 Athina Dritsoula
Athina Dritsoula Carlotta Camilli
Carlotta Camilli Stephen E. Moss
Stephen E. Moss John Greenwood
John Greenwood- UCL Institute of Ophthalmology, University College London, London, United Kingdom
The establishment of new blood vessels, and their subsequent stabilization, is a critical process that facilitates tissue growth and organ development. Once established, vessels need to diversify to meet the specific needs of the local tissue and to maintain homeostasis. These processes are tightly regulated and fundamental to normal vessel and tissue function. The mechanisms that orchestrate angiogenesis and vessel maturation have been widely studied, with signaling crosstalk between endothelium and perivascular cells being identified as an essential component. In disease, however, new vessels develop abnormally, and existing vessels lose their specialization and function, which invariably contributes to disease progression. Despite considerable research into the vasculopathic mechanisms in disease, our knowledge remains incomplete. Accordingly, the identification of angiocrine and angiopathic molecules secreted by cells within the vascular microenvironment, and their effect on vessel behaviour, remains a major research objective. Over the last decade the secreted glycoprotein leucine-rich α-2 glycoprotein 1 (LRG1), has emerged as a significant vasculopathic molecule, stimulating defective angiogenesis, and destabilizing the existing vasculature mainly, but not uniquely, by altering both canonical and non-canonical TGF-β signaling in a highly cell and context dependent manner. Whilst LRG1 does not possess any overt homeostatic role in vessel development and maintenance, growing evidence provides a compelling case for LRG1 playing a pleiotropic role in disrupting the vasculature in many disease settings. Thus, LRG1 has now been reported to damage vessels in various disorders including cancer, diabetes, chronic kidney disease, ocular disease, and lung disease and the signaling processes that drive this dysfunction are being defined. Moreover, therapeutic targeting of LRG1 has been widely proposed to re-establish a quiescent endothelium and normalized vasculature. In this review, we consider the current status of our understanding of the role of LRG1 in vascular pathology, and its potential as a therapeutic target.
Introduction
The formation of a vascular network is a fundamental prerequisite in serving the needs of the surrounding tissue and enabling normal tissue function. The process of angiogenesis, whether during development or postnatally as during reproduction and wound healing, has been studied extensively and many of the controlling elements have been defined (1, 2). Vascular endothelial growth factor (VEGF) has emerged as the master regulator, but other factors also contribute to vessel growth and the subsequent stabilization and maturation processes, which are necessary for the establishment of functional vessels (3). The vasculature, however, is highly heterogeneous with vessel structure and function depending on position in the vascular hierarchy and on the physiological requirements of the surrounding tissue. To achieve such diverse functionality, different local signaling factors are required during development and throughout life to maintain this site-specific specialization. Nevertheless, some ubiquitous signaling interactions are deemed essential for vessel maturation, most notably the crosstalk between the endothelium and perivascular mural cells, which are critical to vascular homeostasis. Indeed, disruption of this cellular interplay is recognised as a major factor in the destabilization of existing vessels in several diseases.
Aside from the disruption of existing vessels, the formation of new vessels in disease has attracted enormous attention as they are usually abnormal, often forming a chaotic and immature network that may be poorly perfused, leaky, and fragile (2). Whilst many of the factors driving new vessel growth in disease are also those responsible for developmental and physiological angiogenesis, it is clear that differences must exist. Thus, it has been proposed that the balance of expression between pro- and anti-angiogenic factors may be disturbed, while additional disease-specific players may corrupt the normal angiogenic and maturation processes. Although our understanding of these factors, and the associated signaling pathways that disrupt both new and existing vascular structure and function is considerable, there remain significant gaps in our knowledge as highlighted by the large number of patients who fail to respond favourably to standard of care therapies aimed at alleviating vascular dysfunction (2, 4).
The interaction between the endothelium and perivascular mural cells is an imperative and extremely complex process that is central to normal blood vessel development and long-term homeostasis. Abnormalities in this relationship are implicated in numerous vascular diseases, such as diabetic retinopathy, pulmonary hypertension, kidney disease and cancer, where decreased mural cell coverage or abnormal recruitment results in destabilized leaky microvessels and vascular rarefication (5–9). In addition, activation or de-differentiation of vascular smooth muscle cells and pericytes can lead to vascular remodeling and medial thickening in larger vessels (5). Also emerging is the disruptive role played by endothelial to mesenchymal transition (EndMT) (10), a process similar to epithelial to mesenchymal transition (EMT), in which TGF-β plays a predominant role. Irrespective of cause, when vessels become dysfunctional, they not only fail to provide their selective barrier and delivery functions but can also negatively influence the behaviour of surrounding tissue. Designing therapeutic strategies, therefore, that aim to normalize the vasculature has received increasing attention, especially in cancer (11), but further insight into our understanding of interactions that drive vascular dysfunction in disease is needed. Through such improved insight, new therapeutic targets will be identified for which activation or inhibition will enable repair of dysfunctional vessels and the facilitation of physiological revascularization during disease. One recently identified candidate, the angiocrine factor leucine-rich α-2 glycoprotein 1 (LRG1), is emerging as a ubiquitous and potentially critical player in driving vascular pathology, and therefore offers substantial potential as a new therapeutic target in many different diseases. This review aims to present the diverse and emerging vasculopathic roles that LRG1 has on the vasculature and its influence on the local microenvironment.
LRG1 in homeostasis and disease
LRG1 is a member of the conserved family of leucine-rich repeat glycoproteins that are involved in numerous physiological functions, including protein-protein interactions and innate immune responses (12, 13). Under normal conditions LRG1 is expressed almost exclusively by liver hepatocytes, from where it is secreted into the circulation and maintained at an approximate concentration of 20–50 μg/ml in the plasma (14) (Figure 1). Neutrophils, which are powerful mediators of innate immunity, are also known to express LRG1 constitutively and are likely to contribute to LRG1-mediated pathogenic effects in inflamed tissues and diseased vasculature (Figure 1). In these cells, LRG1 is stored within granules, and its expression has been associated with accelerated neutrophilic granulopoiesis, suggesting a potential role for LRG1 in myeloid cell differentiation (15, 16). Moreover, LRG1 has been shown to be involved in the formation of neutrophil extracellular traps (NETs), a process known as NETosis, via a signaling pathway involving ALK5 and AKT (17). NETosis is an innate immune response initiated against pathogens but is also frequently associated with vascular dysfunction (18). These findings, together with increased LRG1 levels in serum in response to microbial or viral infections and other inflammatory stimuli, as well as the high binding affinity of LRG1 for cytochrome c (Cyt c) following its release upon apoptotic signals, have led to the proposal that LRG1 functions as an acute phase protein involved in the innate immune response (19, 20). Aside from these major sources of LRG1, low levels of expression have been reported under normal conditions in other tissues including lung, kidney, heart, brain, testis, and the vasculature, where LRG1 is predominantly expressed in endothelial cells (21) (Figure 1). Many of these studies, however, use immunohistochemical detection, which might reflect the presence of extracellular, and matrix/endothelial cell sequestered LRG1 derived from the circulation. Nevertheless, a recent transcriptomics study dissecting the molecular heterogeneity of blood vascular endothelial cells from skin has shown that LRG1 transcript is exclusively found in the blood vessels and more specifically in healthy skin enriched in the postcapillary venules (22).
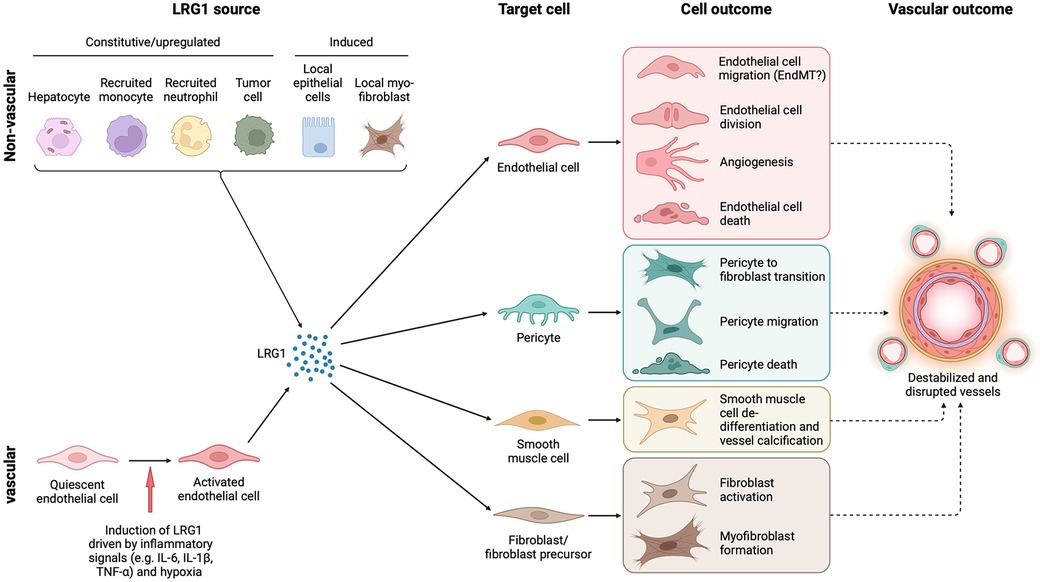
Figure 1. Cell source, target cells and outcome of LRG1 in the vasculature. LRG1 is expressed constitutively by only a few cell types, including hepatocytes and neutrophils, but during disease can be induced in various other cells. In the vasculature, endothelial cells are the only source of LRG1, but other local cells may also be induced to express LRG1 such as fibroblasts and tumor cells. Irrespective of the source, LRG1 affects the function of all cellular components of blood vessels resulting in vascular damage. Vascular dysfunction is closely associated with fibrosis, and both may influence each other through LRG1 signaling. EndMT, endothelial to mesenchymal transition. Created with BioRender.com.
Although current evidence suggests that LRG1 expression may be important to the functions of different organs and processes, its primary physiological role remains poorly defined, with Lrg1-deficient mice presenting with normal development and fertility, and no obvious phenotype indicating a non-essential role. In recent years, however, accumulating evidence points to the involvement of LRG1 in a wide range of diseases (23, 24). Since 2013, when LRG1 was initially identified as a key vasculopathic molecule in abnormal angiogenesis (25), more causative roles have been attributed. Among others, LRG1 has been found to drive IL-6-dependent pathological angiogenesis through the STAT3 signaling pathway (26, 27), destabilize tumor vasculature and promote tumor growth and metastasis (28–32), drive chronic kidney and lung disease (33–40), stimulate fibrosis (33, 34, 41–48), contribute to diabetes-related pathology (17, 49–57), and regulate pathological placental angiogenesis (58). High circulating LRG1 levels have been reported in many cancers, where LRG1 has been proposed, or used, as a prognostic and diagnostic marker. Indeed, high LRG1 levels in cancer patients' plasma correlate with poor prognosis and survival (59–61), and resistance to standard of care therapy (62). The involvement and roles of LRG1 in key vascular processes are the scope of this review and will be discussed in detail.
Regulation of LRG1 expression
Several transcriptomic and post-transcriptional regulatory studies have shown that the IL-6/STAT3 signaling pathway is a major activator of LRG1 expression (26, 29, 31, 63). Indeed, it was recently reported that LRG1 transcriptional activation is abolished upon deletion of the STAT3 binding site on the LRG1 promoter (26). Inflammation seems to be an important driver of LRG1 expression, with many different cytokines activating LRG1 in a range of cells and disease settings (Figure 2). These include IL-6, IL-1β, TNF-α, IL-17, IL-4, IL-10 and IL-33 and they may act either alone or synergistically (36, 64–68). In addition, PPARβ/δ is able to bind the LRG1 promoter to activate transcription as reported in a chromatin immunoprecipitation assay (47), and similarly, both ELK1 and ELK4 transcription factors have been shown to initiate LRG1 transcription upon mechanical strain (41) and through cooperation with Sp1/Sp3 complex (69), respectively. FOS-like 1 was also identified as a novel activator of LRG1 in a transcriptomics study (39). There is also evidence that hypoxia can induce LRG1 expression (70), consistent with the presence of potentially active HIF-1α binding elements in the LRG1 promoter. At the post-transcriptional and post-translational levels, microRNAs, long non-coding RNAs, and histone modifications have also been reported to regulate LRG1 expression (23) (Figure 2). Furthermore, different glycosylation patterns have been identified and these might influence LRG1 function (23). Overall, while experimental evidence that unravels the pleiotropic functions of LRG1 continues to accumulate, further work is required to establish the key regulatory mechanisms that control LRG1 expression and function, some of which will be discussed in detail later.
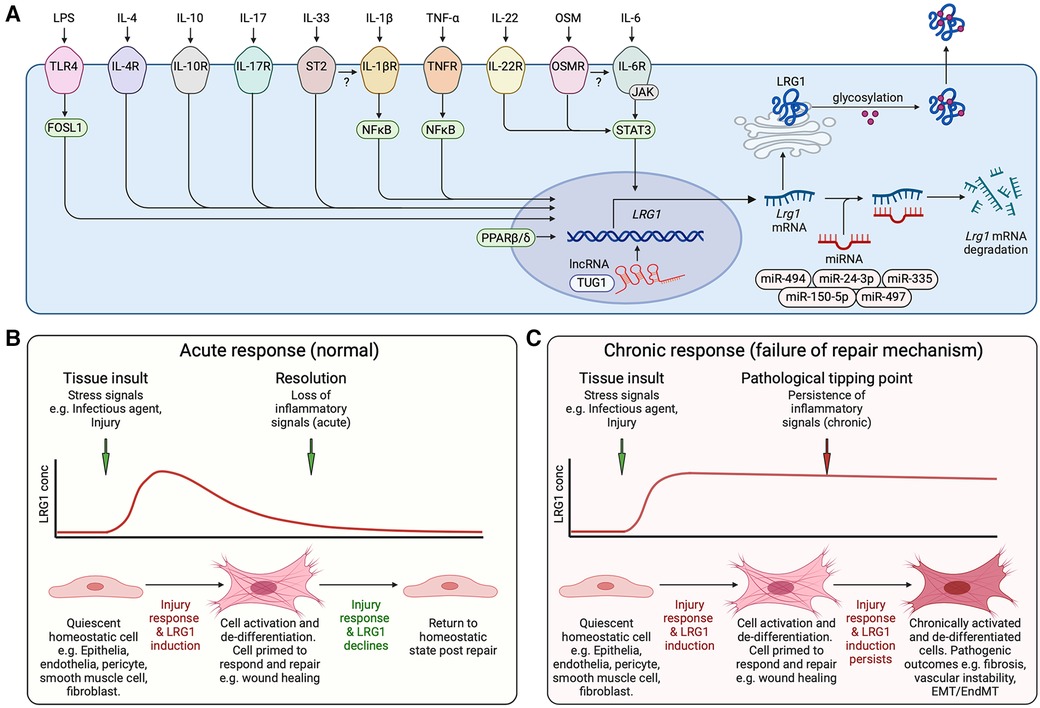
Figure 2. Induction of LRG1. (A) Schematic representation of inflammatory factors reported to activate LRG1 gene expression and known transcriptional and post-transcriptional regulatory mechanisms. Cytokines and lipopolysaccharide (LPS) acting through their cognate receptors and downstream transcription factors drive LRG1 gene induction. lncRNA TUG1 facilitates LRG1 transcription while miR-335, miR-494, miR-497, miR-150-5p and miR-24-3p promote the degradation of LRG1 mRNA. LRG1 protein is differentially glycosylated in a cell- and function-specific manner that may affect its activity. The secreted protein may combine to form dimers and trimers. OSM, oncostatin M; IL, interleukin; TLR4, toll-like receptor 4; lncRNA, long non-coding RNA; miRNA, microRNA. (B) LRG1 is induced during cell damage and infection as part of the repair mechanism. In the presence of LRG1 and TGF-β, we propose that cells are activated and de-differentiated into a less mature state to enable wound healing. As inflammatory signals subside, LRG1 expression diminishes allowing resolution of tissue damage and cells to return to a quiescent state. (C) Under chronic conditions, inflammatory signals maintain LRG1 expression perpetuating the unstable cell state and preventing completion of the healing process. EMT, epithelial to mesenchymal transition; EndMT, endothelial to mesenchymal transition. Created with BioRender.com.
