 Jente R. A. Boen1,2
Jente R. A. Boen1,2 Andreas B. Gevaert1,3
Andreas B. Gevaert1,3 Amélie Dendooven4,5Dustin Krüger2
Amélie Dendooven4,5Dustin Krüger2 Michiel Tubeeckx2Jens Van Fraeyenhove2
Michiel Tubeeckx2Jens Van Fraeyenhove2 Tine Bruyns2
Tine Bruyns2 Vincent F. M. Segers2,3,†
Vincent F. M. Segers2,3,† Emeline M. Van Craenenbroeck1,3*†
Emeline M. Van Craenenbroeck1,3*†
- 1Research Group Cardiovascular Diseases, GENCOR Department, University of Antwerp, Wilrijk, Belgium
- 2Laboratory of Physiopharmacology, GENCOR Department, University of Antwerp, Wilrijk, Belgium
- 3Department of Cardiology, Antwerp University Hospital (UZA), Edegem, Belgium
- 4Department of Pathology, Ghent University Hospital, Gent, Belgium
- 5Laboratory of Experimental Medicine and Pediatrics, University of Antwerp, Wilrijk, Belgium
Aims: MiR-181c-5p overexpression associates with heart failure (HF) and cardiac damage, but the underlying pathophysiology remains unclear. This study investigated the effect of miR-181c-5p inhibition on cardiac function and fibrosis in a rodent model of diastolic dysfunction, and evaluated additional effects on kidney as relevant comorbid organ.
Methods and results: Diastolic dysfunction was induced in male C57/BL6J mice (n = 20) by combining high-fat diet, L-NG-nitroarginine methyl ester, and angiotensin II administration, and was compared to sham controls (n = 18). Mice were randomized to subcutaneous miR-181c-5p antagomiR (INH) or scrambled antagomiR injections (40 mg/kg/week). HF mice demonstrated diastolic dysfunction and increased fibrosis, which was attenuated by INH treatment. Remarkably, HF + INH animals had a threefold higher mortality rate (60%) compared to HF controls (20%). Histological examination revealed increased glomerular damage in all INH treated mice, and signs of thrombotic microangiopathy (TMA) in mice who died prematurely. Quantitative polymerase chain reaction demonstrated a miR-181c-5p-related downregulation of cardiac but not renal Tgfbr1 in HF + INH mice, while INH treatment reduced renal but not cardiac Vegfa expression in all mice.
Conclusion: This study demonstrates cardiac anti-fibrotic effects of miR-181c-5p inhibition in a rodent HF model through targeting of Tgfbr1 in the heart. Despite improved diastolic function, HF + INH mice had higher mortality due to increased predisposition for TMA, increased renal fibrosis and glomerular damage, associated with Vegfa downregulation in kidneys.
1 Introduction
Heart failure (HF) is a global health problem affecting 1%–3% of the adult population, with a 5-year mortality rate of 50%–75% (1–3). HF prevalence continues to rise due to an ageing population (1, 3–5), and leads to high hospitalization rates, high healthcare expenditures, and a low quality of life. It has been estimated that 50% of HF cases present with preserved ejection fraction (HFpEF; EF ≥ 50%), but a treatment that improves mortality is still not available for these patients (6, 7). HFpEF requires a personalized and highly organized care, including treatment of risk factors [hypertension, diabetes, hyperlipidemia, chronic kidney disease (CKD)] (7). Currently, the molecular mechanisms of HFpEF are incompletely understood. For instance, the driving mechanisms of left ventricular hypertrophy and myocardial fibrosis, and the links with non-cardiac comorbidities such as CKD remain largely undetermined (8, 9).
MicroRNAs (miRNAs) are small non-coding RNAs with a length of 18–24 nucleotides. They modulate cell–cell communication by transcriptional and post-transcriptional gene silencing/activation (10–13). MiRNA–target interactions depend on the abundance and accessibility of miRNAs and target mRNAs, timing, cell type and state (11, 14). MiRNAs control expression of over 60% of protein coding genes, highlighting their impact on cell physiology. Several miRNA-based therapies are under development in the field of cardiovascular disease (15). Specific miRNAs can potentially be used as biomarkers for noncommunicable diseases (16–18).
Recent studies demonstrate a role for miR-181c-5p in cardiovascular physiology, but its multi-organ crosstalk is understudied (19–23). In the heart, overexpression of miR-181c-5p is associated with enhanced propensity for HF development and cardiac damage (24–28). Increased levels of circulating miR-181c-5p are associated with diabetes-related HFpEF in elderly (29) and poor treatment response in HFpEF patients (30). Some studies suggest a cardioprotective effect of miR-181c-5p inhibition. Potential underlying mechanisms include improvement of endothelial and mitochondrial function, attenuation of myocardial fibrosis, and cardiorenal interaction, but these pathophysiologic mechanisms remain understudied. In this study we evaluate (1) the protective and anti-fibrotic role of systemic miR-181c-5p inhibition in HF progression using a rodent model with mild diastolic dysfunction, and (2) consider multi-organ effects at the level of the kidney as related comorbid organ in HF.
2 Materials and methods
2.1 Animals and experimental design
Diastolic dysfunction was induced in male C57/BL6J mice (Charles River Laboratories, n = 20) aged 15 weeks by combining three frequently used stimuli (31–33). For a period of six weeks animals were fed a 60% kcal high-fat diet (HFD, Research Diets Inc., D12492) together with L-NG-Nitro arginine methyl ester (L-NAME, Merck, N5751) administration in drinking water (0.5 g/L). In week five, angiotensin II (AngII, 1000 ng/kg/min, Merck; A9525) was co-administered for two weeks by osmotic minipumps (Alzet 1004, Charles River Laboratories) implanted subcutaneously under general anesthesia (4%–5% Sevoflurane). Control animals (n = 18) were fed normal diet and drinking water, and underwent sham-surgery at week five with PBS-filled vehicles. Two weeks after minipump implantation, invasive hemodynamic measurements were performed, and mice were euthanized at the age of 21 weeks by exsanguination.
All animals were housed in a facility with 12 h dark-light cycle and both food and water ad libitum (Figure 1). Animals that died prematurely without completion of all measurements (n = 7) were not included for analyses. 2 animals (INH group) were excluded because of fighting injuries during the study. All animal experiments were performed according to the latest Guide for the Care and Use of Laboratory Animals (National Institute of Health) and were approved by the Ethical Committee for Animal Testing at University of Antwerp (LA1100154), and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
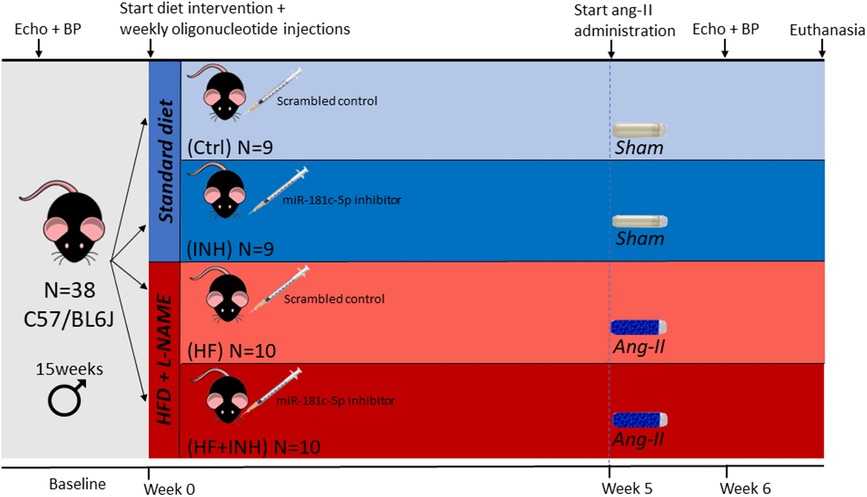
Figure 1. Experimental design of the study. The study consisted of four treatment groups: healthy controls with scrambled (Ctrl) or inhibitor injections (INH), and scrambled (HF) or inhibitor treated (HF + INH) animals with HF phenotype. We induced HF with diastolic dysfunction by administering NO synthase inhibitor L-NG-Nitro arginine methyl ester (L-NAME, 0.5 g/L) via drinking water and high fat diet (HFD, 60% fat) for six weeks, while angiotensin II (ang-II, 1,000 ng/kg/min) was co-administered in the final two weeks by osmotic minipumps. Healthy controls were given normal chow and drinking water, and sham-surgery was performed with PBS-vehicles. Subcutaneous injections (40 mg/kg) with either mmu-miR-181c-5p miRCURY power inhibitor or scrambled antagomiR occurred weekly for the duration of the study. After six weeks, we sacrificed the animals at the age of 21 weeks. BP, blood pressure.
