 Eng Leng Saw
Eng Leng Saw Martin Fronius
Martin Fronius Rajesh Katare
Rajesh Katare Yoshihiko Kakinuma
Yoshihiko Kakinuma- 1Whitaker Cardiovascular Institute, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, United States
- 2Department of Physiology, HeartOtago, University of Otago, Dunedin, New Zealand
- 3Department of Bioregulatory Science, Graduate School of Medicine, Nippon Medical School, Tokyo, Japan
Diabetic heart disease remains the leading cause of death in individuals with type-2 diabetes mellitus (T2DM). Both insulin resistance and metabolic derangement, hallmark features of T2DM, develop early and progressively impair cardiovascular function. These factors result in altered cardiac metabolism and energetics, as well as coronary vascular dysfunction, among other consequences. Therefore, gaining a deeper understanding of the mechanisms underlying the pathophysiology of diabetic heart disease is crucial for developing novel therapies for T2DM-associated cardiovascular disease. Cardiomyocytes are equipped with the cholinergic machinery, known as the non-neuronal cardiac cholinergic system (NNCCS), for synthesizing and secreting acetylcholine (ACh) as well as possessing muscarinic ACh receptor for ACh binding and initiating signaling cascade. ACh from cardiomyocytes regulates glucose metabolism and energetics, endothelial function, and among others, in an auto/paracrine manner. Presently, there is only one preclinical animal model – diabetic db/db mice with cardiac-specific overexpression of choline transferase (Chat) gene - to study the effect of activated NNCCS in the diabetic heart. In this mini-review, we discuss the physiological role of NNCCS, the connection between NNCCS activation and cardiovascular function in T2DM and summarize the current knowledge of S-Nitroso-NPivaloyl-D-Penicillamine (SNPiP), a novel inducer of NNCCS, as a potential therapeutic strategy to modulate NNCCS activity for diabetic heart disease.
1 Introduction
Type-2 diabetes mellitus (T2DM), a chronic metabolic disorder characterized by hyperglycemia and insulin resistance, continues to grow in incidence and prevalence worldwide (1). Diabetic heart disease, referred to a collection of cardiovascular disease such as coronary artery disease, myocardial infarction, diabetic cardiomyopathy, and heart failure, is a major cause of mortality among individuals with T2DM [reviewed in (2)]. While the pathophysiology of diabetic heart disease is complex and yet to be clearly defined, insulin resistance and metabolic derangements contribute significantly [reviewed in (3)]. Both factors impair cardiac glucose metabolism (4–7) and coronary perfusion (8–11), as well as promotes myocardial fibrosis (12–14), contributing to cardiovascular dysfunction in diabetic individuals.
The parasympathetic nervous system exerts negative chronotropic, inotropic, and dromotropic effects on the heart, primarily mediated through acetylcholine (ACh) produced by cholinergic neurons [reviewed in (15)]. Subsequent studies have revealed that cardiomyocytes possess a cholinergic machinery capable of synthesizing and secreting ACh. This machinery, referred to as the non-neuronal cardiac cholinergic system (NNCCS), is crucial for auto/paracrine regulation of the heart [reviewed in (16)]. The presence of NNCCS is considered complementary to the parasympathetic nervous system, given the sparse cholinergic innervation in the ventricular myocardium (17–21). In the heart, other non-neuronal cardiac cells such as endothelial cells similarly possess this cholinergic machinery (22, 23), while smooth muscle cells, pericytes and fibroblasts have not been characterized or shown to possess cholinergic machinery (24). The primary source of non-neuronal ACh in the heart is likely cardiomyocytes, given their prevalence among cardiac cells (25).
NNCCS comprises key components such as the ACh-synthesizing enzyme (choline acetyltransferase, ChAT), vesicular ACh transporter (VAChT), choline transporter 1 (CHT1) and ACh-degrading enzyme (acetylcholinesterase, AChE). Furthermore, cardiomyocytes express multiple isoforms of muscarinic ACh receptor (MAChR), with the type-2 isoform (M2AChR) being predominantly expressed (26), enabling ACh binding and initiation of signaling transduction. The current understanding of the role of NNCCS in the heart includes the regulation of glucose metabolism, induction of angiogenesis, regulation of heart rate, anti-hypertrophy, and immunomodulation (27–33).
Dysregulation of NNCCS has previously been demonstrated in the left ventricle of patients with non-ischemic dilated cardiomyopathy (33) and diabetic heart disease (34). In this mini-review, we specially focus on the role of NNCCS in the diabetic heart in T2DM. We first discuss the physiological role of NNCCS and evidence of NNCCS dysregulation in the diabetic heart, followed by a discussion about the effect of activated NNCCS on the cardiac and vascular function demonstrated in diabetic db/db mice with cardiac-specific overexpression of Chat gene. Lastly, we discuss the therapeutic potential of S-Nitroso-NPivaloyl-D-Penicillamine (SNPiP), a novel nitric oxide donor and an inducer of NNCCS, to modulate NNCCS activity for treating diabetic heart.
2 Role of NNCCS in regulating normal physiology of the heart
The NNCCS maintains efficient energy metabolism in cardiomyocytes by promoting glucose-dependent metabolism and suppressing mitochondrial oxygen consumption and activity (28, 29, 35–37). Previous studies have shown that this energy regulatory role is mediated by the pro-survival phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt)/hypoxia-inducible factor 1 (HIF)-α non-hypoxic signaling cascade induced by ACh, which results in an upregulation of glucose transporter (GLUT)-4 to enhance glucose uptake (28, 29, 38). GLUT-4 is noted as the predominantly expressed isoform of GLUT in the adult heart [reviewed in (39)]. Although cardiomyocytes primarily utilize free fatty acid for energy production, glucose is more energy-efficient, requiring few oxygen molecules for oxidation (phosphate/oxygen ratio = 2.58) compared to free fatty acid (phosphate/oxygen ratio = 2.33) [reviewed in (40)].
