 Bilal Bashir1,2,3
Bilal Bashir1,2,3 Jonathan Schofield2Paul Downie4Michael France5Darren M. Ashcroft1,6Alison K. Wright1,6
Jonathan Schofield2Paul Downie4Michael France5Darren M. Ashcroft1,6Alison K. Wright1,6 Stefano Romeo7,8,9
Stefano Romeo7,8,9 Ioanna Gouni-Berthold10
Ioanna Gouni-Berthold10 Akhlaq Maan2Paul N. Durrington1Handrean Soran1,2,3*
Akhlaq Maan2Paul N. Durrington1Handrean Soran1,2,3*
- 1Faculty of Biology Medicine and Health, University of Manchester, Manchester, United Kingdom
- 2Department of Endocrinology, Diabetes & Metabolism, Manchester University NHS Foundation Trust, Manchester, United Kingdom
- 3NIHR/Wellcome Trust Clinical Research Facility, Manchester, United Kingdom
- 4Department of Clinical Biochemistry, Bristol Royal Infirmary, Bristol, United Kingdom
- 5Department of Clinical Biochemistry, Central Manchester University Hospitals, NHS Foundation Trust, Manchester, United Kingdom
- 6Centre for Pharmacoepidemiology and Drug Safety, Division of Pharmacy and Optometry, School of Health Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, United Kingdom
- 7Department of Molecular and Clinical Medicine, University of Gothenburg, Gothenburg, Sweden
- 8Clinical Nutrition Unit, Department of Medical and Surgical Sciences, Magna Graecia University, Catanzaro, Italy
- 9Cardiology Department, Sahlgrenska University Hospital, Gothenburg, Sweden
- 10Centre for Endocrinology, Diabetes and Preventive Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
Aims: Historically, atherosclerotic cardiovascular disease (ASCVD) risk profile mitigation has had a predominant focus on low density lipoprotein cholesterol (LDL-C). In this narrative review we explore the residual ASCVD risk profile beyond LDL-C with a focus on hypertriglyceridaemia, recent clinical trials of therapeutics targeting hypertriglyceridaemia and novel modalities addressing other residual ASCVD risk factors.
Findings: Hypertriglyceridaemia remains a significant ASCVD risk despite low LDL-C in statin or proprotein convertase subtilisin/kexin type 9 inhibitor-treated patients. Large population-based observational studies have consistently demonstrated an association between hypertriglyceridaemia with ASCVD. This relationship is complicated by the co-existence of low high-density lipoprotein cholesterol. Despite significantly improving atherogenic dyslipidaemia, the most recent clinical trial outcome has cast doubt on the utility of pharmacologically lowering triglyceride concentrations using fibrates. On the other hand, purified eicosapentaenoic acid (EPA), but not in combination with docosahexaenoic acid (DHA), has produced favourable ASCVD outcomes. The outcome of these trials suggests alternate pathways involved in ASCVD risk modulation. Several other pharmacotherapies have been proposed to address other ASCVD risk factors targeting inflammation, thrombotic and metabolic factors.
Implications: Hypertriglyceridaemia poses a significant residual ASCVD risk in patients already on LDL-C lowering therapy. Results from pharmacologically lowering triglyceride are conflicting. The role of fibrates and combination of EPA and DHA is under question but there is now convincing evidence of ASCVD risk reduction with pure EPA in a subgroup of patients with hypertriglyceridaemia. Clinical guidelines should be updated in line with recent clinical trials evidence. Novel agents targeting non-conventional ASCVD risks need further evaluation.
1 Introduction
Atherosclerotic Cardiovascular Disease (ASCVD) remains the leading cause of morbidity and mortality worldwide despite new mechanistic insights and preventative strategies to mitigate ASCVD risk. Increased prevalence of conditions that predispose to ASCVD events i.e., obesity, diabetes, hypertension and atherogenic dyslipidaemia contributes to the increasing burden of care attributed to ASCVD that costs >200 billion US dollars annually for US and comparable figure for Europe (1, 2). Low-density lipoprotein cholesterol (LDL-C) has been demonstrated in large genetic, epidemiological, and clinical studies as a leading cause of atherosclerosis and ASCVD (3, 4). A meta-regression analysis of 26 randomised controlled trials (RCTs) has demonstrated a stepwise reduction in ASCVD risk with 22% relative risk reduction with each 1 mmol/L reduction in LDL-C (5). Patients with higher pre-treatment LDL-C benefit more (6) and there is no limit below which further LDL-C lowering ceases to confer ASCVD protection (7). Despite this, there remains significant residual risk in statin treated patients (8–13). Addition of pharmacotherapies like proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition can reduce LDL-C to a very low level. Despite achieving LDL-C of <30 mg/dl (0.7 mmol/L) a substantial number of individuals still experience ASCVD events (14). In this narrative review, we delve into residual cardiovascular risks that extend beyond LDL-C with a focus on the role of triglyceride rich lipoproteins (TRL) as a residual ASCVD risk factor and on the impact of triglyceride (TG) lowering pharmacotherapy on residual ASCVD risk.
2 Methods
We conducted a comprehensive search across multiple electronic databases, including AMED, Embase, HMIC, Pubmed, Ovid Emcare, Ovid MEDLINE and other relevant papers of interest collected by the authors. Our search strategy utilized the following terms: “HYPERTRIGLYCERIDAEMIA”, “FIBRATES”, “RESIDUAL RISK”, “RANDOMISED CONTROLLED TRIALS”, “OMEGA 3 FATTY ACID”, “CARDIOVASCULAR”, “TRIGLYCERDE RICH LIPOPROTEINS” and “TRIGLYCERIDE”. Boolean operators “AND” and “OR” were employed to combine and separate search terms effectively. Only articles published in the English language were considered for inclusion in this review. Exclusion criteria encompassed articles published in languages other than English, conference abstracts, and case reports. Additionally, only studies involving human participants were included. To supplement our database search, we manually scrutinized the reference lists of identified trials, review articles, and previous meta-analyses to identify any additional relevant data.
3 Residual atherosclerotic cardiovascular disease risk
3.1 Residual ASCVD risk in clinical trials
The Further cardiovascular OUtcomes Research with PCSK9 Inhibition subjects with Elevated Risk (FOURIER) trial evaluated patients with known ASCVD with pre-treatment LDL-C of 2.4 mmol/L (92 mg/dl) that was reduced to 0.7 mmol/L (30 mg/dl) with evolocumab, 9.8% of drug recipients still experienced ASCVD events over median followup period of 2.2 years (14). Similarly, in the Evaluation of Cardiovascular outcomes after Acute coronary syndrome during treatment with Alirocumab (ODYSSEY Outcomes), despite achieving LDL-C as low as 1.7 mmol/L (66 mg/dl), probability estimate of alirocumab recipients to have ASCVD event was 12.8% at 4 years (15). There were similar results in the Studies of PCSK9 Inhibition and the Reduction of Vascular Events (SPIRE) trials, though the relative percentage of ASCVD events was lower than FOURIER and ODYSSEY Outcomes (2.1% in SPIRE 1 at median followup of 7 months and 3.4% in SPIRE 2 at median followup of 12 months) (16) In the landmark statin trials where LDL-C lowering reduced the relative risk of ASCVD, participants who received statins still exhibited a significant residual cardiovascular risk. This was 22.4% at 2 years in the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI) study (achieved LDL-C 62 mg/dl, 1.6 mmol/L) (17), 9.3% over median follow up of 4.8 years in the Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) study (achieved LDL-C 81 mg/dl, 2.1 mmol/L) (18), and 8.7% over a median follow up of 4.9 years in the Treating to New Targets (TNT) study (achieved LDL-C 77 mg/dl, 1.9 mmol/L) (19). That a heightened incidence of ASCVD events persists despite attainment of low levels of LDL-C prompted the conceptualisation of residual cardiovascular risk due to additional metabolic, inflammatory, and thrombotic risk factors. The mitigation of ASCVD events necessitates a comprehensive and multifaceted approach addressing these diverse components.
3.2 Lipoproteins in atherosclerosis—limitations of LDL-C calculation
The key initial event in the genesis of atherosclerosis is the entrapment of lipoproteins in vascular intima followed by the engulfment by macrophages. Lipoprotein subfractions other than LDL can be preferentially entrapped and engulfed without the need to be chemically modified and may enhance the process of atherosclerosis. While the conventional clinical approach employs LDL-C as a marker for atherosclerotic risk, many atherogenic particles are relatively deficient in cholesterol and so their atherogenicity is underestimated by cholesterol measurement. In most clinical laboratories, LDL-C is an estimated through the Friedwald formula, dependant on knowledge of the total cholesterol (TC), High density lipoprotein cholesterol (HDL-C) and TG concentration. With TG >4.5 mmol/L (400 mg/dl) an estimate for LDL-C cannot be provided by this formula and even modest excursions in TG concentration, will result in underestimation of LDL-C concentration. Alternative calculators are available that offer LDL-C estimations up to TG levels of 10 mmol/L (20).
3.3 Apolipoprotein B100 and lipoprotein sub-fractions as a marker for ASCVD risk estimation
Apolipoprotein B100 (ApoB) serves as a comprehensive metric for the total atherogenic particle count. Each atherogenic lipoprotein particle, such as LDL, very low-density lipoprotein (VLDL), and Intermediate Density Lipoprotein (IDL), contains a single ApoB molecule. Consequently, the blood ApoB level provides a direct reflection of the overall number of atherogenic particles, irrespective of their size or density. This establishes ApoB as a more precise indicator of ASCVD risk compared to traditional lipid measurements, which fail to consider particle number or size. The Apolipoprotein-related Mortality Risk (AMORIS) study found that ApoB levels and the ApoB/Apolipoprotein A1 ratio were stronger predictors of ASCVD than LDL-C, whilst TG was found to be an independent risk factor for ASCVD (21). In the post hoc analysis of the TNT trial, higher levels of TRL (VLDL, IDL and chylomicron remnants) were associated with an increased risk of major ASCVD events independent of LDL-C concentration (22). Mendelian randomisation studies, epidemiological observations and RCTs of lipid-lowering drugs have implicated cholesterol-rich ApoB particles in addition to LDL i.e., VLDL, IDL and Lipoprotein (a) [Lp(a)] as being directly causal in ASCVD (4). In a prospective observational study of 4,932 individuals from the Jackson Heart Study and the Framingham Offspring Cohort Study, free of coronary heart disease (CHD) at baseline followed-up for 8 years, remnant lipoprotein cholesterol (RLP-C) was linked to the onset of CHD. After adjusting for other ASCVD risk factors and HDL-C, this association was driven by IDL-C which significantly elevated CHD risk by 25% (HR 1.25, 95% CI 1.07–1.46, P < 0.001) (23).
Variations in the Lipoprotein lipase (LPL) gene that augment LPL activity are correlated with reduced TG levels and a concomitant decrease in ApoB concentration. Variations in the low-density lipoprotein receptor (LDLR) gene that enhance the activity of the LDLR are linked to decreased LDL-C concentration and a corresponding reduction in ApoB. For every 10 mg/dl decline in plasma ApoB concentration attributable to LPL score-associated variants, a parallel decrease of 0.8 mmol/L (69.9 mg/dl) in TG concentration is observed, with no discernible alteration in LDL-C, and a diminished risk of CHD (odds ratio (OR), 0.771 [95% CI, 0.741–0.802]). An equivalent 10 mg/dl decrease in plasma ApoB concentration associated with LDLR score-related variants corresponds to a 0.4 mmol/L (14.1 mg/dl) reduction in LDL-C concentration, no alteration in TG, and a similarly decreased risk of CHD [OR 0.773 (95% CI, 0.747–0.801)]. Consequently, despite inducing modifications in distinct lipid profiles, both LPL and LDLR scores exhibit analogous reductions in CHD risk for the same decrement in plasma ApoB concentration (24). This underscores that in hytriglyceridaemic populations ApoB is a better predictor of cardiovascular risk than cholesterol-based parameters, and a pivotal treatment target (25). There has been accumulating evidence recently which suggest that the risk attributed to an incremental rise in TRL/remnant cholesterol surpasses that of an equivalent increase in LDL-C (26). This was elaborated more recently in a Bjornsen et al. in a well characterised population from the UK Biobank. The authors investigated 502,460 participants in the UK Biobank, examining all single nucleotide polymorphisms (SNPs) associated with TRL and LDL-C identified via genome wide association studies and standard lipid profiles, including ApoB. These SNPs were divided into 2 clusters. Cluster 1 included SNPs affecting receptor mediated clearance and hence LDL-C more than TRL/remnant cholesterol, while cluster 2 had SNPs with a stronger impact on lipolysis and hence TRL/remnant cholesterol. The OR for CHD per standard deviation (SD) increase in ApoB was 1.76 (95% CI: 1.58–1.96) in cluster 2, which was significantly higher than the OR in cluster 1 [1.33 (95% CI: 1.26–1.40)]. These findings suggest that the association of ApoB with CHD risk varies depending on the type of particle harbouring ApoB and in this study, TRL/remnant particles demonstrated significantly greater atherogenicity per particle compared to LDL (27).
Despite low ApoB concentration, rare cases of familial dysbetalipoproteinaemia (FDBL) with ApoE2 homozygosity exhibited heightened ASCVD risk (28). This is attributed to impaired liver processing of chylomicron remnants, leading to prolonged circulation and abnormal cholesterol enrichment due to cholesteryl ester transfer protein (CETP) mediated lipid exchanges. Generating atherogenic small-dense LDL particles, which does not figure well in LDL-C measurements, in patients with high TG level as well as TG's strong association with atherogenic components of the metabolic syndrome, high-sensitivity C-reactive protein (hsCRP), coagulation are other factors contribute to increased ASCVD risk in hypertriglyceridaemia (29–34). It is now accepted that using ApoB to assess ASCVD risk in hypertriglyceridaemia (2–10 mmol/L) better reflects the total number of atherogenic particles than do LDL-C or non-HDL-C, particularly in patients with hypertriglyceridemia (25, 29–36).
3.4 Severe hypertriglyceridaemia and ASCVD
There is an apparent paradox that hypertriglyceridaemia >10 mmol/L (885 mg/dl) association with ASCVD risk is less than less severe hypertriglyceridaemia. In the CALIBER study, Patel and colleagues, found no increased risk of myocardial infarction (MI) in individuals with TG >10 mmol/L (885 mg/dl), while increased risk was found in mild to moderate hypertriglyceridaemia (1.7–10.0 mmol/L, 150–885 mg/dl) that persisted despite statin and/or fibrate treatment (37). This is consistent with the notion that small but numerous TG depleted particles are more atherogenic that large TG rich particles such as chylomicrons. TG >10 mmol/L (885 mg/dl) is often associated with chylomicronaemia with particles enriched in TG relative to ApoB. Monogenic disorders causing severe hypertriglyceridemia have increased chylomicron concentrations with a heightened risk of acute pancreatitis, but generally not of premature atherosclerosis, likely due to the limited ability of chylomicrons to traverse the vascular endothelial barrier (38). Additionally, in severe hypertriglyceridemia (TG >10.0 mmol/L; 885 mg/dl) ApoB immunoassays are compromised by analytical interference in blood samples due to turbidity caused by large chylomicron and VLDL particles (39). TG measurement does not, therefore, reflect an increased number of atherogenic particles in these cases. Indeed, chylomicron associated ApoB generally contributes very little to total plasma ApoB.
