 Jordan M. Bobek
Jordan M. Bobek Gage M. Stuttgen
Gage M. Stuttgen Daisy Sahoo
Daisy Sahoo- 1Department of Biochemistry, Medical College of Wisconsin, Milwaukee, WI, United States
- 2Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI, United States
- 3Division of Endocrinology & Molecular Medicine, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI, United States
Introduction: Recent findings demonstrate that high density lipoprotein (HDL) function rather than HDL-cholesterol levels themselves may be a better indicator of cardiovascular disease risk. One mechanism by which HDL can become dysfunctional is through oxidative modification by reactive aldehydes. Previous studies from our group demonstrated that HDL modified by reactive aldehydes alters select cardioprotective functions of HDL in macrophages. To identify mechanisms by which dysfunctional HDL contributes to atherosclerosis progression, we designed experiments to test the hypothesis that HDL modified by reactive aldehydes triggers endoplasmic reticulum (ER) stress in primary murine macrophages.
Methods and results: Peritoneal macrophages were harvested from wild-type C57BL/6J mice and treated with thapsigargin, oxLDL, and/or HDL for up to 48 hours. Immunoblot analysis and semi-quantitative PCR were used to measure expression of BiP, p-eIF2α, ATF6, and XBP1 to assess activation of the unfolded protein response (UPR). Through an extensive set of comprehensive experiments, and contrary to some published studies, our findings led us to three novel discoveries in primary murine macrophages: (i) oxLDL alone was unable to induce ER stress; (ii) co-incubation with oxLDL or HDL in the presence of thapsigargin had an additive effect in which expression of ER stress markers were significantly increased and prolonged as compared to cells treated with thapsigargin alone; and (iii) HDL, in the presence or absence of reactive aldehydes, was unable blunt the ER stress induced by thapsigargin in the presence or absence of oxLDL.
Conclusions: Our systematic approach to assess the role of native and modified HDL in mediating primary macrophage ER stress led to the discovery that lipoproteins on their own require the presence of thapsigargin to synergistically increase expression of ER stress markers. We further demonstrated that HDL, in the presence or absence of reactive aldehydes, was unable to blunt the ER stress induced by thapsigargin in the presence or absence of oxLDL. Together, our findings suggest the need for more detailed investigations to better understand the role of native and modified lipoproteins in mediating ER stress pathways.
1 Introduction
Cardiovascular disease (CVD) remains the leading cause of death globally. Atherosclerosis, one of the major causes of CVD (1), is an inflammatory disease defined by the progressive buildup of plaques consisting of lipid-laden macrophages, cellular debris, cholesterol, lipids, and lipoproteins in the arteries that increases the likelihood of myocardial infarction and stroke (2). Low-density lipoprotein (LDL) and high-density lipoprotein (HDL) play critical roles in the pathogenesis of atherosclerosis due to their roles in transporting cholesterol between tissues and cell types, including macrophages (3).
Macrophages play an important role in the development of atherosclerosis as they regulate inflammatory responses and promote plaque formation (4). The expression of scavenger receptor CD36 on the macrophage cell surface facilitates the uptake of oxidized LDL (oxLDL). The accumulation of esterified cholesterol from oxLDL in the macrophage initiates foam cell formation in the arterial walls that eventually progresses to atherosclerotic plaque formation [reviewed in (5–7)].
A major cardio-protective function of HDL is its role in reverse cholesterol transport (RCT) where it functions to extract cholesterol from lipid-laden macrophages in peripheral tissues and deliver it to the liver for total body cholesterol excretion (8, 9). This process, combined with other anti-atherogenic properties of HDL [reviewed in (10, 11)], helps to reduce or prevent the formation of atherosclerotic plaque. Epidemiological studies have shown that high levels of HDL-cholesterol are correlated with decreased risk for CVD (12). However the failure of HDL-cholesterol raising therapeutics to show clinical benefit (13, 14) suggests that assessing HDL function may be a more relevant indicator of CVD risk than HDL-cholesterol levels themselves (15).
Under conditions of oxidative stress or inflammation, HDL can be susceptible to oxidation and become dysfunctional (16, 17). HDL dysfunction has been studied in many diseases such as coronary artery disease (18) and chronic kidney disease (19), where HDL isolated from these patients had a decreased ability to perform its normal anti-inflammatory and antioxidant functions. Dysfunctional HDL has also been described in diabetes, obesity, and metabolic syndrome (20–22). While the processes that lead to HDL dysfunction are not fully understood, one possible mechanism is by oxidative modification by reactive aldehydes that have been found to alter lipoprotein function in circulation in disease states such as atherosclerosis (23–26). Previous studies from our group have demonstrated that, unlike native HDL, incubation of macrophages with HDL modified by reactive aldehydes inhibits cell migration, promotes lipid accumulation, and increases reactive oxygen species generation (27, 28), all critical steps in initiating the atherosclerotic process. A better understanding of additional pathways by which dysfunctional HDL promotes pathways that lead to atherosclerosis is critical to preventing this disease.
Numerous studies have linked CVD to endoplasmic reticulum (ER) stress (29–38). Protein misfolding and ER stress can be induced by various factors such as hypoxia, calcium deprivation, impaired glycosylation, oxidative stress, and other pathological conditions (39, 40). These events can activate the unfolded protein response (UPR) which, in turn, stimulates signaling pathways to help eliminate ER stress to maintain homeostasis. Three UPR pathways are activated under conditions of ER stress: PERK, IRE1, and ATF6 [reviewed in (41)]. Each branch of the UPR possesses a unique signal transduction mechanism, but all branches work in parallel to upregulate genes encoding ER chaperones and proteins associated with protein degradation and downregulation of protein synthesis (41). Conditions such as inflammation associated with CVD impose high demands on protein-folding machinery in the ER, thereby inducing ER stress (35, 42). On the other hand, ER stress can exert negative effects on CVD progression as it can induce states of inflammation and oxidative stress (30, 34). It has also been demonstrated that ER stress regulates lipid metabolism in macrophages by modulating the expression of membrane cholesterol transporters, specifically, by increasing expression of the oxLDL receptor, CD36 (43), decreasing expression of cholesterol efflux receptors (44), and increasing de novo cholesterol synthesis (45, 46), thus resulting in increased lipid accumulation and foam cell generation [reviewed in (47)]. Additionally, expression of the UPR marker, phosphorylated IRE1α, was observed in atherosclerotic lesions which also contained high levels of 4-hydroxynonenal (HNE) adducts, a reactive aldehyde produced from lipid peroxidation (48, 49).
Studies examining the role of lipoproteins in mediating ER stress responses remain limited. OxLDL has been shown to upregulate expression of various UPR markers in vascular cells and macrophages, thereby inducing ER stress (49–54). On the other hand, HDL was shown to blunt oxLDL-induced ER stress in the same cell types (55, 56). Based on these findings, the initial goal of this study was to investigate whether modification of HDL by reactive aldehydes would impair HDL's ability to blunt the ER stress response in macrophages. Interestingly, through an extensive set of comprehensive experiments, and contrary to several published studies, we demonstrate that physiological levels of oxLDL do not induce ER stress in murine peritoneal macrophages. Furthermore, ER stress was only induced when oxLDL was co-incubated with the ER stress inducer, thapsigargin. Additionally, we found that the addition of HDL or modified forms of HDL did not protect from ER stress induced by thapsigargin in the presence or absence of oxLDL. Rather both native and modified forms of HDL had similar effects on ER stress as oxLDL.
2 Materials and methods
2.1 Materials
Ammonium-Chloride-Potassium (ACK) lysis buffer, DNase I, high-capacity cDNA reverse transcriptase kit, Roswell Park Memorial Institute (RPMI) 1640, TaKaRa Ex Taq, thapsigargin (TG), thioglycolate medium, and tunicamycin (TM) were purchased from Thermo Fisher Scientific (Waltham, MA). Human HDL, phosphatase inhibitor cocktail, Sandoz 58-035 (ACAT inhibitor) and 1,1,3,3-Tetramethoxypropane for malondialdehyde (MDA) generation were purchased from Sigma-Aldrich (St. Louis, MO). 4-hydroxynonenal was from Cayman Chemical (Ann Arbor, MI) and acrolein was from Ultra Scientific (North Kingstown, RI). The Total RNA Purification Kit was purchased from Norgen Biotek Corp (Thorold, ON, Canada). Rabbit monoclonal antibody against eIF2α (1:1,000), phospho-eIF2α (1:600) and BiP (1:1,000) were from Cell Signaling (Danvers, MA). Human acetylated low-density lipoprotein, mouse monoclonal antibodies against ATF6 (1:1,000) and beta-Actin (1:2,500), and ProLong Gold Antifade Mountant with DNA stain DAPI, were purchased from Thermo Fisher Scientific (Waltham, MA). [1,2-3H (N)]-Cholesterol was purchased from PerkinElmer (Shelton, CT). Human oxLDL was from Lee Biosolutions, Inc (Maryland Heights, MO). Horseradish peroxidase (HRP)-conjugated donkey-anti-rabbit-IgG (1:10,000) and sheep-anti-mouse IgG secondary (1:10,000) antibodies were purchased from Cytiva (Amersham, UK). All other reagents were of analytical grade.