LRG1 and angiogenesis
In the early stages of development, blood vessels are formed through a tightly orchestrated process called vasculogenesis, where mesoderm-derived angioblasts, the precursor of endothelial cells, establish angioblastic cords that advance into a primitive vascular plexus and subsequently into blood vessels. At later stages of development, and postnatally in the reproductive system and during physiological tissue repair, angiogenesis occurs in response to tissue growth and hypoxia. The angiogenic mechanism is tightly regulated and influenced by pro-angiogenic factors, with VEGF being a key master regulator, essential for the development and maintenance of the vascular system (3). Other growth factors with established pro-angiogenic activities include the FGFs, PDGF, angiopoietins, HGF and the TGF-β superfamily. In particular, PDGF and the angiopoietins exert their functions on the endothelium in a paracrine manner through their binding to, or their expression by, perivascular mural cells. In addition, angiopoietins regulate vascular homeostasis and promote vessel stability through the recruitment of mural cells and mediate their interactions with the endothelium (71). Among other potent angiogenic molecules are angiopoietin-like 4 (72), apelin (73), Frizzled A (74, 75), thrombomodulin (76), AGGF1 (77), Slit3 (78), as well as a plethora of pro-angiogenic peptides, the therapeutic potential of which is reviewed elsewhere (79). Aside from the established regulators of angiogenesis, there are many ancillary factors that influence vessel growth and maturation and which, under certain circumstances, may even circumvent the angiogenic dependency on VEGF. As will be seen in this review, LRG1 is now considered to be one of these adjunct pro-angiogenic factors and hence can be added to this extensive list. However, contrary to many of the factors mentioned above, LRG1 does not play a role in developmental or physiological angiogenesis as knock-out mice exhibit no overt phenotype, breed successfully and live a normal life span (21). In the context of disease, however, LRG1 is now recognised as a potent pro-neoangiogenic factor as it has been shown to promote pathological angiogenesis in numerous diseases and de novo growth in in vitro experimental settings (Table 1). It is worth noting, however, that angiogenesis under in vitro/ex vivo conditions is unlikely to represent normal angiogenesis, as the environment is more aligned to a pathogenic than homeostatic state. As outlined in more detail in the sections below, and with the caveat above, several studies have investigated the pro-angiogenic properties of LRG1 and have shown that it can act directly on endothelial cells, inducing cell proliferation and tube formation in vitro as well as on other cells impacting proliferation, migration and viability (17, 27, 34, 41, 80–84, 86). In addition, LRG1 promotes vessel growth and sprouting in ex vivo mouse metatarsal bone and aortic ring angiogenesis assays (25, 26). This is attenuated in explant tissues derived from Lrg1−/− mice or when a function-blocking anti-LRG1 antibody is used (25, 26, 84, 94). Similarly, knocking down LRG1 through siRNA silencing decreased the tube formation capacity of HUVEC co-cultured with pancreatic cancer cells (81). Vessels that develop in response to LRG1 stimulation, in the absence of other exogenous drivers, exhibit a pathological phenotype. In particular, LRG1 treatment of metatarsal bones and aortic rings resulted in vessels with reduced coverage of αSMA+ and NG2+ mural cells (26). Although the mechanisms behind this remain unknown, it seems likely that both autocrine and paracrine mechanisms affect the recruitment, proliferation, migration and differentiation state of mural cells in a LRG1-dependent manner. These studies raise the question whether LRG1 is a true angiogenic factor or whether it affects the stability of existing vessels permitting them to be more permissive to other angiogenic stimuli. Indeed, similar to VEGF, LRG1 can destabilize existing vessels and this may promote the angiogenic process. In addition, as LRG1 is ubiquitously present in the circulation it remains unclear what the relative contribution of systemic LRG1 vs. locally produced LRG1 is to vascular pathology. As with TGF-β, the effect of LRG1 on cell function is dependent on multiple factors one of which appears to be its local concentration, indicating a possible threshold effect achieved through local production. Moreover, the differential effect of luminal vs. abluminal exposure has yet to be determined as this may also dictate outcome.
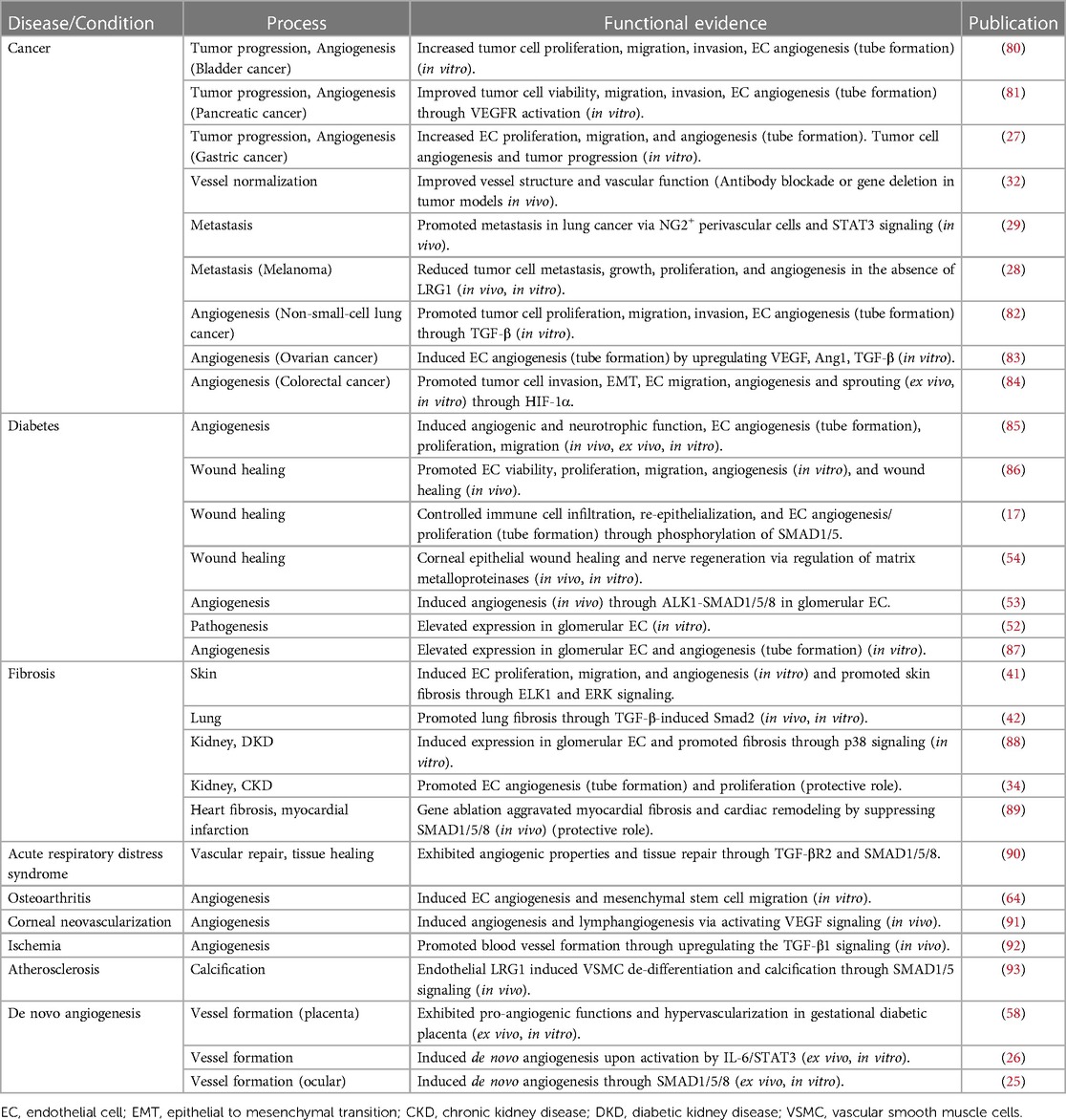
Table 1. Evidence for the functional role of LRG1 in disease pathogenesis and vascular processes.
The data above support the idea that in contrast to many angiogenic factors with homeostatic roles in the maintenance of a healthy vasculature, LRG1 expression is induced locally in response to pathogenic cues to drive the formation of a destabilized vasculature (Figure 3). These cues include inflammatory signals, hypoxia and vessel destabilizing factors that not only induce LRG1 expression but may also act on the vasculature independently creating an overall disruptive milieu. Acute inflammation, for example, causes dysfunction to existing vessels and often precedes neovascularization. Indeed, when inflammation becomes chronic and unresolved, it can promote the sustained activation of downstream mechanisms that trigger endothelial dysfunction, vascular remodeling, and abnormal neovascularization all of which serve as major risk factors for the development of cardiovascular disease (95). Our studies, and other recent evidence, demonstrate that endothelial cells themselves represent a key source of LRG1 in the diseased milieu (Figure 1). For instance, single cell RNA sequencing revealed Lrg1 as one of the most upregulated genes in disease-associated endothelial cells in murine models of liver cancer (96) and atherosclerosis (97), while a seminal study demonstrated that LRG1 originating from endothelial cells contributes primarily, in an autocrine fashion, to vessel malfunction and disease severity in a model of emphysema (37). Interestingly, transcriptomics studies also showed higher Lrg1 expression in endothelial cells isolated from LPS-treated lungs, pointing to inflammation-induced LRG1 being responsible for aberrant vasculogenesis (39). Similarly, Lrg1 was found upregulated in diseased endothelium from inflamed retinal vessels in a mouse model of experimental autoimmune uveitis, suggesting a role for LRG1 in retinal inflammation and angiogenesis (98).
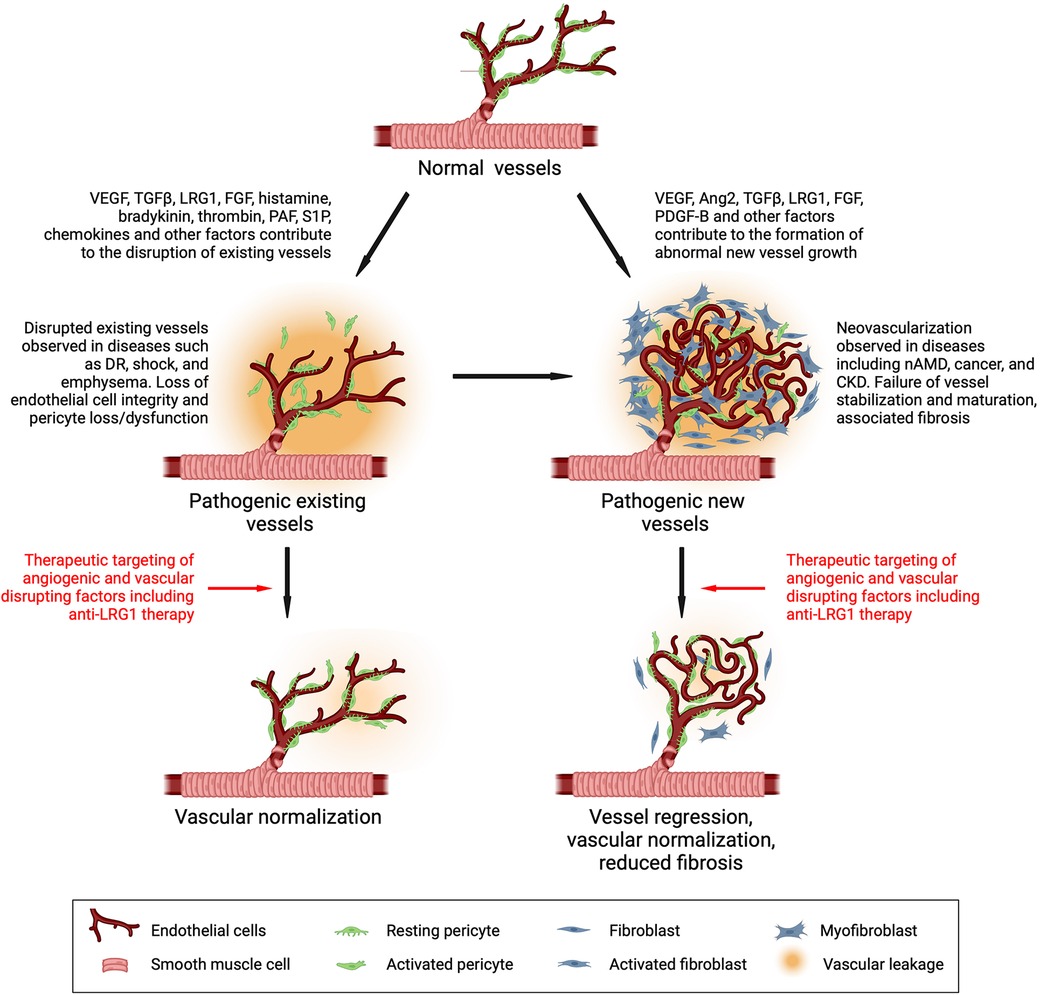
Figure 3. Drivers of vascular dysfunction and its therapeutic reversal. Vessels under normal conditions are stabilized and maintained in a mature functional state through cross-talk between the endothelial cell and mural cells (pericytes and smooth muscle cells). During disease, vasoactive factors are released that destabilize existing vessels and stimulate new dysfunctional vessel growth, both of which contribute to disease progression. Targeting factors that alter vascular function in disease is a common therapeutic objective. Inhibiting LRG1 activity has been shown to normalize vessels and may therefore be a promising therapeutic approach. DR, diabetic retinopathy; nAMD, neovascular age-related macular degeneration; CKD, chronic kidney disease. Created with BioRender.com.
Abnormal angiogenesis is associated with numerous conditions, and the role of LRG1 in this pathology has become evident. Thus, as discussed in more detail below, it has been reported to be involved in neovascular dysfunction in ocular disease, diabetes, kidney disease, lung disease, impaired wound healing, inflammatory conditions, gestational diabetes and cancer. In addition, LRG1 may also contribute to destabilization of existing vessels by impacting on endothelial cell adhesion and permeability (Figure 3). Such emerging evidence highlights the potential of targeting LRG1 with novel therapeutic strategies to attenuate vascular disruption in disease. In support of this, and as highlighted elsewhere in this review, deletion of the Lrg1 gene or blocking LRG1 function has been shown to partially reverse the vascular pathology in many conditions. Thus, therapeutic blockade of the angiopathic properties of LRG1 will therefore not only permit vessels to revascularize tissue in a more physiological manner but has the potential to re-establish a quiescent endothelial state in existing disrupted vessels.
In order to devise therapeutic strategies targeting LRG1, it is important to understand the mode of action through which LRG1 promotes defective angiogenesis or impairs the established vasculature. To date, it is evident that TGF-β signaling is a key downstream mediator of LRG1 activity. In endothelial cells, TGF-β signals through binding to the TGF-βRII receptor followed by initiation of signaling either via the ALK1 or ALK5 kinase. In homeostasis, TGF-β signaling maintains endothelial cell quiescence by regulating ALK5 and the downstream SMAD2/3 transcription factors (99). However, high levels of LRG1 can shift the balance of endothelial TGF-β signaling towards the ALK1 kinase, which associates with Endoglin (ENG) to activate the pro-angiogenic SMAD1/5/8 arm of transcription factors leading to endothelial cell proliferation, migration and pathological angiogenesis (25, 26, 53, 99) (Figure 4). Indeed, ENG has been proposed as being essential to allow the interactions between LRG1, TGF-β and ALK1 (25, 100) but evidence from other cell types demonstrates that it can still elicit TGF-βRII signaling in the absence of ENG. Nevertheless, this so called TGFβ angiogenic switch is not a binary response as outcome is more likely due to the relative balance between these two canonical signaling arms that dictates the context-dependent nuanced response. Moreover, LRG1 also activates the non-canonical TGF-β pathway in various cells and settings (23), which may influence vascular structure and function (Figure 4). Merely promoting the pro-angiogenic TGF-β switch, therefore, is unlikely to fully explain the complex angiopathic effects of LRG1. There also exists the distinct possibility that LRG1 can modify BMP signaling although this remains to be determined. Outside the TGF-β axis, an alternative mechanism has been noted, whereby LRG1 binds to the Latrophilin-2 receptor to promote LRG1-dependent angioneurin effects in diabetes, where angiogenic and neurotrophic processes are in place (85). Additionally, recent studies have provided evidence that LRG1 disrupts vascular homeostasis by altering the fine balance between endothelial cells and pericytes causing leaky and destabilized vessels (26, 32). How this imbalance is achieved needs to be further explored, but autocrine and paracrine signaling mechanisms between the endothelium and pericytes appear to be critical. Those mechanisms may involve the release of angiocrine factors including Ang1 and Ang2, or LRG1-driven signaling in pericytes.