In addition to various lipid subfractions, other metabolic, inflammatory contribute to and thrombotic pathways also fuel residual ASCVD risk as summarised in Figure 1.
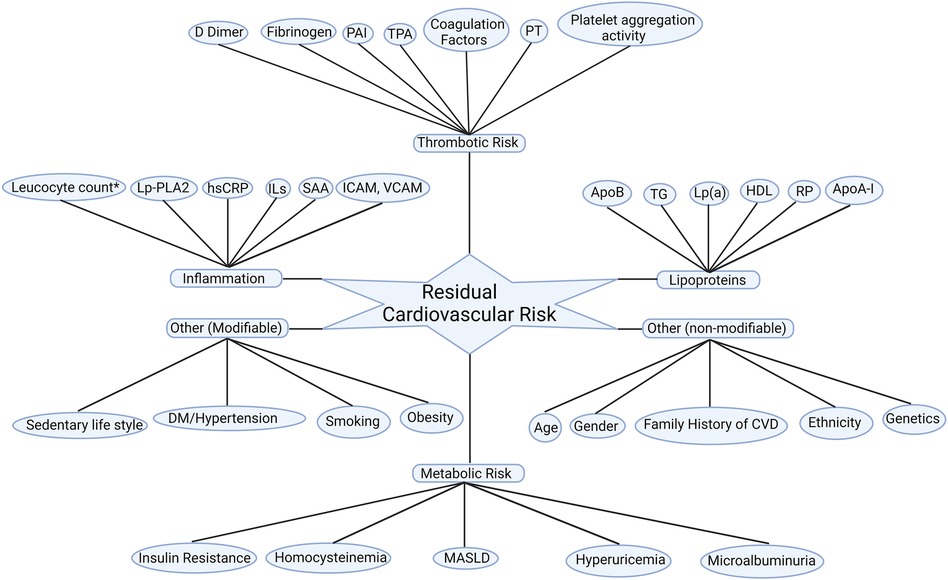
Figure 1. Residual cardiovascular risk factors. Apo A-I, apolipoprotein A-I; ApoB, apolipoprotein B100; CVD: cardiovascular disease; DM, diabetes mellitus; HDL-C; high-density lipoprotein cholesterol; hsCRP, high sensitivity C reactive protein; ICAM, intercellular adhesion molecule; ILs, interleukins; Lp(a), lipoprotein (a); Lp-PLA2, lipoprotein-associated phospholipase A2; MASLD: metabolic dysfunction-associated steatotic liver disease; PAI, plasminogen activator inhibitor; PT, prothrombin time; RP, remanent particles; SAA, serum amyloid A; TG, triglycerides; TPA, tissue plasminogen activator; VCAM; vascular cell adhesion molecule. *Leucocyte count after acute myocardial infarction.
4 Triglycerides as a residual ASCVD risk factor
4.1 Definition and measurement of triglycerides
Despite heterogeneity in the definition of hypertriglyceridaemia, normal levels of fasting TG have been defined at <1.7 mmol/L (<150 mg/dl) (40–42). Persistent hypertriglyceridaemia is defined as a fasting TG ≥1.7 mmol/L (≥150 mg/dl) following a minimum of 4–12 weeks of lifestyle intervention, a stable dose of maximally tolerated statin when indicated as well as evaluation and management of secondary causes of hypertriglyceridaemia (41). More recently a more stringent criteria of TG <1.2 mmol/L (100 mg/dl) has been proposed to define optimal TG concentration (43). There has been discrepancy in recommendations between different guidelines regarding fasting or non-fasting lipid measurements for ASCVD risk assessment (43–46). Non-fasting and fasting samples provide comparable results for TC, LDL-C and HDL-C. The concentration of TG is elevated during the postprandial phase (40) though the increment is modest in majority of patients, between 0.14–0.3 mmol/L (12–27 mg/dl) (41) Non-fasting rather than fasting TG concentration is independently associated with atherosclerosis and incident future ASCVD events independent of other ASCVD risk factors, lipid parameters and insulin resistance (45, 47, 48). Using non-fasting samples of 6,391 participants in the Women's Health Study, a cut-off of 1.98 mmol/L (175 mg/dl) has been proposed to predict future ASCVD events (49). Fasting and non-fasting TG was found to be in good agreement in the Anglo-Scandinavian Cardiac Outcome Trial (ASCOT-LLT) with no difference in ASCVD outcomes between both groups (50). Most of the lipid modification clinical trials in last couple of decades used fasting lipid samples, though the implication of postprandial lipaemia and delayed clearance of TRL in postprandial state on ASCVD risk was conceptualised as early as 1979 (51) and has been subsequently tested in several clinical studies where postprandial TG was better predictor of ASCVD risk (52–54). This could be due to remnant particles that contribute to atherogenesis and are better captured in non-fasting samples. Humans are in a postprandial state most of the time during the day and therefore, a postprandial lipid profile may prove to be a more reliable and physiological marker of future ASCVD risk.
4.2 Hypertriglyceridaemia and ASCVD—mechanism and implications
With the rising prevalence of obesity, diabetes, insulin resistance and metabolic syndrome, evidence to suggest a causal relationship between hypertriglyceridemia and ASCVD has been accumulating. Catabolism of TRL leads to the liberation of remnant particles, small dense LDL particles (sdLDL), HDL3 and free fatty acids (FFA) (55, 56). FFA have a multidimensional role that triggers endothelial dysfunction through oxidative stress, impaired nitric oxide (NO) production, inflammation, and endothelial cell apoptosis (57–59) (Figure 2). Remnant particles, which are lipolytic products of chylomicrons and VLDL, vary in size and composition. They are smaller than their parent molecule and have a greater cholesterol-to-TG ratio. Increased production of VLDL and slower clearance of remnant particles and VLDL due to reduced LPL activity delays their conversion to downstream lipoprotein particles (38) thereby increasing their circulatory time. Similarly, inability of hepatic receptors to clear them from the circulation e.g., in individuals harbouring homozygous Apolipoprotein E2 isoform (FDBL) increases the time spent in the systemic circulation. With increased circulatory time, they are more likely to be entrapped in vascular intima, and in contrast to LDL, can be taken up by macrophages without chemical modifications, facilitating the process of atherogenesis (61). Apolipoprotein B48, a lipoprotein-associated with gut-derived chylomicron particles and chylomicron remnants has been found in atherosclerotic plaques derived from human aortic, carotid, and femoral endarterectomy specimens (62, 63).
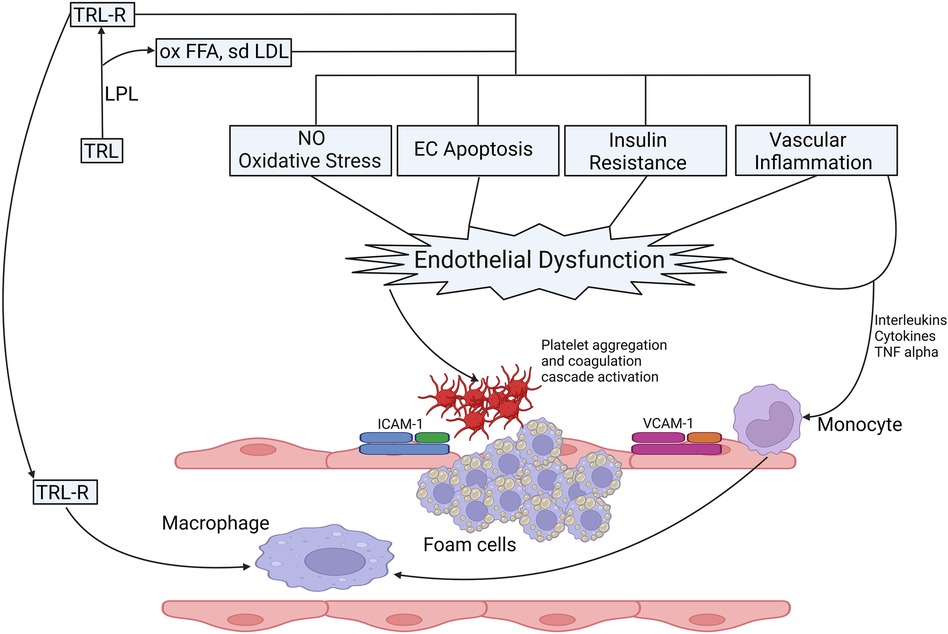
Figure 2. Possible mechanisms of TRLs in the process of the onset and progression of atherosclerosis. Catabolism of TRL leads to the production of FFA, sdLDL and their oxidized products, oxidized FFA and ox-sdLDL and remnant particles. Catabolic products of triglycerides increase the production of ROS, increase oxidative stress, reduce NO production, induce EC apoptosis, effects insulin signalling leading to IR, vascular inflammation the expression of proinflammatory cytokines and upregulate endothelial expression adhesion molecules (ICAM-1 and VCAM-1) facilitating the migration of proinflammatory leucocytes to enhance inflammatory response (55). Retention of TRL lipoproteins, remanent particles and breakdown products in vascular intima attract monocytes that differentiate into macrophages leading to the production of foam cells after engulfing TRL and remanent particles, which form the core of atherosclerotic plaque. The proinflammatory milieu leads to aggregation and activation of platelets and coagulation cascade, thereby inducing a pro-coagulant state and clot formation (55, 56, 60). CAM, intercellular adhesion molecule; EC, endothelial cells; FFA, free fatty acid; IR, insulin resistance; ICAM, inter-cellular adhesion molecule; LPL, lipoprotein lipase; NO: nitric oxide; ox, oxidised; sdLDL, small dense LDL; TNF, Tumour necrosis factor; TRL, triglyceride rich lipoproteins; TRLR, triglyceride rich lipoprotein remnants; VCAM, vascular cell adhesion molecule.
4.3 Complex association between hypertriglyceridemia and atherosclerotic cardiovascular disease: the confounding role of HDL-C
Despite large-scale epidemiological and population-based studies suggesting the association of hypertriglyceridaemia with ASCVD (Table 1), unlike LDL-C, it has been a challenge to establish the causal role of TG with ASCVD. The difficulty in establishing this causal link partly stems from the inverse relationship between TG and HDL-C (73) and progressively increasing levels of RLP-C and density of LDL with increasing TG: HDL-C ratio (74) thereby confounding the independent effect of hypertriglyceridaemia on ASCVD. Recent epidemiological studies have attempted to address this by studying the effect of hypertriglyceridaemia in patients with LDL-C <2.6 mmol/L (100 mg/dl), a routinely accepted target in people who are at moderate risk of future ASCVD event (75). In a population-based study of 27,953 statin-treated patients from the Pacific Northwest and Southern California, with LDL-C 1.0–2.6 mmol/L (40–100 mg/dl), hypertriglyceridaemia (200–499 mg/dl, 2.2–5.6 mmol/L) was found to independently increase the risk of nonfatal MI and coronary revascularisation over an average follow-up period of 5.3 years (70). Similar findings were observed in a primary prevention cohort with diabetes (n = 28,318) and LDL-C <2.6 mmol/L (100 mg/dl) where the risk of CHD was found to be higher in the cohort with hypertriglyceridaemia (TG >150 mg/dl, 1.7 mmol/L) and low HDL-C (≤50 mg/dl, 1.3 mmol/L and ≤40 mg/dl, 1.0 mmol/L for women and men respectively), [Women: HR 1.35 (1.14–1.60), Men: HR 1.62 (1.43–1.83)]. This difference was significant only in women in the cohort with low HDL-C and high TG and highest in men in the same group when compared to low HDL-C, normal TG or normal HDL-C, high TG (76). Similarly, hypertriglyceridaemia (>150 mg/dl, 1.7 mmol/L) has been demonstrated to be associated with subclinical atherosclerosis regardless of baseline LDL-C levels, [LDL-C <100 mg/dl, 2.6 mmol/L OR: 1.85 (1.08–3.18), LDL-C >100 mg/dl, 2.6 mmol/L OR: 1.42 (1.11–1.80)] and vascular inflammation (12).
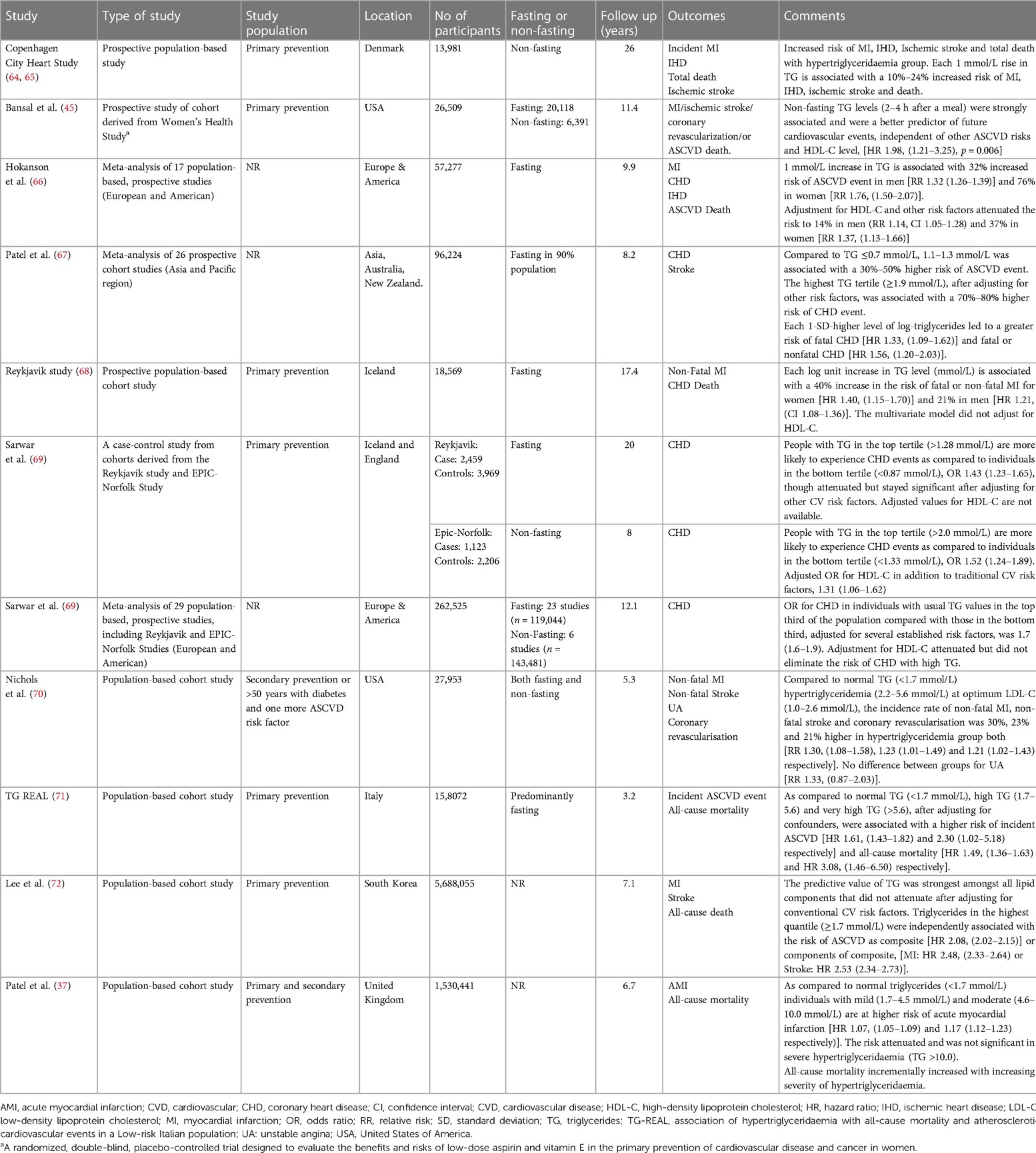
Table 1. Major studies evaluating the effect of hypertriglyceridaemia on ASCVD outcomes.