Furthermore, NNCCS regulates angiogenesis and maintains coronary vasculature in the heart. This role is similarly mediated by HIF-1α non-hypoxic signaling, leading to the upregulation of pro-angiogenic vascular endothelial growth factor (VEGF) in cardiomyocytes (27–29, 38). The secreted VEGF induces an angiogenic response in endothelial cells in a paracrine manner.
Additionally, NNCCS plays a crucial physiological role in modulating heart rate and contraction force by counteracting sympathetic stimulation (31, 33). While the underlying mechanism remains to be clearly elucidated, it is proposed that ACh released from cholinergic neurons (parasympathetic nervous system) stimulates ACh synthesis in cardiomyocytes through a positive feedback mechanism (35). The cardiac-derived ACh then acts on neighboring cardiomyocytes and other cardiac cells such as sinoatrial and atrioventricular node cells, facilitating the propagation of the neuronal cholinergic effect despite the sparse cholinergic innervation in the ventricular myocardium. Moreover, NNCCS is known to counteract sympathetic-induced cardiac hypertrophy and remodeling in response to stress (31–33, 41).
Multiple studies have demonstrated that NNCCS, similar to vagal nerve stimulation (42), helps maintain gap junctions by regulating the expression and localization of connexin 43 and β-catenin in cardiomyocytes (28, 37). Functional gap junctions are crucial for cell-cell communication and, most importantly, for propagation of action potential in the heart (43). This suggests that NNCCS is essential for maintaining the electrophysiological function of the heart. However, the precise mechanism by which NNCCS regulates connexin 43 and β-catenin has not yet been demonstrated.
The immunomodulatory role of NNCCS is a newly discovered physiological function. NNCCS decreases the expression of pro-inflammatory mediators, including c-c motif chemokine ligand (CCL)-2 and -7, thereby preventing the influx of proinflammatory c-c motif chemokine receptor (CCR)-2-positive monocytes to the site of injury in the heart (32). This suggests that cardiomyocytes, at least in part, modulates local inflammatory responses through NNCCS and mediates paracrine interactions with inflammatory cells. However, the precise mechanisms by which NNCCS regulates expression of pro-inflammatory mediators remain unclear and warrant further investigation. A schematic illustration of the role of NNCCS in regulating normal physiology of heart is shown in Figure 1.
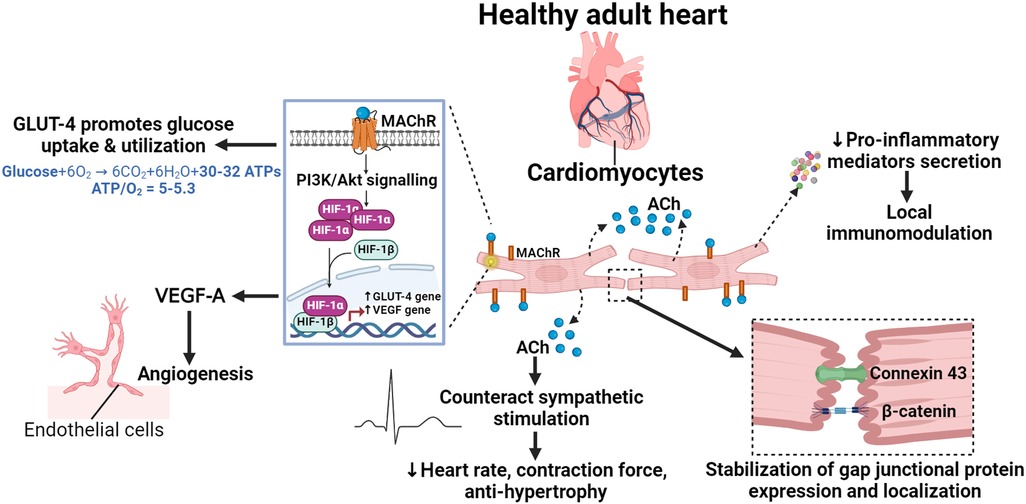
Figure 1. The physiological role of NNCCS in the healthy heart. Acetylcholine (ACh) released from cardiomyocytes initiates pro-survival PI3K/Akt/HIF-1α non-hypoxic signalling in an auto/paracrine manner, inducing the expression of glucose transporter (GLUT)-4 and the pro-angiogenic vascular endothelial growth factor (VEGF)-A. GLUT-4 promotes glucose uptake/utilization, leading to efficient energy metabolism, while VEGF-A induces angiogenic activity in endothelial cells. ACh from cardiomyocytes helps amplify neuronal cholinergic effects to modulate heart rate and contraction force, as well as to counteract sympathetic-induced cardiac hypertrophy and remodelling. ACh from cardiomyocytes maintains functional gap junctions by regulating the expression and localization of connexin 43 and β-catenin. Additionally, it also modulates the local inflammatory response by regulating the expression of pro-inflammatory mediators.
3 NNCCS in T2DM
A time-course study using diabetic db/db mice of different ages was conducted to assess the putative role of NNCCS during disease progression (34). In early stage of T2DM, characterized by hyperglycemia and insulin resistance in db/db mice (12–16 weeks old) (44), cardiac ACh levels (i.e., measured in the left ventricular lysate) remained unchanged despite decreased ChAT protein expression in the diabetic heart. In established (20–24 weeks old) and progressed stages of T2DM (28–32 weeks old) with deteriorating cardiovascular function (11, 45), cardiac ACh levels decreased along with reduced ChAT protein expression in the diabetic heart. This suggests that NNCCS dysregulation may play a role in the pathogenesis of cardiovascular dysfunction in diabetic hearts.
