 Mingxuan Che
Mingxuan Che Fuhai Li1
Fuhai Li1 Yaning Jia
Yaning Jia Jidong Zhang
Jidong Zhang Shiguo Liu
Shiguo Liu- 1Cardiovascular Medicine Department, The Affiliated Hospital of Qingdao University, Qingdao, China
- 2Medical Genetic Department, The Affiliated Hospital of Qingdao University, Qingdao, China
- 3Prenatal Diagnosis Center, The Affiliated Hospital of Qingdao University, Qingdao, China
Barth syndrome (BTHS) is a rare X-linked recessive genetic disorder characterized by a broad spectrum of clinical features including cardiomyopathy, skeletal myopathy, neutropenia, growth delay, and 3-methylglutaconic aciduria. This disease is caused by loss-of-function mutations in the TAFAZZIN gene located on chromosome Xq28, resulting in cardiolipin deficiency. Most patients are diagnosed in childhood, and the mortality rate is highest in the early years. We report a case of acute, life-threatening metabolic decompensation occurring one day after birth. A novel TAFAZZIN splice site mutation was identified in the patient, marking the first reported case of such a mutation in BTHS identified in China. The report aims to expand our understanding of the spectrum of TAFAZZIN mutations in BTHS.
1 Introduction
BTHS (MIM 302060) was initially identified as a rare X-linked recessive genetic disorder primarily affecting the mitochondrial function in neutrophils, as well as cardiac and skeletal muscles. The syndrome is characterized by cardiomyopathy, neutropenia, developmental delays, skeletal muscle dysfunction, and 3-methylglutaconic aciduria (1). The causative factor of BTHS has been traced to loss-of-function mutations in the TAFAZZIN gene located on the Xq28 region of the X chromosome (2). This study reports a Chinese pediatric case of BTHS, which is the first reported case of this specific novel splice site mutation. This discovery enhances our understanding of the molecular mechanisms underlying the disease.
2 Case presentation
The male neonate was delivered via cesarean section at 36 weeks of gestation due to fetal cardiac anomalies and umbilical cord entanglement. He weighed 2,780 g at birth, with Apgar scores of 9 at 1, 5, and 10 min. His condition deteriorated rapidly after delivery, with severe cyanosis, frothing, and reduced vitality. Initial vital signs demonstrated no fever, low blood pressure (65/29 mmHg), tachycardia (heart rate 128–152 beats/min), or rapid breathing (respiratory rate 52–62 breaths/min), with oxygen saturation at 91%–95%. Chest radiography revealed cardiomegaly (Figure 1A). Concurrent echocardiography showed left atrial and ventricular enlargement with thickening of the ventricular wall and septum, with a left ventricular internal diameter of 1.9 cm and a wall thickness of 0.5 cm. The left ventricular ejection fraction was 36%, indicating decreased contractility, but there was no noncompaction of the LV myocardium (Figure 1B). Blood tests revealed metabolic acidosis (pH 7.21, PCO2 59 mmHg, HCO3–0.60 mmol/L), coagulation abnormalities (activated partial thromboplastin time 3.12), creatine kinase levels at 438 U/L, and brain natriuretic peptide levels at 15,638 pg/ml, with normal troponin levels. The neutrophil count was 1.36*109/L. Based on these clinical manifestations, BTHS was suspected. Treatment included continuous low-flow nasal cannula oxygen and anti-infective therapy with piperacillin sodium, tazobactam sodium, sodium creatine phosphate, and oral digoxin to enhance myocardial function. The patient's cardiac function improved significantly, and he was eventually discharged. Subsequent whole-exome sequencing was performed to identify mutations in the couple and their children, revealing a novel DNA splicing site mutation in intron 10 of TAFAZZIN (c.778 -30_778del) (Figure 1C). The mutation was inherited from a mother with no cardiac disease history. According to the American College of Medical Genetics and Genomics criteria (Pathogenic very strong 1 and Moderate 2), this novel variant was likely pathogenic.