Despite the strong association between hypertriglyceridaemia and ASCVD this relationship is not straightforward partly due to other lipid abnormalities, most commonly low HDL-C. Although this has been accounted for in some studies, when the confounding factor of low HDL-C is reflected, the link between hypertriglyceridaemia and ASCVD weakens (66, 69). Although ApoB is associated with proatherogenic lipoprotein particles, Apolipoprotein A1 (ApoA1) is found in anti-atherogenic particles e.g., HDL and its subfractions. The ratio of ApoB/ApoA1 have been suggested as a better predictor of ASCVD events as compared to the individual concentration of pro- and anti-atherogenic molecules (21).
4.3.1 Lessons learnt from Mendelian randomisation
Epidemiological studies, while crucial for identifying associations, often face limitations in establishing causation, particularly in complex phenomena like the relationship between raised TRL and ASCVD. Confounders such as low HDL-C and others factors can obscure direct causative links. Mendelian randomization studies leveraging human genetics present a promising avenue. By exploiting genetic variants as proxies for lifelong exposure to elevated TRL, in a study of 73,513 individuals of Danish descent, genetic variants affecting levels of non fasting remnant cholesterol alone, non-fasting remnant cholesterol combined with HDL-C, HDL-C alone, and LDL-C were investigated for their impact on ischemic heart disease (IHD). The findings revealed a substantial causal risk increase for elevated nonfasting remnant cholesterol, independent of HDL-C levels, suggesting a causal association with a 2.8-fold increased risk of ASCVD. Similarly, the non fasting remnant cholesterol to HDL-C ratio showed a 2.9-fold causal risk increase. Conversely, while observational estimates indicated a 1.6-fold increased risk of ASCVD for each 1-mmol/L decrease in HDL-C, the causal estimate was inconclusive at 0.7-fold. Furthermore, for LDL-C, a 1.5-fold causal risk increase was observed, supporting its known association with ASCVD (77). These findings underscore the causal role of elevated remnant cholesterol in ASCVD development, independent of low HDL-Clevels. Similar observations were made by Do et al. (78) and others where SNPs in TG raising alleles e.g., ANGPTL3, APOC2, APOA5, GPIHBP1, LMF1 were found to increase the risk of ASCVD, while conversely, in APOC3 loss of function heterozygosity led to reduction in ASCVD events (79–81).
5 Triglycerides as residual ASCVD risk factor in statin-treated patients
Landmark statin trials that shaped our understanding of cardiovascular risk reduction with intensive LDL-C reduction still displayed residual ASCVD risk (22.4% at 2 years, 9.3% at 4.3 years, and 8.7% at 4.9 years in PROVE IT TIMI, IDEAL and TNT respectively) despite intensive statin treatment and achieving LDL-C of 1.6 mmol/L (62 mg/dl), 2.1 mmol/L (81 mg/dl) and 2.0 mmol/L (77 mg/dl) respectively (17–19). Even though efficacy of statins in primary prevention of ASCVD in type 2 diabetes mellitus (T2DM) was well demonstrated in the Collaborative Atorvastatin Diabetes Study (CARDS), 12.5% of statin recipients had an ASCVD event despite achieving median on-treatment LDL-C 2.0 mmol/L (77 mg/dl) during median follow up of 3.9 years (13). Clearly, residual ASCVD risk persists even after achieving optimal reductions in LDL-C levels in large statin trials, irrespective of the dosage. The factors that may contribute to residual cardiovascular risk are outlined in Figure 1.
A post hoc analysis of the PROVE IT TIMI trial focused on secondary prevention. LDL-C levels below 1.8 mmol/L (70 mg/dl) and TG levels below 2.2 mmol/L (200 mg/dl) demonstrated a 40% reduced risk of subsequent ASCVD events when compared to individuals with TG levels exceeding 2.2 mmol/L (200 mg/dl). TG <1.7 mmol/L (150 mg/dl) was independently associated with a 20% reduction in relative risk of CHD after adjustment for LDL-C and other covariates. A combination of TG <1.7 mmol/L (150 mg/dl) and LDL-C <1.8 mmol/L (70 mg/dl) was associated with the lowest ASCVD events, and each 0.1 mmol/L (10 mg/dl) lower TG concentration led to a decline in the rate of recurrent acute coronary syndrome (ACS), MI and death by 1.6% independent of LDL-C concentrations (8). Similar results were found in the intEnsive statin therapy for hypercholesteroleMic Patients with diAbetic retinopaTHY (EMPATHY) study where serum TG was associated with ASCVD regardless of the intensity of statin therapy. Each 0.1 mmol/L (10 mg/dl) increase in TG was associated with a 2.1% increased risk of ASCVD event and TG >1.5 mmol/L (135 mg/dl) was found to be an independent risk factor for developing ASCVD (82). In post hoc analyses of TNT and IDEAL study, non-HDL-C and ApoB were found to have a stronger association with future ASCVD events as compared to LDL-C alone. Patients with TG in the highest quantile were predicted to be at 63% increased risk as compared with the lowest quantile. This effect was attenuated but not abolished after adjustment for HDL-C. The probability of ASCVD event was 30% higher in individuals with TG levels >1.7 mmol/L (150 mg/dl), as opposed to those with levels <1.7 mmol/L (150 mg/dl). This association between elevated TG levels and an increased risk of new ASCVD events persisted in individuals with an LDL-C cholesterol concentration below 2.6 mmol/L (100 mg/dl) (9, 10). A post hoc analysis of dal-OUTCOMES and Myocardial Ischemia Reduction with Acute Cholesterol Lowering (MIRACL) study reported similar results, where in statin-treated patients, fasting TG was found to be independently associated with short- and long-term risk of developing ASCVD independent of LDL-C. Similar to PROVE IT-TIMI, each 0.1 mmol/L (10 mg/dl) increase in TG was associated with a 1.4%–1.6% increase in the risk of a ASCVD event (11). In all major statin trials, there is a significant residual ASCVD risk in statin recipients, which is due to atherogenic dyslipidaemia and non-LDL lipoprotein subfractions along with other contributors that constitute residual ASCVD risk profile (Figure 1).
6 Triglycerides and residual ASCVD risk in PCSK9 inhibitor treated patients
The FOURIER and ODYSSEY OUTCOMES trials have demonstrated significant LDL-C reduction that has translated into ASCVD risk reduction. However, despite achieving very low LDL-C, a residual risk of 9.8% and 9.5% respectively remained at 2.2 and 2.8 years respectively. Participants in both trials received moderate or high intensity statin in addition to PCSK9 monoclonal antibodies. In FOURIER, mean LDL-C at the end of the trial period was as low as 0.8 mmol/L (30 mg/dl) in the evolocumab arm. Despite this, a significant proportion of patients had ASCVD events. 9.8% of evolocumab recipients had at least one ASCVD event and 6.1% had a subsequent ASCVD event (14, 83). In a prespecified secondary analysis of the FOURIER trial, a monotonic relationship between LDL-C at 4 weeks and ASCVD events was observed, where while high LDL-C was associated with heightened risk, 10.3% of individuals achieving LDL-C <0.5 mmol/L (20 mg/dl) experienced an ASCVD event (84). TG of 1.3 mmol/L (112.3 mg/dl) at the end of the trial period along with other metabolic and inflammatory factors (Figure 1) might explain the residual ASCVD risk in this group who achieved very low LDL-C. ApoB, which constitutes a composite of all major atherogenic lipoproteins, inclusive of LDL, VLDL, IDL, remnant particles and Lp(a) would be a better therapeutic target to minimize ASCVD risk. This is supported by a recent analysis of the ODYSSEY outcome database by Hagstrom et al. where LDL-C was found to underestimate the ASCVD risk and ApoB levels were found to be a better predictor of future ASCVD events independent of LDL-C levels in a cohort of alirocumab treated patients (85). Similarly, intensive LDL-C lowering with evolocumab and statins in the “Global Assessment of Plaque Regression With a PCSK9 Antibody as Measured by Intravascular Ultrasound” (GLAGOV) study achieved a significant reduction in plaque atheroma volume (PAV) and total atheroma volume (TAV) with very low LDL-C (36.6 mg/dl, 0.9 mmol/L). Nevertheless, not all the patients achieved plaque regression. In a subgroup of participants with baseline LDL-C of 1.8 mmol/L (70 mg/dl), with a further 50%–55% reduction after treatment, 20% did not have regression in atheroma volume despite achieving very low LDL-C (86), thereby suggesting a role of TG-rich lipoproteins and other factors (Figure 1) in atheroma development and progression (87). In addition, 12.2% of evolocumab recipients had ASCVD events suggestive of residual factors other than LDL-C contributing to atherogenesis (86).
Residual ASCVD risk in statin and PCSK9-treated patients is not confined to non-LDL subfractions. Pradhan et al. have demonstrated a 62% increase in the risk of future ASCVD events (3.6% annual event rate) in statin and PCSK9-treated patients who have achieved median LDL-C of 1.07 mmol/L (41.7 mg/dl) but have raised high-sensitivity C-reactive protein (>3 mg/L) suggesting complex interplay of multiple residual ASCVD risk factors in the pathogenesis of ASCVD (88).
7 Therapeutic targets
The role of TG in ASCVD is well established from clinical trials (8), epidemiological (69) and Mendelian randomisation studies (24, 89). Serum TG levels are very sensitive to diet, lifestyle, and secondary factors. Three classes of drugs that preferentially reduce serum TG levels are fibrates, omega-3 fatty acids (FA) and niacin. Despite genetic studies (78) and post hoc analysis of landmark statin (8) and PCSK9 trials (85) suggesting a lower risk of ASCVD with reduced TG, the results from pharmacologically achieved lower TG levels with niacin, fibrates and omega-3 FA have been inconsistent. Major clinical trials of fibrates and Omega-3 FA evaluating ASCVD outcomes are summarised in Tables 2, 3 respectively.
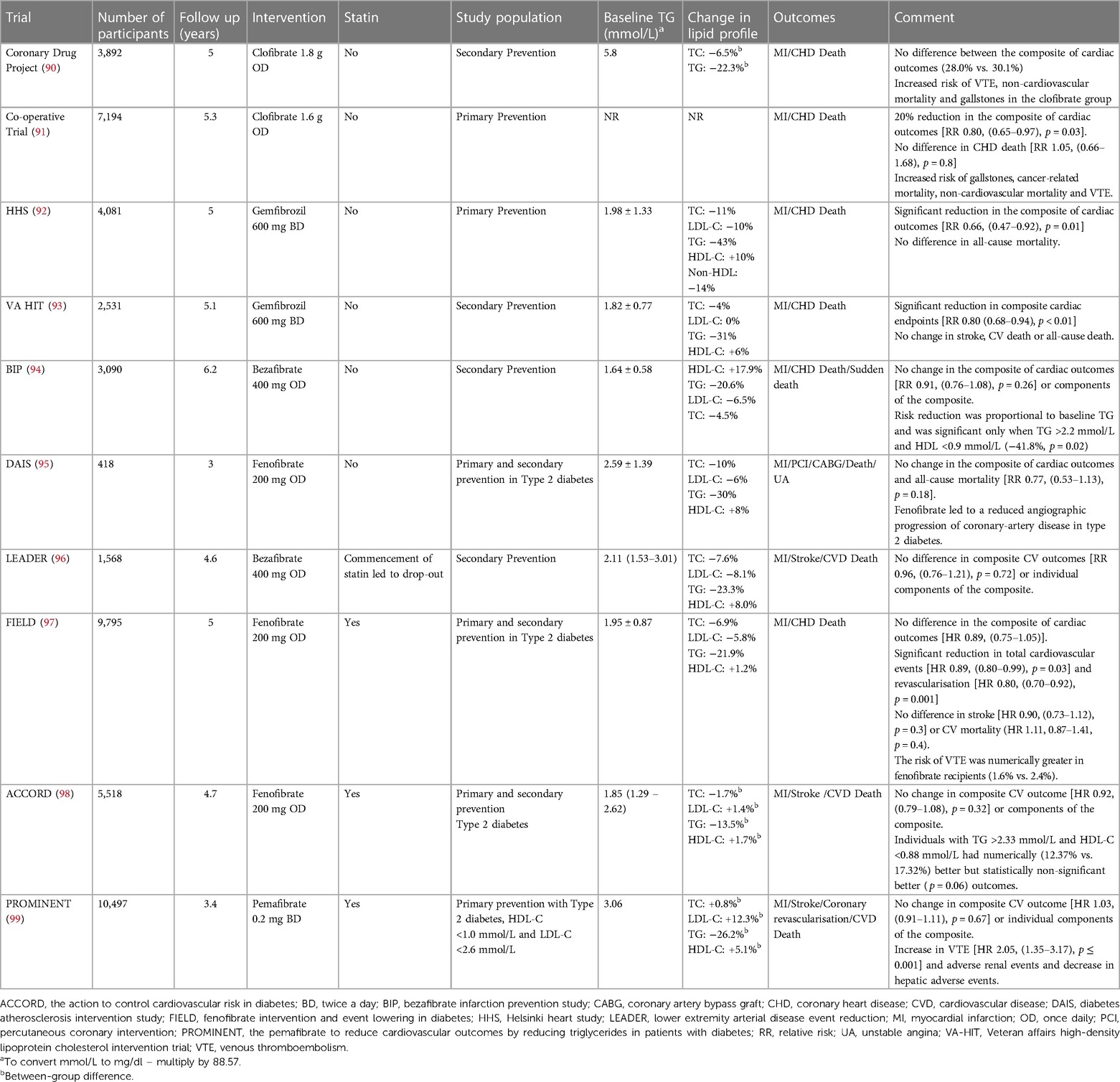
Table 2. Major fibrate trials, effect on lipid profile and ASCVD outcomes.