2.2 Oligonucleotide administration
Both mice assigned to the HFD/l-NAME and the standard diet group were at baseline randomly allocated to either weekly injections with mmu-miR-181c-5p miRCURY power inhibitor (Qiagen, 339203, YCI0201911-FZA) or scrambled antagomiR (Qiagen, 339204, YCI0202244-FZA) (Supplementary Table S1). Injections were performed subcutaneously once weekly for the duration of the study at a dose of 40 mg/kg (Figure 1). This dosage was determined in previous dose response experiments, which also demonstrate inhibitor efficiency (Supplementary Figure S1). The study consisted of four treatment groups: (1) Ctrl: healthy controls with scrambled antagomiR injections; (2) INH: healthy controls with miR-181c-5p inhibitor injections; (3) HF: HF animals with scrambled antagomiR injections; (4) HF + INH: HF animals with miR-181c-5p inhibitor injections.
2.3 Blood pressure measurements
Blood pressure was measured in conscious mice at baseline and at week six with a CODA electronic sphygmomanometer, using a tail-cuff system (Kent Scientific Corporation). For each mouse, five acclimatization cycles followed by 20 consecutive blood pressure measurements were obtained. Data were extracted with CODA v4.1 Software. For each animal, the average of at least eight successful measurements was required.
2.4 Echocardiography
Systolic and diastolic function were analyzed with cardiac ultrasound using a Vevo 2100 Ultrasound imaging system (VisualSonics) with 55-MHz transducer (MS550D). Mice were held under light anesthesia [1.5%–2.5% v/v isoflurane (Forene Abbvie)] during imaging, temperature was held constant at 37 ± 1°C by a heating pad. For examination of left ventricular (LV) diameters the transducer was placed in a parasternal long axis position. Measurement was performed in M-mode, with the cursor position set at the level of the papillary muscles. Ejection fraction, LV mass and fractional shortening were calculated from these parameters. Left atrial (LA) diameter was measured near the aortic valve. Diastolic function was evaluated via apical four-chamber view using pulse wave doppler and tissue doppler imaging. Pulse wave doppler signals were obtained at the level of the LV inflow, tissue doppler measurements were obtained at the septal mitral annulus. E/e’ and E/A were derived from these parameters. Three images were taken from every parameter per animal and analyzed with Vevo Lab software (Version 3.2.0, VisualSonics) by a blinded observer.
2.5 Invasive hemodynamics
Mice were anesthetized using sevoflurane at concentrations of 8% v/v for induction and 4%–5% v/v for maintenance. The animals were placed on a heating pad (Kent Scientific) to maintain body temperature at approximately 37°C while free breathing. Throughout the experimental procedure, warm saline at 37°C was regularly applied to the open wound to prevent fluid loss. A pressure-volume catheter (Millar SPR-839) was inserted into the left ventricle through the right carotid artery using a closed-chest approach. To calculate parallel conductance (or parallel volume, Vp) and estimate the absolute ventricular volume, a catheter connected to a syringe containing hypertonic saline (15% NaCl) was introduced and secured in the right jugular vein, and a 10 µl-bolus injection of hypertonic saline was repeated three times. The end-diastolic pressure-volume relationship (EDPVR) was obtained through preload reduction induced by occluding the inferior vena cava, and fitting an exponential curve to end-diastolic pressure-volume points (LabChart v8, AD Instruments).
2.6 Organ weight
Organs were collected for histological and molecular analyses. Both cardiac mass and kidney mass were determined, as well as tibia length. Lung mass was weighted in wet and dry condition before and after a drying period of 72 h in an incubator at 37°C to evaluate lung congestion.
2.7 Histology
Frontal sections of renal and cardiac tissue were fixated in buffered formol for 24 h followed by a dehydration step in 60% isopropanol and embedding in paraffin. Sections of 5 µm were deparaffinized, rehydrated and treated with 0.9% hydrogen peroxide for blocking endogenous peroxidase activity. After antigen retrieval, permeabilization with TSB-tritonX and blocking with diluted serum solution (1/10), primary antibodies were incubated overnight at 4°C. Secondary antibodies were added for 30 min at room temperature after several washing steps. In case of HRP-labeled antibodies, an additional step with 3,3′-diaminobenzidine was required.
Slides were imaged with an Olympus BX43 light microscope equipped with Olympus Stream Motion software or Hamamatsu C10730-12 scanner microscope with NDP2 view software, while for isolectin B4 staining a Nikon Eclipse T1 fluorescence microscope was used with NIS advanced research software. All images were analyzed with ImageJ v1.53, and semiquantitative histological analyses were performed by two observers, all in blinded fashion.
2.7.1 Masson’s trichrome staining
Masson's Trichrome staining was used to visualize renal and cardiac interstitial and perivascular fibrosis. For perivascular fibrosis, 10 blood vessels per animal were randomly analyzed at 20X magnification. For interstitial fibrosis, 6 random images for each animal were analyzed at 20X magnification.
2.7.2 Hematoxylin eosin (H&E) staining and periodic acid-shiff staining (PAS)
H&E and PAS staining were used to evaluate renal morphology. For glomerular investigation, 50 glomeruli on two independent sections of each animal were randomly studied for glomerular size and for the presence of global/segmental glomerulosclerosis and mesangial matrix expansion. The average glomerular size and percentage of damaged glomeruli was calculated.
2.7.3 WT-1 staining
WT-1 staining (Abcam, ab224806) was used to determine podocyte density evaluated by counting the number of podocytes per glomerular area. 25 glomeruli were randomly analyzed for each animal at 20X magnification.
2.7.4 Cd31 staining
CD31 staining (Cell Signaling, 77699) was used to determine endothelial density in global and glomerular renal tissue. 25 glomeruli on two independent images were randomly analyzed for each animal.
2.7.5 Isolectin B4 staining
Isolectin B4 staining (Invitrogen, I21411) was used to determine capillary density in the heart. The number of capillaries was counted from five independent images for each animal at 40X magnification.
2.8 Reverse transcriptase quantitative polymerase chain reaction
The expression of renal and cardiac mRNA of potential target genes (Tgfbr1, Vegfa, Smad7) (Supplementary Table S2) and the expression level of miR-181c-5p was quantified with multiplex reverse transcriptase quantitative polymerase chain reaction (RT-qPCR). Potential targets were determined based on prediction programs, pathway analysis and current literature. Total RNA was homogenized with Precellys Tissue homogenizer (Bertin Instruments) and extracted from snapfrozen cardiac or renal tissue (10–20 mg) with mirVana™ PARIS™ RNA and Native Protein Purification Kit (Invitrogen, AM1556) according to manufacturer guidelines. RNA quality was controlled with Nanodrop ND-1000 spectrophotometer (Thermofisher Scientific). For mRNA targets, cDNA synthesis was performed with the Reverse transcription reagent kit (Applied Biosystems, N8080234) and subsequently used for qPCR using Taqman Universal master mix no UNG (Applied Biosystems, 4440047) and FAM-labeled TaqMan primers (Thermofisher Scientific). For miRNA levels, reverse transcription step started with cDNA-synthesis using TaqMan MicroRNA Reverse Transcription Kit (Thermofisher Scientific, 4366596), followed by a preamplification step with TaqMan™ PreAmp Master Mix (Thermofisher Scientific, 4488593). The qPCR step was performed as described above. Relative expression was analyzed with 2^(-ΔΔcq) method, using Gapdh and β-actin for renal and Ywhaz and Rplpo for cardiac mRNA reference genes (Supplementary Table S3). For miR-181c-5p expression, Sno142 and Sno202 were applied as normalization genes (Supplementary Table S4). No-reverse transcriptase control and no-template controls were used to assess sample or product contamination.
2.9 Statistics
Data were analyzed with GraphPad Prism 9.0. Graphs are represented with mean ± SD or in case of non-Gaussian distribution with median ± IQR. For comparison of two groups, a nonpaired two-tailed Student's T-test was performed. For statistical testing in multiple groups with several independent between-factors (i.e., treatment and phenotype) with or without a within-factor (i.e., time), a two-way ANOVA or mixed model ANOVA with Tukey's test or Šidák test for multiple comparisons was performed. Survival rate was analyzed with Kaplan Meier using a log-rank test. For end diastolic pressure-volume (EDPVR) curves, non-linear regression with least squares fit was performed. Alpha level was set at 0.05. Normality of data was tested with Shapiro-Wilk test. In case of skewed data, nonparametric testing or log-transformation was performed where possible.
3 Results
3.1 MiR-181c-5p inhibition mildly attenuated diastolic dysfunction
HF mice showed impaired diastolic function, partly attenuated by inhibitor treatment (Figure 2).
