
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 09 May 2024
Sec. Cardiovascular Pharmacology and Drug Discovery
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1392816
This article is part of the Research Topic Exploring Lymphatic Vasculature's Role in Cardiovascular and Metabolic Diseases View all articles
 Summer Simeroth1,2
Summer Simeroth1,2 Pengchun Yu1,2*
Pengchun Yu1,2*
Lymphatic endothelial cells (LECs) line lymphatic vessels, which play an important role in the transport of lymph fluid throughout the human body. An organized lymphatic network develops via a process termed “lymphangiogenesis.” During development, LECs respond to growth factor signaling to initiate the formation of a primary lymphatic vascular network. These LECs display a unique metabolic profile, preferring to undergo glycolysis even in the presence of oxygen. In addition to their reliance on glycolysis, LECs utilize other metabolic pathways such as fatty acid β-oxidation, ketone body oxidation, mitochondrial respiration, and lipid droplet autophagy to support lymphangiogenesis. This review summarizes the current understanding of metabolic regulation of lymphangiogenesis. Moreover, it highlights how LEC metabolism is implicated in various pathological conditions.
Lymphatic vessels (LVs) absorb interstitial fluid from the surrounding tissues and transport it as lymph fluid, which contains immune cells, proteins, and dietary fats, back to the venous circulation. If LVs are impaired or dysfunctional, excessive interstitial fluid cannot be properly drained, causing accumulation in tissues leading to painful swelling, a disease termed lymphedema. Additionally, LVs are also involved in inflammation, graft rejection, myocardial infarction, and many other disease conditions (1–6). Therefore, elucidating how LVs are formed will further the understanding of physiological and pathological processes regulated by lymphatics. Moreover, it will facilitate the development of new treatments for lymphatics-associated diseases.
The primary constituents of the lymphatic vasculature are lymphatic endothelial cells (LECs), which line lymphatic capillaries and collecting LVs (7). Absorption of initial fluid from surrounding tissue is achieved by lymphatic capillaries, which are blunt-ended vessels composed of a single layer of LECs (8). These LECs are interconnected by button-like junctions with a discontinuous basement membrane, thus allowing anchoring filaments to facilitate fluid uptake into lymphatic capillaries via interaction with the extracellular matrix (8, 9). Upon entering the lymphatic vasculature, the fluid absorbed from surrounding tissues is termed “lymph” (10). Lymph carries immune cells and pathogens and is involved in adaptive immune response in the lymph node (11). Moreover, dietary lipids, cholesterol, and macromolecules are taken up by lymphatic capillaries and transported as part of lymph in LVs (12). Therefore, in addition to immune response, LVs are involved in several important physiological and pathological processes (12) (Figure 1). For example, in the intestine, lymphatic capillaries (lacteals) absorb dietary fats incorporated into chylomicrons for transport to the bloodstream (13). Thus, lymphatic dysfunction is associated with obesity and insulin resistance (14). LVs also play a role in reverse cholesterol transport, a process that can ameliorate atherosclerosis (15). In the lung, lymphatic drainage is required during neonatal development of the lung allowing for proper inflation (16). Additionally, meningeal LVs assist in clearing macromolecules in cerebrospinal fluid (17, 18).
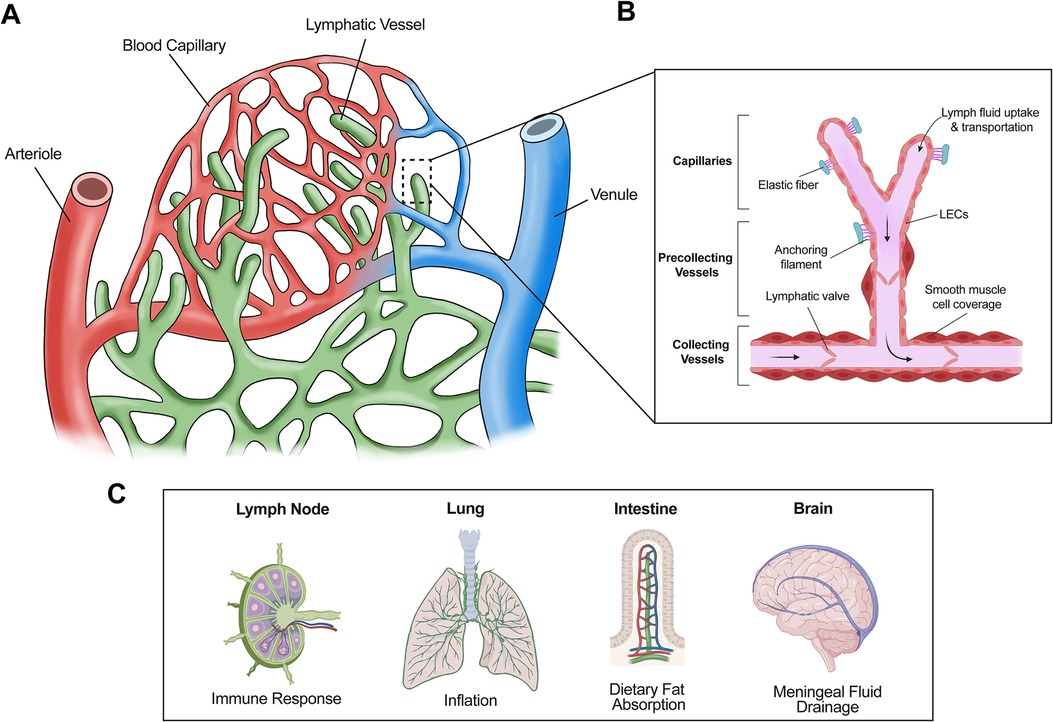
Figure 1. Structure and general functions of lymphatic vessels. (A) The lymphatic system absorbs interstitial fluid from the surrounding tissue bed for transport as lymph. (B) Initial fluid uptake is achieved through lymphatic capillaries through button like junctions that separate to allow fluid entry. The lymph is then passed to the precollecting and collecting lymphatic vessels, which have smooth muscle cell coverage and lymphatic valves, thus facilitating unidirectional transport. (C) Lymphatic vessels are present in multiple tissues and play important roles in several physiological processes. For example, lymphatic vessels facilitate immune response via immune cell trafficking in the lymph nodes, aid in inflation of the lung, absorb dietary fats via lacteals in the intestine, and assist with cerebrospinal fluid drainage in the meninges.
After the interstitial fluid enters lymphatic capillaries via button junctions, it flows towards collecting LVs (Figure 1), which possess zipper junctions in contrast to button junctions (8). These unique zipper junctions form a tighter, more continuous structure with a complete basement membrane to limit the improper entry or exit of lymph fluid (8). Collecting LVs serve a special purpose within the context of fluid transport. Specialized smooth muscle cell coverage and lymphatic valves allow collecting LVs to act as the motor unit of the lymphatic system, propelling lymph fluid unidirectionally towards the lymph nodes where it will eventually converge in the thoracic duct before returning to the circulatory system via the subclavian vein (19).
