
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 05 July 2024
Sec. Cardiovascular Genetics and Systems Medicine
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1424551
 Seung Woo Ryu1,†
Seung Woo Ryu1,† Won Chan Jeong1,†
Won Chan Jeong1,† Geu Ru Hong2
Geu Ru Hong2 Jung Sun Cho3
Jung Sun Cho3 Soo Yong Lee4Hyungseop Kim5Jeong Yoon Jang6Sun Hwa Lee7
Soo Yong Lee4Hyungseop Kim5Jeong Yoon Jang6Sun Hwa Lee7 Dae-Hwan Bae8Jae Yeong Cho9
Dae-Hwan Bae8Jae Yeong Cho9 Ji Hee Kim10Kyung-Hee Kim11Jang Won Son12Beomman Han1Go Hun Seo1
Ji Hee Kim10Kyung-Hee Kim11Jang Won Son12Beomman Han1Go Hun Seo1 Hane Lee1*
Hane Lee1*
Background: The alpha-protein kinase 3 (ALPK3) gene (OMIM: 617608) is associated with autosomal recessive familial hypertrophic cardiomyopathy-27 (CMH27, OMIM: 618052). Recently, several studies have shown that monoallelic premature terminating variants (PTVs) in ALPK3 are associated with adult-onset autosomal dominant hypertrophic cardiomyopathy (HCMP). However, these studies were performed on patient cohorts mainly from European Caucasian backgrounds.
Methods: To determine if this finding is replicated in the Korean HCMP cohort, we evaluated 2,366 Korean patients with non-syndromic HCMP using exome sequencing and compared the cohort dataset with three independent population databases.
Results: We observed that monoallelic PTVs in ALPK3 were also significantly enriched in Korean patients with HCMP with an odds ratio score of 10–21.
Conclusions: We suggest that ALPK3 PTV carriers be considered a risk group for developing HCMP and be monitored for cardiomyopathies.
Hypertrophic cardiomyopathy (HCMP) affects approximately 1 in 500 to 1 in 200 people worldwide (1–3). It is characterized by the thickening of myocardial ventricular walls and is both phenotypically and genetically heterogeneous. While it is commonly inherited in an autosomal dominant fashion within families with incomplete penetrance and variable expressivity, there are also autosomal recessive forms, and non-Mendelian forms may exist (4). To date, there are 51 genes described to be associated with HCMP (1, 5–9). Identifying the genetic basis is important for more precise clinical management and family testing, as clinical prognosis and the possibility of intervention can vary depending on the underlying cause (9–12). With the advent of next-generation sequencing (NGS), it has become readily accessible to sequence many genes simultaneously as panels or exome/genome sequencing. The diagnostic yield from various genetic testing averages around 30%, suggesting that there are still more monogenic gene–disease associations and/or complex non-Mendelian forms yet to be discovered (3, 5, 13).
While most genes associated with HCMP are sarcomere-related genes such as MYBPC3 and MYH7, few non-sarcomere-related genes have been identified (14). One of these non-sarcomere-related genes is the ALPK3 gene coding for alpha-protein kinase 3 (ALPK3), which is a 1,705 amino acid long atypical protein kinase predicted to play an essential role as a transcription factor during cardiomyocyte differentiation (15–17). Biallelic premature terminating variants (PTVs), resulting in loss of function in ALPK3, are associated with autosomal recessive, familial hypertrophic cardiomyopathy 27 (CMH27, OMIM: 618052) characterized by early-onset severe dilated cardiomyopathy in infants that progresses to HCMP over time (18). Studies using mouse models have shown that ALPK3 is expressed early in cardiogenesis and remains expressed in cardiomyocytes (16). ALPK3-null (−/−) mice develop both ventricular hypertrophy and dilation suggesting the underlying pathomechanism of the disease is loss of function of the ALPK3 gene (19). However, despite the available evidence, the relationship and pathogenic mechanism between ALPK3 and HCMP remain unclear.
Recent studies have suggested autosomal dominant inheritance of ALPK3-associated HCMP. The original study by Almomani et al. (18) observed that family members of patients with biallelic pathogenic ALPK3 were at risk of developing later-onset HCMP. In their study, they identified 2 out of 10 family members of autosomal recessive patients who were heterozygous carriers and presented with HCMP, suggesting a possible risk association (18). A subsequent study from Herkert et al. (20) reported that monoallelic rare ALPK3 variants accounted for approximately 2.5% of the unexplained HCMP in 1,548 Dutch patients and 10% of US patients with unexplained HCMP. Lopes et al. (21) reported that heterozygous ALPK3 PTV carriers were enriched in their HCMP cohort. They discovered that 12 out of 770 HCMP patients in their discovery cohorts and 32 out of 2,047 HCMP patients in the validation cohort had monoallelic ALPK3 PTVs such as non-sense, frameshift, and canonical splice site variants. They reported an odds ratio (OR) of 16 when compared to presumed healthy controls in gnomAD v3.1.2 (21). Dai et al. (22) demonstrated that 4 out of 793 HCM cases of East Asians have monoallelic PTVs in ALPK3 with an odds ratio of 5.72 compared to the gnomAD v2.1.1 control.
These previous studies were predominantly performed on European Caucasian descent, except for one study performed on the Chinese population. Here, using a large East Asian HCMP cohort, we sought to replicate the previous studies by assessing the genetic burden of ALPK3 PTVs in Korean HCMP patients. A total of 2,366 Korean patients with non-syndromic HCMP were compared to matched controls. Heterozygous PTVs in ALPK3 were significantly enriched with an odds ratio between 10 and 21, which is consistent with previous studies.
