence suggests that—in addition to lipoprotein quantitative parameters—certain lipoprotein component concentrations and functional alterations are relevant to the pathology of ischemic stroke, and particularly to the atherothrombotic subtype.
3 Ischemic stroke and qualitative lipoprotein changes
Despite the association between plasma lipoprotein levels and ASCVD (including atherothrombotic stroke), mounting evidence points to the fact that lipoprotein concentration is not always the key, as the presence of atherosclerosis is not exclusively determined by quantitative lipid parameters such as high LDL-C levels (66). The importance of other lipoprotein-related factors is suggested by subjects with normal LDL-C still having residual CV risk and by a diminished association with LDL-C when adjusting for other lipoproteins (67, 68). Those observations are likely based on the fact that circulating lipoproteins comprise subclasses that are heterogeneous in size, density, composition, and function, and with different involvement in ASCVD.
Alterations in HDL and LDL particle size in ischemic stroke occur parallel to changes in chemical composition, as reported in several studies (69–74). Just beginning to be deciphered is the specific lipid and protein composition of lipoproteins involved in atherothrombotic stroke, with interesting preliminary data obtained from lipidomic and proteomic studies of lipoproteins isolated from ischemic patients. Lepedda et al. (72) showed increased acute-phase serum amyloid A (SAA) levels in all lipoprotein fractions obtained from the patients in their study, while a proteomic study by Finamore et al. (71) revealed that mainly LDL, but also HDL, showed a higher content of proteins associated with inflammation, immunity, and coagulation, and specific protein signatures for patients with hypoechoic plaques. Regarding lipids, a preliminary lipidomic study in lipoproteins isolated from patients who had undergone carotid endarterectomy showed lipid alterations, particularly in specific phospholipids in the LDL of patients with hypoechoic plaques (73). It is very feasible to consider that such physical and chemical alterations in LDL might lead to a higher prevalence of modified LDL particles in ischemic stroke. The role of modified LDL and the influence of altered qualitative properties of HDL, specifically in relation to atherothrombotic stroke, are discussed in the following sections.
3.1 Qualitative LDL changes: modified LDL
The presence of modified LDL is pivotal to determine susceptibility to atherosclerosis and vulnerable lesions. Alterations in LDL catabolism or other chemical processes favor the LDL modification and abnormal LDL particle formation in terms of composition, size, and electric charge (Figure 2). Specifically referring to atherothrombotic stroke patients, Yatsu et al. (75) reported that their monocyte-derived macrophages displayed a reduced ability to scavenge modified LDL. This fact, together with increased levels of modified LDL forms and the impaired protective effects of HDL, may contribute to rapid disease progression. In this context, while the role of oxidized LDL (oxLDL) in ischemic stroke has been extensively evaluated (76), less studied is the importance of a modified form of LDL with a negative charge, called electronegative LDL [LDL(-), also known as L5], found in circulation (77). The roles played by modified LDL in the form of oxLDL and LDL(-), and also by small dense LDL (sdLDL), are discussed below.
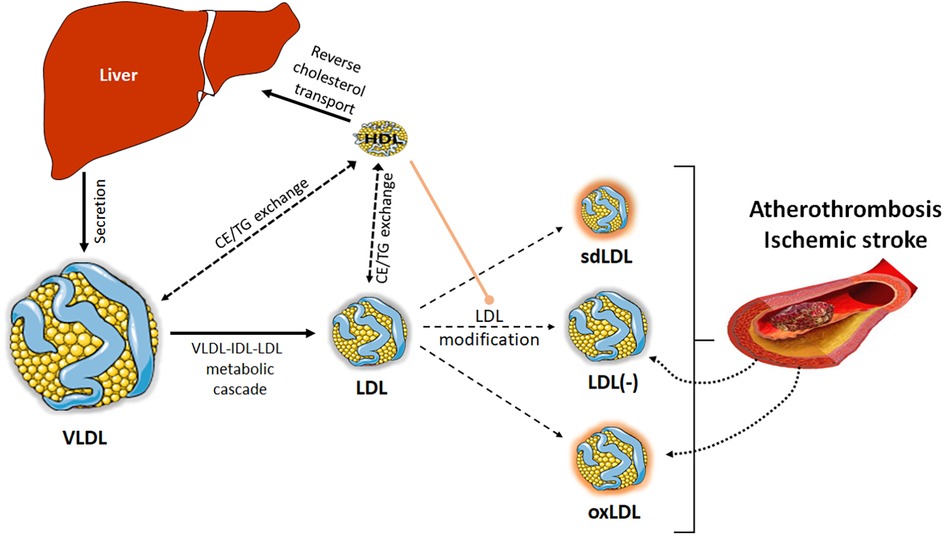
Figure 2. LDL modification. Besides the modification of LDL in the arterial wall, LDL particles can also suffer different alterations in blood, leading to modified LDL particles. Alterations in VLDL-IDL-LDL catabolic cascade or in the lipid transfer processes among lipoproteins can favor the formation of sdLDL or LDL(-). Other processes, such as overload of non-esterified fatty acids or oxidative phenomena occurring in blood can also lead to the formation of LDL(-) or oxLDL. All these modified LDL particles will contribute to the development of atherosclerosis. In turn, the rupture of the atherosclerotic plaque after an ischemic event can release its content of modified lipoproteins, thereby contributing to the plasma pool of LDL(-) and oxLDL. For its part, HDL plays an antiatherogenic role, preventing the formation of modified LDL, through its antioxidant capacity or the ability to capture non-esterified fatty acids.
3.1.1 Oxidized LDL
It is widely accepted that LDL can be modified by oxidation in the subendothelial space, where it is exposed to free radicals and oxidative enzymes released by environment cells. This has led to thinking that oxLDL detected in plasma comes from the subendothelial space after plaque rupture. Accordingly, most studies analyzing oxLDL association with ischemic stroke have reported that oxLDL levels in blood are higher in patients in the acute ischemic phase than in healthy controls (78–81), and are reduced by post-event statin therapy (80, 82). Moreover, for patients with acute ischemic stroke, Wang et al. (83) found a relationship between oxLDL plasma levels and the National Institute of Health Stroke Scale score. Blood concentrations of oxLDL are reported to be particularly increased in patients with large-artery atherosclerosis compared with other stroke subtypes (79, 80, 84), and have also been associated with lacunar stroke in small vessels (85, 86). The concentration of oxLDL in plasma and in the atherosclerotic plaque has been associated with the vulnerability of carotid plaque (87), although no association was found in a recent study (78).
Those studies carried out in the acute ischemic phase do not clarify whether the elevated oxLDL levels are a consequence or cause of the stroke. However, several prospective studies have suggested that, as well as being associated with the acute stroke phase, oxLDL plays a role in the future occurrence of vascular events (88–91) and in 1-year recurrence (92), and is also associated with poorer patient cognitive and functional evolution (93, 94). In symptomatic patients, some studies have related oxLDL with transient ischemic attack (89, 95). High levels of oxLDL and high-sensitivity C-reactive protein (CRP), when combined, are associated with increased risk of recurrent stroke, combined vascular events, and poor functional outcome in patients with minor stroke or transient ischemic attack (94).
Taken together, those studies not only demonstrate a seemingly solid relationship between elevated oxLDL levels and ischemic stroke onset and recurrence, but also point to plasma oxLDL as a putative predictor of stroke in asymptomatic cases.
3.1.2 Electronegative LDL
LDL(-), a heterogeneous entity that encompasses several LDL forms modified by different mechanisms and having an increased negative charge in common, is a minor plasma form of modified LDL with atherogenic properties (96–100). Among other atherogenic properties, LDL(-) has increased susceptibility to aggregate and to bind to proteoglycans, inducing inflammation, apoptosis, and cell proliferation in several arterial wall cell types (101–103). LDL(-), which constitutes about 3%–5% of total LDL in healthy subjects, is present in higher proportions in pathologies associated with vascular risk, including dyslipidemia and diabetes (104), and in subclinical atherosclerosis in association with the degree of carotid stenosis (105, 106). In metabolic syndrome, LDL(-) levels were higher in men than in premenopausal women, which can contribute to their higher predisposition to CV risk (107). LDL(-) is also increased during the acute phase of vascular events such as myocardial infarction (108) and ischemic stroke (78, 109).
