
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 13 November 2024
Sec. Cardiovascular Genetics and Systems Medicine
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1461899
This article is part of the Research Topic Case Reports in Cardiovascular Genetics and Systems Medicine: 2024 View all 12 articles
 Chen Liang1,2
Chen Liang1,2 Jie-Yuan Jin3
Jie-Yuan Jin3 Hai-Hong Shi2
Hai-Hong Shi2 Hao-Xian Li2
Hao-Xian Li2 Lin-Lin Chen4Yang-Hui Zhang5Qin Wang6Qiu-Li Li2Rui-Man Li7*
Lin-Lin Chen4Yang-Hui Zhang5Qin Wang6Qiu-Li Li2Rui-Man Li7*
Factor XI deficiency is a rare inherited coagulation disorder with an estimated prevalence of affecting 1 in 1 million. It is characterized by mild and variable bleeding phenotypes, including bruises, nosebleeds, hematuria, and postpartum hemorrhage. It can be caused by either allelic or biallelic variants in coagulation factor XI (F11). Coagulation factor XI is a glycoprotein that circulates in plasma as a non-covalent complex with high-molecular-weight kininogen. It is converted to an active protease, coagulation factor XIa, which participates in blood coagulation as a catalyst. In this study, we recruited a family with Factor XI deficiency and identified two F11 variants using whole-exome sequencing. One (NM_000128.4: c.841C>T, p.Q281X) was a known variant, and the other (NM_000128.4: c.1832T>G, p.V611G) had not been reported. In addition, we compiled the characteristics of known missense variants in coagulation factor XI. Our findings enriched the variant spectrum of Factor XI deficiency and contributed to the genetic counseling and molecular diagnostics of Factor XI deficiency.
Hemophilia is a group of classical coagulation disorders that can result in excessive bleeding, requiring intervention to restore hemostasis (1). Hemophilia can be categorized into hemophilia A (Factor VIII deficiency, OMIM_306700), resulting from coagulation factor VIII (F8) variants; hemophilia B (Factor IX deficiency, OMIM_306900), caused by coagulation factor IX (F9) variants; and Factor XI deficiency (OMIM_612416), associated with coagulation factor XI (F11) variants (2–4). Compared with Factor VIII and IX deficiencies, Factor XI deficiency is notably rare, with a prevalence of approximately one in one million (1). It typically presents with mild and variable bleeding phenotypes, such as bruises, epistaxis, hematuria, and postpartum hemorrhage, and severe spontaneous bleeding is infrequent (5). This disease is usually found in the following conditions: trauma or surgery-induced excessive bleeding, routine pre-surgical laboratory evaluation, and genetic screening of a probands’ family members (6).
Factor XI deficiency is associated with biallelic variants of F11, whereas certain heterozygous variants in F11 can also result in this disorder, with milder phenotypes and incomplete penetrance (7). Coagulation factor XI is a glycoprotein that circulates in plasma as a non-covalent complex with high-molecular-weight kininogen. It is converted to an active protease, coagulation factor XIa, in the presence of calcium ions, participating in blood coagulation as a catalyst (8, 9). Options for treatment with coagulation factor XI include fresh frozen plasma, coagulation factor XI concentrates, and low-dose recombinant coagulation factor VIIa, with antifibrinolytic agents as an adjunctive therapy (10).
In this study, we reported a family with Factor XI deficiency caused by heterozygous variants or compound heterozygous variants in F11 (NM_000128.4: c.841C>T, p.Q281X; c.1832T>G, p.V611G). Our identification enriched the variant spectrum of Factor XI deficiency. We compiled the features of known missense variants in coagulation factor XI and provided evidence to support genetic counseling and molecular diagnostics in Factor XI deficiency.