The HCMP patient cohort comprised 2,366 Korean patients who underwent exome sequencing at a single reference laboratory in South Korea between 2019 and 2023. The inclusion criteria for the cohort included exhibiting left ventricular hypertrophy (LVH) as a primary phenotype and being predicted as an East Asian from the genetic data. Patients who did not meet these criteria were excluded. Clinical evaluations were conducted by clinicians from 16 different hospitals and clinics across the country. Since the study was conducted in a diagnostic setting and all the samples and data were de-identified throughout, Institutional Review Board (IRB) approval was not required.
Exome sequencing was performed using high molecular weight genomic DNA extracted from whole blood samples using QIAamp DNA Blood Mini Kit (Qiagen) or buccal swab samples using AccuBuccal DNA Preparation Kit (Accugene). Exome capture was performed using xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, IA, USA), and sequencing was performed using the NovaSeq 6000 platform (Illumina, San Diego, CA, USA) as 150 bp paired-end reads. Alignment to the GRCh37 human reference genome was performed using BWA-MEM2 (v2.2.1), and SAMtools v1.18 was used for bam file sorting and marking duplicates (23, 24). Recalibration and variant calling for single nucleotide variants (SNVs) and small insertion/deletion variants (indels) were performed using GATK v4.2.14. Copy number variants (CNVs) were identified using CoNIFER v0.2.2 and 3bCNV, an internally developed tool that uses depth-of-coverage information of each exon (25, 26). High-quality variants (allele depth ≥ 20, variant allele fraction ≥ 0.25 for heterozygous variants) were annotated, filtered, and classified using EVIDENCE v4, which incorporates Ensembl Variant Effect Predictor (VEP) for annotation and the American College of Medical Genetics and Genomics (ACMG) guideline for variant classification (27–29). The splicing prediction was performed using SpliceAI and SpliceVi (https://splicevi.io/), a publicly available in-house developed SpliceAI visualization tool (30). The filtered and classified variants were manually reviewed by medical geneticists and physicians. The variants that can most likely explain the patient's phenotype were selected for reporting.
The age distribution of the Korean HCMP patients was 2–99 years old, with a median age of 61 years (mean 59 years). Sixty-six percent were male patients (1,564/2,366). Pathogenic variants in the cardiovascular disorder genes were identified in 20.4% of patients (482/2,366). In 18 of these patients, more than one variant/gene was reported as likely causal for the cardiovascular phenotype. As in a previous study (6), MYBPC3 and MYH7 variants accounted for the majority of findings, each accounting for 40.7% (196/482) and 24.9% (120/482) of the total diagnoses, respectively. The third most frequently identified gene TNNI3 accounted for 12.2% of the total diagnoses (59/482).
A total of 36 unrelated HCMP patients out of 2,366 patients (1.5%) carried rare pathogenic or likely pathogenic (P/LP) PTVs in ALPK3, classified based on the ACMG guidelines (Figure 1A, Table 1). The median age of these patients was 52 years, with a mean of 53 (+/- 17.1) years. The youngest patient was 17 years old. All patients exhibited left ventricular hypertrophy, which is a hallmark of HCMP. Arrhythmia and syncope were the second most common phenotypes (Table 1). With 7 of the variants recurring, a total of 14 unique variants were identified: 7 non-sense (stop gain) variants, 5 frameshift variants, and 2 splicing variants located in the essential canonical splice site. In silico splicing prediction algorithm, SpliceAI predicted the consequence of the two splicing variants, namely, NM_020778.5:c.3965 + 2T>G (p.?) and NM_020778.5:c.4724-1G>A (p.?), to introduce a new stop codon (Supplementary Figures S1A,B). Of the 14 unique variants, 6 variants were novel variants that have not been described in any database or literature to our knowledge (Table 2). None of the HCMP patients had a copy number variant (CNV) spanning at least three consecutive exons in ALPK3. For one patient, the variant was inherited from an affected parent, who was tested later by exome sequencing and therefore not included in the 36 patients, consistent with an autosomal dominant inheritance pattern.
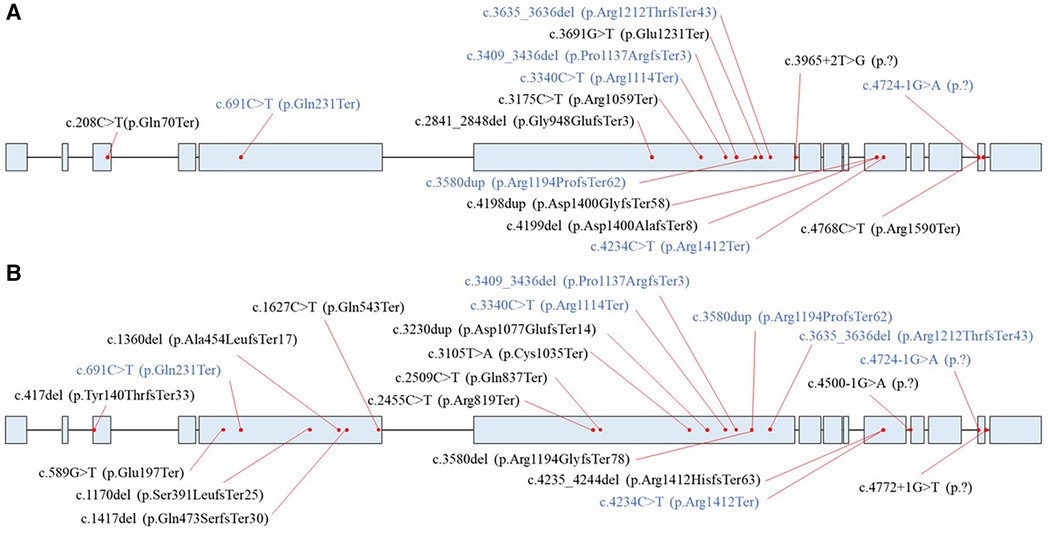
Figure 1 Distribution of ALPK3 (NM_020778.5) premature terminating variants. (A) Variants identified in the HCMP patient cohort. (B) Variants identified in the control databases. Blue represents variants that were present in both the HCMP cohort and control data sets.