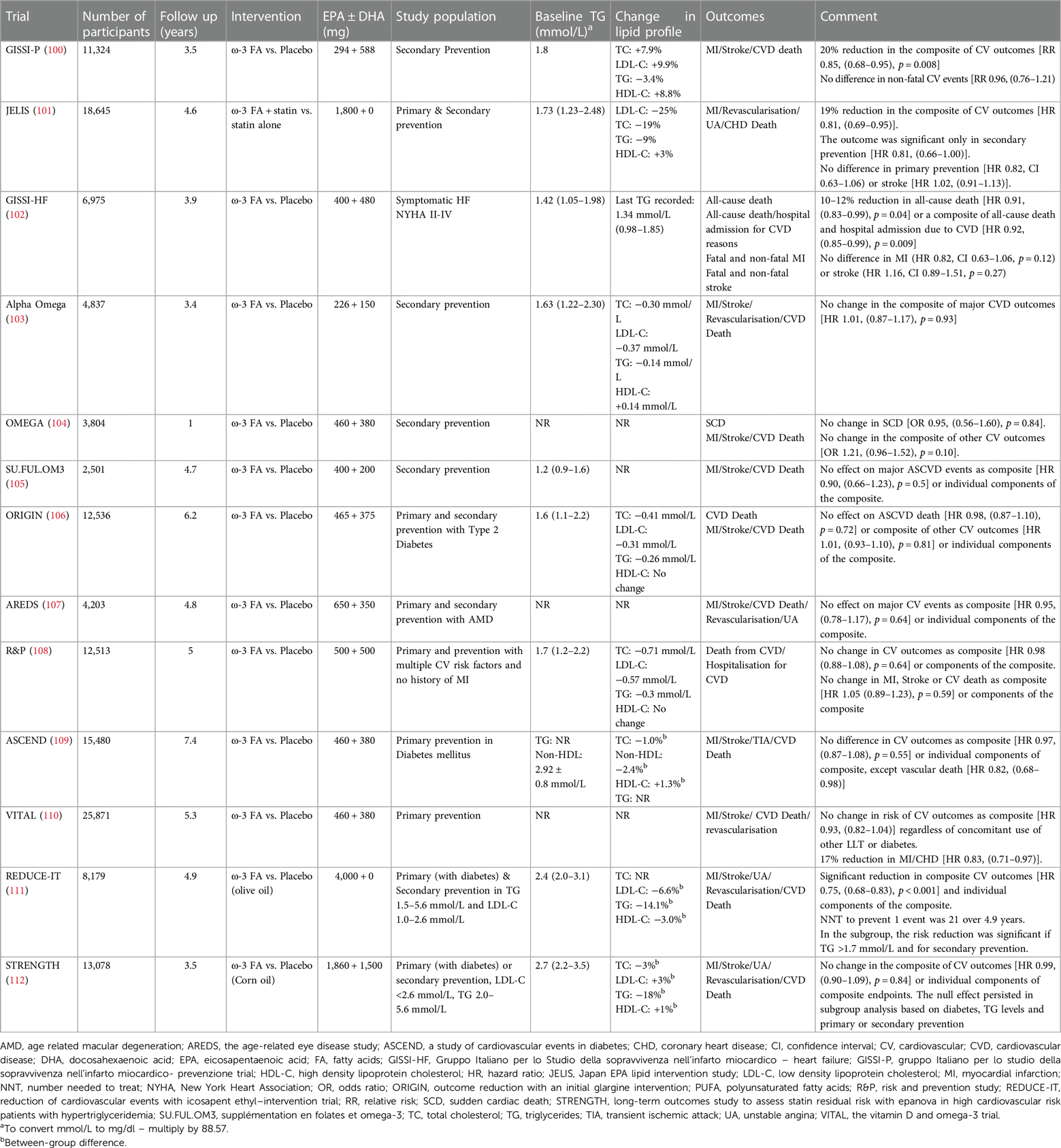
Table 3. Major omega 3 fatty acid trials, effect on lipid profile and ASCVD outcomes.
7.1 Diet and lifestyle
TG levels are highly responsive to dietary interventions and physical activity; therefore, the first line of intervention is often diet and lifestyle modifications (38). Epidemiological and clinical trial data substantiate the correlation between the Mediterranean-style dietary pattern and reduction in TG levels (113, 114). Modification of macronutrient composition through dietary interventions, adopting low-carbohydrate diets, and implementing caloric restriction have shown efficacy in improving TG (115). Notably, the Mediterranean diet emerges as the dietary pattern with the most consistent and robust evidence supporting its efficacy in addressing hypertriglyceridaemia (113). Additionally, among dietary components, the consumption of omega-3 FA has been the subject of a substantial number of RCTs that have consistently demonstrated their effectiveness in reducing TG levels (116, 117). Sea food and oily fish are a rich source of polyunsaturated fatty acids (PUFA) that not only lead to a significant decrease in TG but also improve blood pressure, systemic inflammation and increase HDL-C (118). In the Framingham Heart Study Offspring Cohort, individuals in the highest quintile for the Mediterranean-style diet exhibited the lowest TG levels over a 7-year follow-up (119). Additionally, these interventions offer ancillary benefits such as weight loss and reduced waist circumference. A comprehensive approach to lifestyle modification, encompassing dietary strategies with an emphasis on reduced carbohydrate and saturated fat intake, regular physical activity, and weight management, can result in substantial TG reductions ranging from 20% to 50%.
7.2 Niacin
While there exists evidence substantiating the correlation between elevated TG and a heightened susceptibility to ASCVD events, a multitude of clinical trials assessing pharmacological interventions aimed at reducing TG levels have not demonstrated reduction in ASCVD events. The Atherothrombosis Intervention in Metabolic Syndrome with Low HDL-C/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) study investigated the impact of niacin on individuals on intensive statin therapy with elevated TG levels and low HDL-C levels. Despite niacin achieving a 31% reduction in TG and 21% increase in HDL-C in a cohort with baseline LDL-C <1.8 mmol/L (70 mg/dl), no difference in the composite primary cardiovascular endpoint emerged between the niacin-administered cohort and the control group (120). Similar results were replicated later by the Heart Protection Study 2–Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE), which investigated the combined administration of niacin and laropiprant, a prostaglandin D2 receptor 1 antagonist, with simvastatin and/or ezetimibe (121). A systematic review and metanalysis spanning the preceding six decades and 17 RCTs failed to show ASCVD risk reduction with niacin treatment across all patient cohorts, including those on statin treatment (122). Consequently, niacin has been withdrawn from the European market.
7.3 Fibrates
Fibrates are synthetic ligands for peroxisome proliferator-activated receptor (PPAR) alpha receptors, through which they exert lipid-lowering and pleiotropic effects via increasing lipolysis by upregulating LPL activity, hepatic FA uptake, increased production of ApoA1, inhibition of apolipoprotein C3 and reduced expression of various pro-inflammatory cytokines and adhesion molecules (123). Whilst hypertriglyceridaemia is deemed as a risk factor for ASCVD and current guidelines recommend employing additional measures to reduce TG <1.7 mmol/L (150 mg/dl) to mitigate cardiovascular risk (75, 124), the addition of fibrates to statins to achieve this has not shown any additional benefit (Table 2). The history of employing fibrates to reduce cardiovascular risk dates to the 1950s when a group of farm workers were found to have low cholesterol after being exposed to an insecticide (phenyl ethyl acetic acid) that led to the synthesis of its analogue, clofibrate (125, 126). When clofibrate was employed for the first time in a primary prevention cohort, although showing a significant reduction in nonfatal MI, it failed to demonstrate a significant benefit in cardiovascular mortality. Furthermore, it increased the risk of gallstones and non-cardiovascular mortality (91) which precluded its use in the modern era. Whilst the outcomes from clofibrate were disappointing, primary, and secondary prevention trials towards the end of 20th century with another fibrate, gemfibrozil demonstrated significant benefits in reducing cardiovascular events (92, 127). The secondary prevention trial (VA HIT) included patients only with HDL-C <1.0 mmol/L (39 mg/dl) thereby signalling the benefit of fibrates in individuals with atherogenic dyslipidaemia (high TG, low HDL-C). Intriguingly, the ASCVD benefit was attributed to increased HDL-C rather than a reduction in TG levels. Subgroup analysis of the Helsinki Heart Study (HHS) trial echoed similar results where the greatest benefit was derived in individuals with low HDL-C (<1.08 mmol/L, 42 mg/dl) and high TG (>2.3 mmol/L, 204 mg/dl) (128). The predominant effect of fibrates in ameliorating ASCVD risk in atherogenic dyslipidaemia, characterized by high TG and low HDL-C, has been replicated in subsequent trials with bezafibrate and fenofibrate where, although these drugs failed to demonstrate significant ASCVD risk reduction across the whole cohort, participants with atherogenic dyslipidaemia derived maximum benefit (94, 98) (Table 2). Several meta-analyses have demonstrated ASCVD risk reduction with fibrates only in the setting of atherogenic dyslipidaemia (129, 130). In addition to ASCVD risk reduction, fibrates reduce the progression of diabetic retinopathy (131, 132).
More recently, a selective PPAR alpha receptor modulator, pemafibrate, has been evaluated for ASCVD risk reduction in hypertriglyceridaemia in a subset of patients with LDL-C <1.8 mmol/L (70 mg/dl) whilst on statins or <2.6 mmol/L (100 mg/dl) in cases of statin intolerance. In addition, the study was focused on individuals with atherogenic dyslipidaemia i.e., with T2DM, TG >2.2 mmol/L (195 mg/dl) and HDL-C <1.0 mmol/L (38 mg/dl). Two-thirds of the study population had prior ASCVD events. This cohort was representative of modern-day residual ASCVD risk profile where intensive LDL-C reduction and background statin therapy have been employed. Pemafibrate is distinct from other fibrates as it is a selective PPAR receptor modulator. Though the lipid-modifying effect of pemafibrate is comparable with fenofibrate, pemafibrate has superior pleiotropic effects and increases HDL's cholesterol efflux capacity in vitro (133). Nevertheless, despite better pharmacokinetics and achieving a significant reduction in TG, VLDL-C and remnant particles and a comparable HDL-C increase compared to earlier fibrate trials, no significant reduction in major ASCVD events was demonstrated, thereby casting doubt on the utility of TG reduction via fibrates on ASCVD events in statin-treated patients with adequately lowered LDL-C. Safety analysis of pemafibrate also revealed that drug recipients were twice as likely to suffer deep vein thrombosis (DVT) and pulmonary embolism (PE) compared to placebo. Similar findings have been reported with fenofibrate [FIELD trial, RR for venous thromboembolism (VTE) 1.5 (1.1–2.0)] (97) and clofibrate [Coronary Drug Project, RR for PE 1.8 (1.1–2.8)] (90). The association of fibrates with VTE has been supported by a French pharmacovigilance database and other case-control studies (134–137). The reason for the increased risk of VTE is not known, though an increased level of homocysteine with or without other contributing factors might explain the increased risk (137).
Kim and colleagues have recently conducted a meta-analysis and meta-regression analysis of 12 RCTs, including Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) trial, employing fibrates for ASCVD risk reduction. While authors demonstrated overall reduction in ASCVD risk, mainly in secondary prevention group, it was found that reduction in ASCVD events was significantly associated with reduction in LDL-C. Each 1 mmol/L reduction in LDL-C level was associated with a reduction in major adverse cardiovascular events (MACE) with a relative risk (RR) 0.71 (95% CI 0.49–0.94, p = 0.01). On the other hand, a reduction in TG concentration was not associated with a significant reduction in MACE. Change in LDL-C was found to be the main driver of heterogeneity between the studies (138). Fibrates can reduce LDL-C, however this LDL-C lowering potential is inconsistent (139) and is significantly dampened with concomitant use of statins (140). In the meta-analysis by Kim et al. both baseline and LDL-C change were inversely correlated with the year of publication, suggesting better lipid control post-statin era and hence diminished efficacy of fibrates in reducing LDL-C (138). There also appears a negative correlation between change in LDL-C and baseline LDL-C with Pemafibrate (141). sdLDL are more atherogenic than large buoyant LDL particles. Using Sampson formula to calculate sdLDL-C, no difference has been shown in its concentration between pemafibrate and placebo group (142). Likely, low baseline LDL-C levels diminish the role of TG in sdLDL formation. Additionally, Pemafibrate stimulates hepatic TG lipase, potentially enhancing sdLDL production, which could offset the decrease in sdLDL attributed to lower TG levels. In the PROMENENT study Pemafibrate reduced remnant cholesterol and TG, however increased levels of ApoB, a surrogate indicator for LDL particle count. Given the strong correlation between ApoB levels and sdLDL-C levels, the lack of reduction in estimated sdLDL-C levels in the PROMINET trial is not unexpected (142). The beneficial impact of Pemafibrate on ASCVD might be restricted to individuals with hypertriglyceridemia those with relatively higher LDL-C and would be interesting to investigate the effect of Pemafibrate in a sub-cohort of PROMINENT trial who had higher baseline LDL-C. Moreover, recent advancements in the treatment of diabetes and hypertension, along with the growing utilization of cardioprotective anti-hyperglycaemic medications, high-intensity statins, along with addition of other potent LDL-C lowering drugs may have reduced the remaining cardiovascular risk to such an extent that it becomes challenging to discern notable differences in outcomes solely through triglyceride lowering strategies.
In summary, the use of fibrates to mitigate ASCVD risk was supported by early trials with the greatest benefit being derived for patients with atherogenic dyslipidaemia. However, results from the PROMINENT study (99) along with an increased propensity to develop VTE with fibrates have cast doubt over their clinical utility for ASCVD risk reduction and they should be administered cautiously in patients, particularly in individuals at high risk of VTE.
7.4 Omega 3 fatty acids
Omega 3 FA are PUFA that cannot be synthesised by humans. They are found in abundance in seafood and hence are also called marine fatty acids. Our understanding of the relationship between increased consumption of PUFA, favourable lipid profile and reduce incidence of CHD dates to the 1970s when Greenland Inuit whose diet was rich in seafood were found to have favourable metabolic profiles as compared to Danish controls (143). Subsequently, several potential mechanisms via which omega-3 FA can reduce the burden of ASCVD independent of its lipid-lowering potential have been proposed (144). Despite this, clinical studies have produced divergent results for ASCVD outcomes with omega-3 FA supplementation (Table 3).
