 Paul León1
Paul León1 Vanessa I. Romero
Vanessa I. Romero
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 13 June 2024
Sec. Cardiovascular Genetics and Systems Medicine
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1387063
This article is part of the Research Topic Real-World Data in Cardiovascular Medicine and Genomics View all 5 articles
This case report details the identification of a novel likely pathogenic splicing variant in the TTN gene, associated with dilated cardiomyopathy (DCM), in a 42-year-old male patient presenting with early-onset heart failure and reduced ejection fraction. DCM is a nonischemic heart condition characterized by left biventricular dilation and systolic dysfunction, with approximately one-third of cases being familial and often linked to genetic mutations. The TTN gene, encoding the largest human protein essential for muscle contraction and sarcomere structure, is implicated in about 25% of DCM cases through mutations, especially truncating variants. Our investigation revealed a previously unreported G > C mutation at the splice acceptor site in intron 356 of TTN, confirmed by Sanger sequencing and not found in population databases, suggesting a novel contribution to the understanding of DCM etiology. The case emphasizes the critical role of the TTN gene in cardiac function and the genetic complexity underlying DCM. A comprehensive literature review highlighted the prevalence and significance of splice variants in the TTN gene, particularly those affecting the titin A-band, which is known for its role in muscle contraction and stability. This variant's identification underscores the importance of genetic screening in patients with DCM, offering insights into the disease's familial transmission and potential therapeutic targets. Our findings contribute to the expanding knowledge of genetic factors in DCM, demonstrating the necessity of integrating genetic diagnostics in cardiovascular medicine. This case supports the growing evidence linking splicing mutations in specific regions of the TTN gene to DCM development and underscores the importance of genetic counseling and testing in managing heart disease.
Dilated cardiomyopathy (DCM—OMIM: 604145) is a nonischemic heart condition characterized by structural and functional abnormalities, primarily marked by left biventricular dilation and systolic dysfunction. Unlike other heart issues, DCM occurs without coronary artery disease, hypertension, valvular, or congenital abnormalities (1). Its etiology encompasses idiopathic factors, exposure to toxins, infections, or metabolic agents; idiopathic cases, lacking identified causes, are presumed to have a genetic contribution. Approximately one-third of cases are familial DCM (FDCM), necessitating genetic counseling, especially when relatives exhibit unexplained heart failure or premature sudden cardiac death (2). In adults, DCM follows an autosomal dominant inheritance pattern, characterized by incomplete penetrance and age-related variable expression (3). Clinically, manifestations vary, ranging from asymptomatic cases to symptoms like dyspnea at rest/exertion, fatigue, abdominal/ankle edema, and angina pectoris. DCM is associated with over 50 genes, with Titin (TTN) gene mutations being the most common, accounting for approximately 25% of cases, particularly truncating variants (4–7). These genetic insights are crucial for understanding the complex landscape of DCM and guiding appropriate management strategies.
The TTN gene encodes a virtual sarcomere component crucial for striated muscle contraction and holds the distinction of being the largest human protein, composed of 364 exons (7–9). TTN's extensive size supports sarcomere structure, aiding contraction (8), facilitating myofibrillogenesis by attaching vital proteins, and mediating cellular processes via its TK domain (9). Seven human TTN isoforms have been identified, comprising approximately a million splice variants theoretically capable of generating from its 364 exons. Among these, cardiomyocytes express three isoforms: N2BA and N2B in adults, and fetal cardiac titin in infants (8, 9).
We report a case involving a family with history of FDCM. Our investigation included panel sequencing, which revealed the presence of a novel likely pathogenic variant within the TTN gene, analyzing its impact will expand our understanding of splicing variants in the TTN gene. We conducted a thorough search in databases to identify splicing variants in the TTN gene that had been previously documented, categorized them based on the specific clinical characteristics they presented.