2.2 Animal care
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. Wild type C57BL6/J mice [originally purchased from Jackson Laboratories (Bar Harbor, ME)] were housed and bred in a pathogen-free barrier facility in accordance with federal and institutional guidelines under a normal light-dark cycle and fed a standard chow diet.
2.3 Isolation and maintenance of peritoneal murine macrophages
Male and female wild type C57BL6/J mice between 8 and 12 weeks of age received intraperitoneal injections of 4% thioglycolate medium (2 ml per mouse). The inflammatory response was allowed to proceed for 4 days prior to euthanasia by CO2 inhalation followed by cervical dislocation. Cold RPMI media containing 10% FBS + 1% Pen/Strep was injected into the peritoneal cavity and the abdomen was gently massaged before the peritoneal fluid was extracted. The peritoneal exudate cells (PECs) were pelleted by centrifugation at 400 × g for 10 min at 4°C. The PECs were resuspended with ACK lysis buffer and incubated on ice for 10 min to lyse contaminating red blood cells. PECs were pelleted again by centrifugation and resuspended in RPMI media containing 10% FBS + 1% Pen/Strep. Macrophages were counted using an automated cell counter (BioRad) and plated for experiments. Cells were maintained in RPMI media containing 10% FBS + 1% Pen/Strep and incubated at 37°C/5% CO2 for at least 24 h prior to treatment. The medium was replaced with serum-free RPMI media during treatments for up to 48 h.
2.4 Cell Lysis
Whole cell lysates were harvested from cells by incubating with radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors (aprotinin, leupeptin, phenylmethylsulfonylfluoride and pepstatin) and phosphatase inhibitor cocktail on ice for 10 min. Lysates were clarified by centrifugation at 8,000 × g for 10 min at 4°C. Total protein concentration of lysates was determined by the Lowry method (57).
2.5 Isolation and modification of human lipoproteins
Human LDL (d = 1.063 g/ml) and HDL (d = 1.210 g/ml) were isolated from human plasma via density gradient ultracentrifugation (58). Lipoproteins were dialyzed overnight against PBS at 4°C and purity was verified using fast protein liquid chromatography (FPLC) analysis. To generate oxLDL, isolated LDL was oxidized by dialysis against 5 µM copper sulfate (CuSO4) in PBS for 6 h at 37°C, and the reaction was stopped by dialysis in PBS containing 0.02% (0.54 nM) EDTA overnight at 4°C. An additional dialysis against PBS for 6 h at 4°C was performed to remove any remaining EDTA (59). Thiobarbituric acid reactive substance (TBAR) and electrophoretic mobility shift (EMSA) assays were performed to verify oxidation levels and changes in charge, respectively. Lipoproteins were used within three weeks of isolation and stored under argon in 4°C to prevent oxidation. To generate the modified forms of HDL, HDL was incubated with 500 µM HNE, 250 µM acrolein, or 15 µM MDA at 37°C for 18 h as previously described (28). MDA was generated prior to each reaction (60). Each reaction was terminated with 20-fold molar excess of aminoguanidine hydrochloride to scavenge excess aldehyde. Modified forms of HDL were used in experimentation immediately and modification was verified using EMSA assays and immunoblot analyses as previously described (28).
2.6 Immunoblot analysis
Cellular lysates (25 µg) combined with an equal volume of sample treatment buffer containing 5% 2-mercaptoethanol were boiled for 5 min at 95°C, separated by 10% SDS-PAGE, and wet-transferred to a nitrocellulose membrane. The membranes were blocked in 5% milk or BSA in tris-buffered saline with tween (TBST) at room temperature and then incubated with the primary antibody in 1% milk or BSA overnight at 4°C. Membranes were washed 3 times in TBST at room temperature and then incubated with horseradish peroxidase-conjugated secondary antibody in 1% milk at room temperature for 1 h. SuperSignal West Pico or Fempto Chemiluminescent Substrate (Thermofisher) was allowed to react with membranes for 5 min before blots were imaged using a ChemiDoc MP. The intensity/optical density values of the bands were measured using Bio Rad Image Lab Software and normalized by a house-keeping protein (β-actin).
2.7 RNA extraction and semi-quantitative reverse transcriptase PCR analysis
Total RNA was extracted using the Total RNA Purification Kit (Norgen Biotek Corp.) following the manufacturer's instructions. Contaminating DNA was digested with DNase I (ThermoFisher) before 1 µg total RNA was reverse-transcribed into single-stranded cDNA using the high-capacity cDNA reverse transcriptase kit (ThermoFisher). The following primers were designed and purchased from IDT Technologies (Coralville, IA): XBP1 forward primer: 5’-AGA GGT GGA GGC CAA GGG GAG T-3’, reverse primer: 5’-GGG TCC AAC TTG TCC AGA ATG CCC-3’ (length of 200 bp for unspliced product and 174 bp for spliced product). The primers were synthesized by TaKaRa Ex Taq (Fisher) and the products were separated on a 3% agarose gel containing ethidium bromide and imaged using a ChemiDoc MP.
2.8 Free cholesterol efflux to HDL
Bone-marrow derived macrophages were plated at a density of 1.0 × 106 cells in a 12-well plate and incubated at 37°C/5% CO2 overnight. Cells were labeled with [3H]cholesterol for 24 h and then allowed to equilibrate for 24 h in RMPI containing 0.2% BSA before adding 50 μg/ml HDL for 4 h. Free cholesterol efflux to HDL was calculated by dividing the radioactive counts in the media by the total radioactivity in the media and cells. All incubations were performed in the presence of ACAT inhibitor.
2.9 Macrophage foam cell formation
Peritoneal macrophages were seeded in a 6-well plate on a cover slip at a density of 0.75 × 106 cells per well and incubated at 37°C/5% CO2 overnight. Media was replaced with serum-free RMPI containing 0–25 μg/ml oxLDL or acetylated LDL (acLDL) for 24 h at 37°C/5% CO2. The cells were washed, fixed in 4% paraformaldehyde, and then treated with a 0.156% Oil Red O (ORO) stain for 5 min. After washing the cells, the cover slips were mounted onto a glass slide with ProLong Gold Antifade Mountant with DAPI and sealed with clear nail polish. The slides were imaged using a Keyence All-in-One Fluorescence Microscope (BZ-X800) and ImageJ was used to quantify the total number of “foam cells” and total number of cells stained with DAPI.
2.10 Electrophoretic mobility shift assay
Lipoproteins (10 μg) were separated on a 1% agarose gel for 30 min at 100 V. Gels were stained with Coomassie Blue for 30 min and de-stained overnight. The charge differences between the native and oxidized forms of lipoproteins were calculated by comparing migration distances on the gel.
2.11 Data normalization and statistical analyses
Each experiment was repeated at least three times, with macrophages harvested from three independent isolations. Densitometry data was normalized to the housekeeping gene and expressed as either relative intensity or further normalized to the positive control to serve as the baseline. Statistical analyses were performed using GraphPad Prism (San Diego, CA), with details of each analysis provided in Figure Legends.