Of 2 studies focused on LDL(-) in ischemic stroke, only one was specifically conducted in patients with carotid atherosclerosis (78). Despite a normal lipid profile, that population with recent ischemic stroke showed higher blood levels of LDL(-) and oxLDL than control subjects. The favorable lipid profile may be attributable to the known drop in lipid levels and to the administration of high-dose statins after the ischemic event. Importantly, the proportion of LDL(-), but not of oxLDL, has been associated with carotid plaque features ascribed to vulnerability, particularly the degree of carotid stenosis, hypoechogenicity, and diffuse intraplaque neovascularization (78). This would suggest that LDL(-) may be a marker of plaque vulnerability in ischemic stroke associated with carotid atherosclerosis, as proposed by Shen et al. (109); in the latter study, conducted in patients with acute ischemic stroke, higher values of LDL(-) than in the study of Puig et al. (78) were reported, presumably owing to differences in the population, time of blood extraction, and the chromatographic method of LDL(-) isolation. Interestingly, that study suggested that LDL(-) triggers ischemic stroke by promoting thrombosis, through the induction of platelet aggregation and hemostasis via lectin-like oxidized LDL receptor-1 (LOX-1) and IκB kinase 2 (IKK2)/nuclear factor–κB (NF-κB) signaling (109). The authors suggest that LDL(-)-induced platelet activation promotes their aggregation and αβ amyloid peptide release, leading to increased platelet reactivity and stroke complications.
Interestingly, the roles of LDL(-) and Lp(a) have recently been compared in regard to ASCVD development in a study (110), which indicates that both atherogenic lipoproteins contribute to residual CV risk through different mechanisms.
3.1.3 Small dense LDL
As compared with large LDL particles, prevalence of sdLDL particles is strongly associated with early development of CV disease. While sdLDL, as generated by metabolic alterations in subjects with hypertriglyceridemia (111), cannot be considered as a modified form of LDL, it is closely related to modified LDLs such as oxLDL, glycosylated LDL, and LDL(-) for 2 reasons. First, sdLDL is prone to modification by oxidation or glycosylation, favoring the formation of oxLDL and glycosylated LDL (112, 113). And second, sdLDL has a slightly increased electronegative charge compared with large and intermediate LDL particles, so a part of LDL(-) is made up of sdLDL particles. In fact, LDL(-) is smaller and more dense in normolipidemic and hypertriglyceridemic subjects (114, 115).
In the field of stroke, several studies have associated the prevalence of sdLDL particles with an increased risk of ischemic stroke (74, 116, 117) and with neuroimaging markers of cerebral small vessel disease (118), an association shown to be particularly robust in a large prospective study conducted in the general population (119). Moreover, a recent study has reported that higher sdLDL cholesterol (sdLDL-C) levels are associated with an increased risk of incident carotid plaques and especially vulnerable plaques, even in patients with normal LDL-C values (120). sdLDL has also been associated with poor prognosis after stroke (121).
3.2 Qualitative HDL changes
While the inverse association between HDL-C and coronary heart disease is widely accepted, the causal relation between HDL and atherosclerosis has not yet been fully elucidated. Therapeutic approaches to raising HDL-C have not been as effective as expected in lowering CV risk (122). The hypothesis that HDL-C concentration is the only factor determining the beneficial role of HDL is now considered questionable, and is gradually being replaced by the hypothesis that HDL functionality encompasses several physiological functions beyond cholesterol efflux that are essential to determining protection against atherosclerosis (123).
For pathological and inflammatory conditions, it is well established that biochemical changes in HDL are associated with dysfunctional HDL and the development of ASCVD (124, 125). The main alterations in HDL qualitative properties described for patients with ischemic stroke are summarized in Table 1. A number of studies have described the presence of large HDL particles in patients with ischemic stroke (69, 70, 126), with some patients also showing biochemical HDL alterations leading to impaired functionality at different levels: cholesterol efflux ability (126), endothelial cell protection (69), antioxidant capacity (70, 127), and anti-inflammatory potential (70).

Table 1. Altered HDL features in patients with ischemic stroke.
The loss of an anti-atherosclerotic function in HDL in ischemic patients has been associated with alterations in the protein cargo, which includes several apos, but also proteins that participate in acute phase response and platelet activation, among them: apoE, apoA-IV, apoJ, apoF, apoL1, apoM, apoC-IV, α-1-antitrypsin, inter-α-trypsin inhibitor, paraoxonase-1, anthrax toxin receptor-2, serpina1, prenylcysteine oxidase-1, and SAA (72, 126). Decreased apoA-I in HDL from ischemic patients probably accounts for loss of functionality, according to Ortiz-Munoz et al. (69), who suggested that, in parallel with diminished apoA-I content, HDL displays fewer protective actions on endothelial cells. HDL particle size seems to impact on HDL function and on stroke outcome, with large HDL particles associated with both diminished anti-oxidant and anti-inflammatory properties and unfavorable outcomes (70).
Other authors have found associations between HDL subspecies and both specific apo content patterns (apoA-I, apoC-III, ApoE, apoJ) and vascular brain injury, including both covert and overt brain infarcts (128). HDL protein alterations in ischemic stroke patients have been attributed to proteome remodeling, owing to the existing inflammatory milieu in the acute phase (126). This interesting study by Plubell et al. describes that changes in HDL proteins in the early acute phase are associated with stroke recovery.
Besides protein cargo, it is well stablished that lipid composition has impact on the quality of HDL. For instance, the relative composition in triglycerides and cholesterol esters of the lipid core affects the conformation of apoA-I and the ensuing antioxidant activity of HDL (129, 130). Regarding lipids in the surface of HDL, the content of non-esterified cholesterol modulates the fluidity of lipoprotein surface, which has a direct effect on the oxidizability of HDL (129). Also, phospholipids and their fatty acid composition have an impact on anti-inflammatory activity of HDL (131). Sphingomyelin content influences HDL's efflux capacity (132). Ceramides, independently, or as precursors of sphingosine 1 phosphate (S1P), also have a relevant role in HDL function (133). S1P confers to HDL atheroprotective properties, including its capacity against apoptosis (134), inflammation (135) and vasodilatation (136, 137). However, contrariwise to LDL, little is known about lipid alterations in HDL in ischemic stroke, and further research is needed. Only the study by Nieddu et al. (73) has analyzed HDL lipidomics in the context of ischemic stroke, but the main differences were observed in LDL (as discussed before), not in HDL.
As occurs with LDL, an increased negative electric charge of HDL has also been also associated with HDL functionality (138), with a number of studies reporting the presence of HDL with increased negative charge, named H5 or HDL(-), in inflammation-related diseases (139–141). These negative HDL particles have been shown to impair cholesterol efflux and anti-inflammatory and anti-apoptotic actions, and to even promote inflammation and foam cell formation (140). An increased presence of oxidized HDL (oxHDL) has also been reported in both atherosclerotic plaques and blood circulation in several diseases, including acute myocardial infarction (141, 142). Accordingly, any increase in H5 or oxHDL in ischemic stroke, and particularly in atherothrombotic stroke, merits further investigation.
4 Lipoprotein-based therapies to prevent ischemic stroke
The main lipid-related therapy to reduce ischemic stroke risk is statin administration, whose main beneficial effects—derived from their inhibition of HMG-CoA reductase—are to reduce cholesterol biosynthesis and modulate lipid metabolism. Statins can also improve the biological characteristics of lipoproteins, by changing their chemical composition and decreasing oxLDL and LDL(-) levels (143–145). The additional fact that statins exert pleiotropic and anti-atherosclerotic effects, independently of their hypolipidemic action, significantly contributes to reduce CV event and mortality rates, with greater benefits in patients at high risk (146). Regarding ischemic stroke, several studies [summarized in (147)] have reported statins to reduce the ischemic stroke risk without increasing the hemorrhagic stroke risk.