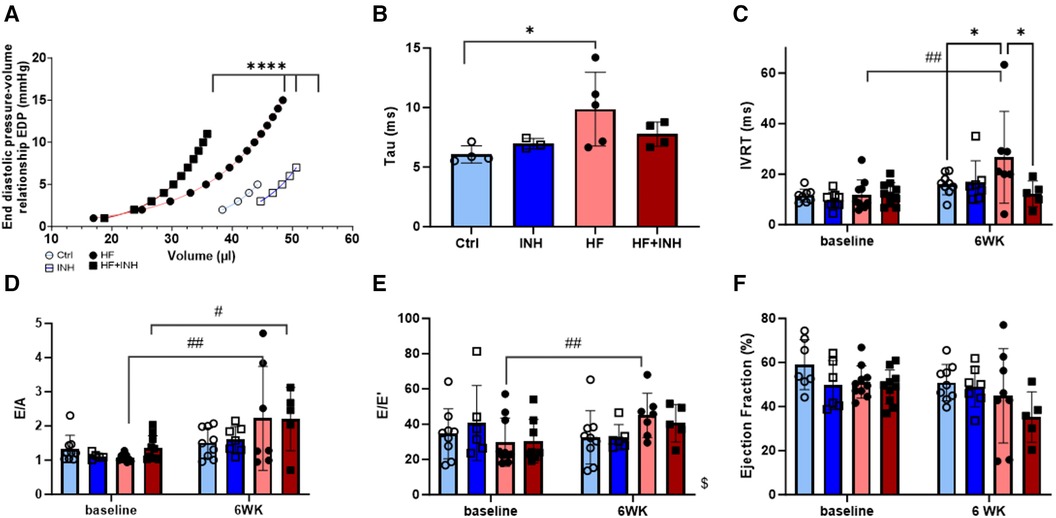
Figure 2. Cardiac function determined by invasive hemodynamics and echocardiography. Invasive hemodynamics validate diastolic dysfunction in HF mice with (A) leftwards shift of end diastolic pressure volume curve, and (B) increase of relaxation constant tau. Echocardiographic measures of diastolic dysfunction in HF mice illustrate an increase of (C) isovolumic relaxation time (IVRT), (D) E/A ratio over a period of six weeks— E/A represents the early (E) and late (A) ventricular filling velocities— and (E) E/e’ ratio over a period of six weeks — E/e’ represents the early mitral inflow over early mitral annular diastolic velocity. (F) Ejection fraction. N = 3-5 per group for invasive hemodynamics, N = 5-10 per group for echocardiography. Mixed-effect model for repeated measurements over time with Tukey's test or Šidák test for multiple comparisons. Two-way ANOVA with Šidák test for multiple corrections for single measurements. Non-linear regression with least square fit for edpvr curves. Mean ± SD, *p < 0.05, ****p < 0.0001, #p < 0.05 with baseline, ##p < 0.002 with baseline, $p < 0.05 interaction time × treatment. WK, week; IVRT, isovolumic relaxation time; EDP, end diastolic pressure; LV, left ventricular.
EDPVR curves demonstrate increased LV stiffness in HF mice represented by a leftward shift of the curve (Ctrl vs. HF p < 0.001). Inhibitor treatment in HF mice partially prevented this shift (p < 0.001), indicating a lower grade of LV stiffness. Similarly, HF mice show higher values for isovolumic relaxation constant tau (Ctrl 6.0 ± 0.6 ms, HF 9.3 ± 3.1 ms, p = 0.03), which are slightly reduced in HF + INH mice (HF + INH 7.8 ± 1.0 ms, p = 0.4 vs. HF). Echocardiography demonstrated a prolonged isovolumic relaxation time (IVRT) in HF mice (Ctrl 16 ± 4 ms, HF 27 ± 18 ms, p = 0.04), while inhibitor treatment normalized these values (HF + INH 14 ± 7 ms, p = 0.03 vs. HF). E/A and E/e’ did not differ between HF group and HF + INH group (E/A: HF 2.2 ± 1.5, HF + INH 2.2 ± 0.9, p > 0.99; E/e’: HF 45 ± 13, HF + INH 41 ± 11, p = 0.7). Ejection fraction was not significantly altered between groups over time, though a numerical drop was seen in the HF + INH group (42 ± 10%). None of the groups had evidence of pulmonary congestion.
3.2 MiR-181c-5p inhibition partly attenuated cardiac remodeling
HF mice displayed altered cardiac structure, which was partly attenuated by miR-181c-5p inhibition (Figure 3).
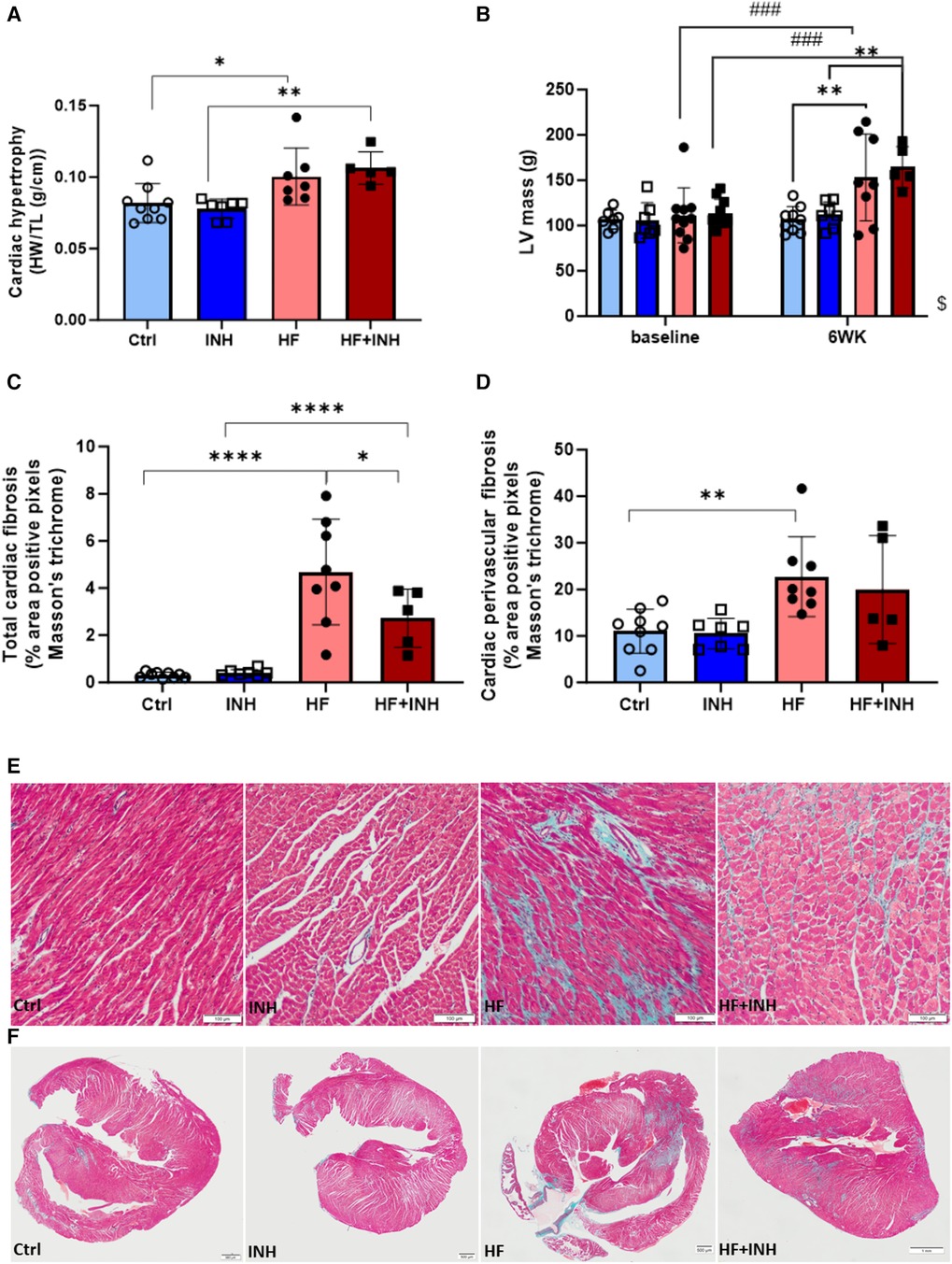
Figure 3. Cardiac morphology. (A) Heart weight over tibia length. (B) Left ventricular mass validated by echocardiography, (C–F) Total cardiac fibrosis and perivascular fibrosis determined by average percentage positive pixels with Masson's trichrome staining. N = 5-10 per group, 2-way ANOVA, mean ± SD. Mixed-effect model for repeated measurements over time with Tukey's test or Šidák test for multiple comparisons.*p < 0.05, **p < 0.002, ****p < 0.0001, ###p < 0.0002 with baseline, $p < 0.05 interaction time × treatment. HW, heart weight; TL, tibia length; LV, left ventricular; WK, week.
