 Yan Sun
Yan Sun Frank A. Dinenno
Frank A. Dinenno Peiyang Tang
Peiyang Tang Maria I. Kontaridis
Maria I. Kontaridis- 1Department of Biomedical Research and Translational Medicine, Masonic Medical Research Institute, Utica, NY, United States
- 2Department of Medicine, Division of Cardiology, Beth Israel Deaconess Medical Center, Boston, MA, United States
- 3Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA, United States
Protein Tyrosine Phosphatase 1B (PTP1B) has emerged as a significant regulator of metabolic and cardiovascular disease. It is a non-transmembrane protein tyrosine phosphatase that negatively regulates multiple signaling pathways integral to the regulation of growth, survival, and differentiation of cells, including leptin and insulin signaling, which are critical for development of obesity, insulin resistance, type 2 diabetes, and cardiovascular disease. Given PTP1B's central role in glucose homeostasis, energy balance, and vascular function, targeted inhibition of PTP1B represents a promising strategy for treating these diseases. However, challenges, such as off-target effects, necessitate a focus on tissue-specific approaches, to maximize therapeutic benefits while minimizing adverse outcomes. In this review, we discuss molecular mechanisms by which PTP1B influences metabolic and cardiovascular functions, summarize the latest research on tissue-specific roles of PTP1B, and discuss the potential for PTP1B inhibitors as future therapeutic agents.
Introduction
Obesity has reached epidemic proportions worldwide, with over 70% of the US population classified as overweight, obese, or morbidly obese. Projections from the World Health Organization indicate that by 2030, half of the US population will be considered obese, with children being the most vulnerable demographic (1). Obesity significantly increases the risk of developing several diseases, including certain cancers, type 2 diabetes (T2D), and cardiovascular disease (CVD) (2). Obesity adversely affects glucose and lipid levels, raises arterial blood pressure, induces inflammation, and impairs pulmonary function, which can lead to cardiac hypertrophy, atrial fibrillation, and heart failure if untreated (3–6). Despite understanding the contributing factors, the molecular mechanisms linking obesity to CVD remain poorly understood.
Primarily driven by high-calorie diets, sedentary lifestyles, and stress, insulin resistance is linked to the development of T2D, metabolic syndrome and chronic low-grade inflammation (7, 8). Indeed, the global prevalence of T2D, which accounts for 90%–95% of all diabetic cases, has surged by approximately 90% from 1990 to 2021 and is expected to increase an additional 60% by 2050 to affect more than 1.31 billion people (9, 10). This rapid rise in T2D, characterized by the dysregulation of glucose homeostasis, poses significant challenges for global health systems.
Insulin resistance is also a primary risk factor for CVD, non-alcoholic fatty liver disease (NAFLD), and hypertension (11, 12). CVD accounted for 18 million deaths in 2019 and is projected to exceed 22 million by the year 2030 (13). Atherosclerosis, characterized by lipid plaque accumulation in arterial walls, underlies most of these cardiovascular events, causing myocardial infarctions (MIs) and strokes (14). Chronic low-grade inflammation also plays a crucial role in the pathogenesis of CVD and can exacerbate systemic insulin resistance (15).
The interconnection between obesity, insulin resistance, and CVD underscores the need for comprehensive prevention and management strategies, including lifestyle changes and targeted interventions to enhance metabolic and cardiovascular health. In this regard, enzymes that modulate protein signaling pathways are crucial to maintaining cellular homeostasis; their controlled function is instrumental to preventing the development of insulin resistance, T2D, and CVD.
Protein Tyrosine Phosphatase 1B (PTP1B), encoded by the PTPN1 gene, is a non-receptor protein tyrosine phosphatase (PTP) that has been extensively studied due to its regulatory function in insulin and leptin signaling (16, 17). As such, PTP1B plays a critical role in metabolic regulation, making it a potential target for therapeutic intervention in obesity and diabetes, as well as in CVD pathophysiology. Here, in this review, we will discuss the significance of the biochemical, functional, and mechanistic roles for PTP1B, and how its hyperactivation can lead to the development of metabolic dysfunction and CVD. We will also discuss the current and emerging therapeutic strategies for treating obesity and diabetes, including use of PTP1B inhibitors.
PTP1B structure and function
PTP1B is a 50kDA protein initially discovered by biochemical purification methods targeting enzymes involved in protein tyrosine dephosphorylation (16, 18, 19). The C-terminus of the enzyme contains two proline-rich domains that interact with Src-homology 3 (SH3) domain-containing proteins to recruit binding substrates (19), as well as a hydrophobic domain that anchors to the endoplasmic reticulum (ER) to allow access to intracellular targets (20, 21). The N-terminal domain of PTP1B houses the catalytic domain, which includes the essential cysteine (Cys215) and arginine (Arg221) residues of the phosphatase signature motif [I/V]HCXXGXXR[S/T], a sequence that defines all protein tyrosine phosphatases (17).
The crystal structure of PTP1B has also revealed its biochemical function; it is a PTP that forms a catalytic pocket with high specificity for phosphotyrosine-containing substrates (22). Upon substrate binding, PTP1B undergoes a conformation change, closing its WPD loop around the phosphotyrosine residue to stabilize and ready the invariant aspartic acid (Asp181) for hydrolytic catalysis, a process that proceeds in two steps (23). First, a nucleophilic attack by the cysteine residue forms a phosphocysteine intermediate, which is hydrolyzed by glutamine (Gln262) and Asp181 (23). Next, the closed WPD loop sequesters the phosphocysteine intermediate to prevent the transfer of phosphate to extraneous acceptors. This process is critical to PTP1B's function, making it an integral regulator of signaling pathways involved in metabolic regulation.
PTP1B and RTK signaling
As a PTP, PTP1B catalyzes the dephosphorylation of receptor tyrosine kinases (RTKs) to modulate multiple spatiotemporally complex signaling processes, including those mediated by epidermal growth factor (EGF), leptin, and insulin (17). RTKs are single-span transmembrane receptors that facilitate communication between cells and their extracellular environment. Tyrosine phosphorylation (pTyr), a crucial post-translational modification, regulates essential biological processes that include cellular proliferation, migration, and invasion (24). The process is mediated by the coordinated actions of both protein tyrosine kinases (PTKs), which add phosphate (PO4) groups, and PTPs, which remove these phosphate groups (25, 26). Disruption of this delicate balance between PTKs and PTPs can result in aberrant pTyr. While most previous research historically focused on the role of PTKs, recent studies have emphasized the critical functions of PTPs in maintaining this balance. As PTPs can act as both initiators and terminators of pTyr signaling, their importance in maintaining cellular homeostasis is paramount (27, 28); absence or dysfunction of PTPs can lead to a myriad of pathological conditions, including autoimmunity, cancer, CVD, and obesity-related metabolic disorders (29, 30).
Insulin signaling and the role of PTP1B
In healthy individuals, food intake elevates blood glucose and stimulates insulin release from the pancreas, inducing a feedback mechanism that increases glucose uptake by peripheral tissues to regain normal blood glucose concentrations (31, 32). Insulin resistance occurs when tissues such as skeletal muscle, adipose tissue, and the liver become less responsive to insulin, leading to reduced glucose uptake, hyperglycemia, and hyperinsulinemia, and eventually, T2D (33, 34).
Insulin signaling, a key RTK pathway, and PTP1B regulation are integral mediators of this biological process. Briefly, when insulin is released into the bloodstream by the pancreas, it binds to cells through the insulin receptor (IR), inducing autophosphorylation and activation of its intrinsic kinase activity (35). The activated receptor then phosphorylates its downstream effectors, the insulin receptor substrates (IRS1/2), which mediate activation of phosphatidylinositol (PI) 3′-kinase (PI3K), a lipid kinase complex consisting of a regulatory p85 subunit and a catalytic p110 subunit that phosphorylates the 3′-hydroxyl group of phosphoinositides. Specifically, with respect to insulin signaling, phosphorylated IRS proteins interact with the regulatory p85 subunit of PI3K through their SH2 domains, inducing a conformational change that relieves the inhibition on the p110 catalytic subunit, recruiting the now activated PI3K complex from the cytosol to the plasma membrane (PM). Once at the membrane, PI3K can phosphorylate its substrate, phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2, also known as PIP2] to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3), driving downstream signaling and activation of protein kinase B (PKB; also known as AKT) (36). AKT then further promotes activation of downstream effectors involved in protein synthesis and survival, as well as induces translocation of glucose transporters to the cell membrane, resulting in increased glucose uptake by the cells (37) (Figure 1).
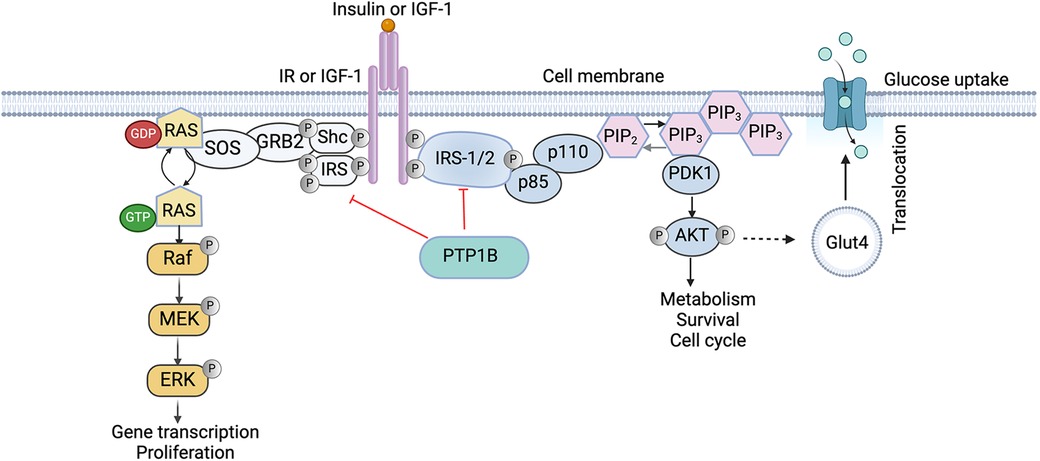
Figure 1. Role of PTP1B in the insulin signaling cascade. Binding of Insulin receptor substrates (IRS) activates the phosphatidylinositol-4,5-biphosphate-3-kinase (PI3K)/AKT pathway which is crucial for cellular survival and the mitogen-activated protein kinase (MAPK) pathway that is responsible for cell differentiation and cell growth. PTP1B negatively regulates insulin signaling by directly dephosphorylating both the IR and IRS1/2, reducing the activity of downstream effectors, including the PI3K/AKT pathway, reducing cellular glucose uptake. IR, insulin receptor; IGF, insulin-like growth factor; PTP1B, protein tyrosine phosphatase 1B; MEK, MAPK ERK kinase; ERK, extracellular signal-regulated kinases; p85 and p110, subunits of PI3K; PIP, phosphatidylinositol phosphate; PDK, protein dependent kinase. Image created with BioRender.com.