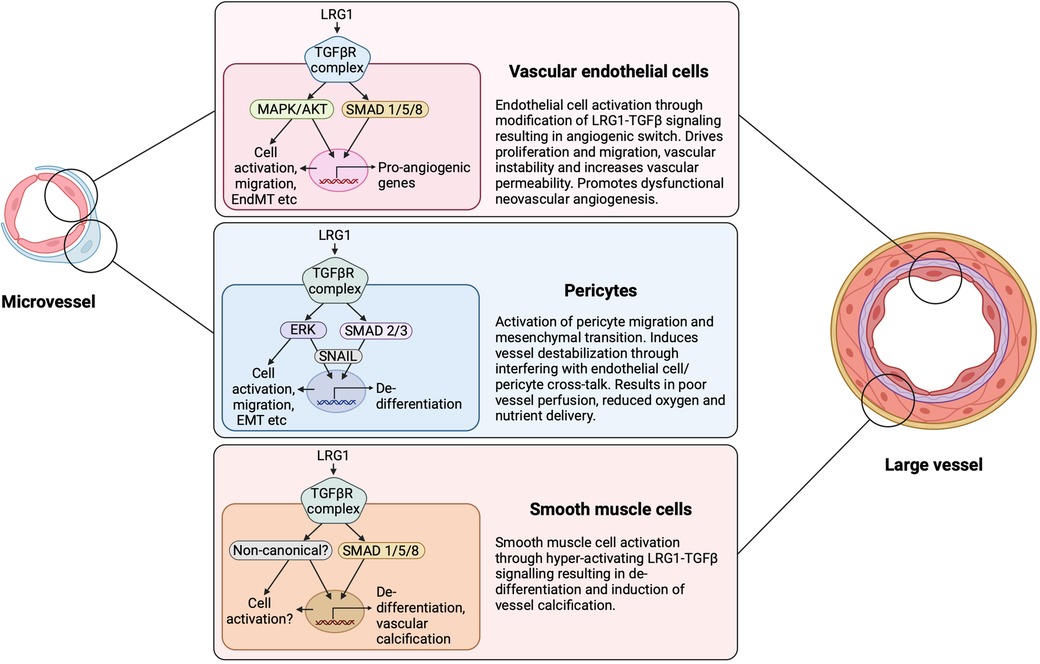
Figure 4. LRG1 cell-dependent signaling and outcome. LRG1 can induce signaling in all cellular constituents of the vasculature. Depending on the configuration of signaling receptors and downstream influences, LRG1 can cause the TGF-β angiogenic switch through acting on the endothelial cell, pericyte and smooth muscle cell. LRG1 has been shown to alter the canonical and non-canonical TGF-β signaling pathways in a cell-dependent manner. EndMT, endothelial to mesenchymal transition; EMT, epithelial to mesenchymal transition. Created with BioRender.com.
LRG1, tissue repair and wound healing
Postnatally, angiogenesis is an important part of the repair and wound healing process. This involves a tightly regulated sequence of events including the recruitment of many different cell types driven in part by overlapping phases of inflammation and tissue remodeling (101). Failure of wound healing can lead to chronic wounds, which can be observed in many conditions such as eye disease, lung and heart disease, and diabetes (102). Upregulation of LRG1 in various cell types during the normal wound healing process clearly suggests its involvement, possibly through the promotion of new vessels and cell de-differentiation and migration (Figure 1). Whilst the role of LRG1 in normal blood vessel formation during wound healing is still unknown, reduced blood vessel density has been observed in the wound bed of Lrg1−/− mice (17), suggesting its involvement in post-developmental vessel formation. In a murine skin wound healing model, LRG1 drives keratinocyte migration and re-epithelization by improving HIF-1α stability through ELK3 (103), whereas in a similar setting, LRG1 promoted endothelial cell proliferation, angiogenesis and EMT (17). In the latter study, LRG1 expression within the wound was also observed in bone-marrow-derived cells such as neutrophils, monocytes, macrophages and dendritic cells. The presence of LRG1 was shown to be essential for timely wound repair as Lrg1−/− mice exhibited a significant delay in wound closure, which was reversed when bone marrow-derived LRG1-expressing stem cells were transfused into the knock-out animals (17). However, in wounds from diabetic mice, as well as from patients with diabetes, LRG1 expression was markedly enhanced and in mice was shown to result in delayed wound closure. LRG1 was also associated with the adverse inflammatory response by stimulating excessive neutrophil attraction and promoting NETosis in a TGF-β/ALK5/ΑKT-dependent manner (17). The delay in wound healing in diabetic mice was reversed in Lrg1−/− mice, demonstrating its context dependent activity. In diabetic wounds in the rat, LRG1 has also been shown to increase blood vessel density through activating the WNT/β-catenin pathway, which, counterintuitively, may exacerbate the wound healing process (86).
Whilst not entirely consistent, these studies suggest that under normal conditions LRG1 promotes physiological tissue repair by promoting angiogenesis and cell migration, but in a chronic inflammatory setting, as seen in diabetes, LRG1 remains induced prolonging the initiating stage of wound healing (Figure 2). Our current understanding, therefore, is that LRG1 is induced as part of the acute innate immune response mechanism that sequesters Cyt c, and through modification of TGF-β signaling activates endothelial cells and pericytes resulting in destabilized vessels and stimulation of sprouting angiogenesis. We propose that through other switches in the TGF-β signaling, LRG1 also drives EMT that permits loss of epithelial junctional integrity, cell division and migration required for wound closure. As the inflammatory response resolves under normal conditions LRG1 expression is attenuated allowing for cellular re-differentiation, wound stabilization, and maturation. However, in the presence of a continuing chronic inflammatory stimulus LRG1 expression is sustained, and even increased, which maintains cells in a de-differentiated less mature state and thus prevents wound resolution and tissue homeostasis (Figure 2).
LRG1, ischemia and stroke
A role for LRG1 has been reported to be part of the repair mechanism in cerebral damage due to ischaemic injury. Interestingly, high circulating LRG1 levels have been found positively associated with stroke severity and poor functional outcomes (104–106), as well as with poor cognitive impairment and neurological function (107). In the brain, following ischemic stroke injury a single cell transcriptomics study revealed the emergence of a LRG1-positive endothelial cell subpopulation in the brain infarct area, the expression profile of which suggests its involvement in the complex regulation of angiogenesis, with both pro- and anti-angiogenic factors expressed (108). In fact, in a mouse cerebral artery occlusion model, LRG1 increased apoptosis and autophagy through TGF-β and SMAD1/5 exacerbating the ischemic injury (109). Similarly, increased LRG1 was found in brain endothelial cells in a rat brain ischemia model with expression correlating positively with VEGF, Ang2 and TGF-β (92), indicating a protective role for LRG1 by promoting de novo formation of blood vessels, possibly through TGF-β (92). De novo vessel formation was confirmed, with CD34 staining showing significantly increased microvessel density after stroke, which again correlated with LRG1 expression (92). However, whilst new vessels were observed which may confer survival benefit, the likelihood is that these penumbral vessels are dysfunctional compromising their beneficial effect (110). A more recent study employing a cerebral ischemia-reperfusion injury model in the mouse demonstrated that in the Lrg1−/− mouse there was reduced cerebral oedema and infarct size, which was accompanied by improvement in neurological function (111). Crucially, this study showed that following cerebral ischaemia, cells of the blood-brain barrier were able to retain the expression of barrier function related genes, such as claudin 11, integrin β5, protocadherin 9, and annexin A2, more effectively in the absence of LRG1 (111). This demonstrates that LRG1 has a detrimental effect on the blood-brain barrier and thus contributes to vasogenic cerebral oedema. In its wider role, Lrg1 knockout also permitted a more anti-inflammatory and tissue-repairing environment and reduced neuronal cell death. In a different ischaemic setting, tissue remodeling after myocardial infarction involved the infiltration of LRG1 expressing myeloid cells that were reported to exert a cardioprotective role by promoting signaling through the pro-angiogenic TGF-β/SMAD1/5/8 axis and contributing to post-infarct vessel formation (89).
LRG1 and vascular pathology of the eye
Retinal neovascularization
Vascular problems of the eye, such as in diabetic retinopathy or neovascular (exudative or wet) age-related macular degeneration (nAMD), are characterized by vessel remodeling and angiogenesis and can occur intraretinally, subretinally or at the vitreal interface. Irrespective of the ocular disease, vascular dysfunction is often a major contributing factor to loss of vision in the developed world (112). The first evidence that LRG1 is actively implicated in pathogenic neovascularization emerged from its identification as the most up-regulated gene in a transcriptomics analysis of retinal microvessels isolated from three different animal models (retinal dystrophy 1, VLDLR knock out, Grhl13ct/J curly tail), each of which showed marked retinal vascular remodeling and pathology (25). This study went on to show that the Lrg1 transcript is almost undetectable in the healthy mouse retina but in experimental retinal vascular diseases such as experimental choroidal neovascularization (CNV) (113) and oxygen-induced retinopathy (OIR) (114) that model choroidal and retinal neovascularization respectively, expression of LRG1 is induced in endothelial cells (25). In OIR, the abnormal retinal neovascularization that occurs at the vitreous interface is believed to be driven largely by hypoxia, whereas in CNV the laser burn to the retinal pigment epithelium (RPE) results in vessels sprouting from the choriocapillaris through the breached RPE into the subretinal space as a result of injury-induced hypoxic and inflammatory cues. Irrespective of the model system employed, genetic deletion of Lrg1 led to a diminished pathogenic neovascular response coupled with a reduction in vascular leakage as assessed by fundus fluorescein angiography. Of note, however, revascularization of the hyperoxia-induced vessel ablated region in OIR proceeds in a more physiological patterned manner and is not affected by Lrg1 deletion (25, 94), further demonstrating that LRG1 contributes to pathogenic, but not physiological, neovascularization. Similar findings were seen when LRG1 activity was blocked by intravitreal delivery of a specific function-blocking antibody, the application of which significantly reduced OIR and CNV lesion size (25, 94) and lends credence to its potential as a therapeutic target.
In addition to the disease-associated induction of endothelial LRG1 expression, myeloid cells have also been found to express LRG1 in choroidal neovascularization. A recent study exploring the transcriptional profiling of immunosenescence in myeloid cells during the development of CNV showed that aged senescent myeloid cells of CNV-induced animals exhibited an upregulated angiogenic transcriptional profile with significantly elevated Lrg1 and Arg1 expression compared to young cells (115). This is the first study to provide evidence of upregulated Lrg1 expression in cells other than the endothelium contributing to choroidal neovascularization. Although it is not clear yet whether this is an epiphenomenon or there is a direct causal role for LRG1 in CNV-related immunosenescence, the data suggest a potential role for LRG1 and myeloid cells in the immunoregulation of CNV that merits further investigation.
Corroborating the mouse studies above, elevated expression of LRG1 has also been found in the aqueous and vitreous humour and in retinal choroidal neovascular membranes of nAMD patients (116–119), as well as in dry (atrophic) AMD (120). Mundo et al., showed that in ocular sections of nAMD membranes LRG1 is co-localized not only with endothelial cells but also with myofibroblasts, suggesting an alternative cell source through which LRG1 may exert its pathological effects (118). Thus, myofibroblast-derived LRG1 may contribute to endothelial dysfunction in the retina in a paracrine manner, but it may also be involved in promoting retinal fibrosis through autocrine mechanisms (118). This suggests that, two-way LRG1 cross talk between endothelial cells and fibroblasts may drive both vascular dysfunction and fibrosis. This is in accordance with other findings showing that LRG1 promotes fibrosis in various tissues and conditions including the skin (41), idiopathic pulmonary fibrosis (42), renal fibrosis (34) as well as diabetic nephropathy (33, 121), by directly affecting the physiology and activity of fibroblast-like tissue resident cells (Figure 1). Interestingly, a recent study showed that exogenous LRG1 promotes the EMT of RPE cells as evidenced through high levels of αSMA and fibronectin and low levels of ZO-1 (122). The concept that vascular dysfunction can contribute to fibrosis is not new (123) and it raises the interesting possibility that endothelial derived LRG1 may also trigger the fibrotic response in the eye. Similarly, endothelial cells have been shown to contribute to matrix deposition when they undergo phenotypic differentiation via EndMT, a process predominantly regulated by TGF-β. EndMT is a hallmark of cardiovascular disease and has been studied in a variety of experimental models (124). Although there is no known link between EndMT and LRG1, unpublished evidence from our laboratory suggests that LRG1 is overexpressed in a cytokine induced EndMT model system and may therefore play a role in the eye.
Diabetic retinopathy
In diabetic retinopathy, where retinal vascular dysfunction, dropout and neovascularization are prominent, LRG1 has been reported to be upregulated not only in the vitreous humour and in ocular tissues (25, 50, 125, 126), but also systemically in the plasma of patients with proliferative diabetic retinopathy (PDR) (50, 127). Diabetic retinopathy is the most common complication of diabetes, leading to impaired vision and ultimately to vision loss (128). The first stage of the disease is non-proliferative and is characterized by microvascular abnormalities leading to macular oedema, microaneurysms, micro-haemorrhages, and poorly perfused, occluded, and de-endothelialized capillaries. In PDR, the later stage of the disease, loss of vessels and subsequent hypoxia results in the growth of abnormal neovessels leading to vitreous haemorrhage, tractional detachment and eventually blindness. Since LRG1 is almost undetectable in a healthy human eye, its elevated expression in the vitreous in diabetes is most likely to be due to activated local production in response to disease, but it may also derive from the plasma as a consequence of vascular leakage. Whether the upregulated expression drives the vascular complications in the eye or contributes to other diabetes-associated pathology remains to be elucidated. Indeed, accumulating evidence indicates that LRG1 is upregulated in the plasma (56, 129) and urine (130) of people with diabetes, hence it can serve as a biomarker for PDR (131). However, LRG1 did not offer a significant improvement when used in a risk prediction model for PDR (57). Nevertheless, the greater evidence so far implicates LRG1 in retinal neovascularization, with its genetic deletion or inhibition of function through antibody blockade ameliorating vascular pathology, lesion size and leakage in animal models (25, 94). In addition, unpublished data from our lab shows that LRG1 can promote the phenotypic and functional differentiation of pericytes towards a fibrotic state and that this contributes to vascular dysfunction in diabetic retinopathy through both canonical and non-canonical TGF-β signaling (20) (Figures 1, 4). This evidence highlights the possibility that endothelial derived LRG1 may also impact retinal function in a paracrine manner. Although our understanding of diabetic retinopathy has increased substantially, and anti-VEGF therapeutics have revolutionized diabetic macular oedema and PDR treatment, a substantial number of patients remain or become refractive which clearly implicates the involvement of other players and highlights the need for further research. The evidence to date suggests that LRG1 may be a contributing factor to resistance to anti-VEGF strategies and thus LRG1 blockade could improve outcome.
Corneal neovascularization
The angiogenic properties of LRG1 have also been described in the context of corneal neovascularization in a corneal alkali burn mouse model, where LRG1 promoted a significant outgrowth not only of blood vessels, but also of lymphatic vessels (91). The normal cornea is avascular in order to maintain transparency but under pathological conditions, such as inflammation or trauma, new vessels invade the avascular tissue causing visual impairment. In Lrg1 knock down studies using siRNA, a limited angiogenic and lymphangiogenic response was observed compared to control mice (91). In this study, different members of the VEGF family were implicated in LRG1-driven corneal angiogenesis and lymphangiogenesis, with VEGF-A/B and their receptors VEGFR-1/2 regulating the former and VEGF-C/D together with VEGFR-2/3 regulating the latter (91). How LRG1 impacts on this remains unclear but it raises the possibility of cross regulation between these pathways. Apart from neovascularization, LRG1 was also found to promote corneal fibrosis by increased deposition of αSMA, collagen type I and CTGF in the corneal epithelium (48). This LRG1-driven effect was mediated by neutrophil infiltration at the site of corneal injury through the phosphorylation of STAT3 and the upregulation of IL-6/STAT3 signaling (48). These data point to LRG1 as a potential therapeutic target to ameliorate pathogenesis in corneal disease.
LRG1 and chronic kidney disease
The role of LRG1 in kidney disease, in particular diabetic nephropathy, is gaining interest (132) with growing evidence that LRG1 contributes to vascular rarefaction and abnormal neovascularization. In normal kidney, LRG1 expression is found in glomerular endothelial cells co-localized with that of CD31, as well as in the tubulointerstitium (53). The kidney is a highly vascularized organ, where maintenance of normal blood flow is crucial for renal function which, following pathological complications such as vessel loss and fibrosis, may become seriously compromised. Endothelial dysfunction coupled with abnormal angiogenesis have long been known to contribute to the pathogenesis of diabetic nephropathy and other chronic kidney conditions, although the underlying molecular mechanisms are poorly understood (33, 53, 133). Several studies show that in a variety of chronic kidney disease (CKD) models LRG1 gene and protein expression is significantly increased. Indeed, a transcriptomics study has shown that in a model of diabetic nephropathy, Lrg1 was upregulated in glomerular endothelial cells, where it mediated high glucose-induced pathological angiogenesis (87). Similarly, other studies have also shown that in experimental diabetic kidney disease (DKD) Lrg1 gene expression induced in glomerular endothelial cells is involved in vascular rarefication and subsequent neovascularization and fibrosis, partly via activation of the p38 and TGF-β-SMAD1/5/8 pathway (52, 53, 87, 88). Consistent with these reports, LRG1 overexpression has also been shown to result in exacerbation of disease (33, 53). What is interesting, but needs further corroboration, is the suggestion that the initiating mechanism driving vascular dysfunction is mediated by LRG1 and is independent of VEGF. This is based on the finding that LRG1 expression precedes the expression of VEGF and its receptor VEGFR-2 in diabetic nephropathy (52) and that VEGF is expressed mainly in the injured podocytes following endothelial injury (134, 135). These data prompted the authors to suggest that glomerular endothelial LRG1 may be an initiating factor in vascular pathology in the diabetic kidney.