During early development, blood vascular endothelial cells differentiate in situ from a population of endothelial cell progenitors known as angioblasts and form the primitive vascular plexus, a process termed vasculogenesis (20). This primary vessel network further expands through angiogenesis, during which new blood vessels sprout from existing vessels (20). Under the direction of signaling pathways such as Notch and vascular endothelial growth factor (VEGF) signaling, blood vascular endothelial cells differentiate into arterial and venous endothelial cells, which respectively form arteries and veins that function together to support oxygen exchange and transportation of nutrients and waste (21). In parallel with blood vascular development, lymphatic vascular formation begins with LEC differentiation (22). The first lymphatic structures, the primary lymph sacs, develop from the embryonic cardinal veins (23). After the development of the primary lymph sacs, new LVs continue sprouting, branching, and maturing to form a more extensive LV network (24). LEC differentiation and subsequent LV expansion are controlled by two critical drivers—PROX1 and VEGF receptor 3 (VEGFR3) (23, 25). The transcription factor PROX1 is a master driver of LEC differentiation, and its expression is regulated by COUP-TFII and SOX18 signaling (26, 27). PROX1 promotes the expression of VEGFR3 in LECs, which mediates the effect of VEGF-C in stimulating LEC sprouting, migration, and proliferation during LV development (28, 29).
In addition to PROX1–VEGFR3 signaling, several other molecules, particularly cellular metabolism regulators, have recently been identified as crucial drivers of LV formation. In this review, we will summarize our understanding of LV formation from the perspective of cellular metabolism and explore how this understanding may offer new therapeutic strategies for human pathological conditions.
LECs generate nearly 70% of ATP via glycolysis instead of other metabolic processes such as oxidative phosphorylation, a unique characteristic differentiating LECs from many other cell types (30, 31). In addition to energy generation, glycolysis can also be exploited as a means of biomass production, which supports cell proliferation and growth (32). The distinctive reliance of LECs on glycolysis even in the presence of oxygen is mirrored by the Warburg Effect in cancer cells (32).
The rate-limiting steps in glycolysis are under the control of specific enzymes that aid in the regulation of metabolism, such as hexokinases (HKs) which convert glucose to glucose-6-phosphate in the first step of glycolysis (33, 34). In mammals, there are four HK isozymes: HK1, HK2, HK3, and HK4 (30). These HK isozymes differ in their regulatory properties, localizations, and enzymatic activities. More specifically, the activity of HK1, HK2, and HK3 is generally feedback inhibited by the presence of high glucose-6-phosphate levels whereas HK4 activity does not commonly exhibit this limitation under similar conditions (33). Furthermore, HK1 and HK2 are physically linked to the mitochondria in contrast to HK3 and HK4 (33). The association with the mitochondria is necessary for HK2-mediated proliferation of hepatocellular carcinoma cells, as disruption of this binding inhibits tumor growth and induces apoptosis (35). Moreover, the binding of HK2 to the mitochondria aids in the protection against oxidative stress and cell death (36, 37). Despite these studies, it remains unclear whether the physical association between HK2 and the mitochondria is important for LEC proliferation and survival.
Functional roles of HK2 in LV development have been recently implicated using genetic mouse models. Genetic deletion of Hk2 in mice results in impaired formation of the primitive lymphatic vascular plexus during early embryonic development (38). More specifically, embryos that underwent LEC-specific Hk2 knockout displayed attenuated LV sprouting and migration towards the midline in the dorsal skin as early as embryonic day (E) 15.5 (38). Consistently, Hk2 knockout in LECs suppresses fibroblast growth factor 2 (FGF2)-induced lymphangiogenesis in the adult mouse cornea (38). Moreover, pan-endothelial deletion of Hk2 produced embryos with impeded arterial development and angiogenesis in the embryonic skin, supporting that HK2 contributes to both angiogenesis and lymphangiogenesis (38). Another regulator of endothelial cell metabolism and angiogenesis is 6-Phosphofructo-2-kinase/fructose-2, 6-bisphosphatase, isoform 3 (PFKFB3) which acts to synthesize fructose-2,6-bisphospate (F2,6P2), an important allosteric activator of 6-phosphofructo-1-kinase (PFK-1) (31). During glycolysis, PFK-1 controls the rate-limiting conversion step of fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F1,6P2) (31). When PFKFB3 is ablated in murine models, developmental angiogenesis in the retina and the hindbrain as well as pathological retinal neovascularization are all impaired, demonstrating the importance of glycolysis in endothelial cell growth and vessel formation (31, 39). In addition to PFKFB3 and HK2, recent findings have also demonstrated the importance of pyruvate kinase type M2 (PKM2) for lymphangiogenesis (40). PKM2 converts phosphoenol pyruvate to pyruvate, generating ATP at the end of the glycolytic pathway (41). Thus, as a main driver of glycolytic ATP production, PKM2 has been linked to lymphangiogenic processes of LECs, and treatment with shikonin, which non-specifically inhibits PKM2 and several other molecules, alleviates lymphatic lesions induced by lipopolysaccharides in rats (40).
Furthermore, glycolysis has been shown to play a functional role in pathologies relating to abnormal pericyte function (42). Pericytes, also defined as mural cells, are a specialized cell type that envelops the endothelial cells that make up the capillary system (43, 44). Pericytes embed themselves within the basement membrane of endothelial cells, facilitating a close interaction between the microvasculature and mural cells (43). Pericytes are recruited to the microvasculature during development via cell signaling pathways, notably utilizing factors such as platelet-derived growth factor B (PDGF-B) and its receptor, PDGF receptor β (PDGFRβ) (45–48). Specifically, PDGF-B is secreted by endothelial tip cells along the angiogenic front during vessel formation, which then attracts pericytes expressing PDGFRβ, aiding in vasculature maturation (43). Functionally, the recruitment of pericytes is important in mediating blood flow as they contribute to regulating both vasoconstriction and vasodilation (49). Pericytes have recently become a topic of interest in the study of pathological tumor growth as vasculature within tumors has demonstrated dysregulation in pericyte-endothelial cell interactions leading to abnormal blood vessel sprouting (42, 50). Additionally, abnormal pericyte death contributes to diabetic retinopathy, as altered blood flow in the retina contributes to vascular leakage resulting in decreased vision acuity (51–56). Interestingly, HK2 has proven to be critical for pericyte contractility during tumor angiogenesis (42). HK2 is a main driver of tumor pericyte glycolysis, which (via the ROCK2-MLC2 pathway) increases pericyte contractility, resulting in impaired blood flow in tumors (42). Thus, inhibition of HK2 activity using a non-specific inhibitor remodels tumor vasculature and enhances the delivery and efficacy of a chemotherapy drug (42). However, it is still unknown what instigates aberrant pericyte glycolysis in tumors and whether genetic ablation of HK2 in pericytes can impact tumor vasculature.