Interestingly, additional evidence suggested a compensatory effect in response to reduced cardiac ACh levels, as demonstrated by decreased AChE protein expression and increased CHT1 protein expression in the diabetic heart, specifically in progressed stage of T2DM (34). M2AChR protein expression was consistently reduced in the diabetic heart across early, established, and progressed stages of T2DM, indicating likely diminished NNCCS-mediated signaling.
Moreover, reduced cardiac ACh levels were accompanied by decreased cardiac glucose contents and GLUT-4 protein expression in the heart of db/db mice in established and progressed stages (34). This suggests that changes in cardiac ACh levels and/or NNCCS activity is associated with impaired glucose metabolism and may contribute to the development of cardiovascular dysfunction in T2DM. Besides these changes in db/db mice, reduced ChAT and CHT1 protein expressions were observed in the left ventricle biopsies from patients with T2DM (34), underscoring the putative clinical significance of NNCCS in T2DM-related cardiovascular disease.
While the mechanism behind NNCCS dysregulation in T2DM remains to be fully elucidated, in vitro studies showed that insulin upregulates ChAT expression in cultured human cardiomyocytes. Interestingly, this effect was abolished in cardiomyocytes exposed to palmitate to induce insulin-resistance [data published in supplemental results by Saw et al. (34)]. This study was the first to demonstrate insulin's effect on ChAT expression in cardiomyocytes, as was previously known only in neuronal cells (46–48). Further, these findings also suggest a potential link between hyperlipidemia and NNCCS dysregulation in T2DM, as hyperlipidemia can lead to excessive intracellular lipid accumulation and subsequently lipotoxicity, which is detrimental to cardiovascular function (49).
4 NNCCS and cardiac function in T2DM
In T2DM, cardiac dysfunction often remains asymptomatic and undetected in the early stage, marked by left ventricular (LV) hypertrophy and diastolic dysfunction [review in (3)]. As the disease progresses, cardiac dysfunction deteriorates further, leading to LV dilation and systolic dysfunction. This progression is accompanied by significant alterations in cardiac energy metabolism (4–7) and accelerated myocardial fibrosis (12–14) in diabetic hearts, contributing to cardiac dysfunction. Activation of NNCCS in diabetic hearts, as demonstrated in unique diabetic db/db mice with cardiac-specific overexpression of Chat gene (db/db-ChAT-tg mice), has been shown to improve cardiac function by mitigating alterations in cardiac energy metabolism and myocardial fibrosis (Figure 2) (34). This improvement is associated with augmented cardiac ACh levels as well as activation of ACh-mediated pro-survival PI3K/Akt/HIF-α non-hypoxic signaling cascade through MAChR in the heart of db/db-ChAT-tg mice (28, 29, 38).
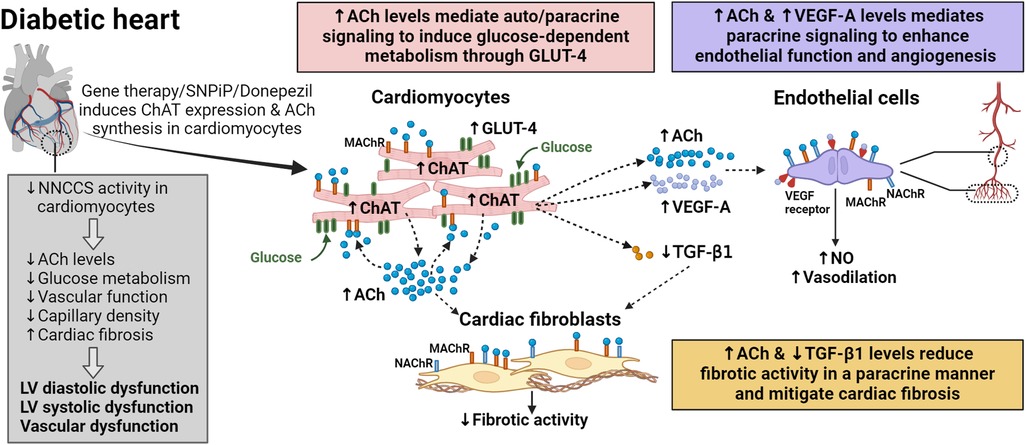
Figure 2. Auto/paracrine function of NNCCS in the diabetic heart. Reduced NNCCS activity is associated with diabetes mellitus-induced cardiovascular dysfunction. NNCCS activity in diabetic heart can be induced through gene therapy (cardiac-specific overexpression of the Chat gene) or pharmacological agents such as SNPiP and donepezil. Activated NNCCS leads to elevated acetylcholine (ACh) levels in diabetic cardiomyocytes, inducing the expression of glucose transporter (GLUT)-4, pro-angiogenic vascular endothelial growth factor (VEGF)-A, while diminishing the expression of pro-fibrotic transforming growth factor (TGF)-β1. In diabetic cardiomyocytes with activated NNCCS, GLUT-4 promotes glucose uptake/utilization, improving cardiac efficiency. Diabetic cardiomyocytes mediate paracrine signalling in endothelial cells through VEGF-A and ACh to enhance endothelial function and promote angiogenesis of coronary vessels and capillaries. Similarly, diabetic cardiomyocytes mediate paracrine signalling through ACh to attenuate the fibrotic activity of cardiac fibroblasts. Reduced TGF-β1 levels also diminish fibrotic activity in the diabetic heart.