3 Results
3.1 Thapsigargin induces ER stress in murine peritoneal macrophages
We applied a highly systematic approach to assess the role of lipoproteins in mediating ER stress. A key first step in this process was to establish a good positive control for ER stress in our primary peritoneal macrophage model system. Thapsigargin is a known inducer of ER stress due to its role in inhibiting the sarco-/endoplasmic reticulum calcium ATPase and disrupting calcium homeostasis (61). While it is a commonly-used positive control in ER stress experiments, to our knowledge, there are no studies that address appropriate concentration ranges of thapsigargin for use in primary macrophages. We treated peritoneal macrophages from C57BL/6 wild-type mice with increasing concentrations of thapsigargin ranging from 0.1 to 1 µM for 8 and 24 h and analyzed BiP expression. BiP, also referred to as GRP78, is a central regulator for ER stress and can control the activation of the three ER stress pathways [reviewed in (62)]. While all tested concentrations of thapsigargin induced BiP expression, the lowest concentration of 0.1 µM was sufficient to induce BiP expression at both 8 and 24 h (Figures 1A,B). Additionally, we examined the splicing of XBP-1 at the mRNA level to ensure the IRE1 UPR pathway responded in a similar fashion. Comparable findings were observed in which all concentrations of thapsigargin induced splicing of XBP-1, with 0.1 µM thapsigargin being sufficient at both time points to induce splicing (Figure 1C). We therefore decided to use 0.1 µM thapsigargin (unless otherwise specified) as a positive control for ER stress in all experiments described in this study.
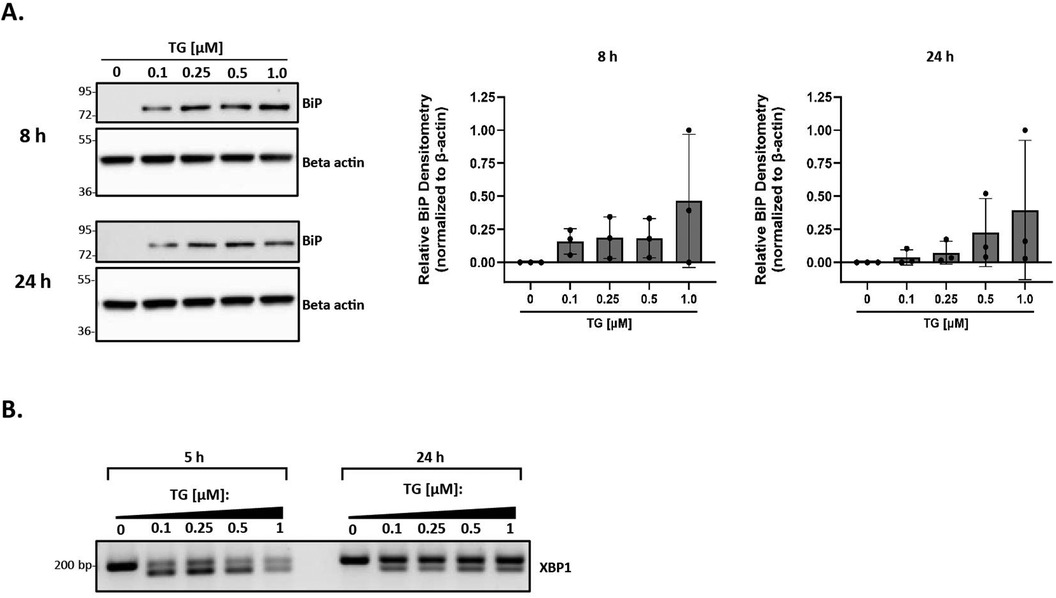
Figure 1. Thapsigargin induces ER stress in murine peritoneal macrophages. Murine peritoneal macrophages were treated with 0–1 µM thapsigargin (TG) (in a final concentration of 0.5% DMSO) for 8 h (A) or 24 h (B) Whole cell lysates were harvested and BiP expression was analyzed by immunoblot analyses. Densitometry was used to calculate changes in band intensity normalized to the housekeeping gene, beta-actin. Blots shown are representative of n = 3 independent macrophage isolations. (C) RNA was isolated and reverse transcribed to cDNA. Primers designed for the unspliced (200 bp) and spliced (174 bp) forms of XBP-1 were used in a semi-quantitative PCR reaction and products were separated on a 3% agarose gel. The gel shown is representative of n = 3 independent macrophage isolations.
3.2 OxLDL alone does not induce ER stress in murine peritoneal macrophages at physiological concentrations
The levels of oxLDL in the human body range from 6 µg/ml to 33 µg/ml, with a concentration above 15 µg/ml being common in patients with coronary artery disease (63). However, most reports demonstrating oxLDL-induced ER stress in RAW 264.7 and THP-1 macrophages (43, 52, 54, 56) used supra-physiological levels of 100 µg/ml oxLDL or above. As such, our initial goal was to demonstrate that we could reproduce ER stress in the presence of oxLDL as observed in previous studies (43, 52, 54), but at physiological concentrations. Peritoneal macrophages were treated with oxLDL ranging from 25 to 100 µg/ml (total protein concentration) for up to 24 h and UPR markers, BiP and spliced XBP-1, were analyzed to assess ER stress. We repeatedly found that treatment with oxLDL alone at all concentrations was unable to induce BiP expression (Figure 2A) or splicing of XBP-1 (Figure 2B) at all tested time points. However, we were able to demonstrate that co-incubation of macrophages with thapsigargin and oxLDL for 8 h induced BiP expression up to 5.5-fold for all concentrations of oxLDL as compared to macrophages treated with thapsigargin alone (Figure 2A). Furthermore, co-incubation of macrophages with thapsigargin and oxLDL for 24 h induced 10- to 13-fold higher levels of BiP expression as compared to thapsigargin-treated macrophages (Figure 2A). Additionally, splicing of XBP-1 was evident in macrophages co-treated with 25–100 oxLDL µg/ml and thapsigargin at all concentrations at 5 and 24 h (Figure 2B). Based on these findings, we chose to use 25 µg/ml oxLDL for subsequent experiments to keep concentrations of this lipoprotein within physiological levels.
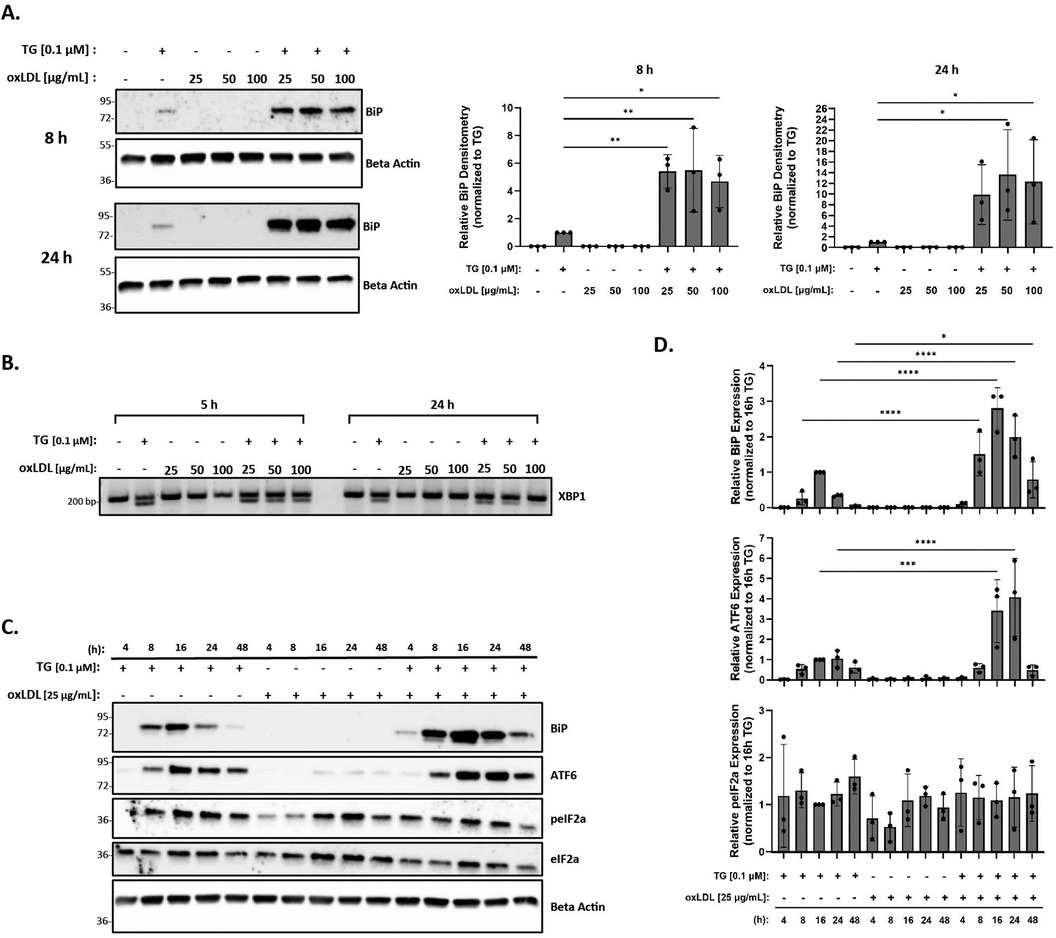
Figure 2. OxLDL does not induce ER stress in murine peritoneal macrophages at physiological concentrations. Murine peritoneal macrophages were treated with 25–100 µg/ml oxLDL in the presence or absence of 0.1 µM thapsigargin (TG). (A) Whole cell lysates were harvested and analyzed for BiP expression by immunoblot analyses. (B) RNA was isolated, and reverse transcribed to cDNA. Primers designed for the unspliced (200 bp) and spliced (174 bp) forms of XBP-1 were used in a semi-quantitative PCR reaction and products were separated on a 3% agarose gel. (C) Peritoneal macrophages were treated with 25 µg/ml oxLDL in the presence or absence of 0.1 µM thapsigargin for 4–48 h. Whole cell lysates were harvested and analyzed for expression of several ER stress markers by immunoblot analyses. (D) Densitometry was performed to calculate changes in band intensity. Data were first normalized to beta-actin and then normalized to thapsigargin expression at 16 h. Statistical analyses were determined by one-way ANOVA with Sidak's multiple comparison post hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Immunoblots and agarose gels are representative of n = 3 independent isolations.