HK2 is regulated by MYC expression, which in turn is regulated by FGF signaling in LECs (38). Therefore, FGF signaling activation elevates, while its inhibition suppresses, glycolysis in LECs (38). Moreover, genetic ablation of FGF receptor 1/3 or MYC attenuates retinal angiogenesis and the formation of the primitive LV network in the embryonic skin, similar to the effect of Hk2 knockout in LECs (38, 57). Interestingly, although MYC regulates the expression of multiple glycolytic enzymes in cancer cells (58), FGF-MYC signaling preferentially controls the expression of HK2, not HK1 or other rate-limiting glycolytic enzymes in LECs (38). Based on these results, it can be concluded that the decrease in FGF signaling results in a reduction of MYC, which markedly reduces HK2-driven glycolysis, causing subsequent defects in lymphangiogenesis (38). Despite this progress, many important questions remain to be addressed. For example, how FGF signaling activation upregulates MYC expression in LECs is unknown. Moreover, it is also unclear how FGF-MYC signaling selectively controls HK2 over the other rate-limiting glycolytic enzymes in LECs.
FAO is a biochemical process in which fatty acids are converted to acetyl-CoA, which is then oxidized via the tricarboxylic acid (TCA) cycle and the electron transport chain (ETC) (59). First, fatty acids enter the cell via cell surface transport molecules (59). Fatty acid translocase (FAT/CD36), membrane-bound fatty acid binding proteins (FABP), and fatty acid transport proteins (FATP) that are specific to the tissue are cell surface proteins that allow fatty acids to enter (59). Once inside the cell, fatty acyl-CoA synthase (FACS) adds a CoA group to the fatty acid, thus forming long-chain acyl-CoA (60). Carnitine palmitoyltransferase (CPT)1, which resides on the outer mitochondrial membrane, then converts long-chain acyl-CoA to long-chain acylcarnitine (60). This step is followed by transportation of long-chain acylcarnitine into the mitochondrial matrix, which is mediated by the action of carnitine-acylcarnitine translocase (60). CPT2, located on the inner mitochondrial membrane, then converts long-chain acylcarnitine back to long-chain acyl-CoA, which goes through β-oxidation to generate acetyl-CoA (60).
The importance of FAO for LV development has been revealed using mice deficient in CPT1A, which is the most abundant CPT1 isoform in LECs, and the CPT1 inhibitor etomoxir (61). Cpt1a deletion in LECs results in impaired and disorganized lymphatic growth towards the dorsal midline and a reduction in filopodia number, coupled with edema and blood-filled lymphatics in mouse embryos (61). Pharmacological inhibition of CPT1 via etomoxir inhibited FAO during early-stage development, which decreased the number of PROX1-expressing cells in the cardinal vein during early LEC differentiation, leading to defective dermal LV formation, reduced filopodia formation at the lymphangiogenic front, and a reduction in LEC proliferation (61). However, it is important to note that the activity of etomoxir may be independent of CPT1 activity under certain biological contexts, as displayed in cancer cell proliferation and T cell formation (62, 63). Additionally, fatty acid transporter CD36 has been shown to be expressed in the lacteals of intestinal lymphatics, which act to transport dietary lipids through the lymphatic system for subsequent entry to the subclavian vein (64, 65). Deletion of Cd36 in a murine model caused discontinuous VE-cadherin junctions in lacteals (65), a process that has been shown to play a key role in chylomicron transport and diet-induced obesity (66). Thus, Cd36 deficiency leads to LV leakage, late-onset obesity, and an increased risk of developing type 2 diabetes (65). Metabolically, silencing CD36 in LECs causes a reduction in FAO corresponding to an increase in glycolysis (65).
FAO exerts epigenetic control through histone acetylation (67). Histone acetylation occurs via the acetylation of lysine residues present on histones, during which acetyl groups act on the charged lysine residues to reduce the interaction between the histones and DNA, thus resulting in an open conformation that allows for the recruitment of various effectors that influence gene expression (68). Acetyl-CoA produced during FAO can be utilized as a substrate for histone acetylation (69). Histone acetyltransferases rely on acetyl-CoA levels produced via FAO and glycolysis to exert transcriptional control, and in glucose-limited environments, FAO is the main contributor of acetyl-CoA (67). First reported in 2009, ATP citrate lyase (ACL) converts glucose-derived citrate to acetyl-CoA when the appropriate nutrients are available, and deletion of ACL has been shown to decrease the expression of several glycolytic genes, providing further evidence that acetyl-CoA aids in transcriptional control (70). In this manner, acetyl-CoA has been shown to be an important regulator of target gene epigenetic modification by manner of histone acetylation (67). P300, a member of the histone acetyltransferase family, aids in epigenetic control via acetylation of histones, modifying core histones within the nucleosome allowing for transcriptional activation (71). FAO-derived acetyl-CoA fuels the activity of P300, which is recruited to the VEGFR3 promoter region together with PROX1 (61, 72). Thus, acetyl-CoA is utilized by P300 to exert transcriptional control by interacting with the PROX1-P300 complex, promoting acetylation of PROX1 target genes to control their transcription (61). Additionally, acetyl-CoA/CoA ratios determined by histone acetylation via P300 increased when PROX1 was overexpressed (61). Therefore, acetyl-CoA derived from FAO promotes PROX1-P300 expression which drives VEGFR3 transcription during lymphatic vascular development (61). In addition to supporting epigenetic regulation, acetyl-CoA generated from FAO contributes to the synthesis of deoxyribonucleotide triphosphate (dNTP) in endothelial cells (73). Isotopic labeling demonstrates that fatty acid carbons fuel the TCA cycle, contributing to the building of biomass, including dNTP (73). Therefore, genetic ablation of CPT1A suppresses endothelial proliferation in vitro and during vascular development in mice (73).
As previously mentioned, LECs utilize glycolysis to generate most of their ATP, and it is known that this process is further promoted via FGFR signaling during lymphangiogenesis (38). However, cells can shift their preferred metabolic pathway in response to stimuli that cause changes in energy demands—a term coined “metabolic flexibility” (74). Recently it has been found that inhibition of FGFR signaling-driven glycolysis in LECs upregulates CPT1A expression and FAO (74). As such, FAO, which makes little contribution to LEC ATP production under normal conditions, plays an important role in energy production and compensates for the deficit in energy when glycolysis is deficient (74). This process is mediated by peroxisome proliferator-activated receptor alpha (PPARα), which is upregulated upon FGFR inhibition and in turn, activates CPT1A transcription (74).