A 42-year-old male presented at our genetic outpatient clinic with early-onset heart failure and reduced ejection fraction. He was taking amlodipine, misoprostol, spironolactone, and forxiga. His medical history revealed chest pain during exercise at 18 years old, followed by the onset of dyspnea at 30. By the age of 35, the patient experienced chest pain again, accompanied by severe fluid retention, leading to a hospitalization lasting two weeks. He was treated with diuretics during his hospital stay and was subsequently discharged. However, he discontinued his medication once his condition improved. In June 2022, noticed fluid retention in his feet, a kidney ultrasound was normal and Contrast-Enhanced Urographic Computed Tomography reported prostatic hyperplasia. In November 2022, follow-up laboratory results showed elevated D-dimer (1,818.26 ng/ml), natriuretic peptide (1,843 ng/L), 24 h urine protein (271.25 mg/24 h), aspartate aminotransferase (AST) (57 U/L), total (5.50 mg/dl) direct (3.60 mg/dl) and indirect bilirubin and decreased creatinine clearance (39.18 ml/min). A transthoracic echocardiography revealed a Left Ventricular Ejection Fraction of 24.9%. Remodeling was observed with a tendency toward sphericity, along with a flow gram indicating a restrictive pattern, elevated filling pressures, and reduced systolic function in the right ventricle. The results indicated DCM with severe systolic and diastolic dysfunction of the left ventricle, along with slightly elevated filling pressures and significantly reduced global longitudinal strain. Additionally, the patient exhibited severe biatrial dilatation, mild mitral insufficiency, an intermediate likelihood of pulmonary hypertension, moderate tricuspid insufficiency, and right ventricular systolic dysfunction (Supplementary Figure S1). In December 2022, a 24 h blood pressure monitoring was conducted, revealing nocturnal diastolic arterial hypertension. The father of the patient died at age 40 due to congestive heart failure and the paternal grandfather died of endocarditis at age 54. The patient has 3 children with two different mothers (Figure 1) (Supplementary Figures S1, S2).
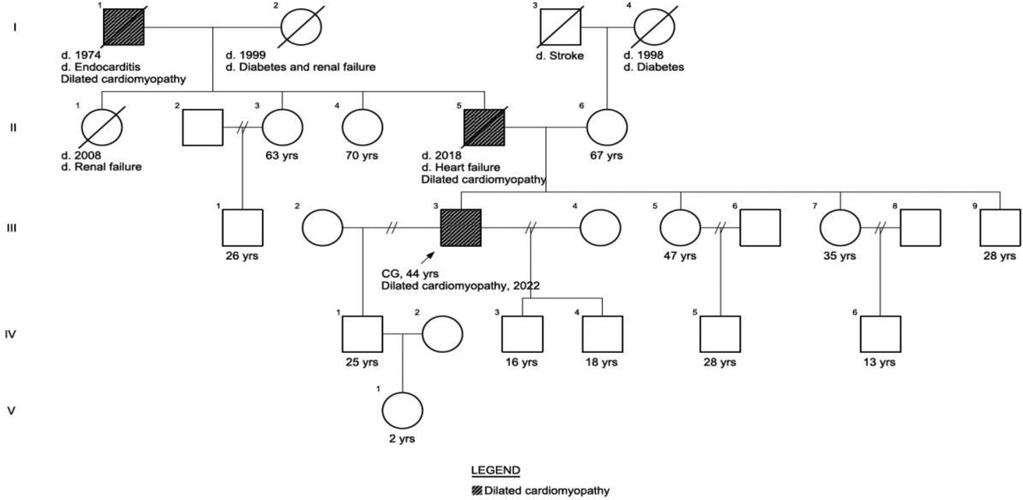
Figure 1 Patient phenotypical pedigree. The patient’s father of the patient died at age 40 due to congestive heart failure and the paternal grandfather died of endocarditis at age 54. The patient has 2 children with different mothers.
We ordered the Cardiomyopathy Comprehensive Panel that analyzes 121 genes and identified a (heterozygous) G > C c.100172-1 located in the splice acceptor; therefore, this variant affects the splicing process and consequently the damages the protein (Supplementary Figure S3). It is categorized as a likely pathogenic variant in intron 356 of TTN. This variant has not been reported in the literature in individuals affected with TTN-related conditions and is not present in population databases (gnomAD no frequency). The splice variant was confirmed by Sanger sequencing. We carried out Sanger sequencing on six family members and identified the variant in the patient's older sister and middle son. However, neither of them exhibited cardiac symptoms. We suggest that the variant exhibits no penetrance in the sister and recommend a cardiac evaluation for the middle son.