The initial landmark trial of omega-3 FA, Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico- Prevenzione trial (GISSI-P) ignited excitement when it showed a 15%–20% reduction in fatal and non-fatal ASCVD events (100). Similar results were reproduced in Japan EPA lipid Intervention Study (JELIS) where Eicosapentaenoic acid (EPA) supplementation led to a 19% reduction in major ASCVD events (101). Nonetheless, this earlier excitement waned when subsequent trials failed to reproduce ASCVD benefits (Table 3). Positive outcomes in GISSI-P and JELIS could be due to the diet and lifestyle of the study population from Italy and Japan respectively who consume seafood on a more regular basis and hence may have higher circulating omega-3 FA levels. Attained level of blood EPA is an important factor and may explain the positive outcome of JELIS trial. This perception is supported by findings from two recent trials, Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial (REDUCE-IT) (111) and Long-Term Outcomes Study to Assess Statin Residual Risk with Epanova in High Cardiovascular Risk Patients with Hypertriglyceridemia (STRENGTH) (112), where outcomes for REDUCE-IT, like JELIS, were positive after using a higher dose of purified icosapent ethyl (IPA) (4 g/day) leading to a greater increment in its blood levels. However, outcomes were neutral for STRENGTH, where the increment in EPA levels was lower as compared to REDUCE-IT. Baseline EPA level in study participants in JELIS was 97 µg/ml which was significantly higher than REDUCE-IT and STRENGTH, (26.1 µg/ml and 21.0 µg/ml respectively) but achieved EPA levels after supplementation with omega-3 FA were comparable between JELIS (1.8 g/day of purified IPA, 70% increase, 169 µg/ml) and REDUCE-IT (4 g/day of purified IPA, 394% increase, 144 µg/ml) which produced positive outcomes. In STRENGTH however, the achieved EPA levels after supplementation [EPA 1,860 mg + docosahexaenoic acid (DHA) 1,500 mg/day] led to a 269% increment yet the absolute achieved EPA levels remained lower (89.6 µg/ml) than the baseline EPA levels of JELIS participants (101, 111, 112). This suggests employing higher doses of EPA may help in achieving an “effective therapeutic level” of circulating EPA to reduce ASCVD events. Moreover, the mean baseline TG level in JELIS participants was 1.7 mmol/L (150 mg/dl) which, according to current guidelines is defined as normal (38). This, along with ASCVD risk reduction disproportionate to the amount of TG reduction and failure of fibrates to reduce ASCVD events despite achieving greater TG reduction suggests independent pathways through which EPA exerts its antiatherogenic effects. Trials demonstrating neutral outcomes employed a combination of EPA and DHA while trials demonstrating positive ASCVD outcomes employed purified EPA. This might suggest that any beneficial effect conferred by EPA is partially neutralised by DHA, though there is no mechanistic data to suggest any proatherosclerotic and/or prothrombotic effects of DHA. One plausible explanation could be the low absolute dose of EPA used in EPA + DHA as compared to the higher one used in EPA monotherapy (without DHA) studies.
EPA and DHA are two distinct molecules that have diverse effects on membrane integrity, stability, and cholesterol distribution (145). While EPA preserves membrane structure, DHA increases membrane fluidity and has fewer antioxidant properties that wane more quickly as compared to EPA (146). The antioxidant properties of EPA exceed quantitatively those of fibrates which might explain positive ASCVD outcomes despite proportionately less TG reduction (146). The differential interaction of EPA and DHA with the cell membrane, cholesterol distribution, formation of cholesterol crystals and atherosclerotic plaque, antioxidant capacity and modulation of endothelial dysfunction (147) might explain the ASCVD protection conferred by EPA-based therapeutics. The application of the INSPIRE biobank registry (formerly known as the Intermountain Heart Collaborative Study) afforded a unique opportunity to explore the relationship between spontaneously acquired levels of omega-3 metabolites and the occurrence of long-term major adverse cardiovascular events (MACE) within a diverse cohort of high-risk individuals encompassing both primary and secondary prevention populations referred for angiography (148). The findings substantiated the observed cardioprotective impact linked to elevated circulating and acquired levels of EPA, as opposed to DHA. Notably, these results suggested that increased DHA levels and a resultant reduced EPA/DHA ratio might diminish the cardiovascular protective effect of EPA. Elevated plasma concentrations of EPA and the combined EPA + DHA demonstrated a protective effect against incident MACE. However, unadjusted DHA alone did not display a correlation with incident MACE or a protective effect. Furthermore, DHA, when adjusted for EPA, exhibited an almost twofold increased risk of MACE for individuals in the highest quartile compared to the lowest quartile of DHA. These findings, in conjunction with reduced attained EPA serum levels, may contribute to understanding the outcomes observed in recent trials, such as the STRENGTH and REDUCE-IT (148). The Randomized Trial for Evaluation in Secondary Prevention Efficacy of Combination Therapy - Statin and Eicosapentaenoic Acid (RESPECT-EPA) was a recent open label trial focussed on patients with secondary prevention on background statin therapy. After 6 years, a marginally significant reduction in the primary cardiovascular outcome (10.9% vs. 14.9%, hazard ratio 0.785, p = 0.0547) and a significant decrease in the composite secondary endpoint (8.0% vs. 11.3%, hazard ratio 0.734, p = 0.0306) was observed. The trial employed same dose of EPA as in JELIS but the baseline EPA level was half of that of JELIS participants (45 µg/ml vs. 97 µg/ml) and focus on patients with higher chronic inflammation suggested by lower EPA:AA (arachidonic acid) ratio (149, 150). Though the details of the study are awaited, like JELIS, higher baseline EPA levels in a Japanese cohort with subsequent higher clinically meaningful levels after treatment might suggest that absolute serum EPA levels govern ASCVD outcomes whereas patients with low baseline EPA might require higher doses of EPA to achieve clinically significant levels. Similar observations had been made in a sub-study of REDUCE-IT where achieved EPA levels in the treatment group were found to be associated with ASCVD events, heart failure and cardiovascular death (151).
7.5 Weight loss and bariatric surgery
The common dyslipidaemia associated with obesity is marked by elevated TG levels and low HDL-C. Most diet and lifestyle interventions accompanied by some degree of weight loss are translated into improved hypertriglyceridaemia (152). Pharmacological interventions with glucagon like peptide 1 (GLP1) and gastric inhibitory polypeptide (GIP) receptor agonists are associated with a 20%–25% reduction in TG accompanied by 15%–20% weight loss (153, 154). Bariatric surgery (BS) offers another option to attain significant and sustained weight loss that not only improves hypertriglyceridaemia but also improves the qualitative composition of lipoprotein particles (155). Incidence of hypertriglyceridaemia was significantly reduced after bariatric surgery during the follow up period of 2 years (156) where TG level remains the strongest univariate predictor of mortality in the Swedish Obese Subject (SOS) study (157). In a metanalysis of 178 studies, recipients of BS demonstrated a significant decrease in mean TG levels as compared to both baseline and non-surgical controls. Reduction in TG varied depending on type of BS employed with the greatest reduction observed with Roux-en-Y gastric bypass (RYGB), but each procedure displayed significant reductions compared to baseline and controls (158). We have demonstrated significant reductions in TG along with other atherogenic lipoproteins, markers of systemic inflammation and insulin resistance after BS in patients with and without diabetes (159–162). In T2DM the susceptibility to ASCVD is significantly increased by the existence of microvascular disease that may manifest as nephropathy, neuropathy, or retinopathy (163). Hypertriglyceridaemia is associated with small nerve fibre damage and cardiac autonomic neuropathy (117, 155, 164, 165). Our findings, along with those of others, indicate evidence of small nerve fibre regeneration post-bariatric surgery. Additionally, we established a correlation between improvements in neuropathic parameters and reductions in TG levels (160, 166, 167). Notably, the beneficial effects on small nerve fibre structure and function extend beyond patients with T2DM. A similar association of hypertriglyceridaemia is noted in relation to retinopathy and nephropathy (38).
7.6 Others
In addition to lipid-modifying therapy targeting serum TG, several other therapeutic agents have been or are in the process of development, targeting various potential mediators of residual cardiovascular risk with variable success (Table 4). Careful selection of patients, after addressing traditional modifiable risk factors based on clinical features, laboratory values, risk of adverse effects, co-morbidities and patient preferences can aid in defining the choice of novel therapy. The absolute benefit gained by these add-on novel therapies largely depends upon the baseline residual risk after addressing conventional modifiable risks.
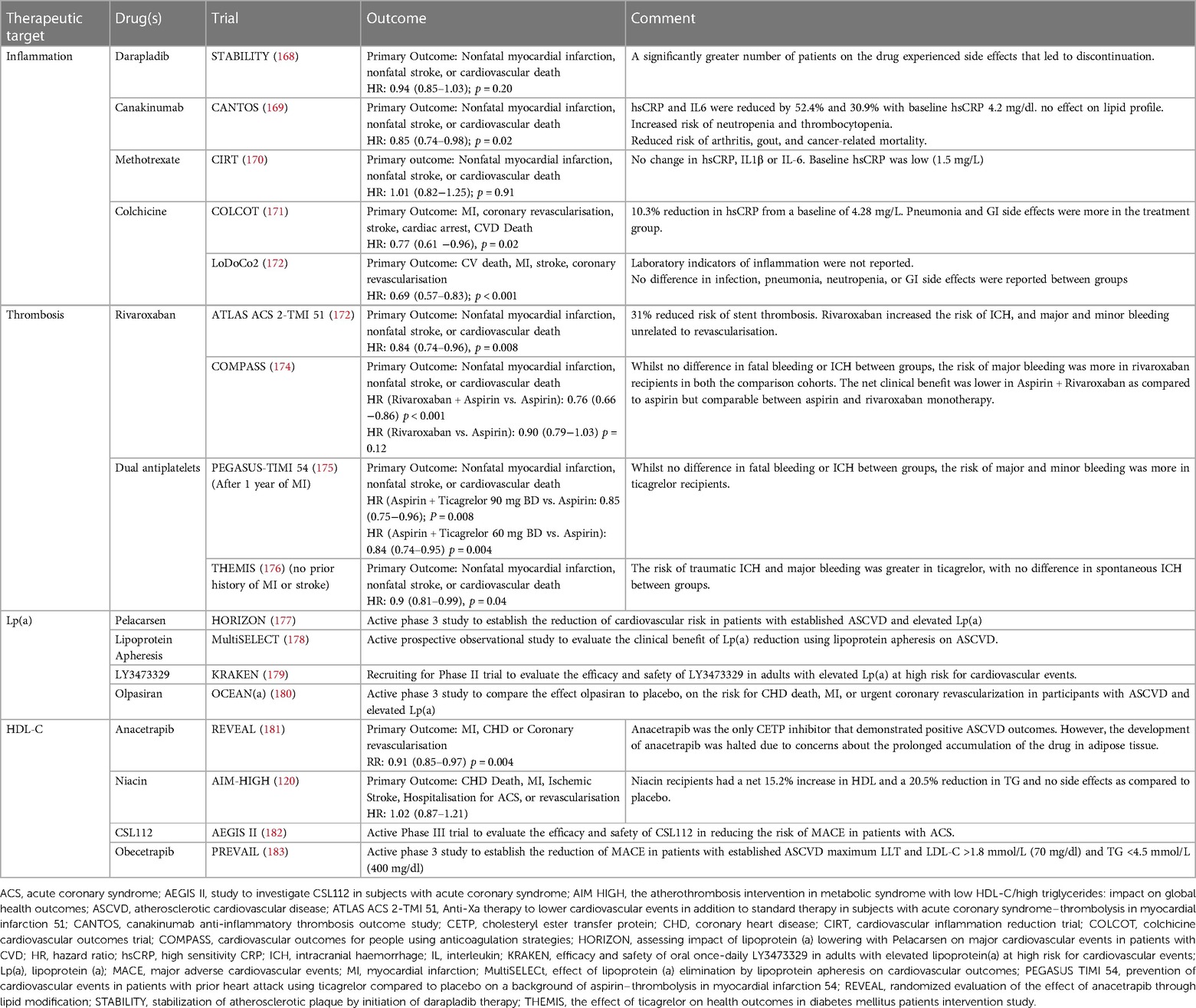
Table 4. Therapeutic targets for reducing atherosclerotic cardiovascular disease (ASCVD) risk: summary of clinical trials and outcomes.
8 Conclusion
Historically ASCVD risk reduction measures have predominantly been LDL-centric. Despite significant strides in developing LDL-C lowering agents and their proven benefits in reducing ASCVD risk, a substantial portion of the secondary prevention cohort remains undertreated. Factors such as clinical inertia, discrepancies in access to effective lipid-lowering therapies, and challenges in implementing guidelines contribute to this problem. Even with optimal guideline-based treatment, lipid-related but also lipid-independent residual risk remains a significant contributor to recurrent events, emphasizing the need to identify atherogenic targets beyond LDL-C.
TRL as a risk factor for ASCVD have gained much attention recently supported by epidemiological, genetic, and mechanistic studies. Addressing this TRL-associated risk is challenging, given mixed results from clinical outcome studies evaluating various therapeutic approaches. Fibrates had previously been shown to be of benefit in atherogenic dyslipidaemia but recent results from the PROMINENT trial have cast doubt on their utility in ASCVD risk reduction. Further, increased risk of VTE has been reported inconsistently in earlier fibrate trials and therefore merits careful consideration. ASCVD risk reduction from REDUCE-IT and RESPECT EPA but not from STRENGTH and other studies employing combined EPA and DHA suggest TG-independent pathways to mitigate ASCVD risk with purified EPA products. Patients at high risk of recurrent ASCVD events may benefit from employing additional therapeutic agents to target components of the residual cardiovascular risk profile.
Author contributions
BB: Data curation, Writing – original draft, Writing – review & editing. JS: Writing – original draft, Writing – review & editing. PD: Writing – original draft, Writing – review & editing. MF: Writing – original draft, Writing – review & editing. DA: Writing – original draft, Writing – review & editing. AW: Writing – original draft, Writing – review & editing. SR: Writing – original draft, Writing – review & editing. IG-B: Writing – original draft, Writing – review & editing. AM: Writing – original draft, Writing – review & editing. PD: Writing – original draft, Writing – review & editing. HS: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by an unrestricted grant from Amarin.
Acknowledgments
The authors acknowledge the support from Manchester National Institute for Health Research (NIHR)/WELLCOME Trust Clinical Research Facility, Lipid disease fund, Hyperlipidaemia Education & Atherosclerosis Research Trust UK (HEART UK).