KBO is critical in maintaining metabolic homeostasis in organisms, supporting the TCA cycle and thus mitochondrial respiration, especially under nutrient-poor conditions (75). KBO has been identified as a potential therapeutic target for several neurodegenerative diseases such as Alzheimer's and Parkinson's (76). 3-oxoacid CoA-transferase 1 (OXCT1) acts as one of the key regulators of KBO by catalyzing a rate-limiting step. KBO eventually leads to the production of two molecules of acetyl-CoA, which can subsequently enter into the TCA cycle (77). Other key enzymes include BDH1 (3-hydroxybutyrate dehydrogenase 1), mThiolase, and citrate synthase (78). In KBO, ketone body β-hydroxybutyrate (β-OHB) can be converted to acetoacetate via BDH1 (78). Acetoacetate can then be catalyzed to form acetoacetyl-CoA via OXCT1, which will go on to produce acetyl-CoA following mThiolase catalytic cleavage (77, 78). This cleavage event produces two acetyl-CoA molecules for entry into the TCA cycle controlled by citrate synthase (77, 78). Among ketone bodies, β-OHB is the most commonly found in the circulating bloodstream, which LECs can readily access (78). Inhibition of KBO via depletion of OXCT1 or BDH1 suppresses proliferation, migration, and spouting of LECs in vitro (78). Moreover, genetic deletion of Oxct1 in LECs impedes LV development in the dorsal skin of mouse embryos (78). These data suggest that KBO is an important mechanism regulating LV growth (78). Furthermore, OXCT1 knockdown lowers the acetyl-CoA/CoA ratio and the amount of TCA cycle intermediates produced (78), therefore revealing the metabolic mechanisms by which KBO impacts LV development.
The mitochondrial respiratory chain is made up of various complexes that shuttle electrons across the mitochondrial membrane to produce a proton energy gradient as hydrogen atoms are pumped into the intermembrane space, eventually fueling the phosphorylation of ATP from ADP by complex V, also known as ATPase (79). Among these complexes is complex III, which shuttles electrons to the inner mitochondrial-associated protein cytochrome c, which then delivers the electrons to complex IV (79). Complex III is made up of various subunits that assist in this trafficking process (80). One such subunit is ubiquinol-cytochrome c reductase complex III subunit IV (QPC) encoded by the Uqcrq gene (80). Mutant QPC mice display a downregulation of lymphatic markers leading to an attenuation in LV development (81). Chemical inhibition of complex III also causes reduced methylation at the genetic loci of Vegfr3 and Prox1 (81). The resulting downregulation of PROX1 in LECs causes a reduction in LEC fate specification, which is further exacerbated by the reduction of VEGFR3 that is mediated via PROX1 in the feedback-loop system necessary to retain LEC fate identity (81). Mitochondrial respiration also plays a role in regulating the NAD+ (oxidized nicotinamide adenine dinucleotide)/NADH (reduced nicotinamide adenine dinucleotide) ratio, nucleotide synthesis, and levels of several TCA cycle intermediates, such as citrate, fumarate, and malate (81). Moreover, inhibition of complex III changes H3K4 methylation and H3K27 acetylation at the VEGFR3 locus (81), offering evidence that epigenetic modifications of Vegfr3 are critically mediated by complex III of the mitochondria (81).
Autophagy, a conserved lysosomal degradation and cellular recycling pathway, has been implicated as playing an important role in the blood vasculature and the maintenance of cellular metabolic homeostasis (82–84). Specifically, autophagy allows the cell to recycle metabolites to use as fuel for metabolism or to support biosynthesis (82, 83). Similarly, autophagy has recently been identified as an active mechanism in supporting lymphangiogenesis (85). This is accomplished via the autophagy of LDs, a process known as lipophagy (85). LDs are unique organelles found in most cell types that store neutral lipids surrounded by a phospholipid monolayer which can interact and associate with various organelles including the mitochondria to influence cellular metabolism (86). Furthermore, LDs sequester fatty acids in the form of triacylglycerol to be used as metabolic fuel during cell growth or when nutrient levels are depleted (87). In LECs, lipophagy supports the trafficking and release of lipids to the mitochondria to maintain FAO (85). Not only does lipophagy influence LEC metabolism, but it also promotes the expression of lymphangiogenic markers such as PROX1 and VEGFR3 (85). Furthermore, lipophagy has also been identified as playing a role in pathological lymphangiogenesis, as genetic depletion of essential autophagy gene Atg5 in mice LECs caused reduced lymphangiogenesis in a corneal wound healing model (85). Thus, autophagy of LDs in LECs represents a critical molecular mechanism supporting metabolic homeostasis, lymphangiogenesis, and its related gene expression (85).
Lymphedema is characterized by the accumulation of interstitial fluid resulting in impaired movement and often painful swelling in patients and can occur in various locations in the human body, most notably the limbs (88). It is often characterized as a chronic disease and is classified as either primary lymphedema or secondary lymphedema (88). Primary lymphedema is an inherited condition often caused by a genetic mutation that causes lymphatic formation defects, affecting 1 in every 100,000 people (88). More commonly, lymphedema arises as a result of damage, infection, or injury to the lymphatic system (88, 89). This is known as secondary lymphedema and affects 1 in 1,000 people in the United States alone (88). Notably, individuals who have undergone treatment for breast cancer or other gynecological cancers have an increased risk of developing secondary lymphedema, as 1 in 5 breast cancer survivors have been reported to develop this disease due to lymph node dissection and other cancer therapeutic techniques (90, 91). Although lymphedema is prevalent in the population, progressive, and often persists throughout the patient's life, therapeutic measures primarily consist of lymph massage, compression, exercise, and various microsurgical techniques with limited success (88, 92, 93). Thus, the need for drug therapies that target lymphedema is critical.
A tail injury mouse model of lymphedema that recapitulates human secondary lymphedema can be used as a method to explore what may influence the pathology of the disease (94). This is achieved by surgically removing LVs within the tail and then examining edema post-injury (94–96). As aforementioned, KBO has been found to promote lymphangiogenesis as LVs can readily access circulating β-OHB (78). Accordingly, it was found that mice that were fed the high-fat low-carb ketogenic diet (HFLC-KD) displayed less swelling in a tail injury model, and that ketone bodies aid in anti-inflammatory mechanisms by affecting leukocytes directly via β-OHB supplementation (78). Furthermore, mice fed the HFLC-KD also displayed reduced dermal thickening, decreased lymphatic dilation, and improved transport of lymph to the lymph node (78). Thus, targeting KBO shows promise in ameliorating lymphedema and promoting lymphangiogenesis as a potential future therapeutic measure.
The cornea, the outer clear layer of the eye that serves to refract light, is largely avascular (97). The absence of blood and lymphatic vasculature from the cornea maintains transparency, which is crucial for proper vision functionality (98, 99). The maintenance of this avascularity is established early in life, and under normal conditions is maintained throughout adulthood (100). However, in pathological conditions inflammation can cause the expansion of LVs and blood vessels into the avascular cornea (101). This aberrant lymphangiogenesis and angiogenesis can cause a decrease in the transparency of the cornea and loss of visual acuity (101). Furthermore, lymphangiogenesis into the cornea increases the risk of corneal graft rejection, a process that is mediated by VEGF-C/VEGFR3 signaling (4, 102–107). As many as ∼20% of patients who receive a corneal graft will face symptoms associated with rejection, with endothelial rejection being the most common cause (108).