In various other in vivo models of CKD, including the albumin overload model (136), unilateral ureteral obstruction (UUO) model (34), and the aristolochic acid-induced nephrotoxicity (AAN) model (33), LRG1 protein levels have also been shown to be higher, often correlating with increased pro-inflammatory and pro-fibrotic cytokines. Such findings support the contention that LRG1 is not only induced by inflammatory cues but that it also promotes the renal inflammatory response including the activation of macrophages in a TGF-βR1-dependent manner (137).
In the kidney, endothelial cells are not the only source of LRG1, as it has also been shown that renal tubular epithelial cells (33) and HK-2 human proximal tubular epithelial cells (34) can be induced in vitro to express LRG1. Tubular epithelial cell derived LRG1 has been shown to activate the TGF-β-SMAD3 pathway in fibroblasts resulting in increased renal fibrosis (33). This additional cellular source of LRG1 may not only trigger fibrosis but also contribute further to endothelial cell dysfunction. Indeed, it has also been proposed that LRG1 mediated microvascular dysfunction in the kidney may facilitate the onset and progression of fibrosis (34), supporting the notion that these two phenomena are interrelated (123). What is clear from these studies is that in the kidney LRG1 not only stimulates vessel loss and the formation of abnormal neovessels, it also drives the fibrotic response, all of which combine to reduce glomerular filtration rates (121, 138). In accordance with the studies above, increased renal LRG1 expression is reflected in the urine, where significantly elevated levels correlate closely with the degree of renal tubular dysfunction (136). Consistent with these findings, Lrg1 knockout reduces the deterioration of kidney function (33, 53). Similarly, suppression of LRG1 and ALK1-dependent angiogenesis by metformin showed significant renoprotective effects in a diabetic rat model (139).
Clinical evidence also points to LRG1 playing a role in the pathogenesis of CKD. Thus, it has been shown that in people with type 2 diabetes and DKD, plasma LRG1 levels predict both albuminuria and CKD progression beyond traditional risk factors (56, 140). Similarly, in children with type 1 diabetes a clear relationship between plasma LRG1 and estimated glomerular filtration rate (eGFR) decline suggests that LRG1 may be an early marker of DKD progression (49). In separate studies, using human DKD tissue, LRG1 gene expression has been found to be increased in glomerular endothelial cells (53). Moreover, in other forms of CKD, including lupus nephritis (36), IgA nephropathy (34), and end-stage kidney disease dialysis patients (35), increased LRG1 plasma and biopsy tissue levels correlate with worse outcome, increased inflammatory markers, and greater fibrosis. Of note, LRG1 levels correlated positively with IL-6, a known activator of LRG1 gene expression, as well as with a more advanced state of T cell differentiation and the presence of cardiovascular disease and peripheral arterial occlusive disease (35) demonstrating its potential systemic involvement.
In addition to plasma, increased urine LRG1 levels in diabetes is also associated with an increased risk of progression to end stage kidney disease independent of traditional cardiorenal risk factors (55), and in kidney transplant recipients LRG1 has been considered a potential kidney injury marker that correlates with other tubular injury markers and functional deterioration (141). These human data support the evidence from experimental studies that LRG1 is an important factor in driving CKD through initial effects on the kidney vasculature and the subsequent fibrotic response. One of the most compelling pieces of clinical evidence that LRG1 contributes to DKD, however, was a recently reported GWAS study in people with type 2 diabetes and CKD, where a 5'UTR variant (rs4806985) in the promoter region of LRG1 was found to influence its gene expression resulting in elevated plasma LRG1 and a robust association with increased risk of rapid decline in kidney function (51). This is the first study describing a polymorphism risk to LRG1 circulating levels, suggesting a potential use for LRG1 in stratifying patients with diabetes into subsets based on their genetic predisposition. Additionally, genetically influenced plasma LRG1 levels were also associated with lower cognitive function, further supporting a role for LRG1 as a novel biomarker for cognitive decline in type 2 diabetes mellitus (142). The conclusion drawn in many of these studies is that LRG1 is a potentially important therapeutic target as it is seen as a master upstream orchestrator of pathogenic TGF-β signaling. It is well established that the TGF-β pathway has a critical role in neovascular and fibrotic processes and that targeting constituents of this pathway continues to be considered an attractive therapeutic strategy. However, the need to retain homeostatic TGF-β signaling remains a challenge but one which may be overcome by targeting LRG1 as this is a key upstream factor in causing the switch from quiescent housekeeping to pathogenic disruptive signaling.
LRG1 and lung disease
Inflammation, tissue repair, endothelial dysfunction and increased interstitial pressure are common phenomena in pulmonary disease leading to prominent vascular-related complications such as pulmonary embolism, abnormal microthrombi, and microvascular damage (143). Alveolar epithelial and endothelial permeability are also compromised with impaired gas exchange and vascular leakage due to loss or destabilization of cell junctions. In addition, the damaged endothelium may disrupt vascular tone and cause dysregulation of anti-inflammatory and anti-thrombogenic endothelial properties, and together with damaged epithelium can trigger the tissue repair process (144). To date, evidence shows that LRG1 is involved in pulmonary vascular dysfunction with increased expression seen in lung disease, including chronic obstructive pulmonary disease (COPD), interstitial pneumonia, airway inflammation in asthma, and active tuberculosis, with LRG1 levels serving as a biomarker for early diagnosis, progression, and prognosis (38, 66, 145–148).
Several reports show that LRG1 is upregulated in lung epithelial and endothelial cells, mainly but not exclusively in inflammation-induced pathology (37, 39, 66, 90). In particular, in human COPD tissue, upregulated LRG1 was localized specifically to the endothelium and correlated positively with marked airflow obstruction, decline in lung function and severity of emphysema (37). COPD is a heterogeneous long term lung disease characterized by persistent airway inflammation, microvascular dysfunction, dysregulated angiogenesis, and endothelial apoptosis (149). Specific endothelial deletion of LRG1 in a murine elastase model of COPD protected against severe parenchymal destruction, highlighting a critical role for LRG1 in promoting the development of maladaptive lung vasculature (37). Although the exact mechanism that mediates this process has not yet been defined, it is possible that LRG1 impacts angiogenic responses following endothelial injury either by promoting defective angiogenesis or through the development of fibrosis. Indeed, LRG1 has been shown to trigger a pro-fibrotic response in the lung by activating lung fibroblasts and the subsequent production and deposition of extracellular matrix via TGF-β signaling and the phosphorylation of SMAD2 and SMAD3 in mouse bleomycin models (42, 44). Strikingly, in a recent single cell transcriptomics study, LRG1 was described as an extracellular matrix coding gene and its expression was found increased and maintained at high levels in aging lung, which associated LRG1 with age-related inflammation and tissue stiffness (150). However, LRG1-mediated fibroblast activation and proliferation was not regulated by SMAD2 or 3, the levels of which were either unchanged or repressed, respectively, implying that other signaling mechanisms, such as activation of alternative canonical or non-canonical pathways, are in place (150). Indeed, consistent with previously published data (25, 26, 53), in viral-induced lung injury, LRG1 upregulation drives endothelial cell proliferation and the angiogenic responses required for tissue repair by activating SMAD1/5 signaling (90).
Elevated levels of circulating LRG1 have also been reported in severe COVID-19 patients in blood, plasma, and tissue proteomic studies (151–156). Although most of these studies associate elevated LRG1 expression with an early immune and inflammatory response, it is possible that LRG1 exerts an angiopathic role in the pulmonary microvasculature related to COVID-19. In fact, a dysregulated cytokine immune response, known as a “cytokine storm”, has been established and studied extensively in patients with COVID-19 (157). This includes highly elevated expression of pro-inflammatory cytokines that correlates with COVID-19 disease severity and requires immediate attention, as excessive activation of immune cells can lead to complicated and potentially lethal medical syndromes (158). Among others, IL-6 is a key player in this cytokine response with significantly elevated circulating levels in the plasma of patients with COVID-19, and consequently it has been reported to contribute to the related vascular pathology (159, 160). Blocking IL-6 signaling as a therapeutic intervention has been extensively studied in many diseases (161, 162), and COVID-19 randomized controlled clinical trials with biologics targeting the IL-6 receptor, including the Tocilizumab antibody, have shown evidence of clinical benefit (163–166). Elevation of both IL-6 and LRG1 in patients with COVID-19 suggests that circulating pro-inflammatory IL-6 may induce systemic and local upregulation of angiopathic LRG1 in the pulmonary microvasculature. Indeed, IL-6 upregulates LRG1 expression in human pulmonary microvascular endothelial cells, and this effect is reversed when IL-6 signaling is blocked by the tocilizumab antibody (26). Upregulated LRG1 in turn may contribute to the development of a destabilized vasculature with prominent endothelial dysfunction and vascular leakage during impaired lung tissue wound healing (Figures 1, 4). This LRG1-dependent microangiopathy may be mediated by the pro-angiogenic TGF-β-SMAD1/5 signaling arm, as evidenced in other endothelial-related disease settings (25, 26, 90). As reported in experimental CKD, and described above, dysregulated microvascular endothelial cells in the lung in COVID may also be a driver of fibrosis (37), linking LRG1 to these two key pathogenic processes.
LRG1 and inflammation-associated disease
Over the past decade several studies have provided evidence that LRG1 is involved in various inflammatory and autoimmune diseases, and may act as a useful clinical and diagnostic biomarker (23, 167–174). In such conditions, levels of LRG1 are upregulated at the site of inflammation, with expression induced by pro-inflammatory cytokines secreted by various cell types, followed by the initiation of a series of downstream vascular events. During inflammation, one of the key vascular responses is an increase in vessel permeability that, alongside other inflammatory changes, facilitates the extravasation of immune molecules and cells to the site of injury (175). In addition to tissue resident cells, recruited immune cells provide an additional source of angiogenic factors that play a part in the inflammatory and reparative response. Over time this response resolves but under certain chronic conditions unresolved inflammation, including the persistence of LRG1 expression, continues through an imbalance of stimulatory, inhibitory and disruptive factors, and can give rise to long-term vasculopathic outcomes (176) (Figure 2).
Osteoarthritis (OA) is the most common inflammatory joint disorder, with aberrant endothelial cell proliferation, vascular penetration, and synovial fibrosis being the main disease leading mechanisms that contribute to structural damage and pain (177). Lrg1 transcript has been found to be upregulated in chondrocytes upon IL-6 stimulation (178). In OA the pro-inflammatory cytokine TNF-α, a key player in the pathophysiology of the disease and major activator of pro-angiogenic factors (179, 180), was also shown to induce LRG1 expression in the subchondral bone and articular cartilage (64). In this setting it promoted angiogenesis, mesenchymal stem cell migration and aberrant bone formation via MAPK-dependent p38/p65 signaling. This highlights a new potential mechanism through which LRG1 promotes abnormal neovascularization coupled with de novo bone formation. Supplementary to its role in angiogenesis in OA, LRG1 has also been shown to contribute to synovial fibrosis and joint stiffness by promoting secretion of extracellular matrix in synovial cells, cell migration and wound healing (46), further suggesting that LRG1 not only affects endothelial cells but also other cells in the cartilage exerting multiple parallel pathogenic responses.
Although LRG1 has attracted substantial interest with regards to its use as a clinical biomarker in a plethora of inflammatory conditions, little is known about its mechanistic involvement in the development and progression of vascular defects in these conditions. Nevertheless, LRG1 has been implicated in Kawasaki disease, an acute systemic vasculitis causing inflammation of small to medium sized blood vessels resulting in cardiovascular complications (181, 182). The main cytokines responsible for inducing LRG1, TFN-α and IL-6, are elevated in the plasma of patients with Kawasaki disease, and a proteomic analysis of serum exosomes of patients with coronary artery aneurysms caused by Kawasaki disease showed upregulated LRG1 levels, although no causal link with pathology was shown (183). Similarly, another study on Kawasaki disease in children identified LRG1 as a potential trigger of endothelial cell activation and cardiac remodeling that closely associated with IL-1β signaling (184). The cell source contributing to increased circulating LRG1 levels in these conditions has not yet been identified but evidence from other conditions suggests that upon inflammatory stimulation LRG1 is most likely produced by endothelial cells, where it exerts its vasculopathic effects via autocrine and paracrine pathways on the endothelium and the adjacent mural cells, respectively. Although the exact vascular pathogenesis in Kawasaki disease is not well understood, vascular complications include necrotizing arteritis associated with neutrophilic and immune cell infiltration, the release of pro-inflammatory cytokines, luminal myofibroblast proliferation and progressive obstruction of the coronary lumen have all been linked with LRG1 in other conditions and so it is likely that it plays a role in this and other vasculitides. In support of this, LRG1 has also been found to be a promising serum biomarker for large vessel vasculitis (LVV) (185), and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) (186, 187).
Inflammation is a hallmark of cardiovascular disease and destabilized vasculature, and frequently serves as a trigger during the early stages of disease, while increased expression of inflammatory cytokines is associated with a higher risk of cardiovascular diseases (188). Thus, LRG1 may be anticipated to initiate or mediate inflammatory responses to some extent in conditions with cardiovascular risk. In fact, LRG1 has been shown to promote cardiovascular disease by regulating endothelial dysfunction and inflammation through TGF-β and SMAD1/5/8 signaling in endothelial cells, interrupting normal endothelium-dependent vasodilation and availability of nitric oxide (129). Furthermore, in a recent study employing the Western diet apolipoprotein E knockout (ApoE−/−) mouse model of atherosclerosis, LRG1 was detected within the atherosclerotic plaque, particularly in calcified regions (93). The cell source of LRG1 was found to be endothelial cells that had been activated by inflammatory mediators. Furthermore, this study demonstrated that LRG1 was responsible for inducing vascular smooth muscle cell activation and vessel calcification via a SMAD1/5 signaling pathway (Figure 4). The authors conclude that LRG1 is a significant contributor to the development of plaque complications and therefore a potential therapeutic target. On the other hand, and in contrast to most other studies, in arterial stenosis it was suggested that endothelial LRG1 could serve as a negative regulator of inflammation in response to TNF-α by inhibiting expression of ICAM1 and VCAM1 and thus blocking monocyte recruitment, offering a significant atheroprotective effect (189). Interestingly, this is consistent with what has been observed in cancer (see below) where endothelial anergy, as indicated by ICAM1 and VCAM1 expression, is reversed upon LRG1 inhibition.
Exactly how LRG1 mediates vascular inflammation is not clear although there is growing evidence for LRG1 regulating a pro-inflammatory accumulation of immune cells. In particular, LRG1 is involved in neutrophil function modulating NETosis, which is closely associated with inflammatory processes (17). NETosis has been described as a form of necrosis and is associated with many diseases including COVID, thrombosis and CKD, as well as wound healing and other vascular processes (190–192). As mentioned above, a recent study suggested that LRG1 mediates NETosis and contributes to poor wound healing in diabetic mice (17). Although future work is required to unravel the role of LRG1 in regulating neutrophil function, we speculate that NETosis might be an important additional mechanism through which LRG1 may impact on vascular structure and function.
LRG1 and cancer
LRG1 expression has been studied in a range of malignancies where it has been shown to be elevated and to associate with poor prognosis and survival (23, 61, 62, 193, 194). In addition, raised blood LRG1 levels have been established as a tumor biomarker with potential clinical value and also as a predictive marker for cancer onset (30, 59, 60, 62, 193, 195–200). A growing number of studies support an integral role for LRG1 in cancer, where it has been shown to control cell viability and apoptosis, and promote epithelial cells to undergo EMT, a crucial step in tumor progression and metastasis (27, 28, 30, 31, 80–84, 201–203). In particular, LRG1 acts directly on tumor cell proliferation, migration, and invasion contributing to tumor growth and survival (Table 1), and these functions have been described in detail elsewhere (23, 29, 193). Consistent with LRG1 playing a role in tumor progression, LRG1 blockade in different tumor models inhibits growth and improves survival and thus has been proposed as a potentially beneficial therapeutic target (29, 32). There is also accumulating evidence that LRG1 has important angiocrine and angiopathic functions in cancer, not only by promoting the development of destabilized and immature neo-vessels, but also by impairing already established co-opted vasculature (Figure 3). As widely acknowledged, growing tumors require a constant supply of oxygen and nutrients, and serving these needs often relies on a concomitant developing vascular network. The tumor vasculature, however, is typically abnormal exhibiting impaired structure and function. In particular, tumor blood vessels are immature, tortuous and chaotic in organisation, with an abnormal vessel wall characterized by a discontinuous endothelium, incomplete coverage of mural cells, and atypical basement membrane structures leading to poor perfusion and leakiness (204). These characteristics also create a hypoxic and acidic environment within the tumor tissue that favours malignancy and metastasis and combine to reduce immune cell infiltration and effective immune responses, and restrict the delivery of therapeutics and effectiveness of radiotherapy.