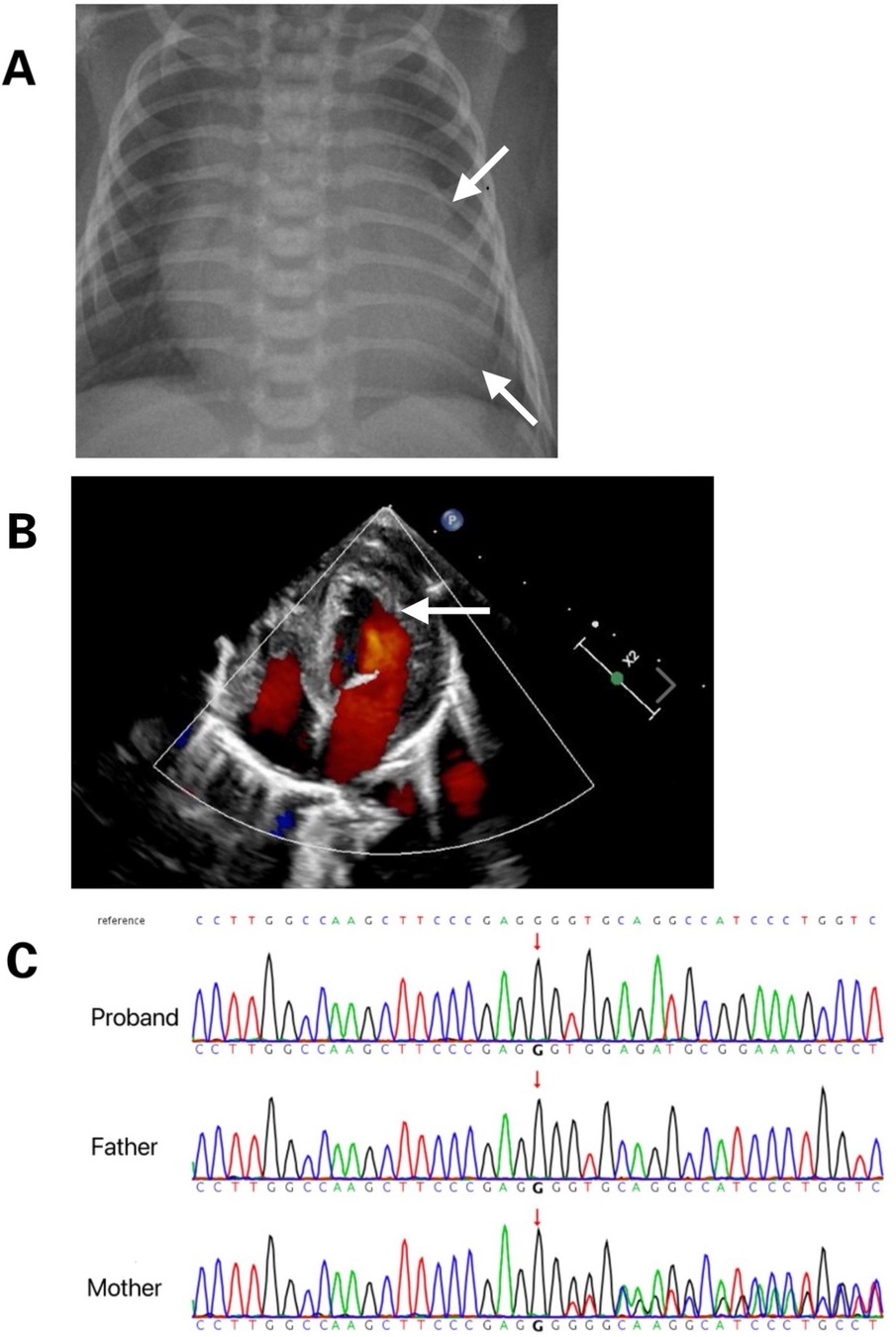
Figure 1. (A) Chest radiograph displays marked cardiomegaly. (B) Cardiac ultrasound reveals myocardial hypertrophy. (C) Tafazzin sequencing electropherogram displays that the patient is hemizygous for the in-frame deletion, c.778- 30_778del (arrow). The variant is not detected in his mother, indicating that it was a de novo mutation.
3 Discussion
BTHS is an X-linked recessive genetic disorder first described by Barth et al. (3) characterized by cardiomyopathy, skeletal myopathy, neutropenia, growth delay, and increased urinary excretion of 3-methylglutaconic acid (3). Additional features include dilation, hypertrophy, and incomplete compaction of the left ventricle, leading to congestive heart failure. Early studies indicated high mortality rates in infants and children (4). Molecular genetic testing for mutations in TAFAZZIN has been used to diagnose BTHS, with over 120 pathogenic mutations identified across all 11 exons and introns of the gene (5). Nicola et al. described younger cousins with dilated cardiomyopathy including myocardial thickening, excessive trabeculation, mild-to-moderate mitral valve insufficiency, and impaired left ventricular contractile function with an ejection fraction of 38%. Older cousins exhibited mild clinical features. A hemizygous DNA splice site mutation (c.777+1G>A) was identified in intron 10 of TAFAZZIN (6) among these cousins. TAE et al. also reported a 13-month-old boy with refractory heart failure attributed to dilated cardiomyopathy due to hemi-frame shift deletion of nine amino acids within exon 10 of TAFAZZIN (c.725_751del, p.Pro242_Glu250del) (7). Similar to the results of our study, these reports indicate that TAFAZZIN mutations may affect cardiac metabolism and dysfunction. Laure et al. reported the first confirmed case of BTHS in a female patient who experienced severe heart failure at one month old diagnosed with dilated, hypo contractile, and hypertrophic cardiomyopathy, with incomplete contraction of the left ventricle (8). Then, a large deletion encompassing the first five exons of TAFAZZIN was confirmed through genetic analysis. BTHS is caused by mutations in TAFAZZIN located on chromosome Xq28, which encodes tafazzin, a protein involved in the remodeling of cardiolipin, a phospholipid vital for mitochondrial membrane integrity. Tafazzin, a phospholipid transacylase, is involved in the remodeling of phosphatidylglycerol and cardiolipin within the inner mitochondrial membrane. Defects in tafazzin enzyme activity lead to cardiolipin loss, disrupting the stability of respiratory chain supercomplexes, and impairing electron transport chain function (9–11). Recent studies have demonstrated that the genetic background of a mouse model of BTHS influenced the phenotypic expression of tafazzin, potentially altering mitochondrial quality control (12). A new mutation identified in China, a DNA splicing site mutation in intron 10 of TAFAZZIN (c.778-30_778del), aids in establishing the genotype-phenotype relationship of tafazzin induced BTHS and improves our understanding of its function.