Because of the close relationship between lymphangiogenesis and corneal graft rejection, corneal models of injury-induced lymphangiogenesis have become a valuable way of investigating the role of lymphatic metabolism in pathological conditions (109). As such, FAO has emerged as one contributing metabolic pathway (61). It has been found that in a corneal injury mouse model the inhibition of the key FAO enzyme CPT1 via etomoxir reduced lymphangiogenesis (61). Likewise, glycolysis has also been implicated in the control of corneal lymphangiogenesis (38). When pellets that released FGF2 were implanted into the cornea, mice that underwent LEC-specific deletion of the key glycolytic enzyme HK2 displayed decreased lymphangiogenesis in the cornea (38). Additionally, galectin-8, a carbohydrate-binding protein that promotes adhesion and cell motility of LECs, has been found to promote pathological lymphangiogenesis in the cornea (110). Specifically, galectin-8 is upregulated in both mouse and human corneas following corneal inflammation, and increased galectin-8 escalates the rate of graft rejection in a mouse model of corneal transplantation (110). Consequently, inhibition or knockout of galectin-8 has shown promise in ameliorating lymphangiogenesis in the cornea in vivo (110). These results point towards potential therapeutic targets that may be investigated further for their role in inflammation-induced lymphangiogenesis contributing to graft rejection.
Recent discoveries in LEC metabolism have expanded current scientific knowledge on the lymphatic vasculature and lymphangiogenesis. Glycolysis, FAO, KBO, mitochondrial respiration, and LD autophagy are all utilized by LECs to support necessary processes underlying lymphatic development (Figure 2). Specifically, glycolysis is the main metabolic pathway utilized by LECs to generate ATP, driving lymphatic development. FAO has also been shown to support energy production when glycolysis is diminished, indicating the role of metabolic flexibility in maintaining energy homeostasis in LECs. Furthermore, FAO supports the generation of acetyl-CoA, which can be utilized as a substrate for epigenetic modification of VEGFR3 in LECs via histone acetylation, thus driving lymphatic development. Additionally, KBO supports lymphatic development and is of particular interest as a potential therapeutic strategy to address lymphedema. Mitochondrial respiration plays a role in regulating LEC fate identity via epigenetic modification, and aids in the production of TCA cycle intermediates. Lastly, LD autophagy can be utilized to support FAO in LECs during lymphangiogenesis. Although it is established that LECs utilize the aforementioned metabolic pathways to support lymphatic development, several knowledge gaps remain to be addressed. For example, how are these metabolic processes regulated by different growth factors or cytokines during development and pathological conditions? How do different metabolic pathways interact and coordinate to enable LV growth? Under which physiological and/or pathological conditions do LECs shift their energy sources? Is there any metabolic crosstalk between LECs and their surrounding environment? Although much remains to be discovered mechanistically and clinically, enzymes involved in LEC metabolism are promising targets in the development of new treatments for human pathological conditions related to the lymphatic system.
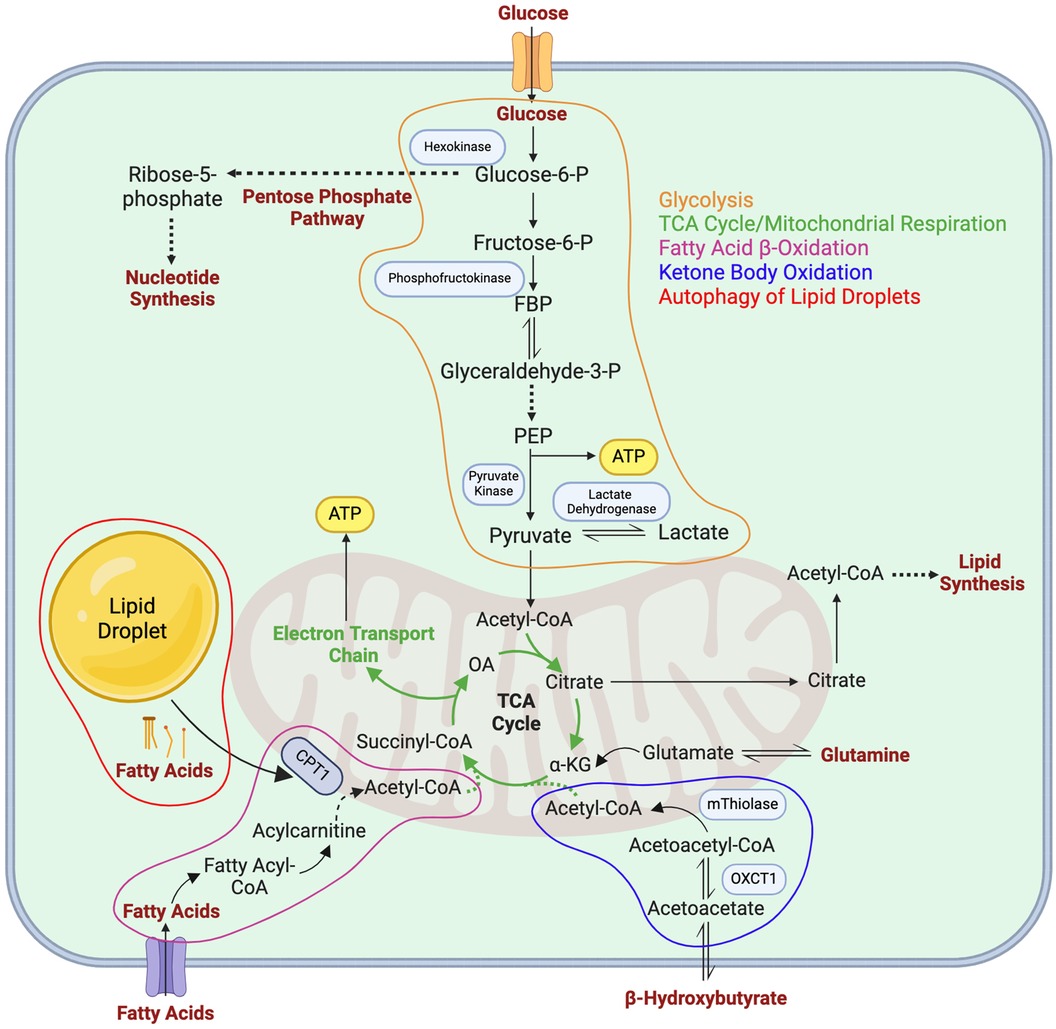
Figure 2. Metabolic pathways in LECs. Major metabolic pathways utilized by LECs as described in detail in the main text are depicted in orange (glycolysis), green (mitochondrial respiration/the TCA cycle), purple (fatty acid β-oxidation), blue (ketone body oxidation), and red (autophagy of lipid droplets). FBP, fructose 1,6-bisphosphate; PEP, phosphoenolpyruvate carboxylase; α-KG, α-ketoglutarate; OXCT1, 3-oxoacid CoA-transferase 1; CPT1, carnitine palmitoyltransferase 1; OA, oxaloacetate.
SS: Visualization, Writing – original draft, Writing – review & editing. PY: Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work is funded by OMRF research funding, the American Heart Association (23IPA1054589), and the National Institutes of Health (GM139763 and HL162985).