Other more recently developed lipid-lowering agents demonstrate strong efficacy and are useful to prevent ASCVD. For patients at very high or high CV risk who are not responsive to or are intolerant of statins, ezetimibe, alone or in combination with statins, and PCSK9 inhibitors may reduce stroke risk (148). In the FOURIER study of patients with established atherosclerosis, PCSK9 inhibition with evolocumab added to statins reduced the risk of ischemic stroke (149). Bempedoic acid is a new lipid-lowering medication for the prevention and treatment of CV disease; currently in Phase III clinical trials (150), its effect on ischemic stroke is still pending of evaluation. Likewise, not yet tested for ischemic stroke is icosapent ethyl, an omega-3 fatty acid with triglyceride-lowering action that has shown promising results in reducing plasma triglyceride levels and major adverse CV events (151).
Given that lipoprotein function more than concentration plays a key role in ischemic stroke, future studies should address the qualitative properties of LDL and HDL and strategies to mitigate the generation of oxLDL and LDL(-). In this regard, the ability of statins to normalize alterations in lipoprotein composition and size and to reduce elevated modified lipoprotein levels is well known (152, 153). A recent study has reported that PCSK9 inhibition lowers LDL aggregation susceptibility, an LDL modification associated with future CV-related death (154). Antibodies against LDL(-) with athero-protective action have been detected in human and murine model blood and atherosclerotic plaques (155, 156), and based on one of those antibodies, a peptide with inflammatory properties has been designed that has potential to generate vaccines to immunize against LDL(-) and prevent atherosclerosis (157).
In sum, further investigations focused on the qualitative properties of lipoproteins and overcoming their impaired functioning are essential to understanding and preventing ischemic stroke, and particularly the atherothrombotic stroke subtype.
5 Conclusions
The particularities of ischemic stroke of atherothrombotic origin make it necessary to further explore alterations in lipoprotein functioning in stroke patients. In contrast with coronary disease, where these alterations have been extensively studied, for ischemic stroke, relatively few studies have explored abnormal lipoprotein functioning. The fact that quantitative alterations in the lipid profile are less frequent in patients with ischemic stroke than in patients with coronary atherosclerosis would suggest that in the former qualitative lipoprotein alterations may play a key role. In this context, to decipher the contribution of such alterations to this disease and to determine the molecular mechanisms involved poses a challenge in terms of designing new therapies addressed at preventing ischemic stroke.
Author's note
SB and JS-Q are members of the CIBER of Diabetes and Metabolic Diseases (CIBERDEM; CB07/08/0016). S.B. is member of EpiLipidNET COST action. SB and PC-R are members of RICORS-ICTUS (RD21/0006/0006). SB, JS-Q and NP are members of the Quality Research Group 2017-SGR–1149 of the Generalitat de Catalunya, and of the Vascular Biology Working Group of the Spanish Society of Atherosclerosis. IR Sant Pau is accredited by CERCA Programme/Generalitat de Catalunya.
Author contributions
SB: Writing – original draft. NP: Writing – review & editing. PC-R: Writing – review & editing. JS-Q: Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Fundació La Marató TV3 (201716.10) and the Instituto de Salud Carlos III (cofinanced by the European Regional Development Fund; FIS PI19/00421, PI20/00334, FI20/00252).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1470364/full#supplementary-material
References
1. Mead GE, Sposato LA, Sampaio Silva G, Yperzeele L, Wu S, Kutlubaev M, et al. A systematic review and synthesis of global stroke guidelines on behalf of the world stroke organization. Int J Stroke. (2023) 18(5):499–531. doi: 10.1177/17474930231156753
2. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340(2):115–26. doi: 10.1056/NEJM199901143400207
3. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114(12):1852–66. doi: 10.1161/CIRCRESAHA.114.302721
4. Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. (2015) 161(1):161–72. doi: 10.1016/j.cell.2015.01.036
5. Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. (2009) 50(Suppl):S376–381. doi: 10.1194/jlr.R800087-JLR200
6. Jiang F, Gibson AP, Dusting GJ. Endothelial dysfunction induced by oxidized low-density lipoproteins in isolated mouse aorta: a comparison with apolipoprotein-E deficient mice. Eur J Pharmacol. (2001) 424(2):141–9. doi: 10.1016/S0014-2999(01)01140-2
7. Yang Z, He Y, Wu D, Shi W, Liu P, Tan J, et al. Antiferroptosis therapy alleviated the development of atherosclerosis. MedComm. (2024) 5(4):e520. doi: 10.1002/mco2.520
8. Williams KJ, Tabas I. Atherosclerosis and inflammation. Science. (2002) 297(5581):521–2. doi: 10.1126/science.297.5581.521
9. Lhotak S, Gyulay G, Cutz JC, Al-Hashimi A, Trigatti BL, Richards CD, et al. Characterization of proliferating lesion-resident cells during all stages of atherosclerotic growth. J Am Heart Assoc. (2016) 5(8):e003945. doi: 10.1161/JAHA.116.003945
10. Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. (2014) 371(25):2383–93. doi: 10.1056/NEJMoa1409065
11. Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. (2009) 50(Suppl):S189–194. doi: 10.1194/jlr.R800088-JLR200
12. Navab M, Berliner JA, Subbanagounder G, Hama S, Lusis AJ, Castellani LW, et al. HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arterioscler Thromb Vasc Biol. (2001) 21(4):481–8. doi: 10.1161/01.ATV.21.4.481
13. Malekmohammad K, Bezsonov EE, Rafieian-Kopaei M. Role of lipid accumulation and inflammation in atherosclerosis: focus on molecular and cellular mechanisms. Front Cardiovasc Med. (2021) 8:707529. doi: 10.3389/fcvm.2021.707529
14. Morris PB, Ballantyne CM, Birtcher KK, Dunn SP, Urbina EM. Review of clinical practice guidelines for the management of LDL-related risk. J Am Coll Cardiol. (2014) 64(2):196–206. doi: 10.1016/j.jacc.2014.05.015
15. Rader DJ, Hovingh GK. HDL and cardiovascular disease. Lancet. (2014) 384(9943):618–25. doi: 10.1016/S0140-6736(14)61217-4
16. Saenger AK, Christenson RH. Stroke biomarkers: progress and challenges for diagnosis, prognosis, differentiation, and treatment. Clin Chem. (2010) 56(1):21–33. doi: 10.1373/clinchem.2009.133801
17. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation. (2015) 131(4):e29–322. doi: 10.1161/CIR.0000000000000152