PTP1B negatively regulates this process by directly dephosphorylating both the IR and its downstream effectors, IRS1 and IRS2, reducing their activity and downregulating PI3K/AKT signaling (38). Indeed, whole-body PTP1B knockout (KO) mice exhibit enhanced insulin sensitivity, elevated phosphorylation of IR and IRS-1, and resistance to high-fat diet (HFD)-induced weight gain and obesity (27). Functionally, PTP1B KO mice have elevated basal metabolic rates and increased energy expenditures (39). Interestingly, the effects of reduced adiposity are not consequent to decreased fat cell numbers; rather, they are mediated by having decreased fat cell sizes (39). Collectively, these findings demonstrate a critical role for PTP1B in the regulation of metabolism in vivo.
PTP1B regulation of leptin signaling
Leptin, an adipocyte-secreted hormone, acts in the mediobasal hypothalamic nuclei to suppress food intake and increase energy expenditure, regulating energy balance and body mass (40). As such, circulating leptin levels correlate with adiposity (41). Mechanistically, leptin binds to the leptin (LR), a type I cytokine receptor that is expressed in the hypothalamus, and activates its effector JAK2, a protein that promotes the growth and division of cells (42). Once activated, JAK2 induces activation of the IRS proteins, which like insulin signaling, can lead to the activation of the PI3K/AKT pathway (43) (Figure 2). Activation of JAK2 also directly phosphorylates the LR itself, allowing for recruitment of the SH2 domain-containing protein-tyrosine phosphatase 2 (SHP2), another critical PTP that promotes activation of the extracellular signal-regulated kinase 1/2 (ERK1/2)-mitogen activated protein kinases (MAPK) pathway, which is critical for the proliferation and growth of cells (44, 45). In parallel, JAK2-mediated LR phosphorylation also drives activation of the signal transducer and activator of transcription factor 3 (STAT3) pathway, which suppresses transcription of both the hypothalamic neuropeptide Y (Npy) and agouti-related protein (Agrp), co-expressive neuropeptides that stimulate food intake and repress energy expenditure (46).
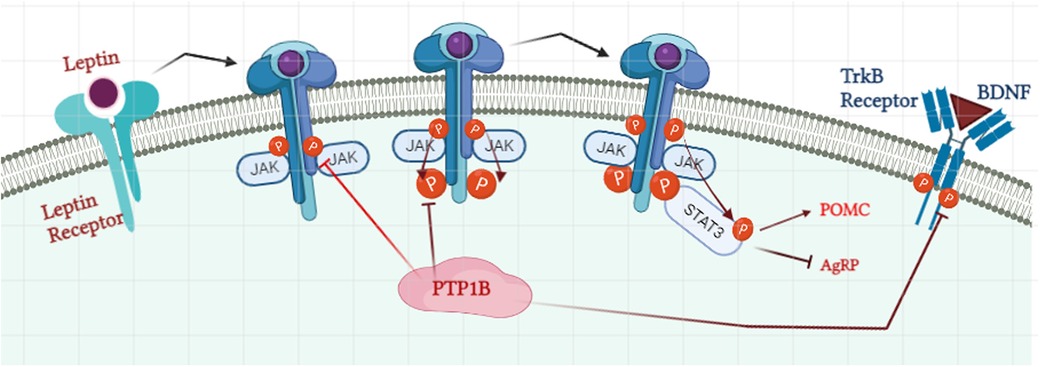
Figure 2. Role of PTP1B in regulation of the leptin signaling pathway. Binding of leptin to the leptin receptor initiates the recruitment of Janus Kinase 2 (JAK2) and their auto-phosphorylation process. Phosphorylated JAK2 then phosphorylates the tyrosine residue on the receptor which allows the docking of signal transducer and activator of transcription 3 (STAT3). STAT3 promotes the activation of Proopiomelanocortin (POMC) and inhibits the expression of agouti-related protein (AgRP). PTP1B suppresses the phosphorylation level of JAK2 and leptin receptors and henceforth STAT3 docking. PTP1B: protein tyrosine phosphatase 1B. Image created with BioRender.com.
Like with insulin, PTP1B is a negative regulator of leptin signaling by directly dephosphorylating JAK2, thereby blocking the phosphorylation and activities of both the receptor and STAT3 (47). PTP1B KO mice have increased leptin sensitivity, lower leptin/body fat ratios, and enhanced hypothalamic signaling, leading to reduced adiposity and increased energy expenditure (48).
Effects of PTP1B in metabolism
Located on chromosome 20q13.1-13.2, several single nucleotide polymorphisms (SNPs) in the PTPN1 gene have been linked to obesity and diabetes in various populations. In French Canadians, for example, at least six PTP1B SNPs have been associated with these conditions (49, 50). PTP1B SNPs have also been identified in individuals with early onset diabetes in Danes, Canadians, and Italians (10, 51, 52). Moreover, the PTP1B SNP rs3787348 is linked to a poorer weight reduction outcome in obese Japanese patients (53). Based on this, and the PTP1B global KO mouse data, it is clear that PTP1B influences glucose homeostasis and energy balance through its actions on insulin and leptin pathways. Inhibition of PTP1B enhances signaling to these pathways, improving glucose uptake and energy expenditure, and thus highlights the crucial role for PTP1B in metabolic health.
However, despite these overall positive outcomes, global, as well as myeloid-specific, PTP1B KO mice can develop acute myeloid leukemia and have been shown to have reduced lifespans (27, 54), suggesting differential tissue-specific effects of PTP1B. As such, global therapeutic targeting of PTP1B, particularly for treatment of metabolic diseases, appears to be rather complicated. To therefore circumvent this issue and to better elucidate the precise molecular, functional, and biological regulations of PTP1B, tissue-specific deletion models for PTP1B have been generated and studied (Table 1).
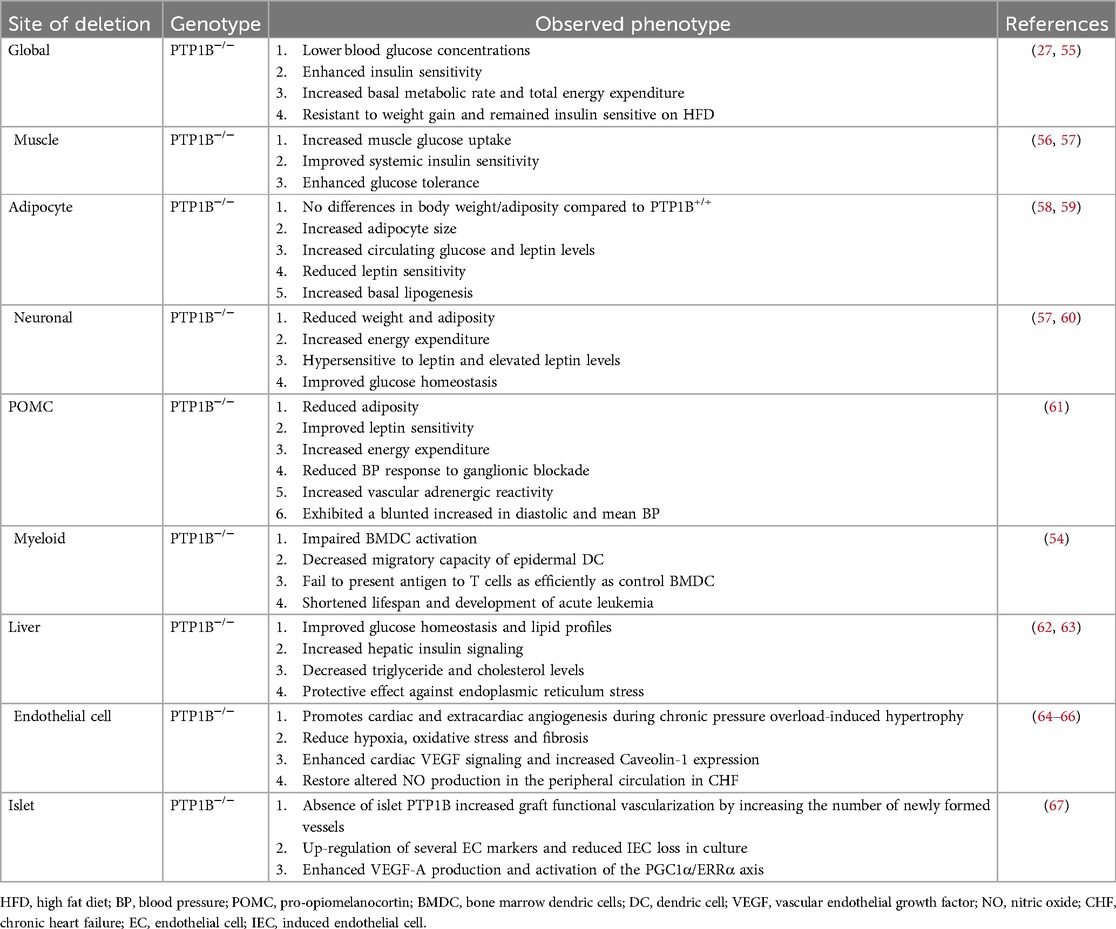
Table 1. Effects of global and tissue specific deletion of PTP1B in rodents.
Skeletal muscle
PTP1B deletion in skeletal muscle cells enhances systemic insulin sensitivity and increases glucose uptake in the muscle, independent of body weight and adiposity (56). These mice also exhibit increased insulin signaling, as indicated by enhanced phosphorylation of the IR and its downstream effector IRS1, as compared to controls (56). PI3K activity and phosphorylation of both AKT and ERK1/2 are also elevated in these mice. Despite these effects, however, deficiency of PTP1B in skeletal muscle does not affect lipid profiles, leptin levels, leptin sensitivity, body weight, or adiposity (56, 68), suggesting that these mechanisms for PTP1B regulation reside in other tissues and/or cell types.
