 Yukiomi Tsuji1*
Yukiomi Tsuji1* Masatoshi Yamazaki2
Masatoshi Yamazaki2 Masafumi Shimojo1
Masafumi Shimojo1 Satoshi Yanagisawa1Yasuya Inden1
Satoshi Yanagisawa1Yasuya Inden1 Toyoaki Murohara1
Toyoaki Murohara1
- 1Departments of Cardiovascular Research and Innovation, Cardiology and Advanced Cardiovascular Therapeutics, Nagoya University Graduate School of Medicine, Nagoya, Japan
- 2Department of Cardiology, Nagano Hospital, Soja and Medical Device Development and Regulation Research Center and Department of Precision Engineering, The University of Tokyo, Tokyo, Japan
Torsades de Pointes (TdP) refers to a polymorphic ventricular tachycardia (VT) with undulating QRS axis that occurs in long QT syndrome (LQTS), although the term has been used to describe polymorphic ventricular tachyarrhythmias in which QT intervals are not prolonged, such as short-coupled variant of TdP currently known as short-coupled ventricular fibrillation (VF) and Brugada syndrome. Extensive works on LQTS-related TdP over more than 50 years since it was first recognized by Dessertennes who coined the French term meaning “twisting of the points”, have led to current understanding of the electrophysiological mechanism that TdP is initiated by triggered activity due to early afterdepolarization (EAD) and maintained by reentry within a substrate of inhomogeneous repolarization. While a recently emerging notion that steep voltage gradients rather than EADs are crucial to generate premature ventricular contractions provides additions to the initiation mode, the research to elucidate the maintenance mechanism hasn't made much progress. The reentrant activity that produces the specific form of VT is not well characterized. We have conducted optical mapping in a rabbit model of electrical storm by electrical remodeling (QT prolongation) due to chronic complete atrioventricular block and demonstrated that a tissue-island with prolonged refractoriness due to enhanced late Na+ current (INa−L) contributes to the generation of drifting rotors in a unique manner, which may explain the ECG characteristic of TdP. Moreover, we have proposed that the neural Na+ channel NaV1.8-mediated INa−L may be a new player to form the substrate for TdP. Here we discuss TdP mechanisms by comparing the findings in electrical storm rabbits with recently published studies by others in simulation models and human and animal models of LQTS.
1 Introduction
Torsades de Pointes (TdP) is a polymorphic ventricular tachycardia (VT) with distinctive fluctuating morphology of QRS complexes that seem to magically twist around the baseline that occurs in congenital and acquired long QT syndrome (LQTS). Although the term has been used to describe polymorphic ventricular tachyarrhythmias in which QT intervals are not prolonged, such as short-coupled variant of TdP currently known as short-coupled ventricular fibrillation (VF) (1, 2) and Brugada syndrome (3), those have different mechanistic and therapeutic entity. TdP is better confined to polymorphic VT with marked QT interval prolongation (4). There have been extensive clinical and experimental studies on LQTS-related TdP [see a scientific statement from AHA/ACC (4) and a comprehensive review paper by Nattel (5)] since it was first described in an elderly patient with heart block by Dessertennes who coined the French term meaning “twisting of the points” (6). Current understanding of the electrophysiological mechanism of TdP is that it is initiated by a triggered beat due to early afterdepolarization (EAD) and maintained by reentry within a substrate of inhomogeneous repolarization. The issue, however, remains attractive because of existence of debates and controversies. Where are premature ventricular contractions (PVCs) that induce TdP originated from? How reentrant sources differ between TdP and other types of polymorphic VT? What predisposes TdP to degeneration into VF?
We have conducted optical mapping in a rabbit model of electrical storm by electrical remodeling (QT prolongation) due to chronic complete atrioventricular block (CAVB) and recently demonstrated that a tissue-island with prolonged refractoriness due to enhanced late Na+ current (INa−L) contributes to the generation of drifting rotors in a unique manner, which may explain the undulating QRS morphology characteristic of TdP (7). Moreover, we have proposed that neural Na+ channel NaV1.8-mediated late Na+-current (INa−L) may be a new player to form the substrate for TdP (7). This review article discusses TdP mechanisms by comparing the findings in electrical storm rabbits with recently published studies by others in simulation models and human and animal models of LQTS.
2 Consensus of arrhythmia mechanisms in LQTS
EAD-induced triggered activity is associated with both congenital and acquired LQTS, in which arrhythmias occur pause- or bradycardia-dependently. Prolongation of ventricular action potential duration (APD) prolongs the time window during which L-type calcium channel remains open and facilitates the generation of EADs. The channel blocker verapamil eliminates or reduces EADs, abolishes PVCs and suppresses TdP in congenital LQTS type 1 (LQT1) or type 2 (LQT2) caused by mutations in KCNQ1 or KCNH2 encoding KV7.1 or KV11.1 subunit responsible for the slowly or rapidly activating delayed rectifier potassium currents (IKs or IKr) respectively (8, 9) and in experimental models of LQT1 and 2 (10, 11) as well as type 3 (LQT3) (12) caused by mutations in SCN5A encoding sodium channel subunit NaV1.5. In addition, there is experimental evidence that some but not all drugs designated as IKr blockers can generate arrhythmias by augmenting INa−L (13). Significance of INa−L in the genesis of arrhythmogenic EADs is supported by the fact that class IB sodium channel blockers lidocaine and mexiletine are effective to suppress TdP in patients with LQT1, 2, and 3 (9, 14).
Delayed afterdepolarization (DAD)-induced triggered activity is also implicated in the mechanisms of LQTS. An increase in cytosolic Ca2+ concentrations due to prolongation of action potential causes DADs that are facilitated by rapid pacing and catecholamines. In patients who are mostly children with a much severe form of congenital LQTS, the onset of the arrhythmia is typically not bradycardia- or pause-dependent (15).
Cardiac Purkinje system plays an important role in the onset of TdP. Compared with ventricular myocytes, Purkinje cells are particularly susceptible to EAD and DAD because of the unique cellular electrophysiological property in human Purkinje fibers (16). APD is longer in Purkinje cells than in ventricular myocytes, which is attributed to larger INa−L and smaller inward rectifier potassium current (IK1) (16, 17). Repolarization reserve reduction in LQTS causes more enhanced APD prolongation in Purkinje cells. IKr blockade with quinidine induces EADs, occurring in 80% of canine Purkinje fibers, but not in ventricular myocardium (18). Focal activity generated in subendocardial Purkinje tissue is the primary trigger of TdP in a canine model of LQT3 (19). Catheter ablation targeting Purkinje potentials is useful in some congenital LQTS patients (20). Lower expression of Na+/K+-ATPase pump (16), together with larger INa−L, causes intracellular Na+ accumulation followed by secondary Ca2+-load elevation through Na+/Ca2+-exchanger, promoting DADs in Purkinje cells. Moreover, Purkinje fibers have 3 times higher transcript expression of CALM1 encoding calmodulin, a Ca2+-binding protein, than ventricular myocardium (16), potentially promoting activation of Ca2+/calmodulin-dependent kinase II (CaMKII), a Ca2+-activating signal molecule involved in the generation of EAD/DAD (21). Besides Purkinje fibers, EAD-induced triggered activity occurs in myocardium with remarkable APD prolongation. EADs and subsequent TdP events originate from epi- and mid-myocardial layers (10, 22) as well as subendocardial myocardium (12, 23, 24).