18. Petty GW, Brown RD Jr, Whisnant JP, Sicks JD, O'Fallon WM, Wiebers DO. Ischemic stroke subtypes: a population-based study of incidence and risk factors. Stroke. (1999) 30(12):2513–6. doi: 10.1161/01.STR.30.12.2513
19. Adams HP Jr, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of org 10172 in acute stroke treatment. Stroke. (1993) 24(1):35–41. doi: 10.1161/01.STR.24.1.35
20. Chaturvedi S, Bruno A, Feasby T, Holloway R, Benavente O, Cohen SN, et al. Carotid endarterectomy–an evidence-based review: report of the therapeutics and technology assessment subcommittee of the American academy of neurology. Neurology. (2005) 65(6):794–801. doi: 10.1212/01.wnl.0000176036.07558.82
21. Derdeyn CP. Cerebral hemodynamics in carotid occlusive disease. AJNR Am J Neuroradiol. (2003) 24(8):1497–9.13679257
22. Gonzalez-Hernandez A, Fabre-Pi O, Lopez-Fernandez JC. Vascular risk factors in patients with ischaemic stroke. Rev Neurol. (2008) 47(3):167.18654974
23. Iemolo F, Martiniuk A, Steinman DA, Spence JD. Sex differences in carotid plaque and stenosis. Stroke. (2004) 35(2):477–81. doi: 10.1161/01.STR.0000110981.96204.64
24. Sangiorgi G, Roversi S, Biondi Zoccai G, Modena MG, Servadei F, Ippoliti A, et al. Sex-related differences in carotid plaque features and inflammation. J Vasc Surg. (2013) 57(2):338–44. doi: 10.1016/j.jvs.2012.07.052
25. Hellings WE, Pasterkamp G, Verhoeven BA, De Kleijn DP, De Vries JP, Seldenrijk KA, et al. Gender-associated differences in plaque phenotype of patients undergoing carotid endarterectomy. J Vasc Surg. (2007) 45(2):289–96. discussion 296–287. doi: 10.1016/j.jvs.2006.09.051
26. Lavallee PC, Charles H, Albers GW, Caplan LR, Donnan GA, Ferro JM, et al. Effect of atherosclerosis on 5-year risk of major vascular events in patients with transient ischaemic attack or minor ischaemic stroke: an international prospective cohort study. Lancet Neurol. (2023) 22(4):320–9. doi: 10.1016/S1474-4422(23)00067-4
27. North American Symptomatic Carotid Endarterectomy Trial C, Barnett HJM, Taylor DW, Haynes RB, Sackett DL, Peerless SJ, et al. Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med. (1991) 325(7):445–53. doi: 10.1056/NEJM199108153250701
28. Mendez I, Hachinski V, Wolfe B, Serum lipids after stroke. Neurology. (1987) 37(3):507–11. doi: 10.1212/WNL.37.3.507
29. Amarenco P, Labreuche J, Elbaz A, Touboul PJ, Driss F, Jaillard A, et al. Blood lipids in brain infarction subtypes. Cerebrovasc Dis. (2006) 22(2–3):101–8. doi: 10.1159/000093237
30. Hindy G, Engstrom G, Larsson SC, Traylor M, Markus HS, Melander O, et al. Role of blood lipids in the development of ischemic stroke and its subtypes. Stroke. (2018) 49(4):820–7. doi: 10.1161/STROKEAHA.117.019653
31. Ong CT, Wong YS, Sung SF, Wu CS, Hsu YC, Su YH, et al. progression of mild to moderate stenosis in the internal carotid arteries of patients with ischemic stroke. Front Neurol. (2018) 9:1043. doi: 10.3389/fneur.2018.01043
32. Furberg CD, Adams HP Jr, Applegate WB, Byington RP, Espeland MA, Hartwell T, et al. Effect of lovastatin on early carotid atherosclerosis and cardiovascular events. Asymptomatic carotid artery progression study (ACAPS) research group. Circulation. (1994) 90(4):1679–87. doi: 10.1161/01.CIR.90.4.1679
33. Amarenco P, Hobeanu C, Labreuche J, Charles H, Giroud M, Meseguer E, et al. Carotid atherosclerosis evolution when targeting a low-density lipoprotein cholesterol concentration <70mg/dl after an ischemic stroke of atherosclerotic origin. Circulation. (2020) 142(8):748–57. doi: 10.1161/CIRCULATIONAHA.120.046774
34. Amarenco P, Kim JS, Labreuche J, Charles H, Abtan J, Bejot Y, et al. A comparison of two LDL cholesterol targets after ischemic stroke. N Engl J Med. (2020) 382(1):9. doi: 10.1056/NEJMoa1910355
35. Cordero A, Rodriguez-Manero M, Facila L, Fernandez-Olmo MR, Gomez-Martinez MJ, Valle A, et al. Prevention of myocardial infarction and stroke with PCSK9 inhibitors treatment: a metanalysis of recent randomized clinical trials. J Diabetes Metab Disord. (2020) 19(2):759–65. doi: 10.1007/s40200-020-00557-6
36. Goldstein LB, Toth PP, Dearborn-Tomazos JL, Giugliano RP, Hirsh BJ, Pena JM, et al. Aggressive LDL-C lowering and the brain: impact on risk for dementia and hemorrhagic stroke: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. (2023) 43(10):e404–42. doi: 10.1161/ATV.0000000000000164
37. Sniderman AD, Navar AM, Thanassoulis G. Apolipoprotein B vs low-density lipoprotein cholesterol and non–high-density lipoprotein cholesterol as the primary measure of apolipoprotein B lipoprotein-related risk. JAMA Cardiol. (2022) 7(3):257–8. doi: 10.1001/jamacardio.2021.5080
38. Johannesen CDL, Mortensen MB, Langsted A, Nordestgaard BG. Apob and non-HDL cholesterol versus LDL cholesterol for ischemic stroke risk. Ann Neurol. (2022) 92(3):379–89. doi: 10.1002/ana.26425
39. Blaha MJ, Bhatia HS. Lipoprotein(a), residual cardiovascular risk, and the search for targeted therapy. J Am Coll Cardiol. (2024) 83(16):1540–2. doi: 10.1016/j.jacc.2024.03.370
40. Melita H, Manolis AA, Manolis TA, Manolis AS. Lipoprotein(a) and cardiovascular disease: a missing link for premature atherosclerotic heart disease and/or residual risk. J Cardiovasc Pharmacol. (2022) 79(1):e18–35. doi: 10.1097/FJC.0000000000001160
41. Erqou S, Thompson A, Di Angelantonio E, Saleheen D, Kaptoge S, Marcovina S, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. (2010) 55(19):2160–7. doi: 10.1016/j.jacc.2009.10.080
42. Qi Y, Duan Y, Deng Q, Yang N, Sun J, Li J, et al. Independent relationship of lipoprotein(a) and carotid atherosclerosis with long-term risk of cardiovascular disease. J Am Heart Assoc. (2024) 13(9):e033488. doi: 10.1161/JAHA.123.033488
43. Undas A, Slowik A, Wolkow P, Szczudlik A, Tracz W. Fibrin clot properties in acute ischemic stroke: relation to neurological deficit. Thromb Res. (2010) 125(4):357–61. doi: 10.1016/j.thromres.2009.11.013
44. Lackova A, Gdovinova Z, Kozarova M, Koren D, Lacko M. Lipoprotein (a) concentration as a risk factor for ischaemic stroke and its subtypes. Neurol Neurochir Pol. (2024) 58(3):316–22. doi: 10.5603/pjnns.98343
45. Scipione CA, Sayegh SE, Romagnuolo R, Tsimikas S, Marcovina SM, Boffa MB, et al. Mechanistic insights into lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a). J Lipid Res. (2015) 56(12):2273–85. doi: 10.1194/jlr.M060210
46. Boffa MB, Koschinsky ML. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol. (2019) 16(5):305–18. doi: 10.1038/s41569-018-0153-2
47. Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin MJ, et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. (2008) 49(10):2230–9. doi: 10.1194/jlr.M800174-JLR200
48. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. (2016) 134(8):611–24. doi: 10.1161/CIRCULATIONAHA.116.020838