Adipose
Adipose-specific PTP1B KO mice maintain similar body weight to controls when fed a HFD, but have larger adipocytes, increased circulating leptin levels, and increased basal lipogenesis (58). These mice have decreased phosphorylation of IR and its downstream effector AKT and have overall increased expression of lipogenic genes (58). Moreover, pyruvate kinase M2 (PKM2) is a newly identified substrate for PTP1B in adipocytes, but its mechanism of regulation in these cells remains unclear (69). In addition, PTP1B's role in different adipose depots is variable, influencing adipogenesis and insulin sensitivity differently between white and brown adipocytes; specifically, PTP1B knockdown in 3T3-L1 adipocytes inhibits adipogenesis (69), whereas PTP1B deficiency in brown adipocytes accelerates adipogenesis and enhances insulin sensitivity through activation of AKT and increased resistance to TNFα-induced apoptosis (70, 71).
Liver
Mice with liver-specific PTP1B deletion show increased insulin sensitivity and suppressed hepatic gluconeogenesis and lipogenic genes, without affecting weight gain (62, 63). In addition, these mice have alleviated endoplasmic reticulum (ER) stress following HFD, with decreased phosphorylation of downstream p38-MAPK and c-Jun N-terminal kinase (JNK)-MAPK signaling pathways, as well as reduced ER-resident proteins, PERK and eukaryotic initiation factor 2 α (eIF2α) factors (62, 63). This decreased signaling also leads to lowered expression of stress-related transcription factors and improved lipid profiles (62, 63). Liver-specific PTP1B KO, however, does not impact leptin levels, leptin sensitivity, body weight, or adiposity (62, 63), underscoring the liver-specific benefits of PTP1B inhibition in metabolic regulation.
Neuronal
Neuronal deletion of PTP1B in mice show decreased body mass and adiposity, as well as improved glucose homeostasis (57). These mice also exhibit increased physical activity and energy expenditure and are hypersensitive to leptin with elevated leptin levels (57). Moreover, deletion of PTP1B in arcuate pro-opiomelanocortin (POMC), a specific regulatory neuronal subpopulation that controls metabolism and is expressed only in hypothalamus and regions of the spinal cord and brain stem, reduces adiposity, improves leptin sensitivity, and increases energy expenditure (61, 72). These mice are also protected against leptin- and sympathetically mediated increases in blood pressure. Collectively, these data suggest that PTP1B modulates leptin production and resistance primarily through activation of pathways within the central nervous system (CNS) (61). In addition, POMCs induce brain-derived neurotrophic factor (BDNF) signaling in the hypothalamus, mediating neural connectivity and neurite outgrowth (73). In this regard, PTP1B modulates this process by acting as a direct negative regulator of the downstream BDNF effector, tropomyosin receptor kinase B (TrkB) (60, 73, 74), thus increasing both AKT and ERK activation. These effects highlight the critical role of the CNS in PTP1B-mediated metabolic regulation and the control of food intake and energy expenditure.
PTP1B in CVDs
As previously discussed, obesity is a major risk factor for CVD, increasing mortality and morbidity, particularly in individuals with central adiposity (75). Obesity is also associated with the development of high blood pressure, a leading risk factor for stroke, cardiac hypertrophy, and heart failure (76, 77). Several studies have established a significant link between PTPs, PTKs and cardiac pathophysiology (78). Indeed, increased activation of PTP1B in the heart is associated with development of heart failure in both rats and humans (78, 79). Cardiac contractile dysfunction and intracellular calcium dysregulation are also linked to elevated levels of PTP1B (80). Thus, there is precedence for a critical role for PTP1B in CVD (81).
PTP1B and endothelial cells (ECs)
Endothelial dysfunction is a primary event in the progression of CVD and is a known predictor of diabetes- and obesity-related CVD (82). Increased PTP1B expression in ECs impairs macro- and micro-vascular systems and increases the risk of having an MI or ischemic stroke (64, 83–87). Mechanistically, enhanced PTP1B-mediated signaling in ECs inhibits activation of AKT and blocks endothelial nitric oxide synthase (eNOS), an enzyme responsible for producing nitric oxide (NO), a potent vasodilator and regulator of glucose uptake (64, 88). Inhibition of PTP1B enhances serine phosphorylation of eNOS and improves NO-mediated vasodilation in mice with chronic heart failure induced by coronary ligation (64). These data suggest that PTP1B inhibition could potentially be considered for patients with CVD and/or endothelial dysfunction.
Vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2) and its ligand VEGF-A are key mediators of angiogenesis, the process by which existing blood vessels grow (89). Binding of VEGF-A to VEGFR2 induces autophosphorylation of the receptor and, subsequently, leads to activation of multiple crucial downstream signaling pathways necessary for survival, proliferation, and migration of ECs (90). To drive angiogenesis, VEGFR signaling activates Src, which induces phosphorylation of VE-Cadherin, as well as P38-MAPK, to mediate EC migration (91). In addition, VEGFR activation induces signaling to both ERK1/2 and PI3K/AKT, to promote EC proliferation and EC survival, respectively (92) (Figure 3).
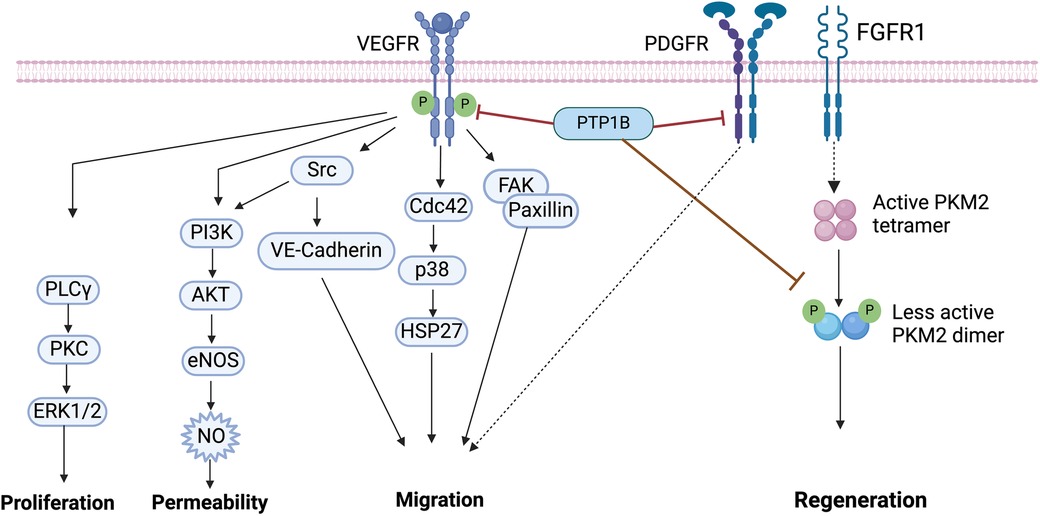
Figure 3. Vascular endothelial growth factor (VEGF) signaling. Binding of VEGF to VEGFR induces its tyrosine phosphorylation, which is followed by the activation of downstream effectors including PLCγ/PKC/ERK1/2, PI3K/AKT, Src, p38 and FAK/Paxillin. PTP1B binds to VEGFR and directly dephosphorylates VEGFR. PTP1B directly binds to platelet-derived growth factor receptor (PDGFR), inducing motility in smooth muscle cells (SMCs). The fibroblast growth factor receptor (FGFR1) directly phosphorylates pyruvate kinase M2 (PKM2), PTP1B dephosphorylates PKM2 and regulates myocyte regeneration. VEGF, vascular endothelial growth factor; PTP1B, protein tyrosine phosphatase 1B; FAK, focal adhesion kinase; PI3K, phosphatidylinositol 3-kinases; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; PLCy, phospho-lipase C gamma; PKC, protein kinase C; ERK1/2, extracellular signal-regulated kinase 1/2; PKM2, pyruvate kinase M2. Image created with BioRender.com.
Here too, PTP1B has a prominent role in CVD; it directly dephosphorylates VEGFR2 to inhibit angiogenesis (66). Mice with endothelial-specific deletion of PTP1B, however, have improved angiogenesis, enhanced wound healing, and are protected against development of chronic heart failure post-MI (66, 93). Primary ECs isolated from these mice reveal that the mechanism of this regulation is mediated by enhanced VEGF-A-induced VEGFR2 activity, which leads to increased ERK signaling to induce angiogenesis (66). Similar effects on angiogenesis, revascularization and survival are also noted in PTP1B-deleted pancreatic islets (67), as well as in human umbilical vein ECs, both in vitro and in vivo (94).
PTP1B and vascular smooth muscle cells (VSMCs)
The motility and proliferation of VSMCs is critical for neointima formation and remodeling in response to vascular injury (95). Elevated levels of platelet-derived growth factor (PDGF) and/or fibroblast growth factor-2 (FGF2) contribute to this remodeling event, mediating autophosphorylation and activation of PDGF receptor β (PDGF-βR) and increased downstream signaling (96). Here, induction of NO can increase PTP1B activity, whose direct binding to the PDGF-βR can then inhibit downstream signaling and suppress proliferation and motility in cultured SMCs (96–98).
Indeed, adenoviral overexpression of PTP1B blocks insulin-mediated activation of IR and decreases cell motility, whereas dominant-negative expression of PTP1B attenuates NO-mediated inhibition of cell motility (98). Treatment of injured arteries with dominant negative PTP1B adenovirus, however, increases cell proliferation, intimal cell number, and neointima formation, without affecting apoptosis. Moreover, primary isolated newborn rat aortic SMCs treated with an antisense oligonucleotide against PTP1B enhances cell motility and increases pTyr of several adhesion molecules, including p130cas, paxillin and focal adhesion kinase (FAK) (99) (Figure 3). Together, these findings demonstrate that PTP1B is an important regulatory protein involved in motility and proliferation, and that it may be a good potential therapeutic target for treating VSMC pathophysiology.
PTP1B and cardiac myocytes
Cardiomyocytes (CMs) are contractile muscle cells that allow the heart to maintain proper pumping function (100). CMs are also the major source of VEGF signaling in the heart (101, 102). Here, activation of P38-MAPK promotes secretion of VEGF-A, which in turns acts as a feedback mechanism to stimulate binding and activation of the VEGFR (103). CM-mediated VEGF signaling also mediates cardiac morphogenesis, transforming cardiac ECs into mesenchymal cells in mice (104, 105). However, the mechanisms for how PTP1B is involved in this process in CMs requires further study.
Interestingly, research on PTP1B activity in CMs has demonstrated both protective and deleterious effects. For example, PTP1B directly binds PKM2, increasing its activity in CMs (106). Since PKM2 is a key cell cycle regulator, increasing its activity induces CM cell division, improves cardiac function, and extends long-term survival after acute or chronic MI (107) (Figure 3). Conversely, PTP1B overexpression mediates age-induced CM contractile dysfunction and other myocardial anomalies (80, 108).