This study received ethical approval from the Review Board of Jiangmen Maternal & Child Health Care Hospital [No. 112 (2022)]. A pregnant woman diagnosed with Factor XI deficiency and her family members were recruited. All of the participants (Han ethnic group) provided written informed consent for their participation in this study and the publication of related data. This study was conducted in accordance with the ethical standards of the 1964 Declaration of Helsinki and its subsequent amendments.
Peripheral blood samples of subjects were collected, and their genomic DNAs were extracted. Berry Genomics Company Limited (Beijing, China) performed whole-exome sequencing (WES) of the proband following protocols as previously described (11). Given that the proband was diagnosed with Factor XI deficiency, we specifically analyzed variants in F8, F9, and F11. Variant detection rates were annotated according to GnomAD (http://gnomad.broadinstitule.org) and the Chinese Millionome Database (CMDB; http://cmdb.bgi.com/cmdb/). MutationTaster (http://www.mutationtaster.org), SIFT (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), Combined Annotation Dependent Depletion (CADD) (https://cadd.gs.washington.edu/snv), and MetaDome (https://stuart.radboudumc.nl/metadome/dashboard) were utilized to predict the pathogenicity of the variants. The pathogenicity classification of the variants was confirmed in adherence with the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) (12].
The variants in all of the participants were verified using Sanger sequencing. Gene sequencing of F11 was obtained from the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/gene/2160). Primer pairs (F11-841f: 5′-GCTAGCATGAGCTGACTTTACT-3′, F11-841r: 5′-TCTCAGCCAGAATGCAGAAC-3′; F11-1832f: 5′-GAAGCGTCTGAGTTGATCTGT-3′, F11-1832r: 5′-TTCAGCGTGTTACTGTGGAG-3′) were synthesized by Sangon Biotech Company Limited (Shanghai, China).
The three-dimensional models of coagulation factor XI were downloaded from the AlphaFold database (https://alphafold.ebi.ac.uk/entry/P03951), and PyMOL was used to establish the mutant protein models. The hydrophobicity of coagulation factor XI and its mutant proteins was predicted using Expasy (https://web.expasy.org/protscale/).
Missense variants were documented based on the Human Gene Mutation Database (HGMD) (https://www.hgmd.cf.ac.uk/ac/gene.php?gene=F11) and the PubMed database (https://pubmed.ncbi.nlm.nih.gov/?term=F11+mutation&size=200). Compared with polar amino acids, methionine and glycine are hydrophobic and they have higher hydrophilicity than other non-polar amino acids. The disulfide bond structures of the variants were determined by referring to the AlphaFold database.
The proband (II: 2; Figure 1A) was a 31-year-old woman at 29 weeks of gestation (first pregnancy). According to her self-statement, 4 years ago, she was diagnosed with Factor XI deficiency [her activated partial thromboplastin time was 106.0 s (normal reference range: 28–38 s) and coagulation factor XI level was below 1%] during a preoperative examination of uterine fibroids. Except for a plasma transfusion after the operation (conducted without major bleeding), she did not receive any other treatment. Recently, she had observed an ecchymosis had appeared on her right hand again. She requested that our department assess the risk and severity of Factor XI deficiency in her baby.
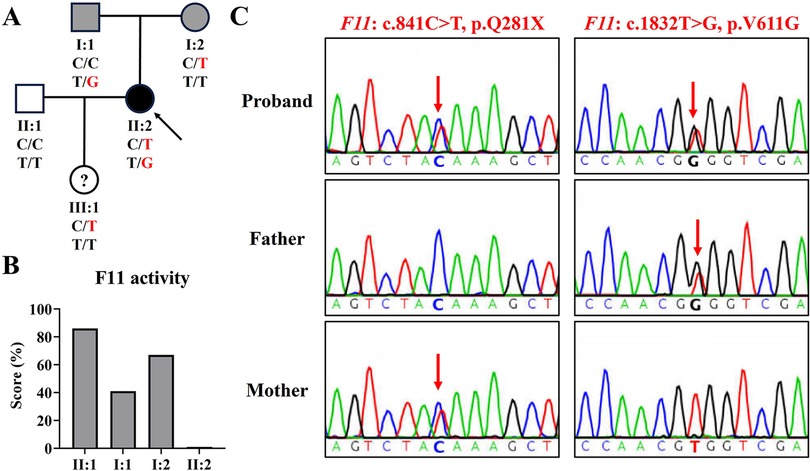
Figure 1. The family genogram, F11 activity, and sanger sequencing of the family. (A) The family pedigree of the family. The black symbol represents the affected member, gray symbols represent mild phenotype, “?” represents phenotype unknown, the arrow indicates the proband, and red text represents the variant. (B) The activity levels of coagulation factor IX in the family. (C) Sequence chromatograms of the proband and her parents. Red arrows indicate variant sites.