The angiogenic potential of LRG1 has been assessed and described in various types of tumors, including colorectal, gastric, pancreatic, ovarian, and non-small-cell lung cancer (193). Specifically, LRG1 has been proposed to enhance the angiogenic process through acting directly on endothelial cells to induce proliferation and migration, but also indirectly through stimulating proangiogenic factors such as VEGFA (84). Moreover, LRG1 has been associated with increased microvessel density, suggesting that it impacts tumor vascular growth (26). As in other diseases, the direct effect of LRG1 on vessel function is believed to be mediated primarily through modification of canonical TGF-β and SMAD signaling, but in all likelihood also through hyperactivation of non-canonical TGF-β pathways. Alternative mechanisms for LRG1-driven angiogenesis have been proposed including regulation through HIF-1α, which is associated with resistance to cancer chemotherapy and increased patient mortality (205). HIF-1α knockdown was shown to block LRG1-mediated angiogenesis, EMT, and tumor invasiveness, and is consistent with LRG1 being induced in response to hypoxia (84). In another study, ERK mediated phosphorylation of ELK4 in a human colorectal cell line resulted in complex formation with SP1/3 and the induction of LRG1 gene expression (69). This, it was argued, results in enhanced tumor angiogenesis through activation of the TGF-β-SMAD1/5 pathway in endothelial cells. Whatever the mechanism, the evidence is clear that LRG1 plays a central role in driving abnormal vessel formation in solid tumors.
In line with LRG1 driving vessel abnormalization, strong evidence indicates that vessel structure and function in tumors can be improved by knocking out Lrg1 or by its inhibition. Indeed, restoring vessel function, a process referred to as vascular normalization, represents a promising strategy to facilitate drug delivery, enhance cytotoxic T cell function, and increase the tumor response to standards of care and immunotherapies (206, 207). In this context, Lrg1 gene deletion, or functional blockade of the protein, has been shown to improve tumor vascular function as manifested by better perfusion, reduced tumor hypoxia and reduced vascular leakage (32). In particular, vessel size, basement membrane and perivascular mural cell coverage of the endothelium were all significantly increased in the absence of LRG1 (32). As a likely consequence of vascular normalization, LRG1 inhibition not only led to significant improvements in the delivery and efficacy of anti-tumor therapies, but also improved immune-cell infiltration (32). This may partly be explained by re-activation of anergic endothelial cells to allow leukocyte infiltration, seen for example by increased ICAM-1 and VCAM-1 expression. Collectively, these data show that the angiopathic functions of LRG1 not only promote a pro-oncogenic vascular microenvironment in primary and metastatic tumors, but also contribute to immune modulation. Unpublished data from our lab show that in addition to the vascular normalization effects, inhibition of LRG1 also promotes tumor infiltration of T-cells by modulating the immunosuppressive tumor microenvironment, thereby supporting a switch from being immunologically “cold” to “hot”.
Angiogenesis is not the only means through which tumors obtain a vascular supply. Vessel co-option is a surrogate mechanism whereby tumor cells employ the established vasculature to support growth, survival, and metastasis. In a recent study where, in the presence of sunitinib, tumor growth escapes from VEGF-dependent angiogenesis through vessel co-option, single cell transcriptomics revealed a surprisingly similar signature between tumor co-opted endothelial cells and pericytes and their healthy non-tumor bearing counterparts (208). This finding was further confirmed in other vessel co-opted tumor metastatic models. The similarity in the cell transcriptome was predominantly due to the lack of genes associated with angiogenesis and pericyte activation that are observed in angiogenic tumors. Intriguingly, however, Lrg1 was found to be one of the top 10 genes that were differentially expressed in co-opted postcapillary vein endothelial cells compared to normal endothelium (208). This suggests that Lrg1 is one of the few genes to be expressed in both tumor angiogenic and co-opted endothelial cells. Further studies, however, will be required to determine whether Lrg1 induction in co-opted vessels exerts similar vasculopathic effects as observed in angiogenic tumor vessels and in vessels of other diseases.
Aside from endothelial cells, a major cell source of LRG1 in cancer is frequently the tumor cells (Figure 1) but this is not always the case. As in other diseases, there is evidence that LRG1 is also expressed by other cell types including fibroblasts, and immune cells. In all experimental studies conducted thus far, however, LRG1 expression has been shown to co-localize with vessel markers, such as CD31 and CD34 (60, 209) illustrating its ubiquitous presence in tumor endothelial cells. Recent work on the role of LRG1 in cancer showed that in some cancer models Lrg1 expression was mostly restricted to the vascular endothelium, with no expression detected in the perivascular mural cell population or the cancer cells themselves (29, 32). Nevertheless, this was sufficient to impact on tumor growth as Lrg1 knock-out or antibody blockade were still effective in reducing tumor growth. Interestingly, using similar tumor models it has been shown that the primary tumor induces systemic vascular LRG1 expression and that this primes the vascular metastatic niche and promotes tumor metastasis (29). This priming was also associated with an expansion of NG2+ perivascular mural cells, which have been described as effective mediators of metastasis (210). LRG1 induction in the tumor mass and systemically in cancer is most probably through IL-6 and STAT3, with contributions from other signaling pathways. Indeed, in metastasis models the STAT3 signaling pathway has been shown to mediate LRG1-driven tumor metastasis, and that this can be significantly reduced in Lrg1 deficient mice or following LRG1 antibody blockade (28, 29, 31). Through a different mechanism, liver endothelium-derived LRG1 has been shown to promote tumor growth and metastasis of colorectal cancer in a paracrine manner through binding to the HER3 receptor, leading to its phosphorylation and activation (202). If evidence from other diseases translates to cancer, other cell sources of LRG1 are likely to impact the tumor microenvironment. In particular, fibroblasts and neutrophils are a key source in other disease settings and their contribution to cancer merits further investigation.
Conclusion
Over the last 10 years our understanding of how the secreted glycoprotein LRG1 contributes to physiological and pathological processes has grown exponentially and, in the latter case, demonstrates beyond doubt that it plays a significant contributing role in disease. Whilst we are only just beginning to appreciate the extensive biological role of LRG1, it is clear that much of its activity is mediated through its switching effect on the ubiquitous and complex TGF-β signaling network. Not surprisingly, therefore, the biological effect of LRG1 is wide ranging and highly cell and context dependent, reflecting the biological diversity of TGF-β activity. Accordingly, LRG1 exerts pleiotropic effects depending on the cell target and the influence of other environmental cues, affecting not only the vasculature but also other cell types that are under the influence of TGF-β including epithelial cells, cancer cells, immune cells, and fibroblasts, that in turn may also feedback to affect vascular function. Whilst much speculation remains surrounding the normal physiological role of LRG1, the weight of evidence that it exerts pathological effects is now compelling even though its acceptance as a mainstream pathogenic effector molecule is only just gaining traction. Amongst its effects, those it has on the vasculature are likely to be of substantial clinical importance in a wide range of diseases including cancer, chronic kidney disease, diabetic retinopathy, and emphysema. Indeed, its potential role as a major pathogenic mediator of systemic cardiovascular disease is only just beginning to be considered.
LRG1 has been found to be over-expressed in many disease tissues, where its local production, especially under chronic inflammatory conditions, appears to exacerbate pathological cell dysfunction. This is in line with its likely physiological role as a component of the repair mechanism. Thus, LRG1 can induce de-differentiation of epithelial, endothelial and pericyte cells to support the wound healing process, but under chronically stressed cell conditions, LRG1 is not switched off and its persistence has destabilizing effects resulting in aberrant pathological responses (Figure 2). In the context of vascular function, LRG1 can act on endothelial cells and mural cells affecting the fine interactive balance needed for a stable and mature vasculature (Figure 4). In its sustained and heightened presence, existing and new vessels become unstable and reactive resulting in vascular leakage, fragility, and the failure of new vessels to mature (Figure 3). It presents, therefore, an intriguing and potentially valuable therapeutic target. Critically, LRG1 is particularly attractive in the context of therapeutic targeting of TGF-β signaling as this has been fraught with setbacks, predominantly because TGF-β and its receptors all have critical housekeeping roles. To date no essential homeostatic role for LRG1 has been described rendering it a potentially more suitable therapeutic target. Thus, we reason that inhibiting LRG1 will block the pathogenic activity of TGF-β without disturbing these key homeostatic functions. We anticipate, therefore, that over the next decade our understanding of LRG1 biology will be substantially enhanced and that its therapeutic targeting in multiple indications will be well advanced.
Author contributions
AD: Visualization, Writing – original draft, Writing – review & editing. CC: Writing – review & editing. SEM: Writing – review & editing. JG: Conceptualization, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
Supported by grants awarded to JG and SEM from The British Heart Foundation (PG/16/50/32182), the Wellcome Trust (Investigator Awards 206413/Z/17/Z and 206413/B/17/Z), the UK Research and Innovation/Medical Research Council UK project grants G1000466 and MR/L002973/1; DPFS/DCS award G0902206 and MR/N006410/1; the Confidence in Concept award MC/PC/14118, the Diabetes UK grant 18/0005856, the UCL Proof of Concept fund and the UCL Technology Fund. JG and SEM were also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Conflict of interest
JG and SEM are founders of a company spun out by UCL Business to commercialize a LRG1 function-blocking therapeutic antibody developed through the UK Medical Research Council DPFS funding scheme. JG and SEM are members of the scientific advisory board and are shareholders of this company and named inventors on three patents related to LRG1 as a therapeutic target.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eelen G, Treps L, Li X, Carmeliet P. Basic and therapeutic aspects of angiogenesis updated. Circ Res. (2020) 127(2):310–29. doi: 10.1161/circresaha.120.316851
2. Dudley AC, Griffioen AW. Pathological angiogenesis: mechanisms and therapeutic strategies. Angiogenesis. (2023) 26(3):313–47. doi: 10.1007/s10456-023-09876-7
3. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. (2019) 176(6):1248–64. doi: 10.1016/j.cell.2019.01.021
4. Wu JB, Tang YL, Liang XH. Targeting VEGF pathway to normalize the vasculature: an emerging insight in cancer therapy. Onco Targets Ther. (2018) 11:6901–9. doi: 10.2147/ott.S172042
5. Zhuge Y, Zhang J, Qian F, Wen Z, Niu C, Xu K, et al. Role of smooth muscle cells in cardiovascular disease. Int J Biol Sci. (2020) 16(14):2741–51. doi: 10.7150/ijbs.49871
6. van Splunder H, Villacampa P, Martínez-Romero A, Graupera M. Pericytes in the disease spotlight. Trends Cell Biol. (2024) 34(1):58–71. doi: 10.1016/j.tcb.2023.06.001
7. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. (2005) 97(6):512–23. doi: 10.1161/01.RES.0000182903.16652.d7
8. Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. (2011) 21(2):193–215. doi: 10.1016/j.devcel.2011.07.001
9. von Tell D, Armulik A, Betsholtz C. Pericytes and vascular stability. Exp Cell Res. (2006) 312(5):623–9. doi: 10.1016/j.yexcr.2005.10.019
10. Xu Y, Kovacic JC. Endothelial to mesenchymal transition in health and disease. Annu Rev Physiol. (2023) 85:245–67. doi: 10.1146/annurev-physiol-032222-080806
11. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. (2011) 91(3):1071–121. doi: 10.1152/physrev.00038.2010
12. Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. (2001) 11(6):725–32. doi: 10.1016/S0959-440X(01)00266-4
13. Ng A, Xavier RJ. Leucine-rich repeat (LRR) proteins: integrators of pattern recognition and signaling in immunity. Autophagy. (2011) 7(9):1082–4. doi: 10.4161/auto.7.9.16464
14. Weivoda S, Andersen JD, Skogen A, Schlievert PM, Fontana D, Schacker T, et al. Elisa for human serum leucine-rich alpha-2-glycoprotein-1 employing cytochrome C as the capturing ligand. J Immunol Methods. (2008) 336(1):22–9. doi: 10.1016/j.jim.2008.03.004
15. O'Donnell LC, Druhan LJ, Avalos BR. Molecular characterization and expression analysis of leucine-rich alpha2-glycoprotein, a novel marker of granulocytic differentiation. J Leukocyte Biol. (2002) 72(3):478–85. doi: 10.1189/jlb.72.3.478
16. Ai J, Druhan LJ, Hunter MG, Loveland MJ, Avalos BR. LRG-accelerated differentiation defines unique G-CSFR signaling pathways downstream of PU.1 and C/EBPepsilon that modulate neutrophil activation. J Leukocyte Biol. (2008) 83(5):1277–85. doi: 10.1189/jlb.1107751
17. Liu C, Teo MHY, Pek SLT, Wu X, Leong ML, Tay HM, et al. A multifunctional role of leucine-rich Α-2-glycoprotein 1 in cutaneous wound healing under normal and diabetic conditions. Diabetes. (2020) 69(11):2467–80. doi: 10.2337/db20-0585
18. Bonaventura A, Liberale L, Carbone F, Vecchié A, Diaz-Cañestro C, Camici GG, et al. The pathophysiological role of neutrophil extracellular traps in inflammatory diseases. Thromb Haemostasis. (2018) 118(1):6–27. doi: 10.1160/th17-09-0630