In response to HFD, global deletion of PTP1B in mice reduces cardiac pathologies, including hypertrophy (108). Our own laboratory also recently demonstrated that CM-specific deletion of PTP1B in mice ameliorates HFD-induced cardiomyopathy and cardiac steatosis (106). Furthermore, metabolomics data revealed that CM-specific deletion of PTP1B elevates fatty acid oxidation and lipolysis, but reduces glucose metabolism (106). In response to hypoxia-reoxygenation of isolated primary neonatal mouse CMs, PTP1B expression increases; ablation of PTP1B in these cells increases AKT activity and decreases hypoxia-associated apoptosis (109). These findings indicate that PTP1B inhibition may mitigate cardiac abnormalities and improve heart function.
While these data all suggest a protective role for PTP1B deletion in the heart, Coulis et al. have found that CM-specific deletion of PTP1B in mice may also be pathological, inducing a hypertrophic phenotype that is exacerbated by pressure overload (110). Specifically, they found that argonaute 2 (AGO2), a critical component of the RNA-induced silencing complex, is inactivated in response to CM-specific deletion of PTP1B, thereby preventing miR-208b-mediated inhibition of mediator complex subunit 13 (MED13) and leads to thyroid hormone-associated pathological cardiac hypertrophy (110). However, in this regard, inhibition of miR-208b has previously been shown to improve, not induce, cardiac dysfunction in titin-induced dilated cardiomyopathy (111). Moreover, upregulation of MED13 is protective in the heart, conferring resistance to obesity (112). Regardless, it remains possible that different pathological stimuli may lead to different outcomes and/or that too much or too little PTP1B can lead to similar, but distinct, pathological outcomes (106). Exactly if and how PTP1B is involved in modulating these various physiological vs. pathological responses in the heart remains to be determined (106).
PTP1B as a potential therapeutic target
Overall, given PTP1B's critical role in insulin and leptin signaling, it is considered an excellent target for treating obesity and diabetes (17). However, targeting PTP1B, in a cellular and tissue-specific manner, has been challenging, particularly because of its positively charged active site, its selectivity, and its bioavailability (78, 113). Consequently, only a few PTP1B inhibitors have been tested in clinical trials, and most have, unfortunately, been discontinued due to insufficient efficiency, lack of specificity and detrimental side effects (114–116) (Table 2). Therefore, more research is needed to fully elucidate the effects of PTP1B inhibition in cardiometabolic diseases.
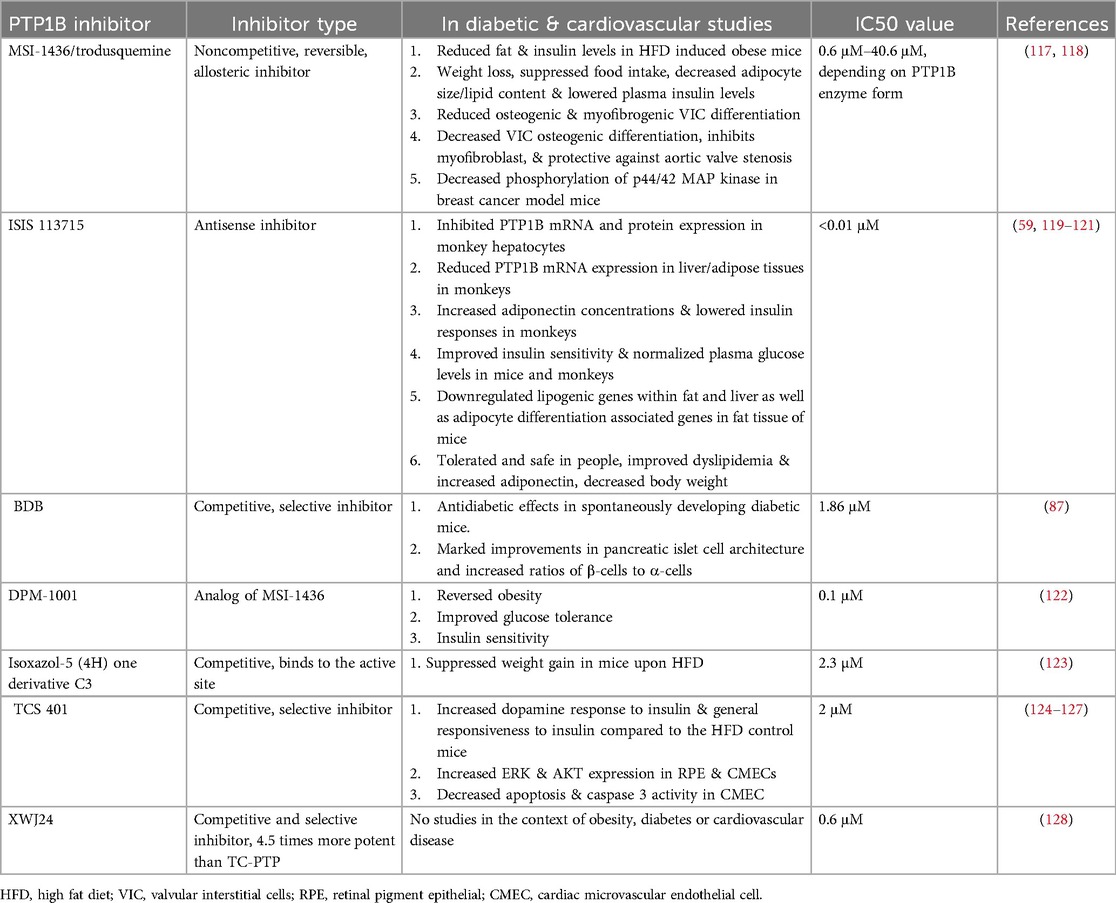
Table 2. An overview of PTP1B inhibitors.
Ertiprotafib, a monocarboxylic acid mimetic, is a noncompetitive multiple-action inhibitor (129). It was the first PTP1B inhibitor to be tested in clinical trials for the treatment of diabetes (130). This inhibitor has an atypical mechanism of action; while most small molecules bind to and increase their target's melting temperature (Tm) to stabilize their interaction, ertiprotafib lowers the Tm of PTP1B, destabilizing it (131, 132). Indeed, the mechanism by which ertiprotafib inhibits PTP1B activity is mediated by inducing protein aggregation (133). However, the selectivity of this compound is very low, with IC50 values of 1.6–29 µM, depending on assay conditions (129). Therefore, development of newer and more specific inhibitors was needed.
Trodusquemine, also known as MSI-1436, is a natural aminosterol cholestane that is a noncompetitive, reversible, and allosteric inhibitor of PTP1B (117, 134). In terms of specificity, this small molecule targets a novel allosteric binding site located within the C-terminus of PTP1B, allowing it to bind with affinity values that range from 0.6 μM to 40 6 μM, depending on the enzyme form (117). Because of its high specificity, trodusquemine was initially considered to be the more effective inhibitor with fewer off-target side effects (135). Indeed, in HFD-fed mice, MSI-1436 significantly decreased obesity by reducing insulin levels, decreasing body weight, lowering lipid content, decreasing adipocyte size, and suppressing food intake (118). In addition, MSI-1436 reduced osteogenic and myofibrogenic valvular interstitial cell (VIC) differentiation, alleviating aortic valve fibro-calcification and stenosis in a diet-induced mouse model of calcific aortic valve disease (136). Unfortunately, though effective in these mouse studies, this inhibitor was discontinued for use in human clinical trials due to its low activity and poor bioavailability in patients (19).
JTT-551, a mixed-type PTP1B inhibitor, was shown to have good selectivity. Like trodusquemine, chronic administration of JTT-551 in mice with diet-induced obesity demonstrated anti-obesity properties (137). In addition, JTT-551 reduced blood glucose and exhibited antidiabetic effects, without changes in body weight, in diabetic mice (137). Taken together, these data indicated that JTT-551 was an effective inhibitor for treatment of T2D and obesity; however, trials here too were discontinued due to insufficient efficacy and increased adverse effects in patients (138).
New therapies on the horizon for PTP1B inhibition
Fortunately, several new PTP1B inhibitors have been recently generated and are in process of being tested for efficacy against a myriad of cardiometabolic diseases, including T2D. The hope is that one (or several) will succeed in patient trials and provide future therapeutic benefits for these devastating diseases.
IONIS-PTP-1BRx, also known as ISIS-113715, is an antisense PTP1B inhibitor with an IC50 of less than 0.01 μM (59). In obese insulin-resistant monkeys, this inhibitor improved insulin sensitivity, increased adiponectin concentration, and lowered insulin responses (59). Similarly, in insulin-resistant diabetic mice, IONIS-PTP-1BRx improved insulin sensitivity, normalized plasma glucose levels, and decreased expression of lipogenic genes in both fat and liver, downregulating adipocyte differentiation (120). In 2018, IONIS-PTP-1BRx made its way to clinical trials, and preliminary data indicate that treated T2D diabetic patients have reduced body weight and decreased mean HbA1c levels (121), all without adverse interactions with other antidiabetic drugs (119). However, as of the date of this submission, no further updates have been posted about this study.
BDB [3-bromo-4,5-bis(2,3-dibromo-4,5-dihydroxybenzyl)-1,2-benzenediol] is a recently developed competitive inhibitor of PTP1B with demonstrated high selectivity (87). In vitro, BDB is cell-permeable and enhances insulin signaling. Moreover, oral administration of BDB leads to antidiabetic effects in spontaneously developing diabetic mice, with marked improvements in pancreatic islet cell architecture and increases in ratios of β-cells to α-cells (87). No information, however, is yet available about its progression to clinical trials.
DPM-1001, an analog of MSI-1436 with an IC50 of 0.1 μM, reverses obesity and improves insulin sensitivity and glucose tolerance in HFD mice (139). In May of 2022, DepYmed released a statement that it received FDA orphan drug designation for the clinical candidate DPM-1001, for the treatment of Wilson Disease, a rare genetic disorder that prevents the body from removing copper, causing the metal to build up in the liver, brain, and corneas (122) (https://www.iospace.com/article/releases/depymed-receives-fda-orphan-drug-designation-for-clinical-candidate-dpm-1001-for-the-treatment-of-wilson-disease/). This classification for DPM-1001 is an important milestone, as it furthers the development and progression of PTP1B inhibitors for use in diseased patients.