HF mice had an increased heart weight (Ctrl 82 ± 14 mg/mm, HF 100 ± 20 mg/mm, p = 0.03), which was confirmed by echocardiography (LV mass: Ctrl 107 ± 14 mg, HF 153 ± 48 mg, p = 0.002). Inhibitor administration did not prevent the development of cardiac hypertrophy (heart weight: HF + INH 106 ± 11 mg/mm, p = 0.7 vs. HF; LV mass: HF + INH 165 ± 23 mg, p = 0.9 vs. HF).
Histological analysis demonstrated increased cardiac fibrosis in HF mice (Ctrl 0.31 ± 0.12%, HF 4.96 ± 1.84%, p < 0.0001), which was significantly reduced by inhibitor treatment (HF + INH 2.72 ± 1.24%, p = 0.02 vs. HF). Looking at the location of fibrosis, increased perivascular fibrosis was seen in HF mice compared to healthy mice (Ctrl 11.02 ± 4.70% vs. HF 22.748 ± 8.55%, p = 0.005). However, perivascular fibrosis was not attenuated by inhibitor injection in HF animals (19.97 ± 11.56%, p = 0.7 vs. HF). Capillary density was similar between groups.
3.3 MiR-181c-5p inhibition induced additional renal damage in HF mice
HF mice had signs of renal damage (Figure 4), and inhibitor treatment induced additional renal damage in both healthy and HF mice.
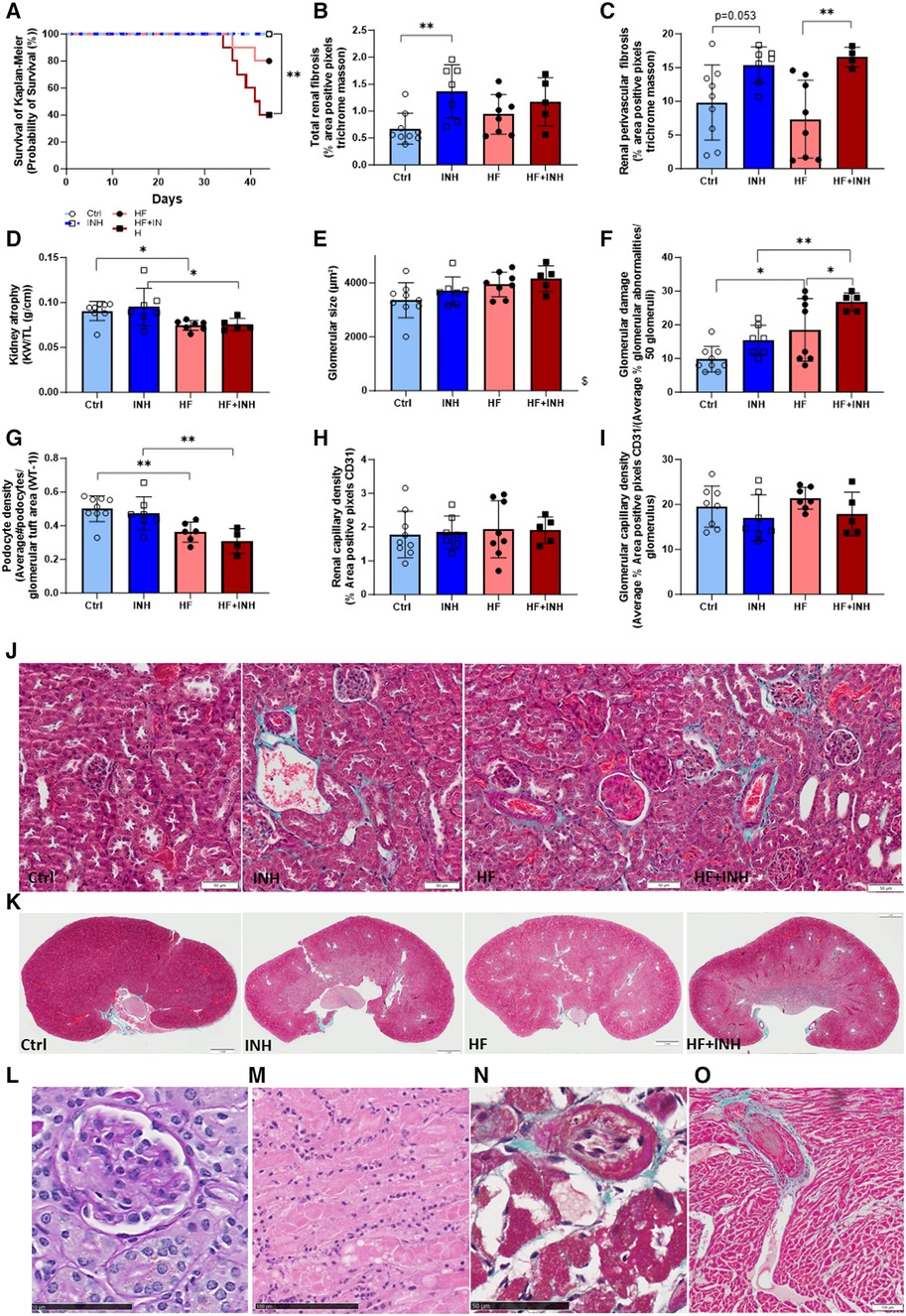
Figure 4. Renal morphology and thrombotic microangiopathy (TMA)-associated phenotype. (A) Survival analysis with Kaplan-Meier, (B,C,J,K) total and perivascular renal fibrosis determined by average percentage positive pixels with Masson's trichrome staining. (D) Kidney weight. Glomerular structure evaluated by (E) glomerular size determined on PAS staining, (F) glomerular damage determined on PAS staining, (G) podocyte density determined with WT-1 staining. (H,I) Renal and glomerular capillary density evaluated on CD31 staining. Premature deaths showed characteristic features of TMA in renal tissue, with (L) glomerular endothelial swelling and fibrine clots (*) on PAS staining, (M) tubular necrosis with inflammatory cells on H&E, while also extended to cardiac tissue showing (N) intimal thickening of cardiac artery and (O) thrombus formation (*) in necrotic region with inflammatory cells on Masson's trichrome staining. N = 5-9 per group. Kaplan Meier analysis with log-rank test. Two-way ANOVA with Šidák test for multiple comparisons. Mean ± SD, *p < 0.05, **p < 0.002, $p < 0.05 interaction time × treatment. KW, kidney weight; TL, tibia length.
HF mice had a reduced kidney weight compared to healthy mice, which was not affected by inhibitor administration (Ctrl 91 ± 11 mg/mm, HF 74 ± 5 mg/mm, p = 0.04; INH 95 ± 21 mg/mm, HF + INH 75 ± 7 mg/mm, p = 0.03). Likewise, podocyte density (which drives glomerulosclerosis and CKD) was lower in HF mice compared to healthy counterparts (Ctrl 0.501 ± 0.077/µm2, HF 0.362 ± 0.060/µm2, p = 0.007), but without any major effects of inhibitor treatment.
Renal interstitial and perivascular fibrosis was similar between Ctrl and HF mice (interstitial fibrosis: Ctrl 0.68 ± 0.29%, HF 0.94 ± 0.37%, p = 0.33; perivascular fibrosis: Ctrl 9.80 ± 5.56%, HF 7.35 ± 5.76%, p = 0.5). However, interstitial fibrosis significantly increased by inhibitor treatment in healthy mice, and to a lesser extent in HF conditions (interstitial: INH 1.37 ± 0.49%, p = 0.0036 vs. Ctrl; HF + INH 1.17 ± 0.45%, p = 0.5 vs. HF). We found large areas of perivascular fibrosis in all inhibitor-treated groups (INH 15.38 ± 2.70%, p = 0.053 vs. Ctrl; HF + INH 16.56 ± 1.46%, p = 0.008 vs. HF).
Histological H&E staining and PAS staining illustrated glomerular damage in HF animals, which was more pronounced in HF + INH group. HF animals had an enlarged glomerular size (p = 0.019) compared to healthy littermates, which is an indicator of glomerular hyperfiltration. HF mice also showed a higher percentage of additional glomerular damage— segmental and global glomerulosclerosis, mesangial matrix expansion and onset phase of sclerosis —(Ctrl 9.78 ± 3.80%, HF 18.50 ± 3.90%, p = 0.01; INH 15.43 ± 4.43%, HF + INH 26.80 ± 2.68%, p = 0.006), which was significantly increased when miR-181c-5p was inhibited (HF vs. HF + INH p = 0.04). We did not observe any significant differences in renal or glomerular capillary density.
3.4 MiR-181c-5p inhibition increased mortality through renal damage
HF mice had a higher mortality rate when administered with miR-181c-5p inhibitor (Figure 4, Supplementary Figure S2), likely due to a higher predisposition to thrombotic microangiopathy (TMA).