49. Small AM, Pournamdari A, Melloni GEM, Scirica BM, Bhatt DL, Raz I, et al. Lipoprotein(a), C-reactive protein, and cardiovascular risk in primary and secondary prevention populations. JAMA Cardiol. (2024) 9(4):385–91. doi: 10.1001/jamacardio.2023.5605
50. Aronis KN, Zhao D, Hoogeveen RC, Alonso A, Ballantyne CM, Guallar E, et al. Associations of lipoprotein(a) levels with incident atrial fibrillation and ischemic stroke: the ARIC (atherosclerosis risk in communities) study. J Am Heart Assoc. (2017) 6(12):e00737270. doi: 10.1161/JAHA.117.007372
51. Berman AN, Blankstein R. Current and future role of lipoprotein(a) in preventive cardiology. Curr Opin Cardiol. (2019) 34(5):514–8. doi: 10.1097/HCO.0000000000000661
52. Glader CA, Stegmayr B, Boman J, Stenlund H, Weinehall L, Hallmans G, et al. Chlamydia pneumoniae antibodies and high lipoprotein(a) levels do not predict ischemic cerebral infarctions. Results from a nested case-control study in Northern Sweden. Stroke. (1999) 30(10):2013–8. doi: 10.1161/01.STR.30.10.2013
53. Lange KS, Nave AH, Liman TG, Grittner U, Endres M, Ebinger M. Lipoprotein(a) levels and recurrent vascular events after first ischemic stroke. Stroke. (2017) 48(1):36–42. doi: 10.1161/STROKEAHA.116.014436
54. van Dam-Nolen DHK, van Dijk AC, Crombag G, Lucci C, Kooi ME, Hendrikse J, et al. Lipoprotein(a) levels and atherosclerotic plaque characteristics in the carotid artery: the plaque at RISK (PARISK) study. Atherosclerosis. (2021) 329:22–9. doi: 10.1016/j.atherosclerosis.2021.06.004
55. Duan Y, Zhao D, Sun J, Liu J, Wang M, Hao Y, et al. Lipoprotein(a) is associated with the progression and vulnerability of new-onset carotid atherosclerotic plaque. Stroke. (2023) 54(5):1312–9. doi: 10.1161/STROKEAHA.122.042323
56. Kumar P, Swarnkar P, Misra S, Nath M. Lipoprotein (a) level as a risk factor for stroke and its subtype: a systematic review and meta-analysis. Sci Rep. (2021) 11(1):15660. doi: 10.1038/s41598-021-95141-0
57. Lindenstrom E, Boysen G, Nyboe J. Influence of total cholesterol, high density lipoprotein cholesterol, and triglycerides on risk of cerebrovascular disease: the Copenhagen city heart study. BMJ. (1994) 309(6946):11–5. doi: 10.1136/bmj.309.6946.11
58. Li W, Huang Z, Fang W, Wang X, Cai Z, Chen G, et al. Remnant cholesterol variability and incident ischemic stroke in the general population. Stroke. (2022) 53(6):1934–41. doi: 10.1161/STROKEAHA.121.037756
59. Wu Z, Jiang Y, Guo Z, Li P, Zheng Y, Wang Y, et al. Remnant cholesterol traits and risk of stroke: a multivariable Mendelian randomization study. PNAS Nexus. (2024) 3(2):pgae033. doi: 10.1093/pnasnexus/pgae033
60. Qian S, You S, Sun Y, Wu Q, Wang X, Tang W, et al. Remnant cholesterol and common carotid artery intima-media thickness in patients with ischemic stroke. Circ Cardiovasc Imaging. (2021) 14(4):e010953. doi: 10.1161/CIRCIMAGING.120.010953
61. Gronholdt ML, Nordestgaard BG, Nielsen TG, Sillesen H. Echolucent carotid artery plaques are associated with elevated levels of fasting and postprandial triglyceride-rich lipoproteins. Stroke. (1996) 27(12):2166–72. doi: 10.1161/01.STR.27.12.2166
62. Kofoed SC, Gronholdt ML, Bismuth J, Wilhjelm JE, Sillesen H, Nordestgaard BG. Echolucent, rupture-prone carotid plaques associated with elevated triglyceride-rich lipoproteins, particularly in women. J Vasc Surg. (2002) 36(4):783–92. doi: 10.1016/S0741-5214(02)00134-9
63. Alam R, Yatsu FM, Kasturi R, Bui G. Low and high density lipoprotein metabolism in atherothrombotic brain infarction. Stroke. (1992) 23(9):1265–70. doi: 10.1161/01.STR.23.9.1265
64. Tirschwell DL, Smith NL, Heckbert SR, Lemaitre RN, Longstreth WT Jr, Psaty BM. Association of cholesterol with stroke risk varies in stroke subtypes and patient subgroups. Neurology. (2004) 63(10):1868–75. doi: 10.1212/01.WNL.0000144282.42222.DA
65. Singh K, Chandra A, Sperry T, Joshi PH, Khera A, Virani SS, et al. Associations between high-density lipoprotein particles and ischemic events by vascular domain, sex, and ethnicity. Circulation. (2020) 142(7):657–69. doi: 10.1161/CIRCULATIONAHA.120.045713
66. Fernandez-Friera L, Fuster V, Lopez-Melgar B, Oliva B, Garcia-Ruiz JM, Mendiguren J, et al. Normal LDL-cholesterol levels are associated with subclinical atherosclerosis in the absence of risk factors. J Am Coll Cardiol. (2017) 70(24):2979–91. doi: 10.1016/j.jacc.2017.10.024
67. Vanuzzo D. The epidemiological concept of residual risk. Intern Emerg Med. (2011) 6(Suppl 1):45–51. doi: 10.1007/s11739-011-0669-5
68. Matsuura Y, Kanter JE, Bornfeldt KE. Highlighting residual atherosclerotic cardiovascular disease risk. Arterioscler Thromb Vasc Biol. (2019) 39(1):e1–9. doi: 10.1161/ATVBAHA.118.311999
69. Ortiz-Munoz G, Couret D, Lapergue B, Bruckert E, Meseguer E, Amarenco P, et al. Dysfunctional HDL in acute stroke. Atherosclerosis. (2016) 253:75–80. doi: 10.1016/j.atherosclerosis.2016.08.035
70. Varela LM, Meseguer E, Lapergue B, Couret D, Amarenco P, Meilhac O. Changes in high-density lipoproteins related to outcomes in patients with acute stroke. J Clin Med. (2020) 9(7):2269. doi: 10.3390/jcm9072269
71. Finamore F, Nieddu G, Rocchiccioli S, Spirito R, Guarino A, Formato M, et al. Apolipoprotein signature of HDL and LDL from atherosclerotic patients in relation with carotid plaque typology: a preliminary report. Biomedicines. (2021) 9(9):1156. doi: 10.3390/biomedicines9091156
72. Lepedda AJ, Nieddu G, Zinellu E, De Muro P, Piredda F, Guarino A, et al. Proteomic analysis of plasma-purified VLDL, LDL, and HDL fractions from atherosclerotic patients undergoing carotid endarterectomy: identification of serum amyloid A as a potential marker. Oxid Med Cell Longev. (2013) 2013:385214. doi: 10.1155/2013/385214
73. Nieddu G, Michelucci E, Formato M, Ciampelli C, Obino G, Signore G, et al. Molecular characterization of plasma HDL, LDL, and VLDL lipids cargos from atherosclerotic patients with advanced carotid lesions: a preliminary report. Int J Mol Sci. (2022) 23(20):12449. doi: 10.3390/ijms232012449
74. Zeljkovic A, Vekic J, Spasojevic-Kalimanovska V, Jelic-Ivanovic Z, Bogavac-Stanojevic N, Gulan B, et al. LDL and HDL subclasses in acute ischemic stroke: prediction of risk and short-term mortality. Atherosclerosis. (2010) 210(2):548–54. doi: 10.1016/j.atherosclerosis.2009.11.040
75. Yatsu FM, Alam R, Alam S. Scavenger activity in monocyte-derived macrophages from atherothrombotic strokes. Stroke. (1986) 17(4):709–13. doi: 10.1161/01.STR.17.4.709
76. Gao S, Liu J. Association between circulating oxidized low-density lipoprotein and atherosclerotic cardiovascular disease. Chronic Dis Transl Med. (2017) 3(2):89–94. doi: 10.1016/j.cdtm.2017.02.008