4 Conclusions
This report shows a splicing site mutation in intron 10 of TAFAZZIN (c.778-30_778del), which is the first genetically confirmed case of BTHS in a Chinese child.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by The Affiliated Hospital of Qingdao University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
MC: Writing – original draft, Writing – review & editing. FL: Software, Writing – original draft. YJ: Methodology, Writing – original draft. QL: Formal Analysis, Writing – review & editing. JH: Validation, Writing – original draft. JZ: Supervision, Writing – original draft. SL: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.This work was supported by the National Natural Science Foundation of China (30971586) and the National Key Research and Development Program of China under Grant No. 2016YFC100307-6.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N. Barth syndrome. Orphanet J Rare Dis. (2013) 8:23. doi: 10.1186/1750-1172-8-23
2. Aprikyan AA, Khuchua Z. Advances in the understanding of Barth syndrome. Br J Haematol. (2013) 161:330–8. doi: 10.1111/bjh.12271
3. Barth PG, Scholte HR, Berden JA, Vanderkleivanmoorsel JM, Luythouwen IEM, Vantveerkorthof ET, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. (1983) 62:327–55. doi: 10.1016/0022-510X(83)90209-5
4. Barth PG, Valianpour F, Bowen VM, Lam J, Duran M, Vaz FM, et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A. (2004) 126:349–54. doi: 10.1002/ajmg.a.20660
5. Taylor C, Rao ES, Pierre G, Chronopoulou E, Hornby B, Heyman A, et al. Clinical presentation and natural history of Barth syndrome: an overview. J Inherit Metab Dis. (2022) 45:7–16. doi: 10.1002/jimd.12422
6. Tovaglieri N, Russo S, Micaglio E, Corcelli A, Lobasso S. Case report: variability in clinical features as a potential pitfall for the diagnosis of Barth syndrome. Front Pediatr. (2023) 11:1250772. doi: 10.3389/fped.2023.1250772
7. Yoo TY, Kim MR, Son JS, Lee R, Bae SH, Chung S, et al. Identification of a novel de novo mutation of the TAZ gene in a Korean patient with Barth syndrome. J Cardiovasc Ultrasound. (2016) 24(2):153–7. doi: 10.4250/jcu.2016.24.2.153
8. Cosson L, Toutain A, Simard G, Kulik W, Matyas G, Guichet A, et al. Barth syndrome in a female patient. Mol Genet Metab. (2012) 106(1):115–20. doi: 10.1016/j.ymgme.2012.01.015
9. Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. (2006) 281:39217–24. doi: 10.1074/jbc.M606100200
10. Houtkooper RH, Turkenburg M, Poll-The BT, Karall D, Perez-Cerda C, Morrone A, et al. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim Biophys Acta. (2009) 1788:2003–14. doi: 10.1016/j.bbamem.2009.07.009
11. Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. (2007) 292(1):C33–C44. doi: 10.1152/ajpcell.00243.2006
Keywords: Barth syndrome, cardiomyopathy, tafazzin, rare x-linked disease, neutropenia
Citation: Che M, Li F, Jia Y, Liu Q, Hu J, Zhang J and Liu S (2024) Case Report: A Chinese child with Barth syndrome caused by a novel TAFAZZIN mutation. Front. Cardiovasc. Med. 11:1465912. doi: 10.3389/fcvm.2024.1465912
Received: 17 July 2024; Accepted: 26 August 2024;
Published: 6 September 2024.
Edited by:
Neil Morgan, University of Birmingham, United KingdomReviewed by:
Feng Jiang, Stanford University, United StatesAtsuhito Takeda, Hokkaido University Hospital, Japan
Copyright: © 2024 Che, Li, Jia, Liu, Hu, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jidong Zhang, MTg2NjE4MDE1NjZAMTYzLmNvbQ==; Shiguo Liu, bGl1c2hpZ3VvQHFkdS5lZHUuY24=