Our patient variant was reported as likely pathogenic; however, our findings suggest a pathogenic effect. The variant is not present in population databases (PS4 PM2 BA1 BS1 BS2), the domain is known to be critical to the protein function (PM1), there is evidence of pathogenicity in other studies (PM4 BP3), the patient has a well-defined clinical characterization including a possible consistent mode of inheritance- dominant (PP4).
We conducted a comprehensive literature review including reported cases of splice variants in TTN, focusing on adults with early-onset heart failure. After filtering, we identified 35 documented cases. Our findings align with what Patel et al. (10) suggested when they mentioned that mutations occurring in nucleotides flanking either the 5′ exon's end (donor site) or the 3′ exon's start (acceptor site) are characterized as likely pathogenic or pathogenic with mutations on the 3′ acceptor site being more prevalent. The splice donor site consists of nine base pairs that encode the invariant dinucleotide (GT) found at the 5′ end of nearly all introns. The splice acceptor site, spanning 23 base pairs, includes an invariant dinucleotide (AG). Mutations impacting the GT or AG dinucleotides within highly expressed exons are identified as likely pathogenic or pathogenic, leading to truncation variants of the TTN gene. Mutations occurring within the remaining seven base pairs of the donor site, or the 21 base pairs of the acceptor site may potentially disrupt splicing and are classified as variants of unknown significance (10). A significant proportion of pathogenic splice variants were identified within exons or introns located in the titin A-band (Figure 2). This region is characterized by a high frequency of constitutively expressed exons and consists of alternating fibronectin and immunoglobulin (Ig) regions, which serve as anchors for myosin binding during muscle contraction (8). However, most mutations that lead to DCM are found within the fibronectin type III domain of the A-band (11).
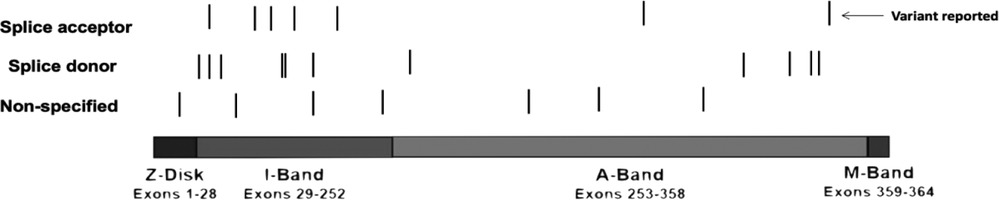
Figure 2 Graphical description of the TTN variants. We identified 35 reported cases of TTN variants in the splicing regions (Table 1) and constructed a visual representation of the reported variants along TTN. Splicing variants are the lines below the image, the black line means there was a variation reported in that specific location. TTN bands are shown—Z-disc, I-band, A-band, M-band. There are 1 variant located in the Z-disk, 14 variants in the I-band and 11 variants in the A-band. The arrow indicates our reported variant. Furthermore, for 2 variants reported the location its unknown and therefore, are not included in the graph.