77. Sanchez-Quesada JL, Benitez S, Ordonez-Llanos J. Electronegative low-density lipoprotein. Curr Opin Lipidol. (2004) 15(3):329–35. doi: 10.1097/00041433-200406000-00014
78. Puig N, Camps-Renom P, Sole A, Aguilera-Simon A, Jimenez-Xarrie E, Fernandez-Leon A, et al. Electronegative LDL is associated with plaque vulnerability in patients with ischemic stroke and carotid atherosclerosis. Antioxidants (Basel). (2023) 12(2):438. doi: 10.3390/antiox12020438
79. Uno M, Kitazato KT, Nishi K, Itabe H, Nagahiro S. Raised plasma oxidised LDL in acute cerebral infarction. J Neurol Neurosurg Psychiatry. (2003) 74(3):312–6. doi: 10.1136/jnnp.74.3.312
80. Tsai NW, Lee LH, Huang CR, Chang WN, Chang YT, Su YJ, et al. Statin therapy reduces oxidized low density lipoprotein level, a risk factor for stroke outcome. Crit Care. (2014) 18(1):R16. doi: 10.1186/cc13695
81. Kassner SS, Kollmar R, Bonaterra GA, Hildebrandt W, Schwab S, Kinscherf R. The early immunological response to acute ischemic stroke: differential gene expression in subpopulations of mononuclear cells. Neuroscience. (2009) 160(2):394–401. doi: 10.1016/j.neuroscience.2009.02.050
82. Moon GJ, Kim SJ, Cho YH, Ryoo S, Bang OY. Antioxidant effects of statins in patients with atherosclerotic cerebrovascular disease. J Clin Neurol. (2014) 10(2):140–7. doi: 10.3988/jcn.2014.10.2.140
83. Wang A, Cui Y, Meng X, Su Z, Cao Y, Yang Y, et al. The relationship between oxidized low-density lipoprotein and the NIHSS score among patients with acute ischemic stroke: the SOS-stroke study. Atherosclerosis. (2018) 270:21–5. doi: 10.1016/j.atherosclerosis.2018.01.028
84. Vibo R, Korv J, Roose M, Kampus P, Muda P, Zilmer K, et al. Acute phase proteins and oxidised low-density lipoprotein in association with ischemic stroke subtype, severity and outcome. Free Radic Res. (2007) 41(3):282–7. doi: 10.1080/10715760601083235
85. Guldiken B, Guldiken S, Turgut B, Turgut N, Demir M, Celik Y, et al. The roles of oxidized low-density lipoprotein and interleukin-6 levels in acute atherothrombotic and lacunar ischemic stroke. Angiology. (2008) 59(2):224–9. doi: 10.1177/0003319707304134
86. Cuadrado-Godia E, Ois A, Garcia-Ramallo E, Giralt E, Jimena S, Rubio MA, et al. Biomarkers to predict clinical progression in small vessel disease strokes: prognostic role of albuminuria and oxidized LDL cholesterol. Atherosclerosis. (2011) 219(1):368–72. doi: 10.1016/j.atherosclerosis.2011.07.114
87. Nishi K, Itabe H, Uno M, Kitazato KT, Horiguchi H, Shinno K, et al. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol. (2002) 22(10):1649–54. doi: 10.1161/01.ATV.0000033829.14012.18
88. Tsimikas S, Willeit P, Willeit J, Santer P, Mayr M, Xu Q, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. (2012) 60(21):2218–29. doi: 10.1016/j.jacc.2012.08.979
89. Wang A, Yang Y, Su Z, Yue W, Hao H, Ren L, et al. Association of oxidized low-density lipoprotein with prognosis of stroke and stroke subtypes. Stroke. (2017) 48(1):91–7. doi: 10.1161/STROKEAHA.116.014816
90. Meisinger C, Baumert J, Khuseyinova N, Loewel H, Koenig W. Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation. (2005) 112(5):651–7. doi: 10.1161/CIRCULATIONAHA.104.529297
91. Kiechl S, Willeit J, Mayr M, Viehweider B, Oberhollenzer M, Kronenberg F, et al. Oxidized phospholipids, Lipoprotein(a), Lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes. Arterioscler Thromb Vasc Biol. (2007) 27(8):1788–95. doi: 10.1161/ATVBAHA.107.145805
92. Wang A, Xu J, Chen G, Wang D, Johnston SC, Meng X, et al. Oxidized low-density lipoprotein predicts recurrent stroke in patients with minor stroke or TIA. Neurology. (2018) 91(10):e947–55. doi: 10.1212/WNL.0000000000006118
93. Wang A, Liu J, Meng X, Li J, Wang H, Wang Y, et al. Association between oxidized low-density lipoprotein and cognitive impairment in patients with ischemic stroke. Eur J Neurol. (2018) 25(1):185–91. doi: 10.1111/ene.13497
94. Xu Q, Wu Q, Li H, Tian X, Zuo Y, Zhang Y, et al. Joint high level of oxidized low-density lipoprotein and high-sensitivity C-reactive protein are associated with recurrent stroke and poor functional outcome in Minor stroke or transient ischemic attack. J Am Heart Assoc. (2022) 11(20):e027665. doi: 10.1161/JAHA.122.027665
95. Wang A, Dai L, Zhang N, Lin J, Chen G, Zuo Y, et al. Oxidized low-density lipoprotein (LDL) and LDL cholesterol are associated with outcomes of minor stroke and TIA. Atherosclerosis. (2020) 297:74–80. doi: 10.1016/j.atherosclerosis.2020.02.003
96. Mello AP, da Silva IT, Abdalla DS, Damasceno NR. Electronegative low-density lipoprotein: origin and impact on health and disease. Atherosclerosis. (2011) 215(2):257–65. doi: 10.1016/j.atherosclerosis.2010.12.028
97. Estruch M, Sanchez-Quesada JL, Ordonez Llanos J, Benitez S. Electronegative LDL: a circulating modified LDL with a role in inflammation. Mediators Inflamm. (2013) 2013:1. doi: 10.1155/2013/181324
98. Ke LY, Stancel N, Bair H, Chen CH. The underlying chemistry of electronegative LDL’s atherogenicity. Curr Atheroscler Rep. (2014) 16(8):428. doi: 10.1007/s11883-014-0428-y
99. De Castellarnau C, Sanchez-Quesada JL, Benitez S, Rosa R, Caveda L, Vila L, et al. Electronegative LDL from normolipemic subjects induces IL-8 and monocyte chemotactic protein secretion by human endothelial cells. Arterioscler Thromb Vasc Biol. (2000) 20(10):2281–7. doi: 10.1161/01.ATV.20.10.2281
100. Lu J, Yang JH, Burns AR, Chen HH, Tang D, Walterscheid JP, et al. Mediation of electronegative low-density lipoprotein signaling by LOX-1. Circ Res. (2009) 104(5):619–27. doi: 10.1161/CIRCRESAHA.108.190116
101. Sanchez-Quesada JL, Villegas S, Ordonez-Llanos J. Electronegative low-density lipoprotein. A link between apolipoprotein B misfolding, lipoprotein aggregation and proteoglycan binding. Curr Opin Lipidol. (2012) 23(5):479–86. doi: 10.1097/MOL.0b013e328357c933
102. Ke LY, Law SH, Mishra VK, Parveen F, Chan HC, Lu YH, et al. Molecular and cellular mechanisms of electronegative lipoproteins in cardiovascular diseases. Biomedicines. (2020) 8(12):550. doi: 10.3390/biomedicines8120550
103. Bancells C, Benitez S, Jauhiainen M, Ordonez-Llanos J, Kovanen PT, Villegas S, et al. High binding affinity of electronegative LDL to human aortic proteoglycans depends on its aggregation level. J Lipid Res. (2009) 50(3):446–55. doi: 10.1194/jlr.M800318-JLR200
104. Akyol S, Lu J, Akyol O, Akcay F, Armutcu F, Ke LY, et al. The role of electronegative low-density lipoprotein in cardiovascular diseases and its therapeutic implications. Trends Cardiovasc Med. (2017) 27(4):239–46. doi: 10.1016/j.tcm.2016.11.002