19. Jemmerson R. Paradoxical roles of leucine-rich α(2)-glycoprotein-1 in cell death and survival modulated by transforming growth factor-beta 1 and cytochrome C. Front Cell Dev Biol. (2021) 9:744908. doi: 10.3389/fcell.2021.744908
20. Cummings C, Walder J, Treeful A, Jemmerson R. Serum leucine-rich alpha-2-glycoprotein-1 binds cytochrome C and inhibits antibody detection of this apoptotic marker in enzyme-linked immunosorbent assay. Apoptosis. (2006) 11(7):1121–9. doi: 10.1007/s10495-006-8159-3
21. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. (2015) 347(6220):1260419. doi: 10.1126/science.1260419
22. He Y, Tacconi C, Dieterich LC, Kim J, Restivo G, Gousopoulos E, et al. Novel blood vascular endothelial subtype-specific markers in human skin unearthed by single-cell transcriptomic profiling. Cells. (2022) 11(7):1111. doi: 10.3390/cells11071111
23. Camilli C, Hoeh AE, De Rossi G, Moss SE, Greenwood J. LRG1: an emerging player in disease pathogenesis. J Biomed Sci. (2022) 29(1):6. doi: 10.1186/s12929-022-00790-6
24. De Rossi G, Da Vitoria Lobo ME, Greenwood J, Moss SE. LRG1 as a novel therapeutic target in eye disease. Eye (Lond). (2022) 36(2):328–40. doi: 10.1038/s41433-021-01807-4
25. Wang X, Abraham S, McKenzie JAG, Jeffs N, Swire M, Tripathi VB, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature. (2013) 499(7458):306–11. doi: 10.1038/nature12345
26. Dritsoula A, Dowsett L, Pilotti C, O'Connor MN, Moss SE, Greenwood J. Angiopathic activity of LRG1 is induced by the il-6/STAT3 pathway. Sci Rep. (2022) 12(1):4867. doi: 10.1038/s41598-022-08516-2
27. He L, Feng A, Guo H, Huang H, Deng Q, Zhao E, et al. LRG1 mediated by ATF3 promotes growth and angiogenesis of gastric cancer by regulating the SRC/STAT3/VEGFA pathway. Gastric Cancer. (2022) 25(3):527–41. doi: 10.1007/s10120-022-01279-9
28. Kwan YP, Teo MHY, Lim JCW, Tan MS, Rosellinny G, Wahli W, et al. LRG1 promotes metastatic dissemination of melanoma through regulating eGFR/STAT3 signalling. Cancers (Basel). (2021) 13(13):3279. doi: 10.3390/cancers13133279
29. Singhal M, Gengenbacher N, Abdul Pari AA, Kamiyama M, Hai L, Kuhn BJ, et al. Temporal multi-omics identifies LRG1 as a vascular niche instructor of metastasis. Sci Transl Med. (2021) 13(609):eabe6805. doi: 10.1126/scitranslmed.abe6805
30. Yamamoto M, Takahashi T, Serada S, Sugase T, Tanaka K, Miyazaki Y, et al. Overexpression of leucine-rich α2-glycoprotein-1 is a prognostic marker and enhances tumor migration in gastric cancer. Cancer Sci. (2017) 108(10):2052–60. doi: 10.1111/cas.13329
31. Zhong B, Cheng B, Huang X, Xiao Q, Niu Z, Chen YF, et al. Colorectal cancer-associated fibroblasts promote metastasis by up-regulating LRG1 through stromal il-6/STAT3 signaling. Cell Death Dis. (2021) 13(1):16. doi: 10.1038/s41419-021-04461-6
32. O’Connor MN, Kallenberg DM, Camilli C, Pilotti C, Dritsoula A, Jackstadt R, et al. LRG1 destabilizes tumor vessels and restricts immunotherapeutic potency. Med. (2021) 2(11):1231–52.e10. doi: 10.1016/j.medj.2021.10.002
33. Hong Q, Cai H, Zhang L, Li Z, Zhong F, Ni Z, et al. Modulation of transforming growth factor-β-induced kidney fibrosis by leucine-rich ⍺-2 glycoprotein-1. Kidney Int. (2022) 101(2):299–314. doi: 10.1016/j.kint.2021.10.023
34. Liu TT, Luo R, Yang Y, Cheng YC, Chang D, Dai W, et al. LRG1 mitigates renal interstitial fibrosis through alleviating capillary rarefaction and inhibiting inflammatory and pro-fibrotic cytokines. Am J Nephrol. (2021) 52(3):228–38. doi: 10.1159/000514167
35. Yang FJ, Hsieh CY, Shu KH, Chen IY, Pan SY, Chuang YF, et al. Plasma leucine-rich α-2-glycoprotein 1 predicts cardiovascular disease risk in End-stage renal disease. Sci Rep. (2020) 10(1):5988. doi: 10.1038/s41598-020-62989-7
36. Yang Y, Luo R, Cheng Y, Liu T, Dai W, Li Y, et al. Leucine-rich α2-glycoprotein-1 upregulation in plasma and kidney of patients with lupus nephritis. BMC Nephrol. (2020) 21(1):122. doi: 10.1186/s12882-020-01782-0
37. Hisata S, Racanelli AC, Kermani P, Schreiner R, Houghton S, Palikuqi B, et al. Reversal of emphysema by restoration of pulmonary endothelial cells. J Exp Med. (2021) 218(8):e20200938. doi: 10.1084/jem.20200938
38. Li R, Zhao X, Liu P, Wang D, Chen C, Wang Y, et al. Differential expression of serum proteins in chronic obstructive pulmonary disease assessed using label-free proteomics and bioinformatics analyses. Int J Chron Obstruct Pulmon Dis. (2022) 17:2871–91. doi: 10.2147/copd.S383976
39. Nitkin CR, Xia S, Menden H, Yu W, Xiong M, Heruth DP, et al. Fosl1 is a novel mediator of endotoxin/lipopolysaccharide-induced pulmonary angiogenic signaling. Sci Rep. (2020) 10(1):13143. doi: 10.1038/s41598-020-69735-z
40. Chen J, Zhang Z, Feng L, Liu W, Wang X, Chen H, et al. LRG1 silencing attenuates ischemia-reperfusion renal injury by regulating autophagy and apoptosis through the TGFβ1- Smad1/5 signaling pathway. Arch Biochem Biophys. (2024) 753:109892. doi: 10.1016/j.abb.2024.109892
41. Gao Y, Zhou J, Xie Z, Wang J, Ho CK, Zhang Y, et al. Mechanical strain promotes skin fibrosis through LRG-1 induction mediated by ELK1 and ERK signalling. Commun Biol. (2019) 2:359. doi: 10.1038/s42003-019-0600-6
42. Honda H, Fujimoto M, Serada S, Urushima H, Mishima T, Lee H, et al. Leucine-rich α-2 glycoprotein promotes lung fibrosis by modulating TGF-β signaling in fibroblasts. Physiol Rep. (2017) 5(24):e13556. doi: 10.14814/phy2.13556
43. Liu C, Lim ST, Teo MHY, Tan MSY, Kulkarni MD, Qiu B, et al. Collaborative regulation of LRG1 by TGF-β1 and PPAR-β/δ modulates chronic pressure overload-induced cardiac fibrosis. Circ Heart Fail. (2019) 12(12):e005962. doi: 10.1161/circheartfailure.119.005962
44. Nakajima H, Nakajima K, Serada S, Fujimoto M, Naka T, Sano S. The involvement of leucine-rich α-2 glycoprotein in the progression of skin and lung fibrosis in bleomycin-induced systemic sclerosis model. Mod Rheumatol. (2021) 31(6):1120–8. doi: 10.1080/14397595.2021.1883841
45. Park HN, Song MJ, Choi YE, Lee DH, Chung JH, Lee ST. LRG1 promotes ECM integrity by activating the TGF-β signaling pathway in fibroblasts. Int J Mol Sci. (2023) 24(15):12445. doi: 10.3390/ijms241512445
46. Sarkar A, Chakraborty D, Kumar V, Malhotra R, Biswas S. Upregulation of leucine-rich alpha-2 glycoprotein: a key regulator of inflammation and joint fibrosis in patients with severe knee osteoarthritis. Front Immunol. (2022) 13:1028994. doi: 10.3389/fimmu.2022.1028994
47. Sng MK, Chan JSK, Teo Z, Phua T, Tan EHP, Wee JWK, et al. Selective deletion of PPARβ/δ in fibroblasts causes dermal fibrosis by attenuated LRG1 expression. Cell Discov. (2018) 4:15. doi: 10.1038/s41421-018-0014-5
48. Yu B, Yang L, Song S, Li W, Wang H, Cheng J. LRG1 facilitates corneal fibrotic response by inducing neutrophil chemotaxis via STAT3 signaling in alkali-burned mouse corneas. Am J Physiol Cell Physiol. (2021) 321(3):C415–28. doi: 10.1152/ajpcell.00517.2020
49. Altıncık SA, Yıldırımçakar D, Avcı E, Özhan B, Girişgen İ, Yüksel S. Plasma leucine-rich α-2-glycoprotein 1—a novel marker of diabetic kidney disease in children and adolescents with type 1 diabetes Mellitus? Pediatr Nephrol. (2023) 38(12):4043–9. doi: 10.1007/s00467-023-06019-4
50. Chen C, Chen X, Huang H, Han C, Qu Y, Jin H, et al. Elevated plasma and vitreous levels of leucine-rich-α2-glycoprotein are associated with diabetic retinopathy progression. Acta Ophthalmol (Copenh). (2019) 97(3):260–4. doi: 10.1111/aos.13633
51. Gurung RL, Dorajoo R, Yiamunaa M, Liu JJ, Pek SLT, Wang J, et al. Association of genetic variants for plasma LRG1 with rapid decline in kidney function in patients with type 2 diabetes. J Clin Endocrinol Metab. (2021) 106(8):2384–94. doi: 10.1210/clinem/dgab268
52. Haku S, Wakui H, Azushima K, Haruhara K, Kinguchi S, Ohki K, et al. Early enhanced leucine-rich α-2-glycoprotein-1 expression in glomerular endothelial cells of type 2 diabetic nephropathy model mice. BioMed Res Int. (2018) 2018:2817045. doi: 10.1155/2018/2817045
53. Hong Q, Zhang L, Fu J, Verghese DA, Chauhan K, Nadkarni GN, et al. LRG1 promotes diabetic kidney disease progression by enhancing TGF-β-induced angiogenesis. J Am Soc Nephrol. (2019) 30(4):546–62. doi: 10.1681/asn.2018060599
54. Li W, Wang X, Cheng J, Li J, Wang Q, Zhou Q, et al. Leucine-rich α-2-glycoprotein-1 promotes diabetic corneal epithelial wound healing and nerve regeneration via regulation of matrix metalloproteinases. Exp Eye Res. (2020) 196:108060. doi: 10.1016/j.exer.2020.108060
55. Liu JJ, Liu S, Wang J, Pek SLT, Lee J, Gurung RL, et al. Urine leucine-rich α-2 glycoprotein 1 (LRG1) predicts the risk of progression to end-stage kidney disease in patients with type 2 diabetes. Diabetes Care. (2023) 46(2):408–15. doi: 10.2337/dc22-1611
56. Liu JJ, Pek SLT, Ang K, Tavintharan S, Lim SC. Plasma leucine-rich α-2-glycoprotein 1 predicts rapid eGFR decline and albuminuria progression in type 2 diabetes mellitus. J Clin Endocrinol Metab. (2017) 102(10):3683–91. doi: 10.1210/jc.2017-00930
57. Zhang X, Pek SLT, Tavintharan S, Sum CF, Lim SC, Ang K, et al. Leucine-rich α-2-glycoprotein predicts proliferative diabetic retinopathy in type 2 diabetes. J Diabetes Complicat. (2019) 33(9):651–6. doi: 10.1016/j.jdiacomp.2019.05.021
58. Yao J, Chang X, He Q, Li H, Duan T, Wang K. Exosome enriched leucine-rich alpha-2-glycoprotein-1 and extracellular matrix protein 1 proteins induce abnormal placental angiogenesis in pregnant mice. Placenta. (2023) 143:45–53. doi: 10.1016/j.placenta.2023.09.008
59. Wang CH, Li M, Liu LL, Zhou RY, Fu J, Zhang CZ, et al. LRG1 expression indicates unfavorable clinical outcome in hepatocellular carcinoma. Oncotarget. (2015) 6(39):42118–29. doi: 10.18632/oncotarget.5967
60. Sun DC, Shi Y, Wang LX, Lv Y, Han QL, Wang ZK, et al. Leucine-rich alpha-2-glycoprotein-1, relevant with microvessel density, is an independent survival prognostic factor for stage iii colorectal cancer patients: a retrospective analysis. Oncotarget. (2017) 8(39):66550–8. doi: 10.18632/oncotarget.16289
61. Guldvik IJ, Zuber V, Braadland PR, Grytli HH, Ramberg H, Lilleby W, et al. Identification and validation of leucine-rich α-2-glycoprotein 1 as a noninvasive biomarker for improved precision in prostate cancer risk stratification. Eur Urol Open Sci. (2020) 21:51–60. doi: 10.1016/j.euros.2020.08.007
62. Hoefsmit EP, Völlmy F, Rozeman EA, Reijers ILM, Versluis JM, Hoekman L, et al. Systemic LRG1 expression in melanoma is associated with disease progression and recurrence. Cancer Res Commun. (2023) 3(4):672–83. doi: 10.1158/2767-9764.Crc-23-0015
63. Hughes K, Wickenden JA, Allen JE, Watson CJ. Conditional deletion of STAT3 in mammary epithelium impairs the acute phase response and modulates immune cell numbers during post-lactational regression. J Pathol. (2012) 227(1):106–17. doi: 10.1002/path.3961
64. Wang Y, Xu J, Zhang X, Wang C, Huang Y, Dai K, et al. Tnf-α-induced LRG1 promotes angiogenesis and mesenchymal stem cell migration in the subchondral bone during osteoarthritis. Cell Death Dis. (2017) 8(3):e2715. doi: 10.1038/cddis.2017.129
65. Naka T, Fujimoto M. LRG is a novel inflammatory marker clinically useful for the evaluation of disease activity in rheumatoid arthritis and inflammatory bowel disease. Immunol Med. (2018) 41(2):62–7. doi: 10.1080/13497413.2018.1481582
66. Honda H, Fujimoto M, Miyamoto S, Ishikawa N, Serada S, Hattori N, et al. Sputum leucine-rich alpha-2 glycoprotein as a marker of airway inflammation in asthma. PLoS One. (2016) 11(9):e0162672. doi: 10.1371/journal.pone.0162672
67. Shirai R, Hirano F, Ohkura N, Ikeda K, Inoue S. Up-regulation of the expression of leucine-rich alpha(2)-glycoprotein in hepatocytes by the mediators of acute-phase response. Biochem Biophys Res Commun. (2009) 382(4):776–9. doi: 10.1016/j.bbrc.2009.03.104
68. Makita N, Hizukuri Y, Yamashiro K, Murakawa M, Hayashi Y. Il-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. Int Immunol. (2015) 27(3):131–41. doi: 10.1093/intimm/dxu090
69. Zhu Z, Guo Y, Liu Y, Ding R, Huang Z, Yu W, et al. ELK4 promotes colorectal cancer progression by activating the neoangiogenic factor LRG1 in a noncanonical SP1/3-dependent manner. Adv Sci (Weinh). (2023) 10(32):e2303378. doi: 10.1002/advs.202303378
70. Feng J, Zhan J, Ma S. LRG1 promotes hypoxia-induced cardiomyocyte apoptosis and autophagy by regulating hypoxia-inducible factor-1α. Bioengineered. (2021) 12(1):8897–907. doi: 10.1080/21655979.2021.1988368
71. Thorin E, Labbé P, Lambert M, Mury P, Dagher O, Miquel G, et al. Angiopoietin-like proteins: cardiovascular biology and therapeutic targeting for the prevention of cardiovascular diseases. Can J Cardiol. (2023) 39(12):1736–56. doi: 10.1016/j.cjca.2023.06.002
72. Fernández-Hernando C, Suárez Y. ANGPTL4: a multifunctional protein involved in metabolism and vascular homeostasis. Curr Opin Hematol. (2020) 27(3):206–13. doi: 10.1097/moh.0000000000000580
73. Wu L, Chen L, Li L. Apelin/APJ system: a novel promising therapy target for pathological angiogenesis. Clin Chim Acta. (2017) 466:78–84. doi: 10.1016/j.cca.2016.12.023
74. Barandon L, Couffinhal T, Dufourcq P, Ezan J, Costet P, Daret D, et al. Frizzled a, a novel angiogenic factor: promises for cardiac repair. Eur J Cardiothorac Surg. (2004) 25(1):76–83. doi: 10.1016/s1010-7940(03)00506-2
75. Huang A, Huang Y. Role of SFRPS in cardiovascular disease. Ther Adv Chronic Dis. (2020) 11:2040622320901990. doi: 10.1177/2040622320901990
76. Shi CS, Shi GY, Chang YS, Han HS, Kuo CH, Liu C, et al. Evidence of human thrombomodulin domain as a novel angiogenic factor. Circulation. (2005) 111(13):1627–36. doi: 10.1161/01.Cir.0000160364.05405.B5
77. Wang J, Peng H, Timur AA, Pasupuleti V, Yao Y, Zhang T, et al. Receptor and molecular mechanism of AGGF1 signaling in endothelial cell functions and angiogenesis. Arterioscler Thromb Vasc Biol. (2021) 41(11):2756–69. doi: 10.1161/atvbaha.121.316867
78. Zhang B, Dietrich UM, Geng JG, Bicknell R, Esko JD, Wang L. Repulsive axon guidance molecule SLIT3 is a novel angiogenic factor. Blood. (2009) 114(19):4300–9. doi: 10.1182/blood-2008-12-193326
79. Selvaprithviraj V, Sankar D, Sivashanmugam A, Srinivasan S, Jayakumar R. Pro-angiogenic molecules for therapeutic angiogenesis. Curr Med Chem. (2017) 24(31):3413–32. doi: 10.2174/0929867324666170724142641