Other potent inhibitors have also been recently studied in the context of metabolism. A competitive derivative of Isoxazol-5(4H) suppresses weight gain in HFD mice, although at a comparatively reduced potency than other PTP1B inhibitors (123). TCS 401, another competitive and selective inhibitor, increases dopamine responses to insulin in HFD mice (124). Inhibitors, such as XWJ24 and compounds from D. crassirhizomai and M. alba, have recently shown strong PTP1B inhibition, but have yet to be tested in HFD or CVD studies (128, 140, 141). Finally, a new patent application has identified 5-(naphthalen-2-yl)-1,2,5-thiadiazolidin-3-one 1,1-dioxide derivatives (represented by “formula 1”) as a newly garnered PTP1B inhibitor, but its efficacy and specificity have yet to be determined (114).
Navigating the challenges: future opportunities for PTP1B inhibitors in disease treatment
New targeting strategies for PTP1B inhibition
The transcriptional and translational regulation of PTP1B is well studied, providing potential novel sites for therapeutic targeting, particularly for metabolic disorders and cancers. In this regard, characterization of the PTP1B promoter region revealed several binding sites for transcription factors such as Egr-1, Sp1, Sp3 (142), YB-1 (143), NF-κB (144) and HIF (145). These factors can be activated by various stimuli, including glucose, insulin, proinflammatory cytokines, and oncoproteins (146). Additionally, PTP1B expression is regulated by several miRNAs, including miR-338-3p, miR-744, miR-122, miR-193a-3p, miR-135a, miR-146-b, and miR-206 (147–152). Each of these can be considered as additional, indirect PTP1B inhibitors for future studies, as they have the potential to be targeted with high specificity, to reduce downstream PTP1B signaling, and hopefully, to prevent development of insulin resistance, T2D, CVD, as well as onset of other diseases such as cancer. The efficacy of this strategy remains to be determined.
Outside the box approaches for PTP1B inhibition
Over the past few decades, numerous PTP1B inhibitors have been identified, but their lack of specificity and cellular permeability pose significant challenges for clinical use (115). Most inhibitors target the catalytic active site, leading to non-specific inhibition of all PTPs (153). Despite efforts to develop more specific inhibitors, the high conservation of the catalytic site among PTPs, particularly TCPTP, remains a major obstacle. To overcome these challenges, researchers are exploring innovative strategies including high-throughput screening (154), virtual screening (155), DNA-encoded libraries (156), and PROTAC approaches (157) to develop drugs with greater efficacy and increased specificity. Additionally, designing inhibitors with improved membrane permeability and bioavailability is crucial. In this regard, nanoparticle-based delivery systems and graphene quantum dots show promise in enhancing oral bioavailability and specificity (158, 159). Addressing these challenges will be necessary for developing effective and safe PTP1B inhibitors for clinical treatment of various diseases in the near future.
Summary
PTP1B signaling is a critical regulator of glucose homeostasis and energy balance, conducive to mediating the onset of obesity, insulin resistance, T2D, and CVD (49, 160). Consequently, inhibition of PTP1B is an attractive novel strategy for treating these diseases. Though significant progress has been made in understanding the molecular mechanisms of PTP1B regulation in metabolic processes that affect diabetes, obesity and the heart, tissue-specific targeting and selective potency with existing PTP1B inhibitors remains challenging. Future research is promising in this regard, and advancements in this field, through novel and targeted modulation of PTP1B, are likely to yield effective results and therapies for treatment of individuals with these devastating ailments.
Author contributions
YS: Funding acquisition, Investigation, Methodology, Validation, Writing – original draft. FD: Investigation, Methodology, Supervision, Validation, Writing – review & editing. PT: Investigation, Visualization, Writing – original draft. MIK: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the National Institutes of Health (Grants R01-HL122238, R01-HL102368), the American Heart Association Transformation Grant Awards (20TPA35490426, 23TPA1065811), and the Masonic Medical Research Institute to MIK.; and the Halfond Weil Postdoctoral Fellowship to YS.
Acknowledgments
Figures were created using BioRender.com. The manuscript writing was edited and grammatically checked and cross-referenced using Chegg and ChatGPT.
Conflict of interest
MIK has received grant funding from Onconova Therapeutics and is a consultant for BioMarin Pharmaceutical Inc; both funding and consulting projects are independent of the work in this manuscript and have no overlap with the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ward ZJ, Bleich SN, Cradock AL, Barrett JL, Giles CM, Flax C, et al. Projected U.S. State-level prevalence of adult obesity and severe obesity. N Engl J Med. (2019) 381:2440–50. doi: 10.1056/NEJMsa1909301
2. Chawla A, Chawla R, Jaggi S. Microvasular and macrovascular complications in diabetes mellitus: distinct or continuum? Indian J Endocrinol Metab. (2016) 20:546–51. doi: 10.4103/2230-8210.183480
3. Lavie Carl J, Mcauley Paul A, Church Timothy S, Milani Richard V, Blair Steven N. Obesity and cardiovascular diseases. J Am Coll Cardiol. (2014) 63:1345–54. doi: 10.1016/j.jacc.2014.01.022
4. Lavie CJ, Alpert MA, Arena R, Mehra MR, Milani RV, Ventura HO. Impact of obesity and the obesity paradox on prevalence and prognosis in heart failure. JACC Heart Fail. (2013) 1:93–102. doi: 10.1016/j.jchf.2013.01.006
5. Cersosimo E, Defronzo RA. Insulin resistance and endothelial dysfunction: the road map to cardiovascular diseases. Diabetes Metab Res Rev. (2006) 22:423–36. doi: 10.1002/dmrr.634
6. Kolwicz SC JR, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. (2013) 113:603–16. doi: 10.1161/CIRCRESAHA.113.302095
7. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. (2018) 20:12. doi: 10.1007/s11906-018-0812-z
8. Kosmas CE, Silverio D, Tsomidou C, Salcedo MD, Montan PD, Guzman E. The impact of insulin resistance and chronic kidney disease on inflammation and cardiovascular disease. Clin Med Insights Endocrinol Diabetes. (2018) 11:1179551418792257. doi: 10.1177/1179551418792257
9. Ong KL, Stafford LK, Mclaughlin SA, Boyko EJ, Vollset SE, Smith AE, et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the global burden of disease study 2021. Lancet. (2023) 402:203–34. doi: 10.1016/S0140-6736(23)01301-6
10. Echwald SM, Bach H, Vestergaard H, Richelsen B, Kristensen K, Drivsholm T, et al. A P387l variant in protein tyrosine phosphatase-1B (PTP-1B) is associated with type 2 diabetes and impaired serine phosphorylation of PTP-1B in vitro. Diabetes. (2002) 51:1–6. doi: 10.2337/diabetes.51.1.1
11. Gast KB, Tjeerdema N, Stijnen T, Smit JW, Dekkers OM. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: meta-analysis. PLoS One. (2012) 7:e52036. doi: 10.1371/journal.pone.0052036
12. Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. (2010) 56:1113–32. doi: 10.1016/j.jacc.2010.05.034
13. Nawsherwan , Mubarik S, Bin W, Le Z, Sang M, Lin Y, et al. Epidemiological trends in cardiovascular disease mortality attributable to modifiable risk factors and its association with sociodemographic transitions across BRICS-plus countries. Nutrients. (2023) 15(17):3757. doi: 10.3390/nu15173757
14. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. (2012) 487:325–9. doi: 10.1038/nature11260
15. Kosmas CE, Bousvarou MD, Kostara CE, Papakonstantinou EJ, Salamou E, Guzman E. Insulin resistance and cardiovascular disease. J Int Med Res. (2023) 51:3000605231164548. doi: 10.1177/03000605231164548
16. Tonks NK, Diltz CD, Fischer EH. Characterization of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. (1988) 263:6731–7. doi: 10.1016/S0021-9258(18)68703-4
17. Tonks NK. PTP1B: from the sidelines to the front lines!. FEBS Lett. (2003) 546:140–8. doi: 10.1016/S0014-5793(03)00603-3
18. Tonks NK, Diltz CD, Fischer EH. Purification of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. (1988) 263:6722–30. doi: 10.1016/S0021-9258(18)68702-2
19. Liu F, Hill DE, Chernoff J. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the SRC homology 3 domain of p130(CAS). J Biol Chem. (1996) 271:31290–5. doi: 10.1074/jbc.271.49.31290
20. Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. (1992) 68:545–60. doi: 10.1016/0092-8674(92)90190-N
21. Haj FG, Verveer PJ, Squire A, Neel BG, Bastiaens PI. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science. (2002) 295:1708–11. doi: 10.1126/science.1067566
22. Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. (1994) 263:1397–404. doi: 10.1126/science.8128219
23. Pannifer ADB, Flint AJ, Tonks NK, Barford D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography*. J Biol Chem. (1998) 273:10454–62. doi: 10.1074/jbc.273.17.10454
24. Hunter T. The genesis of tyrosine phosphorylation. Cold Spring Harb Perspect Biol. (2014) 6:a020644. doi: 10.1101/cshperspect.a020644
25. Yip SC, Saha S, Chernoff J. PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem Sci. (2010) 35:442–9. doi: 10.1016/j.tibs.2010.03.004