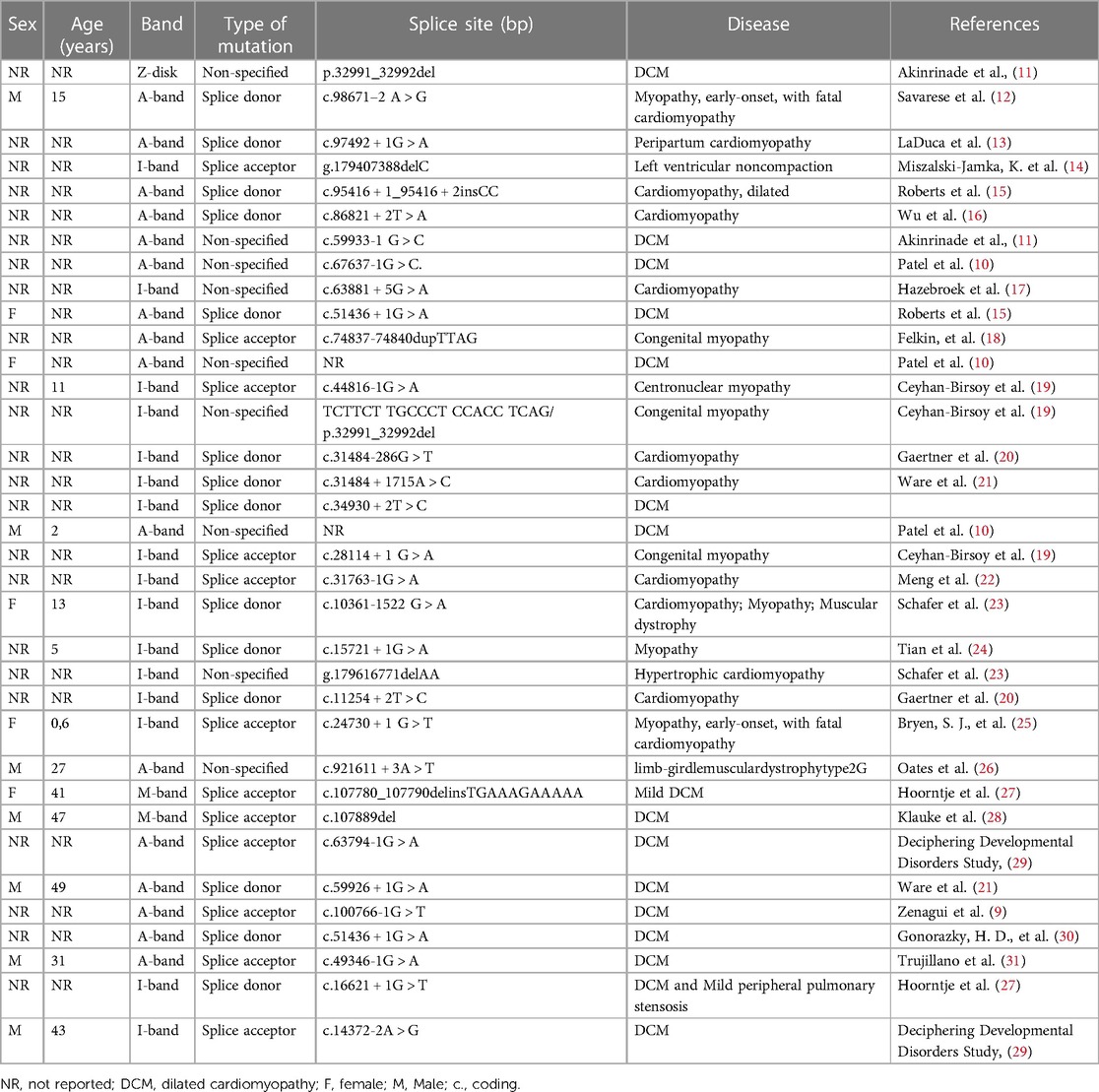
Table 1 TTN splice variants of 35 reported cases.
We present a case of a 42-year-old male patient with severe fluid retention, prostatic hyperplasia, and diastolic arterial hypertension. Genetic sequencing of this patient revealed a novel splice variant located in the A-band intron 356. Akinrinade et al. (11) compiled data from 1,000 Genomes Project, Exome Sequencing Project (ESP) and Exome Aggregation Consortium (ExAC) and found that TTN variants affecting splice sites are linked to early muscle weakness, reduced reflexes, and respiratory issues. These symptoms are similar to the ones exhibited by our patient. This occurs because these types of mutations disrupt exon acceptor sites, leaving transcripts unspliced and leading to truncated titin protein (11). Patel et al. (10) noted that individuals DCM frequently possess TTN variants located a few base pairs away from the donor or acceptor splice sites. In our study, the identified mutation is situated just one base pair from the acceptor splice site of a constitutively expressed exon (Table 1). In fact, exon 356 has a PSI score of 100% meaning that the proportion of TTN transcripts that incorporate this exon based is 100% in DCM, Even though some residues contribute to intron splice guiding, the nucleotide flanking either the 5′ exon at the donor sire or the start of the 3′ exon on the acceptor site are the most comonly mutated in patients with DCM (7). The acceptor site spans for about 23bp at the 3′ exon boundary which encapsulates the AG splice acceptor site. Multiple studies have reported that variants affecting the AG region of the acceptor site in exons with high PSI scores are often categorized as pathogenic TTN variants (2, 7, 15) Huang et al. (32) reported a similar case of DCM with a mutation in the A-band splice variant in exon 343 affecting all isoforms. Most of the TTN truncation variants including splicing leading to DCM exhibit a position-dependent impact with more severe symptoms with mutations closer to the C-terminus (33). Furthermore, TTN variants located in the I-band are more tolerated since many exons on this area are subject to alternative splicing (7, 33). Herman et al. (34) also noticed the absence of DCM-associated mutations in the Z and M-band subunits. Our case emphasizes the significance of the novel A-band splice variant underlining its role in DCM development and progression.