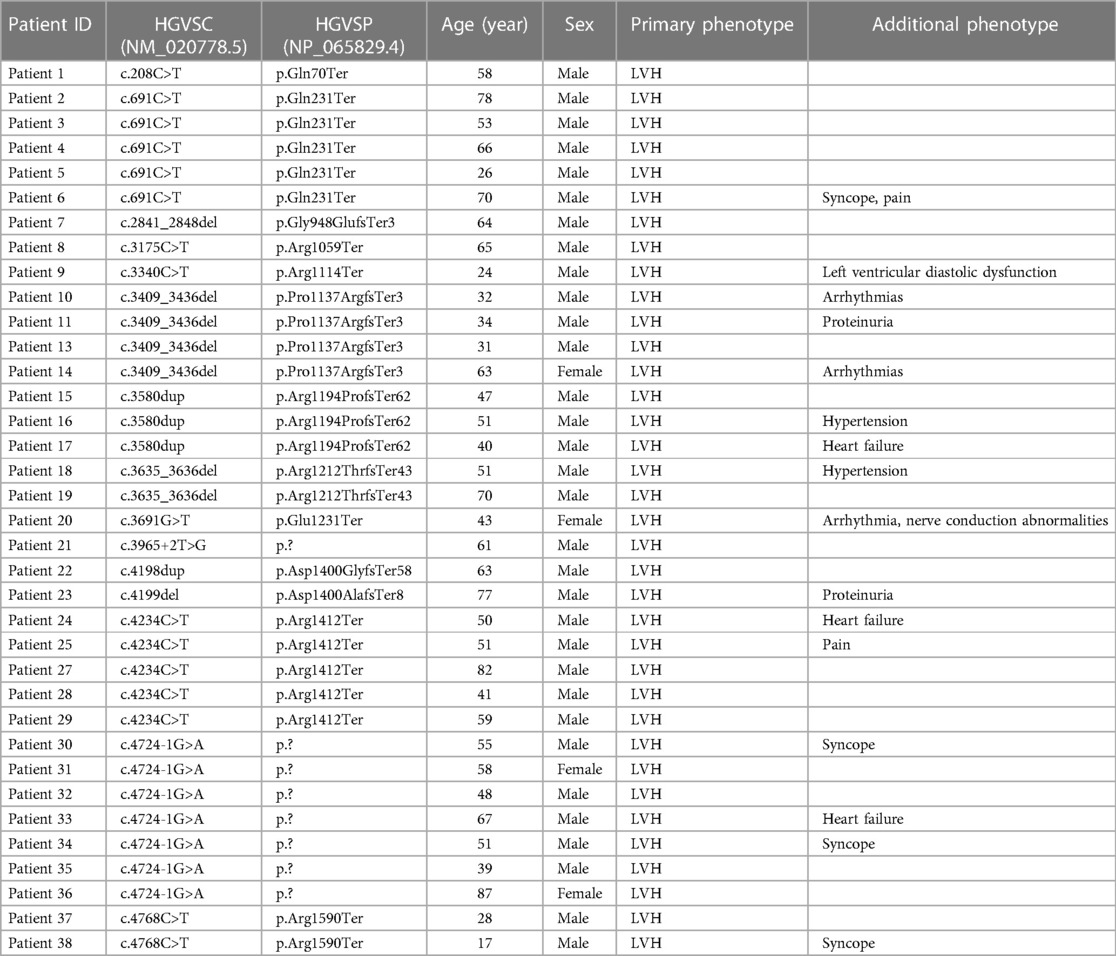
Table 1 List of ALPK3 PTV carrier HCMP patients. Phenotype information provided for the exome sequencing order is shown. NM_020778.5 transcript is used for the cDNA position. LVH, left ventricular hypertrophy.