Next, we investigated the ER stress response over a time course in macrophages treated with oxLDL, but this time, we included additional UPR markers in our assessment. In these studies, peritoneal macrophages were treated with 25 µg/ml oxLDL in the presence or absence of thapsigargin at various time points between 4 and 48 h. BiP, p-eIF2α, and ATF6 protein expression were then quantified to assess ER stress. While we did not observe ER stress in the presence of oxLDL alone at all time points, we did observe an initiation of ER stress by 8 h, a maximal ER stress response around 16 h, and a resolution of ER stress by 48 h in thapsigargin-treated macrophages (Figure 2C). Similar expression patterns of BiP and ATF6 in macrophages co-treated with oxLDL and thapsigargin were observed, with early signs of stress being evident at 4 h. Again, maximal ER stress was observed at 16 h and persisted up to 48 h (Figures 2C,D). However, no apparent pattern was observed in phosphorylated eIF2α across the various treatment groups and time course. Additional time points were also investigated for splicing of XBP-1 and no changes in response to oxLDL treatments alone were observed across time points (Supplementary Figure S1).
3.3 HDL does not blunt the ER stress response induced by thapsigargin in the presence or absence of oxLDL
Previous studies in vascular cells and THP-1 macrophages have shown that HDL reduces ER stress induced by oxLDL in the presence of tunicamycin (an ER stress inducer) (56). Therefore, we hypothesized that the addition of HDL to thapsigargin-treated macrophages in the presence or absence of oxLDL would similarly result in decreased ER stress. In this set of studies, peritoneal macrophages were incubated with 25–100 μg/ml HDL in the presence or absence of thapsigargin for 8 and 24 h. As expected, we found that incubation with HDL alone was unable to trigger ER stress at 8 or 24 h across concentrations (Figure 3A). Contrary to what we expected, we observed that the addition of HDL to thapsigargin-treated peritoneal macrophages resulted in a four- and five-fold increase in BiP expression at 8 and 24 h, respectively, as compared to macrophages treated with thapsigargin only (Figures 3A,D). Furthermore, co-treatment of macrophages with thapsigargin and HDL also resulted in splicing of XBP1 at 5 or 24 h (Figure 3B). Similar to what we observed with oxLDL, co-incubation of HDL with thapsigargin resulted in maximal expression of BiP and ATF6 around 16 h, with resolution of stress occurring after 48 h and variable phosphorylation of eIF2α between treatment groups and times (Figures 3C,D).
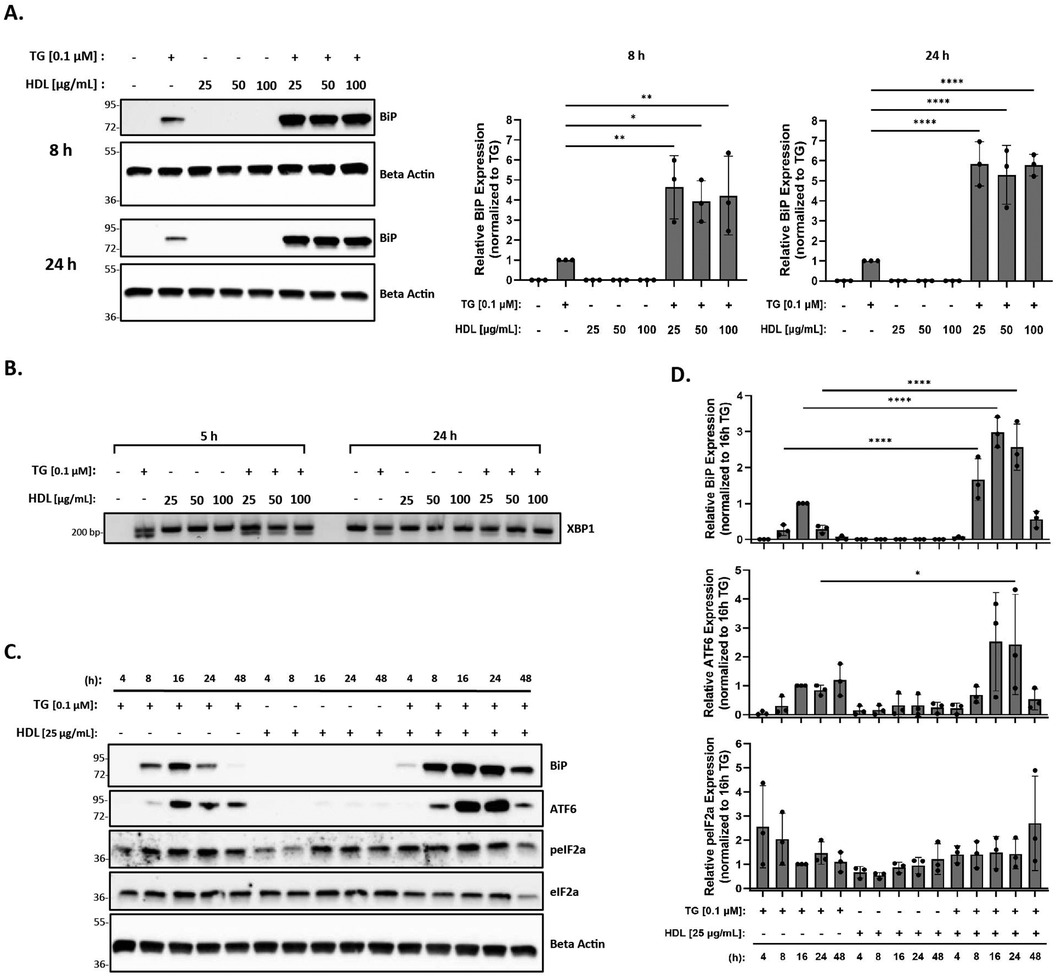
Figure 3. HDL does not blunt the ER stress response induced by thapsigargin. Murine peritoneal macrophages were treated with 25–100 µg/ml HDL in the presence or absence of 0.1 µM thapsigargin (TG). (A) Whole cell lysates were harvested and analyzed for BiP expression by immunoblot analyses. (B) RNA was isolated, and reverse transcribed to cDNA. Primers designed for the unspliced (200 bp) and spliced (174 bp) forms of XBP-1 were used in a semi-quantitative PCR reaction and products were separated on a 3% agarose gel. (C) Peritoneal macrophages were treated with 25 µg/ml HDL in the presence or absence of 0.1 µM thapsigargin for 4–48 h. Whole cell lysates were harvested and analyzed for expression of several ER stress markers by immunoblot analyses. (D) Densitometry was performed to calculate changes in band intensity. Data were first normalized to beta-actin and then normalized to thapsigargin expression at 16 h. Statistical analyses were determined by one-way ANOVA with Sidak's multiple comparison post hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All immunoblots and agarose gels are representative of n = 3 independent isolations.