At this visit to our department, a coagulation factor XI activity test revealed that her coagulation factor XI activity was less than 1% (normal reference range: 72%–130%), her husband's (II: 1) was 86%, her father's (I: 1) was 41%, and her mother's (I: 2) was 67% (Figure 1B). This indicated that the proband was seriously deficient in coagulation factor XI, and her parents also had mild deficiencies. In addition, her platelet count was 232 × 109 L (normal reference range: 100–300 × 109 L) and her plasma fibrinogen concentration was 2.17 g/L (normal reference range: 2.00–4.00 g/L).
Using WES, we identified two F11 variants (NM_000128.4: c.841C>T, p.Q281X; c.1832T>G, p.V611G) in the proband (Table 1). Variant c.1832T>G, p.V611G was novel and variant c.841C>T, p.Q281X has been reported several times (5, 13). Sanger sequencing confirmed that variant c.841C>T was inherited from her mother and variant c.1832T>G was derived from her father (Figure 1C). Her husband tested negative for both variants.
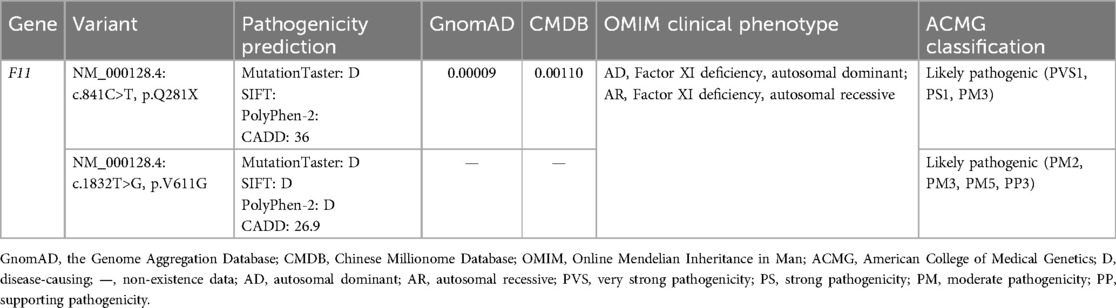
Table 1. The information and pathogenicity classification of the variants identified in the proband.
These two F11 variants were respectively classified as “Pathogenic” and “Likely pathogenic” in adherence with ACMG standards (Table 1). Null variants are one of the known pathogenic mechanisms of Factor XI deficiency, and variant c.841C>T was a nonsense variant (PVS1). This variant had been previously reported (PS1). The two F11 variants were confirmed to be inherited from the patient’s parents (PM3). Variant c.1832T>G was absent from controls in the GnomAD and CMDB (PM2). However, variant p.V611M has been reported, and our variant, c.1832T>G, p.V611G, triggered a change in different residues at the same amino acid site (PM5) (14). p.V611 was highly conserved across evolution and was predicted by MetaDome to be intolerant to functional genetic variation (Figures 2A,B); predictions from MutationTaster, SIFT, PolyPhen-2, and CADD further supported the deleterious impact of variant c.1832T>G (PP3).