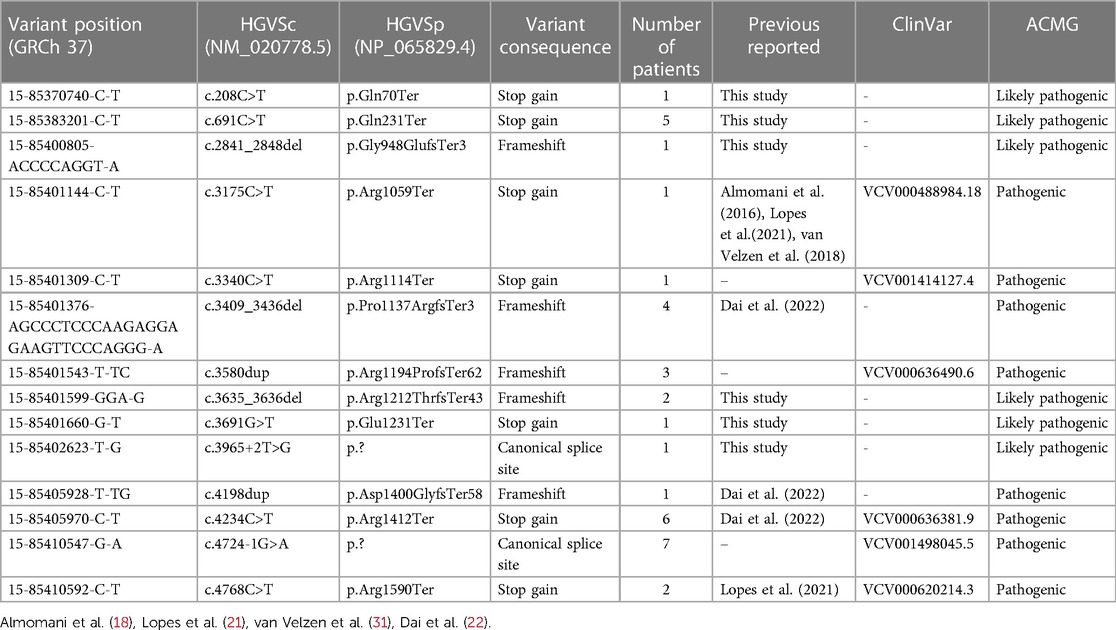
Table 2 ALPK3 variants and their consequences identified in HCMP patients.
Of the 36 unrelated individuals carrying a PTV in ALPK3, additional clinical features were collected for 24 individuals (Table 3). For the 18 patients with the age-of-onset information available, the mean age of onset was 42 years old (SD, 16.3) for 18 individuals. The average max left ventricular thickness was 19 mm (SD, 5.3) with an average left ventricular ejection fraction (LVEF) of 64.1% (SD, 11.4). Of the 24 patients, 11 patients had atypical left ventricular morphology, 8 were asymmetric, and 5 were concentric. Seven patients had experienced heart failure.
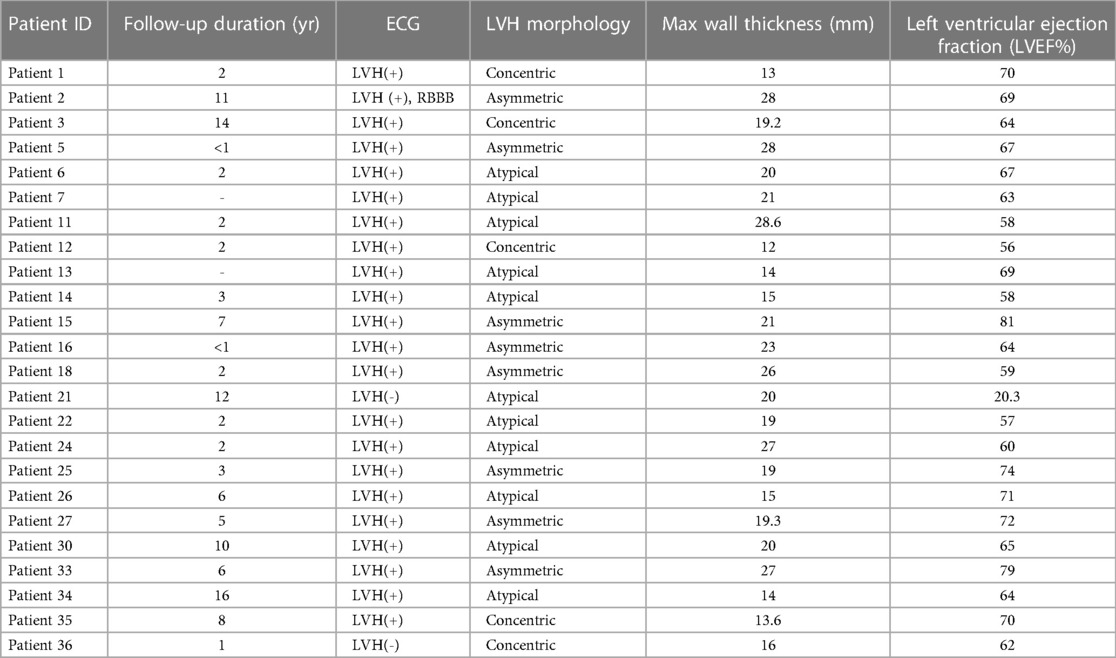
Table 3 Clinical LVH features of ALPK3 variant carriers.
We investigated if ALPK3 PTVs are enriched in our Korean HCMP patient cohort by comparing our HCMP cohort with three independent control cohorts as follows.
The first comparison was made to the 22,448 East Asian population dataset within the Genome Aggregation Database (gnomAD v4.0, https://gnomad.broadinstitute.org/), the largest publicly available control database providing 807,162 exomes/genome sequencing data from the general population assumed to be healthy (32, 33). There were 14 unique ALPK3 PTVs across 16 individuals out of 22,448 individuals (0.07%) (Table 4). All 14 variants were extremely rare with minor allele frequency (MAF) of <0.1% in the East Asian population. All 16 variants were heterozygous and classified as likely pathogenic or pathogenic based on the same classification algorithm used for classifying variants identified in our cohort. Eight variants were frameshift, five non-sense, and one essential splice site. SpliceAI predicted the consequence of the splicing variant c.4772+1G>T (p.?) to introduce a new stop codon (Supplementary Figure S1C). The odds ratio (OR) of this comparison was 21.66 (36/2,330 vs. 16/22,432; 95% CI: 12.00–39.10) (Figure 2).