80. Zhao X, Chen J, Zhang C, Xie G, Othmane B, Kuang X, et al. LncRNA AGAP2-AS1 interacts with IGF2BP2 to promote bladder cancer progression via regulating LRG1 mRNA stability. Cell Signal. (2023) 111:110839. doi: 10.1016/j.cellsig.2023.110839
81. Cai D, Chen C, Su Y, Tan Y, Lin X, Xing R. LRG1 in pancreatic cancer cells promotes inflammatory factor synthesis and the angiogenesis of Huvecs by activating VEGFR signaling. J Gastrointest Oncol. (2022) 13(1):400–12. doi: 10.21037/jgo-21-910
82. Li Z, Zeng C, Nong Q, Long F, Liu J, Mu Z, et al. Exosomal leucine-rich-Alpha2-glycoprotein 1 derived from non-small-cell lung cancer cells promotes angiogenesis via TGF-β signal pathway. Mol Ther Oncolytics. (2019) 14:313–22. doi: 10.1016/j.omto.2019.08.001
83. Fan M, Li C, He P, Fu Y, Li M, Zhao X. Knockdown of long noncoding RNA-taurine-upregulated gene 1 inhibits tumor angiogenesis in ovarian cancer by regulating leucine-rich α-2-glycoprotein-1. Anticancer Drugs. (2019) 30(6):562–70. doi: 10.1097/cad.0000000000000734
84. Zhang J, Zhu L, Fang J, Ge Z, Li X. LRG1 modulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via HIF-1α activation. J Exp Clin Cancer Res. (2016) 35:29. doi: 10.1186/s13046-016-0306-2
85. Yin GN, Kim DK, Kang JI, Im Y, Lee DS, Han AR, et al. Latrophilin-2 is a novel receptor of LRG1 that rescues vascular and neurological abnormalities and restores diabetic erectile function. Exp Mol Med. (2022) 54(5):626–38. doi: 10.1038/s12276-022-00773-5
86. Yang P, Li S, Zhang H, Ding X, Tan Q. LRG1 accelerates wound healing in diabetic rats by promoting angiogenesis via the Wnt/β-catenin signaling pathway. Int J Low Extrem Wounds. (2022):15347346221081610. doi: 10.1177/15347346221081610
87. Fu J, Wei C, Zhang W, Schlondorff D, Wu J, Cai M, et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int. (2018) 94(2):326–45. doi: 10.1016/j.kint.2018.02.028
88. Tsuruta H, Yasuda-Yamahara M, Yoshibayashi M, Kuwagata S, Yamahara K, Tanaka-Sasaki Y, et al. Fructose overconsumption accelerates renal dysfunction with aberrant glomerular endothelial-mesangial cell interactions in db/db mice. Biochim Biophys Acta Mol Basis Dis. (2024) 1870(4):167074. doi: 10.1016/j.bbadis.2024.167074
89. Kumagai S, Nakayama H, Fujimoto M, Honda H, Serada S, Ishibashi-Ueda H, et al. Myeloid cell-derived LRG attenuates adverse cardiac remodelling after myocardial infarction. Cardiovasc Res. (2016) 109(2):272–82. doi: 10.1093/cvr/cvv273
90. Zhao G, Xue L, Weiner AI, Gong N, Adams-Tzivelekidis S, Wong J, et al. TGF-βr2 signaling coordinates pulmonary vascular repair after viral injury in mice and human tissue. Sci Transl Med. (2024) 16(732):eadg6229. doi: 10.1126/scitranslmed.adg6229
91. Song S, Cheng J, Yu BJ, Zhou L, Xu HF, Yang LL. LRG1 promotes corneal angiogenesis and lymphangiogenesis in a corneal alkali burn mouse model. Int J Ophthalmol. (2020) 13(3):365–73. doi: 10.18240/ijo.2020.03.01
92. Meng H, Song Y, Zhu J, Liu Q, Lu P, Ye N, et al. LRG1 promotes angiogenesis through upregulating the TGF-β1 pathway in ischemic rat brain. Mol Med Rep. (2016) 14(6):5535–43. doi: 10.3892/mmr.2016.5925
93. Grzesiak L, Amaya-Garrido A, Feuillet G, Malet N, Swiader A, Sarthou MK, et al. Leucine-rich alpha-2 glycoprotein 1 accumulates in complicated atherosclerosis and promotes calcification. Int J Mol Sci. (2023) 24(22):16537. doi: 10.3390/ijms242216537
94. Kallenberg D, Tripathi V, Javaid F, Pilotti C, George J, Davis S, et al. A humanized antibody against LRG1 that inhibits angiogenesis and reduces retinal vascular leakage. J bioRxiv. (2021):2020.07.25.218149. doi: 10.1101/2020.07.25.218149
95. Jeong JH, Ojha U, Lee YM. Pathological angiogenesis and inflammation in tissues. Arch Pharmacal Res. (2021) 44(1):1–15. doi: 10.1007/s12272-020-01287-2
96. Zhao Q, Molina-Portela MDP, Parveen A, Adler A, Adler C, Hock E, et al. Heterogeneity and chimerism of endothelial cells revealed by single-cell transcriptome in orthotopic liver tumors. Angiogenesis. (2020) 23(4):581–97. doi: 10.1007/s10456-020-09727-9
97. Örd T, Lönnberg T, Nurminen V, Ravindran A, Niskanen H, Kiema M, et al. Dissecting the polygenic basis of atherosclerosis via disease-associated cell state signatures. Am J Hum Genet. (2023) 110(5):722–40. doi: 10.1016/j.ajhg.2023.03.013
98. Lipski DA, Foucart V, Dewispelaere R, Caspers LE, Defrance M, Bruyns C, et al. Retinal endothelial cell phenotypic modifications during experimental autoimmune uveitis: a transcriptomic approach. BMC Ophthalmol. (2020) 20(1):106. doi: 10.1186/s12886-020-1333-5
99. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. (2002) 21(7):1743–53. doi: 10.1093/emboj/21.7.1743
100. Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. (2004) 23(20):4018–28. doi: 10.1038/sj.emboj.7600386
101. DiPietro LA. Angiogenesis and wound repair: when enough is enough. J Leukocyte Biol. (2016) 100(5):979–84. doi: 10.1189/jlb.4MR0316-102R
102. Jung K, Covington S, Sen CK, Januszyk M, Kirsner RS, Gurtner GC, et al. Rapid identification of slow healing wounds. Wound Repair Regen. (2016) 24(1):181–8. doi: 10.1111/wrr.12384
103. Gao Y, Xie Z, Ho C, Wang J, Li Q, Zhang Y, et al. LRG1 promotes keratinocyte migration and wound repair through regulation of HIF-1α stability. J Invest Dermatol. (2020) 140(2):455–64.e8. doi: 10.1016/j.jid.2019.06.143
104. Zhang M, Wang Y, Wang J, Li X, Ma A, Pan X. Serum LRG1 as a novel biomarker for cardioembolic stroke. Clin Chim Acta. (2021) 519:83–91. doi: 10.1016/j.cca.2021.04.002
105. Gu J, Liu C, Yao Y. Prognostic potency of plasma LRG1 measurement at multiple time points in acute ischemic stroke patients. Biomark Med. (2024) 18(5):181–90. doi: 10.2217/bmm-2023-0545
106. Savarraj JPJ, McBride DW, Park E, Hinds S, Paz A, Gusdon A, et al. Leucine-rich alpha-2-glycoprotein 1 is a systemic biomarker of early brain injury and delayed cerebral ischemia after subarachnoid hemorrhage. Neurocrit Care. (2023) 38(3):771–80. doi: 10.1007/s12028-022-01652-7
107. Cheng X, Wei H, Liu Y, Sun Y, Ye J, Lu P, et al. Relation between LRG1 and CD4(+) T cells, cognitive impairment and neurological function in patients with acute ischemic stroke. Biomark Med. (2024) 18(1):5–14. doi: 10.2217/bmm-2023-0674
108. Jin C, Shi Y, Shi L, Leak RK, Zhang W, Chen K, et al. Leveraging single-cell RNA sequencing to unravel the impact of aging on stroke recovery mechanisms in mice. Proc Natl Acad Sci U S A. (2023) 120(25):e2300012120. doi: 10.1073/pnas.2300012120
109. Jin J, Sun H, Liu D, Wang H, Liu Q, Chen H, et al. LRG1 promotes apoptosis and autophagy through the TGFβ-smad1/5 signaling pathway to exacerbate ischemia/reperfusion injury. Neuroscience. (2019) 413:123–34. doi: 10.1016/j.neuroscience.2019.06.008
110. Rust R. Insights into the dual role of angiogenesis following stroke. J Cereb Blood Flow Metab. (2020) 40(6):1167–71. 10.1177/0271678×2090681532065073
111. Ruan Z, Cao G, Qian Y, Fu L, Hu J, Xu T, et al. Single-cell RNA sequencing unveils LRG1’s role in cerebral ischemia‒reperfusion injury by modulating various cells. J Neuroinflammation. (2023) 20(1):285. doi: 10.1186/s12974-023-02941-4
112. Campochiaro PA. Ocular neovascularization. J Mol Med (Berl). (2013) 91(3):311–21. doi: 10.1007/s00109-013-0993-5
113. Lambert V, Lecomte J, Hansen S, Blacher S, Gonzalez ML, Struman I, et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat Protoc. (2013) 8(11):2197–211. doi: 10.1038/nprot.2013.135
114. Smith LE, Wesolowski E, McLellan A, Kostyk SK, D'Amato R, Sullivan R, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Visual Sci. (1994) 35(1):101–11. PMID: 7507904.
115. Schlecht A, Thien A, Wolf J, Prinz G, Agostini H, Schlunck G, et al. Immunosenescence in choroidal neovascularization (CNV)-transcriptional profiling of naïve and CNV-associated retinal myeloid cells during aging. Int J Mol Sci. (2021) 22(24):13318. doi: 10.3390/ijms222413318
116. Kim TW, Kang JW, Ahn J, Lee EK, Cho KC, Han BN, et al. Proteomic analysis of the aqueous humor in age-related macular degeneration (AMD) patients. J Proteome Res. (2012) 11(8):4034–43. doi: 10.1021/pr300080s
117. Koss MJ, Hoffmann J, Nguyen N, Pfister M, Mischak H, Mullen W, et al. Proteomics of vitreous humor of patients with exudative age-related macular degeneration. PLoS One. (2014) 9(5):e96895. doi: 10.1371/journal.pone.0096895
118. Mundo L, Tosi GM, Lazzi S, Pertile G, Parolini B, Neri G, et al. LRG1 expression is elevated in the eyes of patients with neovascular age-related macular degeneration. Int J Mol Sci. (2021) 22(16):8879. doi: 10.3390/ijms22168879
119. Nobl M, Reich M, Dacheva I, Siwy J, Mullen W, Schanstra JP, et al. Proteomics of vitreous in neovascular age-related macular degeneration. Exp Eye Res. (2016) 146:107–17. doi: 10.1016/j.exer.2016.01.001
120. Qu SC, Xu D, Li TT, Zhang JF, Liu F. iTRAQ-based proteomics analysis of aqueous humor in patients with dry age-related macular degeneration. Int J Ophthalmol. (2019) 12(11):1758–66. doi: 10.18240/ijo.2019.11.15
121. Zhang A, Fang H, Chen J, He L, Chen Y. Role of VEGF-a and LRG1 in abnormal angiogenesis associated with diabetic nephropathy. Front Physiol. (2020) 11:1064. doi: 10.3389/fphys.2020.01064
122. Zhou L, Shi DP, Chu WJ, Yang LL, Xu HF. LRG1 promotes epithelial-mesenchymal transition of retinal pigment epithelium cells by activating Nox4. Int J Ophthalmol. (2021) 14(3):349–55. doi: 10.18240/ijo.2021.03.03
123. Huang C, Ogawa R. The vascular involvement in soft tissue fibrosis-lessons learned from pathological scarring. Int J Mol Sci. (2021) 22(16):8879. doi: 10.3390/ijms21072542
124. Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol. (2021) 41(9):2357–69. doi: 10.1161/atvbaha.121.313788
125. Gao BB, Chen X, Timothy N, Aiello LP, Feener EP. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J Proteome Res. (2008) 7(6):2516–25. doi: 10.1021/pr800112g
126. Kim T, Kim SJ, Kim K, Kang UB, Lee C, Park KS, et al. Profiling of vitreous proteomes from proliferative diabetic retinopathy and nondiabetic patients. Proteomics. (2007) 7(22):4203–15. doi: 10.1002/pmic.200700745
127. Hase K, Kanda A, Hirose I, Noda K, Ishida S. Systemic factors related to soluble (pro)renin receptor in plasma of patients with proliferative diabetic retinopathy. PLoS One. (2017) 12(12):e0189696. doi: 10.1371/journal.pone.0189696
128. Lee R, Wong TY, Sabanayagam C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis (Lond). (2015) 2:17. doi: 10.1186/s40662-015-0026-2
129. Pek SL, Tavintharan S, Wang X, Lim SC, Woon K, Yeoh LY, et al. Elevation of a novel angiogenic factor, leucine-rich-α2-glycoprotein (LRG1), is associated with arterial stiffness, endothelial dysfunction, and peripheral arterial disease in patients with type 2 diabetes. J Clin Endocrinol Metab. (2015) 100(4):1586–93. doi: 10.1210/jc.2014-3855
130. Singh H, Yu Y, Suh MJ, Torralba MG, Stenzel RD, Tovchigrechko A, et al. Type 1 diabetes: urinary proteomics and protein network analysis support perturbation of lysosomal function. Theranostics. (2017) 7(10):2704–17. doi: 10.7150/thno.19679
131. Frudd K, Sivaprasad S, Raman R, Krishnakumar S, Revathy YR, Turowski P. Diagnostic circulating biomarkers to detect vision-threatening diabetic retinopathy: potential screening tool of the future? Acta Ophthalmol. (2022) 100(3):e648–68. doi: 10.1111/aos.14954
132. Chen C, Zhang J, Yu T, Feng H, Liao J, Jia Y. LRG1 contributes to the pathogenesis of multiple kidney diseases: a comprehensive review. Kidney Dis (Basel). (2024):1. doi: 10.1159/000538443
133. Baaten C, Vondenhoff S, Noels H. Endothelial cell dysfunction and increased cardiovascular risk in patients with chronic kidney disease. Circ Res. (2023) 132(8):970–92. doi: 10.1161/circresaha.123.321752
134. Simon M, Gröne HJ, Jöhren O, Kullmer J, Plate KH, Risau W, et al. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol. (1995) 268(2 Pt 2):F240–50. doi: 10.1152/ajprenal.1995.268.2.F240
135. Tanabe K, Maeshima Y, Sato Y, Wada J. Antiangiogenic therapy for diabetic nephropathy. BioMed Res Int. (2017) 2017:5724069. doi: 10.1155/2017/5724069
136. Lee H, Fujimoto M, Ohkawara T, Honda H, Serada S, Terada Y, et al. Leucine rich α-2 glycoprotein is a potential urinary biomarker for renal tubular injury. Biochem Biophys Res Commun. (2018) 498(4):1045–51. doi: 10.1016/j.bbrc.2018.03.111
137. Jiang WJ, Xu CT, Du CL, Dong JH, Xu SB, Hu BF, et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics. (2022) 12(1):324–39. doi: 10.7150/thno.63735
138. Fadini GP, Albiero M, Bonora BM, Avogaro A. Angiogenic abnormalities in diabetes mellitus: mechanistic and clinical aspects. J Clin Endocrinol Metab. (2019) 104(11):5431–44. doi: 10.1210/jc.2019-00980
139. Mohammad HMF, Galal Gouda S, Eladl MA, Elkazaz AY, Elbayoumi KS, Farag NE, et al. Metformin suppresses LRG1 and TGFβ1/ALK1-induced angiogenesis and protects against ultrastructural changes in rat diabetic nephropathy. Biomed Pharmacother. (2023) 158:114128. doi: 10.1016/j.biopha.2022.114128
140. Chung JO, Park SY, Cho DH, Chung DJ, Chung MY. Relationship between plasma leucine-rich α-2-glycoprotein 1 and urinary albumin excretion in patients with type 2 diabetes. Front Endocrinol (Lausanne). (2023) 14:1232021. doi: 10.3389/fendo.2023.1232021
141. Popova A, Vasiļvolfa A, Rācenis K, Erts R, Šlisere B, Saulīte AJ, et al. Leucine-rich alpha-2-glycoprotein (LRG-1) as a potential kidney injury marker in kidney transplant recipients. Ann Transplant. (2022) 27:e936751. doi: 10.12659/aot.936751
142. Low S, Moh A, Pandian B, Tan XL, Pek S, Zheng H, et al. Association between plasma LRG1 and lower cognitive function in Asians with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2024):dgad768. doi: 10.1210/clinem/dgad768
143. Kropski JA, Richmond BW, Gaskill CF, Foronjy RF, Majka SM. Deregulated angiogenesis in chronic lung diseases: a possible role for lung mesenchymal progenitor cells (2017 Grover conference series). Pulm Circ. (2018) 8(1):2045893217739807. doi: 10.1177/2045893217739807
144. Lucas A, Yasa J, Lucas M. Regeneration and repair in the healing lung. Clin Transl Immunol. (2020) 9(7):e1152. doi: 10.1002/cti2.1152
145. Ishida T, Kotani T, Serada S, Fujimoto M, Takeuchi T, Makino S, et al. Correlation of increased serum leucine-rich α2-glycoprotein levels with disease prognosis, progression, and activity of interstitial pneumonia in patients with dermatomyositis: a retrospective study. PLoS One. (2020) 15(6):e0234090. doi: 10.1371/journal.pone.0234090