Mortality was 60% in the HF + INH group, 20% in the HF group, and 0% in both control and INH groups (log-rank p = 0.003, Figure 4A). All mice with premature death showed a phenotype compatible with TMA: tubular damage (acute tubular necrosis, tubular dilation) and glomerulomegaly, often combined with microvascular thrombi in glomerular capillaries and endothelial swelling. Fibrin clots were also detected in cardiac vessels of mice with renal TMA, and coexisted with necrotic regions and intimal thickening in the heart. At the level of the abdominal aorta, blood clots were detected during dissection in two HF + INH mice. Additionally, these mice demonstrated some degree of chronic renal damage under the form of glomerulosclerosis, interstitial fibrosis and tubular necrosis.
3.5 Tgfbr1 and vegfa as potential targets of miR-181c-5p
HF mice showed increased expression of cardiac Tgfbr1 and renal Vegfa (Figure 5), which was reduced by inhibitor treatment.
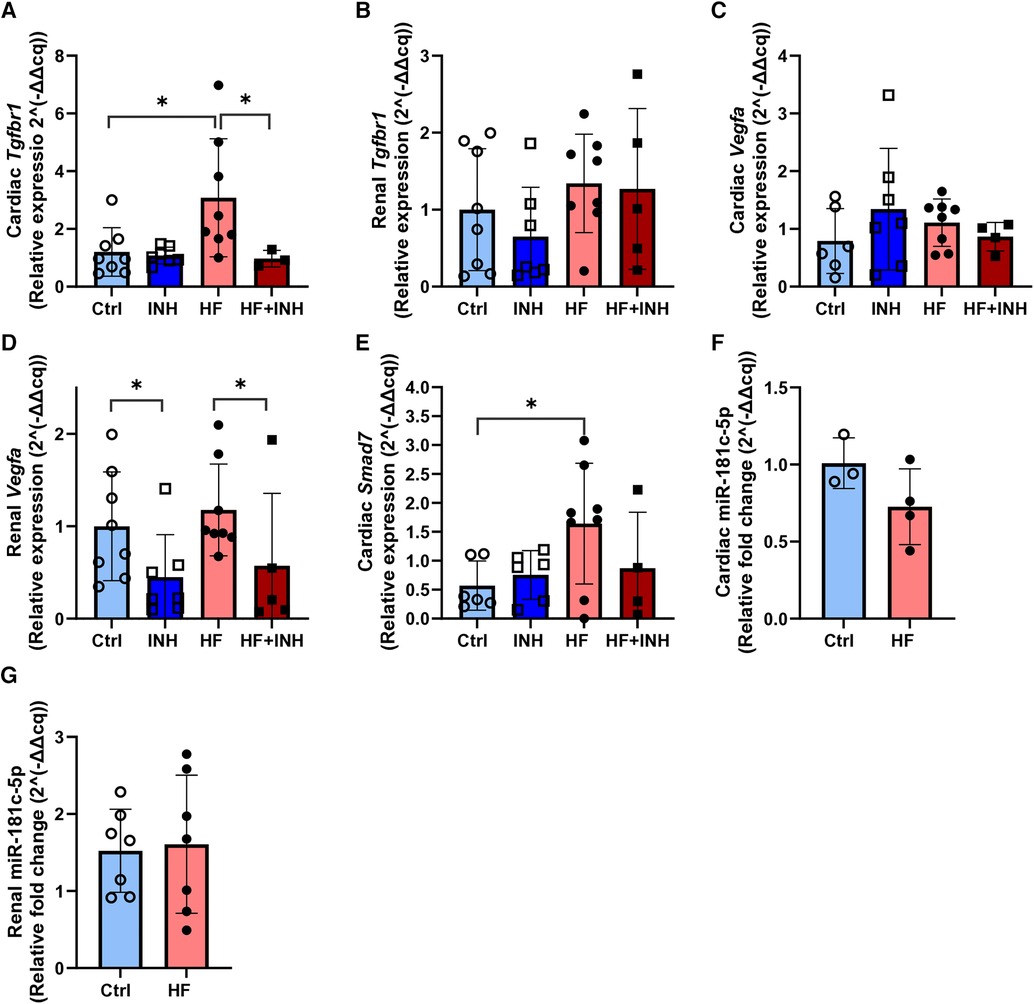
Figure 5. Expression of mRNA targets and miR-181c-5p in cardiac and renal tissue. (A,B) Cardiac and renal target expression of Tgfbr1. (C,D) Cardiac and renal target expression of Vegfa. (E) Cardiac expression of Smad7. (F,G) Cardiac and renal expression of miR-181c-5p. N = 3-8 per group. Evaluated with RT-qPCR and relative quantification according to 2^(-delta delta cq) method with two-way ANOVA followed by Tukey's test for multiple comparisons or unpaired t-test for miR-181c-5p. Mean ± SD, *p < 0.05.
In the heart, we detected higher expression levels of Tgfbr1 mRNA in HF mice compared to healthy animals (Ctrl 1.2 ± 0.8, HF 3.1 ± 2.0, p = 0.01). However, inhibition of miR-181c-5p normalized Tgfbr1 expression in the HF + INH group (HF + INH 0.97 ± 0.29, p = 0.04 vs. HF), which confirms that Tgfbr1 is a direct/indirect target of miR-181c-5p in the heart. Expression of cardiac Vegfa mRNA did not differ between groups. Smad7 mRNA was increased in the HF group (Ctrl 0.46 ± 0.37, HF 1.64 ± 1.04, p = 0.03), and numerically lowered in HF + INH, but without significant effect of inhibitor treatment (HF + INH 0.89 ± 0.97, p = 0.24 vs HF).
In contrast, Vegfa mRNA levels in renal tissue were halved in inhibitor-treated groups compared to scrambled groups, regardless of their phenotype (Ctrl 1 ± 0.59, INH 0.45 ± 0.46, p = 0.01; HF 1.18 ± 0.50, HF + INH 0.57 ± 0.78, p = 0.04). This demonstrates a miR-181c-5p-mediated regulation of Vegfa in kidney. Renal expression of Tgfbr1 was not influenced by treatment or phenotype.
3.6 Model-related effects on miR-181c-5p expression
The HF model did not influence cardiac and renal expression of miR-181c-5p (Figure 5).
Cardiac and renal expression of miR-181c-5p was analyzed in animals injected with scrambled antagomiR. MiR-181c-5p expression in cardiac tissue was slightly lower in HF mice, but without reaching statistical significance (Ctrl 1.01 ± 0.16, HF 0.73 ± 0.25, p = 0.14). Similarly, the HF phenotype did not affect renal expression of miR-181c-5p (Ctrl 1.52 ± 0.54, HF 1.61 ± 0.90, p = 0.83).
4 Discussion
We found that systemic inhibition of miR-181c-5p attenuated the development of myocardial fibrosis in a rodent model of mild diastolic dysfunction, in part by modulating Tgfbr1-signaling. However, the threefold higher mortality rate opposes these beneficial cardiac effects, and shifts the perspective from cardiac hero to renal villain: systemic miR-181c-5p inhibition caused renal damage (fibrosis, glomerular damage)—even in healthy mice— and predisposed to TMA development. These unexpected findings were explained by a miR-181c-5p-mediated reduction in renal Vegfa expression.
Multiple studies have shown that miR-181c-5p overexpression mostly harms the heart, while miR-181c-5p downregulation has cardioprotective effects (24, 25, 27, 34). Potential mechanisms by which increased miR-181c-5p affects cardiac structure and function include mitochondrial dysfunction, ROS production, inflammation, apoptosis and endothelial dysfunction (24–27, 34). A few studies have already proven the clinical value of miR-181c-5p: increased levels of circulating miR-181c-5p were detected in aged HFpEF patients with diabetes (29) and in HFpEF patients with a low response for exercise training (30). Recent in vitro studies in human cardiac fibroblasts identified SMAD7 as a new cardiac target of miR-181c-5p, which suggests a potential role for miR-181c-5p in cardiac fibrosis (29). In this study we now confirm in vivo an effect of miR-181c-5p on cardiac fibrosis through the TGF-β axis.
SMAD7, an inhibitor of TGF-β-signaling, was identified by the research group of Jankauskas et al. as a target of miR-181c-5p in cardiac fibroblasts, which we confirmed during our in vivo dose-finding studies (Supplementary Figure S1) in healthy cardiac tissue. While we expected a synergetic effect of miR-181c-5p inhibition on cardiac fibrosis by targeting both Tgfbr1 and Smad7 in HF conditions, we found that cardiac Smad7 did not significantly differed from the HF scrambled group, and was rather downregulated instead of upregulated as seen in healthy mice. The exact explanation remains unclear, but we assume that the complex miRNA target dynamics (11) —which are sensitive towards internal and external triggers, like disease conditions or stress— might underly our findings. Examination of multiple disease models at different time points and further pathway analysis could provide more insights into the miR-181c-5p/Smad7 axis. However, based on the expression results in this study, most anti-fibrotic effects probably associate with Tgfbr1-targeting. Therefore they clearly show a modulatory role for miR-181c-5p in Tgf-ß-signaling.