Figures were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer. (2014) 14(3):159–72. doi: 10.1038/nrc3677
2. Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dube KN, et al. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. (2015) 522(7554):62–7. doi: 10.1038/nature14483
3. Vieira JM, Norman S, Villa Del Campo C, Cahill TJ, Barnette DN, Gunadasa-Rohling M, et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J Clin Invest. (2018) 128(8):3402–12. doi: 10.1172/JCI97192
4. Chen L, Hamrah P, Cursiefen C, Zhang Q, Pytowski B, Streilein JW, et al. Vascular endothelial growth factor receptor-3 mediates induction of corneal alloimmunity. Nat Med. (2004) 10(8):813–5. doi: 10.1038/nm1078
5. Brouillard P, Witte MH, Erickson RP, Damstra RJ, Becker C, Quere I, et al. Primary lymphoedema. Nat Rev Dis Primers. (2021) 7(1):77. doi: 10.1038/s41572-021-00309-7
6. Dieterich LC, Seidel CD, Detmar M. Lymphatic vessels: new targets for the treatment of inflammatory diseases. Angiogenesis. (2014) 17(2):359–71. doi: 10.1007/s10456-013-9406-1
7. Pepper MS, Skobe M. Lymphatic endothelium: morphological, molecular and functional properties. J Cell Biol. (2003) 163(2):209–13. doi: 10.1083/jcb.200308082
8. Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. (2007) 204(10):2349–62. doi: 10.1084/jem.20062596
9. Yao LC, McDonald DM. Plasticity of airway lymphatics in development and disease. Adv Anat Embryol Cell Biol. (2014) 214:41–54. doi: 10.1007/978-3-7091-1646-3_4
11. Cruz de Casas P, Knopper K, Dey Sarkar R, Kastenmuller W. Same yet different—how lymph node heterogeneity affects immune responses. Nat Rev Immunol. (2023). doi: 10.1038/s41577-023-00965-8
12. Escobedo N, Oliver G. The lymphatic vasculature: its role in adipose metabolism and obesity. Cell Metab. (2017) 26(4):598–609. doi: 10.1016/j.cmet.2017.07.020
13. Petrova TV, Koh GY. Biological functions of lymphatic vessels. Science. (2020) 369(6500). doi: 10.1126/science.aax4063
14. Cao E, Watt MJ, Nowell CJ, Quach T, Simpson JS, De Melo Ferreira V, et al. Mesenteric lymphatic dysfunction promotes insulin resistance and represents a potential treatment target in obesity. Nat Metab. (2021) 3(9):1175–88. doi: 10.1038/s42255-021-00457-w
15. Huang LH, Elvington A, Randolph GJ. The role of the lymphatic system in cholesterol transport. Front Pharmacol. (2015) 6:182. doi: 10.3389/fphar.2015.00182
16. Jakus Z, Gleghorn JP, Enis DR, Sen A, Chia S, Liu X, et al. Lymphatic function is required prenatally for lung inflation at birth. J Exp Med. (2014) 211(5):815–26. doi: 10.1084/jem.20132308
17. Ahn JH, Cho H, Kim JH, Kim SH, Ham JS, Park I, et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature. (2019) 572(7767):62–6. doi: 10.1038/s41586-019-1419-5
18. Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest. (2017) 127(9):3210–9. doi: 10.1172/JCI90603
19. Makinen T, Norrmen C, Petrova TV. Molecular mechanisms of lymphatic vascular development. Cell Mol Life Sci. (2007) 64(15):1915–29. doi: 10.1007/s00018-007-7040-z
20. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. (2000) 6(4):389–95. doi: 10.1038/74651
21. Trimm E, Red-Horse K. Vascular endothelial cell development and diversity. Nat Rev Cardiol. (2023) 20(3):197–210. doi: 10.1038/s41569-022-00770-1
22. Oliver G, Srinivasan RS. Endothelial cell plasticity: how to become and remain a lymphatic endothelial cell. Development. (2010) 137(3):363–72. doi: 10.1242/dev.035360
23. Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. (1999) 98(6):769–78. doi: 10.1016/S0092-8674(00)81511-1
24. Hu Z, Zhao X, Wu Z, Qu B, Yuan M, Xing Y, et al. Lymphatic vessel: origin, heterogeneity, biological functions, and therapeutic targets. Signal Transduct Target Ther. (2024) 9(1):9. doi: 10.1038/s41392-023-01723-x
25. Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. (1996) 15(7):1751. doi: 10.1002/j.1460-2075.1996.tb00521.x
26. Yang Y, Garcia-Verdugo JM, Soriano-Navarro M, Srinivasan RS, Scallan JP, Singh MK, et al. Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood. (2012) 120(11):2340–8. doi: 10.1182/blood-2012-05-428607
27. Srinivasan RS, Geng X, Yang Y, Wang Y, Mukatira S, Studer M, et al. The nuclear hormone receptor coup-TFII is required for the initiation and early maintenance of Prox1 expression in lymphatic endothelial cells. Genes Dev. (2010) 24(7):696–707. doi: 10.1101/gad.1859310
28. Hong YK, Harvey N, Noh YH, Schacht V, Hirakawa S, Detmar M, et al. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev Dyn. (2002) 225(3):351–7. doi: 10.1002/dvdy.10163
29. Srinivasan RS, Escobedo N, Yang Y, Interiano A, Dillard ME, Finkelstein D, et al. The Prox1-Vegfr3 feedback loop maintains the identity and the number of lymphatic endothelial cell progenitors. Genes Dev. (2014) 28(19):2175–87. doi: 10.1101/gad.216226.113
30. Yu P, Wu G, Lee HW, Simons M. Endothelial metabolic control of lymphangiogenesis. Bioessays. (2018) 40(6):e1700245. doi: 10.1002/bies.201700245
31. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. (2013) 154(3):651–63. doi: 10.1016/j.cell.2013.06.037
32. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
33. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. (2003) 206(Pt 12):2049–57. doi: 10.1242/jeb.00241
34. Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and akt. Oncogene. (2006) 25(34):4683–96. doi: 10.1038/sj.onc.1209595
35. DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. (2018) 9(1):446. doi: 10.1038/s41467-017-02733-4
36. Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. (2008) 15(3):521–9. doi: 10.1038/sj.cdd.4402285
37. Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. (2008) 28(3):1007–17. doi: 10.1128/MCB.00224-07
38. Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, et al. FGF-dependent metabolic control of vascular development. Nature. (2017) 545(7653):224–8. doi: 10.1038/nature22322
39. Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler Thromb Vasc Biol. (2014) 34(6):1231–9. doi: 10.1161/ATVBAHA.113.303041
40. Jiang H, Zou Y, Zhao J, Li X, Yang S, Zhou X, et al. Pyruvate kinase M2 mediates glycolysis in the lymphatic endothelial cells and promotes the progression of lymphatic malformations. Am J Pathol. (2021) 191(1):204–15. doi: 10.1016/j.ajpath.2020.10.003
41. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
42. Meng YM, Jiang X, Zhao X, Meng Q, Wu S, Chen Y, et al. Hexokinase 2-driven glycolysis in pericytes activates their contractility leading to tumor blood vessel abnormalities. Nat Commun. (2021) 12(1):6011. doi: 10.1038/s41467-021-26259-y
43. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. (2011) 21(2):193–215. doi: 10.1016/j.devcel.2011.07.001
44. Holm A, Heumann T, Augustin HG. Microvascular mural cell organotypic heterogeneity and functional plasticity. Trends Cell Biol. (2018) 28(4):302–16. doi: 10.1016/j.tcb.2017.12.002
45. Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. (2003) 314(1):15–23. doi: 10.1007/s00441-003-0745-x
46. Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. (2009) 29(5):630–8. doi: 10.1161/ATVBAHA.107.161521
47. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. (2008) 22(10):1276–312. doi: 10.1101/gad.1653708
48. Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. (1997) 277(5323):242–5. doi: 10.1126/science.277.5323.242
49. Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. (2005) 7(4):452–64. doi: 10.1215/S1152851705000232
50. Jiang Z, Zhou J, Li L, Liao S, He J, Zhou S, et al. Pericytes in the tumor microenvironment. Cancer Lett. (2023) 556:216074. doi: 10.1016/j.canlet.2023.216074
51. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. (2009) 9(4):239–52. doi: 10.1038/nrc2618
52. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
53. Abramsson A, Berlin O, Papayan H, Paulin D, Shani M, Betsholtz C. Analysis of mural cell recruitment to tumor vessels. Circulation. (2002) 105(1):112–7. doi: 10.1161/hc0102.101437
54. Nolan-Stevaux O, Truitt MC, Pahler JC, Olson P, Guinto C, Lee DC, et al. Differential contribution to neuroendocrine tumorigenesis of parallel egfr signaling in cancer cells and pericytes. Genes Cancer. (2010) 1(2):125–41. doi: 10.1177/1947601909358722
55. Song N, Huang Y, Shi H, Yuan S, Ding Y, Song X, et al. Overexpression of platelet-derived growth factor-BB increases tumor pericyte content via stromal-derived factor-1alpha/CXCR4 axis. Cancer Res. (2009) 69(15):6057–64. doi: 10.1158/0008-5472.CAN-08-2007
56. Kern TS, Tang J, Mizutani M, Kowluru RA, Nagaraj RH, Romeo G, et al. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. (2000) 41(12):3972–8.11053301
57. Wilhelm K, Happel K, Eelen G, Schoors S, Oellerich MF, Lim R, et al. FOXO1 Couples metabolic activity and growth state in the vascular endothelium. Nature. (2016) 529(7585):216–20. doi: 10.1038/nature16498
58. Dong Y, Tu R, Liu H, Qing G. Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat. Signal Transduct Target Ther. (2020) 5(1):124. doi: 10.1038/s41392-020-00235-2
59. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90(1):207–58. doi: 10.1152/physrev.00015.2009
60. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
61. Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature. (2017) 542(7639):49–54. doi: 10.1038/nature21028
62. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. (2018) 28(3):504–15.e7. doi: 10.1016/j.cmet.2018.06.002
63. Yao CH, Liu GY, Wang R, Moon SH, Gross RW, Patti GJ. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol. (2018) 16(3):e2003782. doi: 10.1371/journal.pbio.2003782
64. Ko CW, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol. (2020) 17(3):169–83. doi: 10.1038/s41575-019-0250-7
65. Cifarelli V, Appak-Baskoy S, Peche VS, Kluzak A, Shew T, Narendran R, et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat Commun. (2021) 12(1):3350. doi: 10.1038/s41467-021-23808-3
66. Zhang F, Zarkada G, Han J, Li J, Dubrac A, Ola R, et al. Lacteal junction zippering protects against diet-induced obesity. Science. (2018) 361(6402):599–603. doi: 10.1126/science.aap9331
67. Etchegaray JP, Mostoslavsky R. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol Cell. (2016) 62(5):695–711. doi: 10.1016/j.molcel.2016.05.029
68. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. (2015) 16(4):258–64. doi: 10.1038/nrm3931
69. Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. (2007) 8(4):284–95. doi: 10.1038/nrm2145
70. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. (2009) 324(5930):1076–80. doi: 10.1126/science.1164097
71. Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. (2013) 152(4):859–72. doi: 10.1016/j.cell.2013.01.032
72. Ichise T, Yoshida N, Ichise H. Ras/MAPK signaling modulates VEGFR-3 expression through ets-mediated p300 recruitment and histone acetylation on the Vegfr3 gene in lymphatic endothelial cells. PLoS One. (2012) 7(12):e51639. doi: 10.1371/journal.pone.0051639
73. Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. (2015) 520(7546):192–7. doi: 10.1038/nature14362
74. Song H, Zhu J, Li P, Han F, Fang L, Yu P. Metabolic flexibility maintains proliferation and migration of FGFR signaling-deficient lymphatic endothelial cells. J Biol Chem. (2021) 297(4):101149. doi: 10.1016/j.jbc.2021.101149
75. Montenegro-Navarro N, Garcia-Baez C, Garcia-Caballero M. Molecular and metabolic orchestration of the lymphatic vasculature in physiology and pathology. Nat Commun. (2023) 14(1):8389. doi: 10.1038/s41467-023-44133-x
76. Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. (2003) 112(6):892–901. doi: 10.1172/JCI200318797
77. Zhang S, Xie C. The role of OXCT1 in the pathogenesis of cancer as a rate-limiting enzyme of ketone body metabolism. Life Sci. (2017) 183:110–5. doi: 10.1016/j.lfs.2017.07.003
78. Garcia-Caballero M, Zecchin A, Souffreau J, Truong ACK, Teuwen LA, Vermaelen W, et al. Role and therapeutic potential of dietary ketone bodies in lymph vessel growth. Nat Metab. (2019) 1(7):666–75. doi: 10.1038/s42255-019-0087-y
79. Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. (2008) 15(4):660–6. doi: 10.1038/sj.cdd.4402307
80. Barel O, Shorer Z, Flusser H, Ofir R, Narkis G, Finer G, et al. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am J Hum Genet. (2008) 82(5):1211–6. doi: 10.1016/j.ajhg.2008.03.020
81. Ma W, Gil HJ, Liu X, Diebold LP, Morgan MA, Oxendine-Burns MJ, et al. Mitochondrial respiration controls the Prox1-Vegfr3 feedback loop during lymphatic endothelial cell fate specification and maintenance. Sci Adv. (2021) 7(18). doi: 10.1126/sciadv.abe7359