Finally, as HDL has been shown to blunt oxLDL-mediated ER stress in vascular cells and THP-1 macrophages (56), we investigated the effects of HDL on macrophage ER stress induced by oxLDL in the presence of thapsigargin. Peritoneal macrophages were treated with thapsigargin and 25 µg/ml of oxLDL and HDL for various time points. Protein expression of BiP, p-eIF2α, and ATF6, along with splicing of XBP-1 were analyzed. We observed that the addition of HDL did not change the expression of BiP, p-eIF2α or ATF6 that was already induced by thapsigargin and oxLDL (Figure 4A). In fact, based on the densitometry of the bands, it appears that expression of ER stress markers may increase, although the changes are not statistically significant when all replicates are combined (Figure 4B). Similar patterns were noted for splicing of XBP1 at 5 h, however, by 24 h, only unspliced XBP1 was observed (Figure 4C).
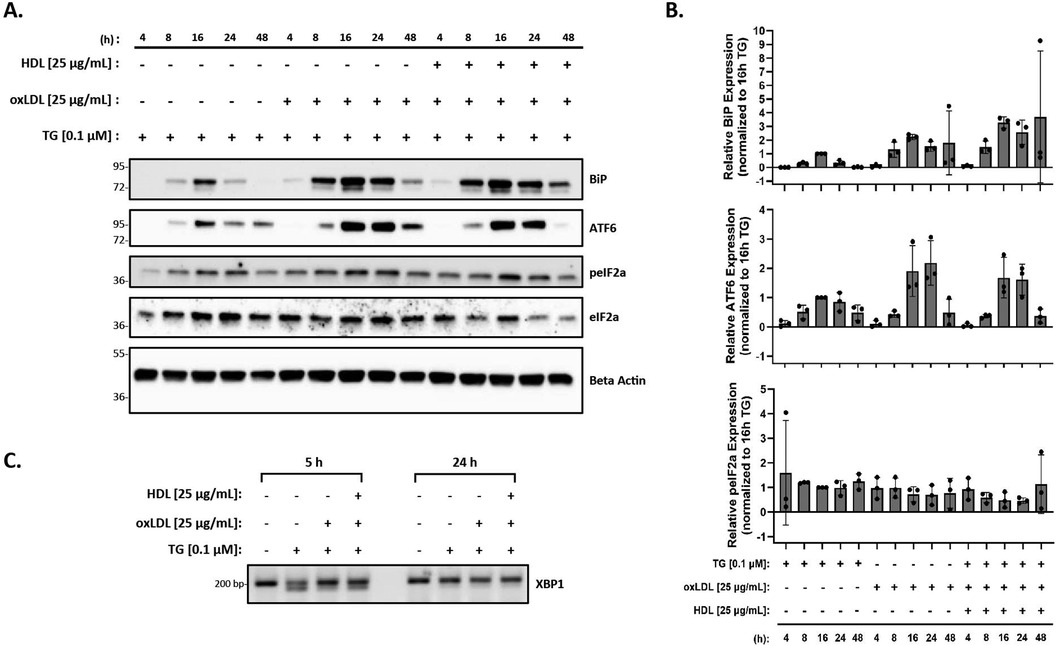
Figure 4. HDL does not blunt the ER stress response induced by oxLDL in the presence of thapsigargin. Murine peritoneal macrophages were treated with 0.1 µM thapsigargin (TG) in the presence or absence of 25 µg/ml oxLDL and/or HDL for indicated treatment times. (A) Whole cell lysates were harvested and analyzed by immunoblot analyses. (B) Densitometry was performed to calculate changes in band intensity. Data were first normalized to beta-actin, and then normalized to thapsigargin expression at 16 h. Statistical analyses were determined by one-way ANOVA with Sidak's multiple comparison post hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (C) RNA was isolated, and reverse transcribed to cDNA. Primers designed for the unspliced (200 bp) and spliced (174 bp) forms of XBP-1 were used in a semi-quantitative PCR reaction and products were separated on a 3% agarose gel. The immunoblots and gel shown are representative of n = 3 independent isolations.
3.4 Observations are independent of lipoprotein source, ER stressor, and cell type
The fact that we were unable to reproduce observations from previous studies raised several questions. First, we wanted to investigate an alternative ER stressor, tunicamycin, to use in our co-incubations. It has been demonstrated that THP-1 macrophages co-treated with oxLDL and tunicamycin had increased ER stress as compared to macrophages treated with tunicamycin; furthermore, HDL was able to reduce this ER stress (56). We tried replicating these findings and observed increased BiP expression in macrophages treated with oxLDL in the presence of tunicamycin as compared to macrophages treated with tunicamycin only (Supplementary Figure S2). However, we did not observe reduced expression of BiP in the presence of tunicamycin and HDL.
Second, we wanted to test whether the quality, source and function of lipoproteins were variables in our studies. In these experiments, we tested two batches of lipoproteins. In the first set, we examined ER stress in the presence of oxLDL and HDL that were purchased from a vendor. In the second set, HDL and LDL were isolated by density gradient ultracentrifugation (DGUC) of human plasma (58), and isolated LDL was then oxidized using the copper sulfate method [adapted from (64)]. As shown in Supplementary Figure S3A, we observed the same ER stress patterns with both sources of oxLDL, where neither batch of oxLDL was able to induce ER stress on its own. Similarly, HDL from either source was unable to blunt ER stress in response to a co-incubation of oxLDL or tunicamycin (Supplementary Figure S3B). We additionally performed an extra purification step during the isolation of HDL by DGUC by performing FPLC on the HDL fraction to ensure the removal of the Lp(a) fraction. Interestingly, this batch of HDL resulted in a greater additive effect on ER stress when incubated with thapsigargin-treated macrophages in the presence or absence of oxLDL (Supplementary Figure S3C). Therefore, the lipoprotein source was excluded as a possible variable in our studies. Furthermore, all data presented in this study relied on lipoproteins isolated by DGUC. Finally, we verified the function of our lipoproteins in primary macrophages. Specifically, we performed a cholesterol efflux assay with HDL and demonstrated that HDL could accept [3H]cholesterol that was effluxed from bone marrow derived macrophages (Supplementary Figure 4A). Additionally, we incubated peritoneal macrophages with oxLDL for 24 h and established that it could generate foam cells as seen by increased Oil red O staining. (Supplementary Figure S4B).
Last, we investigated ER stress in response to lipoproteins in two other cell types. We first tested bone marrow-derived macrophages as an alternate primary macrophage model. However, we observed the same trends in these macrophages such that HDL is unable to blunt oxLDL-induced ER stress in the presence of thapsigargin (Supplementary Figure S5A). We next repeated a similar experiment in human microvascular endothelial cells (HMEC-1s). Unlike what was previously reported (49), we did not observe oxLDL-induced ER stress in this cell line, nor was HDL able to blunt the ER stress response (Supplementary Figure S5B).
3.5 Native HDL and modified HDL exhibit similar ER stress patterns
The initial goal of this study was to investigate whether modification of HDL by reactive aldehydes would impair HDL's ability to blunt the ER stress response in macrophages. Despite our inability to demonstrate that HDL on its own could blunt ER stress induced by thapsigargin in the presence or absence of oxLDL, we moved forward to test whether HDL modified by reactive aldehydes could increase ER stress levels. HDL was modified by acrolein, malondialdehyde (MDA) or 4-HNE and the presence of modifications was verified as previously described (27, 28, 60). In addition, TBAR or EMSA assays were performed to verify oxidation levels and changes in charge, respectively (Supplementary Figures S6). As shown in Figure 5, all three forms of modified HDL exhibited similar ER stress patterns as HDL, suggesting that the presence of reactive aldehydes does not alter HDL's role in ER stress response pathways unless in the presence of thapsigargin.
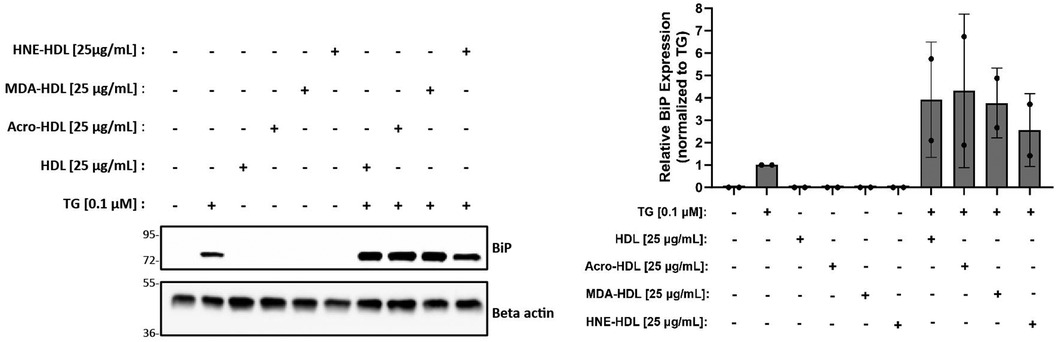
Figure 5. HDL modified by reactive aldehydes does not induce ER stress. Murine peritoneal macrophages were treated with 0.1 µM thapsigargin (TG) in the presence or absence of 25 µg/ml native HDL or HDL modified by acrolein, MDA, or 4-HNE for indicated treatment times. Whole cell lysates were harvested and analyzed by immunoblot analyses. Densitometry was performed to calculate changes in band intensity. Data were first normalized to beta-actin, and then normalized to thapsigargin expression. The immunoblot shown is representative of n = 2 independent isolations.