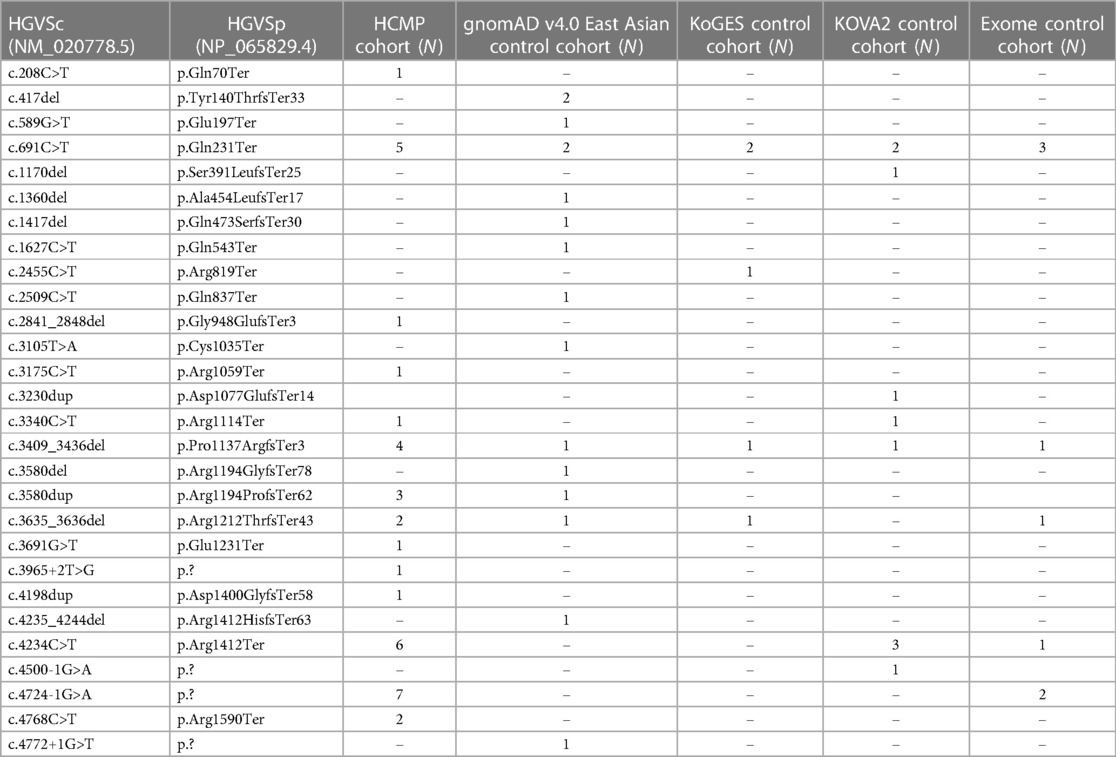
Table 4 ALPK3 variants identified in control cohorts.
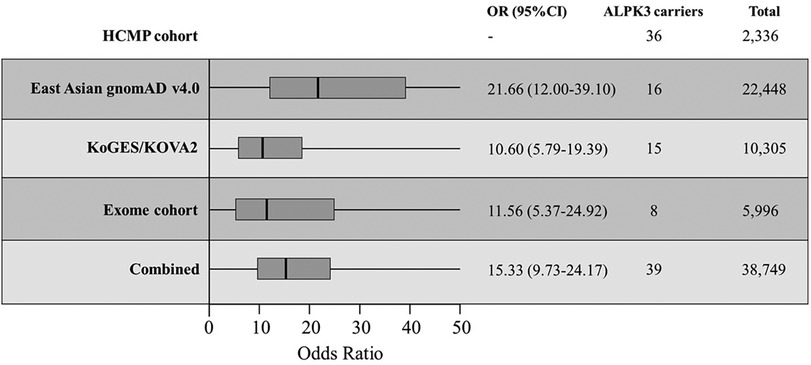
Figure 2 The odds ratios (OR) of different comparisons. Korean HCMP cohort consisted of 2,336 patients and were compared against the East Asian gnomAD v4.0 database, combined KoGES and KOVA2 database, internal exome database, and all combined. The thick vertical line is the calculated OR with the box representing the 95% CI.
The second control dataset used for comparison was combined genomic data obtained through the Korean Genome and Epidemiology Study (KoGES) consisting of 5,000 genome sequencing data from assumed healthy Korean individuals and the Korean Variant Archive (KOVA2) consisting of 3,409 exome sequencing data and 1,896 genome sequencing data from assumed healthy Korean individuals (34, 35). The age distribution of the KoGES samples was 40–69 years, and 2,336 (46.7%) were male (34). Demographic information was not available for the KOVA2 dataset except for all healthy individuals.
In the KoGES database, four unique ALPK3 PTVs were observed from five individuals. In the KOVA2 database, seven unique variants from 10 individuals were identified (Table 3). Together, 15 individuals carried ALPK3 PTVs out of 10,305 individuals (0.15%). Together, four non-sense variants, four frameshift variants, and one essential splice site variant were observed from 15 individuals. SpliceAI predicted the consequence of the splicing variant, c.4500-1G>A (p.?), to introduce a new stop codon (Supplementary Figure S1D). All variants were observed as heterozygous. The odds ratio of this comparison to the combined cohort of KoGES and KOVA2 was 10.60 (36/2,330 vs. 15/10,290; 95% CI: 5.79–19.39) (Figure 2).