105. Chang CY, Chen CH, Chen YM, Hsieh TY, Li JP, Shen MY, et al. Association between negatively charged low-density lipoprotein L5 and subclinical atherosclerosis in rheumatoid arthritis patients. J Clin Med. (2019) 8(2):177. doi: 10.3390/jcm8020177
106. Niccoli G, Baca M, De Spirito M, Parasassi T, Cosentino N, Greco G, et al. Impact of electronegative low-density lipoprotein on angiographic coronary atherosclerotic burden. Atherosclerosis. (2012) 223(1):166–70. doi: 10.1016/j.atherosclerosis.2012.04.005
107. Lee AS, Chen WY, Chan HC, Hsu JF, Shen MY, Chang CM, et al. Gender disparity in LDL-induced cardiovascular damage and the protective role of estrogens against electronegative LDL. Cardiovasc Diabetol. (2014) 13:64. doi: 10.1186/1475-2840-13-64
108. Yang TC, Chang PY, Lu SC. L5-LDL from ST-elevation myocardial infarction patients induces IL-1beta production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am J Physiol Heart Circ Physiol. (2017) 312(2):H265–74. doi: 10.1152/ajpheart.00509.2016
109. Shen MY, Chen FY, Hsu JF, Fu RH, Chang CM, Chang CT, et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood. (2016) 127(10):1336–45. doi: 10.1182/blood-2015-05-646117
110. Akyol O, Yang CY, Woodside DG, Chiang HH, Chen CH, Gotto AM. Comparative analysis of atherogenic lipoproteins L5 and lp(a) in atherosclerotic cardiovascular disease. Curr Atheroscler Rep. (2024) 26(7):317–29. doi: 10.1007/s11883-024-01209-3
111. Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. (2002) 43(9):1363–79. doi: 10.1194/jlr.R200004-JLR200
112. Tribble DL, Rizzo M, Chait A, Lewis DM, Blanche PJ, Krauss RM. Enhanced oxidative susceptibility and reduced antioxidant content of metabolic precursors of small, dense low-density lipoproteins. Am J Med. (2001) 110(2):103–10. doi: 10.1016/S0002-9343(00)00700-2
113. Younis NN, Soran H, Pemberton P, Charlton-Menys V, Elseweidy MM, Durrington PN. Small dense LDL is more susceptible to glycation than more buoyant LDL in type 2 diabetes. Clin Sci (Lond). (2013) 124(5):343–9. doi: 10.1042/CS20120304
114. Sanchez-Quesada JL, Benitez S, Otal C, Franco M, Blanco-Vaca F, Ordonez-Llanos J. Density distribution of electronegative LDL in normolipemic and hyperlipemic subjects. J Lipid Res. (2002) 43(5):699–705. doi: 10.1016/S0022-2275(20)30111-5
115. Gaubatz JW, Gillard BK, Massey JB, Hoogeveen RC, Huang M, Lloyd EE, et al. Dynamics of dense electronegative low density lipoproteins and their preferential association with lipoprotein phospholipase A2. J Lipid Res. (2007) 48(2):348–57. doi: 10.1194/jlr.M600249-JLR200
116. Albers JJ, Slee A, Fleg JL, O'Brien KD, Marcovina SM. Relationship of baseline HDL subclasses, small dense LDL and LDL triglyceride to cardiovascular events in the AIM-HIGH clinical trial. Atherosclerosis. (2016) 251:454–9. doi: 10.1016/j.atherosclerosis.2016.06.019
117. Zhou P, Liu J, Wang L, Feng W, Cao Z, Wang P, et al. Association of small dense low-density lipoprotein cholesterol with stroke risk, severity and prognosis. J Atheroscler Thromb. (2020) 27(12):1310–24. doi: 10.5551/jat.53132
118. Yu X, Yu Y, Wei C, Wang L, Jiang J, Zhang R, et al. Association between small dense low-density lipoprotein cholesterol and neuroimaging markers of cerebral small vessel disease in middle-aged and elderly Chinese populations. BMC Neurol. (2021) 21(1):436. doi: 10.1186/s12883-021-02472-6
119. Balling M, Nordestgaard BG, Varbo A, Langsted A, Kamstrup PR, Afzal S. Small dense low-density lipoprotein cholesterol and ischemic stroke. Ann Neurol. (2023) 93(5):952–64. doi: 10.1002/ana.26598
120. Ma X, Wang Q, Hu X, Wang X, Zhao Y, Liu X, et al. Association of sdLDL-C with incident carotid plaques with stable and vulnerable morphology: a prospective cohort study. Stroke. (2024) 55(3):576–85. doi: 10.1016/j.cdtm.2017.02.008
121. Manabe Y, Morihara R, Matsuzono K, Nakano Y, Takahashi Y, Narai H, et al. Estimation of the presence of small dense lipoprotein cholesterol in acute ischemic stroke. Neurol Int. (2015) 7(1):5973. doi: 10.4081/ni.2015.5973
122. Barter P. Lessons learned from the investigation of lipid level management to understand its impact in atherosclerotic events (ILLUMINATE) trial. Am J Cardiol. (2009) 104(10 Suppl):10E–5E. doi: 10.1016/j.amjcard.2009.09.014
123. Sirtori CR, Corsini A, Ruscica M. The role of high-density lipoprotein cholesterol in 2022. Curr Atheroscler Rep. (2022) 24(5):365–77. doi: 10.1007/s11883-022-01012-y
124. Carnuta MG, Stancu CS, Toma L, Sanda GM, Niculescu LS, Deleanu M, et al. Dysfunctional high-density lipoproteins have distinct composition, diminished anti-inflammatory potential and discriminate acute coronary syndrome from stable coronary artery disease patients. Sci Rep. (2017) 7(1):7295. doi: 10.1038/s41598-017-07821-5
125. Rosenson RS, Brewer HB Jr, Ansell BJ, Barter P, Chapman MJ, Heinecke JW, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. (2016) 13(1):48–60. doi: 10.1038/nrcardio.2015.124
126. Plubell DL, Fenton AM, Rosario S, Bergstrom P, Wilmarth PA, Clark WM, et al. High-density lipoprotein carries markers that track with recovery from stroke. Circ Res. (2020) 127(10):1274–87. doi: 10.1161/CIRCRESAHA.120.316526
127. Damayanthi D, Krishnamoorthy S, Sylaja PN, Gopala S. Increased high-density lipoprotein-oxidant index in ischemic stroke patients. Biomed Rep. (2022) 17(5):87. doi: 10.3892/br.2022.1570
128. Koch M, Aroner SA, Fitzpatrick AL, Longstreth WT Jr, Furtado JD, Mukamal KJ, et al. HDL (high-density lipoprotein) subspecies, prevalent covert brain infarcts, and incident overt ischemic stroke: cardiovascular health study. Stroke. (2022) 53(4):1292–300. doi: 10.1161/STROKEAHA.121.034299
129. Cazzola R, Cassani E, Barichella M, Cestaro B. Impaired fluidity and oxidizability of HDL hydrophobic core and amphipathic surface in dyslipidemic men. Metab Clin Exp. (2013) 62(7):986–91. doi: 10.1016/j.metabol.2013.01.012
130. Curtiss LK, Bonnet DJ, Rye KA. The conformation of apolipoprotein A-I in high-density lipoproteins is influenced by core lipid composition and particle size: a surface plasmon resonance study. Biochemistry. (2000) 39(19):5712–21. doi: 10.1021/bi992902m
131. Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ. Phospholipid composition of reconstituted high density lipoproteins influences their ability to inhibit endothelial cell adhesion molecule expression. J Lipid Res. (2000) 41(8):1261–7. doi: 10.1016/S0022-2275(20)33434-9
132. Marmillot P, Patel S, Lakshman MR. Reverse cholesterol transport is regulated by varying fatty acyl chain saturation and sphingomyelin content in reconstituted high-density lipoproteins. Metab Clin Exp. (2007) 56(2):251–9. doi: 10.1016/j.metabol.2006.09.021