The TTN protein is the largest one in humans and is divided into four functional regions. These are the N-terminal Z-line which anchors to the sarcomeric Z-disk. Whereas the I-band is responsible for providing the protein's elastic properties since it consists of tandem Ig segments of linked Ig-like domains (7). The A-band is inextensible and binds myosin and myosin-binding protein C (MyBP-C) to provide a stabilizer role of the thick filament. Finally, the M-line, embedded in the C-terminal, expresses a kinase that regulate gene expression and cardiac remodeling (7).
DCM is a primary myocardial condition caused by a combination of genetic and non-genetic components with a variable clinical presentation. However, even the non-genetic types of DCM can potentially be influenced by the patient's genetic profile such as suffering from hypertension, valve disease, and inflammatory responses (35). Around 30% of DCM cases have been attributed to a familial genetic origin where mutations in the TTN protein have been shown to be the major cause for DCM (7). The TTN protein has a major role in cell structure, muscle elasticity and interacts with multiple protein partners which means that different mutations could have a significantly different impact leading to diverse clinical manifestations depending on each case (36). For instance, Roberts et al. (15) found that patients having TTN truncation mutations show lower stroke volume, thinner left ventricle walls and more severe impaired left ventricle function in comparison with non-familial cases of DCM. Moreover, patients with TTN truncations variants tend to suffer from sustained ventricular tachycardia (7) but exhibit a good response to medical treatment (37). Furthermore, in a multisite study of patients with DCM, Hofmeyer et al. (38) noted that patients with advanced DCM, meaning that they need a left ventricular assist device or a heart transplant, were twice as likely to carry a TTN truncation variant compared with non-advanced DCM patients. Consequently, the location where the mutation takes place in the TTN protein will determine the biological impact with variants located in the A-band being more likely to result in worst cases of DCM (7).
A limitation for this study was that, even though some databases such as the HGMD and PubMed show a considerable number of TTN splicing variants reported, once we look at every variation mentioned in papers, most of them are either missense or nonsense. This became an issue since the number of true TTN splicing variants reported is lower and we have less cases to compare with ours.
TTN, the largest human protein with significant heart expression, plays a role in elasticity, passive force during diastolic and systolic functions, and sarcomere assembly/length (36). Here, we describe the case of a patient with DCM harboring an intron 356 splice mutation in the TTN gene as the likely cause for his condition, accompanied by a literature review of splicing variants from PubMed and HGMD. Our results further support evidence that links splicing mutations in the A-band encoding region with the development of DCM in comparison with mutations in other regions of the TTN gene.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
This study was approved by the institutional review board of Universidad San Francisco de Quito (USFQ) (CODE 2018-217E) and conducted by the Declaration of Helsinki. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the patient. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
PL: Data curation, Formal Analysis, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. PF: Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. NH: Conceptualization, Data curation, Formal Analysis, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. KT: Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. AM: Conceptualization, Data curation, Visualization, Writing – original draft, Writing – review & editing. VR: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We would like to thank the family of the patient for their collaboration. We would also like to thank the university and the professors who provided us with the necessary knowledge and equipment to complete this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1387063/full#supplementary-material