The third comparison was made to the internal dataset of 5,996 exome sequencing data from Korean patients collected between 2017 and 2023. Unlike the previous two control cohorts used for comparison, the internal exome sequencing data consisted of patients with suspected rare disorders but without HCMP phenotype. These patients had an age distribution between 1 year and 90 years with a median age of 22 years (mean, 28 years), and 3,280 were male (54%). There were 8 out of 5,996 patients (0.13%) with no indication of HCMP or cardiac phenotype identified with five unique heterozygous ALPK3 PTVs (Table 3). The age distribution of these eight patients was between 1 year and 44 years with a median age of 9 years (mean, 14.6 years). Two variants were non-sense, two were frameshift, and one was an essential splice site variant predicted to result in premature termination. All five variants were also observed in the HCMP cohort. The odds ratio (OR) of this comparison was 11.56 (36/2,330 vs. 8/5,988; 95% CI: 5.37–24.92) (Figure 2).
Finally, the comparison was made to the combined cohort of East Asian gnomAD v4.0 data, KoGES, KOVA2, and the internal dataset. Assuming no overlap between different databases, the combined cohort consisted of 38,749 individuals with 39 individuals (0.10%) carrying 21 different ALPK3 PTVs (Figure 1B). The distribution of ALPK3 PTV variants identified in the HCMP patient cohort and the patient control cohort was not significantly different (Figure 1). The final odds ratio was 15.34 (36/2,330 vs. 39/38,710; 95% CI:9.73–24.17) (Figure 2).
HCMP is one of the most commonly inherited cardiovascular diseases with a reported prevalence of 1 in 500 to 1 in 200 people globally and 1 in 300 people in South Korea (3, 36). While it contributes significantly to overall mortality and morbidity worldwide, early intervention and proper management can mediate sudden cardiac death and heart failure (37). Knowing the molecular basis of the disorder can help patients receive more precise interventions. Genetic testing is an efficient way to provide a molecular diagnosis to patients, prospective patients, and/or their family members at risk. That is why many (42%) of the 81 genes on the ACMG recommended secondary finding gene list are for cardiovascular diseases (10). However, despite over 50 different genes having been identified to be associated with HCMP, molecular diagnosis is only established for 30% of cases, suggesting that additional genetic loci and disease mechanisms for known genes remain to be discovered (1, 5, 8).
ALPK3, located on chromosome 15q25.2, is a protein kinase that is associated with autosomal recessive early-onset hypertrophic cardiomyopathy (OMIM: 618052) (15–18). This study replicates the findings that carriers of monoallelic ALPK3 PTVs have a higher risk of developing late-onset HCMP. Out of 2,366 unrelated individuals with HCMP patients studied, heterozygous ALPK3 PTVs were identified in 1.5% of total HCMP cases. Our study is the largest cohort studied so far and has also demonstrated comparable odds ratios of 15.33, which is similar to the odds ratio of 16.11 reported by Lopes et al. Although our cohort was limited to patients from South Korea, previous studies from other ethnic groups, including Turkey, the Dutch, the United States, the United Kingdom, and China, have all demonstrated a risk of acquiring HCMP with an ALPK3 PTVs (13, 16–18). Thus, HCMP-associated risk for ALPK3 does not seem to be limited to specific ethnic groups.
Although the first symptoms of HCMP can present as early as adolescence, the penetrance of HCMP is in general age-dependent with the average age of diagnosis being 41 years (20, 38–40). A study estimated the penetrance of HCMP by ALPK3 heterozygous PTVs to be >95% by the age of 75 years for males and 80% for females (21). In our exome cohort, of the eight patients with ALPK3 PTVs without clinical manifestation of HCMP, six individuals were below the age of 17. If these six individuals are excluded as they could potentially present with HCMP at a later age, the odds ratio becomes 25.07 (36/2,319 vs. 2/3,383; 95% CI:6.02–104.34), higher than the original calculation and higher compared to previous studies that did not account for the age of the control samples: 16.11 from Lopes et al. and 5.72–8.14 from Dai et al. (21, 22). However, because of the small number of cases, the 95% CI is wide, and further studies are needed with larger case numbers.
In our study, one patient (Patient 21), who is 63 years old, also carried a pathogenic variant in MYBPC3 (NM_000256.3:c.2067+1G>A), which segregated with HCMP in the family. The ALPK3 PTV did not segregate with HCMP in the family. There was no obvious difference in phenotype or severity between this patient and other family members. However, more dual-diagnosis cases would be needed to investigate if ALPK3 heterozygous PTV modifies the phenotype.
Two studies by Herkert et al. and Dai et al. reported potential associations between HCMP and monoallelic deleterious ALPK3 missense variants (20, 22). Although pathogenic assessment of missense variants is difficult and is often limited to in silico prediction, missense variants were suggested to represent a significant proportion of ALPK3-associated HCMP in both studies. In a study by Herkert et al. (20), 26 out of 36 identified variants in the Dutch population, and 6 out of 15 variants were missense in the US population were missense. Dai et al. (22) presented odds ratios of 3.17–3.61 for ALPK3 missense variants. Our HCMP cohort did not have patients with a deleterious missense variant in ALPK3 predicted by in silico tools REVEL (>0.6) or 3Cnet (>0.6). Further studies would be needed to define which missense variants are deleterious in ALPK3 and determine their contribution to unexplained HCMP.
Long-term follow-up for HCMP patients with monoallelic ALPK3 PTVs has suggested that the clinical course is more similar to patients with pathogenic variants in one of the sarcomere genes than to patients without (21). Given that patients with pathogenic variants in the sarcomere genes often have a more severe clinical prognosis than patients without, ALPK3 PTV carriers are expected to have a relatively more severe clinical prognosis and therefore should be monitored for proper clinical management. Interestingly, a previous study found that 20% of the ALPK3 PTV carriers had a subclinical increase in serum creatine kinases which is a feature associated with skeletal myopathy (21). Because our ALPK3 PTV-positive patients have not been followed up for a long term yet or have been tested for serum creatine kinase level, further studies are needed to see if similar findings are observed.