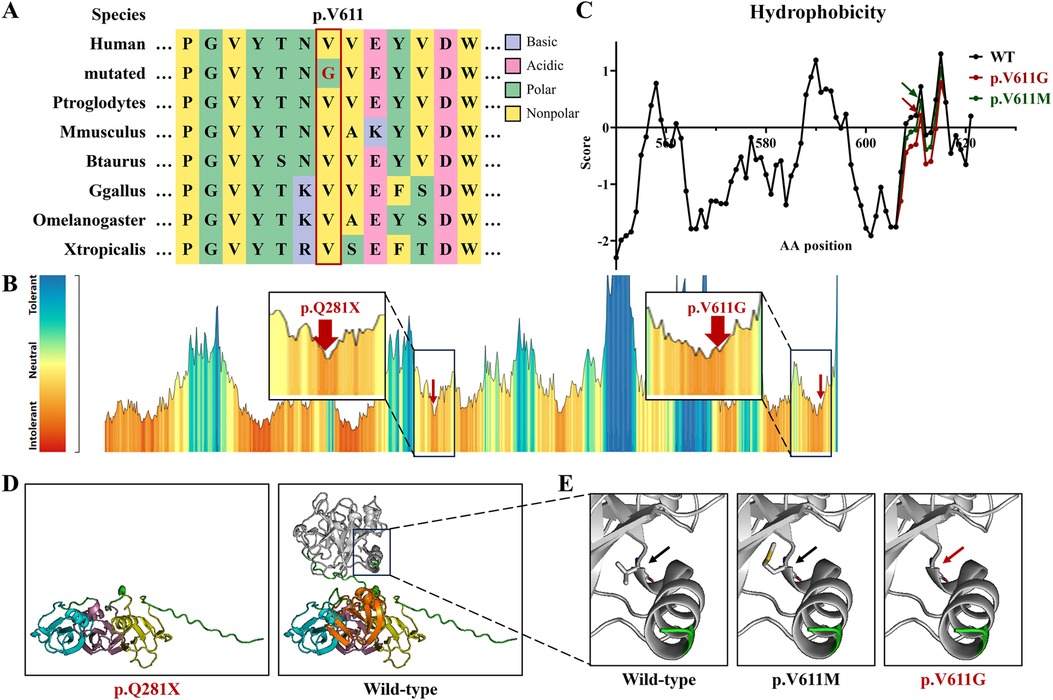
Figure 2. The bioinformatics analysis of the F11 variants in this study. (A) The peptide sequences surrounding the mutated residues (p.V611G) with multiple interspecies alignments. Purple highlights basic amino acids, pink highlights acidic amino acids, green highlights polar amino acids, yellow highlights non-polar amino acids, and red text represents variant p.V611G from this study. (B) The intolerant prediction for functional genetic variations of coagulation factor IX. Red arrows indicate the variants in this study. (C) The hydrophobicity prediction of coagulation factor IX fragments with wild type (black), p.V611M (green), and p.V611G (red). (D) Protein models of coagulation factor IX with wild type, p.Q281X, p.V611M, and p.V611G. Yellow represents the Apple 1 domain, purple represents the Apple 2 domain, blue represents the Apple 3 domain, orange represents the Apple 4 domain, white represents the Protease domain, and red text represents the variants in this study.
The hydrophobicity analysis showed that variant p.V611G reduced hydrophobicity at the variant site and its surrounding regions (Figure 2C). Three-dimensional protein modeling showed that variant p.Q281X produced a truncated protein and that variant p.V611G was positioned in an α-helix, without obvious structural changes (Figure 2D).
A patient follow-up showed that a cesarean section had been performed at 38 + 2 weeks of gestation due to the left occiput transverse (LOT) position of the fetus. The maternal blood loss was 630 ml within 2 h after delivery and 880 ml within 24 h, and was treated by the transfusion of two units of red blood cell (RBC) concentrates. The proband reported that she was in good condition and her baby is a girl (III: 1). Sanger sequencing showed that the baby harbored variant c.841C>T, p.Q281X, but her coagulation factor XI activity was not tested.