Insulin resistance and metabolic derangement lead to a shift in substrate supply and utilization in cardiomyocytes, thereby altering cardiac energy metabolism in diabetic conditions. Increased fatty acid and decreased glucose metabolism were previously described, which may predispose the heart to the development of diastolic and systolic dysfunction (4–7). These metabolic changes occur early in T2DM (50), suggesting that early rectification of cardiac metabolism may prevent the development of diabetes-associated cardiac dysfunction (51, 52). In the heart of db/db-ChAT-tg mice and cultured human cardiomyocytes with overexpression of CHAT gene, activated NNCCS increased glucose contents, GLUT-4 protein expression and membrane translocation (34). This indicates a shift towards glucose utilization in diabetic cardiomyocytes, leading to greater cardiac efficiency (i.e., cardiac work/O2 consumed) and improved energy status (53). Upregulation of GLUT-4 protein expression is mediated by HIF1 transcription factor, as PI3K/Akt signaling prevents degradation of HIF-α, allowing it to dimerize with HIF-β to form HIF1 under normoxic conditions.
Accelerated myocardial fibrosis increases ventricular wall stiffness, contributing to diastolic dysfunction in the diabetic heart (12–14). Cardiac fibroblasts are the key extracellular matrix (ECM)-producing cells responsible for myocardial fibrosis (54). Their fibrotic activity can be activated by neighboring cells including cardiomyocytes, immune cells, endothelial cells, and others, through fibrogenic growth factors in the heart [reviewed in (55, 56)]. Activated NNCCS led to a reduction in pro-fibrotic transforming growth factor (TGF)-β1 protein expression and fibrotic area in heart of db/db-ChAT-tg mice (34). While various cell types can produce TGF-β1 (57), cardiomyocytes are likely the major source under pathological condition (58). However, ACh may also directly act on cardiac fibroblasts since these cells express MAChR and nicotinic ACh receptor (NAChR) (59), and a previous study showed that activation of M3AChR in cardiac fibroblasts led to anti-fibrotic effect through microRNA-29b/beta-site app cleaving enzyme 1 pathway (60).
5 NNCCS and vascular function in T2DM
In diabetic hearts, impaired endothelium-dependent vasodilation and capillary rarefaction disrupt coronary microcirculation, limiting the delivery of oxygen and nutrients to cardiomyocytes (8–11). Cardiac endothelial cells regulate vascular tone by producing the potent vasodilator nitric oxide (NO) in response to ACh binding on MAChR and NAChR (23, 61). Their proliferative activity can be mediated by pro-angiogenic factors including VEGF-A, which is essential for angiogenesis [reviewed in (62)].
Activation of NNCCS resulted in preserved endothelial function as well as an increase in the number of second order coronary vessels and capillary density, along with a concurrent increase in VEGF-A protein expression in the heart of db/db-ChAT-tg mice (Figure 2) (34). Elevated ACh levels from cardiomyocytes stimulate the synthesis of vasoactive factors in endothelial cells, including NO, that trigger vasodilation (23, 61), and also induce endothelial cell migration, a key event in angiogenesis, through NAChR (63). Upregulation of VEGF-A is induced by HIF1 transcription factor through ACh-mediated PI3K/Akt signalling cascade in cardiomyocytes. Notably, VEGF-A serves a dual role in angiogenesis induction and enhanced NO production by upregulating endothelial NO synthase (eNOS) expression in endothelial cells (64, 65). Moreover, since endothelial cells also possess cholinergic machinery like cardiomyocytes (22, 23), it is plausible that ACh from cardiomyocytes further stimulate endothelial cells to synthesize ACh in a positive feedback mechanism (35), thereby contributing to improved vascular function in the diabetic heart.
6 Targeting NNCCS as a possible therapy for diabetic heart disease
Despite emerging evidence demonstrating the beneficial effects of ACh against cardiovascular disease, there is currently no clinical therapy available to deliver or augment cardiac ACh levels in the heart. This makes NNCCS as a potential therapeutic target to achieve this goal. Possible approaches, such as vagal nerve stimulation, stem cell therapy, AChE inhibitors, gene therapy and microRNA therapy, have been summarized previously [reviewed in (16)].
Indirect evidence linking NNCCS with cardioprotection in clinical settings was previously demonstrated (66–69). These studies reported that patients with Alzheimer's disease treated with AChE inhibitors, including donepezil, experienced an improved survival rate, prognosis, and a reduced risk in cardiovascular events. Donepezil, an AChE inhibitor, was demonstrated to augment ACh synthesis in cultured primary rat cardiomyocytes (35), suggesting that donepezil can play a role in inducing NNCCS activation. Based on these basic and clinical results, donepezil may protect the heart from cardiovascular diseases, partly through NNCCS activation.
S-Nitroso-NPivaloyl-D-Penicillamine (SNPiP), a novel NNCCS inducer and also a NO donor (patent number is JP7338877 and available from FUJIFILM Wako Chemicals Corporation as product code 197-19151), has previously been shown to induce NNCCS activity in cellular models and mice (70). SNPiP activated NNCCS through NO/cyclic guanosine monophosphate (cGMP) signalling, leading to increased ChAT protein expression and cardiac ACh levels in the heart (70, 71). This resulted in enhanced cardiac output and diastolic function in healthy mice. A recent transcriptome analysis revealed that SNIPiP also activated various signalling pathways, namely hypoxic response pathway, cGMP pathway, and oxytocin signalling (71), and further, upregulated atrial natriuretic peptide and modulated calcium handling, known to have a cardioprotective effect [reviewed in (72)].