Our cardiac findings align with a study that reported reduced cardiac fibrosis after systemic miR-181c-5p inhibition in a mouse model with coronary artery disease through SIRT1-targeting (34). Interestingly, SIRT1 can inhibit Tgfbr1 levels and affect fibrosis-related TGF-β-signaling (35). We assume that this pathway might substantiate our findings; we identified an atypical downregulation of cardiac Tgfbr1 mRNA upon miR-181c-5p inhibition, which might refer to indirect targeting. However, the exact mechanism remains unclear; the research group of He X et al. identified TGFBR1 as a direct target of miR-181c-5p with luciferase reporter assay in glioblastoma cells (36). More research is required to unravel the exact Tgfbr1-interaction in cardiac tissue.
The effects of cardiomyocyte-specific Tgfbr1 knockdown have been studied in a murine model (37) and correspond to our study outcomes: cardiac alterations were restricted to cardiac fibrosis, with minimal impact on cardiac hypertrophy or function. This is in contrast to observations of Roman et al, who described antihypertrophic effects of transgenic miR-181c/d deletion in a murine model of obesity (24). We expect that differences in timing of miR-181c modulation, incomplete suppression of miR-181c by antagomiR, or supplementary effects of miR-181d might have led to this discrepancy (38).
The ability of microRNAs to target multiple mRNAs in a tissue-dependent manner challenges current research and requires precaution for data interpretation (39). However, most studies only focus on the organ of interest. A multi-organ approach can provide more insights about ongoing events in comorbid organs, certainly in case of systemic miRNA interventions. In this study we observed severe renal damage in mice treated with miR-181c-5p inhibitor, in both healthy and HF conditions, which can influence heart-kidney crosstalk. As a result, these multi-organ effects can bias our understanding of miR-181c-5p in the isolated heart and underestimate the effect on cardiac function. A targeted approach for cardiac-specific delivery of miRNAs could eliminate the influence of these multi-organ effects (40).
In kidney, one paper reported a protective effect of miR-181c mimics against cyclosporine-associated renal toxicity in vitro and in vivo by attenuating renal fibrosis and improving renal function (41). Wenjuan et al. illustrated that ectopic miR-181c administration suppressed the immune response in sepsis-induced acute kidney injury (42). Our results confirm these deleterious renal effects of low miR-181c-5p levels, and they demonstrate for the first time the adverse renal alterations, particularly interstitial fibrosis, in healthy mice. We demonstrate that these outcomes partially relate to miR-181c-5p-mediated effect on Vegfa-signaling. However, it is likely that other unrecognized pathways participate (like inflammation, ROS) (41, 42).
An earlier study postulated that miR-181c-5p indirectly targets Vegfa via HIF1ɑ in endothelial cells validated with a luciferase reporter assay focused on 3′ UTR (43). Similarly, sequence complementarity between miR-181c-5p seed sequence and Vegfa 5′UTR is limited, which supports an indirect regulation. Yet, the upstream target in this study remains unidentified. Although this miR-181c-5p/Vegfa -axis has not been reported in kidney earlier, endothelial modulation has been described in diabetic conditions, with contrasting effects across different tissues (19). In our study, we did not detect endothelial effects (such as renal capillary rarefaction) in mice treated with miR-181c-5p inhibitor.
Balanced expression of Vegfa is essential for renal physiology, where podocytes/pericytes are the exclusive source for its renal expression (44–46). Interestingly, clinical and preclinical data observed glomerular injury and renal TMA as a consequence of reduced renal Vegfa expression, for instance in patients treated with a Vegfa antibody (bevacizumab) and Vegfa knockout animal models (45, 47, 48). Patients treated with a less potent Vegfa antibody (ranibizumab) show less renal toxicity, which refers to dose-dependent effects (49). These observations strongly correspond with our histological findings in HF mice that died prematurely, and underpin the threefold higher predisposition for TMA development. The variable onset of TMA in this study likely associates with expression variation of Vegfa, model-dependent factors, and reflects the variable clinical onset (50). These findings support a recent study that emphasizes the regulatory role of miRNAs in different disease mechanisms of TMA (51).
In line with Vegfa knockdown studies, we identified very mild glomerular damage in healthy mice (52), but the extended areas of interstitial fibrosis were less common in literature. A few studies reported similar findings with increased levels of glomerulosclerosis and tubulointerstitial fibrosis after VEGF-receptor inhibition (53, 54). However, the underlying mechanism is unclear. Recent evidence points out to different direct and indirect pathways (Figure 6), while researchers also report the involvement of a pericyte-to-myofibroblast activation (54).
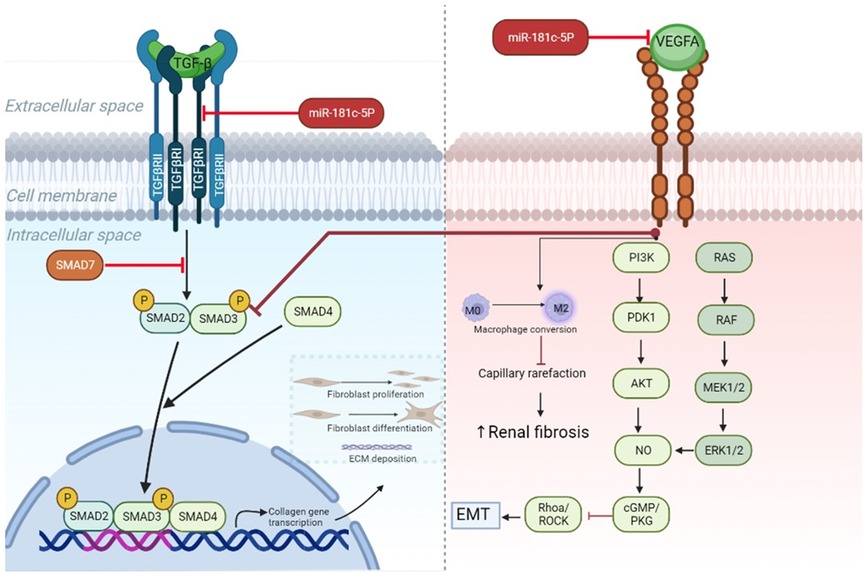
Figure 6. MiR-181c-5p-modulated pathways in heart and kidney. TGF-β downstream signaling induces activation of SMAD2/SMAD3 proteins and subsequent complex formation with SMAD4. By entering the nucleus, the protein complex can initiate transcription of genes involved in fibrosis, such as collagen, which after translation leads to extracellular matrix deposition, fibroblast proliferation, and differentiation from resting fibroblasts into myofibroblasts. All these processes drive fibrogenesis. In cardiac tissue miR-181c-5p inhibition reduces the levels of Tgfbr1— which is a type 1 receptor of TGF-β—and cooperates in this way to block myocardial fibrosis. Recent research demonstrates anti-fibrotic effects of Vegfa signaling in kidney (55). This can occur via direct targeting of TGF-β signaling by blocking SMAD3, while endothelial mesenchymal transition (EMT) is hampered through PI3K/AKT and ERK1/2 signaling activation. Furthermore, preventing capillary rarefaction by its proangiogenic properties, like conversion of resting M0 in activated M2 macrophages, hinders renal fibrosis. Inhibition of miR-181c-5p in renal tissue results in lowered expression levels of Vegfa, which reduces downstream signaling and could promote increased fibrotic activity. ECM, extracellular matrix; EMT, endothelial mesenchymal transition; Tgf-β, transforming growth factor β; Vegfa, vascular endothelial growth factor A; TgfbrI, transforming growth factor β receptor I.
4.1 Limitations
This study must be interpreted in the light of the following limitations. Unforeseen death, particularly in HF + INH group, resulted in fewer data points and limited collection of plasma and urine samples, especially in the most severely affected animals. Therefore we lack clear functional data of the kidney in this group (Supplementary Figure S3), and the study must be assessed in the light of survivorship bias in the HF + INH group (only the least affected animals were analyzed in full). While we acknowledge that ideally both sexes should be examined, we only studied male mice due to practical considerations. Results obtained in mice might not fully translate to humans due to interspecies variations. Finally, this short-term study might not capture the long-term effects of miR-181c-5p modulation on HF progression, and diastolic dysfunction was only mildly affected in the mouse model on echocardiography (E/A and E/e’ did not reach statistical significance).