4 Discussion
Our initial goal of investigating modified HDL's role in mediating macrophage ER stress was challenged by our inability to replicate data that would serve as controls in our originally planned experiments. However, our observations did lead us to systematically determine the role of oxLDL and HDL in mediating ER stress responses in thioglycolate-elicited murine peritoneal macrophages. Our comprehensive studies led us to three novel discoveries in macrophages: (i) oxLDL alone was unable to induce ER stress; (ii) co-incubation with oxLDL HDL, or modified forms of HDL in the presence of thapsigargin had an additive effect on expression of ER stress markers that was prolonged as compared to cells treated with thapsigargin alone; and (iii) HDL, in the presence or absence of reactive aldehydes, was unable blunt the ER stress induced by thapsigargin in the presence or absence of oxLDL.
Our experimental conditions were modeled after numerous studies that have shown that oxLDL induces ER stress in macrophages (43, 52, 54, 56). Most studies report the use of 0.5 μM–1 μM thapsigargin to induce ER stress (61), and we questioned whether this range was too high, and possibly explained why we were unable to observe a decrease in ER stress when HDL was added to cells in some of our initial studies (data not shown). After testing a range of concentrations, we found that as little as 0.1 μM thapsigargin alone or in the presence of oxLDL was sufficient to induce ER stress in macrophages (Figures 1, 2). However, even at this lower concentration, HDL was still unable to decrease ER stress induced by oxLDL co-incubated with thapsigargin (Figure 4).
Several reports also used supra-physiological concentrations of lipoproteins in their studies that may not be suitable for cell culture treatments (63, 65). In our studies, our goal was to identify a range of lipoproteins concentrations that would be physiologically relevant and sufficient to demonstrate ER stress in the presence of thapsigargin (Figure 2). Based on our data, we chose to use 25 ug/ml for both oxLDL and HDL as it is on the lower end of physiological concentrations. Further, 25 ug/ml oxLDL is also sufficient to trigger phenotypic switching in murine primary peritoneal macrophages (66), a process that could be a trigger for the initiation of atherosclerosis.
Based on previous studies (55, 56), we hypothesized that the addition of HDL to oxLDL- and thapsigargin-treated macrophages would result in decreased ER stress. It has been proposed that HDL reduces ER stress by removing excess cholesteryl esters that were likely deposited into macrophages by oxLDL. However, we did not observe a decrease in ER stress induced by oxLDL in the presence of thapsigargin with the addition of HDL (Figure 4). Like Muller et al. (55), we tried pre-incubating macrophages with HDL prior to treatment with oxLDL in the presence or absence of thapsigargin. We also performed experiments where macrophages were first lipid-loaded with oxLDL in the presence or absence of thapsigargin for up to 24 h, prior to treatment with HDL (56). However, both methods resulted in the same observations where oxLDL did not induce ER stress and HDL could not reduce the additive ER stress induced by the co-incubation of thapsigargin and oxLDL (data not shown). Even more surprising was that HDL co-incubated with thapsigargin resulted in the same additive ER stress response as observed with oxLDL. While the mechanisms driving this synergistic effect are unknown, we speculate that oxLDL and HDL may mediate independent signaling pathways that lead to the ER stress response. Another explanation could be due to the fact that HDL particles are quite heterogeneous as shown by proteomic analyses (67), and it is possible that unique proteins within the HDL proteome that may be able to enhance the amount of ER stress when stress is already present in the cell. Further, while we layer HDL with argon to prevent oxidation, we cannot exclude the possibility that the HDL we use may harbor certain modifications (other than reactive aldehydes) that could be responsible for altering HDL's ability to blunt thapsigargin- or oxLDL-induced ER stress. Further investigations are required to better understand HDL's role in the ER stress response.
Given that HDL itself was unable to blunt ER stress, it was difficult to test our original hypothesis that modification of HDL by reactive aldehydes would prevent HDL's ER stress blunting functions. In fact, our observations suggested that HDL, whether modified by acrolein, HNE or MDA, behaved just like unmodified HDL as demonstrated by similar levels of BiP expression in the presence of thapsigargin (Figure 5). The finding that modified forms of HDL, on their own, did not induce ER stress in the macrophages was somewhat surprising as we have previously demonstrated that modified HDL increases lipid accumulation in macrophages (27), a process that has been shown to be accompanied by ER stress (50). In fact, it has also been shown that copper-modified HDL causes intracellular lipid accumulation (51, 68) in which promotes pro-atherogenic mechanisms such as ER stress-induced apoptosis (51) and inflammation (68).
The unfolded protein response is an adaptive cellular response that helps to decrease ER stress. As such, the expression of ER stress markers should start to decrease after homeostasis is restored. This made us question whether we were missing the time point at which oxLDL begins to trigger ER stress in our macrophage model. Therefore, we assessed ER stress markers at various time points ranging from 4 to 48 h. In doing so, we were able to capture time points with our positive control, thapsigargin, where we observed no ER stress markers expression (4 h), a maximal ER stress response (16–24 h), and the resolution of ER stress (48 h). Similar expression patterns in the rise and fall of ER stress markers were observed when macrophages were incubated with oxLDL or HDL in the presence of thapsigargin. Interestingly, in our hands, oxLDL on its own was unable to trigger ER stress. These observations are remarkably similar to those made by the Tabas group where oxLDL alone was unable to induce apoptosis in primary peritoneal macrophages. Rather, it was co-incubation of macrophages with oxLDL and thapsigargin that synergistically induced apoptosis (69). We speculate that oxLDL alone may not deliver enough free cholesterol into the macrophages to have the cytotoxic effects reported in Feng et al. (50) where macrophages were treated with acetylated LDL in the presence of an ACAT inhibitor. We also investigated whether treatment with acLDL alone could induce ER stress in our macrophages, however, we still found that similar to oxLDL, co-incubation with thapsigargin was needed to induce expression of BiP (Supplementary Figure S7). We speculate that the small amount of free cholesterol delivered to the cells by oxLDL (or acLDL) may result in an ER stress response in the resilient macrophage only in the presence of another source of stress in the system, such as treatment with thapsigargin or tunicamycin. Further studies are required to reveal the mechanisms by which oxLDL cooperates with thapsigargin to increase the ER stress response compared to thapsigargin treatment alone.
While our experimental conditions were similar to previously published studies, we searched for a rational explanation for why we were unable to demonstrate oxLDL-induced ER stress, as well as the blunting effect of HDL on ER stress. Towards these efforts, we took several different approaches: we tested the effects of tunicamycin as a different ER stressor, we compared different sources of lipoproteins, as well as their purity. We also tested a range of lipoprotein concentrations in our experiments. In the end, none of these factors appeared to be important variables in our assessment of ER stress. These observations led us to postulate that there could be an inherent difference in ER stress patterns when using primary macrophages as a model system as we used in our studies compared to RAW 264.7 or THP-1 cells. Very few studies have demonstrated the expression of UPR and apoptosis markers in primary macrophages in response to modified forms of LDL or HDL (50, 51, 70), although we were unsuccessful at reproducing these data. On the other hand, while THP-1 or RAW 264.7 macrophages are commonly used for their relative ease of culturing and assay flexibility, THP-1 cells are a human leukemia monocytic cell line, while RAW 264.7 cells are monocyte/macrophage-like cells that originated from a tumor in a mouse induced with the Abelson murine leukemia virus. As such, primary macrophages are considered a physiologically relevant model system for in vitro studies, which is an important consideration when studying specific disease mechanisms. Further investigations will be needed to distinguish differences in ER stress responses between cancer-derived macrophage lines and primary macrophages.