Factor XI deficiency is primarily caused by F11 variants and is inherited in autosomal recessive or autosomal dominant patterns (7). Genetic screening is used to identify its etiologies and carry out molecular diagnosis. However, Sanger sequencing is only used to screen for variants in specific sequences, and there is a possibility that F11 variants are not detected, for example, when checking whether a disease is triggered by copy number variations (CNVs) or other gene variants. In contrast, WES can screen for variants in the exons of all genes, rather than a certain gene, which is a more rigorous analysis strategy. Furthermore, WES can also be used to analyze CNVs, although this is not a gold-standard strategy. For these reasons, the proband chose WES, and Sanger sequencing was performed to detect the identified variants in all her family members. She harbored compound heterozygous variants in F11 (NM_000128.4: c.841C>T, p.Q281X; c.1832T>G, p.V611G), resulting in severe Factor XI deficiency, while each parent carried one F11 variant respectively and had a mild deficiency. Typically, homozygous variant carriers exhibit extremely low coagulation factor XI activity (0%–20%), and heterozygous variant carriers display partial deficiency (30%–70%) (15, 16). Our results align with these observations. The individual variants led to coagulation factor XI activity levels of 67% and 41%, respectively, while their compound heterozygous variants resulted in only 1% activity. In addition, we recommended that the proband be careful not to injure the fetus in the womb and talk to her attending doctor about her disease in advance to prepare for the possibility of needing a blood transfusion during her delivery. In fact, she did receive the transfusion of RBC concentrates in her cesarean section. The baby (III: 1) carried the variant c.841C>T, p.Q281X, but it was not tested whether she had a mild Factor XI deficiency.
Coagulation factor XI consists of four N-terminal domains, namely, plasminogen module (PAN)/Apple domains (Apple 1–4), and a serine proteases/trypsin domain (Protease) (9, 17). Apple domains can directly bind to kininogen, coagulation factor XIIa, platelets, coagulation factor IX, and heparin (9, 18, 19). Variant p.V611G is situated in the Protease domain, and a known variant, p.V611M, occurs in the same amino acid site (Figure 3A) (14). They both augment the hydrophilicity of the mutant region, without the destruction of the α-helix, but the increase in hydrophilicity due to our variant was greater than that of p.V611M (Figures 2C,D). The impacts triggered by p.V611G and p.V611M should be verified in vitro and/or in vivo.