In conclusion, we replicated previous studies suggesting that monoallelic PTVs in ALPK3 increase the risk for late-onset HCMP. Although the odds ratio of 15.33 is lower than the odds ratio of MYBPC3 PTVs, which are known to have odds ratios of >100, we believe that ALPK3 should also be considered for inclusion in the secondary finding gene list and diagnostic laboratories should consider reporting monoallelic ALPK3 PTVs when the patient is suspected of having a non-syndromic HCMP phenotype; negative carriers of ALPK3 PTVs should be routinely monitored (10, 41). Although the disease mechanism is different, MYH7 missense variants and TNNI3 missense variants have similar OR as ALPK3 with odds ratios of 14.4 and 14.3, respectively (41).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethical approval was not required for the studies involving humans because the study was conducted in a diagnostic setting and as all the samples and data were de-identified throughout, Institutional Review Board (IRB) approval was not required. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
SR: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing. WJ: Conceptualization, Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. GH: Resources, Validation, Writing – review & editing. JSC: Resources, Validation, Writing – review & editing. SYL: Resources, Validation, Writing – review & editing. HK: Resources, Validation, Writing – review & editing. JJ: Resources, Validation, Writing – review & editing. SHL: Resources, Validation, Writing – review & editing. D-HB: Resources, Validation, Writing – review & editing. JYC: Resources, Validation, Writing – review & editing. JK: Resources, Validation, Writing – review & editing. K-HK: Resources, Validation, Writing – review & editing. JS: Resources, Validation, Writing – review & editing. BH: Data curation, Methodology, Writing – review & editing. GS: Conceptualization, Writing – review & editing. HL: Conceptualization, Writing – review & editing.
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Authors SWR, WCJ, BH, GHS, HL were employed by company 3billion, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1424551/full#supplementary-material
1. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. Authors/Task Force members, Elliott PM, Anastasakis A, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. (2014) 35(39):2733–79. doi: 10.1093/eurheartj/ehu284
2. McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. (2015) 21(2):174–82. doi: 10.1016/j.cmet.2015.01.013
3. Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol. (2014) 64(1):83–99. doi: 10.1016/j.jacc.2014.05.003
4. Lipov A, Jurgens SJ, Mazzarotto F, Allouba M, Pirruccello JP, Aguib Y, et al. Exploring the complex spectrum of dominance and recessiveness in genetic cardiomyopathies. Nat Cardiovasc Res. (2023) 2(11):1078–94. doi: 10.1038/s44161-023-00346-3
5. Melas M, Beltsios ET, Adamou A, Koumarelas K, McBride KL. Molecular diagnosis of hypertrophic cardiomyopathy (HCM): in the heart of cardiac disease. J Clin Med. (2022) 12(1):225. doi: 10.3390/jcm12010225
6. Cirino AL, Ho C. Hypertrophic cardiomyopathy overview. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al. editors. GeneReviews®. Seattle: University of Washington (1993). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1768/ (Accessed January 9, 2024).
7. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. Circulation. (2011) 124(24):2761–96. doi: 10.1161/CIR.0b013e318223e230
8. Bonaventura J, Polakova E, Vejtasova V, Veselka J. Genetic testing in patients with hypertrophic cardiomyopathy. Int J Mol Sci. (2021) 22(19):10401. doi: 10.3390/ijms221910401
9. Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med. (2019) 12(2):e002460. doi: 10.1161/CIRCGEN.119.002460
10. Miller DT, Lee K, Abul-Husn NS, Amendola LM, Brothers K, Chung WK, et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2023) 25(8):100866. doi: 10.1016/j.gim.2023.100866
11. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Rhythm Society. Circulation. (2018) 138(13):e210–71. doi: 10.1161/CIR.0000000000000548
12. Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. (2013) 15(7):565–74. doi: 10.1038/gim.2013.73
13. Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. (2003) 107(17):2227–32. doi: 10.1161/01.CIR.0000066323.15244.54
14. Walsh R, Offerhaus JA, Tadros R, Bezzina CR. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. (2022) 19(3):151–67. doi: 10.1038/s41569-021-00608-2
15. Middelbeek J, Clark K, Venselaar H, Huynen MA, van Leeuwen FN. The alpha-kinase family: an exceptional branch on the protein kinase tree. Cell Mol Life Sci. (2010) 67(6):875–90. doi: 10.1007/s00018-009-0215-z
16. Hosoda T, Monzen K, Hiroi Y, Oka T, Takimoto E, Yazaki Y, et al. A novel myocyte-specific gene Midori promotes the differentiation of P19CL6 cells into cardiomyocytes. J Biol Chem. (2001) 276(38):35978–89. doi: 10.1074/jbc.M100485200
17. Phelan DG, Anderson DJ, Howden SE, Wong RCB, Hickey PF, Pope K, et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur Heart J. (2016) 37(33):2586–90. doi: 10.1093/eurheartj/ehw160
18. Almomani R, Verhagen JMA, Herkert JC, Brosens E, van Spaendonck-Zwarts KY, Asimaki A, et al. Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol. (2016) 67(5):515–25. doi: 10.1016/j.jacc.2015.10.093
19. Van Sligtenhorst I, Ding ZM, Shi ZZ, Read RW, Hansen G, Vogel P. Cardiomyopathy in α-kinase 3 (ALPK3)–deficient mice. Vet Pathol. (2012) 49(1):131–41. doi: 10.1177/0300985811402841
20. Herkert JC, Verhagen JMA, Yotti R, Haghighi A, Phelan DG, James PA, et al. Expanding the clinical and genetic spectrum of ALPK3 variants: phenotypes identified in pediatric cardiomyopathy patients and adults with heterozygous variants. Am Heart J. (2020) 225:108–19. doi: 10.1016/j.ahj.2020.03.023
21. Lopes LR, Garcia-Hernández S, Lorenzini M, Futema M, Chumakova O, Zateyshchikov D, et al. Alpha-protein kinase 3 (ALPK3) truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur Heart J. (2021) 42(32):3063–73. doi: 10.1093/eurheartj/ehab424
22. Dai J, Li K, Huang M, Sun Y, Liu H, Li Z, et al. The involvement of ALPK3 in hypertrophic cardiomyopathy in East Asia. Front Med (Lausanne). (2022) 9:915649. doi: 10.3389/fmed.2022.915649
23. Vasimuddin M, Misra S, Li H, Aluru S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). IEEE (2019). p. 314–24. doi: 10.1109/IPDPS.2019.00041
24. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. (2009) 25(16):2078–9. doi: 10.1093/bioinformatics/btp352
25. van der Auwera G, O’Connor BD. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. 1st ed. Sebastopol, CA: O'Reilly Media, Incorporated (2020).