Conflict of interest
HS: Received personal fees from Amgen, Akcea, Synageva, NAPP, Novartis, Takeda, Sanofi, Pfizer and Kowa & received research grants and donations from Akcea, Pfizer, MSD, AMGEN, Genzyme-Sanofi, Synageva, Amryt, Synageva and Alexion. DA: reports research grants from Boehringer-Ingelheim, Bristol Myers Squibb, Janssen, and the LEO Foundation. JS: received grants and research support from Astra Zeneca, Daiichi-Sankyo, Eli Lilly and Company and Novo Nordisk; speaker fees from Novartis, Astra Zeneca, Daiichi-Sankyo and Sanofi; and consultancy fees from Amgen, Boehringer Ingelheim, Eli Lilly and Company and Sanofi. PD: Received honoraria from Sobi, Novartis, Amgen, Daiichi Sankyo, Sanofi and Amarin. SR: Received honoraria from NOVARTIS, AMGEN, Astra Zeneca, Ultragenyx, Sanofi, Foresite Labs, and research grants from Astra Zeneca. IG-B: received personal honoraria for consulting from Amgen, Regeneron, Aegereon, Akcea Therapeutics, Daiichi-Sankyo, Novartis, Ultragenyx, Sanofi and Amarin.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. European Heart Network. European Cardiovascular Disease Statistics 2017 [Internet]. (2017). [cited 2022 Dec 13]. Available online at: https://ehnheart.org/cvd-statistics.html
2. Centres for Disease Control and Prevention. Health Topics—Heart Disease and Heart Attack [Internet]. (2021). [cited 2022 Dec 13]. Available online at: https://www.cdc.gov/policy/polaris/healthtopics/heartdisease/index.html#print
3. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. (2017) 38(32):2459–72. doi: 10.1093/eurheartj/ehx144
5. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. (2010) 376(9753):1670–81. doi: 10.1016/S0140-6736(10)61350-5
6. Soran H, Schofield JD, Durrington PN. Cholesterol, not just cardiovascular risk, is important in deciding who should receive statin treatment. Eur Heart J. (2015) 36(43):2975–83. doi: 10.1093/eurheartj/ehv340
7. Soran H, Kwok S, Adam S, Ho JH, Durrington PN. Evidence for more intensive cholesterol lowering. Curr Opin Lipidol. (2017) 28(4):291–9. doi: 10.1097/MOL.0000000000000433
8. Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. (2008) 51(7):724–30. doi: 10.1016/j.jacc.2007.10.038
9. Kastelein JJP, van der Steeg WA, Holme I, Gaffney M, Cater NB, Barter P, et al. Lipids, apolipoproteins, and their ratios in relation to cardiovascular events with statin treatment. Circulation. (2008) 117(23):3002–9. doi: 10.1161/CIRCULATIONAHA.107.713438
10. Faergeman O, Holme I, Fayyad R, Bhatia S, Grundy SM, Kastelein JJP, et al. Plasma triglycerides and cardiovascular events in the treating to new targets and incremental decrease in end-points through aggressive lipid lowering trials of statins in patients with coronary artery disease. Am J Cardiol. (2009) 104(4):459–63. doi: 10.1016/j.amjcard.2009.04.008
11. Schwartz GG, Abt M, Bao W, DeMicco D, Kallend D, Miller M, et al. Fasting triglycerides predict recurrent ischemic events in patients with acute coronary syndrome treated with statins. J Am Coll Cardiol. (2015) 65(21):2267–75. doi: 10.1016/j.jacc.2015.03.544
12. Raposeiras-Roubin S, Rosselló X, Oliva B, Fernández-Friera L, Mendiguren JM, Andrés V, et al. Triglycerides and residual atherosclerotic risk. J Am Coll Cardiol. (2021) 77(24):3031–41. doi: 10.1016/j.jacc.2021.04.059
13. Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HAW, Livingstone SJ, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the collaborative atorvastatin diabetes study (CARDS): multicentre randomised placebo-controlled trial. Lancet. (2004) 364(9435):685–96. doi: 10.1016/S0140-6736(04)16895-5
14. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. (2017) 376(18):1713–22. doi: 10.1056/NEJMoa1615664
15. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. (2018) 379(22):2097–107. doi: 10.1056/NEJMoa1801174
16. Ridker PM, Revkin J, Amarenco P, Brunell R, Curto M, Civeira F, et al. Cardiovascular efficacy and safety of bococizumab in high-risk patients. N Engl J Med. (2017) 376(16):1527–39. doi: 10.1056/NEJMoa1701488
17. Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. (2004) 350(15):1495–504. doi: 10.1056/NEJMoa040583
18. Pedersen TR, Faergeman O, Kastelein JJP, Olsson AG, Tikkanen MJ, Holme I, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. (2005) 294(19):2437–45. doi: 10.1001/jama.294.19.2437
19. LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. (2005) 352(14):1425–35. doi: 10.1056/NEJMoa050461
20. Sampson M, Ling C, Sun Q, Harb R, Ashmaig M, Warnick R, et al. A new equation for calculation of low-density lipoprotein cholesterol in patients with normolipidemia and/or hypertriglyceridemia. JAMA Cardiol. (2020) 5(5):540–8. doi: 10.1001/jamacardio.2020.0013
21. Walldius G, Jungner I, Holme I, Aastveit AH, Kolar W, Steiner E. High apolipoprotein B, low apolipoprotein A-I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): a prospective study. Lancet. (2001) 358(9298):2026–33. doi: 10.1016/S0140-6736(01)07098-2
22. Vallejo-Vaz AJ, Fayyad R, Boekholdt SM, Hovingh GK, Kastelein JJ, Melamed S, et al. Triglyceride-rich lipoprotein cholesterol and risk of cardiovascular events among patients receiving statin therapy in the TNT trial. Circulation. (2018) 138(8):770–81. doi: 10.1161/CIRCULATIONAHA.117.032318
23. Joshi PH, Khokhar AA, Massaro JM, Lirette ST, Griswold ME, Martin SS, et al. Remnant lipoprotein cholesterol and incident coronary heart disease: the Jackson heart and Framingham offspring cohort studies. J Am Heart Assoc. (2016) 5(5):e002765. doi: 10.1161/JAHA.115.002765
24. Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, et al. Association of triglyceride-lowering LPL variants and LDL-C–lowering LDLR variants with risk of coronary heart disease. JAMA. (2019) 321(4):364. doi: 10.1001/jama.2018.20045
25. Soran H, France MW, Kwok S, Dissanayake S, Charlton-Menys V, Younis NN, et al. Apolipoprotein B100 is a better treatment target than calculated LDL and non-HDL cholesterol in statin-treated patients. Ann Clin Biochem. (2011) 48(Pt 6):566–71. doi: 10.1258/acb.2011.010277
26. Quispe R, Martin SS, Michos ED, Lamba I, Blumenthal RS, Saeed A, et al. Remnant cholesterol predicts cardiovascular disease beyond LDL and ApoB: a primary prevention study. Eur Heart J. (2021) 42(42):4324–32. doi: 10.1093/eurheartj/ehab432
27. Björnson E, Adiels M, Taskinen MR, Burgess S, Rawshani A, Borén J, et al. Triglyceride-rich lipoprotein remnants, low-density lipoproteins, and risk of coronary heart disease: a UK biobank study. Eur Heart J. (2023) 44(39):4186–95. doi: 10.1093/eurheartj/ehad337
28. Bea AM, Cenarro A, Marco-Bened V, Laclaustra M, Martn C, Ibarretxe D, et al. Diagnosis of familial dysbetalipoproteinemia based on the lipid abnormalities driven by APOE2/E2 genotype. Clin Chem. (2023) 69(2):140–8. doi: 10.1093/clinchem/hvac213
29. de Graaf J, Hendriks JC, Demacker PN, Stalenhoef AF. Identification of multiple dense LDL subfractions with enhanced susceptibility to in vitro oxidation among hypertriglyceridemic subjects. Normalization after clofibrate treatment. Arterioscler Thromb. (1993) 13(5):712–9. doi: 10.1161/01.ATV.13.5.712
30. Durrington PN, Mackness MI, Bhatnagar D, Julier K, Prais H, Arrol S, et al. Effects of two different fibric acid derivatives on lipoproteins, cholesteryl ester transfer, fibrinogen, plasminogen activator inhibitor and paraoxonase activity in type IIb hyperlipoproteinaemia. Atherosclerosis. (1998) 138(1):217–25. doi: 10.1016/S0021-9150(98)00003-3
31. Charlton-Menys V, Durrington P. Apolipoproteins AI and B as therapeutic targets. J Intern Med. (2006) 259(5):462–72. doi: 10.1111/j.1365-2796.2006.01646.x
32. Durrington PN, Bhatnagar D, Mackness MI, Morgan J, Julier K, Khan MA, et al. An omega-3 polyunsaturated fatty acid concentrate administered for one year decreased triglycerides in simvastatin treated patients with coronary heart disease and persisting hypertriglyceridaemia. Heart. (2001) 85(5):544–8. doi: 10.1136/heart.85.5.544
33. Durrington PN. Triglycerides are more important in atherosclerosis than epidemiology has suggested. Atherosclerosis. (1998) 141 Suppl:S57–62. doi: 10.1016/S0021-9150(98)00219-6
34. Soran H, Adam S, Ho JH, Durrington PN. The Reaven syndrome: an historical perspective. Diab Vasc Dis Res. (2019) 16(2):116–7. doi: 10.1177/1479164119828899
35. Langlois MR, Sniderman AD. Non-HDL cholesterol or apoB: which to prefer as a target for the prevention of atherosclerotic cardiovascular disease? Curr Cardiol Rep. (2020) 22(8):67. doi: 10.1007/s11886-020-01323-z
36. Ballantyne CM, Pitt B, Loscalzo J, Cain VA, Raichlen JS. Alteration of relation of atherogenic lipoprotein cholesterol to apolipoprotein B by intensive statin therapy in patients with acute coronary syndrome [from the limiting UNdertreatment of lipids in ACS with rosuvastatin (LUNAR) trial]. Am J Cardiol. (2013) 111(4):506–9. doi: 10.1016/j.amjcard.2012.10.037
37. Patel RS, Pasea L, Soran H, Downie P, Jones R, Hingorani AD, et al. Elevated plasma triglyceride concentration and risk of adverse clinical outcomes in 1.5 million people: a CALIBER linked electronic health record study. Cardiovasc Diabetol. (2022) 21(1):102. doi: 10.1186/s12933-022-01525-5
38. Bashir B, Ho JH, Downie P, Hamilton P, Ferns G, Datta D, et al. Severe hypertriglyceridaemia and chylomicronaemia syndrome—causes, clinical presentation, and therapeutic options. Metabolites. (2023) 13(5):621. doi: 10.3390/metabo13050621
39. Langlois MR, Nordestgaard BG, Langsted A, Chapman MJ, Aakre KM, Baum H, et al. Quantifying atherogenic lipoproteins for lipid-lowering strategies: consensus-based recommendations from EAS and EFLM. Clin Chem Lab Med. (2020) 58(4):496–517. doi: 10.1515/cclm-2019-1253
40. Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J. (2016) 37(39):2999–3058. doi: 10.1093/eurheartj/ehw272
41. Virani SS, Morris PB, Agarwala A, Ballantyne CM, Birtcher KK, Kris-Etherton PM, et al. 2021 ACC expert consensus decision pathway on the management of ASCVD risk reduction in patients with persistent hypertriglyceridemia: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. (2021) 78(9):960–93. doi: 10.1016/j.jacc.2021.06.011
42. Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, et al. Evaluation and treatment of hypertriglyceridemia: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2012) 97(9):2969–89. doi: 10.1210/jc.2011-3213
43. Ginsberg HN, Packard CJ, Chapman MJ, Borén J, Aguilar-Salinas CA, Averna M, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. (2021) 42(47):4791–806. doi: 10.1093/eurheartj/ehab551
44. Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. (2014) 63(25 Pt B):2889–934. doi: 10.1016/j.jacc.2013.11.002
45. Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. (2007) 298(3):309–16. doi: 10.1001/jama.298.3.309
46. Langsted A, Nordestgaard BG. Nonfasting lipids, lipoproteins, and apolipoproteins in individuals with and without diabetes: 58,434 individuals from the Copenhagen general population study. Clin Chem. (2011) 57(3):482–9. doi: 10.1373/clinchem.2010.157164
47. Boquist S, Ruotolo G, Tang R, Björkegren J, Bond MG, de Faire U, et al. Alimentary lipemia, postprandial triglyceride-rich lipoproteins, and common carotid intima-media thickness in healthy, middle-aged men. Circulation. (1999) 100(7):723–8. doi: 10.1161/01.CIR.100.7.723
48. Teno S, Uto Y, Nagashima H, Endoh Y, Iwamoto Y, Omori Y, et al. Association of postprandial hypertriglyceridemia and carotid intima-media thickness in patients with type 2 diabetes. Diabetes Care. (2000) 23(9):1401–6. doi: 10.2337/diacare.23.9.1401
49. White KT, Moorthy MV, Akinkuolie AO, Demler O, Ridker PM, Cook NR, et al. Identifying an optimal cutpoint for the diagnosis of hypertriglyceridemia in the nonfasting state. Clin Chem. (2015) 61(9):1156–63. doi: 10.1373/clinchem.2015.241752
50. Mora S, Chang CL, Moorthy MV, Sever PS. Association of nonfasting vs fasting lipid levels with risk of Major coronary events in the anglo-scandinavian cardiac outcomes trial-lipid lowering arm. JAMA Intern Med. (2019) 179(7):898–905. doi: 10.1001/jamainternmed.2019.0392
51. Zilversmit DB. Atherogenesis: a postprandial phenomenon. Circulation. (1979) 60(3):473–85. doi: 10.1161/01.CIR.60.3.473
52. Groot PH, van Stiphout WA, Krauss XH, Jansen H, van Tol A, van Ramshorst E, et al. Postprandial lipoprotein metabolism in normolipidemic men with and without coronary artery disease. Arterioscler Thromb. (1991) 11(3):653–62. doi: 10.1161/01.ATV.11.3.653
53. Ginsberg HN, Jones J, Blaner WS, Thomas A, Karmally W, Fields L, et al. Association of postprandial triglyceride and retinyl palmitate responses with newly diagnosed exercise-induced myocardial ischemia in middle-aged men and women. Arterioscler Thromb Vasc Biol. (1995) 15(11):1829–38. doi: 10.1161/01.ATV.15.11.1829
54. Karpe F, de Faire U, Mercuri M, Gene Bond M, Hellénius ML, Hamsten A. Magnitude of alimentary lipemia is related to intima-media thickness of the common carotid artery in middle-aged men. Atherosclerosis. (1998) 141(2):307–14. doi: 10.1016/S0021-9150(98)00184-1
55. Peng J, Luo F, Ruan G, Peng R, Li X. Hypertriglyceridemia and atherosclerosis. Lipids Health Dis. (2017) 16(1):233. doi: 10.1186/s12944-017-0625-0
56. Toth PP. Triglycerides and atherosclerosis: bringing the association into sharper focus. J Am Coll Cardiol. (2021) 77:3042–5. doi: 10.1016/j.jacc.2021.04.058
57. Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK. Role of free fatty acids in endothelial dysfunction. J Biomed Sci. (2017) 24(1):50. doi: 10.1186/s12929-017-0357-5
58. Kwaifa IK, Bahari H, Yong YK, Noor SM. Endothelial dysfunction in obesity-induced inflammation: molecular mechanisms and clinical implications. Biomolecules. (2020) 10(2). doi: 10.3390/biom10020291
59. Sena CM. Endothelial dysfunction in type 2 diabetes: targeting inflammation. In: Carrilho F, editor. Rijeka: IntechOpen (2018). p. 10.