146. Fujimoto M, Matsumoto T, Serada S, Tsujimura Y, Hashimoto S, Yasutomi Y, et al. Leucine-rich alpha 2 glycoprotein is a new marker for active disease of tuberculosis. Sci Rep. (2020) 10(1):3384. doi: 10.1038/s41598-020-60450-3
147. Tan DBA, Ito J, Peters K, Livk A, Lipscombe RJ, Casey TM, et al. Protein network analysis identifies changes in the level of proteins involved in platelet degranulation, proteolysis and cholesterol metabolism pathways in AECOPD patients. COPD. (2020) 17(1):29–33. doi: 10.1080/15412555.2019.1711035
148. Schiff HF, Walker NF, Ugarte-Gil C, Tebruegge M, Manousopoulou A, Garbis SD, et al. Integrated plasma proteomics identifies tuberculosis-specific diagnostic biomarkers. JCI Insight. (2024):e173273. doi: 10.1172/jci.insight.173273
149. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. (2022) 399(10342):2227–42. doi: 10.1016/S0140-6736(22)00470-6
150. He M, Borlak J. A genomic perspective of the aging human and mouse lung with a focus on immune response and cellular senescence. Immun Ageing. (2023) 20(1):58. doi: 10.1186/s12979-023-00373-5
151. Messner CB, Demichev V, Wendisch D, Michalick L, White M, Freiwald A, et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. (2020) 11(1):11–24.e4. doi: 10.1016/j.cels.2020.05.012
152. Demichev V, Tober-Lau P, Nazarenko T, Lemke O, Kaur Aulakh S, Whitwell HJ, et al. A proteomic survival predictor for COVID-19 patients in intensive care. PLoS Digit Health. (2022) 1(1):e0000007. doi: 10.1371/journal.pdig.0000007
153. Nie X, Qian L, Sun R, Huang B, Dong X, Xiao Q, et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell. (2021) 184(3):775–91.e14. doi: 10.1016/j.cell.2021.01.004
154. Mohammed Y, Goodlett DR, Cheng MP, Vinh DC, Lee TC, McGeer A, et al. Longitudinal plasma proteomics analysis reveals novel candidate biomarkers in acute COVID-19. J Proteome Res. (2022) 21(4):975–92. doi: 10.1021/acs.jproteome.1c00863
155. Park J, Kim H, Kim SY, Kim Y, Lee J-S, Dan K, et al. In-depth blood proteome profiling analysis revealed distinct functional characteristics of plasma proteins between severe and non-severe COVID-19 patients. Sci Rep. (2020) 10(1):22418. doi: 10.1038/s41598-020-80120-8
156. Pagani L, Chinello C, Risca G, Capitoli G, Criscuolo L, Lombardi A, et al. Plasma proteomic variables related to COVID-19 severity: an untargeted nLC-MS/MS investigation. Int J Mol Sci. (2023) 24(4):3570. doi: 10.3390/ijms24043570
157. Henderson LA, Canna SW, Schulert GS, Volpi S, Lee PY, Kernan KF, et al. On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheumatol. (2020) 72(7):1059–63. doi: 10.1002/art.41285
158. Mangalmurti N, Hunter CA. Cytokine storms: understanding COVID-19. Immunity. (2020) 53(1):19–25. doi: 10.1016/j.immuni.2020.06.017
159. Santa Cruz A, Mendes-Frias A, Oliveira AI, Dias L, Matos AR, Carvalho A, et al. Interleukin-6 is a biomarker for the development of fatal severe acute respiratory syndrome coronavirus 2 pneumonia. Front Immunol. (2021) 12:613422. doi: 10.3389/fimmu.2021.613422
160. Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect. (2020) 9(1):1123–30. doi: 10.1080/22221751.2020.1770129
161. Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. (2018) 17(6):395–412. doi: 10.1038/nrd.2018.45
162. Rose-John S. Blocking only the bad side of IL-6 in inflammation and cancer. Cytokine. (2021) 148:155690. doi: 10.1016/j.cyto.2021.155690
163. Kaye AG, Siegel R. The efficacy of il-6 inhibitor tocilizumab in reducing severe COVID-19 mortality: a systematic review. PeerJ. (2020) 8:e10322. doi: 10.7717/peerj.10322
164. Campochiaro C, Dagna L. The conundrum of interleukin-6 blockade in COVID-19. Lancet Rheumatol. (2020) 2(10):e579–e80. doi: 10.1016/S2665-9913(20)30287-3
165. Rubin EJ, Longo DL, Baden LR. Interleukin-6 receptor inhibition in COVID-19—cooling the inflammatory soup. N Engl J Med. (2021) 384(16):1564–5. doi: 10.1056/NEJMe2103108
166. Murthy S, Lee TC. Il-6 blockade for COVID-19: a global scientific call to arms. Lancet Respir Med. (2021) 9(5):438–40. doi: 10.1016/S2213-2600(21)00127-2
167. Serada S, Fujimoto M, Ogata A, Terabe F, Hirano T, Iijima H, et al. iTRAQ-based proteomic identification of leucine-rich alpha-2 glycoprotein as a novel inflammatory biomarker in autoimmune diseases. Ann Rheum Dis. (2010) 69(4):770–4. doi: 10.1136/ard.2009.118919
168. Muruganandam M, Ariza-Hutchinson A, Patel RA, Sibbitt WL Jr. Biomarkers in the pathogenesis, diagnosis, and treatment of systemic sclerosis. J Inflamm Res. (2023) 16:4633–60. doi: 10.2147/jir.S379815
169. Bălănescu P, Bălănescu E, Băicuș C, Bălănescu A. Circulatory cytokeratin 17, marginal zone B1 protein and leucine-rich α2-glycoprotein-1 as biomarkers for disease severity and fibrosis in systemic sclerosis patients. Biochem Med (Zagreb). (2022) 32(3):030707. doi: 10.11613/bm.2022.030707
170. Kawanami T, Kawanami-Iwao H, Takata T, Ishigaki Y, Tomosugi N, Takegami T, et al. Comprehensive analysis of protein-expression changes specific to immunoglobulin G4-related disease. Clin Chim Acta. (2021) 523:45–57. doi: 10.1016/j.cca.2021.08.025
171. Sun Y, Wang F, Zhou Z, Teng J, Su Y, Chi H, et al. Urinary proteomics identifying novel biomarkers for the diagnosis of adult-onset still’s disease. Front Immunol. (2020) 11:2112. doi: 10.3389/fimmu.2020.02112
172. Shimizu M, Inoue N, Mizuta M, Nakagishi Y, Yachie A. Serum leucine-rich α2-glycoprotein as a biomarker for monitoring disease activity in patients with systemic juvenile idiopathic arthritis. J Immunol Res. (2019) 2019:3140204. doi: 10.1155/2019/3140204
173. Fujimoto M, Serada S, Suzuki K, Nishikawa A, Ogata A, Nanki T, et al. Leucine-rich α2 -glycoprotein as a potential biomarker for joint inflammation during anti-interleukin-6 biologic therapy in rheumatoid arthritis. Arthritis Rheumatol. (2015) 67(8):2056–60. doi: 10.1002/art.39164
174. Tintor G, Jukić M, Šupe-Domić D, Jerončić A, Pogorelić Z. Diagnostic utility of serum leucine-rich α-2-glycoprotein 1 for acute appendicitis in children. J Clin Med. (2023) 12(7):2455. doi: 10.3390/jcm12072455
175. Aguilar-Cazares D, Chavez-Dominguez R, Carlos-Reyes A, Lopez-Camarillo C, Hernadez de la Cruz ON, Lopez-Gonzalez JS. Contribution of angiogenesis to inflammation and cancer. Front Oncol. (2019) 9:1399. doi: 10.3389/fonc.2019.01399
176. Alfaro S, Acuña V, Ceriani R, Cavieres MF, Weinstein-Oppenheimer CR, Campos-Estrada C. Involvement of inflammation and its resolution in disease and therapeutics. Int J Mol Sci. (2022) 23(18):10719. doi: 10.3390/ijms231810719
177. Mapp PI, Walsh DA. Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat Rev Rheumatol. (2012) 8(7):390–8. doi: 10.1038/nrrheum.2012.80
178. Liu X, Liu R, Croker BA, Lawlor KE, Smyth GK, Wicks IP. Distinctive pro-inflammatory gene signatures induced in articular chondrocytes by oncostatin M and il-6 are regulated by suppressor of cytokine signaling-3. Osteoarthritis Cartilage. (2015) 23(10):1743–54. doi: 10.1016/j.joca.2015.05.011
179. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. (2011) 7(1):33–42. doi: 10.1038/nrrheum.2010.196
180. Marrelli A, Cipriani P, Liakouli V, Carubbi F, Perricone C, Perricone R, et al. Angiogenesis in rheumatoid arthritis: a disease specific process or a common response to chronic inflammation? Autoimmun Rev. (2011) 10(10):595–8. doi: 10.1016/j.autrev.2011.04.020
181. Yanagimachi M, Fukuda S, Tanaka F, Iwamoto M, Takao C, Oba K, et al. Leucine-rich alpha-2-glycoprotein 1 and angiotensinogen as diagnostic biomarkers for Kawasaki disease. PLoS One. (2021) 16(9):e0257138. doi: 10.1371/journal.pone.0257138
182. Kimura Y, Yanagimachi M, Ino Y, Aketagawa M, Matsuo M, Okayama A, et al. Identification of candidate diagnostic serum biomarkers for Kawasaki disease using proteomic analysis. Sci Rep. (2017) 7:43732. doi: 10.1038/srep43732
183. Xie XF, Chu HJ, Xu YF, Hua L, Wang ZP, Huang P, et al. Proteomics study of serum exosomes in Kawasaki disease patients with coronary artery aneurysms. Cardiol J. (2019) 26(5):584–93. doi: 10.5603/CJ.a2018.0032
184. Kessel C, Koné-Paut I, Tellier S, Belot A, Masjosthusmann K, Wittkowski H, et al. An immunological axis involving interleukin 1β and leucine-rich-α2-glycoprotein reflects therapeutic response of children with Kawasaki disease: implications from the Kawakinra trial. J Clin Immunol. (2022) 42(6):1330–41. doi: 10.1007/s10875-022-01301-w
185. Umezawa N, Mizoguchi F, Maejima Y, Kimura N, Hasegawa H, Hosoya T, et al. Leucine-rich alpha-2 glycoprotein as a potential biomarker for large vessel vasculitides. Front Med (Lausanne). (2023) 10:1153883. doi: 10.3389/fmed.2023.1153883
186. Ishizaki J, Takemori A, Suemori K, Matsumoto T, Akita Y, Sada KE, et al. Targeted proteomics reveals promising biomarkers of disease activity and organ involvement in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Res Ther. (2017) 19(1):218. doi: 10.1186/s13075-017-1429-3
187. Kronbichler A, Lee KH, Denicolò S, Choi D, Lee H, Ahn D, et al. Immunopathogenesis of ANCA-associated vasculitis. Int J Mol Sci. (2020) 21(19):7319. doi: 10.3390/ijms21197319
188. Sorriento D, Iaccarino G. Inflammation and cardiovascular diseases: the most recent findings. Int J Mol Sci. (2019) 20(16):3879. doi: 10.3390/ijms20163879
189. Pang KT, Ghim M, Liu C, Tay HM, Fhu CW, Chia RN, et al. Leucine-rich α-2-glycoprotein 1 suppresses endothelial cell activation through Adam10-mediated shedding of TNF-α receptor. Front Cell Dev Biol. (2021) 9:706143. doi: 10.3389/fcell.2021.706143
190. Behzadifard M, Soleimani M. NETosis and SARS-COV-2 infection related thrombosis: a narrative review. Thromb J. (2022) 20(1):13. doi: 10.1186/s12959-022-00375-1
191. Vorobjeva NV, Chernyak BV. NETosis: molecular mechanisms, role in physiology and pathology. Biochemistry (Mosc). (2020) 85(10):1178–90. doi: 10.1134/s0006297920100065
192. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETS) in disease: potential anti-nets therapeutics. Clin Rev Allergy Immunol. (2021) 61(2):194–211. doi: 10.1007/s12016-020-08804-7
193. Lin M, Liu J, Zhang F, Qi G, Tao S, Fan W, et al. The role of leucine-rich alpha-2-glycoprotein-1 in proliferation, migration, and invasion of tumors. J Cancer Res Clin Oncol. (2022) 148(2):283–91. doi: 10.1007/s00432-021-03876-0
194. Göbel A, Rachner TD, Hoffmann O, Klotz DM, Kasimir-Bauer S, Kimmig R, et al. High serum levels of leucine-rich α-2 glycoprotein 1 (LRG-1) are associated with poor survival in patients with early breast cancer. Arch Gynecol Obstet. (2024). doi: 10.1007/s00404-024-07434-0
195. Andersen JD, Boylan KL, Jemmerson R, Geller MA, Misemer B, Harrington KM, et al. Leucine-rich alpha-2-glycoprotein-1 is upregulated in sera and tumors of ovarian cancer patients. J Ovarian Res. (2010) 3:21. doi: 10.1186/1757-2215-3-21
196. Choi YJ, Yoon W, Lee A, Han Y, Byun Y, Kang JS, et al. Diagnostic model for pancreatic cancer using a multi-biomarker panel. Ann Surg Treat Res. (2021) 100(3):144–53. doi: 10.4174/astr.2021.100.3.144
197. Ladd JJ, Busald T, Johnson MM, Zhang Q, Pitteri SJ, Wang H, et al. Increased plasma levels of the APC-interacting protein MAPRE1, LRG1, and IGFBP2 preceding a diagnosis of colorectal cancer in women. Cancer Prev Res (Phila). (2012) 5(4):655–64. doi: 10.1158/1940-6207.Capr-11-0412
198. Lee DH, Yoon W, Lee A, Han Y, Byun Y, Kang JS, et al. Multi-biomarker panel prediction model for diagnosis of pancreatic cancer. J Hepatobiliary Pancreat Sci. (2023) 30(1):122–32. doi: 10.1002/jhbp.986
199. Walker MJ, Zhou C, Backen A, Pernemalm M, Williamson AJ, Priest LJ, et al. Discovery and validation of predictive biomarkers of survival for non-small cell lung cancer patients undergoing radical radiotherapy: two proteins with predictive value. EBioMedicine. (2015) 2(8):841–50. doi: 10.1016/j.ebiom.2015.06.013
200. Wu J, Yin H, Zhu J, Buckanovich RJ, Thorpe JD, Dai J, et al. Validation of LRG1 as a potential biomarker for detection of epithelial ovarian cancer by a blinded study. PLoS One. (2015) 10(3):e0121112. doi: 10.1371/journal.pone.0121112
201. Luan L, Hu Q, Wang Y, Lu L, Ling J. Knockdown of lncRNA NEAT1 expression inhibits cell migration, invasion and EMT by regulating the miR-24-3p/LRG1 axis in retinoblastoma cells. Exp Ther Med. (2021) 21(4):367. doi: 10.3892/etm.2021.9798
202. Rathore M, Wright M, Martin D, Huang W, Taylor D, Miyagi M, et al. LRG1 is a novel Her3 ligand to promote growth in colorectal cancer. J bioRxiv. (2023):2023.02.18.529070. doi: 10.1101/2023.02.18.529070
203. Zhang Y, Luo Q, Wang N, Hu F, Jin H, Ge T, et al. LRG1 suppresses the migration and invasion of hepatocellular carcinoma cells. Med Oncol. (2015) 32(5):146. doi: 10.1007/s12032-015-0598-7
204. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. (2013) 31(17):2205–18. doi: 10.1200/jco.2012.46.3653
205. Wicks EE, Semenza GL. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest. (2022) 132(11):e159839. doi: 10.1172/jci159839
206. Magnussen AL, Mills IG. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br J Cancer. (2021) 125(3):324–36. doi: 10.1038/s41416-021-01330-z
207. Martin JD, Seano G, Jain RK. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu Rev Physiol. (2019) 81:505–34. doi: 10.1146/annurev-physiol-020518-114700
208. Teuwen LA, De Rooij L, Cuypers A, Rohlenova K, Dumas SJ, García-Caballero M, et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep. (2021) 35(11):109253. doi: 10.1016/j.celrep.2021.109253
209. Amer R, Tiosano L, Pe'er J. Leucine-rich α-2-glycoprotein-1 (LRG-1) expression in retinoblastoma. Invest Ophthalmol Visual Sci. (2018) 59(2):685–92. doi: 10.1167/iovs.17-22785
Keywords: LRG1, TGF-β, vascular dysfunction, inflammation, fibrosis, angiogenesis, vessel normalization, vascular
Citation: Dritsoula A, Camilli C, Moss SE and Greenwood J (2024) The disruptive role of LRG1 on the vasculature and perivascular microenvironment. Front. Cardiovasc. Med. 11:1386177. doi: 10.3389/fcvm.2024.1386177
Received: 14 February 2024; Accepted: 17 April 2024;
Published: 30 April 2024.
Edited by:
Margreet R. De Vries, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Kyung Lee, Icahn School of Medicine at Mount Sinai, United StatesRoberta Soares, University of Miami, United States
© 2024 Dritsoula, Camilli, Moss and Greenwood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Athina Dritsoula YXRoaW5hLmRyaXRzb3VsYS4wOUB1Y2wuYWMudWs=