4.2 Conclusions and future perspectives
Briefly, this study provided new insights in the dual role of miR-181c-5p inhibition in heart and kidney and (1) identifies Tgfbr1-signaling as direct/indirect target of miR-181c-5p in the failing heart, (2) confirms that miR-181c-5p inhibition mildly attenuates HF progression by reducing cardiac fibrosis, which makes it an interesting target for fibrotic treatment, (3) establishes miR-181c-5p as key player for renal structure in healthy and HF conditions, (4) identifies renal Vegfa-signaling as target of miR-181c-5p, direct or indirect, and (5) emphasizes the additional value of multi-organ approach in systemic miRNA interventions to monitor comorbid organs.
Based on the promising therapeutic potential of miR-181c-5p inhibition for cardiovascular diseases as stated in literature, these findings are of considerate interest regarding the comorbid nature of cardiovascular pathology. Despite the beneficial effects in the heart, miR-181c-5p downregulation has detrimental effects on the kidney. Future research might reveal if cardiac-specific inhibition of miR-181c-5p could function as new anti-fibrotic target in the heart and could improve cardiac function to a larger extent when adverse effects from multi-organ crosstalk are eliminated. Further validation of Vegfa and Tgfbr1 as organ-specific targets of miR-181c-5p in renal and cardiac tissue is required, but miR-181c-5p seems a valid target for further research on (cardio)-renal dysfunction and TMA.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Ethical Committee for Animal Testing at University of Antwerp. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JB: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. AG: Conceptualization, Investigation, Methodology, Project administration, Supervision, Visualization, Writing – review & editing. AD: Investigation, Methodology, Writing – review & editing. DK: Investigation, Methodology, Software, Writing – review & editing. MT: Investigation, Writing – review & editing. JV: Investigation, Writing – review & editing. TB: Investigation, Methodology, Writing – review & editing. VS: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – review & editing. EV: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by Special Research Fund (BOF-DOCPRO4) PhD fellowship [grant number 180182]; Geconcerteerde onderzoeksactie grant [GOA, grant number PID36444] from University of Antwerp; Fund for Scientific Research Flanders [Application number G021420N; VLIR/iBOF Grant 20-VLIR-iBOF-027 to VS.]; and senior clinical investigator grant from the Fund for Scientific Research (FWO) Flanders [grant numbers 1804320N, 1842219N to VS and EV].
Acknowledgments
The authors gratefully thank Mandy Vermont for her expert technical assistance in histological analysis, and Antwerp Centre for Advanced Microscopy (ACAM) for the use of Nikon T1 fluorescence microscope.
Conflict of interest
AG reports lecture/advisory board fees paid to his institution by Abbott, astrazeneca, Boehringer Ingelheim, Johnson and Johnson, Novartis, and Menarini, and research grants paid to his institution by Abbott and Boehringer Ingelheim; all not related to the work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1383046/full#supplementary-material
Abbreviations
Ang II, angiotensin II; CKD, chronic kidney disease; EDPVR, end diastolic pressure volume relationship; HF, heart failure; HFD, high fat diet; HFpEF, heart failure with preserved ejection fraction; IVRT, isovolumic relaxation time; L-NAME, L-NG-nitro arginine methyl ester; LA, left atrial; LV, left ventricular; RT-qPCR, reverse transcriptase quantitative polymerase chain reaction; Tgfbr1, TGF-β receptor I; TMA, thrombotic microangiopathy; VEGFA, vascular endothelial growth factor A.
References
1. Ponikowski P, Anker SD, AlHabib KF, Cowie MR, Force TL, Hu S, et al. Heart failure: preventing disease and death worldwide. ESC Heart Failure. (2014) 1(1):4–25. doi: 10.1002/ehf2.12005
2. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics—2021 update. Circulation. (2021) 143(8):e254–743. doi: 10.1161/CIR.0000000000000950
3. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. (2022) 118(17):3272–87. doi: 10.1093/cvr/cvac013
4. Roger VL. Epidemiology of heart failure. Circ Res. (2021) 128(10):1421–34. doi: 10.1161/CIRCRESAHA.121.318172
5. Chandra A, Polanczyk CA, Claggett BL, Vaduganathan M, Packer M, Lefkowitz MP, et al. Health-related quality of life outcomes in PARAGON-HF. Eur J Heart Fail. (2022) 24(12):2264–74. doi: 10.1002/ejhf.2738
6. Rahimi G, Tecson KM, Elsaid O, McCullough PA. Role of ischemic heart disease in Major adverse renal and cardiac events among individuals with heart failure with preserved ejection fraction (from the TOPCAT trial). Am J Cardiol. (2021) 142:91–6. doi: 10.1016/j.amjcard.2020.11.034
7. Gevaert AB, Kataria R, Zannad F, Sauer AJ, Damman K, Sharma K, et al. Heart failure with preserved ejection fraction: recent concepts in diagnosis, mechanisms and management. Heart. (2022) 108(17):1342–50. doi: 10.1136/heartjnl-2021-319605
8. Heinzel FR, Hohendanner F, Jin G, Sedej S, Edelmann F. Myocardial hypertrophy and its role in heart failure with preserved ejection fraction. J Appl Physiol (1985). (2015) 119(10):1233–42. doi: 10.1152/japplphysiol.00374.2015
9. Joslin JR, Lioudaki E, Androulakis E. Interrelation between heart failure with preserved ejection fraction and renal impairment. Rev Cardiovasc Med. (2022) 23(2):69. doi: 10.31083/j.rcm2302069
10. Xu L, Yang BF, Ai J. MicroRNA transport: a new way in cell communication. J Cell Physiol. (2013) 228(8):1713–9. doi: 10.1002/jcp.24344
11. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). (2018) 9:402. doi: 10.3389/fendo.2018.00402
12. Boen JRA, Gevaert AB, De Keulenaer GW, Van Craenenbroeck EM, Segers VFM. The role of endothelial miRNAs in myocardial biology and disease. J Mol Cell Cardiol. (2020) 138:75–87. doi: 10.1016/j.yjmcc.2019.11.151
13. Segers VFM, Gevaert AB, Boen JRA, Van Craenenbroeck EM, De Keulenaer GW. Epigenetic regulation of intercellular communication in the heart. Am J Physiol Heart Circ Physiol. (2019) 316(6):H1417–25. doi: 10.1152/ajpheart.00038.2019
14. Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. (2007) 23(1):175–205. doi: 10.1146/annurev.cellbio.23.090506.123406
15. Jalink EA, Schonk AW, Boon RA, Juni RP. Non-coding RNAs in the pathophysiology of heart failure with preserved ejection fraction. Front Cardiovasc Med. (2024) 10. doi: 10.3389/fcvm.2023.1300375
16. Wang J, Chen J, Sen S. MicroRNA as biomarkers and diagnostics. J Cell Physiol. (2016) 231(1):25–30. doi: 10.1002/jcp.25056
17. Condrat CE, Thompson DC, Barbu MG, Bugnar OL, Boboc A, Cretoiu D, et al. miRNAs as biomarkers in disease: latest findings regarding their role in diagnosis and prognosis. Cells. (2020) 9(2). doi: 10.3390/cells9020276
18. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discovery. (2017) 16(3):203–22. doi: 10.1038/nrd.2016.246
19. Solly EL, Psaltis PJ, Bursill CA, Tan JTM. The role of miR-181c in mechanisms of diabetes-impaired angiogenesis: an emerging therapeutic target for diabetic vascular complications. Front Pharmacol. (2021) 12:718679. doi: 10.3389/fphar.2021.718679
20. Yang G, Wu Y, Ye S. MiR-181c restrains nitration stress of endothelial cells in diabetic db/db mice through inhibiting the expression of FoxO1. Biochem Biophys Res Commun. (2017) 486(1):29–35. doi: 10.1016/j.bbrc.2017.02.083
21. Shen X, Li Y, Sun G, Guo D, Bai X. miR-181c-3p and -5p promotes high-glucose-induced dysfunction in human umbilical vein endothelial cells by regulating leukemia inhibitory factor. Int J Biol Macromol. (2018) 115:509–17. doi: 10.1016/j.ijbiomac.2018.03.173
22. Wu J, Zhang G, Xiong H, Zhang Y, Ding G, Ge J. miR-181c-5p mediates apoptosis of vascular endothelial cells induced by hyperoxemia via ceRNA crosstalk. Sci Rep. (2021) 11(1):16582. doi: 10.1038/s41598-021-95712-1