82. Schaaf MB, Houbaert D, Mece O, Agostinis P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. (2019) 26(4):665–79. doi: 10.1038/s41418-019-0287-8
83. Nussenzweig SC, Verma S, Finkel T. The role of autophagy in vascular biology. Circ Res. (2015) 116(3):480–8. doi: 10.1161/CIRCRESAHA.116.303805
84. Jarc E, Petan T. Lipid droplets and the management of cellular stress. Yale J Biol Med. (2019) 92(3):435–52.31543707
85. Mece O, Houbaert D, Sassano ML, Durre T, Maes H, Schaaf M, et al. Lipid droplet degradation by autophagy connects mitochondria metabolism to Prox1-driven expression of lymphatic genes and lymphangiogenesis. Nat Commun. (2022) 13(1):2760. doi: 10.1038/s41467-022-30490-6
86. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. (2019) 20(3):137–55. doi: 10.1038/s41580-018-0085-z
87. Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. (2017) 18(11):671–84. doi: 10.1038/nrm.2017.76
89. Slater HC, Gambhir M, Parham PE, Michael E. Modelling co-infection with malaria and lymphatic filariasis. PLoS Comput Biol. (2013) 9(6):e1003096. doi: 10.1371/journal.pcbi.1003096
90. DiSipio T, Rye S, Newman B, Hayes S. Incidence of unilateral arm lymphoedema after breast cancer: a systematic review and meta-analysis. Lancet Oncol. (2013) 14(6):500–15. doi: 10.1016/S1470-2045(13)70076-7
91. Hayes SC, Janda M, Ward LC, Reul-Hirche H, Steele ML, Carter J, et al. Lymphedema following gynecological cancer: results from a prospective, longitudinal cohort study on prevalence, incidence and risk factors. Gynecol Oncol. (2017) 146(3):623–9. doi: 10.1016/j.ygyno.2017.06.004
92. Warren AG, Brorson H, Borud LJ, Slavin SA. Lymphedema: a comprehensive review. Ann Plast Surg. (2007) 59(4):464–72. doi: 10.1097/01.sap.0000257149.42922.7e
93. Granzow JW. Lymphedema surgery: the current state of the art. Clin Exp Metastasis. (2018) 35(5-6):553–8. doi: 10.1007/s10585-018-9897-7
94. Schneider M, Ny A, Ruiz de Almodovar C, Carmeliet P. A new mouse model to study acquired lymphedema. PLoS Med. (2006) 3(7):e264. doi: 10.1371/journal.pmed.0030264
95. Tian W, Rockson SG, Jiang X, Kim J, Begaye A, Shuffle EM, et al. Leukotriene B(4) antagonism ameliorates experimental lymphedema. Sci Transl Med. (2017) 9(389). doi: 10.1126/scitranslmed.aal3920
96. Zampell JC, Yan A, Elhadad S, Avraham T, Weitman E, Mehrara BJ. CD4(+) cells regulate fibrosis and lymphangiogenesis in response to lymphatic fluid stasis. PLoS One. (2012) 7(11):e49940. doi: 10.1371/journal.pone.0049940
97. Farag TI, Teebi AS. Autosomal recessive inheritance of a syndrome of hypertelorism, hypospadias, and tetralogy of fallot? Am J Med Genet. (1990) 35(4):516–8. doi: 10.1002/ajmg.1320350414
98. Meek KM, Knupp C. Corneal structure and transparency. Prog Retin Eye Res. (2015) 49:1–16. doi: 10.1016/j.preteyeres.2015.07.001
99. Azar DT. Corneal angiogenic privilege: angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing (an American ophthalmological society thesis). Trans Am Ophthalmol Soc. (2006) 104:264–302.17471348
100. Patnam M, Dommaraju SR, Masood F, Herbst P, Chang JH, Hu WY, et al. Lymphangiogenesis guidance mechanisms and therapeutic implications in pathological states of the cornea. Cells. (2023) 12(2):319. doi: 10.3390/cells12020319
101. Yang JF, Walia A, Huang YH, Han KY, Rosenblatt MI, Azar DT, et al. Understanding lymphangiogenesis in knockout models, the cornea, and ocular diseases for the development of therapeutic interventions. Surv Ophthalmol. (2016) 61(3):272–96. doi: 10.1016/j.survophthal.2015.12.004
102. Cursiefen C, Cao J, Chen L, Liu Y, Maruyama K, Jackson D, et al. Inhibition of hemangiogenesis and lymphangiogenesis after normal-risk corneal transplantation by neutralizing VEGF promotes graft survival. Invest Ophthalmol Vis Sci. (2004) 45(8):2666–73. doi: 10.1167/iovs.03-1380
103. Bachmann BO, Bock F, Wiegand SJ, Maruyama K, Dana MR, Kruse FE, et al. Promotion of graft survival by vascular endothelial growth factor a neutralization after high-risk corneal transplantation. Arch Ophthalmol. (2008) 126(1):71–7. doi: 10.1001/archopht.126.1.71
104. Bachmann BO, Luetjen-Drecoll E, Bock F, Wiegand SJ, Hos D, Dana R, et al. Transient postoperative vascular endothelial growth factor (VEGF)-neutralisation improves graft survival in corneas with partly regressed inflammatory neovascularisation. Br J Ophthalmol. (2009) 93(8):1075–80. doi: 10.1136/bjo.2008.145128
105. Dietrich T, Bock F, Yuen D, Hos D, Bachmann BO, Zahn G, et al. Cutting edge: lymphatic vessels, not blood vessels, primarily mediate immune rejections after transplantation. J Immunol. (2010) 184(2):535–9. doi: 10.4049/jimmunol.0903180
106. Yin N, Zhang N, Xu J, Shi Q, Ding Y, Bromberg JS. Targeting lymphangiogenesis after islet transplantation prolongs islet allograft survival. Transplantation. (2011) 92(1):25–30. doi: 10.1097/TP.0b013e31821d2661
107. Yan H, Yuan J, Peng R, Wang T, Deng J, Li W, et al. The blockade of vascular endothelial growth factor C effectively inhibits corneal lymphangiogenesis and promotes allograft survival. J Ocul Pharmacol Ther. (2015) 31(9):546–54. doi: 10.1089/jop.2015.0007
108. Gurnani B, Czyz CN, Mahabadi N, Havens SJ. Corneal Graft Rejection. Treasure Island, FL: StatPearls (2024).
109. Detry B, Blacher S, Erpicum C, Paupert J, Maertens L, Maillard C, et al. Sunitinib inhibits inflammatory corneal lymphangiogenesis. Invest Ophthalmol Vis Sci. (2013) 54(5):3082–93. doi: 10.1167/iovs.12-10856
Keywords: lymphatic vessel, lymphatic endothelial cell, cellular metabolism, metabolic enzyme, lymphatic disease, lymphatic development, lymphangiogenesis
Citation: Simeroth S and Yu P (2024) The role of lymphatic endothelial cell metabolism in lymphangiogenesis and disease. Front. Cardiovasc. Med. 11:1392816. doi: 10.3389/fcvm.2024.1392816
Received: 28 February 2024; Accepted: 18 April 2024;
Published: 9 May 2024.
Edited by:
Xiaofeng Yang, Temple University, United StatesReviewed by:
Xiaolei Liu, Temple University, United States© 2024 Simeroth and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengchun Yu cGVuZ2NodW4teXVAb21yZi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.