26. Shenolikar S. Chapter 105 - Serine/Threonine phosphatase inhibitor proteins. In: Ralph A. Bradshaw, Edward A, editors. Handbook of Cell Signaling, 2nd ed. San Diego: Academic Press (2023). p. 627–29. doi: 10.1016/B978-012124546-7/50466-6
27. Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. (1999) 283:1544–8. doi: 10.1126/science.283.5407.1544
28. Charbonneau H, Tonks NK, Kumar S, Diltz CD, Harrylock M, Cool DE, et al. Human placenta protein-tyrosine-phosphatase: amino acid sequence and relationship to a family of receptor-like proteins. Proc Natl Acad Sci U S A. (1989) 86:5252–6. doi: 10.1073/pnas.86.14.5252
29. Villamar-Cruz O, Loza-Mejía MA, Arias-Romero LE, Camacho-Arroyo I. Recent advances in PTP1B signaling in metabolism and cancer. Biosci Rep. (2021) 41(11):BSR20211994. doi: 10.1042/BSR20211994
30. Montalibet J, Kennedy BP. Therapeutic strategies for targeting PTP1B in diabetes. Drug Discovery Today: Therapeutic Strategies. (2005) 2:129–35. doi: 10.1016/j.ddstr.2005.05.002
31. Röder PV, Wu B, Liu Y, Han W. Pancreatic regulation of glucose homeostasis. Exp Mol Med. (2016) 48:e219. doi: 10.1038/emm.2016.6
32. Roh E, Song DK, Kim MS. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp Mol Med. (2016) 48:e216. doi: 10.1038/emm.2016.4
33. Freeman AM, Acevedo LA, Pennings N. Insulin resistance. In: StatPearls. Treasure Island, FL: StatPearls Publishing (2024). https://www.ncbi.nlm.nih.gov/books/NBK507839/
34. Fazakerley DJ, Krycer JR, Kearney AL, Hocking SL, James DE. Muscle and adipose tissue insulin resistance: malady without mechanism? J Lipid Res. (2019) 60:1720–32. doi: 10.1194/jlr.R087510
35. Haeusler RA, Mcgraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. (2018) 19:31–44. doi: 10.1038/nrm.2017.89
36. Fu Q, Shi Q, West TM, Xiang YK. Cross-talk between insulin signaling and G protein–coupled receptors. J Cardiovasc Pharmacol. (2017) 70(2):74–86. doi: 10.1097/FJC.0000000000000481
37. Schultze SM, Hemmings BA, Niessen M, Tschopp O. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev Mol Med. (2012) 14:e1. doi: 10.1017/S1462399411002109
38. Peterson C, Chandler HL. Insulin facilitates corneal wound healing in the diabetic environment through the RTK-PI3K/akt/mTOR axis in vitro. Mol Cell Endocrinol. (2022) 548:111611. doi: 10.1016/j.mce.2022.111611
39. Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. (2000) 20:5479–89. doi: 10.1128/MCB.20.15.5479-5489.2000
40. Münzberg H, Björnholm M, Bates SH, Myers MG JR. Leptin receptor action and mechanisms of leptin resistance. Cell Mol Life Sci. (2005) 62:642–52. doi: 10.1007/s00018-004-4432-1
41. Seth M, Biswas R, Ganguly S, Chakrabarti N, Chaudhuri AG. Leptin and obesity. Physiol Int. (2020) 107:455–68. doi: 10.1556/2060.2020.00038
42. Sopjani M, Morina R, Uka V, Xuan NT, Dërmaku-Sopjani M. JAK2-mediated intracellular signaling. Curr Mol Med. (2021) 21:417–25. doi: 10.2174/1566524020666201015144702
43. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K Integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. (2005) 115:951–8. doi: 10.1172/JCI24301
44. Bjørbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, et al. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem. (2001) 276:4747–55. doi: 10.1074/jbc.M007439200
45. Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A. (2004) 101:16064–9. doi: 10.1073/pnas.0405041101
46. Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of AGRP and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. (1998) 1:271–2. doi: 10.1038/1082
47. Cook WS, Unger RH. Protein tyrosine phosphatase 1B: a potential leptin resistance factor of obesity. Dev Cell. (2002) 2:385–7. doi: 10.1016/S1534-5807(02)00158-2
48. Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell. (2002) 2:489–95. doi: 10.1016/S1534-5807(02)00148-X
49. Lembertas AV, Pérusse L, Chagnon YC, Fisler JS, Warden CH, Purcell-Huynh DA, et al. Identification of an obesity quantitative trait locus on mouse chromosome 2 and evidence of linkage to body fat and insulin on the human homologous region 20q. J Clin Invest. (1997) 100:1240–7. doi: 10.1172/JCI119637
50. Brown-Shimer S, Johnson KA, Lawrence JB, Johnson C, Bruskin A, Green NR, et al. Molecular cloning and chromosome mapping of the human gene encoding protein phosphotyrosyl phosphatase 1B. Proc Natl Acad Sci USA. (1990) 87:5148–52. doi: 10.1073/pnas.87.13.5148
51. Mok A, Cao H, Zinman B, Hanley AJG, Harris SB, Kennedy BP, et al. A single nucleotide polymorphism in protein tyrosine phosphatase PTP-1B is associated with protection from diabetes or impaired glucose tolerance in Oji-Cree. J Clin Endocrinol Metab. (2002) 87:724–7. doi: 10.1210/jcem.87.2.8253
52. Di Paola R, Frittitta L, Miscio G, Bozzali M, Baratta R, Centra M, et al. A variation in 3′ UTR of hPTP1B increases specific gene expression and associates with insulin resistance. Am J Hum Genet. (2002) 70:806–12. doi: 10.1086/339270
53. Yamakage H, Konishi Y, Muranaka K, Hotta K, Miyamoto Y, Morisaki H, et al. Association of protein tyrosine phosphatase 1B gene polymorphism with the effects of weight reduction therapy on bodyweight and glycolipid profiles in obese patients. J Diabetes Investig. (2021) 12:1462–70. doi: 10.1111/jdi.13492
54. Le Sommer S, Morrice N, Pesaresi M, Thompson D, Vickers MA, Murray GI, et al. Deficiency in protein tyrosine phosphatase PTP1B shortens lifespan and leads to development of acute leukemia. Cancer Res. (2018) 78:75–87. doi: 10.1158/0008-5472.CAN-17-0946
55. Kenner KA, Anyanwu E, Olefsky JM, Kusari J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling. J Biol Chem. (1996) 271:19810–6. doi: 10.1074/jbc.271.33.19810
56. Delibegovic M, Bence KK, Mody N, Hong EG, Ko HJ, Kim JK, et al. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol Cell Biol. (2007) 27:7727–34. doi: 10.1128/MCB.00959-07
57. Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. (2006) 12:917–24. doi: 10.1038/nm1435
58. Owen C, Czopek A, Agouni A, Grant L, Judson R, Lees EK, et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PLoS One. (2012) 7:e32700. doi: 10.1371/journal.pone.0032700
59. Swarbrick MM, Havel PJ, Levin AA, Bremer AA, Stanhope KL, Butler M, et al. Inhibition of protein tyrosine phosphatase-1B with antisense oligonucleotides improves insulin sensitivity and increases adiponectin concentrations in monkeys. Endocrinology. (2009) 150:1670–9. doi: 10.1210/en.2008-0885
60. Ozek C, Kanoski SE, Zhang ZY, Grill HJ, Bence KK. Protein-tyrosine phosphatase 1B (PTP1B) is a novel regulator of central brain-derived neurotrophic factor and tropomyosin receptor kinase B (TrkB) signaling. J Biol Chem. (2014) 289:31682–92. doi: 10.1074/jbc.M114.603621
61. Banno R, Zimmer D, De Jonghe BC, Atienza, M, Rak K, Yang W, et al. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest. (2010) 120:720–34. doi: 10.1172/JCI39620
62. Haj FG, Zabolotny JM, Kim YB, Kahn BB, Neel BG. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) re-expression alters glucose homeostasis of PTP1B-/-mice. J Biol Chem. (2005) 280:15038–46. doi: 10.1074/jbc.M413240200
63. Delibegovic M, Zimmer D, Kauffman C, Rak K, Hong EG, Cho YR, et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. (2009) 58:590–9. doi: 10.2337/db08-0913
64. Vercauteren M, Remy E, Devaux C, Dautreaux B, Henry JP, Bauer F, et al. Improvement of peripheral endothelial dysfunction by protein tyrosine phosphatase inhibitors in heart failure. Circulation. (2006) 114:2498–507. doi: 10.1161/CIRCULATIONAHA.106.630129
65. Gogiraju R, Schroeter MR, Bochenek ML, Hubert A, Münzel T, Hasenfuss G, et al. Endothelial deletion of protein tyrosine phosphatase-1B protects against pressure overload-induced heart failure in mice. Cardiovasc Res. (2016) 111:204–16. doi: 10.1093/cvr/cvw101
66. Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, et al. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation. (2014) 130:902–9. doi: 10.1161/CIRCULATIONAHA.114.009683
67. Figueiredo H, Figueroa ALC, Garcia A, Fernandez-Ruiz R, Broca C, Wojtusciszyn A, et al. Targeting pancreatic islet PTP1B improves islet graft revascularization and transplant outcomes. Sci Transl Med. (2019) 11(497):eaar6294. doi: 10.1126/scitranslmed.aar6294
68. Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. (2000) 6:1401–12. doi: 10.1016/S1097-2765(00)00137-4
69. Bettaieb A, Bakke J, Nagata N, Matsuo K, Xi Y, Liu S, et al. Protein tyrosine phosphatase 1B regulates pyruvate kinase M2 tyrosine phosphorylation. J Biol Chem. (2013) 288:17360–71. doi: 10.1074/jbc.M112.441469
70. Matsuo K, Bettaieb A, Nagata N, Matsuo I, Keilhack H, Haj FG. Regulation of brown fat adipogenesis by protein tyrosine phosphatase 1B. PLoS One. (2011) 6:e16446. doi: 10.1371/journal.pone.0016446
71. Miranda S, González-Rodríguez Á, Revuelta-Cervantes J, Rondinone CM, ValverdE ÁM. Beneficial effects of PTP1B deficiency on brown adipocyte differentiation and protection against apoptosis induced by pro- and anti-inflammatory stimuli. Cell Signal. (2010) 22:645–59. doi: 10.1016/j.cellsig.2009.11.019
72. Bruder-Nascimento T, Butler BR, Herren DJ, Brands MW, Bence KK, Belin De Chantemèle EJ. Deletion of protein tyrosine phosphatase 1b in proopiomelanocortin neurons reduces neurogenic control of blood pressure and protects mice from leptin- and sympatho-mediated hypertension. Pharmacol Res. (2015) 102:235–44. doi: 10.1016/j.phrs.2015.10.012