Functional reentry underlies the maintenance of TdP. LQTS patients have great dispersion of repolarization regions (25), which favor the occurrence of unidirectional block and reentrant activity. There have been proposals of electrophysiological mechanisms how the undulated QRS axis morphology is formed. El-Sherif and colleagues showed in a canine heart model of LQT3 that a transient bifurcation of the single scroll into 2 simultaneous scrolls that involved both right ventricle and left ventricle (LV) separately (26). Jalife and colleagues, based on the findings in experimental and simulation studies (see the Section 4.5) (27–30), have suggested that a drifting rotor(s) is responsible for TdP maintenance. Since the major laboratories reported in 1990s, however, any information on reentrant activation pattern that produces the specific type of VT has not been added.
3 Rabbit model of electrical storm associated with QT prolongation
3.1 Rotors anchored by a refractory island drive TdP
CAVB rabbits show biventricular hypertrophy due to sustained bradycardia and QT prolongation, most of which develop abnormal QTU complex and spontaneous TdP and die suddenly, the electrophysiological features similar to human LQTS (31, 32). When instrumented with implantable cardioverter-defibrillators (ICDs) and followed up for approximately 100 days, the animals have repetitive TdP as non-sustained VT and VF episodes. Electrical storm characterized by clustered VF episodes occurs in ≈50% (Figure 1) (33).
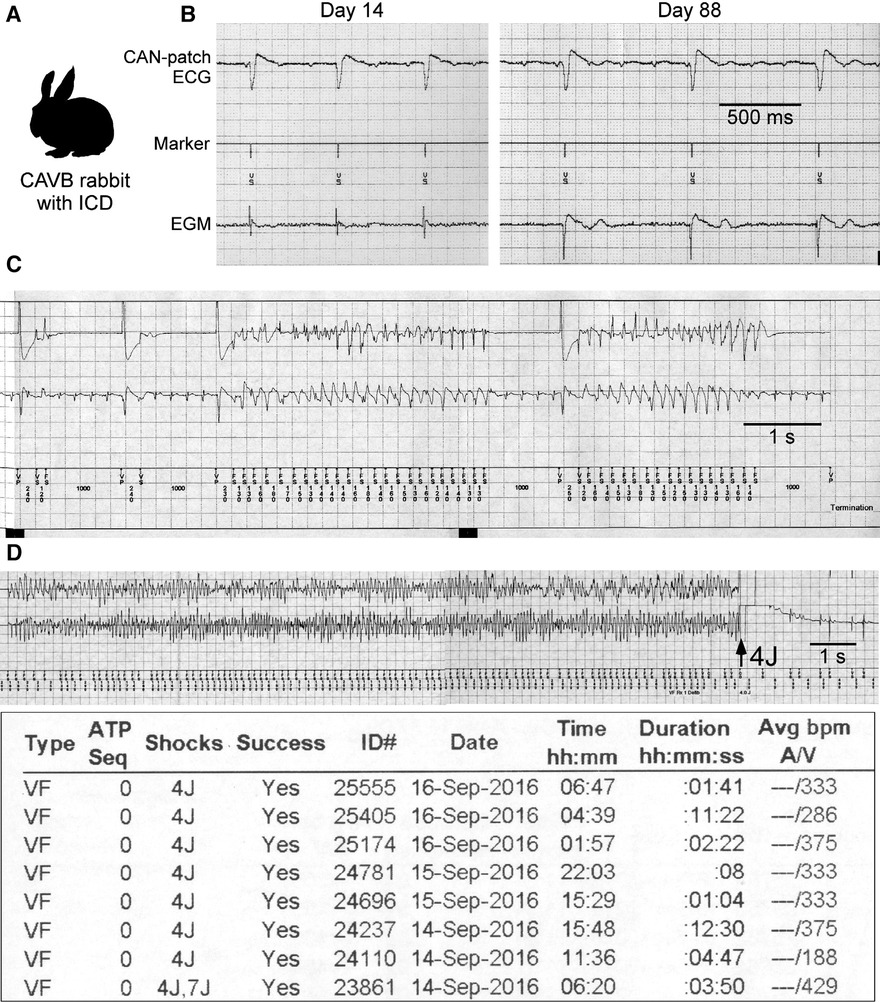
Figure 1. Rabbit model of electrical storm associated with QT prolongation. (A) Complete atrioventricular block (CAVB) creation and implantable cardioverter-defibrillator (ICD) implantation are performed. (B) CAN-patch electrode ECGs and electrograms (EGMs: 2 leads at the epicardial right ventricle) recorded during ICD interrogation at Day 14 and 88. (C,D) Examples of non-sustained VT (NSVT) and ventricular fibrillation (VF) episode and a VF episode report. Modified from Tsuji et al. and Yamazaki et al. (7, 33).
We have recently demonstrated in a perfused heart from an electrical storm rabbit that drifting rotors anchored by a refractory island drive TdP (7). The findings obtained from this animal model are briefly mentioned here with the illustrated summary (Figure 2).
a. Electrical storm hearts had an island-like regions with long APD at the LV base, leading to increased spatial APD dispersion, which were exacerbated by epinephrine (Figure 2A).
b. A long-lasting TdP event was successfully mapped from the initiation until termination. It was initiated by an exogeneous PVC coupled to an escape rhythm following a long pause that failed to enter the tissue-island with refractoriness and formed 5 reentrant waves at the edge of the island. Four disappeared by mutual annihilation and the remaining 1 rotational activity survived, resulting in the 1st beat of TdP. In addition, the PVC that triggered TdP had a spiky QRS complex, suggesting origination from Purkinje fibers (Figure 2B).
c. Phase map analysis of 49 beats during TdP showed that 32 beats (65%) had rotor typed activation including clockwise, counterclockwise, figure-of-8, and dual rotor, and that the remaining 14 (29%) had breakthrough and 3 (6%) one-way propagation patterns. In 46/49 beats (94%), activation patterns could be determined by mapping at restricted regions: inside the recording area of the anterior epicardial surface. The beat-to-beat intervals of 49 beats were almost identical regardless of the type of activation pattern. Note that rotor centers and breakthrough activations all distributed at the periphery of the island. From these observations, we concluded that 1 or 2 drifting rotor(s) around the island is responsible for TdP perpetuation and that the 17 beats with breakthrough and one-way propagation patterns are likely wavefronts propagating form intramural sources (Figures 2B,C).
d. The localized APD prolongation was attributed to INa−L enhancement probably due to upregulation of the neuronal Na+-channel NaV1.8 within myocardium (Figure 2D).