In diabetic db/db mice, SNPiP administration acutely improved diastolic and systolic function with no significant effect in heart rate after a single dose injection for five consecutive days (73). This indicates that SNPiP does not work like inotropic agents but directly improves cardiac relaxation and contraction through the Frank-Starling mechanism (71). However, thorough investigations are needed to evaluate the long-term safety and efficacy of SNPiP. While SNPiP has only been tested in db/db mice, it could potentially protect the heart against other cardiovascular diseases such as myocardial infarction, which is linked to decreased energy metabolism (28, 74), arrhythmia that is linked to electrical remodelling due to decreased connexin 43 (42, 75), thereby potentially slowing the progression of heart failure.
7 Conclusions
Activation of NNCCS offers significant advantages for the diabetic heart. It facilitates auto/paracrine communication among cardiomyocytes, cardiac fibroblasts, and endothelial cells within the diabetic heart, thereby improving cardiac metabolism, mitigating myocardial fibrosis, and preventing vascular rarefaction and endothelial dysfunction (Figure 1). Further research is warranted to delve deeper into these mechanisms and explore their potential therapeutic applications.
Author contributions
ES: Writing – original draft, Writing – review & editing. MF: Writing – review & editing. RK: Writing – review & editing. YK: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (JSPS KAKENHI) (C) Grant Number 16K08560 and the Smoking Research Foundation (2019-20), Japan, awarded to YK; Royal Society of New Zealand Catalyst Seed Funding (18-UOO-011-CSG) and Lottery Health research funding awarded to RK; University of Otago Doctoral Scholarship, research funding from the Department of Physiology, University of Otago and Elizabeth Jean Trotter Postgraduate Research Travelling Scholarship awarded to ELS during his doctoral training. ELS is currently supported by National Heart, Lung and Blood Institute (NHLBI) of the National Institute of Health under award R01HL145985 (to Dr. Flora Sam).
Acknowledgments
Figure was made in BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declared that they were an editorial board member of Frontiers at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc Diabetol. (2018) 17(1):83. doi: 10.1186/s12933-018-0728-6
2. Rajbhandari J, Fernandez CJ, Agarwal M, Yeap BXY, Pappachan JM. Diabetic heart disease: a clinical update. World J Diabetes. (2021) 12(4):383–406. doi: 10.4239/wjd.v12.i4.383
3. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126(11):1501–25. doi: 10.1161/CIRCRESAHA.120.315913
4. Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. (2003) 107(24):3040–6. doi: 10.1161/01.cir.0000072789.89096.10
5. Diamant M, Lamb HJ, Groeneveld Y, Endert EL, Smit JWA, Bax JJ, et al. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes Mellitus. J Am Coll Cardiol. (2003) 42(2):328–35. doi: 10.1016/S0735-1097(03)00625-9
6. How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes. (2006) 55(2):466–73. doi: 10.2337/diabetes.55.02.06.db05-1164
7. Fiorentino TV, Miceli S, Succurro E, Sciacqua A, Andreozzi F, Sesti G. Depressed myocardial mechano-energetic efficiency in subjects with dysglycemia. Diabetes Res Clin Pract. (2021) 177:108883. doi: 10.1016/j.diabres.2021.108883
8. Prior JO, Quiñones MJ, Hernandez-Pampaloni M, Facta AD, Schindler TH, Sayre JW, et al. Coronary circulatory dysfunction in insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus. Circulation. (2005) 111(18):2291–8. doi: 10.1161/01.CIR.0000164232.62768.51
9. Levelt E, Rodgers CT, Clarke WT, Mahmod M, Ariga R, Francis JM, et al. Cardiac energetics, oxygenation, and perfusion during increased workload in patients with type 2 diabetes mellitus. Eur Heart J. (2016) 37(46):3461–9. doi: 10.1093/eurheartj/ehv442
10. Papadogeorgos NÖ, Jörneskog G, Bengtsson M, Kahan T, Kalani M. Severely impaired microvascular reactivity in diabetic patients with an acute coronary syndrome. Cardiovasc Diabetol. (2016) 15(1):66. doi: 10.1186/s12933-016-0385-6
11. Katare R, Pearson JT, Lew JK-S, Wei M, Tsuchimouchi H, Du C-K, et al. Progressive decrease in coronary vascular function associated with type 2 diabetic heart disease. Front Physiol. (2018) 9:696. doi: 10.3389/fphys.2018.00696
12. Salvador DB, Gamba MR, Gonzalez-Jaramillo N, Gonzalez-Jaramillo V, Raguindin PFN, Minder B, et al. Diabetes and myocardial fibrosis: a systematic review and meta-analysis. JACC Cardiovasc Imaging. (2022) 15(5):796–808. doi: 10.1016/j.jcmg.2021.12.008
13. Yang Z, Xu R, Wang J-R, Xu H-Y, Fu H, Xie L-J, et al. Association of myocardial fibrosis detected by late gadolinium-enhanced mri with clinical outcomes in patients with diabetes: a systematic review and meta-analysis. BMJ Open. (2022) 12(1):e055374. doi: 10.1136/bmjopen-2021-055374