23. Yang C, Passos Gibson V, Hardy P. The role of MiR-181 family members in endothelial cell dysfunction and tumor angiogenesis. Cells. (2022) 11(10). doi: 10.3390/cells11101670
24. Roman B, Kaur P, Ashok D, Kohr M, Biswas R, O'Rourke B, et al. Nuclear-mitochondrial communication involving miR-181c plays an important role in cardiac dysfunction during obesity. J Mol Cell Cardiol. (2020) 144:87–96. doi: 10.1016/j.yjmcc.2020.05.009
25. Ge L, Cai Y, Ying F, Liu H, Zhang D, He Y, et al. miR-181c-5p exacerbates hypoxia/reoxygenation-induced cardiomyocyte apoptosis via targeting PTPN4. Oxid Med Cell Longev. (2019) 2019:1957920. doi: 10.1155/2019/1957920
26. Banavath HN, Roman B, Mackowski N, Biswas D, Afzal J, Nomura Y, et al. miR-181c activates mitochondrial calcium uptake by regulating MICU1 in the heart. J Am Heart Assoc. (2019) 8(24):e012919. doi: 10.1161/JAHA.119.012919
27. Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A, et al. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One. (2014) 9(5):e96820. doi: 10.1371/journal.pone.0096820
28. Wang H, Li J, Chi H, Zhang F, Zhu X, Cai J, et al. MicroRNA-181c targets Bcl-2 and regulates mitochondrial morphology in myocardial cells. J Cell Mol Med. (2015) 19(9):2084–97. doi: 10.1111/jcmm.12563
29. Jankauskas SS, Mone P, Avvisato R, Varzideh F, De Gennaro S, Salemme L, et al. miR-181c targets parkin and SMAD7 in human cardiac fibroblasts: validation of differential microRNA expression in patients with diabetes and heart failure with preserved ejection fraction. Mech Ageing Dev. (2023) 212:111818. doi: 10.1016/j.mad.2023.111818
30. Gevaert AB, Witvrouwen I, Van Craenenbroeck AH, Van Laere SJ, Boen JRA, Van de Heyning CM, et al. miR-181c level predicts response to exercise training in patients with heart failure and preserved ejection fraction: an analysis of the OptimEx-Clin trial. Eur J Prev Cardiol. (2021) 28(15):1722–33. doi: 10.1093/eurjpc/zwab151
31. Lim GB. New mouse model reveals nitrosative stress as a novel driver of HFpEF. Nat Rev Cardiol. (2019) 16(7):383. doi: 10.1038/s41569-019-0203-4
32. Withaar C, Meems LMG, Markousis-Mavrogenis G, Boogerd CJ, Silljé HHW, Schouten EM, et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117(9):2108–24. doi: 10.1093/cvr/cvaa256
33. van Ham WB, Kessler EL, Oerlemans M, Handoko ML, Sluijter JPG, van Veen TAB, et al. Clinical phenotypes of heart failure with preserved ejection fraction to select preclinical animal models. JACC Basic Transl Sci. (2022) 7(8):844–57. doi: 10.1016/j.jacbts.2021.12.009
34. Ma C, Zheng X, Wu X, Cheng J, Zhang K. microRNA-181c-5p stimulates the development of coronary artery disease by targeting SIRT1. Hellenic J Cardiol. (2023) 69:31–40. doi: 10.1016/j.hjc.2022.10.001
35. Liu Z-H, Zhang Y, Wang X, Fan X-F, Zhang Y, Li X, et al. SIRT1 Activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition. Biomed Pharmacother. (2019) 118:109227. doi: 10.1016/j.biopha.2019.109227
36. He X, Liu Z, Peng Y, Yu C. MicroRNA-181c inhibits glioblastoma cell invasion, migration and mesenchymal transition by targeting TGF-β pathway. Biochem Biophys Res Commun. (2016) 469(4):1041–8. doi: 10.1016/j.bbrc.2015.12.021
37. Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, et al. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J Clin Invest. (2011) 121(6):2301–12. doi: 10.1172/JCI44824
38. Joshua J, Caswell JL, Monné Rodriguez JM, Kipar A, O’Sullivan ML, Wood G, et al. MicroRNA profiling of the feline left heart identifies chamber-specific expression signatures in health and in advanced hypertrophic cardiomyopathy. J Mol Cell Cardiol Plus. (2023) 4:100037. doi: 10.1016/j.jmccpl.2023.100037
39. Diener C, Keller A, Meese E. The miRNA–target interactions: an underestimated intricacy. Nucleic Acids Res. (2023) 52(4):1544–57. doi: 10.1093/nar/gkad1142
40. Sahoo S, Kariya T, Ishikawa K. Targeted delivery of therapeutic agents to the heart. Nat Rev Cardiol. (2021) 18(6):389–99. doi: 10.1038/s41569-020-00499-9
41. Sun W, Min B, Du D, Yang F, Meng J, Wang W, et al. miR-181c protects CsA-induced renal damage and fibrosis through inhibiting EMT. FEBS Lett. (2017) 591(21):3588–99. doi: 10.1002/1873-3468.12872
42. Yu Y, Li X, Han S, Zhang J, Wang J, Chai J. miR-181c, a potential mediator for acute kidney injury in a burn rat model with following sepsis. Eur J Trauma Emerg Surg. (2022) 49:1035–45. doi: 10.1007/s00068-022-02124-5
43. Hourigan ST, Solly EL, Nankivell VA, Ridiandries A, Weimann BM, Henriquez R, et al. The regulation of miRNAs by reconstituted high-density lipoproteins in diabetes-impaired angiogenesis. Sci Rep. (2018) 8(1):13596. doi: 10.1038/s41598-018-32016-x
44. Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. (2004) 13(1):9–15. doi: 10.1097/00041552-200401000-00002
45. Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF Inhibition and renal thrombotic microangiopathy. N Engl J Med. (2008) 358(11):1129–36. doi: 10.1056/NEJMoa0707330
46. Lin SL, Chang FC, Schrimpf C, Chen YT, Wu CF, Wu VC, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. (2011) 178(2):911–23. doi: 10.1016/j.ajpath.2010.10.012
47. Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol. (2018) 13(2):300–17. doi: 10.2215/CJN.00620117
48. Richards A, Kavanagh D. Pathogenesis of thrombotic microangiopathy: insights from animal models. Nephron Exp Nephrol. (2009) 113(4):e97–103. doi: 10.1159/000235253
49. Hanna RM, Abdelnour L, Hasnain H, Selamet U, Kurtz I. Intravitreal bevacizumab-induced exacerbation of proteinuria in diabetic nephropathy, and amelioration by switching to ranibizumab. SAGE Open Med Case Rep. (2020) 8:2050313×20907033. doi: 10.1177/2050313x20907033
50. Wehrmann F, von Bergwelt-Baildon A, Schönermarck U. Severe hypertension and (renal) thrombotic microangiopathy: solving the puzzle. J Nephrol. (2023) 36(8):2175–7. doi: 10.1007/s40620-023-01659-z
51. Langlo KAR, Silva GJJ, Overrein TS, Adams V, Wisløff U, Dalen H, et al. Circulating microRNAs may serve as biomarkers for hypertensive emergency end-organ injuries and address underlying pathways in an animal model. Front Cardiovasc Med. (2020) 7:626699. doi: 10.3389/fcvm.2020.626699
52. Veron D, Villegas G, Aggarwal PK, Bertuccio C, Jimenez J, Velazquez H, et al. Acute podocyte vascular endothelial growth factor (VEGF-A) knockdown disrupts alphaVbeta3 integrin signaling in the glomerulus. PLoS One. (2012) 7(7):e40589. doi: 10.1371/journal.pone.0040589
53. Hara A, Wada T, Furuichi K, Sakai N, Kawachi H, Shimizu F, et al. Blockade of VEGF accelerates proteinuria, via decrease in nephrin expression in rat crescentic glomerulonephritis. Kidney Int. (2006) 69(11):1986–95. doi: 10.1038/sj.ki.5000439
54. Zhang Y, Nakano D, Guan Y, Hitomi H, Uemura A, Masaki T, et al. A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor–dependent pathway after renal injury in mice. Kidney Int. (2018) 94(3):524–35. doi: 10.1016/j.kint.2018.05.002
Keywords: heart failure, miR-181c-5p, renal dysfunction, thrombotic microangiopathy, fibrosis, diastolic dysfunction
Citation: Boen JRA, Gevaert AB, Dendooven A, Krüger D, Tubeeckx M, Van Fraeyenhove J, Bruyns T, Segers VFM and Van Craenenbroeck EM (2024) Divergent cardiac and renal effects of miR-181c-5p inhibition in a rodent heart failure model. Front. Cardiovasc. Med. 11:1383046. doi: 10.3389/fcvm.2024.1383046
Received: 6 February 2024; Accepted: 12 April 2024;
Published: 25 April 2024.
Edited by:
Marta Focardi, University of Siena, ItalyReviewed by:
Emma Louise Robinson, University of Colorado, United StatesSimona Greco, IRCCS San Donato Polyclinic, Italy
© 2024 Boen, Gevaert, Dendooven, Krüger, Tubeeckx, Van Fraeyenhove, Bruyns, Segers and Van Craenenbroeck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emeline M. Van Craenenbroeck ZW1lbGluZS52YW5jcmFlbmVuYnJvZWNrQHVhbnR3ZXJwZW4uYmU=
†These authors have contributed equally to this work and share last authorship