e. The long APD island and great APD dispersion were present in all of 14 electrical storm rabbit hearts optically mapped.
f. APD dispersion ex vivo corelated with the number of VF episodes in vivo. The association, together with the result of activation patterns of TdP, supports the mechanism described by Jalife and colleagues, whereby either sustained VT or VF results from rapid rotational activity (34, 35). We suggest that the degeneration of TdP to VF occurs when TdP-driving rotors are converted into VF-driving rotors at higher frequency.
g. Another type of a short-lasting non-sustained VT was mapped in a perfused heart from a rabbit without any electrical storm or VF event (Figure 3). Non-electrical storm hearts showed homogeneous APD prolongation with less its dispersion. Multiple breakthrough activations with centrifugal propagation were detected without any reentrant activity. Intriguingly, multiple foci formed membrane voltage oscillation at the center of the recording area (black asterisk shown in the heart picture). The findings suggest that multiple foci associated with homogeneous APD prolongation form a relatively benign form of VT, probably corresponding to short-lasting TdP in human LQTS.
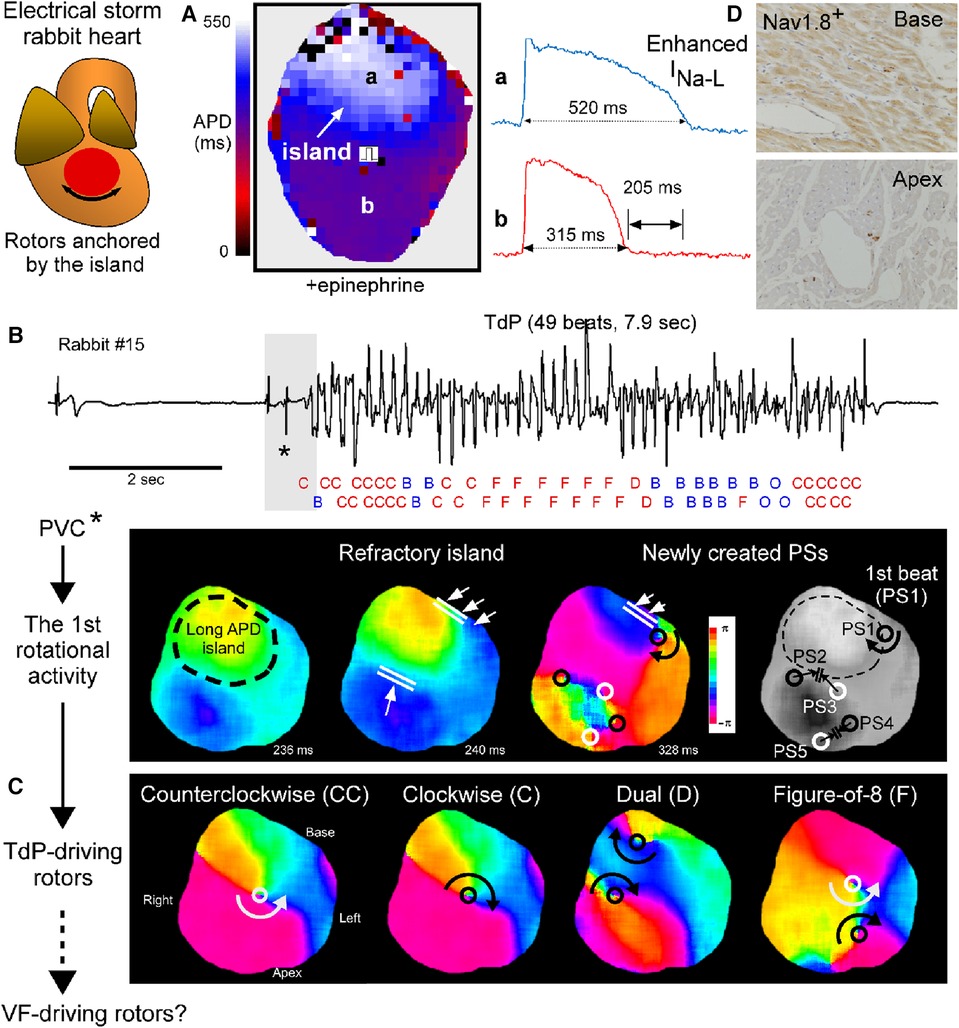
Figure 2. Drifting rotors anchored by a tissue-island drive Tosades de Pointes (TdP). (A) Action potential duration (APD) map at a pacing cycle length of 1,000 msec under the perfusate containing 0.1 μM epinephrine in an electrical storm rabbit heart. Values are APD90 and APD90-dispersion. Regions with prominent APD prolongation look an island. (B) A ECG strip of TdP (top) and phase maps for the initiation (bottom) in electrical storm rabbit heart #15. Activation pattern in each of 49 beats analyzed is labelled with C (clockwise rotor), CC (counterclockwise rotor), D (dual rotor), F (figure-of-8), B (breakthrough) or O (one-way propagation). Black/white circle indicate phase singularity (PS, that is, rotor center) with clockwise/counterclockwise rotation. (C) Phase maps for rotor-typed activation pattern including CC, C, D and F during TdP. (D) Images of immunohistochemistry for NaV1.8 at the left ventricular (LV) tissues. Cross-sections of the base and apex in an electrical storm rabbit heart. NaV1.8-positive staining in myocardium and nerves at the base, but only nerves at the apex. Modified from Yamazaki et al. (7).
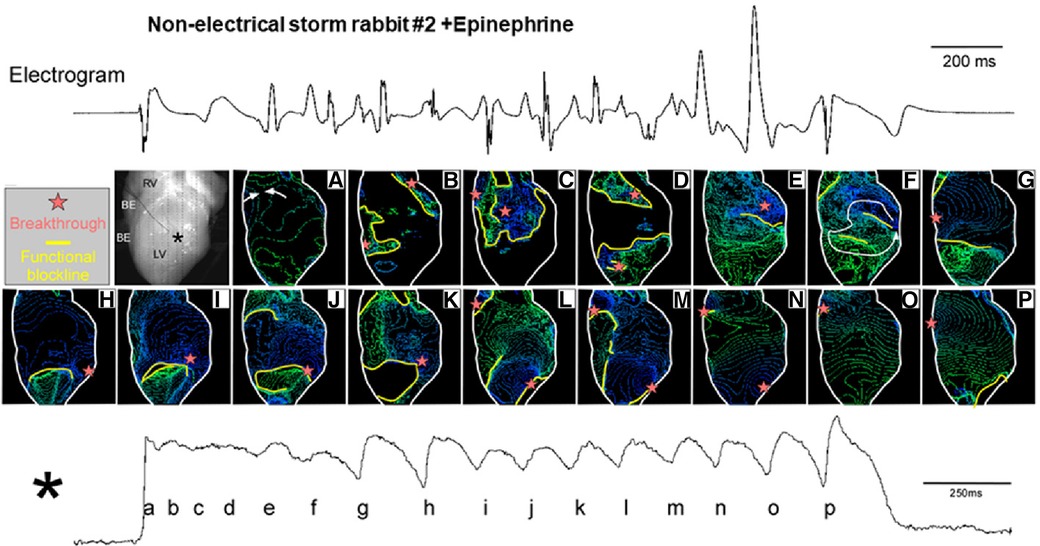
Figure 3. Isochrone maps for non-sustained ventricular tachycardia (NSVT). Mapped heart picture and isochrone maps a-p (upper panel), and the action potential signal at the almost center shown in a black asterisk (lower panel). Isochrone maps for the time points a-p during membrane-voltage oscillation were depicted. Cited from Yamazaki et al. (7).