In conclusion, while the purpose of this study was to define the role of reactive aldehyde-modified HDL in ER stress, our experiments took an unexpected turn. Instead, using a highly systemic approach and through very convincing and reproducible data, we report alternate roles of oxLDL and HDL in mediating ER stress in thioglycolate-elicited murine peritoneal macrophages.
5 Conclusions
This study relied on an in-depth, systematic approach to identify the role of native and modified HDL in mediating primary macrophage ER stress. We discovered that lipoproteins on their own require the presence of thapsigargin to synergistically increase expression of ER stress markers. We further demonstrated that HDL, in the presence or absence of reactive aldehydes, was unable blunt the ER stress induced by thapsigargin in the presence or absence of oxLDL. Together, our findings suggest the need for more detailed investigations to better understand the role of native and modified lipoproteins in mediating ER stress pathways.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Institutional Review Board for Human Research. The studies were conducted in accordance with the local legislation and institutional requirements. The Ethics Committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because Plasma lipoproteins were obtained from non-identified donor blood. The animal study was approved by Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JB: Conceptualization, Methodology, Validation, Writing – original draft, Writing – review & editing, Data curation, Formal Analysis, Investigation, Visualization. GS: Data curation, Methodology, Writing – original draft, Writing – review & editing. DS: Conceptualization, Methodology, Validation, Writing – original draft, Writing – review & editing, Funding acquisition, Project administration, Resources, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health (NIH) Grants HL58012 (DS) and HL165927 (GMS). This work was also partially funded by the Advancing a Healthier Wisconsin Endowment.
Acknowledgments
The authors thank Gage Stuttgen, Emma Tillison, Guadalupe Valdivia Esparza, and Renee Penoske for critical review of this manuscript and helpful discussions. The authors also thank Dr. Mark Castleberry for technical support with lipoprotein isolations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1448607/full#supplementary-material
Abbreviations
ACK, ammonium-chloride-potassium; CVD, cardiovascular disease; DGUC, density gradient ultracentrifugation; EMSA, electrophoretic mobility shift assay; ER, endoplasmic reticulum; FPLC, fast protein liquid chromatography; HMEC-1, human microvascular endothelial cells; HNE, 4-hydroxynonenal; HRP, horseradish peroxidase; LDL, low density lipoprotein; MDA, malondialdehyde; oxLDL, oxidized low density lipoprotein; PEC, peritoneal exudate cells; RCT, reverse cholesterol transport; RIPA, radioimmunoprecipitation assay; RPMI, Roswell Park Memorial Institute; TBAR, thiobarbituric acid reactive substance; TBST, tris-buffered saline with tween; TG, thapsigargin; TM, tunicamycin; UPR, unfolded protein response.
References
1. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics—2021 update. Circulation. (2021) 143(8):e254–743. doi: 10.1161/CIR.0000000000000950
2. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. (2009) 27:165–97. doi: 10.1146/annurev.immunol.021908.132620
3. Lu Y, Cui X, Zhang L, Wang X, Xu Y, Qin Z, et al. The functional role of lipoproteins in atherosclerosis: novel directions for diagnosis and targeting therapy. Aging Dis. (2022) 13(2):491–520. doi: 10.14336/AD.2021.0929
4. Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. (2005) 2(6):309–15. doi: 10.1038/ncpcardio0195
5. Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotre-Taylor K, et al. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann N Y Acad Sci. (2001) 947:214–22. doi: 10.1111/j.1749-6632.2001.tb03943.x
6. Stocker R, Keaney JF Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev. (2004) 84(4):1381–478. doi: 10.1152/physrev.00047.2003
7. Yu X-H, Fu Y-C, Zhang D-W, Yin K, Tang C-K. Foam cells in atherosclerosis. Clin Chim Acta. (2013) 424:245–52. doi: 10.36660/abc.20220659
8. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. (1996) 271(5248):518–20. doi: 10.1126/science.271.5248.518
9. Ji Y, Wang N, Ramakrishnan R, Sehayek E, Huszar D, Breslow JL, et al. Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J Biol Chem. (1999) 274(47):33398–402. doi: 10.1074/jbc.274.47.33398
10. Eren E, Yılmaz N, Aydin O, Ellidağ HY. Anticipatory role of high density lipoprotein and endothelial dysfunction: an overview. Open Biochem J. (2014) 8:100–6. doi: 10.2174/1874091X01408010100
11. Schaefer EJ, Anthanont P, Asztalos BF. High-density lipoprotein metabolism, composition, function, and deficiency. Curr Opin Lipidol. (2014) 25(3):194–9. doi: 10.1097/MOL.0000000000000074
12. Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The framingham study. Am J Med. (1977) 62(5):707–14. doi: 10.1016/0002-9343(77)90874-9
13. Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. (2007) 357(21):2109–22. doi: 10.1056/NEJMoa0706628
14. Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. (2012) 367(22):2089–99. doi: 10.1056/NEJMoa1206797
15. Rader DJ, Tall AR. The not-so-simple HDL story: is it time to revise the HDL cholesterol hypothesis? Nat Med. (2012) 18(9):1344–6. doi: 10.1038/nm.2937
16. Rosenson RS, Brewer HB Jr, Ansell BJ, Barter P, Chapman MJ, Heinecke JW, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. (2016) 13(1):48–60. doi: 10.1038/nrcardio.2015.124
17. Smith JD. Myeloperoxidase, inflammation, and dysfunctional high-density lipoprotein. J Clin Lipidol. (2010) 4(5):382–8. doi: 10.1016/j.jacl.2010.08.007
18. Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, Shih DM, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. (2011) 121(7):2693–708. doi: 10.1172/JCI42946
19. Kaseda R, Jabs K, Hunley TE, Jones D, Bian A, Allen RM, et al. Dysfunctional high-density lipoproteins in children with chronic kidney disease. Metab Clin Exp. (2015) 64(2):263–73. doi: 10.1016/j.metabol.2014.10.020
20. Srivastava RAK. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol Cell Biochem. (2018) 440(1):167–87. doi: 10.1007/s11010-017-3165-z
21. Vollenweider P, von Eckardstein A, Widmann C. HDLs, diabetes, and metabolic syndrome. Handb Exp Pharmacol. (2015) 224:405–21. doi: 10.1007/978-3-319-09665-0_12
22. Wong NKP, Nicholls SJ, Tan JTM, Bursill CA. The role of high-density lipoproteins in diabetes and its vascular complications. Int J Mol Sci. (2018) 19(6):1680. doi: 10.3390/ijms19061680
23. Conklin DJ, Barski OA, Lesgards JF, Juvan P, Rezen T, Rozman D, et al. Acrolein consumption induces systemic dyslipidemia and lipoprotein modification. Toxicol Appl Pharmacol. (2010) 243(1):1–12. doi: 10.1016/j.taap.2009.12.010
24. Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther. (2007) 115(1):13–24. doi: 10.1016/j.pharmthera.2007.03.015
25. Srivastava S, Conklin DJ, Liu SQ, Prakash N, Boor PJ, Srivastava SK, et al. Identification of biochemical pathways for the metabolism of oxidized low-density lipoprotein derived aldehyde-4-hydroxy trans-2-nonenal in vascular smooth muscle cells. Atherosclerosis. (2001) 158(2):339–50. doi: 10.1016/s0021-9150(01)00454-3
26. Tanaga K, Bujo H, Inoue M, Mikami K, Kotani K, Takahashi K, et al. Increased circulating malondialdehyde-modified LDL levels in patients with coronary artery diseases and their association with peak sizes of LDL particles. Arterioscler Thromb Vasc Biol. (2002) 22(4):662–6. doi: 10.1161/01.atv.0000012351.63938.84
27. Chadwick AC, Holme RL, Chen Y, Thomas MJ, Sorci-Thomas MG, Silverstein RL, et al. Acrolein impairs the cholesterol transport functions of high density lipoproteins. PLoS One. (2015) 10(4):e0123138. doi: 10.1371/journal.pone.0123138