133. Malajczuk CJ, Mancera RL. Unravelling the influence of surface lipids on the structure, dynamics and interactome of high-density lipoproteins. Biochim Biophys Acta Biomembr. (2023) 1865(8):184201. doi: 10.1016/j.bbamem.2023.184201
134. Feuerborn R, Becker S, Poti F, Nagel P, Brodde M, Schmidt H, et al. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis. (2017) 257:29–37. doi: 10.1016/j.atherosclerosis.2016.12.009
135. Ruiz M, Frej C, Holmer A, Guo LJ, Tran S, Dahlback B. High-density lipoprotein-associated apolipoprotein M limits endothelial inflammation by delivering sphingosine-1-phosphate to the sphingosine-1-phosphate receptor 1. Arterioscler Thromb Vasc Biol. (2017) 37(1):118–29. doi: 10.1161/ATVBAHA.116.308435
136. Tolle M, Klockl L, Wiedon A, Zidek W, van der Giet M, Schuchardt M. Regulation of endothelial nitric oxide synthase activation in endothelial cells by S1P1 and S1P3. Biochem Biophys Res Commun. (2016) 476(4):627–34. doi: 10.1016/j.bbrc.2016.06.009
137. Okajima F. Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: is this an atherogenic mediator or an anti-atherogenic mediator? Biochim Biophys Acta. (2002) 1582(1-3):132–7. doi: 10.1016/S1388-1981(02)00147-6
138. Hsieh JY, Chang CT, Huang MT, Chang CM, Chen CY, Shen MY, et al. Biochemical and functional characterization of charge-defined subfractions of high-density lipoprotein from normal adults. Anal Chem. (2013) 85(23):11440–8. doi: 10.1021/ac402516u
139. Chan HC, Ke LY, Lu HT, Weng SF, Chan HC, Law SH, et al. An increased plasma level of ApoCIII-rich electronegative high-density lipoprotein may contribute to cognitive impairment in Alzheimer’s disease. Biomedicines. (2020) 8(12):542. doi: 10.3390/biomedicines8120542
140. Chang CK, Cheng WC, Ma WL, Chen PK, Chen CH, Shen PC, et al. The potential role of electronegative high-density lipoprotein H5 subfraction in RA-related atherosclerosis. Int J Mol Sci. (2021) 22(21):11419. doi: 10.3390/ijms222111419
141. Sawada N, Obama T, Koba S, Takaki T, Iwamoto S, Aiuchi T, et al. Circulating oxidized LDL, increased in patients with acute myocardial infarction, is accompanied by heavily modified HDL. J Lipid Res. (2020) 61(6):816–29. doi: 10.1194/jlr.RA119000312
142. Itabe H, Sawada N, Makiyama T, Obama T. Structure and dynamics of oxidized lipoproteins in vivo: roles of high-density lipoprotein. Biomedicines. (2021) 9(6):655 doi: 10.3390/biomedicines9060655
143. Jamialahmadi T, Baratzadeh F, Reiner Z, Mannarino MR, Cardenia V, Simental-Mendia LE, et al. The effects of statin therapy on oxidized LDL and its antibodies: a systematic review and meta-analysis. Oxid Med Cell Longev. (2022) 2022:1. doi: 10.1155/2022/7850659
144. Benitez S, Ordonez-Llanos J, Franco M, Marin C, Paz E, Lopez-Miranda J, et al. Effect of simvastatin in familial hypercholesterolemia on the affinity of electronegative low-density lipoprotein subfractions to the low-density lipoprotein receptor. Am J Cardiol. (2004) 93(4):414–20. doi: 10.1016/j.amjcard.2003.10.034
145. Zhang B, Miura S, Yanagi D, Noda K, Nishikawa H, Matsunaga A, et al. Reduction of charge-modified LDL by statin therapy in patients with CHD or CHD risk factors and elevated LDL-C levels: the SPECIAL study. Atherosclerosis. (2008) 201(2):353–9. doi: 10.1016/j.atherosclerosis.2008.02.024
146. Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for prevention of cardiovascular disease in adults. JAMA. (2016) 316(19):2008–24. doi: 10.1001/jama.2015.15629
147. Hackam DG, Hegele RA. Lipid-modifying therapies and stroke prevention. Curr Neurol Neurosci Rep. (2022) 22(7):375–82. doi: 10.1007/s11910-022-01197-4
148. Khan SU, Yedlapati SH, Lone AN, Hao Q, Guyatt G, Delvaux N, et al. PCSK9 inhibitors and ezetimibe with or without statin therapy for cardiovascular risk reduction: a systematic review and network meta-analysis. Br Med J. (2022) 377:e069116. doi: 10.1136/bmj-2021-069116
149. Giugliano RP, Pedersen TR, Saver JL, Sever PS, Keech AC, Bohula EA, et al. Stroke prevention with the PCSK9 (proprotein convertase subtilisin-kexin type 9) inhibitor evolocumab added to statin in high-risk patients with stable atherosclerosis. Stroke. (2020) 51(5):1546–54. doi: 10.1161/STROKEAHA.119.027759
150. Parham JS, Goldberg AC. Major concepts in treatment with bempedoic acid and inclisiran that clinicians need to know. Curr Atheroscler Rep. (2022) 24(8):619–25. doi: 10.1007/s11883-022-01036-4
151. Burger PM, Bhatt DL, Dorresteijn JAN, Koudstaal S, Mosterd A, Martens F, et al. Effects of icosapent ethyl according to baseline residual risk in patients with atherosclerotic cardiovascular disease: results from REDUCE-IT. Eur Heart J Cardiovasc Pharmacother. (2024) 10(6):488–99. doi: 10.1093/ehjcvp/pvae030
152. Rosenson RS, Brewer HB Jr. New challenges for HDL-modifying therapies as a strategy to lower cardiovascular disease events in statin-treated patients. Cardiovasc Drugs Ther. (2015) 29(1):1–3. doi: 10.1007/s10557-015-6576-7
153. Rosenson RS, Underberg JA. Systematic review: evaluating the effect of lipid-lowering therapy on lipoprotein and lipid values. Cardiovasc Drugs Ther. (2013) 27(5):465–79. doi: 10.1007/s10557-013-6477-6
154. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. (2018) 39(27):2562–73. doi: 10.1093/eurheartj/ehy319
155. Damasceno NR, Sevanian A, Apolinario E, Oliveira JM, Fernandes I, Abdalla DS. Detection of electronegative low density lipoprotein (LDL-) in plasma and atherosclerotic lesions by monoclonal antibody-based immunoassays. Clin Biochem. (2006) 39(1):28–38. doi: 10.1016/j.clinbiochem.2005.09.014
156. Grosso DM, Ferderbar S, Wanschel AC, Krieger MH, Higushi ML, Abdalla DS. Antibodies against electronegative LDL inhibit atherosclerosis in LDLr-/- mice. Braz J Med Biol Res. (2008) 41(12):1086–92. doi: 10.1590/S0100-879X2008001200007
Keywords: ischemic stroke, oxidized LDL, small dense LDL, electronegative LDL, HDL, carotid atherosclerosis
Citation: Benitez S, Puig N, Camps-Renom P and Sánchez-Quesada JL (2024) Atherogenic circulating lipoproteins in ischemic stroke. Front. Cardiovasc. Med. 11:1470364. doi: 10.3389/fcvm.2024.1470364
Received: 25 July 2024; Accepted: 22 November 2024;
Published: 6 December 2024.
Edited by:
Chu-Huang Chen, Texas Heart Institute, United StatesReviewed by:
Lisa Kristina Dannenberg, University Hospital of Düsseldorf, GermanyCorina Rosales, Houston Methodist Research Institute, United States
Copyright: © 2024 Benitez, Puig, Camps-Renom and Sánchez-Quesada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonia Benitez, c2Jlbml0ZXpAc2FudHBhdS5jYXQ=; José Luis Sánchez-Quesada, anNhbmNoZXpxQHNhbnRwYXUuY2F0
†ORCID:
Núria Puig
orcid.org/0000-0002-4699-3162