3.2 Nav1.8 may be a candidate for new drug targeting against tachyarrhythmias
NaV1.8-mediated INa−L has been implicated as the substrate for TdP in this model. High concentration tetrodotoxin (30 μM)-resistant INa−L was increased in the LV myocytes and NaV1.8 was upregulated in the LV tissues and expressed within myocardium corresponding to the island location in optically mapped electrical storm rabbit hearts, whereas NaV1.8-positive nerves were present similarly at the base and apex in electrical storm rabbit tissues (7). Indeed, the selective NaV1.8 blocker A-803467 (10 mg/kg, i.v.) attenuated QT prolongation and suppressed TdP evoked by epinephrine in electrical storm rabbits (7). We show here 2 cases supporting the agent to possess antiarrhythmic actions. In a rabbit with 2 VF episodes, epinephrine infusion evoked repetitive TdP pause-dependently when the pacing was turned off, a provocative measure that was ineffective after A-803467 injection (Figure 4A). In another rabbit, epinephrine-reinfusion after treatment with A-803467 did not evoke any ventricular arrhythmia (Figure 4B).
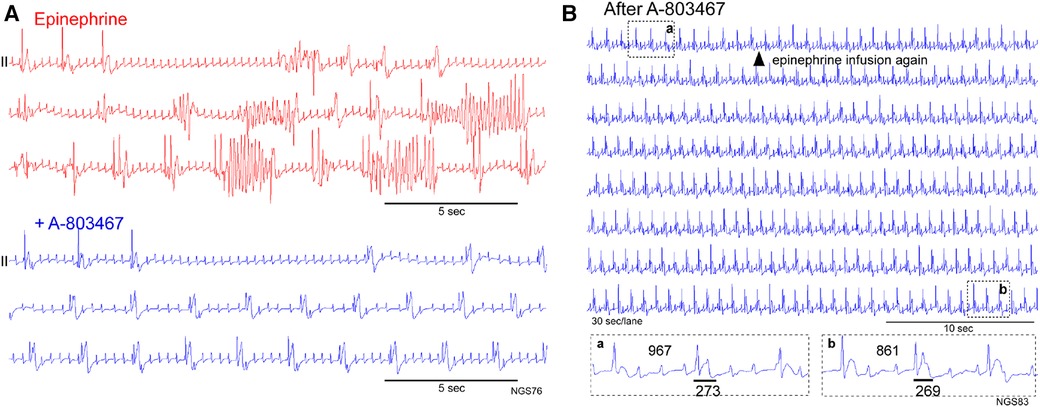
Figure 4. Antiarrhythmic action of the selective NaV1.8 blocker A-803467. (A) Pause- and bradycardia-dependent TdP appeared during epinephrine infusion (red), but not after intravenous A-803467 injection (blue) in a rabbit at Day 65. (B) Epinephrine reinfusion after A-803467 administration in an electrical storm rabbit (Day 56). Epinephrine was infused again from the time point (shown in a filled triangle) in an electrical storm rabbit treated with A-803467. Three-beats ECG traces before (a) and after epinephrine (b) are shown in Boxes. Epinephrine unaffected the QT-interval and did not evoke premature ventricular contraction or ventricular tachyarrhythmia. QT-intervals are shown by horizontal bars.
NaV1.8 encoded by SCN10A is strongly expressed in nociceptive sensory neurons of the dorsal root ganglia and cranial sensory ganglia and much less abundant in hearts than NaV1.5 by SCN5A that mediates cardiac conduction velocity (36). A series of work following genome-wide association studies provides evidence for NaV1.8-mediated INa−L in mouse and rabbit cardiomyocytes perfused with the selective NaV1.8 blocker A-803467 and SCN10A−/− mice (36, 37). NaV1.8 is upregulated in human hypertrophied and failing hearts and A-803467 reduces INa−L, abbreviates APDs and reduces Ca2+-spark frequencies and DADs (38, 39). These findings suggest that NaV1.8 blockade is a more attractive strategy for treating ventricular tachyarrhythmias than NaV1.5-selective INa−L blockers, preventing arrhythmia without relevant effects on peak INa. Sossalla and colleague has recently reported that NaV1.8-driven INa−L is CaMKII-dependent in human failing cardiomyocytes (40). Since electrical storm is associated with striking CaMKII activation (33), it would be interesting to assess involvement of NaV1.8 phosphorylation by CaMKII in enhanced INa−L in our model.
Some investigators provide mechanistic insights into NaV1.8 interaction with NaV1.5 in cardiomyocytes. NaV1.8 functions as a transcription factor for NaV1.5. The C-terminal portion of NaV1.8 encoded by SCN10A-short transcripts which appears to be genetic variants in and around SCN10A, increases NaV1.5 expression and channel current in mouse cardiomyocytes (41). In culture cells, the Na+ current increases 2-fold in HEK cells coexpressing wild type SCN10A and SCN5A, compared to those expressing SCN5A alone, and the Na+ current decreases in HEK cells coexpressing SCN10A with Brugada mutation and wild type SCN5A (42). Further studies are needed in our model to investigate whether NaV1.8 overexpressed in electrical storm hearts is a full-length or a C-terminal portion.
While the potential targeting of NaV1.8 against ventricular arrhythmias in failing and hypertrophic hearts is intriguing, the expression and function of NaV1.8 in structurally normal hearts, where TdP can occur, remain debatable. More studies would be highly required to clarify whether and how NaV1.8 blockade with A-804367 affects peak and late components of Na+ currents in myocytes from electrical storm rabbits and other models of LQTS.
4 Relationship to previous studies in related animal models and patients
Studies in related animal models and patients with congenital LQTS are briefly reviewed here. The key findings are shown in Figure 5.