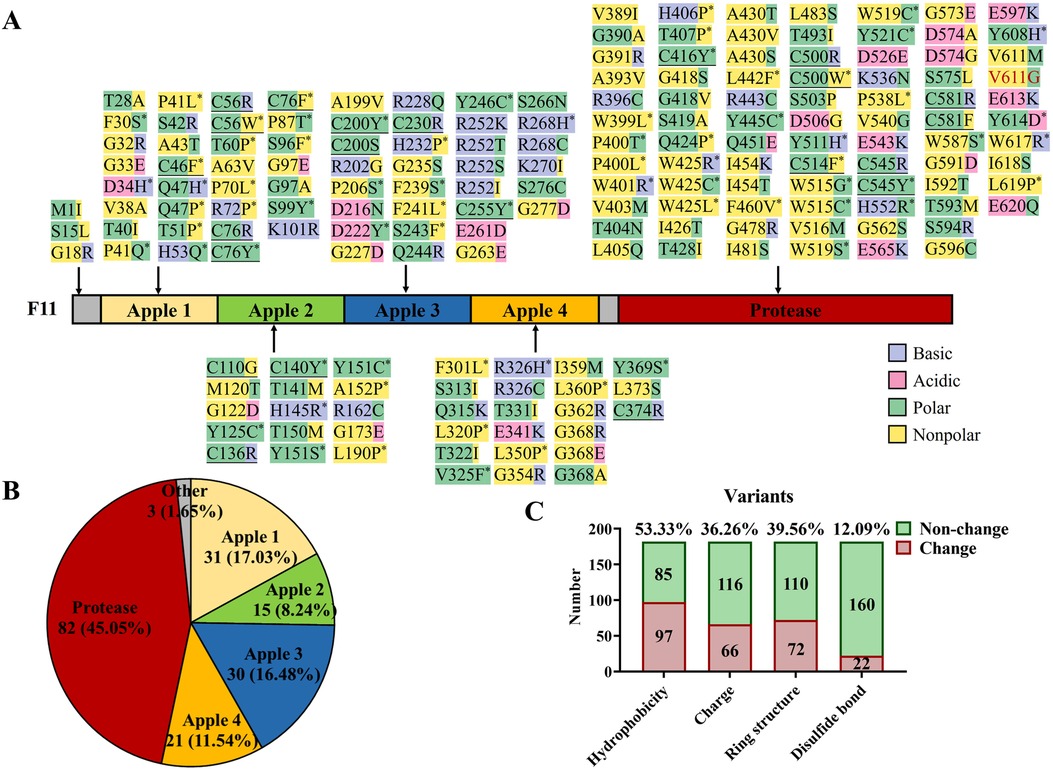
Figure 3. Known missense variants in F11 reported in the literature and this report. (A) Protein structure domains and missense variant collection of coagulation factor IX. Purple highlights basic amino acids, pink highlights acidic amino acids, green highlights polar amino acids, yellow highlights non-polar amino acids, “*” represents the disappearance or acquisition of ring structure in residues, underline represents the destruction of disulfide bonds, and red text represent a variant in this study. (B) The proportion of F11 missense variants in different domains. (C) A general illustration of the effects caused by F11 missense variants.
Variant p.Q281X is positioned in the tail end of the Apple 3 domain, causing a loss of the Apple 4 domain and Protease domain (Figure 2D). According to data from the CMDB, the frequency of this variant in the Chinese population is 1.1 × 10−3 (Table 1) and it may be a founder variant. If half of the heterozygotes of this variant could trigger the lack of coagulation factor IX, the morbidity of Factor XI deficiency in the Chinese population would be far higher than the currently speculated (0.1–246.2 × 10−6) (10). In fact, Asselta et al. estimated that disease-causing F11 heterozygote frequency was 0.0058 in the global population and 0.0090 in the East Asian population (20). This evidence suggests that the incidence of Factor XI deficiency may be severely underestimated. F11 variant screening can help us understand the widespread presence of Factor XI deficiency and prevent related complications.
To gain insight into the characteristics of F11 variants, we mapped 183 known missense variants (the data were from HGMD and literature sources; Figure 3A) (9, 20–26). Of these, 45.05% were located in the Protease domain, the four Apple domains shared 53.29% of the variants, and the other variants only occupied 1.65% in all variants (Figures 3A,B). After further analyzing the effects of these variants, more than half of the variants (53.33%), including our variant p.V611G, were found to change the hydrophobicity, and an electrical charge change (36.26%) and the disappearance or acquisition of a ring structure (39.56%) also were common pathogenic mechanisms. In addition, 12.09% of the variants broke disulfide bonds (Figures 3A,C). Our data can assist clinicians in the assessment of the pathogenicity of novel variants.