1. Schultheiss H-P, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, et al. Dilated cardiomyopathy. Nat Rev Dis Primers. (2019) 5(1):32. doi: 10.1038/s41572-019-0084-1
2. Rosenbaum AN, Agre KE, Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol. (2020) 17:286–97. doi: 10.1038/s41569-019-0284-0
3. Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MRG. Genetic causes of dilated cardiomyopathy. Prog Pediatr Cardiol. (2014) 37(1-2):13–8. doi: 10.1016/j.ppedcard.2014.10.003
4. Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. (2010) 12(11):655–67. doi: 10.1097/GIM.0b013e3181f2481f
5. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic evaluation of cardiomyopathy-A heart failure society of America practice guideline. J Card Fail. (2018) 24(5):281–302. doi: 10.1016/j.cardfail.2018.03.004
6. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. (2016) 37(23):1850–8. doi: 10.1093/eurheartj/ehv727
7. Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MRG, et al. A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front Cardiovasc Med. (2016) 3:21. doi: 10.3389/fcvm.2016.00021
8. Tharp CA, Haywood ME, Sbaizero O, Taylor MRG, Mestroni L. The giant protein titin’s role in cardiomyopathy: genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Front Physiol. (2019) 10:1436. doi: 10.3389/fphys.2019.01436
9. Zenagui R, Lacourt D, Pegeot H, Yauy K, Morales RJ, Theze C, et al. A reliable targeted next-generation sequencing strategy for diagnosis of myopathies and muscular dystrophies, especially for the giant titin and nebulin genes. J Mol Diagn. (2018) 20(4):533–49. doi: 10.1016/j.jmoldx.2018.04.001
10. Patel PN, Ito K, Willcox JAL, Haghighi A, Jang MY, Gorham JM, et al. Contribution of noncanonical splice variants to TTN truncating variant cardiomyopathy. Circ Genomic Precis Med. (2021) 14(5):e003389. doi: 10.1161/CIRCGEN.121.003389
11. Akinrinade O, Koskenvuo JW, Alastalo TP. Prevalence of titin truncating variants in general population. PLoS One. (2015) 10(12):e0145284. doi: 10.1371/journal.pone.0145284
12. Savarese M, Vihola A, Oates EC, Barresi R, Fiorillo C, Tasca G, et al. Genotype-phenotype correlations in recessive titinopathies. Genet Med. (2020) 22(12):2029–40. doi: 10.1038/s41436-020-0914-2
13. LaDuca H, Farwell KD, Vuong H, Lu H-M, Mu W, Shahmirzadi L, et al. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PLoS One. (2017) 12(2):e0170843. doi: 10.1371/journal.pone.0170843
14. Miszalski-Jamka K, Jefferies JL, Mazur W, Głowacki J, Hu J, Lazar M, et al. Novel genetic triggers and genotype-phenotype correlations in patients with left ventricular noncompaction. Circ Cardiovasc Genet. (2017) 10(4):e001763. doi: 10.1161/CIRCGENETICS.117.001763
15. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. (2015) 7(270):270ra6. doi: 10.1126/scitranslmed.3010134
16. Wu L, Brady L, Shoffner J, Tarnopolsky MA. Next-generation sequencing to diagnose muscular dystrophy, rhabdomyolysis, and HyperCKemia. Can J Neurol Sci. (2018) 45(3):262–8. doi: 10.1017/cjn.2017.286
17. Hazebroek MR, Krapels I, Verdonschot J, van den Wijngaard A, Vanhoutte E, Hoos M, et al. Prevalence of pathogenic gene mutations and prognosis do not differ in isolated left ventricular dysfunction compared with dilated cardiomyopathy. Circ Heart Fail. (2018) 11(3):e004682. doi: 10.1161/CIRCHEARTFAILURE.117.004682
18. Felkin LE, Walsh R, Ware JS, Yacoub MH, Birks EJ, Barton PJ, et al. Recovery of cardiac function in cardiomyopathy caused by titin truncation. JAMA Cardiol. (2016) 1(2):234–5. doi: 10.1001/jamacardio.2016.0208
19. Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, Swanson LC, et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. (2013) 81(14):1205–14. doi: 10.1212/WNL.0b013e3182a6ca62