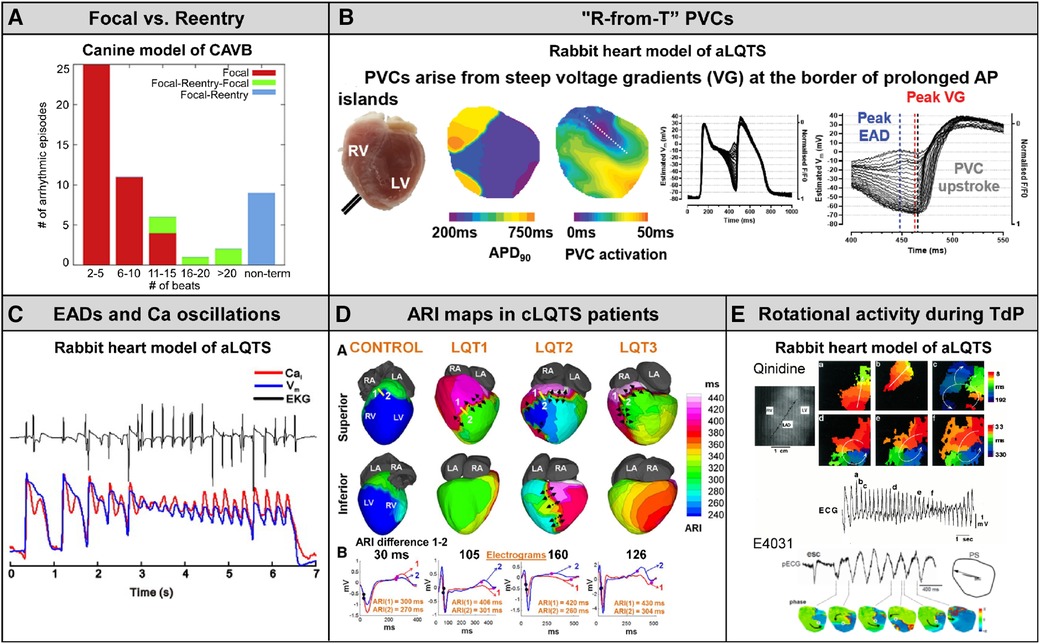
Figure 5. The key findings in related animal models and congenital LQTS patients. (A) Canine model of chronic complete AV block (CAVB). Nonterminal episodes of TdP are always maintained by reentry. Cited from Vandersickel et al. (43). (B) An emerging concept “R-from-T” verified in normal rabbit hearts under the condition of QT interval prolongation (E-4031 0.5 μM and 50% K+/Mg2+). Cited from Alexander et al. (44). (C) Burst of EADs coupled to Ca2+ oscillation form TdP in normal rabbit hearts under the condition of QT interval prolongation (bradycardia, dofetilide 0.5 μM and reduced K+/Mg2+). Cited from Nemec et al. (11). (D) Maps of activation recovery interval (ARI), surrogate maker for action potential duration, in genotyped congenital LQTS syndrome (LQT1, LQT2 and LQT3). Cited from Vijayakumar et al. (25). (E) Rotational activity during TdP detected in normal rabbit hearts under the condition of QT interval prolongation (top: quinidine 5 μM and reduced K+, bottom: E-4031 0.5 μM and reduced K+/Mg2+). Cited from Asano et al. and Maruyama et al. (27, 45).
4.1 Canine model of CAVB
Spatial dispersion of repolarization has been implicated as a substrate of TdP in CAVB dogs. In studies by Vos and colleagues (46, 47), using 56 needle electrodes distributed transmurally to record 224 unipolar electrograms simultaneously, TdP was initiated at the site with maximal heterogeneity of repolarization in the LV and the selective INa−L inhibitor GS967 abolishes TdP through reduced spatial dispersion of repolarization. These findings in CAVB dogs are supported by our results showing that the first beat of TdP following a triggering PVC was a reentrant wave arising in the periphery of the long APD island (Figure 2). In addition, Vos's group originally reported in CAVB dogs that more than 90% of the beats during TdP were focal (48), but a most recent study by his collaborators has developed a novel methodology to detect reentry loops by using graph search algorithms and elegantly demonstrated that TdP can be driven by focal activity as well as reentry depending on the duration of the episode: longer-lasting TdP is maintained by reentry (Figure 5A) (43). Nonterminal TdP that lasts >10 s was all perpetuated by reentry.
4.2 A unifying mechanism of trigger and substrate in simulation models
A mixture of focal activity and reentry has been reported to underlie EAD-mediated arrhythmias in rat/rabbit hearts exposed to H2O2 (49) or hypokalemia in the absence or presence of dofetilide (50) and a transgenic LQT2 rabbit model (51) as well as in the rabbit ventricle simulation model (52). Repetitive focal activations from single or multiple foci from EAD zones, together with regional APD gradients by areas with EADs next to regions without EADs leading to localized conduction block and initiation of reentry, account for the combination of mechanisms underlying EAD-induced TdP and polymorphic VT (53). Moreover, recent simulation studies (54), together with experiments in transgenic LQT2 rabbit hearts (51) and normal rabbit hearts during conditions with IKr blockade with E4031 (0.5 μM) plus 50% K+/Mg2+ (44), have strengthened the idea that steep APD gradients rather than EADs give rise to both the triggering PVC and the substrate for reentrant tachyarrhythmia, the mechanism termed “R-from-T” (Figure 5B) (54). The TdP event we detected in an electrical storm rabbit heart was triggered by a PVC via classical “R-on-T” mechanism in which an exogenous PVC encounters a refractory region (Figure 2), but not “R-from-T”. Since the number of tachyarrhythmia events optically mapped and analyzable was limited, we could not detect ventricular tachyarrhythmias with the “R-from-T” initiation mode. Some PVCs certainly originated at regions adjacent to the long APD island in electrical storm hearts, suggesting that “R-from-T” may be involved in TdP in this model. However, roles of “R-from-T” vs. “R-on-T” in arrhythmogenicity remain unclear.
4.3 Rabbit heart models of acquired LQTS
There are differences in the observed phenomena at the onset and maintenance of arrhythmias between rabbit models. Studies in normal rabbit hearts subjected to the condition of QT interval prolongation by bradycardia, IKr blockade with E-4031 or dofetilide and reduced K+/Mg2+ showed that bursts of EADs coupled to Ca2+ oscillations near the base (area of long APD) occurring in zones as small as 1 mm2 correspond to TdP (Figure 5C) (11, 55). In electrical storm rabbit hearts, EADs were observed in 5/14 electrical storm hearts and some PVCs occurred from phase 2 of the preceding action potential in electrical storm hearts, but neither reentrant excitation wavefronts propagating from the phase 2 PVC nor a burst of EADs were detected. Membrane voltage oscillation resulting from multiple foci in a non-electrical storm rabbit heart with homogeneous APD prolongation (Figure 3) is an etiology different from bursts of EADs observed in those models (11, 55). The high concentration (0.5 μM) of E-4031 or dofetilide used in the acute model produces prominent APD prolongation, to an extent much greater than that occurring spontaneously in our chronic model, possibly explaining some of the discrepancy. A recently published study in a guinea-pig surrogate model of LQT3 demonstrating that voltage/calcium uncoupling predates sustained ventricular tachyarrhythmia provides evidence of reentrant excitation responsible for TdP potentially degenerating into VF (56).