In this study, we recruited a Chinese family with Factor XI deficiencies and identified two F11 variants (NM_000128.4: c.841C>T, p.Q281X [known]; c.1832T>G, p.V611G [novel]) in the patients. Our report again revealed that heterozygous variants were responsible for mild Factor XI deficiency and biallelic variants caused severe type. Furthermore, we conducted an extensive review of the features of F11 missense variants. Our findings not only expand the variant map of F11 but also contribute to genetic counseling and molecular diagnostics in Factor XI deficiency.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Review Board of Jiangmen Maternal & Child Health Care Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
CL: Investigation, Writing – original draft. J-YJ: Conceptualization, Writing – review & editing. H-HS: Data curation, Writing – original draft. H-XL: Investigation, Writing – original draft. L-LC: Data curation, Writing – original draft. Y-HZ: Investigation, Writing – original draft. QW: Funding acquisition, Investigation, Writing – original draft. Q-LL: Data curation, Writing – original draft. R-ML: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82200776), the Natural Science Foundation of Guangdong Province, China (2023A1515012741 and 2022A15150121394), the Medical Science and Technology Research Foundation of Guangdong Province (B2023407), the Youth Talent Support Program of the Jiangmen Association for Science and Technology, and Natural Science Foundation of Hunan Province (2023JJ40994 and 2024JJ6693).
We thank the patients and their family members for their participation in this study and the medical workers for their assistance in the clinical examination and blood specimen collection.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lewandowska MD, Connors JM. Factor XI deficiency. Hematol Oncol Clin North Am. (2021) 35:1157–69. doi: 10.1016/j.hoc.2021.07.012
2. Yazdian M, Groeben H, Ataseven B, Schneider S, Baert T, Bommert M, et al. The role of factor XIII in surgery for advanced stage of epithelial ovarian cancer. Arch Gynecol Obstet. (2022) 305:1311–8. doi: 10.1007/s00404-021-06308-z
3. Mullard A. FDA approves first haemophilia B gene therapy. Nat Rev Drug Discov. (2023) 22:7. doi: 10.1038/d41573-022-00199-8
4. Barg AA, Livnat T, Kenet G. Factor XI deficiency: phenotypic age-related considerations and clinical approach towards bleeding risk assessment. Blood. (2024) 143:1455–64. doi: 10.1182/blood.2023020721
5. Sato E, Kawamata N, Kato A, Oshimi K. A novel mutation that leads to a congenital factor XI deficiency in a Japanese family. Am J Hematol. (2000) 63:165–9. doi: 10.1002/(SICI)1096-8652(200004)63:4%3C165::AID-AJH1%3E3.0.CO;2-Q
6. Wheeler AP, Hemingway C, Gailani D. The clinical management of factor XI deficiency in pregnant women. Expert Rev Hematol. (2020) 13:719–29. doi: 10.1080/17474086.2020.1772745
7. Dhaha Y, El Borgi W, Elmahmoudi H, Achour M, Fekih Salem S, Ben Lakhal F, et al. Factor XI deficiency: about 20 cases and literature review. Tunis Med. (2022) 100:60–5.35822334
8. Asakai R, Davie EW, Chung DW. Organization of the gene for human factor XI. Biochemistry. (1987) 26:7221–8. doi: 10.1021/bi00397a004
9. Moellmer SA, Puy C, Mccarty OJT. Biology of factor XI. Blood. (2024) 143:1445–54. doi: 10.1182/blood.2023020719
10. Zhang X, Lewandowska M, Aldridge M, Iglay K, Wolford E, Shapiro A. Global epidemiology of factor XI deficiency: a targeted review of the literature and foundation reports. Haemophilia. (2023) 29:423–34. doi: 10.1111/hae.14687
11. Liang C, Xiang R, Chang SH, Liu MW, Jin JY. Familial congenital heart disease caused by a frameshift variant in glyoxylate reductase 1 homolog (GLYR1). QJM. (2024) 117:297–9. doi: 10.1093/qjmed/hcad281