28. Schill RL, Knaack DA, Powers HR, Chen Y, Yang M, Schill DJ, et al. Modification of HDL by reactive aldehydes alters select cardioprotective functions of HDL in macrophages. Febs J. (2020) 287(4):695–707. doi: 10.1111/febs.15034
29. Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. (2006) 99(3):275–82. doi: 10.1161/01.RES.0000233317.70421.03
30. Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol. (2013) 75:49–67. doi: 10.1146/annurev-physiol-030212-183707
31. Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. (2010) 16(4):396–9. doi: 10.1038/nm0410-396
32. Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. (2021) 18(7):499–521. doi: 10.1038/s41569-021-00511-w
33. Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. (2006) 98(9):1186–93. doi: 10.1161/01.RES.0000220643.65941.8d
34. Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. (2007) 101(10):975–84. doi: 10.1161/CIRCRESAHA.107.161273
35. Wang S, Binder P, Fang Q, Wang Z, Xiao W, Liu W, et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br J Pharmacol. (2018) 175(8):1293–304. doi: 10.1111/bph.13888
36. Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. (2007) 116(11):1226–33. doi: 10.1161/CIRCULATIONAHA.106.682054
37. Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. (2009) 284(43):29735–45. doi: 10.1074/jbc.M109.018036
38. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. (2010) 107(7):839–50. doi: 10.1161/CIRCRESAHA.110.224766
39. Treiman M. Regulation of the endoplasmic reticulum calcium storage during the unfolded protein response–significance in tissue ischemia? Trends Cardiovasc Med. (2002) 12(2):57–62. doi: 10.1016/s1050-1738(01)00147-5
40. Yoshida H. ER stress and diseases. Febs J. (2007) 274(3):630–58. doi: 10.1111/j.1742-4658.2007.05639.x
41. Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. (2004) 14(1):20–8. doi: 10.1016/j.tcb.2003.11.001
42. Wang X, Xu L, Gillette TG, Jiang X, Wang ZV. The unfolded protein response in ischemic heart disease. J Mol Cell Cardiol. (2018) 117:19–25. doi: 10.1016/j.yjmcc.2018.02.013
43. Yao S, Miao C, Tian H, Sang H, Yang N, Jiao P, et al. Endoplasmic reticulum stress promotes macrophage-derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression. J Biol Chem. (2014) 289(7):4032–42. doi: 10.1074/jbc.M113.524512
44. Guo C, Ma R, Liu X, Chen T, Li Y, Yu Y, et al. Silica nanoparticles promote oxLDL-induced macrophage lipid accumulation and apoptosis via endoplasmic reticulum stress signaling. Sci Total Environ. (2018) 631-632:570–9. doi: 10.1016/j.scitotenv.2018.02.312
45. Maruyama R, Kamoshida Y, Shimizu M, Inoue J, Sato R. ATF6α stimulates cholesterogenic gene expression and de novo cholesterol synthesis. Biosci Biotechnol Biochem. (2013) 77(8):1734–8. doi: 10.1271/bbb.130295
46. McAlpine CS, Werstuck GH. Protein kinase R-like endoplasmic reticulum kinase and glycogen synthase kinase-3α/β regulate foam cell formation. J Lipid Res. (2014) 55(11):2320–33. doi: 10.1194/jlr.M051094
47. Sukhorukov VN, Khotina VA, Bagheri Ekta M, Ivanova EA, Sobenin IA, Orekhov AN. Endoplasmic reticulum stress in macrophages: the vicious circle of lipid accumulation and pro-inflammatory response. Biomedicines. (2020) 8(7):210. doi: 10.3390/biomedicines8070210
48. Jürgens G, Chen Q, Esterbauer H, Mair S, Ledinski G, Dinges HP. Immunostaining of human autopsy aortas with antibodies to modified apolipoprotein B and apoprotein(a). Arterioscler Thromb. (1993) 13(11):1689–99. doi: 10.1161/01.atv.13.11.1689
49. Sanson M, Augé N, Vindis C, Muller C, Bando Y, Thiers J-C, et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells. Circ Res. (2009) 104(3):328–36. doi: 10.1161/CIRCRESAHA.108.183749
50. Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. (2003) 5(9):781–92. doi: 10.1038/ncb1035
51. Yao S, Tian H, Zhao L, Li J, Yang L, Yue F, et al. Oxidized high density lipoprotein induces macrophage apoptosis via toll-like receptor 4-dependent CHOP pathway. J of Lipid Res. (2017) 58(1):164–77. doi: 10.1194/jlr.M071142
52. Yao S, Yang N, Song G, Sang H, Tian H, Miao C, et al. Minimally modified low-density lipoprotein induces macrophage endoplasmic reticulum stress via toll-like receptor 4. Biochim Biophys Acta. (2012) 1821(7):954–63. doi: 10.1016/j.bbalip.2012.03.003
53. Yao S, Zong C, Zhang Y, Sang H, Yang M, Jiao P, et al. Activating transcription factor 6 mediates oxidized LDL-induced cholesterol accumulation and apoptosis in macrophages by up-regulating CHOP expression. J Atheroscler Thromb. (2013) 20(1):94–107. doi: 10.5551/jat.13425
54. Yao ST, Sang H, Yang NN, Kang L, Tian H, Zhang Y, et al. Oxidized low density lipoprotein induces macrophage endoplasmic reticulum stress via CD36. Sheng Li Xue Bao. (2010) 62(5):433–40. doi: 10.1161/JAHA.120.015878
55. Muller C, Salvayre R, Nègre-Salvayre A, Vindis C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. (2011) 18(5):817–28. doi: 10.1038/cdd.2010.149
56. Niculescu LS, Sanda GM, Sima AV. HDL inhibits endoplasmic reticulum stress by stimulating apoE and CETP secretion from lipid-loaded macrophages. Biochem Biophys Res Commun. (2013) 434(1):173–8. doi: 10.1016/j.bbrc.2013.03.050
57. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. (1951) 193(1):265–75. doi: 10.1016/S0021-9258(19)52451-6
58. Chapman MJ, Goldstein S, Lagrange D, Laplaud PM. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J Lipid Res. (1981) 22(2):339–58. doi: 10.1016/S0022-2275(20)35376-1
59. Sparks DL, Phillips MC. Quantitative measurement of lipoprotein surface charge by agarose gel electrophoresis. J Lipid Res. (1992) 33(1):123–30. doi: 10.1016/S0022-2275(20)41889-9
60. Shao B, Pennathur S, Pagani I, Oda MN, Witztum JL, Oram JF, et al. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J Biol Chem. (2010) 285(24):18473–84. doi: 10.1074/jbc.M110.118182
61. Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. (2011) 490:71–92. doi: 10.1016/B978-0-12-385114-7.00004-0
62. Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. (2005) 35(4):373–81. doi: 10.1016/j.ymeth.2004.10.010
63. Holvoet P, Mertens A, Verhamme P, Bogaerts K, Beyens G, Verhaeghe R, et al. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler Thromb Vasc Biol. (2001) 21(5):844–8. doi: 10.1161/01.atv.21.5.844
64. Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. (2000) 105(8):1095–108. doi: 10.1172/JCI8574
65. Sloop CH, Dory L, Roheim PS. Interstitial fluid lipoproteins. J Lipid Res. (1987) 28(3):225–37. doi: 10.1016/S0022-2275(20)38701-0
66. Chen Y, Yang M, Huang W, Chen W, Zhao Y, Schulte ML, et al. Mitochondrial metabolic reprogramming by CD36 signaling drives macrophage inflammatory responses. Circ Res. (2019) 125(12):1087–102. doi: 10.1161/CIRCRESAHA.119.315833
67. Kontush A, Lindahl M, Lhomme M, Calabresi L, Chapman MJ, Davidson WS. Structure of HDL: particle subclasses and molecular components. Handb Exp Pharmacol. (2015) 224:3–51. doi: 10.1007/978-3-319-09665-0_1
68. Soumyarani VS, Jayakumari N. Oxidized HDL induces cytotoxic effects: implications for atherogenic mechanism. J Biochem Mol Toxicol. (2014) 28:481–9. doi: 10.1002/jbt.21588
69. Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. (2010) 12(5):467–82. doi: 10.1016/j.cmet.2010.09.010
Keywords: HDL, modified HDL, oxLDL, primary macrophages, endoplasmic reticulum stress, thapsigargin, lipoprotein, atherosclerosis
Citation: Bobek JM, Stuttgen GM and Sahoo D (2024) A comprehensive analysis of the role of native and modified HDL in ER stress in primary macrophages. Front. Cardiovasc. Med. 11:1448607. doi: 10.3389/fcvm.2024.1448607
Received: 13 June 2024; Accepted: 26 August 2024;
Published: 12 September 2024.
Edited by:
Catherine Martel, Université de Montréal, CanadaReviewed by:
Chieko Mineo, University of Texas Southwestern Medical Center, United StatesRyan Temel, University of Kentucky, United States
Copyright: © 2024 Bobek, Stuttgen and Sahoo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daisy Sahoo, ZHNhaG9vQG1jdy5lZHU=