26. Krumm N, Sudmant PH, Ko A, O'Roak BJ, Malig M, Coe BP, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. (2012) 22(8):1525–32. doi: 10.1101/gr.138115.112
27. Seo GH, Kim T, Choi IH, Park J, Lee J, Kim S, et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin Genet. (2020) 98(6):562–70. doi: 10.1111/cge.13848
28. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The Ensembl variant effect predictor. Genome Biol. (2016) 17(1):122. doi: 10.1186/s13059-016-0974-4
29. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–23. doi: 10.1038/gim.2015.30
30. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176(3):535–548.e24. doi: 10.1016/j.cell.2018.12.015
31. van Velzen HG, Schinkel AFL, Baart SJ, Oldenburg RA, Frohn-Mulder IME, van Slegtenhorst MA, et al. Outcomes of contemporary family screening in hypertrophic cardiomyopathy. Circ Genom Precis Med. (2018) 11(4):e001896. doi: 10.1161/CIRCGEN.117.001896
32. Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. (2024) 625(7993):92–100. doi: 10.1038/s41586-023-06045-0
33. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
34. Kim Y. Han BG, The KoGES Group. Cohort profile: the Korean Genome and Epidemiology Study (KoGES) consortium. Int J Epidemiol. (2017) 46(4):1350. doi: 10.1093/ije/dyx105
35. Lee J, Lee J, Jeon S, Lee J, Jang I, Yang JO, et al. A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population. Exp Mol Med. (2022) 54(11):1862–71. doi: 10.1038/s12276-022-00871-4
36. Moon I, Lee S-Y, Kim H-K, Han K-D, Kwak S, Kim M, et al. Trends of the prevalence and incidence of hypertrophic cardiomyopathy in Korea: a nationwide population-based cohort study. PLoS One. (2020) 15(1):e0227012. doi: 10.1371/journal.pone.0227012
37. Dellefave-Castillo LM, Cirino AL, Callis TE, Esplin ED, Garcia J, Hatchell KE, et al. Assessment of the diagnostic yield of combined cardiomyopathy and arrhythmia genetic testing. JAMA Cardiol. (2022) 7(9):966. doi: 10.1001/jamacardio.2022.2455
38. Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. (2002) 105(4):446–51. doi: 10.1161/hc0402.102990
39. Topriceanu CC, Pereira AC, Moon JC, Captur G, Ho CY. Meta-analysis of penetrance and systematic review on transition to disease in genetic hypertrophic cardiomyopathy. Circulation. (2024) 149(2):107–23. doi: 10.1161/CIRCULATIONAHA.123.065987
40. Lorenzini M, Norrish G, Field E, Ochoa JP, Cicerchia M, Akhtar MM, et al. Penetrance of hypertrophic cardiomyopathy and outcome in sarcomeric mutation carriers. Eur Heart J. (2020) 41(Supplement_2):ehaa946.2074. doi: 10.1093/ehjci/ehaa946.2074
41. Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. (2019) 11(1):5. doi: 10.1186/s13073-019-0616-z
Keywords: ALPK3, premature terminating variant, hypertrophic cardiomyopathy, whole exome sequencing, Korean HCMP population
Citation: Ryu SW, Jeong WC, Hong GR, Cho JS, Lee SY, Kim H, Jang JY, Lee SH, Bae D-H, Cho JY, Kim JH, Kim K-H, Son JW, Han B, Seo GH and Lee H (2024) High prevalence of ALPK3 premature terminating variants in Korean hypertrophic cardiomyopathy patients. Front. Cardiovasc. Med. 11:1424551. doi: 10.3389/fcvm.2024.1424551
Received: 11 May 2024; Accepted: 18 June 2024;
Published: 5 July 2024.
Edited by:
Keiichi Hirono, University of Toyama, JapanReviewed by:
Silin Pan, Qingdao University, Qingdao, China© 2024 Ryu, Jeong, Hong, Cho, Lee, Kim, Jang, Lee, Bae, Cho, Kim, Kim, Son, Han, Seo and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hane Lee, aGxlZUAzYmlsbGlvbi5pbw==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.