73. Vanevski F, Xu B. Molecular and neural bases underlying roles of BDNF in the control of body weight. Front Neurosci. (2013) 7:37. doi: 10.3389/fnins.2013.00037
74. Ohira K, Hayashi M. A new aspect of the TrkB signaling pathway in neural plasticity. Curr Neuropharmacol. (2009) 7:276–85. doi: 10.2174/157015909790031210
75. Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. (2006) 444:875–80. doi: 10.1038/nature05487
76. Wyatt SB, Winters KP, Dubbert PM. Overweight and obesity: prevalence, consequences, and causes of a growing public health problem. Am J Med Sci. (2006) 331:166–74. doi: 10.1097/00000441-200604000-00002
77. Din-Dzietham R, Liu Y, Bielo MV, Shamsa F. High blood pressure trends in children and adolescents in national surveys, 1963 to 2002. Circulation. (2007) 116:1488–96. doi: 10.1161/CIRCULATIONAHA.106.683243
78. Schramm C, Fine DM, Edwards MA, Reeb AN, Krenz M. The PTPN11 loss-of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am J Physiol Heart Circ Physiol. (2012) 302:H231–43. doi: 10.1152/ajpheart.00665.2011
79. Nguyen TD, Schwarzer M, Schrepper A, Amorim PA, Blum D, Hain C, et al. Increased protein tyrosine phosphatase 1B (PTP1B) activity and cardiac insulin resistance precede mitochondrial and contractile dysfunction in pressure-overloaded hearts. J Am Heart Assoc. (2018) 7:e008865. doi: 10.1161/JAHA.118.008865
80. Fang CX, Doser TA, Yang X, Sreejayan N, Ren J. Metallothionein antagonizes aging-induced cardiac contractile dysfunction: role of PTP1B, insulin receptor tyrosine phosphorylation and AKT. Aging Cell. (2006) 5:177–85. doi: 10.1111/j.1474-9726.2006.00201.x
81. Wade F, Belhaj K, Poizat C. Protein tyrosine phosphatases in cardiac physiology and pathophysiology. Heart Fail Rev. (2018) 23:261–72. doi: 10.1007/s10741-018-9676-1
82. Hamilton SJ, Watts GF. Endothelial dysfunction in diabetes: pathogenesis, significance, and treatment. Rev Diabet Stud. (2013) 10:133–56. doi: 10.1900/RDS.2013.10.133
83. Shi Y, Vanhoutte PM. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes. (2017) 9:434–49. doi: 10.1111/1753-0407.12521
84. Unemori EN, Ferrara N, Bauer EA, Amento EP. Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J Cell Physiol. (1992) 153:557–62. doi: 10.1002/jcp.1041530317
85. Hu GJ, Feng YG, Lu WP, Li HT, Xie HW, Li SF. Effect of combined VEGF(165)/SDF-1 gene therapy on vascular remodeling and blood perfusion in cerebral ischemia. J Neurosurg. (2017) 127:670–8. doi: 10.3171/2016.9.JNS161234
86. Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci (Lond). (2005) 109:143–59. doi: 10.1042/CS20050025
87. Luo J, Zheng M, Jiang B, Li C, Guo S, Wang L, et al. Antidiabetic activity in vitro and in vivo of BDB, a selective inhibitor of protein tyrosine phosphatase 1B, from rhodomela confervoides. Br J Pharmacol. (2020) 177:4464–80. doi: 10.1111/bph.15195
88. Kingwell BA. Nitric oxide as a metabolic regulator during exercise: effects of training in health and disease. Clin Exp Pharmacol Physiol. (2000) 27:239–50. doi: 10.1046/j.1440-1681.2000.03232.x
89. Braile M, Marcella S, Cristinziano L, Galdiero MR, Modestino L, Ferrara AL, et al. VEGF-A in cardiomyocytes and heart diseases. Int J Mol Sci. (2020) 21(15):5294. doi: 10.3390/ijms21155294
90. Peach CJ, Mignone VW, Arruda MA, Alcobia DC, Hill SJ, Kilpatrick LE, et al. Molecular pharmacology of VEGF-A isoforms: binding and signalling at VEGFR2. Int J Mol Sci. (2018) 19(4):1264. doi: 10.3390/ijms19041264
91. Zhou Y, Zhu X, Cui H, Shi J, Yuan G, Shi S, et al. The role of the VEGF family in coronary heart disease. Front Cardiovasc Med. ( 2021) 8:738325. doi: 10.3389/fcvm.2021.738325
92. Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. (2012) 2:a006502. doi: 10.1101/cshperspect.a006502
93. Besnier M, Galaup A, Nicol L, Henry JP, Coquerel D, Gueret A, et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. Faseb J. (2014) 28:3351–61. doi: 10.1096/fj.13-245753
94. Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, et al. Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res. (2008) 102:1182–91. doi: 10.1161/CIRCRESAHA.107.167080
95. Louis SF, Zahradka P. Vascular smooth muscle cell motility: from migration to invasion. Exp Clin Cardiol. (2010) 15:e75–85.21264073
96. Markova B, Herrlich P, Rönnstrand L, Böhmer FD. Identification of protein tyrosine phosphatases associating with the PDGF receptor. Biochemistry. (2003) 42:2691–9. doi: 10.1021/bi0265574
97. Chang Y, Ceacareanu B, Zhuang D, Zhang C, Pu Q, Ceacareanu AC, et al. Counter-regulatory function of protein tyrosine phosphatase 1B in platelet-derived growth factor- or fibroblast growth factor-induced motility and proliferation of cultured smooth muscle cells and in neointima formation. Arterioscler Thromb Vasc Biol. (2006) 26:501–7. doi: 10.1161/01.ATV.0000201070.71787.b8
98. Sreejayan N, Lin Y, Hassid A. NO attenuates insulin signaling and motility in aortic smooth muscle cells via protein tyrosine phosphatase 1B–mediated mechanism. Arterioscler Thromb Vasc Biol. (2002) 22:1086–92. doi: 10.1161/01.ATV.0000020550.65963.E9
99. Hassid A, Huang S, Yao J. Role of PTP-1B in aortic smooth muscle cell motility and tyrosine phosphorylation of focal adhesion proteins. Am J Physiol. (1999) 277:H192–8. doi: 10.1152/ajpheart.1999.277.1.H192
100. Zhou P, Pu WT. Recounting cardiac cellular composition. Circ Res. (2016) 118:368–70. doi: 10.1161/CIRCRESAHA.116.308139
101. Giordano FJ, Gerber HP, Williams SP, Vanbruggen N, Bunting S, Ruiz-Lozano P, et al. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci U S A. (2001) 98:5780–5. doi: 10.1073/pnas.091415198
102. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. Faseb J. (1999) 13:9–22. doi: 10.1096/fasebj.13.1.9
103. Curry JM, Eubank TD, Roberts RD, Wang Y, Pore N, Maity A, et al. M-CSF signals through the MAPK/ERK pathway via Sp1 to induce VEGF production and induces angiogenesis in vivo. PLoS One. (2008) 3:e3405. doi: 10.1371/journal.pone.0003405
104. Dor Y, Camenisch TD, Itin A, Fishman GI, Mcdonald JA, Carmeliet P, et al. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. (2001) 128:1531–8. doi: 10.1242/dev.128.9.1531
105. Hsieh PC, Davis ME, Lisowski LK, Lee RT. Endothelial-cardiomyocyte interactions in cardiac development and repair. Annu Rev Physiol. (2006) 68:51–66. doi: 10.1146/annurev.physiol.68.040104.124629
106. Sun Y, Chanrasekhar V, Kessinger CW, Tang P, Gao Y, Kamli-Salino S, et al. Cardiomyocyte-specific deletion of PTP1B protects against HFD-induced cardiomyopathy through direct regulation of cardiac metabolic signaling. bioRxiv [Preprint] (2023). Available at: 10.1101/2023.05.19.541546
107. Magadum A, Singh N, Kurian AA, Munir I, Mehmood T, Brown K, et al. Pkm2 regulates cardiomyocyte cell cycle and promotes cardiac regeneration. Circulation. (2020) 141:1249–65. doi: 10.1161/CIRCULATIONAHA.119.043067
108. Kandadi MR, Panzhinskiy E, Roe ND, Nair S, Hu D, Sun A. Deletion of protein tyrosine phosphatase 1B rescues against myocardial anomalies in high fat diet-induced obesity: role of AMPK-dependent autophagy. Biochim Biophys Acta. (2015) 1852:299–309. doi: 10.1016/j.bbadis.2014.07.004
109. Song H, Zhang Z, Wang L. Small interference RNA against PTP-1B reduces hypoxia/reoxygenation induced apoptosis of rat cardiomyocytes. Apoptosis. (2008) 13:383–93. doi: 10.1007/s10495-008-0181-1
110. Coulis G, Londhe AD, Sagabala RS, Shi Y, Labbé DP, Bergeron A, et al. Protein tyrosine phosphatase 1B regulates miR-208b-argonaute 2 association and thyroid hormone responsiveness in cardiac hypertrophy. Sci Signal. (2022) 15:eabn6875. doi: 10.1126/scisignal.abn6875
111. Zhou Q, Schötterl S, Backes D, Brunner E, Hahn JK, Ionesi E, et al. Inhibition of miR-208b improves cardiac function in titin-based dilated cardiomyopathy. Int J Cardiol. (2017) 230:634–41. doi: 10.1016/j.ijcard.2016.12.171
112. Grueter CE, Van Rooij E, Johnson BA, Deleon SM, Sutherland LB, Qi X, et al. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. (2012) 149:671–83. doi: 10.1016/j.cell.2012.03.029
113. Zhang S, Zhang ZY. PTP1B as a drug target: recent developments in PTP1B inhibitor discovery. Drug Discov Today. (2007) 12:373–81. doi: 10.1016/j.drudis.2007.03.011
114. Abdel-Magid AF. The inhibitors of protein tyrosine phosphatase nonreceptor type 2 (PTPN2) as potential enhancers of cancer immunotherapy and type 1 (PTPN1) as treatment of metabolic diseases. ACS Med Chem Lett. (2022) 13:19–21. doi: 10.1021/acsmedchemlett.1c00678
115. Liu R, Mathieu C, Berthelet J, Zhang W, Dupret JM, Rodrigues Lima F. Human protein tyrosine phosphatase 1B (PTP1B): from structure to clinical inhibitor perspectives. Int J Mol Sci. (2022) 23(13):7027. doi: 10.3390/ijms23137027
116. Sharma B, Xie L, Yang F, Wang W, Zhou Q, Xiang M, et al. Recent advance on PTP1B inhibitors and their biomedical applications. Eur J Med Chem. (2020) 199:112376. doi: 10.1016/j.ejmech.2020.112376
117. Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat Chem Biol. (2014) 10:558–66. doi: 10.1038/nchembio.1528
118. Lantz KA, Hart SG, Planey SL, Roitman MF, Ruiz-White IA, Wolfe HR, et al. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity (Silver Spring). (2010) 18:1516–23. doi: 10.1038/oby.2009.444