14. Pua CJ, Loo G, Kui M, Moy WL, Hii A-A, Lee V, et al. Impact of diabetes on myocardial fibrosis in patients with hypertension: the remodel study. Circ Cardiovasc Imaging. (2023) 16(7):545–53. doi: 10.1161/CIRCIMAGING.123.015051
15. Olshansky B, Sabbah HN, Hauptman PJ, Colucci WS. Parasympathetic nervous system and heart failure: pathophysiology and potential implications for therapy. Circulation. (2008) 118(8):863–71. doi: 10.1161/CIRCULATIONAHA.107.760405
16. Saw EL, Kakinuma Y, Fronius M, Katare R. The non-neuronal cholinergic system in the heart: a comprehensive review. J Mol Cell Cardiol. (2018) 125:129–39. doi: 10.1016/j.yjmcc.2018.10.013
17. Kent KM, Epstein SE, Cooper T, Jacobowitz DM. Cholinergic innervation of the canine and human ventricular conducting system anatomic and electrophysiologic correlations. Circulation. (1974) 50(5):948–55. doi: 10.1161/01.CIR.50.5.948
18. Kawano H, Okada R, Yano K. Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels. (2003) 18(1):32–9. doi: 10.1007/s003800300005
19. Hoover DB, Ganote CE, Ferguson SM, Blakely RD, Parsons RL. Localization of cholinergic innervation in Guinea pig heart by immunohistochemistry for high-affinity choline transporters. Cardiovasc Res. (2004) 62(1):112–21. doi: 10.1016/j.cardiores.2004.01.012
20. Mabe AM, Hoard JL, Duffourc MM, Hoover DB. Localization of cholinergic innervation and neurturin receptors in adult mouse heart and expression of the neurturin gene. Cell Tissue Res. (2006) 326(1):57–67. doi: 10.1007/s00441-006-0215-3
21. Clavijo LC, Pinto TL, Kuchulakanti PK, Torguson R, Chu WW, Satler LF, et al. Metabolic syndrome in patients with acute myocardial infarction is associated with increased infarct size and in-hospital complications. Cardiovasc Revasc Med. (2006) 7(1):7–11. doi: 10.1016/j.carrev.2005.10.007
22. Kirkpatrick CJ, Bittinger F, Nozadze K, Wessler I. Expression and Function of the Non-Neuronal Cholinergic System in Endothelial Cells. (0024-3205 (Print)).
23. Wilson C, Lee MD, McCarron JG. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J Physiol. (2016) 594(24):7267–307. doi: 10.1113/jp272927
24. Kawashima K, Fujii T. Basic and clinical aspects of non-neuronal acetylcholine: overview of non-neuronal cholinergic systems and their biological significance. J Pharmacol Sci. (2008) 106(2):167–73. doi: 10.1254/jphs.FM0070073
25. Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. (2020) 588(7838):466–72. doi: 10.1038/s41586-020-2797-4
26. Wang H, Han H, Zhang L, Shi H, Schram G, Nattel S, et al. Expression of multiple subtypes of muscarinic receptors and cellular distribution in the human heart. Mol Pharmacology. (2001) 59(5):1029–36. doi: 10.1124/mol.59.5.1029
27. Kakinuma Y, Furihata M, Akiyama T, Arikawa M, Handa T, Katare RG, et al. Donepezil, an acetylcholinesterase inhibitor against Alzheimer’s dementia, promotes angiogenesis in an ischemic hindlimb model. J Mol Cell Cardiol. (2010) 48(4):680–93. doi: 10.1016/j.yjmcc.2009.11.010
28. Kakinuma Y, Tsuda M, Okazaki K, Akiyama T, Arikawa M, Noguchi T, et al. Heart-specific overexpression of choline acetyltransferase gene protects murine heart against ischemia through hypoxia-inducible factor-1α-related defense mechanisms. J Am Heart Assoc. (2013) 2(1):e004887. doi: 10.1161/JAHA.112.004887
29. Oikawa S, Kai Y, Mano A, Ohata H, Kurabayashi A, Tsuda M, et al. Non-neuronal cardiac acetylcholine system playing indispensable roles in cardiac homeostasis confers resiliency to the heart. J Physiol Sci. (2021) 71(1):2. doi: 10.1186/s12576-020-00787-6
30. Rocha-Resende C, Roy A, Resende R, Ladeira MS, Lara A, de Morais Gomes ER, et al. Non-neuronal cholinergic machinery present in cardiomyocytes offsets hypertrophic signals. J Mol Cell Cardiol. (2012) 53(2):206–16. doi: 10.1016/j.yjmcc.2012.05.003
31. Roy A, Fields WC, Rocha-Resende C, Resende RR, Guatimosim S, Prado VF, et al. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. (2013) 27(12):5072–82. doi: 10.1096/fj.13-238279
32. Rocha-Resende C, Weinheimer C, Bajpai G, Adamo L, Matkovich SJ, Schilling J, et al. Immunomodulatory role of non-neuronal cholinergic signaling in myocardial injury. JCI Insight. (2019) 5(14). doi: 10.1172/jci.insight.128961
33. Roy A, Dakroub M, Tezini GC, Liu Y, Guatimosim S, Feng Q, et al. Cardiac acetylcholine inhibits ventricular remodeling and dysfunction under pathologic conditions. Faseb J. (2016) 30(2):688–701. doi: 10.1096/fj.15-277046
34. Saw EL, Pearson JT, Schwenke DO, Munasinghe PE, Tsuchimochi H, Rawal S, et al. Activation of the cardiac non-neuronal cholinergic system prevents the development of diabetes-associated cardiovascular complications. Cardiovasc Diabetol. (2021) 20(1):50. doi: 10.1186/s12933-021-01231-8