4.4 ECGi mapping in patients with congenital LQTS
Spatially heterogeneous prolongation of activation recovery interval (ARI), a surrogate marker of APD, of ventricular epicardium has been demonstrated in genotyped LQTS patients, using non-invasive ECG imaging (ECGi) technique (25). Repolarization gradients resulting from the localized ARI prolongation are found steeper in patients with than without history of syncope or sudden cardiac arrest and ICD-defibrillation (Figure 5D). However, ionic mechanisms of heterogeneous ARI prolongation in congenital LQTS, the syndrome monogenic but with variable phenotypes, remain unclear. The findings in electrical storm rabbits agree with the human data: the boundary of the long APD island likely corresponds to regions with steep repolarization gradients. Our proposal of NaV1.8-mediated INa−L enhancement provides insights into the pathophysiology of LQTS. Acquired regulation/modification of cardiac Na+-channels by NaV1.8 may be one of the important factors involved in the heterogeneous substrate in LQTS.
4.5 Drifting rotors with a unique behavior may produce undulating QRS morphology
Studies in simulation models have reported that the polymorphism of VT in the ECG, including TdP, can be determined by a Doppler effect resulting from drift of a single spiral wave (28–30). In agreement with the modeling studies, some investigators reported a meandering spiral wave in normal rabbit hearts subjected to hypokalemia plus quinidine (27) or dofetilide (45) and in a transgenic LQTS type-2 rabbit model (57), but rotational activation was detected in only a few beats during TdP (Figure 5E) and none one has characterized the dynamics of spiral waves. Our study in electrical storm rabbits is the first to report drifting rotors in a unique manner. The detection of a clockwise, counterclockwise, figure-of-8 and dual rotor, all whose cores swirl in the periphery of the long APD island, suggests 1 or 2 scroll source(s) with various types of filaments, potentially contributing to undulating QRS axis formation.
5 Future direction
Overall, the findings from clinical and experimental studies currently available suggest that a PVC arising from steep repolarization gradients (R-from-T) or remote regions (R-on-T) propagates unidirectionally toward regions with abnormally delay repolarization termed a refractory island, resulting in the generation of rotational activity responsible for the 1st beat of TdP followed by drifting rotors around the island that perpetuate TdP. The long-lasting TdP-driven rotors has potential to degenerate into VF. Future studies are needed to characterize the 3-D structure as well as to explore the electrophysiological and molecular features of the torsadogenic island.
Our attention moves to the issue on why electrical storm rabbits have liability to VF. According to the mother rotor hypothesis, APD shortening and increased excitability stabilize rotors at higher frequency (58). Ion channels potentially involved in the conversion from TdP- to VF-driving rotors might include the ATP-sensitive K+ (IK−ATP) channel, Ca2+-activated potassium (SK) channel and IK1 channel. TdP can manifest as acutely decreased pump function and hemodynamic instability (5), leading to myocardial ischemia and ATP depletion that activates IK−ATP channel. In ICD patients with ischemic or non-ischemic cardiomyopathy, myocardial ATP depletion detected by phosphorous magnetic resonance spectroscopy is a predictor for appropriate ICD therapies against VT/VF (59). Preliminary data on proteome in electrical storm heart tissues shows that VF storm is associated with mitochondrial dysfunction and oxidative phosphorylation inactivation (unpublished). In failing rabbit hearts, SK channel activation underlies the post-shock APD shortening leading to recurrent VF (60). In cardiomyocytes from CaMKII-overexpressing rabbits and CAVB rabbits show an increase in IK1 (31, 61). Future studies are needed to identify and characterize VF-driving rotors as well as to explore molecular mechanisms by which the conversion is promoted in a rabbit model of electrical storm, which might open a new avenue to develop novel therapeutic strategies to VF.
Author contributions
YT: Writing – original draft, Writing – review & editing. MY: Data curation, Methodology, Writing – review & editing, Investigation. MS: Writing – review & editing. SY: Writing – review & editing. YI: Writing – review & editing. TM: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
The authors’ work is supported by a research grant 2022 from Suzuken Memorial Foundation and a grant 2023 from SENSHIN Medical Research Foundation to YT and by Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (C) 23K07547 to MY.
Conflict of interest
YT and MS are affiliated with a department sponsored by BIOTRONIK Japan Inc., Japan Lifeline Co., Ltd., Fukuda Denshi Co., Ltd., and Suzuken Co., Ltd., and SY with a department sponsored by Medtronic Japan Co., Ltd. The remaining authors have no relationships relevant to the contents of this paper to disclose.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Leenhardt A, Glaser E, Burguera M, Nurnberg M, Maison-Blanche P, Coumel P. Short-coupled variant of torsade de pointes. A new electrocardiographic entity in the spectrum of idiopathic ventricular tachyarrhythmias. Circulation. (1994) 89(1):206–15. doi: 10.1161/01.cir.89.1.206
2. Steinberg C, Davies B, Mellor G, Tadros R, Laksman ZW, Roberts JD, et al. Short-coupled ventricular fibrillation represents a distinct phenotype among latent causes of unexplained cardiac arrest: a report from the CASPER registry. Eur Heart J. (2021) 42(29):2827–38. doi: 10.1093/eurheartj/ehab275
3. El-Sherif N, Turitto G. The long QT syndrome and torsade de pointes. Pacing Clin Electrophysiol. (1999) 22(1 Pt 1):91–110. doi: 10.1111/j.1540-8159.1999.tb00305.x
4. Drew BJ, Ackerman MJ, Funk M, Gibler WB, Kligfield P, Menon V, et al. Prevention of torsade de pointes in hospital settings: a scientific statement from the American heart association and the American college of cardiology foundation. J Am Coll Cardiol. (2010) 55(9):934–47. doi: 10.1016/j.jacc.2010.01.001
5. Farkas AS, Nattel S. Minimizing repolarization-related proarrhythmic risk in drug development and clinical practice. Drugs. (2010) 70(5):573–603. doi: 10.2165/11535230-000000000-00000