119. Geary RS, Bradley JD, Watanabe T, Kwon Y, Wedel M, Van Lier JJ, et al. Lack of pharmacokinetic interaction for ISIS 113715, a 2′-0-methoxyethyl modified antisense oligonucleotide targeting protein tyrosine phosphatase 1B messenger RNA, with oral antidiabetic compounds metformin, glipizide or rosiglitazone. Clin Pharmacokinet. (2006) 45:789–801. doi: 10.2165/00003088-200645080-00003
120. Waring JF, Ciurlionis R, Clampit JE, Morgan S, Gum RJ, Jolly RA, et al. PTP1B antisense-treated mice show regulation of genes involved in lipogenesis in liver and fat. Mol Cell Endocrinol. (2003) 203:155–68. doi: 10.1016/S0303-7207(03)00008-X
121. Digenio A, Pham NC, Watts LM, Morgan ES, Jung SW, Baker BF, et al. Antisense inhibition of protein tyrosine phosphatase 1B with IONIS-PTP-1BRx improves insulin sensitivity and reduces weight in overweight patients with type 2 diabetes. Diabetes Care. (2018) 41:807–14. doi: 10.2337/dc17-2132
122. Krishnan N, Felice C, Rivera K, Pappin DJ, Tonks NK. DPM-1001 decreased copper levels and ameliorated deficits in a mouse model of Wilson’s disease. Genes Dev. (2018) 32:944–52. doi: 10.1101/gad.314658.118
123. Kafle B, Aher NG, Khadka D, Park H, Cho H. Isoxazol-5(4H)one derivatives as PTP1B inhibitors showing an anti-obesity effect. Chemi Asian J. (2011) 6:2073–9. doi: 10.1002/asia.201100154
124. Fordahl SC, Jones SR. High-fat-diet-induced deficits in dopamine terminal function are reversed by restoring insulin signaling. ACS Chem Neurosci. (2017) 8:290–9. doi: 10.1021/acschemneuro.6b00308
125. Du ZD, Hu LT, Zhao GQ, Li Y, Ma ZZ. Protein tyrosine phosphatase 1B regulates the activity of retinal pigment epithelial cells. Mol Vis. (2015) 21:523–31.25999679
126. Du J, Dong Z, Tan L, Tan M, Zhang F, Zhang K, et al. Tubeimoside I inhibits cell proliferation and induces a partly disrupted and cytoprotective autophagy through rapidly hyperactivation of MEK1/2-ERK1/2 cascade via promoting PTP1B in melanoma. Front Cell Dev Biol. (2020) 8:607757. doi: 10.3389/fcell.2020.607757
127. Ponnusamy M, Ma L, Zhuang S. Necrotic renal epithelial cell inhibits renal interstitial fibroblast activation: role of protein tyrosine phosphatase 1B. Am J Physiol Renal Physiol. (2013) 304:F698–709. doi: 10.1152/ajprenal.00564.2012
128. Xue W, Tian J, Wang XS, Xia J, Wu S. Discovery of potent PTP1B inhibitors via structure-based drug design, synthesis and in vitro bioassay of norathyriol derivatives. Bioorg Chem. (2019) 86:224–34. doi: 10.1016/j.bioorg.2019.01.059
129. Erbe DV, Wang S, Zhang YL, Harding K, Kung L, Tam M, et al. Ertiprotafib improves glycemic control and lowers lipids via multiple mechanisms. Mol Pharmacol. (2005) 67:69–77. doi: 10.1124/mol.104.005553
130. Zhang ZY, Lee SY. PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. Expert Opin Investig Drugs. (2003) 12:223–33. doi: 10.1517/13543784.12.2.223
131. Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. (2005) 44:5258–66. doi: 10.1021/bi048135v
132. Weber PC, Salemme FR. Applications of calorimetric methods to drug discovery and the study of protein interactions. Curr Opin Struct Biol. (2003) 13:115–21. doi: 10.1016/S0959-440X(03)00003-4
133. Kumar GS, Page R, Peti W. The mode of action of the protein tyrosine phosphatase 1B inhibitor ertiprotafib. PLoS One. (2020) 15:e0240044. doi: 10.1371/journal.pone.0240044
134. Rao MN, Shinnar AE, Noecker LA, Chao TL, Feibush B, Snyder B, et al. Aminosterols from the dogfish shark squalus acanthias. J Nat Prod. (2000) 63:631–5. doi: 10.1021/np990514f
135. Julien SG, Tremblay ML. Chapter 99—insulin receptor PTP: pTP1B. In: Bradshaw RA, Dennis EA, editors. Handbook of Cell Signaling, 2nd ed. San Diego: Academic Press (2010).
136. Liu F, Chen J, Hu W, Gao C, Zeng Z, Cheng S, et al. PTP1B inhibition improves mitochondrial dynamics to alleviate calcific aortic valve disease via regulating OPA1 homeostasis. JACC Basic Transl Sci. (2022) 7:697–712. doi: 10.1016/j.jacbts.2022.03.002
137. Fukuda S, Ohta T, Sakata S, Morinaga H, Ito M, Nakagawa Y, et al. Pharmacological profiles of a novel protein tyrosine phosphatase 1B inhibitor, JTT-551. Diabetes Obes Metab. (2010) 12:299–306. doi: 10.1111/j.1463-1326.2009.01162.x
138. Qian S, Zhang M, He Y, Wang W, Liu S. Recent advances in the development of protein tyrosine phosphatase 1B inhibitors for type 2 diabetes. Future Med Chem. (2016) 8:1239–58. doi: 10.4155/fmc-2016-0064
139. Krishnan N, Konidaris KF, Gasser G, Tonks NK. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J Biol Chem. (2018) 293:1517–25. doi: 10.1074/jbc.C117.819110
140. Phong NV, Oanh VT, Yang SY, Choi JS, Min BS, Kim JA. PTP1B inhibition studies of biological active phloroglucinols from the rhizomes of dryopteris crassirhizoma: kinetic properties and molecular docking simulation. Int J Biol Macromol. (2021) 188:719–28. doi: 10.1016/j.ijbiomac.2021.08.091
141. Ha MT, Seong SH, Nguyen TD, Cho WK, Ah KJ, Ma JY, et al. Chalcone derivatives from the root bark of morus alba L. Act as inhibitors of PTP1B and α-glucosidase. Phytochemistry. (2018) 155:114–25. doi: 10.1016/j.phytochem.2018.08.001
142. Fukada T, Tonks NK. The reciprocal role of Egr-1 and Sp family proteins in regulation of the PTP1B promoter in response to the p210 Bcr-abl oncoprotein-tyrosine kinase. J Biol Chem. (2001) 276:25512–9. doi: 10.1074/jbc.M101354200
143. Fukada T, Tonks NK. Identification of YB-1 as a regulator of PTP1B expression: implications for regulation of insulin and cytokine signaling. EMBO J. (2003) 22:479–93. doi: 10.1093/emboj/cdg067
144. Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. (2008) 283:14230–41. doi: 10.1074/jbc.M800061200
145. Suwaki N, Vanhecke E, Atkins KM, GraF M, Swabey K, Huang P, et al. A HIF-regulated VHL-PTP1B-src signaling axis identifies a therapeutic target in renal cell carcinoma. Sci Transl Med. (2011) 3:85ra47. doi: 10.1126/scitranslmed.3002004
146. Rehman K, Akash MS. Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked? J Biomed Sci. (2016) 23:87. doi: 10.1186/s12929-016-0303-y
147. Zhang X, Han X, Tang Y, Wu Y, Qu B, Shen N. miR-744 enhances type I interferon signaling pathway by targeting PTP1B in primary human renal mesangial cells. Sci Rep. (2015) 5:12987. doi: 10.1038/srep12987
148. Yang YM, Seo SY, Kim TH, Kim SG. Decrease of microRNA-122 causes hepatic insulin resistance by inducing protein tyrosine phosphatase 1B, which is reversed by licorice flavonoid. Hepatology. (2012) 56:2209–20. doi: 10.1002/hep.25912
149. Sun F, Yu M, Yu J, Liu Z, Zhou X, Liu Y, et al. miR-338-3p functions as a tumor suppressor in gastric cancer by targeting PTP1B. Cell Death Dis. (2018) 9:522. doi: 10.1038/s41419-018-0611-0
150. Yu M, Liu Z, Liu Y, Zhou X, Sun F, Liu Y, et al. PTP1B markedly promotes breast cancer progression and is regulated by miR-193a-3p. FEBS J. (2019) 286:1136–53. doi: 10.1111/febs.14724
151. Wang S, Cheng Z, Chen X, Xue H. microRNA-135a protects against myocardial ischemia-reperfusion injury in rats by targeting protein tyrosine phosphatase 1B. J Cell Biochem. (2019) 120:10421–33. doi: 10.1002/jcb.28327
152. Xu J, Zhang Z, Chen Q, Yang L, Yin J . miR-146b regulates cell proliferation and apoptosis in gastric cancer by targeting PTP1B. Dig Dis Sci. (2020) 65:457–63. doi: 10.1007/s10620-019-05771-8
153. Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT, Puryer MA, Meng TC, Tonks NK, et al. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol Cell Biol. (2003) 23:2096–108. doi: 10.1128/MCB.23.6.2096-2108.2003
154. Gorgulla C, Boeszoermenyi A, Wang ZF, Fischer PD, Coote PW, Padmanabha Das KM, et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature. (2020) 580:663–8. doi: 10.1038/s41586-020-2117-z
155. Adams J, Thornton BP, Tabernero L. A new paradigm for KIM-PTP drug discovery: identification of allosteric sites with potential for selective inhibition using virtual screening and LEI analysis. Int J Mol Sci. (2021) 22(22):12206. doi: 10.3390/ijms222212206
157. Burslem GM, Crews CM. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. (2020) 181:102–14. doi: 10.1016/j.cell.2019.11.031
158. Feng B, Dong Y, Shang B, Zhang B, Crans DC, Yang X. Convergent protein phosphatase inhibitor design for PTP1B and TCPTP: exchangeable vanadium coordination complexes on graphene quantum dots. Adv Funct Mater. (2022) 32:2108645. doi: 10.1002/adfm.202108645
159. Gunasekaran T, Haile T, Nigusse T, Dhanaraju MD. Nanotechnology: an effective tool for enhancing bioavailability and bioactivity of phytomedicine. Asian Pac J Trop Biomed. (2014) 4:S1–7. doi: 10.12980/APJTB.4.2014C980
Keywords: protein tyrosine phosphatase 1B (PTP1B), cardiovascular disease, insulin resistance, diabetes, obesity, therapeutics
Citation: Sun Y, Dinenno FA, Tang P and Kontaridis MI (2024) Protein tyrosine phosphatase 1B in metabolic and cardiovascular diseases: from mechanisms to therapeutics. Front. Cardiovasc. Med. 11:1445739. doi: 10.3389/fcvm.2024.1445739
Received: 11 June 2024; Accepted: 5 August 2024;
Published: 22 August 2024.
Edited by:
Venkatesh Sundararajan, West Virginia University, United StatesReviewed by:
Marcello Rota, New York Medical College, United StatesSujith Rajan, New York University, United States
Copyright: © 2024 Sun, Dinenno, Tang and Kontaridis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria I. Kontaridis, bWtvbnRhcmlkaXNAbW1yaS5lZHU=