20. Gaertner A, Bloebaum J, Brodehl A, Klauke B, Sielemann K, Kassner A, et al. The combined human genotype of truncating TTN and RBM20 mutations is associated with severe and early onset of dilated cardiomyopathy. Genes (Basel). (2021) 12(6):2–4. doi: 10.3390/genes12060883
21. Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. (2016) 374(3):233–41. doi: 10.1056/NEJMoa1505517
22. Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. (2017) 171(12):e173438. doi: 10.1001/jamapediatrics.2017.3438
23. Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. (2017) 49:46–53. doi: 10.1038/ng.3719
24. Tian X, Liang W-C, Feng Y, Wang J, Zhang VW, Chou C-H, et al. Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS. Neurol Genet. (2015) 1(2):e14. doi: 10.1212/NXG.0000000000000015
25. Bryen SJ, Ewans LJ, Pinner J, MacLennan SC, Donkervoort S, Castro D, et al. Recurrent TTN metatranscript-only c.39974-11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat. (2020) 41(2):403–11. doi: 10.1002/humu.23938
26. Oates EC, Jones KJ, Donkervoort S, Charlton A, Brammah S, Smith JE 3rd, et al. Congenital titinopathy: comprehensive characterization and pathogenic insights. Ann Neurol. (2018) 83(6):1105–24. doi: 10.1002/ana.25241
27. Hoorntje ET, van Spaendonck-Zwarts KY, Te Rijdt WP, Boven L, Vink A, van der Smagt JJ, et al. The first titin (c.59926 + 1G > A) founder mutation associated with dilated cardiomyopathy. Eur J Heart Fail. (2018) 20(4):803–6. doi: 10.1002/ejhf.1030
28. Klauke B, Gaertner-Rommel A, Schulz U, Kassner A, Zu Knyphausen E, Laser T, et al. High proportion of genetic cases in patients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation. PLoS One. (2017) 12(12):e0189489. doi: 10.1371/journal.pone.0189489
29. Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. (2017) 542(7642):433–8. doi: 10.1038/nature21062
30. Gonorazky HD, Naumenko S, Ramani AK, Nelakuditi V, Mashouri P, Wang P, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare mendelian disease. Am J Hum Genet. (2019) 104(3):466–83. doi: 10.1016/j.ajhg.2019.01.012
31. Trujillano D, Bertoli-Avella AM, Kandaswamy KK, Weiss MER, Köster J, Marais A, et al. Clinical exome sequencing: results from 2,819 samples reflecting 1,000 families. Eur J Hum Genet. (2017) 25(2):176–82. doi: 10.1038/ejhg.2016.146
32. Huang S, Ma Y, Zhang Y, Xiong H, Chang X. Centronuclear myopathy due to a de novo nonsense variant and a maternally inherited splice-site variant in TTN: a case report. Clin Case Rep. (2021) 9(7):e04478. doi: 10.1002/ccr3.4478
33. Kellermayer D, Smith JE 3rd, Granzier H. Titin mutations and muscle disease. Pflugers Arch Eur J Physiol. (2019) 471(5):673–82. doi: 10.1007/s00424-019-02272-5
34. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. (2012) 366(7):619–28. doi: 10.1056/NEJMoa1110186
35. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. (2017) 121(7):731–48. doi: 10.1161/CIRCRESAHA.116.309396
36. Chauveau C, Rowell J, Ferreiro A. A rising titan: TTN review and mutation update. Hum Mutat. (2014) 35(9):1046–59. doi: 10.1002/humu.22611
37. Nomura S. Genetic and non-genetic determinants of clinical phenotypes in cardiomyopathy. J Cardiol. (2019) 73(3):187–90. doi: 10.1016/j.jjcc.2018.11.001
Keywords: dilated cardiomyopathy, TTN gene, Ecuador, splicing, truncated variants, case report
Citation: León P, Franco P, Hinojosa N, Torres K, Moreano A and Romero VI (2024) TTN novel splice variant in familial dilated cardiomyopathy and splice variants review: a case report. Front. Cardiovasc. Med. 11:1387063. doi: 10.3389/fcvm.2024.1387063
Received: 16 February 2024; Accepted: 3 May 2024;
Published: 13 June 2024.
Edited by:
Neil Morgan, University of Birmingham, United KingdomReviewed by:
Thiago Britto-Borges, Heidelberg University Hospital, Germany© 2024 León, Franco, Hinojosa, Torres, Moreano and Romero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanessa I. Romero, dnJvbWVyb0B1c2ZxLmVkdS5lYw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.