6. Dessertenne F. Ventricular tachycardia with 2 variable opposing foci. Arch Mal Coeur Vaiss. (1966) 59(2):263–72.4956181
7. Yamazaki M, Tomii N, Tsuneyama K, Takanari H, Niwa R, Honjo H, et al. Rotors anchored by refractory islands drive torsades de pointes in an experimental model of electrical storm. Heart Rhythm. (2022) 19(2):318–29. doi: 10.1016/j.hrthm.2021.10.012
8. Shimizu W, Ohe T, Kurita T, Kawade M, Arakaki Y, Aihara N, et al. Effects of verapamil and propranolol on early afterdepolarizations and ventricular arrhythmias induced by epinephrine in congenital long QT syndrome. J Am Coll Cardiol. (1995) 26(5):1299–309. doi: 10.1016/0735-1097(95)00313-4
9. Shimizu W. The long QT syndrome: therapeutic implications of a genetic diagnosis. Cardiovasc Res. (2005) 67(3):347–56. doi: 10.1016/j.cardiores.2005.03.020
10. Aiba T, Shimizu W, Inagaki M, Noda T, Miyoshi S, Ding WG, et al. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. J Am Coll Cardiol. (2005) 45(2):300–7. doi: 10.1016/j.jacc.2004.09.069
11. Nemec J, Kim JJ, Gabris B, Salama G. Calcium oscillations and T-wave lability precede ventricular arrhythmias in acquired long QT type 2. Heart Rhythm. (2010) 7(11):1686–94. doi: 10.1016/j.hrthm.2010.06.032
12. Milberg P, Reinsch N, Osada N, Wasmer K, Monnig G, Stypmann J, et al. Verapamil prevents torsade de pointes by reduction of transmural dispersion of repolarization and suppression of early afterdepolarizations in an intact heart model of LQT3. Basic Res Cardiol. (2005) 100(4):365–71. doi: 10.1007/s00395-005-0533-8
13. Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C, et al. Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation. (2014) 130(3):224–34. doi: 10.1161/CIRCULATIONAHA.113.007765
14. Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol. (2016) 67(9):1053–8. doi: 10.1016/j.jacc.2015.12.033
15. Viskin S, Fish R, Zeltser D, Belhassen B, Heller K, Brosh D, et al. Arrhythmias in the congenital long QT syndrome: how often is torsade de pointes pause dependent? Heart. (2000) 83(6):661–6. doi: 10.1136/heart.83.6.661
16. Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. (2007) 582(Pt 2):675–93. doi: 10.1113/jphysiol.2006.126714
17. Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. (2002) 90(9):939–50. doi: 10.1161/01.res.0000018627.89528.6f
18. Nattel S, Quantz MA. Pharmacological response of quinidine induced early afterdepolarisations in canine cardiac Purkinje fibres: insights into underlying ionic mechanisms. Cardiovasc Res. (1988) 22(11):808–17. doi: 10.1093/cvr/22.11.808
19. Caref E B, Boutjdir M, Himel HD, El-Sherif N. Role of subendocardial Purkinje network in triggering torsade de pointes arrhythmia in experimental long QT syndrome. Europace. (2008) 10(10):1218–23. doi: 10.1093/europace/eun248
20. Haissaguerre M, Extramiana F, Hocini M, Cauchemez B, Jais P, Cabrera JA, et al. Mapping and ablation of ventricular fibrillation associated with long-QT and Brugada syndromes. Circulation. (2003) 108(8):925–8. doi: 10.1161/01.CIR.0000088781.99943.95
21. Rokita AG, Anderson ME. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation. (2012) 126(17):2125–39. doi: 10.1161/CIRCULATIONAHA.112.124990
22. Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. (1998) 98(21):2314–22. doi: 10.1161/01.cir.98.21.2314
23. Bonatti V, Rolli A, Botti G. Monophasic action potential studies in human subjects with prolonged ventricular repolarization and long QT syndromes. Eur Heart J. (1985) 6(suppl_D):131–43. doi: 10.1093/eurheartj/6.suppl_d.131
24. Vos MA, Gorenek B, Verduyn SC, van der Hulst FF, Leunissen JD, Dohmen L, et al. Observations on the onset of torsade de pointes arrhythmias in the acquired long QT syndrome. Cardiovasc Res. (2000) 48(3):421–9. doi: 10.1016/s0008-6363(00)00192-9
25. Vijayakumar R, Silva JNA, Desouza KA, Abraham RL, Strom M, Sacher F, et al. Electrophysiologic substrate in congenital long QT syndrome: noninvasive mapping with electrocardiographic imaging (ECGI). Circulation. (2014) 130(22):1936–43. doi: 10.1161/CIRCULATIONAHA.114.011359
26. El-Sherif N, Chinushi M, Caref EB, Restivo M. Electrophysiological mechanism of the characteristic electrocardiographic morphology of torsade de pointes tachyarrhythmias in the long-QT syndrome: detailed analysis of ventricular tridimensional activation patterns. Circulation. (1997) 96(12):4392–9. doi: 10.1161/01.cir.96.12.4392
27. Asano Y, Davidenko JM, Baxter WT, Gray RA, Jalife J. Optical mapping of drug-induced polymorphic arrhythmias and torsade de pointes in the isolated rabbit heart. J Am Coll Cardiol. (1997) 29(4):831–42. doi: 10.1016/s0735-1097(96)00588-8
28. Davidenko JM, Pertsov AV, Salomonsz R, Baxter W, Jalife J. Stationary and drifting spiral waves of excitation in isolated cardiac muscle. Nature. (1992) 355(6358):349–51. doi: 10.1038/355349a0
29. Gray RA, Jalife J, Panfilov A, Baxter WT, Cabo C, Davidenko JM, et al. Nonstationary vortexlike reentrant activity as a mechanism of polymorphic ventricular tachycardia in the isolated rabbit heart. Circulation. (1995) 91(9):2454–69. doi: 10.1161/01.cir.91.9.2454
30. Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. (1993) 72(3):631–50. doi: 10.1161/01.res.72.3.631
31. Tsuji Y, Opthof T, Yasui K, Inden Y, Takemura H, Niwa N, et al. Ionic mechanisms of acquired QT prolongation and Torsades de Pointes in rabbits with chronic complete atrioventricular block. Circulation. (2002) 106(15):2012–8. doi: 10.1161/01.cir.0000031160.86313.24
32. Tsuji Y, Zicha S, Qi XY, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. (2006) 113(3):345–55. doi: 10.1161/CIRCULATIONAHA.105.552968
33. Tsuji Y, Hojo M, Voigt N, El-Armouche A, Inden Y, Murohara T, et al. Ca2+-related signaling and protein phosphorylation abnormalities play central roles in a new experimental model of electrical storm. Circulation. (2011) 123(20):2192–203. doi: 10.1161/CIRCULATIONAHA.110.016683
34. Samie FH, Mandapati R, Gray RA, Watanabe Y, Zuur C, Beaumont J, et al. A mechanism of transition from ventricular fibrillation to tachycardia: effect of calcium channel blockade on the dynamics of rotating waves. Circ Res. (2000) 86(6):684–91. doi: 10.1161/01.res.86.6.684
35. Samie FH, Jalife J. Mechanisms underlying ventricular tachycardia and its transition to ventricular fibrillation in the structurally normal heart. Cardiovasc Res. (2001) 50(2):242–50. doi: 10.1016/s0008-6363(00)00289-3
36. Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. (2012) 111(3):322–32. doi: 10.1161/CIRCRESAHA.112.265173
37. Stroud DM, Yang T, Bersell K, Kryshtal DO, Nagao S, Shaffer C, et al. Contrasting Nav1.8 activity in Scn10a-/- ventricular myocytes and the intact heart. J Am Heart Assoc. (2016) 5(11):e002946. doi: 10.1161/JAHA.115.002946
38. Dybkova N, Ahmad S, Pabel S, Tirilomis P, Hartmann N, Fischer TH, et al. Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc Res. (2018) 114(13):1728–37. doi: 10.1093/cvr/cvy152
39. Ahmad S, Tirilomis P, Pabel S, Dybkova N, Hartmann N, Molina CE, et al. The functional consequences of sodium channel NaV 1.8 in human left ventricular hypertrophy. ESC Heart Fail. (2019) 6(1):154–63. doi: 10.1002/ehf2.12378
40. Bengel P, Dybkova N, Tirilomis P, Ahmad S, Hartmann N, Mohamed B, et al. Detrimental proarrhythmogenic interaction of Ca2+/calmodulin-dependent protein kinase II and NaV1.8 in heart failure. Nat Commun. (2021) 12(1):6586. doi: 10.1038/s41467-021-26690-1
41. Man JCK, Bosada FM, Scholman KT, Offerhaus JA, Walsh R, van Duijvenboden K, et al. Variant intronic enhancer controls SCN10A-short expression and heart conduction. Circulation. (2021) 144(3):229–42. doi: 10.1161/CIRCULATIONAHA.121.054083
42. Hu D, Barajas-Martinez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. (2014) 64(1):66–79. doi: 10.1016/j.jacc.2014.04.032
43. Vandersickel N, Bossu A, De Neve J, Dunnink A, Meijborg VMF, van der Heyden MAG, et al. Short-lasting episodes of Torsade de Pointes in the chronic atrioventricular block dog model have a focal mechanism, while longer-lasting episodes are maintained by re-entry. JACC Clin Electrophysiol. (2017) 3(13):1565–76. doi: 10.1016/j.jacep.2017.06.016
44. Alexander C, Bishop MJ, Gilchrist RJ, Burton FL, Smith GL, Myles RC. Initiation of ventricular arrhythmia in the acquired long QT syndrome. Cardiovasc Res. (2023) 119(2):465–76. doi: 10.1093/cvr/cvac103
45. Maruyama M, Lin SF, Xie Y, Chua SK, Joung B, Han S, et al. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ Arrhythm Electrophysiol. (2011) 4(1):103–11. doi: 10.1161/CIRCEP.110.959064
46. Bossu A, Houtman MJC, Meijborg VMF, Varkevisser R, Beekman HDM, Dunnink A, et al. Selective late sodium current inhibitor GS-458967 suppresses Torsades de Pointes by mostly affecting perpetuation but not initiation of the arrhythmia. Br J Pharmacol. (2018) 175(12):2470–82. doi: 10.1111/bph.14217
47. Dunnink A, Stams TRG, Bossu A, Meijborg VMF, Beekman JDM, Wijers SC, et al. Torsade de pointes arrhythmias arise at the site of maximal heterogeneity of repolarization in the chronic complete atrioventricular block dog. Europace. (2017) 19(5):858–65. doi: 10.1093/europace/euw087
48. Boulaksil M, Jungschleger JG, Antoons G, Houtman MJ, de Boer TP, Wilders R, et al. Drug-induced torsade de pointes arrhythmias in the chronic AV block dog are perpetuated by focal activity. Circ Arrhythm Electrophysiol. (2011) 4(4):566–76. doi: 10.1161/CIRCEP.110.958991
49. Morita N, Lee JH, Xie Y, Sovari A, Qu Z, Weiss JN, et al. Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol. (2011) 57(3):366–75. doi: 10.1016/j.jacc.2010.07.045
50. Pezhouman A, Singh N, Song Z, Nivala M, Eskandari A, Cao H, et al. Molecular basis of hypokalemia-induced ventricular fibrillation. Circulation. (2015) 132(16):1528–37. doi: 10.1161/CIRCULATIONAHA.115.016217
51. Huang X, Kim TY, Koren G, Choi BR, Qu Z. Spontaneous initiation of premature ventricular complexes and arrhythmias in type 2 long QT syndrome. Am J Physiol Heart Circ Physiol. (2016) 311(6):H1470–H84. doi: 10.1152/ajpheart.00500.2016
52. Sato D, Xie LH, Sovari AA, Tran DX, Morita N, Xie F, et al. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci USA. (2009) 106(9):2983–8. doi: 10.1073/pnas.0809148106
53. Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. (2010) 7(12):1891–9. doi: 10.1016/j.hrthm.2010.09.017
54. Liu MB, Vandersickel N, Panfilov AV, Qu Z. R-From-T as a common mechanism of arrhythmia initiation in long QT syndromes. Circ Arrhythm Electrophysiol. (2019) 12(12):e007571. doi: 10.1161/CIRCEP.119.007571
55. Parikh A, Mantravadi R, Kozhevnikov D, Roche MA, Ye Y, Owen LJ, et al. Ranolazine stabilizes cardiac ryanodine receptors: a novel mechanism for the suppression of early afterdepolarization and torsades de pointes in long QT type 2. Heart Rhythm. (2012) 9(6):953–60. doi: 10.1016/j.hrthm.2012.01.010
56. Himel HD, Cupelli M, Boutjdir M, El-Sherif N. Voltage/calcium uncoupling underlies sustained Torsade de Pointes ventricular tachyarrhythmia in an experimental model of long QT syndrome. Front Physiol. (2021) 12(53):617847. doi: 10.3389/fphys.2021.617847
57. Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, et al. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest. (2008) 118(6):2246–59. doi: 10.1172/JCI33578
58. Filgueiras-Rama D, Jalife J. Structural and functional bases of cardiac fibrillation. Differences and similarities between atria and ventricles. JACC Clin Electrophysiol. (2016) 2(1):1–3. doi: 10.1016/j.jacep.2015.12.011
59. Samuel TJ, Lai S, Schar M, Wu KC, Steinberg AM, Wei AC, et al. Myocardial ATP depletion detected noninvasively predicts sudden cardiac death risk in patients with heart failure. JCI Insight. (2022) 7(12):e157557. doi: 10.1172/jci.insight.157557
60. Wan J, Chen M, Wang Z, Everett THT, Rubart-von der Lohe M, Shen C, et al. Small-conductance calcium-activated potassium current modulates the ventricular escape rhythm in normal rabbit hearts. Heart Rhythm. (2019) 16(4):615–23. doi: 10.1016/j.hrthm.2018.10.033
Keywords: arrhythmia mechanism, animal model of long QT syndrome, torsades de pointes, ventricular fibrillation, electrical storm
Citation: Tsuji Y, Yamazaki M, Shimojo M, Yanagisawa S, Inden Y and Murohara T (2024) Mechanisms of torsades de pointes: an update. Front. Cardiovasc. Med. 11:1363848. doi: 10.3389/fcvm.2024.1363848
Received: 31 December 2023; Accepted: 19 February 2024;
Published: 5 March 2024.
Edited by:
Masahide Harada, Fujita Health University, JapanReviewed by:
Hiroki Takanari, Tokushima University Hospital, JapanJerome Montnach, INSERM U1087 Institut du Thorax, France
© 2024 Tsuji, Yamazaki, Shimojo, Yanagisawa, Inden and Murohara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukiomi Tsuji eXVraW9taXRzdWppQG1lZC5uYWdveWEtdS5hYy5qcA==