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
13. Kim J, Song J, Lyu CJ, Kim YR, Oh SH, Choi YC, et al. Population-specific spectrum of the F11 mutations in Koreans: evidence for a founder effect. Clin Genet. (2012) 82:180–6. doi: 10.1111/j.1399-0004.2011.01732.x
14. Rimoldi V, Paraboschi EM, Menegatti M, Peyvandi F, Salomon O, Duga S, et al. Molecular investigation of 41 patients affected by coagulation factor XI deficiency. Haemophilia. (2018) 24:e50–5. doi: 10.1111/hae.13378
15. Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. (1991) 325:153–8. doi: 10.1056/NEJM199107183250303
16. Gerber GF, Klute KA, Chapin J, Bussel J, Desancho MT. Peri- and postpartum management of patients with factor XI deficiency. Clin Appl Thromb Hemost. (2019) 25:1076029619880262. doi: 10.1177/1076029619880262
17. Shearin S, Venkateswarlu D. Structural insights into the activation of blood coagulation factor XI zymogen by thrombin: a computational molecular dynamics study. Biophys Chem. (2022) 281:106737. doi: 10.1016/j.bpc.2021.106737
18. Hill M, Mcleod F, Franks H, Gordon B, Dolan G. Genetic analysis in FXI deficiency: six novel mutations and the use of a polymerase chain reaction-based test to define a whole gene deletion. Br J Haematol. (2005) 129:825–9. doi: 10.1111/j.1365-2141.2005.05536.x
19. Esteban J, De La Morena-Barrio ME, Salloum-Asfar S, Padilla J, Minano A, Roldan V, et al. High incidence of FXI deficiency in a Spanish town caused by 11 different mutations and the first duplication of F11: results from the Yecla study. Haemophilia. (2017) 23:e488–96. doi: 10.1111/hae.13210
20. Asselta R, Paraboschi EM, Rimoldi V, Menegatti M, Peyvandi F, Salomon O, et al. Exploring the global landscape of genetic variation in coagulation factor XI deficiency. Blood. (2017) 130:e1–6. doi: 10.1182/blood-2017-04-780148
21. Jiang S, Chen Y, Liu M, Zeng M, Yang L, Jin Y, et al. Gene variants in two families with inherited coagulation factor XI deficiency and identification of mutations. Acta Haematol. (2023) 146:106–16. doi: 10.1159/000528583
22. Kato T, Yamada M, Watanabe T, Yamanaka S, Fukuhara S, Nakao K. Congenital factor XI deficiency with multiple tooth extractions (case report). Exp Ther Med. (2023) 26:509. doi: 10.3892/etm.2023.12208
23. Paya I, Rios SJ, Santamaria A. A new mutation associated with severe factor XI deficiency. Med Clin (Barc). (2023) 161:176–7. doi: 10.1016/j.medcli.2023.04.012
24. Sucker C, Geisen C, Litmathe J, Schmitt U. Concomitant hypofibrinogenemia and factor XI deficiency as rare cause of bleeding during urgent dentistry: case report and short review of the literature. Arch Clin Cases. (2023) 10:110–3. doi: 10.22551/2023.39.1002.10253
25. Demoy M, Labrousse J, Grand F, Moyrand S, Tuffigo M, Lamarche S, et al. Factor XI deficiency: actuality and review of the literature. Ann Biol Clin (Paris). (2024) 82:225–36. doi: 10.1684/abc.2024.1884
Keywords: Factor XI deficiency, hemophilia, coagulation factor XI, variant characteristic, missense variant
Citation: Liang C, Jin J-Y, Shi H-H, Li H-X, Chen L-L, Zhang Y-H, Wang Q, Li Q-L and Li R-M (2024) Case Report: Allelic and biallelic variants in coagulation factor XI cause factor XI deficiency. Front. Cardiovasc. Med. 11:1461899. doi: 10.3389/fcvm.2024.1461899
Received: 9 July 2024; Accepted: 21 October 2024;
Published: 13 November 2024.
Edited by:
Neil Morgan, University of Birmingham, United KingdomReviewed by:
Thomas Pierre Lecompte, Université de Lorraine, FranceCopyright: © 2024 Liang, Jin, Shi, Li, Chen, Zhang, Wang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui-Man Li, aHF5eWxybUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.