35. Kakinuma Y, Akiyama T, Sato T. Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J. (2009) 276(18):5111–25. doi: 10.1111/j.1742-4658.2009.07208.x
36. Oikawa S, Iketani M, Kakinuma Y. A non-neuronal cholinergic system regulates cellular atp levels to maintain cell viability. Cell Physiol Biochem. (2014) 34(3):781–9. doi: 10.1159/000363042
37. Kakinuma Y, Akiyama T, Okazaki K, Arikawa M, Noguchi T, Sato T. A non-neuronal cardiac cholinergic system plays a protective role in myocardium salvage during ischemic insults. PLoS One. (2012) 7(11):e50761. doi: 10.1371/journal.pone.0050761
38. Kakinuma Y, Ando M, Kuwabara M, Katare RG, Okudela K, Kobayashi M, et al. Acetylcholine from vagal stimulation protects cardiomyocytes against ischemia and hypoxia involving additive non-hypoxic induction of hif-1alpha. FEBS Lett. (2005) 579(10):2111–8. doi: 10.1016/j.febslet.2005.02.065
39. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol. (2015) 6(1):331–51. doi: 10.1002/cphy.c150016
40. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of mMetabolic flexibility in the failing heart. Front Cardiovasc Med. (2018) 5:68. doi: 10.3389/fcvm.2018.00068
41. Gavioli M, Lara A, Almeida PWM, Lima AM, Damasceno DD, Rocha-Resende C, et al. Cholinergic signaling exerts protective effects in models of sympathetic hyperactivity-induced cardiac dysfunction. PLoS One. (2014) 9(7):e100179. doi: 10.1371/journal.pone.0100179
42. Ando M, Katare RG, Kakinuma Y, Zhang D, Yamasaki F, Muramoto K, et al. Efferent vagal nerve stimulation protects heart against ischemia-induced arrhythmias by preserving Connexin43 protein. Circulation. (2005) 112(2):164–70. doi: 10.1161/CIRCULATIONAHA.104.525493
43. Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. (2004) 62(2):309–22. doi: 10.1016/j.cardiores.2003.11.035
44. Kodama H, Fujita M, Yamaguchi I. Development of hyperglycaemia and insulin resistance in conscious genetically diabetic (C57bl/ksj-db/db) mice. Diabetologia. (1994) 37(8):739–44. doi: 10.1007/bf00404329
45. Rawal S, Nagesh PT, Coffey S, Van Hout I, Galvin IF, Bunton RW, et al. Early dysregulation of cardiac-specific microrna-208a is linked to maladaptive cardiac remodelling in diabetic myocardium. Cardiovasc Diabetol. (2019) 18(1):13. doi: 10.1186/s12933-019-0814-4
46. Kyriakis JM, Hausman RE, Peterson SW. Insulin stimulates choline acetyltransferase activity in cultured embryonic chicken retina neurons. Proc Natl Acad Sci USA. (1987) 84(21):7463–7. doi: 10.1073/pnas.84.21.7463
47. Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis. (2005) 8(3):247–68. doi: 10.3233/JAD-2005-8304
48. Wang H, Wang R, Zhao Z, Ji Z, Xu S, Holscher C, et al. Coexistences of insulin signaling-related proteins and choline acetyltransferase in neurons. Brain Res. (2009) 1249:237–43. doi: 10.1016/j.brainres.2008.10.046
49. van de Weijer T, Schrauwen-Hinderling VB, Schrauwen P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res. (2011) 92(1):10–8. doi: 10.1093/cvr/cvr212
50. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and Contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. (2005) 146(12):5341–9. doi: 10.1210/en.2005-0938
51. Schmitz FJ, Rosen P, Reinauer H. Improvement of myocardial function and metabolism in diabetic rats by the carnitine palmitoyl transferase inhibitor etomoxir. Horm Metab Res. (1995) 27(12):515–22. doi: 10.1055/s-2007-980016
52. Wu CY, Satapati S, Gui W, Wynn RM, Sharma G, Lou M, et al. A novel inhibitor of pyruvate dehydrogenase kinase stimulates myocardial carbohydrate oxidation in diet-induced obesity. J Biol Chem. (2018) 293(25):9604–13. doi: 10.1074/jbc.RA118.002838
53. Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-Hglut4 mice. Am J Physiol Heart Circ Physiol. (2002) 283(3):H976–82. doi: 10.1152/ajpheart.00088.2002
54. Alex L, Tuleta I, Hanna A, Frangogiannis NG. Diabetes induces cardiac fibroblast activation, promoting a matrix-preserving nonmyofibroblast phenotype, without stimulating pericyte to fibroblast conversion. J Am Heart Assoc. (2023) 12(6):e027463. doi: 10.1161/jaha.122.027463
55. Tuleta I, Frangogiannis NG. Fibrosis of the diabetic heart: clinical significance, molecular mechanisms, and therapeutic opportunities. Adv Drug Delivery Rev. (2021) 176:113904. doi: 10.1016/j.addr.2021.113904
56. Cheng Y, Wang Y, Yin R, Xu Y, Zhang L, Zhang Y, et al. Central role of cardiac fibroblasts in myocardial fibrosis of diabetic cardiomyopathy. Front Endocrinol (Lausanne). (2023) 14:1162754. doi: 10.3389/fendo.2023.1162754
57. Kapur NK. Transforming growth factor-Β. Circ Heart Fail. (2011) 4(1):5–7. doi: 10.1161/CIRCHEARTFAILURE.110.960054
58. Wünsch M, Sharma HS, Markert T, Bernotat-Danielowski S, Schott RJ, Kremer P, et al. In situ localization of transforming growth factor Beta 1 in porcine heart: enhanced expression after chronic coronary artery constriction. J Mol Cell Cardiol. (1991) 23(9):1051–62. doi: 10.1016/0022-2828(91)91640-d