60. Wilck N, Ludwig A. Targeting the ubiquitin-proteasome system in atherosclerosis: status quo, challenges, and perspectives. Antioxid Redox Signal. (2014) 21(17):2344–63. doi: 10.1089/ars.2013.5805
61. Basu D, Bornfeldt KE. Hypertriglyceridemia and atherosclerosis: using human research to guide mechanistic studies in animal models. Front Endocrinol (Lausanne). (2020) 11:504. doi: 10.3389/fendo.2020.00504
62. Pal S, Semorine K, Watts GF, Mamo J. Identification of lipoproteins of intestinal origin in human atherosclerotic plaque. Clin Chem Lab Med. (2003) 41(6):792–5. doi: 10.1515/CCLM.2003.120
63. Nakano T, Nakajima K, Niimi M, Fujita MQ, Nakajima Y, Takeichi S, et al. Detection of apolipoproteins B-48 and B-100 carrying particles in lipoprotein fractions extracted from human aortic atherosclerotic plaques in sudden cardiac death cases. Clin Chim Acta. (2008) 390(1–2):38–43. doi: 10.1016/j.cca.2007.12.012
64. Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. (2007) 298(3):299–308. doi: 10.1001/jama.298.3.299
65. Freiberg JJ, Tybjaerg-Hansen A, Jensen JS, Nordestgaard BG. Nonfasting triglycerides and risk of ischemic stroke in the general population. JAMA. (2008) 300(18):2142–52. doi: 10.1001/jama.2008.621
66. Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. (1996) 3(2):213–9. doi: 10.1097/00043798-199604000-00014
67. Patel A, Barzi F, Jamrozik K, Lam TH, Ueshima H, Whitlock G, et al. Serum triglycerides as a risk factor for cardiovascular diseases in the Asia-pacific region. Circulation. (2004) 110(17):2678–86. doi: 10.1161/01.CIR.0000145542.24347.18
68. Jónsdóttir LS, Sigfússon N, Gudnason V, Sigvaldason H, Thorgeirsson G. Do lipids, blood pressure, diabetes, and smoking confer equal risk of myocardial infarction in women as in men? The Reykjavik study. J Cardiovasc Risk. (2002) 9(2):67–76. doi: 10.1097/00043798-200204000-00001
69. Sarwar N, Danesh J, Eiriksdottir G, Sigurdsson G, Wareham N, Bingham S, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 western prospective studies. Circulation. (2007) 115(4):450–8. doi: 10.1161/CIRCULATIONAHA.106.637793
70. Nichols GA, Philip S, Reynolds K, Granowitz CB, Fazio S. Increased residual cardiovascular risk in patients with diabetes and high versus normal triglycerides despite statin-controlled LDL cholesterol. Diabetes Obes Metab. (2019) 21(2):366–71. doi: 10.1111/dom.13537
71. Arca M, Veronesi C, D’Erasmo L, Borghi C, Colivicchi F, De Ferrari GM, et al. Association of hypertriglyceridemia with all-cause mortality and atherosclerotic cardiovascular events in a low-risk Italian population: the TG-REAL retrospective cohort analysis. J Am Heart Assoc. (2020) 9(19):e015801. doi: 10.1161/JAHA.119.015801
72. Lee H, Park JB, Hwang IC, Yoon YE, Park HE, Choi SY, et al. Association of four lipid components with mortality, myocardial infarction, and stroke in statin-naïve young adults: a nationwide cohort study. Eur J Prev Cardiol. (2020) 27(8):870–81. doi: 10.1177/2047487319898571
73. Després JP, Moorjani S, Tremblay A, Ferland M, Lupien PJ, Nadeau A, et al. Relation of high plasma triglyceride levels associated with obesity and regional adipose tissue distribution to plasma lipoprotein-lipid composition in premenopausal women. Clin Invest Med. (1989) 12(6):374–80. PMID: 2612090
74. Quispe R, Manalac RJ, Faridi KF, Blaha MJ, Toth PP, Kulkarni KR, et al. Relationship of the triglyceride to high-density lipoprotein cholesterol (TG/HDL-C) ratio to the remainder of the lipid profile: the very large database of lipids-4 (VLDL-4) study. Atherosclerosis. (2015) 242(1):243–50. doi: 10.1016/j.atherosclerosis.2015.06.057
75. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. (2020) 41(1):111–88. doi: 10.1093/eurheartj/ehz455
76. Rana JS, Liu JY, Moffet HH, Solomon MD, Go AS, Jaffe MG, et al. Metabolic dyslipidemia and risk of coronary heart disease in 28,318 adults with diabetes Mellitus and low-density lipoprotein cholesterol <100 mg/dl. Am J Cardiol. (2015) 116(11):1700–4. doi: 10.1016/j.amjcard.2015.08.039
77. Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. (2013) 61(4):427–36. doi: 10.1016/j.jacc.2012.08.1026
78. Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. (2013) 45(11):1345–52. doi: 10.1038/ng.2795
79. Sarwar N, Sandhu MS. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. (2010) 375(9726):1634–9. doi: 10.1016/S0140-6736(10)60545-4
80. Rosenson RS, Davidson MH, Hirsh BJ, Kathiresan S, Gaudet D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol. (2014) 64(23):2525–40. doi: 10.1016/j.jacc.2014.09.042 doi: 10.1126/science.1161524
81. Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science (1979). (2008) 322(5908):1702–5. doi: 10.1126/science.1161524
82. Tada H, Kawashiri MA, Nomura A, Yoshimura K, Itoh H, Komuro I, et al. Serum triglycerides predict first cardiovascular events in diabetic patients with hypercholesterolemia and retinopathy. Eur J Prev Cardiol. (2018) 25(17):1852–60. doi: 10.1177/2047487318796989
83. Murphy SA, Pedersen TR, Gaciong ZA, Ceska R, Ezhov MV, Connolly DL, et al. Effect of the PCSK9 inhibitor evolocumab on total cardiovascular events in patients with cardiovascular disease: a prespecified analysis from the FOURIER trial. JAMA Cardiol. (2019) 4(7):613–9. doi: 10.1001/jamacardio.2019.0886
84. Giugliano RP, Pedersen TR, Park JG, de Ferrari GM, Gaciong ZA, Ceska R, et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet. (2017) 390(10106):1962–71. doi: 10.1016/S0140-6736(17)32290-0
85. Hagström E, Steg PG, Szarek M, Bhatt DL, Bittner VA, Danchin N, et al. Apolipoprotein B, residual cardiovascular risk after acute coronary syndrome, and effects of alirocumab. Circulation. (2022) 146(9):657–72. doi: 10.1161/CIRCULATIONAHA.121.057807
86. Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJP, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. (2016) 316(22):2373–84. doi: 10.1001/jama.2016.16951
87. Puri R, Nissen SE, Shao M, Elshazly MB, Kataoka Y, Kapadia SR, et al. Non-HDL cholesterol and triglycerides. Arterioscler Thromb Vasc Biol. (2016) 36(11):2220–8. doi: 10.1161/ATVBAHA.116.307601
88. Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual inflammatory risk on treatment with PCSK9 inhibition and statin therapy. Circulation. (2018) 138(2):141–9. doi: 10.1161/CIRCULATIONAHA.118.034645
89. Varbo A, Benn M, Tybjærg-Hansen A, Nordestgaard BG. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. (2013) 128(12):1298–309. doi: 10.1161/CIRCULATIONAHA.113.003008
90. The Coronory Drugy Project Research Group. Clofibrate and niacin in coronary heart disease. JAMA. (1975) 231(4):360–81. doi: 10.1001/jama.1975.03240160024021
91. Report from the Committee of Principal Investigators. A co-operative trial in the primary prevention of ischaemic heart disease using clofibrate. Heart. (1978) 40:1069–118. doi: 10.1136/hrt.40.10.1069
92. Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, et al. Helsinki heart study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. N Engl J Med. (1987) 317(20):1237–45. doi: 10.1056/NEJM198711123172001
93. Robins SJ, Collins D, Wittes JT, Papademetriou V, Deedwania PC, Schaefer EJ, et al. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA. (2001) 285(12):1585–91. doi: 10.1001/jama.285.12.1585
94. Bezafibrate Infarction Prevention (BIP) study. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation. (2000) 102(1):21–7. doi: 10.1161/01.CIR.102.1.21
95. Diabetes Atherosclerosis Intervention Study Investigators. Effect of fenofibrate on progression of coronary-artery disease in type 2 diabetes: the diabetes atherosclerosis intervention study, a randomised study. Lancet. (2001) 357(9260):905–10. doi: 10.1016/S0140-6736(00)04209-4
96. Meade T, Zuhrie R, Cook C, Cooper J. Bezafibrate in men with lower extremity arterial disease: randomised controlled trial. Br Med J. (2002) 325(7373):1139. doi: 10.1136/bmj.325.7373.1139
97. Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9,795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. (2005) 366(9500):1849–61. doi: 10.1016/S0140-6736(05)67667-2
98. The ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes Mellitus. N Engl J Med. (2010) 362(17):1563–74. doi: 10.1056/NEJMoa1001282
99. Das Pradhan A, Glynn RJ, Fruchart JC, MacFadyen JG, Zaharris ES, Everett BM, et al. Triglyceride lowering with pemafibrate to reduce cardiovascular risk. N Engl J Med. (2022) 387(21):1923–34. doi: 10.1056/NEJMoa2210645
100. GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-prevenzione trial. Lancet. (1999) 354(9177):447–55. doi: 10.1016/S0140-6736(99)07072-5
101. Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. (2007) 369(9567):1090–8. doi: 10.1016/S0140-6736(07)60527-3
102. GISSI-HF Investigators. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. (2008) 372(9645):1223–30. doi: 10.1016/S0140-6736(08)61239-8
103. Kromhout D, Giltay EJ, Geleijnse JM. N–3 fatty acids and cardiovascular events after myocardial infarction. N Engl J Med. (2010) 363(21):2015–26. doi: 10.1056/NEJMoa1003603
104. Rauch B, Schiele R, Schneider S, Diller F, Victor N, Gohlke H, et al. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified Omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation. (2010) 122(21):2152–9. doi: 10.1161/CIRCULATIONAHA.110.948562
105. Galan P, Kesse-Guyot E, Czernichow S, Briancon S, Blacher J, Hercberg S. Effects of B vitamins and omega 3 fatty acids on cardiovascular diseases: a randomised placebo controlled trial. Br Med J. (2010) 341:c6273. doi: 10.1136/bmj.c6273
106. The ORIGIN Trial Investigators. N–3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. (2012) 367(4):309–18. doi: 10.1056/NEJMoa1203859
107. Writing Group for the AREDS2 Research Group. Effect of long-chain ω-3 fatty acids and lutein+zeaxanthin supplements on cardiovascular outcomes: results of the age-related eye disease study 2 (AREDS2) randomized clinical trial. JAMA Intern Med. (2014) 174(5):763–71. doi: 10.1001/jamainternmed.2014.328
108. The Risk and Prevention Study Collaborative Group. N–3 fatty acids in patients with multiple cardiovascular risk factors. N Engl J Med. (2013) 368(19):1800–8. doi: 10.1056/NEJMoa1205409
109. The ASCEND Study Collaborative Group. Effects of n−3 fatty acid supplements in diabetes mellitus. N Engl J Med. (2018) 379(16):1540–50. doi: 10.1056/NEJMoa1804989
110. Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, et al. Marine n−3 fatty acids and prevention of cardiovascular disease and cancer. N Engl J Med. (2018) 380(1):23–32. doi: 10.1056/NEJMoa1811403
111. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. (2018) 380(1):11–22. doi: 10.1056/NEJMoa1812792
112. Nicholls SJ, Lincoff AM, Garcia M, Bash D, Ballantyne CM, Barter PJ, et al. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the STRENGTH randomized clinical trial. JAMA. (2020) 324(22):2268–80. doi: 10.1001/jama.2020.22258
113. Esposito K, Marfella R, Ciotola M, Di Palo C, Giugliano F, Giugliano G, et al. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome. JAMA. (2004) 292(12):1440. doi: 10.1001/jama.292.12.1440
114. Georgoulis M, Chrysohoou C, Georgousopoulou E, Damigou E, Skoumas I, Pitsavos C, et al. Long-term prognostic value of LDL-C, HDL-C, lp(a) and TG levels on cardiovascular disease incidence, by body weight status, dietary habits and lipid-lowering treatment: the ATTICA epidemiological cohort study (2002–2012). Lipids Health Dis. (2022) 21(1):141. doi: 10.1186/s12944-022-01747-2
115. Mateo-Gallego R, Marco-Benedí V, Perez-Calahorra S, Bea AM, Baila-Rueda L, Lamiquiz-Moneo I, et al. Energy-restricted, high-protein diets more effectively impact cardiometabolic profile in overweight and obese women than lower-protein diets. Clin Nutr. (2017) 36(2):371–9. doi: 10.1016/j.clnu.2016.01.018
116. Kaur G, Mason RP, Steg PG, Bhatt DL. Omega-3 fatty acids for cardiovascular event lowering. Eur J Prev Cardiol. (2024) 31(8):1005–14. doi: 10.1093/eurjpc/zwae003
117. Bashir B, Iqbal Z, Schofield J, Soran H. In: Krents AJ, Chilton RJ, editors. Cardiovascular Endocrinology & Metabolism Theory and Practice of Cardiometabolic Medicine. 1st ed. Stacy Masucci (2023). p. 97–135.
118. Alhassan A, Young J, Lean MEJ, Lara J. Consumption of fish and vascular risk factors: a systematic review and meta-analysis of intervention studies. Atherosclerosis. (2017) 266:87–94. doi: 10.1016/j.atherosclerosis.2017.09.028
119. Rumawas ME, Meigs JB, Dwyer JT, McKeown NM, Jacques PF. Mediterranean-style dietary pattern, reduced risk of metabolic syndrome traits, and incidence in the Framingham offspring cohort. Am J Clin Nutr. (2009) 90(6):1608–14. doi: 10.3945/ajcn.2009.27908
120. The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. (2011) 365(24):2255–67. doi: 10.1056/NEJMoa1107579
121. HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. (2014) 371(3):203–12. doi: 10.1056/NEJMoa1300955
122. D’Andrea E, Hey SP, Ramirez CL, Kesselheim AS. Assessment of the role of niacin in managing cardiovascular disease outcomes. JAMA Netw Open. (2019) 2(4):e192224. doi: 10.1001/jamanetworkopen.2019.2224
123. Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res. (1996) 37(5):907–25. doi: 10.1016/S0022-2275(20)42003-6
124. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. (2019) 139(25):e1082–143. doi: 10.1016/j.jacc.2018.11.003
125. Oliver M. The clofibrate saga: a retrospective commentary. Br J Clin Pharmacol. (2012) 74(6):907–10. doi: 10.1111/j.1365-2125.2012.04282.x
126. Cottet J, Mathivat A, Redel J. Therapeutic study of a synthetic hypocholesterolemic agent: phenyl-ethyl-acetic acid. Presse Med. (1954) 62(44):939–41. PMID: 13194567
127. Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans affairs high-density lipoprotein cholesterol intervention trial study group. N Engl J Med. (1999) 341(6):410–8. doi: 10.1056/NEJM199908053410604
128. Manninen V, Tenkanen L, Koskinen P, Huttunen JK, Mänttäri M, Heinonen OP, et al. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki heart study. Implications for treatment. Circulation. (1992) 85(1):37–45. doi: 10.1161/01.CIR.85.1.37
129. Lee M, Saver JL, Towfighi A, Chow J, Ovbiagele B. Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: a meta-analysis. Atherosclerosis. (2011) 217(2):492–8. doi: 10.1016/j.atherosclerosis.2011.04.020
130. Bruckert E, Labreuche J, Deplanque D, Touboul PJ, Amarenco P. Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: a systematic review and meta-analysis. J Cardiovasc Pharmacol. (2011) 57(2):267–72. doi: 10.1097/FJC.0b013e318202709f
131. Keech AC, Mitchell P, Summanen PA, O’Day J, Davis TME, Moffitt MS, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. (2007) 370(9600):1687–97. doi: 10.1016/S0140-6736(07)61607-9
132. Emmerich KH, Poritis N, Stelmane I, Klindzane M, Erbler H, Goldsteine J, et al. Wirksamkeit und sicherheit von etofibrat bei patienten mit nicht proliferativer diabetischer retinopathie. Klin Monbl Augenheilkd. (2009) 226(07):561–7. doi: 10.1055/s-0028-1109516
133. Yamashita S, Masuda D, Matsuzawa Y. Pemafibrate, a new selective PPARα modulator: drug concept and its clinical applications for dyslipidemia and metabolic diseases. Curr Atheroscler Rep. (2020) 22(1):5. doi: 10.1007/s11883-020-0823-5
134. Humbert X, Dolladille C, Sassier M, Valnet-Rabier MB, Vial T, Guitton E, et al. Fibrates et risques thrombo-emboliques veineux: étude cas/non-cas dans la base nationale de pharmacovigilance. Therapies. (2017) 72(6):677–82. doi: 10.1016/j.therap.2017.07.002
135. Squizzato A, Galli M, Romualdi E, Dentali F, Kamphuisen PW, Guasti L, et al. Statins, fibrates, and venous thromboembolism: a meta-analysis. Eur Heart J. (2010) 31(10):1248–56. doi: 10.1093/eurheartj/ehp556
136. Delluc A, Tromeur C, le Moigne E, Nowak E, Mottier D, le Gal G, et al. Lipid lowering drugs and the risk of recurrent venous thromboembolism. Thromb Res. (2012) 130(6):859–63. doi: 10.1016/j.thromres.2012.08.296
137. Lacut K, le Gal G, Abalain JH, Mottier D, Oger E. Differential associations between lipid-lowering drugs, statins and fibrates, and venous thromboembolism: role of drug induced homocysteinemia? Thromb Res. (2008) 122(3):314–9. doi: 10.1016/j.thromres.2007.10.014
138. Kim KA, Kim NJ, Choo EH. The effect of fibrates on lowering low-density lipoprotein cholesterol and cardiovascular risk reduction: a systemic review and meta-analysis. Eur J Prev Cardiol. (2024) 31(3):291–301. doi: 10.1093/eurjpc/zwad331
139. Abourbih S, Filion KB, Joseph L, Schiffrin EL, Rinfret S, Poirier P, et al. Effect of fibrates on lipid profiles and cardiovascular outcomes: a systematic review. Am J Med. (2009) 122(10):962.e1–e8. doi: 10.1016/j.amjmed.2009.03.030
140. Koh KK, Quon MJ, Han SH, Chung WJ, Ahn JY, Seo YH, et al. Additive beneficial effects of fenofibrate combined with atorvastatin in the treatment of combined hyperlipidemia. J Am Coll Cardiol. (2005) 45(10):1649–53. doi: 10.1016/j.jacc.2005.02.052
141. Yamashita S, Hirano T, Shimano H, Tsukamoto K, Yoshida M, Yoshida H. Managing hypertriglyceridemia for cardiovascular disease prevention: lessons from the PROMINENT trial. Eur J Clin Invest. (2024). doi: 10.1111/eci.14227. [Epub ahead of print]38662591
142. Hirano T, Ito Y. The influence of triglycerides on small dense low-density lipoprotein cholesterol levels is attenuated in low low-density lipoprotein-cholesterol range: implications for the negative results of the PROMINENT trial. J Diabetes Investig. (2023) 14(7):902–6. doi: 10.1111/jdi.14013
143. Bang HO, Dyerberg J, Nielsen AB. Plasma lipid and lipoprotein pattern in Greenlandic West-Coast Eskimos. Lancet. (1971) 297(7710):1143–6. doi: 10.1016/S0140-6736(71)91658-8
144. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. (2015) 242(1):357–66. doi: 10.1016/j.atherosclerosis.2015.07.035
145. Jacobs ML, Faizi HA, Peruzzi JA, Vlahovska PM, Kamat NP. EPA And DHA differentially modulate membrane elasticity in the presence of cholesterol. Biophys J. (2021) 120(11):2317–29. doi: 10.1016/j.bpj.2021.04.009
146. Mason RP, Sherratt SCR, Jacob RF. Eicosapentaenoic acid inhibits oxidation of ApoB-containing lipoprotein particles of different size in vitro when administered alone or in combination with atorvastatin active metabolite compared with other triglyceride-lowering agents. J Cardiovasc Pharmacol. (2016) 68(1):33–40. doi: 10.1097/FJC.0000000000000379
147. Sherratt SCR, Mason RP. Eicosapentaenoic acid and docosahexaenoic acid have distinct membrane locations and lipid interactions as determined by x-ray diffraction. Chem Phys Lipids. (2018) 212:73–9. doi: 10.1016/j.chemphyslip.2018.01.002
148. Le VT, Knight S, Watrous JD, Najhawan M, Dao K, Mccubrey RO, et al. Higher docosahexaenoic acid levels lower the protective impact of eicosapentaenoic acid on long-term major cardiovascular events. FrontCardiovasc Med. (2023) 10.
149. American College of Cardiology. RESPECT-EPA: Highly Purified EPA Appears to Reduce Risks of CV Events in Japanese CAD Patients on Statins [Internet]. (2022). [cited 2022 Dec 21]. Available online at: https://www.acc.org/Latest-in-Cardiology/Articles/2022/11/01/22/00/sun-7pm-respect-epa-aha-2022
150. Daida H. Randomized trial for evaluation in secondary prevention efficacy of combination therapy–statin and eicosapentaenoic acid—RESPECT-EPA. American Heart Association Scientific Sessions; November 6, 2022; Chicago, IL.
151. American College of Cardiology. REDUCE-IT EPA Trial Shows Association Between Higher EPA Levels, Reduced CV Events [Internet]. (2020). [cited 2022 Dec 21]. Available online at: https://www.acc.org/latest-in-cardiology/articles/2020/03/24/16/41/mon-1045-eicosapentaenoic-acid-levels-in-reduce-it-acc-2020
152. Chan DC, Watts GF, Ng TWK, Yamashita S, Barrett PHR. Effect of weight loss on markers of triglyceride-rich lipoprotein metabolism in the metabolic syndrome. Eur J Clin Invest. (2008) 38(10):743–51. doi: 10.1111/j.1365-2362.2008.02019.x
153. Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. (2021) 384(11):989–1002. doi: 10.1056/NEJMoa2032183
154. Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. (2022) 387(3):205–16. doi: 10.1056/NEJMoa2206038
155. Bashir B, Adam S, Ho JH, Linn Z, Durrington PN, Soran H. Established and potential cardiovascular risk factors in metabolic syndrome: effect of bariatric surgery. Curr Opin Lipidol. (2023) 34(5):221–33. doi: 10.1097/MOL.0000000000000889
156. Sjöström CD, Lissner L, Wedel H, Sjöström L. Reduction in incidence of diabetes, hypertension and lipid disturbances after intentional weight loss induced by bariatric surgery: the SOS intervention study. Obes Res. (1999) 7(5):477–84. doi: 10.1002/j.1550-8528.1999.tb00436.x
157. Sjöström L, Narbro K, Sjöström CD, Karason K, Larsson B, Wedel H, et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med. (2007) 357(8):741–52. doi: 10.1056/NEJMoa066254
158. Heffron SP, Parikh A, Volodarskiy A, Ren-Fielding C, Schwartzbard A, Nicholson J, et al. Changes in lipid profile of obese patients following contemporary bariatric surgery: a meta-analysis. Am J Med. (2016) 129(9):952–9. doi: 10.1016/j.amjmed.2016.02.004
159. Adam S, Liu Y, Siahmansur T, Ho JH, Dhage SS, Yadav R, et al. Bariatric surgery as a model to explore the basis and consequences of the Reaven hypothesis: small, dense low-density lipoprotein and interleukin-6. Diab Vasc Dis Res. (2019) 16(2):144–52. doi: 10.1177/1479164119826479
160. Azmi S, Ferdousi M, Liu Y, Adam S, Iqbal Z, Dhage S, et al. Bariatric surgery leads to an improvement in small nerve fibre damage in subjects with obesity. Int J Obes (Lond). (2021) 45(3):631–8. doi: 10.1038/s41366-020-00727-9
161. Adam S, Ho JH, Liu Y, Siahmansur T, Siddals K, Iqbal Z, et al. Bariatric surgery-induced high-density lipoprotein functionality enhancement is associated with reduced inflammation. J Clin Endocrinol Metab. (2022) 107(8):2182–94. doi: 10.1210/clinem/dgac244
162. Yadav R, Hama S, Liu Y, Siahmansur T, Schofield J, Syed AA, et al. Effect of roux-en-Y bariatric surgery on lipoproteins, insulin resistance, and systemic and vascular inflammation in obesity and diabetes. Front Immunol. (2017) 8:1512. doi: 10.3389/fimmu.2017.01512
163. Brownrigg JRW, Hughes CO, Burleigh D, Karthikesalingam A, Patterson BO, Holt PJ, et al. Microvascular disease and risk of cardiovascular events among individuals with type 2 diabetes: a population-level cohort study. Lancet Diabetes Endocrinol. (2016) 4(7):588–97. doi: 10.1016/S2213-8587(16)30057-2
164. Azmi S, Ferdousi M, Liu Y, Adam S, Siahmansur T, Ponirakis G, et al. The role of abnormalities of lipoproteins and HDL functionality in small fibre dysfunction in people with severe obesity. Sci Rep. (2021) 11(1):12573. doi: 10.1038/s41598-021-90346-9
165. Iqbal Z, Bashir B, Ferdousi M, Kalteniece A, Alam U, Malik RA, et al. Lipids and peripheral neuropathy. Curr Opin Lipidol. (2021) 32(4):249–57. doi: 10.1097/MOL.0000000000000770
166. Adam S, Azmi S, Ho JH, Liu Y, Ferdousi M, Siahmansur T, et al. Improvements in diabetic neuropathy and nephropathy after bariatric surgery: a prospective cohort study. Obes Surg. (2021) 31(2):554–63. doi: 10.1007/s11695-020-05052-8
167. Reynolds EL, Watanabe M, Banerjee M, Chant E, Villegas-Umana E, Elafros MA, et al. The effect of surgical weight loss on diabetes complications in individuals with class II/III obesity. Diabetologia. (2023) 66(7):1192–207. doi: 10.1007/s00125-023-05899-3
168. The STABILITY Investigators. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med. (2014) 370(18):1702–11. doi: 10.1056/NEJMoa1315878
169. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
170. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. (2018) 380(8):752–62. doi: 10.1056/NEJMoa1809798
171. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381(26):2497–505. doi: 10.1056/NEJMoa1912388
172. Nidorf SM, Fiolet ATL, Eikelboom JW, Schut A, Opstal TSJ, Bax WA, et al. The effect of low-dose colchicine in patients with stable coronary artery disease: the LoDoCo2 trial rationale, design, and baseline characteristics. Am Heart J. (2019) 218:46–56. doi: 10.1016/j.ahj.2019.09.011
173. Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. (2011) 366(1):9–19. doi: 10.1056/NEJMoa1112277
174. Eikelboom JW, Connolly SJ, Bosch J, Dagenais GR, Hart RG, Shestakovska O, et al. Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med. (2017) 377(14):1319–30. doi: 10.1056/NEJMoa1709118
175. Bonaca MP, Bhatt DL, Cohen M, Steg PG, Storey RF, Jensen EC, et al. Long-Term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med. (2015) 372(19):1791–800. doi: 10.1056/NEJMoa1500857
176. Steg PG, Bhatt DL, Simon T, Fox K, Mehta SR, Harrington RA, et al. Ticagrelor in patients with stable coronary disease and diabetes. N Engl J Med. (2019) 381(14):1309–20. doi: 10.1056/NEJMoa1908077
177. Clinicaltrial.gov. Assessing the Impact of Lipoprotein (a) Lowering With Pelacarsen (TQJ230) on Major Cardiovascular Events in Patients With CVD (Lp(a)HORIZON) [Internet]. (2022). [cited 2022 Dec 21]. Available online at: https://clinicaltrials.gov/ct2/show/NCT04023552
178. Clinicaltrials.gov. Effect of Lipoprotein(a) Elimination by Lipoprotein Apheresis on Cardiovascular Outcomes (MultiSELECt) [Internet]. (2022). [cited 2022 Dec 21]. Available online at: https://clinicaltrials.gov/ct2/show/NCT02791802
179. Clinicaltrials.gov. A Study of LY3473329 in Adult Participants With Elevated Lipoprotein(a) at High Risk for Cardiovascular Events (KRAKEN) [Internet]. (2022). [cited 2022 Dec 21]. Available online at: https://clinicaltrials.gov/ct2/show/NCT05563246
180. ClinicalTrials.gov. Olpasiran Trials of Cardiovascular Events and Lipoprotein(a) Reduction (OCEAN(a))—Outcomes Trial [Internet]. (2024). [cited 2024 Feb 8]. Available online at: https://clinicaltrials.gov/study/NCT05581303
181. The HPS3/TIMI55–REVEAL Collaborative Group. Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. (2017) 377(13):1217–27. doi: 10.1056/NEJMoa1706444
182. Clinicaltrials.gov. Study to Investigate CSL112 in Subjects With Acute Coronary Syndrome (AEGIS-II) [Internet]. (2022). [cited 2022 Dec 21]. Available online at: https://clinicaltrials.gov/ct2/show/NCT03473223
183. ClinicalTrials.gov. Cardiovascular Outcome Study to Evaluate the Effect of Obicetrapib in Patients With Cardiovascular Disease (PREVAIL) [Internet]. (2024). [cited 2024 Feb 8]. Available online at: https://clinicaltrials.gov/study/NCT05202509
Keywords: hypertriglyceridaemia, cardiovascular risk, fibrates, omega-3 fatty acids, statins, atherosclerosis, residual risk
Citation: Bashir B, Schofield J, Downie P, France M, Ashcroft DM, Wright AK, Romeo S, Gouni-Berthold I, Maan A, Durrington PN and Soran H (2024) Beyond LDL-C: unravelling the residual atherosclerotic cardiovascular disease risk landscape—focus on hypertriglyceridaemia. Front. Cardiovasc. Med. 11:1389106. doi: 10.3389/fcvm.2024.1389106
Received: 20 February 2024; Accepted: 8 July 2024;
Published: 7 August 2024.
Edited by:
Mohamad Navab, UCLA Health System, United StatesReviewed by:
Peter Penson, Liverpool John Moores University, United KingdomChris Packard, University of Glasgow, United Kingdom
© 2024 Bashir, Schofield, Downie, France, Ashcroft, Wright, Romeo, Gouni-Berthold, Maan, Durrington and Soran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Handrean Soran, aGFuZHJlYW4uc29yYW5AbWZ0Lm5ocy51aw==; aHNvcmFuQGFvbC5jb20=