59. Liu J-J, Huang N, Lu Y, Zhao M, Yu X-J, Yang Y, et al. Improving vagal activity ameliorates cardiac fibrosis induced by angiotensin ii: in vivo and in vitro. Sci Rep. (2015) 5(1):17108. doi: 10.1038/srep17108
60. Li W, Yu J, Yang Y, Wang J, Liu Y, Wang J, et al. M(3) subtype of muscarinic acetylcholine receptor inhibits cardiac fibrosis via targeting microrna-29b/Beta-site app cleaving enzyme 1 axis. Cardiovasc Diagn Ther. (2024) 14(1):143–57. doi: 10.21037/cdt-23-309
61. Smedlund K, Tano JY, Margiotta J, Vazquez G. Evidence for operation of nicotinic and muscarinic acetylcholine receptor-dependent survival pathways in human coronary artery endothelial cells. J Cell Biochem. (2011) 112(8):1978–84. doi: 10.1002/jcb.23169
62. Braile M, Marcella S, Cristinziano L, Galdiero MR, Modestino L, Ferrara AL, et al. Vegf-a in cardiomyocytes and heart diseases. Int J Mol Sci. (2020) 21(15):5294. doi: 10.3390/ijms21155294
63. Ng MKC, Wu J, Chang E, Wang B-Y, Katzenberg-Clark R, Ishii-Watabe A, et al. A central role for nicotinic cholinergic regulation of growth factor–induced endothelial cell migration. Arterioscler, Thromb, Vasc Biol. (2007) 27(1):106–12. doi: 10.1161/01.ATV.0000251517.98396.4a
64. Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun C-O, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. (2001) 98(5):2604–9. doi: 10.1073/pnas.041359198
65. Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A, et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci U S A. (2014) 111(35):12865–70. doi: 10.1073/pnas.1408472111
66. Nordström P, Religa D, Wimo A, Winblad B, Eriksdotter M. The use of cholinesterase inhibitors and the risk of myocardial infarction and death: a nationwide cohort study in subjects with Alzheimer’s disease. Eur Heart J. (2013) 34(33):2585–91. doi: 10.1093/eurheartj/eht182
67. Wu P-H, Lin Y-T, Hsu P-C, Yang Y-H, Lin T-H, Huang C-T. Impact of acetylcholinesterase inhibitors on the occurrence of acute coronary syndrome in patients with dementia. Sci Rep. (2015) 5(1):15451. doi: 10.1038/srep15451
68. Isik AT, Soysal P, Stubbs B, Solmi M, Basso C, Maggi S, et al. Cardiovascular outcomes of cholinesterase inhibitors in individuals with dementia: a meta-analysis and systematic review. J Am Geriatr Soc. (2018) 66(9):1805–11. doi: 10.1111/jgs.15415
69. Shahim B, Xu H, Haugaa K, Zetterberg H, Jurga J, Religa D, et al. Cholinesterase inhibitors are associated with reduced mortality in patients with Alzheimer’s disease and previous myocardial infarction. Eur Heart J Cardiovasc Pharmacother. (2024) 10(2):128–36. doi: 10.1093/ehjcvp/pvad102
70. Oikawa S, Kai Y, Mano A, Nakamura S, Kakinuma Y. A novel nitric oxide donor, S-Nitroso-Npivaloyl-D-Penicillamine, activates a non-neuronal cardiac cholinergic system to synthesize acetylcholine and augments cardiac function. Cell Physiol Biochem. (2019) 52(4):922–34. doi: 10.33594/000000064
71. Takenaka Y, Hirasaki M, Bono H, Nakamura S, Kakinuma Y. Transcriptome analysis reveals enhancement of cardiogenesis-related signaling pathways by S-Nitroso-N-Pivaloyl-D-Penicillamine (snpip): implications for improved diastolic function and cardiac performance. J Cardiovasc Pharmacol. (2024) 83(5):433–45. doi: 10.1097/FJC.0000000000001552
72. Jankowski M, Broderick TL, Gutkowska J. The role of oxytocin in cardiovascular protection. Front Psychol. (2020) 11:2139. doi: 10.3389/fpsyg.2020.02139
73. Oikawa S, Kai Y, Mano A, Nakamura S, Kakinuma Y. S-Nitroso-N-Pivaloyl-D-Penicillamine, a novel non-neuronal ach system activator, modulates cardiac diastolic function to increase cardiac performance under pathophysiological conditions. Int Immunopharmacol. (2020) 84:106459. doi: 10.1016/j.intimp.2020.106459
74. Beaumont E, Southerland EM, Hardwick JC, Wright GL, Ryan S, Li Y, et al. Vagus nerve stimulation mitigates intrinsic cardiac neuronal and adverse myocyte remodeling postmyocardial infarction. Am J Physiol Heart Circ Physiol. (2015) 309(7):H1198–206. doi: 10.1152/ajpheart.00393.2015
Keywords: non-neuronal cardiac cholinergic system, acetylcholine, cardiomyocytes, diabetes mellitus, glucose metabolism, angiogenesis
Citation: Saw EL, Fronius M, Katare R and Kakinuma Y (2024) Mini Review: the non-neuronal cardiac cholinergic system in type-2 diabetes mellitus. Front. Cardiovasc. Med. 11:1425534. doi: 10.3389/fcvm.2024.1425534
Received: 29 April 2024; Accepted: 22 August 2024;
Published: 2 September 2024.
Edited by:
Carole Sudre, University College London, United KingdomReviewed by:
Kroekkiat Chinda, Naresuan University, ThailandSae Uchida, Tokyo Metropolitan Institute for Geriatrics and Gerontology, Japan
Copyright: © 2024 Saw, Fronius, Katare and Kakinuma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eng Leng Saw, ZWxzYXdAYnUuZWR1; Yoshihiko Kakinuma, azEyNDE3ODUzQG5tcy5hYy5qcA==